

Abstract
Myeloid-derived suppressor cells (MDSCs) promote tumor-mediated immunosuppression and cancer progression. Gemcitabine (Gem) is a MDSC-depleting chemotherapeutic agent; however, its clinical use is hampered by its drug resistance and inefficient in vivo delivery. Here we describe a strategy to formulate a Gem analogue gemcitabine monophosphate (GMP) into a lipid-coated calcium phosphate (LCP) nanoparticle, and investigate its antitumor immunity and therapeutic effects after systemic administrations. In the syngeneic mouse model of B16F10 melanoma, compared with free Gem, the LCP-formulated GMP (LCP-GMP) significantly induced apoptosis and reduced immunosuppression in the tumor microenvironment (TME). LCP-GMP effectively depleted MDSCs and regulatory T cells, and skewed macrophage polarization towards the antitumor M1 phenotype in the TME, leading to enhanced CD8+ T-cell immune response and profound tumor growth inhibition. Thus, engineering the in vivo delivery of MDSC-depleting agents using nanotechnology could substantially modulate immunosuppressive TME and boost T-cell immune response for enhanced antitumor efficacy.
Keywords: MDSC, Immunosuppression, tumor microenvironment, nanoparticle, gemcitabine
Introduction
Among many pro-tumor mediators, the prominent presence of myeloid-derived suppressor cells (MDSCs), known as myeloid cells with potent suppressive activity, promotes tumor-mediated immunosuppression and correlates with reduced survival.1 Two major subsets of MDSCs are monocytic MDSC (M-MDSC) and polymorphonuclear MDSC (PMN-MDSC). MDSCs in peripheral lymphoid organs are largely represented by PMN-MDSCs, which contribute to tumor-specific T-cell tolerance. In tumors, M-MDSCs are more prominent and have higher suppressive activity than PMN-MDSCs.2 M-MDSCs differentiate to immune suppressive tumor-associated macrophages (TAMs) within the tumor microenvironment (TME),2,3 suggesting that targeting MDSCs may deliver superior antitumor therapeutic benefits. In addition, MDSCs promote the development and accumulation of immunosuppressive Foxp3+ regulatory T cells (Tregs) in tumors,3 which indirectly influences T-cell function and hampers antitumor immunity.4–6 MDSCs regulate PD-L1 expression on tumor cells, decreasing antitumor immune responses mediated by T lymphocytes.7
Chemotherapeutic agent gemcitabine (Gem), a cytidine nucleoside analog, has been shown to inhibit MDSCs in tumor-bearing mice, leading to improved CD8+ T-cell antitumor activity accompanied by the inhibition of tumor growth.8–10 However, repeated administrations of Gem cause drug resistance, presumably due to the dysfunction of nucleoside transporters required for the cellular uptake of Gem.11 The Gem resistance may also result from the dysfunction of intracellular deoxycytidine kinase, which blocks the phosphorylation process of the prodrug Gem to its bioactive form gemcitabine triphosphate (GTP).12 In addition, due to rapid deamination, the elimination half-life of Gem is short, less than 30 min.13 To address these clinically important issues associated with the drug resistance and short blood circulation time of Gem, we designed a lipid-coated calcium phosphate (LCP) nanoparticle platform to encapsulate Gem derivatives for improved pharmacokinetic profile and bioactivity. It is established that addition of the first phosphate group on Gem is the rate-limiting step to form GTP intracellularly.14 We therefore encapsulated gemcitabine monophosphate (GMP) into LCP to promote the Gem-mediated MDSC modulation and antitumor activities. The LCP-formulated GMP (LCP-GMP) could bypass nucleoside transporters and enter into cells by endocytosis before efficient endosome release and sequential phosphorylation to its bioactive form GTP. This LCP nanocarrier could lead to prolonged blood circulation time of the encapsulated Gem derivatives compared with the unformulated drug.15
In this study, we focused on an aggressive B16F10 melanoma model in which the suppressive leukocytes, MDSCs in particular, contribute to the creation of a highly immunosuppressive TME, leading to impaired antitumor immune responses and thereby enhancing tumor progression.16 We evaluated the antitumor responses of LCP-GMP and free Gem on B16F10 tumor-bearing mice after repeated systemic administrations. We assessed the LCP-GMP-mediated modulations of immunosuppressive TME involving myeloid cells and tumor cells, and investigated the immunological effects of LCP-GMP against melanoma.
Materials and Methods
Materials
GMP disodium salt was synthesized by HDH Pharm Inc (Morrisville, NC). 1,2-dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), 1,2-distearoryl-snglycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol-2000) ammonium salt (DSPE-PEG2000) and 25-[N-[(7-nitro-2–1,3-benzoxadiazol-4-yl)methyl]amino]-27-norcholesterol (25-NBD cholesterol) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol, cyclohexane and Igepal CO-520 were from Sigma-Aldrich (St. Louis, MO). DeadEnd Fluorometric TUNEL assay kit was obtained from Promega Corporation (Madison, WI). PD-L1 antibody was purchased from R&D System. Alexa Fluor 647 Phalloidin was obtained from Thermo Fisher Scientific. Other chemicals, unless otherwise specified, were from Sigma-Aldrich (St. Louis, MO). Fluorescent antibodies against mouse CD4 (clone GK1.5), NK1.1 (clone PK136), IFN-γ (clone XMG1.2), TNF-α (clone MP6-XT22), CD16/32 (clone 93), CD25 (clone PC61.5), CD11c (clone N418) and Foxp3 were from eBioscience. Fluorescent antibodies against mouse CD8a (clone 53–6.7), CD11b (clone M1/70), Ly6C (clone HK1.4), Ly6G (clone 1A8), F4/80 (clone BM8), CD80 (clone 16–10A1), CD206 (clone C068C2) were from Biolegend (San Diego, CA).
Cell lines
B16F10 cells were from American Type Culture Collection (ATCC). B16F10 cells were cultured in DMEM (Dulbecco’s Modified Eagle Medium, high glucose) supplemented with 10% fetal bovine serum (FBS) and antibiotics containing penicillin and streptomycin. Growth medium was changed every 2 to 3 days.
Animals and tumor therapy
The experimental handling of mice was conducted under federal, state, and local guidelines and following an approved protocol from the Institutional Animal Care and Use Committee (IACUC) of the University of Rhode Island. C57BL/6 mice (6- to 8- week-old female, the Jackson laboratory) were subcutaneously (s.c.) inoculated with 5×105 B16F10 cells on day 0, and treated on days 8, 10, 12, 14 by intravenous (i.v.) injections of LCP-GMP, free Gem, and control LCP, with 50.4 µmol/Kg GMP (or Gem). Tumor sizes were measured every day with a caliper and calculated by the formula: V (mm3) = 1/2 width^2 × length. Body weights of tumor-bea ring mice were measured, and the relative body weight was calculated by normalizing the body weight to that of day 8 when the initial treatment was given. On day 16, tumors were collected and tumor weights were measured.
Preparation and characterization of nanoparticles
LCP-GMP particles were prepared by a two-step procedure: preparation of LCP cores followed by outer lipid coating. LCP cores were prepared using water-in-oil microemulsions, with the oil phase containing cyclohexane/Igepal CO-520 (71/29, v/v).17 Specifically, 180 µl of GMP (60 mM) was mixed with Na2HPO4 (pH = 9.0) to a total volume of 600 µl with a final concentration of Na2HPO4 being 12.5 mM. This solution was then added dropwise into 20 ml of oil phase under stirring to form the phosphate microemulsion. CaCl2 solution (600 µl, 2.5 M) was added dropwise into a separate 20 ml of oil phase under stirring to form the calcium microemulsion. Phospholipid DOPA in chloroform (8 µmol) was then added into the phosphate microemulsion containing GMP and Na2HPO4, and the two microemulsions were mixed. After stirring for 5 min, another DOPA (8 µmol) was added into the combined microemulsion. This combined microemulsion was continuously stirred for another 20 min before 40 ml of absolute ethanol was added to break the microemulsion. After brief stirring, the ethanol emulsion mixture was centrifuged at 10,000 g for 15 min to pellet LCP cores and the supernatant was discarded to remove the excess salt, untrapped GMP and DOPA. Additional absolute ethanol was added, and after brief vortexing, LCP cores were pelleted again by centrifugation at 10,000 g for 15 min and the supernatant was discarded. This ethanol washing step was repeated once to make a total of two ethanol wash. The pelleted LCP cores were air-dried and then suspended in 2 ml chloroform. The resulting LCP cores in chloroform were stored in a glass vial at −20°C for future use. To prepare the final LCP-GMP with outer lipid coating, 232.2 µl of cholesterol at 10 mg/ml, 168 µl of DOTAP at 25 mg/ml, 576 µl of DSPE-PEG2000 at 25 mg/ml were mixed with the above LCP cores in chloroform. All lipid stock solutions were prepared in chloroform. The above chloroform mixture of LCP cores and lipids was air-dried followed by vacuum drying overnight. The dried lipids were dissolved in 180 µl of tetrahydrofuran (THF) followed by 300 µl of absolute ethanol, and then suspended in 960 µl of distilled water. The THF, ethanol and distilled water were pre-warmed at 50°C before use. The resulting p article solution was dialyzed in distilled water to remove the THF and ethanol. Cytidine monophosphate (CMP), the nucleoside analogue of GMP, has a chemical structure similar to GMP but without any cytotoxic effect. Thus, CMP was loaded into LCP as a control nanoparticle for LCP-GMP in the following experiments. The preparation of control LCP was the same as that of LCP-GMP, except that GMP was replaced by an equal molar amount of CMP.
