

Abstract
Dementia is growing at an alarming rate worldwide. Although Alzheimer disease is the leading cause, over 50% of individuals diagnosed with Alzheimer disease have vascular lesions at autopsy. There has been an increasing appreciation of the pathogenic role of vascular risk factors in cognitive impairment caused by neurodegeneration. Midlife hypertension is a leading risk factor for late-life dementia. Hypertension alters cerebrovascular structure, impairs the major factors regulating the cerebral microcirculation, and promotes Alzheimer pathology. Experimental studies have identified brain perivascular macrophages as the major free radical source mediating neurovascular dysfunction of hypertension. Recent evidence indicates that high dietary salt may also induce cognitive impairment. Contrary to previous belief, the effect is not necessarily associated with hypertension and is mediated by a deficit in endothelial nitric oxide. Collectively, the evidence suggests a remarkable cellular diversity of the impact of vascular risk factors on the cerebral vasculature and cognition. Whereas long-term longitudinal epidemiological studies are needed to resolve the temporal relationships between vascular risk factors and cognitive dysfunction, single-cell molecular studies of the vasculature in animal models will provide a fuller mechanistic understanding. This knowledge is critical for developing new preventive, diagnostic, and therapeutic approaches for these devastating diseases of the mind.
Keywords: Alzheimer disease, cerebrovascular disease, dementia, endothelial dysfunction, neurovascular coupling
Introduction
Dementia is an umbrella term referring to a progressive and irreversible decline in cognitive abilities sufficient to interfere with activities of daily living, typically associated with aging.1 It affects approximately 50 million people worldwide, a number expected to grow by nearly 9.9 million new cases each year due to demographic shifts and lack of effective therapies.2 While Alzheimer disease (AD) is the leading cause, dementia on vascular basis is the second leading cause of cognitive impairment.3 However, up to 50% of clinically diagnosed AD have a mixed pathology at autopsy including cerebrovascular lesions4 (Figure 1). Therefore, vascular pathology once considered to be linked exclusively to vascular cognitive impairment, has emerged as a key contributor also to other forms of dementia.3,5
Figure 1.

Over 50% of cases of clinically diagnosed Alzheimer disease (AD) feature vascular pathology. AD represents 70% of cases of late-life dementia, diagnosed according to clinical criteria. However, post-mortem neuropathological analysis reveals that only 24% of dementia cases feature exclusively AD pathology (amyloid plaques and neurofibrillary tangles), while over 50% feature vascular pathology, either alone (26%) or mixed with AD pathology (27%).
Hypertension (HTN) is the major vascular risk factor of cognitive impairment.3 Based on new diagnostic guidelines, HTN afflicts almost 50% of the population in the US.6 Owing to its key role in vascular cognitive impairment, the World Health Organization has set a global target of 25% relative reduction in the prevalence of HTN by 2025 as a key measure to reduce the risk of cognitive decline.2 High dietary salt is associated with HTN in many patients, but the effects of salt go beyond its role in blood pressure (BP) elevation. Specifically, a high-salt diet has been independently linked to increased risk of cerebrovascular disease and dementia.7,8 Therefore, dietary salt, although not currently identified as one of the risk factors targeted for prevention of dementia,2 may also contribute to cognitive impairment.9
The purpose of this brief review is to evaluate the current evidence identifying potential mechanisms underlying the damaging effects of HTN and dietary salt on the brain. First, we will discuss the importance of maintaining adequate perfusion to the brain, and the link between reduced cerebral blood flow (CBF) and cognitive impairment. Then, we will examine the structural and functional cerebrovascular alterations caused by HTN, as well as the recently emerged connection between dietary salt and cognitive dysfunction. Finally, we will seek to identify outstanding questions that need to be addressed and related challenges to overcome.
CBF regulation and impact on cognitive function
The brain relies upon a continuous blood supply to deliver essential nutrients to support its dynamic and regionally diverse energetic needs, and to clear potentially toxic byproducts of brain activity.10 Vascular, perivascular, and brain cells work together as a unit, termed the “neurovascular unit” (NVU), to assure adequate blood perfusion to the brain and maintain the homeostasis of the brain’s internal milieau.10 Here, we will briefly review the main mechanisms involved in CBF regulation, and provide evidence linking inadequate cerebral perfusion to cognitive impairment.