The particle size and zeta potential of LCPs were determined by dynamic light scattering (DLS) (Malvern ZetaSizer Nano, Westborough, MA). The encapsulation efficiency of GMP in LCP was measured by a UV spectrophotometer (DU 800 spectrophotometer, Beckman Coulter, Brea, CA) at a wavelength of 275 nm. Transmission electron microscope (TEM) images of LCPs were acquired using JEOL 100CX II TEM (Tokyo, Japan).15,18,19 A small amount of LCP solution was dropped onto a 300 mesh carbon coated copper grid (Ted Pella, Redding, CA). Two minutes later, excess fluid was drained with filter paper, and the copper grid was dried and analyzed with TEM.14,20
LCP-GMP was stored at 4°C after preparation. To determine the stability of LCP-GMP, the changes in its particle size and zeta potential were monitored by DLS for a period of 2~3 weeks. The relative particle size was calculated by normalizing the particle size to that of freshly prepared LCP-GMP on day 0. In the freeze-thaw cycle testing, freshly prepared LCP-GMP was diluted with a 20% sucrose solution (1:1, v/v) to a final sucrose concentration of 10%, and the initial particle size and zeta potential were measured by DLS. The sucrose served as a cryoprotectant during the freezing process. The LCP-GMP sucrose solution was freezed at −80°C for 60 min, and thawed at room temperature bef ore DLS measurements. This freeze-thaw cycles were repeated twice for a total of 3 cycles. To investigate the effect of lyophilization on the stability of LCP-GMP, freshly prepared LCP-GMP was diluted with a 10% sucrose solution (1:1, v/v) to a final sucrose concentration of 5%. The LCP-GMP sucrose solution was freezed at −80°C for 60 min before lyophilization. The lyophil ized LCP-GMP powder was reconstituted with distilled water. The DLS measurements were performed before and after lyophilization, and the particle size and zeta potential of reconstituted LCP-GMP were monitored for another 3 weeks. The changes in its particle size and zeta potential over time served as indicators of the LCP-GMP stability under the above testing conditions.
Myeloid cell uptake of LCP-GMP in vivo
To investigate the in vivo myeloid cell uptake of LCP-GMP, fluorescently labeled LCP-GMP was prepared following the preparation procedure, except that a small amount (1%) of cholesterol was replaced with 25-NBD cholesterol in the outer lipid coating step. B16F10 tumor-bearing mice were i.v. injected with NBD-labeled LCP-GMP. Twelve hours after the injection, the peripheral blood, spleen and tumor were collected and processed to single cell suspensions, followed by antibody staining for PMN-MDSCs (CD11b+ Ly6C− Ly6G+), M-MDSCs (CD11b+ Ly6C+ Ly6G−), macrophages (CD11b+ F4/80+) and dendritic cells (DCs) (CD11b− CD11c+) before flow cytometry analysis.
Western blot analysis
B16F10 tumor-bearing mice were given i.v. injections of LCP-GMP, free Gem, and control LCP on days 8, 10, 12, 14 post tumor cell inoculation. On day 16, tumors were collected and tumor lysates were prepared with radioimmunoprecipitation assay (RIPA) buffer supplemented with protease and phosphatase inhibitor cocktails. Protein concentrations were determined using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL). Protein lysates (40 µg) were loaded, separated by SDS-PAGE electrophoresis and then transferred onto polyvinylidenedifluoride (PVDF) membranes. The membranes were blocked for 1 h at room temperature followed by incubation with a primary antibody overnight at 4°C : c-Myc (D84C12), p-STAT1 (Y701), STAT1, TGF-β and β-tubulin (9F3) rabbit monoclonal antibodies (mAbs) (Cell Signaling Technology); survivin (D-8), Bcl-xL (H-5), p-STAT3 (B-7), STAT3 (F-2) and IL-6 (10E5) mouse mAbs (Santa Cruz Biotechnology); IL-10 (JES5–2A5) rat mAb (Biolegend). The PVDF membranes were then washed four times with TBST (Tris-buffered saline, 0.15% Tween 20) and incubated with anti-rabbit, anti-mouse or anti-rat secondary antibodies (1:5000 dilution) at room temperature for 1 h. Finally, the PVDF membranes were washed four times and developed by an enhanced chemiluminescence system (Thermo Scientific, Milford, MA).
Flow cytometry analysis
Mice bearing B16F10 melanoma were given i.v. injections of LCP-GMP, free Gem, and control LCP on days 8, 10, 12 and 14 post tumor cell inoculation. Two days after the last injection, peripheral blood, tumors and spleens were collected and processed to single cell suspensions for antibody staining and flow cytometry analysis on a BD LSRII Cell Analyzer. Intracellular cytokine staining (ICS) was performed using BD Cytofix/Cytoperm Fixation/Permeabilization kit, following the manufacturer’s protocol. Cells for ICS were stimulated in vitro for 6 h with a cell stimulation cocktail containing 12-myristate 13-acetate (PMA) (81 nM) and ionomycin (1.34 µM) in the presence of brefeldin A (5 µg/ml) for the last 4 h to block cytokine secretion.21 Flow cytometry data were analyzed by FlowJo software.
Immunohistochemistry analysis
Mice bearing B16F10 tumors were i.v. injected with LCP-GMP, free Gem or control LCP on days 8, 10, 12, 14 post tumor cell inoculation. Two days later, tumors were dissected, mounted in OCT cryo-embedding medium, and immediately snap freezing on dry ice. Tissue frozen sections (8 µm) were prepared for the following immunostaining procedure. The slides were fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.2% Triton X-100 for 10 min. The slides were then blocked with 5% goat serum for 1 h at room temperature, followed by incubating with PD-L1 antibody overnight at 4°C. Th e slides were then stained with F-actin marker phalloidin for 40 min, followed by incubating with the nuclear dye DAPI for 10 min. After washing with PBS, the slides were covered with mounting medium. The TUNEL staining was performed following the manufacturer’s instructions (Promega). The images were acquired by a Zeiss LSM 700 confocal microscope. At least ten randomly selected microscopic images were acquired for each treatment group, and the quantitative analysis of confocal images was performed using ImageJ software. The percentage (%) of apoptotic cells in the TUNEL assay was obtained by dividing the number of apoptotic cells (TUNEL positive cells) from the number of total cells (DAPI nucleus staining) in the microscopic field. The mean fluorescence intensities (MFI) of PD-L1 signals in confocal images were quantified, as indicators of the expression levels of PD-L1 in tumors after treatments.
In vivo toxicities
Melanoma-bearing C57BL/6 mice were given four every other day i.v. injections of LCP-GMP, free Gem, or control LCP on days 8, 10, 12 and 14 post tumor cell inoculation, with 50.4 µmol/Kg GMP (or Gem). Two days after the last injection, peripheral blood was drawn from mice, and centrifuged at 800 g for 5 min at 4℃, and sera were collected for biochemical analysis. Blood urea nitrogen (BUN), liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured using AST/ALT enzymatic assay kits (Bioo Scientific Corp.) and urea nitrogen (BUN) colorimetric detection kit (Arbor Assays), respectively, following manufacturers’ instructions.
Statistical analysis
Results were expressed as mean ± standard error of mean (SEM). Two-tailed student’s t-test and one-way analysis of variance (ANOVA) test were used to determine statistical significance. A p value of 5% or lower (p≤0.05) was considered to be statistically significant.
Results
Preparation and characterization of nanoparticles
LCP was composed of a lipid-inorganic hybrid core-shell nanostructure. The phosphorylated drug GMP was precipitated in the calcium phosphate (Ca-P) nano-scaffold coated with a lipid bilayer, followed by grafting a high density of polyethylene glycol (PEG) chains. LCP-GMP was spherical in shape and monodispersed, with a particle size of ~30 nm, as measured by TEM (Fig. 1A) and DLS (Fig. 1B). The zeta potential of LCP-GMP was around −20 mV, as measured by DLS. The encapsulation efficiency of GMP in LCP was ~75%, as measured by UV-Vis spectrometry at a wavelength of 275 nm.
Figure 1.
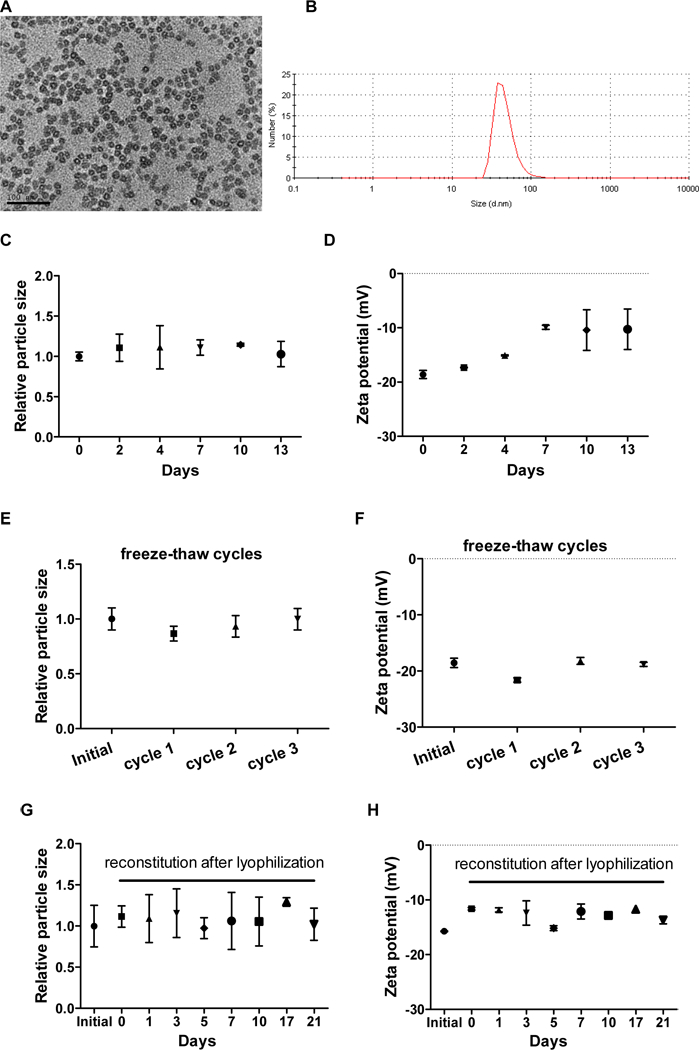
Characterization and stability evaluation of LCP-GMP. (A) TEM picture of LCP-GMP. Scale bar = 100 nm. (B) Particle size of LCP-GMP measured by DLS. LCP-GMP was stored at 4°C after preparation. The particle size and zeta p otential of LCP-GMP were monitored over a period of 2~3 weeks by DLS. (C,D) Freshly prepared LCP-GMP was monitored for 2 weeks to record its particle size (C) and zeta potential (D). (E,F) LCP-GMP was subjected to three freeze-thaw cycles with the presence of 10% sucrose as a cryoprotectant. Its particle size (E) and zeta potential (F) before freeze-thaw and after each freeze-thaw cycle were measured. LCP-GMP sucrose solution was freezed at −80°C for 60 min an d thawed at room temperature before DLS measurements, followed by two additional freeze-thaw process for a total of 3 cycles. Initial: freshly prepared LCP-GMP before freeze-thaw; cycle 1: freeze-thaw once; cycle 2: freeze-thaw twice; cycle 3: freeze-thaw three times. (G,H) LCP-GMP was subjected to lyophilization (freeze-drying) with the presence of 5% sucrose as a cryoprotectant. LCP-GMP sucrose solution was freezed at −80°C for 60 min before lyophilization. The lyophilized LCP-GMP powder was reconstituted with distilled water and monitored for another 3 weeks. Initial: freshly prepared LCP-GMP before lyophilization; days 0–21: reconstituted LCP-GMP after lyophilization.
In the stability testing of LCP-GMP, no significant changes in particle size were observed in 2 weeks (Fig. 1C). The zeta potential of LCP-GMP slightly increased from −20 mV to −10 mV after 3–4 days, and remained stable at −10 mV afterwards (Fig. 1D). Further, freeze-thaw cycle testing was conducted to test the stability of LCP-GMP through a series of extreme and rapid temperature changes by exposing LCP-GMP to a freezing temperature (−80°C) and then thawing it at room temperature, with the presence of 10% sucrose as a cryoprotectant. Compared to freshly prepared LCP-GMP, both particle size and zeta potential remained constant after 3 freeze-thaw cycles (Figs. 1E-F), suggesting that LCP-GMP remained stable despite harsh conditions. Since lyophilization helps lipid nanoparticles achieve long-term stability,22 the stability of reconstituted_LCP-GMP after lyophilization was investigated. As shown in Figs. 1G-H, compared to freshly prepared LCP-GMP, the particle size and zeta potential of reconstituted LCP-GMP remained relatively constant over a period of 3 weeks.
LCP-GMP induced cell apoptosis in tumors
Antiapoptotic proteins survivin and Bcl-xL are involved in the proliferation and survival of MDSCs and tumor cells.23,24 Melanoma-bearing mice were given i.v. injections of LCP-GMP, free Gem or control LCP every other day for six days at a dose of 50.4 µmol/Kg GMP (or Gem) (Fig. 2A). Two days later, tumors were collected and tumor lysates were subjected to western blot analysis. LCP-GMP significantly reduced the expressions of survivin and Bcl-xL in tumors compared with free Gem and control groups (Fig. 2B), suggesting a profound induction of apoptotic and anti-proliferative effects.
Figure 2.

LCP-GMP induced apoptosis in tumors. (A) Timeline of treatments and sample analysis. (B) The expressions of antiapoptotic proteins in tumors were measured by western blot. (C) In vivo tumor cell apoptosis was detected by TUNEL assay (n=4 per group). Blue: nuclei; green: TUNEL positive cell. (D) The percentage (%) of apoptotic cells in the TUNEL assay was analyzed by ImageJ software. ***p<0.0005, Untreated vs. LCP-GMP, LCP-GMP vs. Free Gem.
Gem is known to exert cell killing effect by intercalating into DNA strand and inhibiting processes required for DNA synthesis.25 We next investigated the induction of tumor cell death using TUNEL assay to detect DNA strand breaks in apoptotic cells. Melanoma-bearing mice were treated as above (Fig. 2A). At the low dose of Gem (or GMP) used in this study, mice receiving free Gem showed ~2% apoptotic cells in tumors, only slightly higher than control groups (Fig. 2C-D). However, mice receiving LCP-GMP displayed more than 20% apoptotic cells in tumors, suggesting that LCP significantly enhanced the intratumoral drug delivery and tumor cell killing effect of the loaded drug (Figs. 2C-D). This result coincided with the reduced expressions of antiapoptotic proteins survivin and Bcl-xL in the LCP-GMP-treated tumor (Fig. 2B).
LCP-GMP was taken up by myeloid cells in vivo
B16F10 tumor-bearing mice were i.v. injected with fluorescently labeled LCP-GMP. The fold increase in NBD fluorescence signal compared to corresponding untreated controls, indicated the cellular uptake efficiency of LCP-GMP in different myeloid cell populations. Twelve hours after injection, LCP-GMP was mostly taken up by PMN-MDSCs (CD11b+ Ly6C− Ly6G+) and M-MDSCs (CD11b+ Ly6C+ Ly6G−). Compared to the untreated group, NBD-labeled LCP-GMP induced 20.2-, 31.4- and 6.9- fold increases in NBD fluorescence intensity in PMN-MDSCs as well as 16.0-, 17.0- and 7.7- fold increases in NBD fluorescence intensity in M-MDSCs in the peripheral blood, spleen and tumor, respectively (Figs. 3A-C). Macrophages (CD11b+ F4/80+) took up less LCP-GMP than MDSCs, and DCs (CD11b− CD11c+) displayed the least cellular uptake of LCP-GMP. Compared to the untreated group, NBD-labeled LCP-GMP induced 4.1-, 4.8- and 1.4- fold increases in NBD fluorescence intensity in macrophages as well as 0.9-, 2.4-and 0.09- fold increases in NBD fluorescence intensity in DCs in the peripheral blood, spleen and tumor, respectively (Figs. 3A-C). The representative FACS histograms of the myeloid cell uptake of LCP-GMP in the peripheral blood, spleen and tumor were shown in Figs. 3D-F.
Figure 3.

LCP-GMP was effectively taken up by myeloid cells and significantly eliminated MDSCs in the peripheral blood, spleen and tumor, as well as induced macrophage polarization towards anti-tumor M1 phenotype in tumors. (A-C) The myeloid cell uptake of LCP-GMP in the (A) peripheral blood, (B) spleen and (C) tumor was analyzed at 12 h after i.v. injection of NBD-labeled LCP-GMP into B16F10 tumor-bearing mice. Leukocytes in the peripheral blood, spleen and tumor were stained with antibodies against different myeloid cell populations (macrophage, PMN-MDSC, M-MDSC, DC). Cellular uptake of NBD-labeled LCP-GMP in myeloid cells was analyzed by flow cytometry. The representative FACS histograms of cellular uptake of LCP-GMP in different myeloid cell populations in the (D) peripheral blood, (E) spleen and (F) tumor at 12 h post injection were shown. Gray: Untreated. Red: LCP-GMP. B16F10 tumor-bearing mice were given i.v. injections of LCP-GMP, free Gem and control LCP on days 8, 10, 12, 14 post tumor cell inoculation. On day 16, MDSCs in peripheral blood, spleens and tumors were analyzed by flow cytometry. (G) The frequencies of PMN-MDSCs in peripheral blood after treatments. **p<0.01, Untreated vs. Free Gem; ***p<0.0005, Untreated vs. LCP-GMP. (H) The frequencies of PMN-MDSCs in spleens after treatments. *p<0.05, Untreated vs. LCP-GMP and Untreated vs. Free Gem. (I) The numbers of M-MDSCs in tumors after treatments. Data were normalized to tumor weights. *p<0.05, Untreated vs. Free Gem; **p<0.005, Untreated vs. LCP-GMP. (J) The ratios of anti-tumor M1 to pro-tumor M2 macrophages in tumors after treatments. *p<0.05, Untreated vs. LCP-GMP and LCP-GMP vs. Free Gem. (n=4 per group)
LCP-GMP eliminated MDSCs in vivo
Since PMN-MDSC is the predominant MDSC population in lymphoid organs in tumor-bearing mice, we next determined the frequencies of PMN-MDSCs (CD11b+ Ly6C− Ly6G+) in spleens and peripheral blood following the treatment scheme shown in Fig. 2A.26,27 As shown in Figs. 3G-H, LCP-GMP and free Gem groups decreased the circulating and splenic PMN-MDSCs compared with control groups, and LCP-GMP induced significantly more reduction of circulating and splenic PMN-MDSCs than the free Gem group. In the tumor, M-MDSC (CD11b+ Ly6C+ Ly6G−) is more prominent and its tumor suppressive character is more powerful than PMN-MDSC on a per cell basis.2 We then measured the number of M-MDSCs in tumors after the above treatments. As shown in Fig. 3I, LCP-GMP and free Gem groups decreased the number of M-MDSCs in tumors compared with control groups, and LCP-GMP induced significantly more reduction of M-MDSC in tumors than the free Gem group. These results established that LCP-GMP is more effective in depleting MDSCs in vivo than unformulated Gem.
LCP-GMP promoted phenotypic switch of macrophage in tumors
In tumors, M-MDSC rapidly differentiates into TAM.2 TAM largely acts in a pro-tumor manner and augments the tumor-mediated immunosuppression.2,7 A high ratio of M1/M2 macrophages in the TME is associated with increased survival.28 We then tested whether MDSC depletion could drive TAM polarization towards anti-tumor phenotype in the TME. Two days after the treatments shown in Fig. 2A, LCP-GMP induced the phenotypic switch of TAM from pro-tumor M2 phenotype (CD11b+ F4/80+ CD206+) to anti-tumor M1 phenotype (CD11b+ F4/80+ CD80+), as evidenced by the elevated ratio of M1 to M2 macrophages (M1/M2) in tumors (Fig. 3J). This result suggests that LCP-GMP significantly reduced the immunosuppressive activity of TAM. Though free Gem decreased the tumor-infiltrating M-MDSC, it exerted little effect on TAM polarization compared to control groups (Fig. 3J).
Protein expressions in tumors after treatments
We then tested whether the depletion of MDSCs in tumors and lymphoid organs decreases tumor cell survival and immunosuppressive phenotype in the TME. Following the treatment scheme shown in Fig. 2A, tumor lysates from melanoma-bearing mice were subjected to protein analysis by western blots. As shown in Fig. 4, LCP-GMP reduced the expressions of p-STAT1, p-STAT3 and c-Myc that are associated with MDSC expansion and tumor progression.29,30 LCP-GMP decreased the expressions of immunosuppressive mediators TGF-β, IL-6 and IL-10, suggesting the alleviation of immunosuppression in the TME. IL-6 is important for MDSC generation and survival,31 and the reduced IL-6 in tumors could promote MDSC depletion. S100 calcium-binding protein A8 (S100A8) and S100A9 are known to induce MDSC expansion and are dependent on STAT3 upregulation.3,32 LCP-GMP reduced the expressions of S100A8 and S100A9, consistent with the p-STAT3 downregulation in the LCP-GMP-treated tumor. The downregulations of these transcription factors (such as STAT3 and c-Myc) and anti-inflammatory cytokines (such as IL-10 and TGF-β) in tumors also correlated with the M1 polarization of TAM shown in Fig. 3J.33−36
Figure 4.
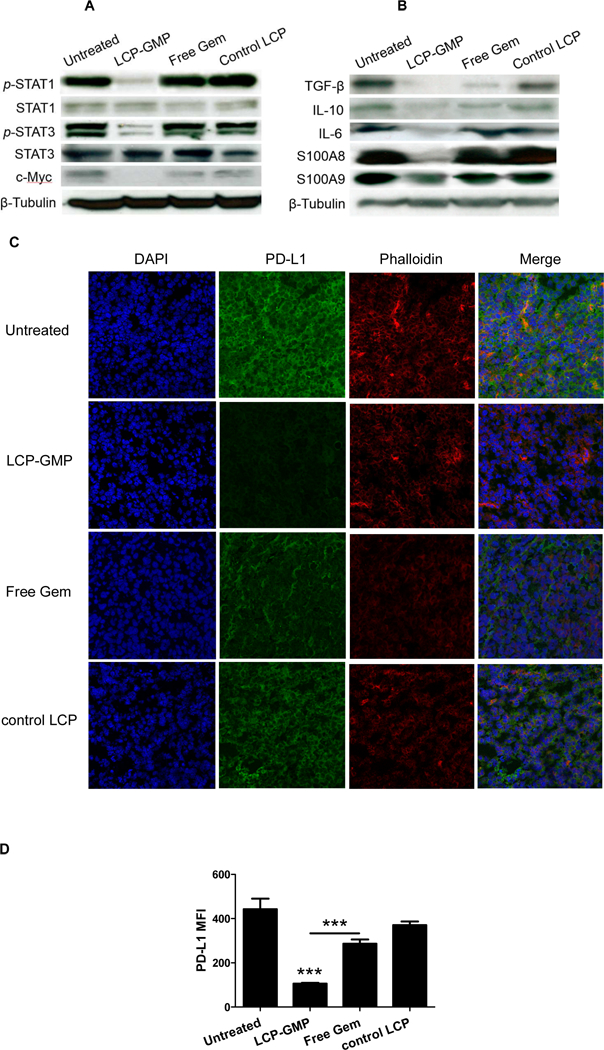
LCP-GMP downregulated the expressions of pro-tumor (A) transcription factors and (B) immunosuppressive mediators as well as (C-D) PD-L1 in tumors after treatments. B16F10 tumor-bearing mice were given i.v. injections of LCP-GMP, free Gem and control LCP on days 8, 10, 12, 14 post tumor cell inoculation. On day 16, tumor lysates were prepared for western blot analysis (A-B), and frozen sections from dissected tumors were prepared for immunostaining (C-D). (C) The PD-L1 expressions in tumors after treatments were detected by immunohistochemistry before observing under a confocal microscope (n=4 per group). Blue: nuclei; green: PD-L1; red: phalloidin. (D) The mean fluorescence intensities (MFI) of PD-L1 signals in confocal images were analyzed by ImageJ software, as indicators of the expression levels of PD-L1 in tumors after treatments. ***p<0.0001, Untreated vs. LCP-GMP and LCP-GMP vs. Free Gem.
LCP-GMP reduced the PD-L1 expression on tumor cells
We next tested whether the reversal of immunosuppression by modulating myeloid cells including MDSCs and TAMs, impacts the immunosuppressive phenotype of tumor cells. It is established that PD-L1 is highly expressed in melanoma and its binding to the inhibitory molecule PD-1 on T cells significantly attenuates the cytotoxic T lymphocyte (CTL)-mediated antitumor immunity. The decreased expression of PD-L1 is often associated with the reversal of immunosuppression.37 After the treatments shown in Fig. 2A, compared with control groups, mice receiving LCP-GMP showed dramatic reduction of PD-L1 expression in tumors (Fig. 4C-D), which makes tumor cells more susceptible to the T-cell attack.38 Mice receiving free Gem displayed minor effect on PD-L1 reduction (Fig. 4C-D). This finding suggests that LCP-GMP effectively alleviated the immunosuppressive TME by decreasing PD-L1 expression and thereby interrupting the PD-1/PD-L1 signaling pathway in tumors. In addition, MYC oncogene regulates the expression of immune checkpoint proteins on the tumor cell surface.39 Decreased PD-L1 expression (Fig. 4C-D) also correlated with the decreased c-Myc expression (Fig. 4A) in the LCP-GMP-treated tumor.
LCP-GMP modulated T cell infiltration in tumors
We further tested whether the LCP-GMP-mediated depletion of MDSCs influences the infiltration patterns of T lymphocytes in tumors, such as CD8+ T cells, conventional CD4+Foxp3− T cells (Tconv) and Tregs. We evaluated the numbers of tumor-infiltrating T cell subsets two days after four systemic administrations (Fig. 2A). Compared with control groups, free Gem reduced the number of tumor-infiltrating CD8+ T cells and Tconv by ~66% and ~55%, respectively; while LCP-GMP did not influence the number of intratumoral CD8+ T cells and Tconv compared to control groups (Figs. 5A-B). Notably, LCP-GMP decreased the amount of intratumoral Tregs by ~90% compared with control groups; while free Gem only caused a reduction of intratumoral Tregs by ~35% (Fig. 5C). Thus, compared with free Gem, LCP-GMP dramatically and selectively depleted Tregs in tumors without compromising the tumor-infiltrating CD8+ T cell and Tconv (Figs. 5A-C). In LCP-GMP group, the dramatic reduction of Tregs led to the significantly increased ratios of CD8+ T cell to Treg (CD8/Treg) and Tconv to Treg (Tconv/Treg) in tumors, indicating a favorable prognosis; whereas free Gem has little effect on the CD8/Treg and Tconv/Treg ratios compared to control groups (Figs. 5D-E).
Figure 5.
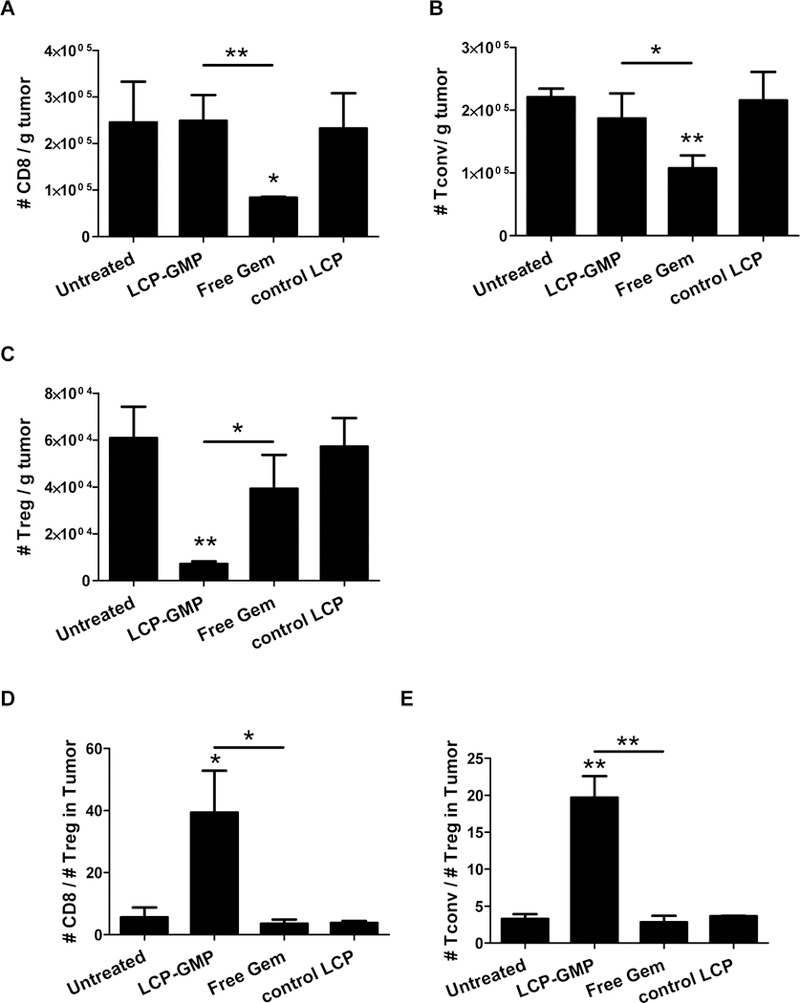
LCP-GMP selectively depleted Tregs without compromising other tumor-infiltrating T cells. B16F10 tumor-bearing mice were given i.v. injections of LCP-GMP, free Gem and control LCP on days 8, 10, 12, 14 post tumor cell inoculation. On day 16, tumors were dissected and processed to single cell suspensions before flow cytometry analysis. The numbers of tumor-infiltrating T cells were normalized to tumor weights. (A) The numbers of tumor-infiltrating CD8+ T cells. *p<0.05, Untreated vs. free Gem; **p<0.01, LCP-GMP vs. free Gem. (B) The numbers of tumor-infiltrating conventional CD4+ Foxp3− T cells (Tconv). *p<0.05, LCP-GMP vs. free Gem; **p<0.005, Untreated vs. free Gem. (C) The numbers of tumor-infiltrating Tregs. *p<0.05, LCP-GMP vs. free Gem; **p<0.01, Untreated vs. LCP-GMP. (D) The ratio of CD8+ T cells to Tregs (CD8/Treg) in tumors. *p<0.05, Untreated vs. LCP-GMP and LCP-GMP vs. free Gem. (E) The ratio of Tconv to Tregs (Tconv/Treg) in tumors. **p<0.005, Untreated vs. LCP-GMP and LCP-GMP vs. free Gem. (n=4 per group).
LCP-GMP induced antitumor CTL responses
We next assessed whether the tumor-infiltrating and systemic CTLs exert their antitumor effector functions by measuring the productions of pro-inflammatory cytokines IFN-γ and TNF-α. Lymphocytes isolated from tumors, peripheral blood and spleens were subjected to intracellular cytokine staining (ICS) after ex vivo polyclonal stimulation. Compared with control groups, LCP-GMP induced 3.6-, 1.4- and 0.5- fold increases of IFN-γ production (Figs. 6A, 6C and 6E) as well as 5.5-, 8.6- and 1.8- fold increases of TNF-α production (Figs. 6B, 6D and 6F) by CD8+ T cells in tumors (Figs. 6A-B), peripheral blood (Figs. 6C-D) and spleens (Figs. 6E-F), respectively; whereas free Gem had little effect on triggering the productions of IFN-γ and TNF-α (Fig. 6). These data suggest that LCP-GMP induced superior CTL effector functions in tumors and lymphoid compartments, activating the adaptive antitumor immune response.
Figure 6.
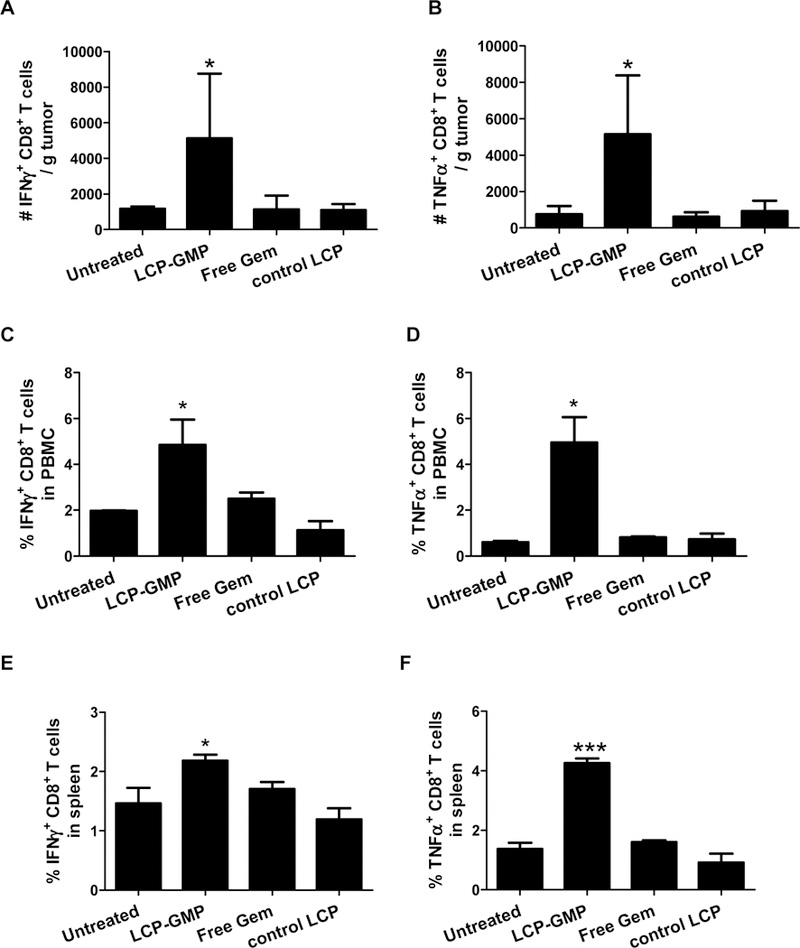
LCP-GMP enhanced T-cell effector functions in tumors and lymphoid compartments. B16F10 tumor-bearing mice were given i.v. injections of LCP-GMP, free Gem and control LCP on days 8, 10, 12, 14 post tumor cell inoculation. On day 16, peripheral blood, tumors and spleens were collected and processed to single cell suspensions. Lymphocytes from (A-B) tumors, (C-D) peripheral blood and (E-F) spleens were polyclonally stimulated with PMA and ionomycin in vitro for 6 h, with the presence of brefeldin A in the last 4 h to block cytokine secretion. The productions of (A, C, E) IFN-γ and (B, D, F) TNF-α by CD8+ T cells in (A-B) tumors, (C-D) peripheral blood and (E-F) spleens were analyzed by intracellular cytokine staining. *p<0.05, ***p<0.0005, Untreated vs. LCP-GMP. (n=4 per group)
LCP-GMP inhibited tumor progression
We further evaluated the tumor growth inhibition after different treatments. The B16F10 tumor-bearing C57BL/6 mice received four i.v. injections of LCP-GMP, Gem or control LCP on days 8, 10, 12, 14 post tumor cell inoculation, with a dose of 50.4 µmol/Kg GMP (or Gem). As shown in Fig. 7A, LCP-GMP significantly retarded the tumor progression compared with control groups; while free Gem only led to a partial tumor growth inhibitory effect. No significant body weight loss was observed during treatments, suggesting all treatments were safe (Fig. 7B). At the end of treatments, tumors were collected and weighed. As shown in Fig. 7C, LCP-GMP treatment led to the lightest tumor weight, only ~15% of the untreated group; while, free Gem reduced the tumor weight to ~65% of the untreated group and control LCP had little effect on the reduction of tumor weight.
Figure 7.
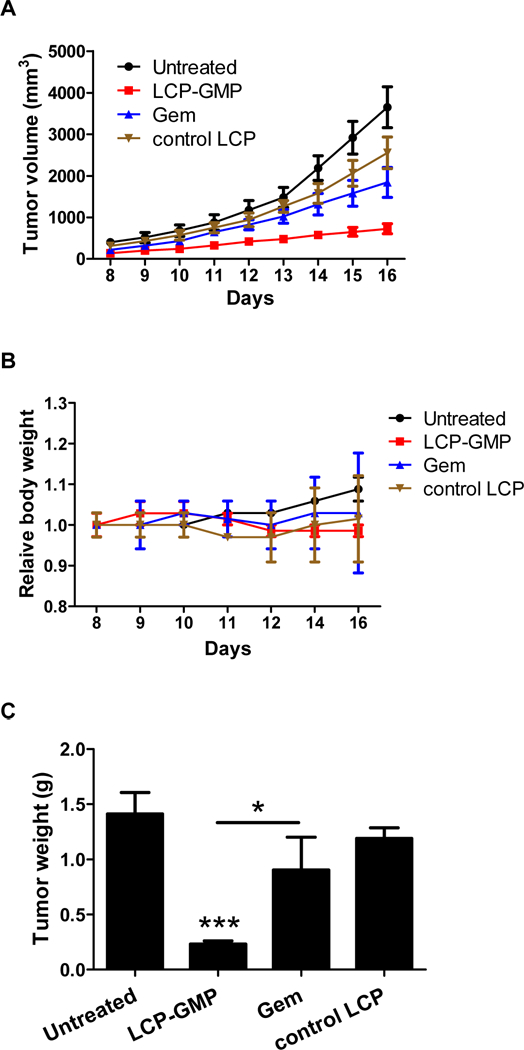
LCP-GMP inhibited melanoma tumor growth. Groups of C57BL/6 mice (n=10 per group) were inoculated with 5× 10 5 B16F10 cells s.c. on day 0. Melanoma-bearing mice received i.v. injections of LCP-GMP, Gem or control LCP on days 8, 10, 12 and 14, with a dose of 50.4 µmol/Kg GMP (or Gem). (A) Tumor volumes and (B) relative body weights were recorded during treatments. (C) Tumor weights on day 16 were shown. ***p<0.001, Untreated vs. LCP-GMP; *p<0.05, LCP-GMP vs. Gem.
LCP-GMP was safe in vivo
We evaluated the serum biochemical parameters as indicators of in vivo toxicities. In serum biochemical assays, BUN is the indicator of renal function, and AST and ALT are indicators of liver damage. Elevation of these parameters correlates with kidney and liver dysfunction. As shown in Supplementary Table 1, all biochemical parameters (BUN, AST, ALT) tested were in the normal range after treatment with LCP-GMP, suggesting that repeated injections of LCP-GMP were safe and did not induce any kidney and liver toxicities. In addition, it was reported that B16 melanoma-bearing mice had elevated levels of liver enzymes AST and ALT which may cause mild liver damage.40 The levels of AST and ALT in LCP-GMP group were lower than those of untreated and control groups, suggesting that LCP-GMP could alleviate the potential melanoma-induced liver dysfunction (Supplementary Table 1).
Discussion
Gem is used to treat a number of types of cancer such as breast cancer, non-small cell lung cancer and pancreatic cancer. While it delivers reasonable efficacy, this chemotherapeutic agent suffers two major disposition barriers. In the plasma, Gem undergoes rapid deamination by cytidine deaminase and deamination represents inactivation. In addition, Gem is hydrophilic and its entrance into cells requires active transporters such as SLC28A1 and SLC29A1.41 After entering the cells, Gem undergoes sequential phosphorylation and becomes monophosphate, diphosphate and triphosphate nucleosides. It is the diphosphate and triphosphate nucleosides that deliver therapeutic activity.42 On the other hand, the formation of monophosphate nucleoside is catalyzed by deoxycytidine kinase. The first phosphorylation appears to be the rate-limiting step and determines the intracellular accumulation of active metabolites of this chemotherapeutic agent.13 In this study, we formulated the monophosphate metabolite into LCP nanoparticles, effectively overcoming the disposition barriers and the rate-limiting step for therapeutic activation.
LCP is a lipid/inorganic hybrid and membrane/core type nanoparticle that can encapsulate phosphorylated drugs, such as nucleoside analogues.15,43,44 In this study, the Ca-P in LCP serves as a carrier scaffold to form microprecipitates with the phosphate group on GMP in aqueous droplets using a water-in-oil microemulsion method. A phospholipid DOPA was then added in the microemulsion to form an inner leaflet lipid layer wrapping around the Ca-P core through the covalent linkage between Ca and DOPA’s phosphate head-group. This microemulsion system thermodynamically stabilizes Ca-P cores entrapping GMP and prevents their agglomeration.45 To facilitate the in vivo delivery of GMP, we further coated the Ca-P core with an asymmetrical lipid bilayer using hydration method. This outer leaflet lipid bilayer is composed of cationic lipid DOTAP, neutral fusogenic lipid cholesterol and amphiphilic DSPE-PEG. LCP-GMP was modified with a high density of amphiphilic DSPE-PEG on the particle surface, which shields the charge of inner layers of lipids. The compact PEGylation and negative surface charge of LCP-GMP alleviate the surface adsorption of serum proteins and reduce non-specific uptake by the reticuloendothelial system, which confers the prolonged in vivo circulation half-life of LCP-GMP.15,46,47 In contrast, the unformulated Gem quickly clears out of the body with a limited time window to exert its bioactivity.15 After cellular endocytosis, the LCP-entrapped GMP could efficiently release from endosomes into cytoplasm by the following mechanisms to prevent endolysosomal degradation. First, the acidic environment in endosomes dissolves the Ca-P in LCP and the elevated ion concentration increases the osmotic pressure in endosomes, which leads to water influx into endosomes and finally endosome disruption.48 Second, the cationic lipid DOTAP in LCP lipid bilayer could form ion pair with the anionic lipids in endosomal membranes, and this electrostatic interaction between cationic and anionic lipids leads to the formation of an inverted hexagonal phase,49 subsequently releasing the entrapped drug to the cytoplasm.47 The released GMP then diffuses into the nucleus to deliver therapeutic activities.
The LCP-GMP induced antitumor responses through the following mechanisms. It effectively triggered tumor cell death by intercalating into DNA strands to inhibit further DNA synthesis.50 This direct tumor cell killing effect was observed by the decreased expressions of antiapoptotic proteins survivin and BcL-xL as well as the profound induction of TUNEL-positive cells in tumors. Some regimens of chemotherapy-induced apoptosis can elicit an effective antitumor immune response.51,52,53 The reduced expressions of antiapoptotic proteins are associated with MDSC depletion in tumor-bearing mice, and result in the loss of tumor-associated Tregs.54,55 MDSCs and Tregs are the major components responsible for tumor-associated immune suppression.27,56 Tumor-derived inflammatory factors, such as TGF-β, IL-6, IL-10, S100A8 and S100A9, initiate immunosuppressive pathways that drive MDSC expansion and further promote the differentiation of MDSCs towards immunosuppressive macrophages.1 Compared with free Gem, LCP-GMP more effectively depleted MDSCs in the peripheral blood, spleen and tumor. These MDSC depletions subsequently alleviated tumor-mediated immunosuppression, as indicated by the decreased expressions of tumor-derived immunosuppressive cytokines TGF-β, IL-10, IL-6 and checkpoint protein PD-L1 as well as significantly reduced intratumoral Tregs and enhanced M1/M2 macrophage ratio in tumors. The reduction of Tregs was associated with the decreased expressions of TGF-β and IL-10.6 The LCP-GMP-mediated M1 polarization of TAM correlated with the downregulations of some transcription factors (i.e. STATs, c-Myc) and the inhibition of anti-inflammatory cytokine signaling (i.e. IL-10, TGF-β).33–36 The reduced S100A8 and S100A9 expressions could inhibit MDSC expansion and its migration to the tumor site.3,57 In addition, the STAT3 level largely dictates the recruitment, activation and immunosuppressive function of MDSCs in the TME.58 In LCP-GMP group, the downregulation of p-STAT3 in tumors suppresses MDSCs and many pro-tumor genes such as survivin, Bcl-xL and c-Myc, leading to decreased tumor cell survival and proliferation.23 c-Myc selectively regulates tumor-infiltrating MDSCs,59 and c-Myc suppression correlated with the reduction of PD-L1 in tumors.39 Further, the MDSC depletion and reversal of immunosuppression correlate with increased adaptive antitumor immune responses and mproved therapeutic outcome of melanoma.60 LCP-GMP elicited strong CTL responses, as indicated by the increased productions of pro-inflammatory cytokines IFN-γ and TNF-α by CD8+ T cells, which contributes to the inhibition of tumor progression. Though free Gem depleted MDSCs as well as partially decreased the PD-L1 expression and Tregs in tumors, it failed to improve the CD8+ T-cell effector function, and only caused a partial effect on tumor growth inhibition. Our data suggest that the expression of PD-L1 in tumors is associated with the presence of MDSCs and Tregs,7,61 and the reduction of immunosuppressive TME by LCP-GMP restores CD8+ T-cell-mediated antitumor immune responses. A schematic illustration of LCP-GMP-mediated immunomodulation is shown in Fig. 8.
Figure 8.
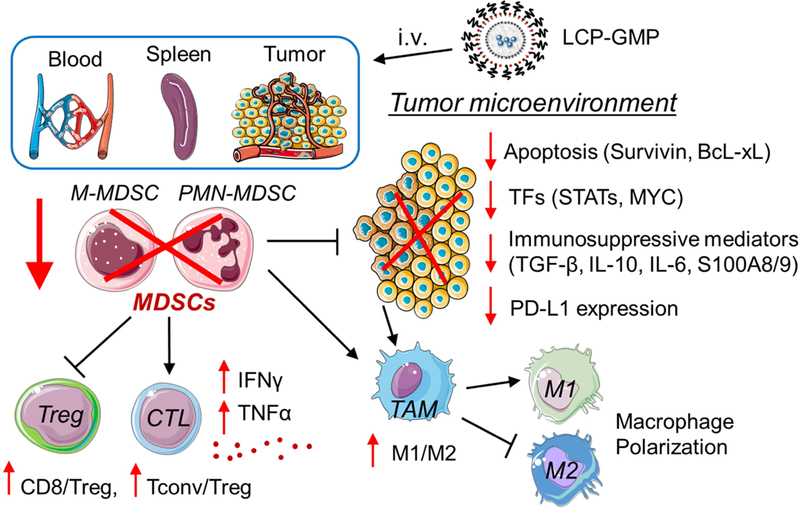
Schematic illustration of LCP-GMP-mediated immunomodulation. After intravenous (i.v.) injections, LCP-GMP triggered significant apoptosis; largely depleted immunosuppressive MDSCs in the peripheral blood, spleen and tumor; downregulated the expressions of pro-tumor transcription factors (TFs), immunosuppressive mediators and checkpoint protein PD-L1 in tumors; induced the polarization of tumor-associated macrophages (TAM) from the pro-tumor M2 phenotype to antitumor M1 phenotype; selectively depleted immunosuppressive Tregs without compromising other tumor-infiltrating T cells; enhanced the production of proinflammatory cytokines in CD8+ T cells and inhibited tumor progression.
Conclusion
In the present study, we formulated chemotherapeutic and MDSC-depleting agent GMP into a lipid-coated calcium phosphate (LCP) nanoparticle. The Ca-P can act as a carrier scaffold for monophosphate metabolite of Gem through the formation of microprecipitates. This is of significance as many anti-cancer and anti-viral agents require intracellular phosphorylation for their therapeutic activity. We have shown that LCP-GMP can (1) trigger significant apoptosis in tumors; (2) largely deplete immunosuppressive MDSCs in tumors and lymphoid compartments; (3) induce macrophage polarization from the pro-tumor M2 phenotype to antitumor M1 phenotype in the TME; (4) downregulate the pro-tumor transcription factors, immunosuppressive mediators and anti-inflammatory cytokines in tumors; (5) significantly decrease the expression of immune checkpoint protein PD-L1 in tumors; (6) selectively deplete immunosuppressive Tregs without compromising other tumor-infiltrating T cells; (7) enhance T-cell-mediated antitumor immunity; (8) effectively inhibit tumor progression without in vivo toxicity. The immunotherapeutic effects shown by LCP-GMP in melanoma suggest that the strategy described in this report can be used for many types of cancer. This is of significance as Gem is commonly used in a few types of cancer only but immunosuppressive TME is common among all types of cancer. Above all, the reduction of tumor-induced immunosuppression by LCP-GMP can boost the endogenous adaptive antitumor immunity and improve antitumor efficacy. These findings point to a simple approach that effectively modulates immunosuppressive TME for enhanced antitumor responses. This strategy of reducing immunosuppressive TME via systemically delivering nanoparticle-formulated MDSC-depleting agents could further combine with other cancer immunotherapies or treatment regimens to achieve improved or synergistic therapeutic outcomes.
Supplementary Material
Acknowledgements
This work was supported by the NIH NIGMS Advance-CTR pilot grant (U54GM115677), Rhode Island Foundation – Medical Research Fund (#20164344) to Y.Z. as well as R01GM61988 and R01ES07965 to B.Y. We would like to thank URI Institute for Immunology and Informatics (iCubed) Cell Analysis and Sorting Core supported by COBRE grant NIH P20 GM104317, Lifespan Molecular Pathology Core, URI Genomic and Sequencing Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
No potential conflicts of interest were disclosed.
Data Availability
All relevant data are available from the authors on request and are included within the manuscript.
Reference
- 1.Gabrilovich DI, Ostrand-Rosenberg S & Bronte V Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12, 253–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar V, Patel S, Tcyganov E & Gabrilovich DI The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol 37, 208–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabrilovich DI & Nagaraj S Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9, 162–174 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maenhout SK, Van Lint S, Emeagi PU, Thielemans K & Aerts JL Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int J Cancer 134, 1077–1090 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Serafini P, Mgebroff S, Noonan K & Borrello I Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 68, 5439–5449 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang B, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 66, 1123–1131 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 66, 124–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Najjar YG & Finke JH Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front Oncol 3, 49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki E, Kapoor V, Jassar AS, Kaiser LR & Albelda SM Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 11, 6713–6721 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Le HK, et al. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol 9, 900–909 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Mackey JR, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58, 4349–4357 (1998). [PubMed] [Google Scholar]
- 12.ocabas NA, et al. Gemcitabine pharmacogenomics: deoxycytidine kinase and cytidylate kinase gene resequencing and functional genomics. Drug Metab Dispos 36, 1951–1959 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veltkamp SA, Beijnen JH & Schellens JH Prolonged versus standard gemcitabine infusion: translation of molecular pharmacology to new treatment strategy. Oncologist 13, 261–276 (2008). [DOI] [PubMed] [Google Scholar]
- 14.hang Y, Schwerbrock NM, Rogers AB, Kim WY & Huang L Codelivery of VEGF siRNA and gemcitabine monophosphate in a single nanoparticle formulation for effective treatment of NSCLC. Mol Ther 21, 1559–1569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Kim WY & Huang L Systemic delivery of gemcitabine triphosphate via LCP nanoparticles for NSCLC and pancreatic cancer therapy. Biomaterials 34, 3447–3458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umansky V, Sevko A, Gebhardt C & Utikal J Myeloid-derived suppressor cells in malignant melanoma. J Dtsch Dermatol Ges 12, 1021–1027 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Li J, Yang Y & Huang L Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Control Release 158, 108–114 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren X, et al. Sulfur-Doped Graphene-Based Immunological Biosensing Platform for Multianalysis of Cancer Biomarkers. ACS Appl Mater Interfaces 9, 37637–37644 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Ren X, et al. Increased electrocatalyzed performance through high content potassium doped graphene matrix and aptamer tri infinite amplification labels strategy: Highly sensitive for matrix metalloproteinases-2 detection. Biosens Bioelectron 94, 694–700 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Peng L, Mumper RJ & Huang L Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy. Biomaterials 34, 8459–8468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Li N, Suh H & Irvine DJ Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat Commun 9, 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, et al. Lyophilization of cationic lipid-protamine-DNA (LPD) complexes. J Pharm Sci 89, 355–364 (2000). [DOI] [PubMed] [Google Scholar]
- 23.De Veirman K, et al. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front Oncol 4, 349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman ML & Czyz M Anti-apoptotic proteins on guard of melanoma cell survival. Cancer Lett 331, 24–34 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Plunkett W, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 22, 3–10 (1995). [PubMed] [Google Scholar]
- 26.Sceneay J, et al. Tracking the fate of adoptively transferred myeloid-derived suppressor cells in the primary breast tumor microenvironment. PLoS One 13, e0196040 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youn JI, Nagaraj S, Collazo M & Gabrilovich DI Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181, 5791–5802 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang M, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res 7, 19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condamine T & Gabrilovich DI Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 32, 19–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hix LM, et al. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem 288, 11676–11688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Umansky V, Blattner C, Gebhardt C & Utikal J The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) 4(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng P, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205, 2235–2249 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tugal D, Liao X & Jain MK Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol 33, 1135–1144 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Wang N, Liang H & Zen K Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 5, 614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pello OM, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 119, 411–421 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Sanmarco LM, et al. IL-6 promotes M2 macrophage polarization by modulating purinergic signaling and regulates the lethal release of nitric oxide during Trypanosoma cruzi infection. Biochim Biophys Acta 1863, 857–869 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Ostrand-Rosenberg S, Horn LA & Haile ST The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol 193, 3835–3841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noman MZ, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211, 781–790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casey SC, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eggert T, et al. Tumor induced hepatic myeloid derived suppressor cells can cause moderate liver damage. PLoS One 9, e112717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SY, et al. Genetic polymorphisms of SLC28A3, SLC29A1 and RRM1 predict clinical outcome in patients with metastatic breast cancer receiving gemcitabine plus paclitaxel chemotherapy. Eur J Cancer 50, 698–705 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Ciccolini J, Serdjebi C, Peters GJ & Giovannetti E Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: an EORTC-PAMM perspective. Cancer Chemother Pharmacol 78, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao J, Zhang Y, Ramishetti S, Wang Y & Huang L Turning an antiviral into an anticancer drug: nanoparticle delivery of acyclovir monophosphate. J Control Release 170, 414–420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srinivas R, et al. Theranostic etoposide phosphate/indium nanoparticles for cancer therapy and imaging. Nanoscale 7, 18542–18551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi L, Ma J & Shen J Synthesis of Copper Nanoparticles in Nonionic Water-in-Oil Microemulsions. J Colloid Interface Sci 186, 498–500 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Li SD & Huang L Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta 1788, 2259–2266 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Satterlee A & Huang L In vivo gene delivery by nonviral vectors: overcoming hurdles? Mol Ther 20, 1298–1304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Chen YC, Tseng YC, Mozumdar S & Huang L Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J Control Release 142, 416–421 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Y & Szoka FC Jr. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 35, 5616–5623 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Mini E, Nobili S, Caciagli B, Landini I & Mazzei T Cellular pharmacology of gemcitabine. Ann Oncol 17 Suppl 5, v7–12 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Bracci L, Schiavoni G, Sistigu A & Belardelli F Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 21, 15–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emens LA & Middleton G The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res 3, 436–443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nowak AK, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol 170, 4905–4913 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Weiss JM, et al. Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/alphaCD40 therapy. J Immunol 192, 5821–5829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, et al. IL-17A weakens the antitumor immuity by inhibiting apoptosis of MDSCs in Lewis lung carcinoma bearing mice. Oncotarget 8, 4814–4825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindau D, Gielen P, Kroesen M, Wesseling P & Adema GJ The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138, 105–115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinha P, et al. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 181, 4666–4675 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dufait I, et al. Signal transducer and activator of transcription 3 in myeloid-derived suppressor cells: an opportunity for cancer therapy. Oncotarget 7, 42698–42715 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aliper AM, Frieden-Korovkina VP, Buzdin A, Roumiantsev SA & Zhavoronkov A Interactome analysis of myeloid-derived suppressor cells in murine models of colon and breast cancer. Oncotarget 5, 11345–11353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fleming V, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol 9, 398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206, 3015–3029 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.