Cerebrovascular autoregulation
Cerebrovascular autoregulation maintains CBF relatively constant in response to BP changes within a certain range, normally from 60 and 150 mmHg.11 Autoregulation ensures adequate perfusion despite changes in perfusion pressure, while protecting downstream microvessels from excessive transmural pressure. Autoregulation is mainly dependent on the property of vascular smooth muscle cells to contract in response to increased transmural pressure (myogenic tone). Thus, a rise in BP results in vasoconstriction, while a decrease in BP leads to vasodilation. This key protective mechanism is mediated by complex molecular events that finely regulate intracellular Ca2+ and the Ca2+ sensitivity of the contractile apparatus to induce assembly of contractile proteins and ultimately induce constriction.12 Bayliss’ myogenic hypothesis of autoregulation13 has been extensively investigated since its original proposal and focuses on mechanoreceptors on vascular smooth muscle cells.14 For example, several members of the transient receptor potential (TRP) channel superfamily are found in vascular smooth muscle cells and have been shown to be mechanosensitive, and TRPC6 and TRPM4 have been shown to have a critical role in cerebral autoregulation. One hypothesis is that TRPC6 increases Ca2+ entry in response to mechanical stress, which in turn activates TRPM4 channels to allow Na+ entry leading to further depolarization, activation of voltage dependent Ca2+ channels, and constriction of the smooth muscle cell.15 Additionally, mechanoreceptors on endothelial cells have been proposed to contribute to autoregulation by modulating the response to shear stress and transmural pressure by releasing both constricting and relaxing-factors.14,16 These mechanisms, in addition to the release of vasoactive metabolites due to reduced cerebral perfusion, may also contribute to the smooth muscle relaxation underlying the lower limit of autoregulation.14
Endothelial cells: Vasoregulatory function and the blood–brain barrier
Endothelial cells regulate vasomotor tone by releasing vasoactive signals which include both constrictor (endothelin-1, thromboxane A2, prostaglandin H2, etc.) and relaxing factors (nitric oxide [NO], bradykinin, endothelium-derived hyperpolarization factor [EDHF], prostacyclin [PGI2], prostaglandin E2, etc.).17 The precise nature of EDHF remains elusive and could be either a chemical or an electrical signal.18 Perhaps the most studied endothelium-dependent mechanism is NO-mediated vasodilation. NO is produced in endothelial cells by endothelial NO synthase (eNOS), and its diffusibility allows it to freely permeate membranes to reach vascular smooth muscle cells.19 Then, NO activates soluble guanylate cyclase (sGC), which in turn leads to the generation of the second messenger cyclic guanosine monophosphate (cGMP).19 Thus, vasodilation is ultimately induced by intracellular cGMP through a reduction in intracellular Ca2+ concentration, activation of K+ channels leading to hyperpolarization and relaxation of the smooth muscle cell, and cGMP-dependent protein kinase 1 activation of myosin light chain phosphatase.20 The endothelium participates in regulating blood flow, as laminar sheer stress and mechanical stretch stimulate production of NO and reactive oxygen species (ROS), major determinants of NO bioavailability.21 Endothelial cells are also involved in the retrograde propagation of the vasodilation evoked by neural activity (discussed in the next section).
The cerebral endothelium is the site of the blood–brain barrier (BBB), which restricts the molecular exchange between blood and brain.22 There are three main mechanisms by which the BBB regulates the entry and removal of molecules into and from the brain: tight junctions, active bidirectional transporters, and vesicular transport (transcytosis). Adjacent endothelial cells are linked by junctional complexes (tight junctions) which prevent paracellular passage of blood borne substances. Astrocytic end-feet play a crucial role in the development and maintenance of integrity of the tight junctions. Cerebral endothelial cells express transporters for the bidirectional transfer of molecules across the BBB.22 Transcytosis is normally very limited in cerebral endothelial cells, which has been attributed the expression of the omega-3 fatty acid transporter Mfsd2a.23 Pericytes, mural cells that replace smooth muscle cells at the level of the capillaries, may also be involved in the regulation of transcytosis.24 Additionally, plasmalemma vesicle-associated protein (plvap), normally associated with high vesicular transport in peripheral vessels, has been found to be negatively correlated with BBB development, so that as the BBB is established during development, the expression of plvap decreases.25
Neurovascular coupling
Neural activity increases local CBF through complex mechanisms involving neurons, astrocytes, and vascular cells known as neurovascular coupling.26 The increase in blood flow is coupled to meet the energetic demands of the brain by increasing the delivery of glucose and oxygen, while also clearing the byproducts of cellular metabolic activity.27 Vasoactive mediators are released during brain activity and act on the cerebral vasculature to induce vasodilation at the activated site and also in upstream vascular segments. This requires a concerted effort involving various cell types along the cerebrovascular tree. We will discuss the contributions of these cells to neurovascular coupling only briefly since the topic has been recently reviewed.10
Synaptic activity is the initiating factor triggering neurovascular coupling. Glutamate binding on both NMDA and AMPA receptors on neurons elicits the release of potent vasodilators, including NO and prostanoids, through the activation of calcium-dependent enzymes such as neuronal NOS (nNOS) and cyclooxygenase (COX) 2. Glutamate could also bind to metabotropic glutamate receptors on neighboring astrocytes to induce vasodilation through the release of arachidonic acid metabolites through COX and cytochrome p450 epoxygenase pathways,26 but this mechanism remains controversial.10 Release K+ ions mediated by calcium-activation of BK channels in astrocytic end-feet has also been implicated in neurovascular coupling.26 K+ ions then induce vascular smooth muscle relaxation through Kir channels.10 However, the contribution of this mechanism also remains unclear since deletion of BK channels does not impair neurovascular coupling.10 Interneurons, cells endowed with powerful vasoactive agents, can also have a profound effect on local CBF, and there is evidence that they could contribute to CBF regulation during neural activity.28 In addition, adenosine, a metabolic messenger produced during ATP hydrolysis, also contributes to the increase in CBF during neuronal activity. Finally, changes in O2 and CO2 concentrations in the brain, as a result of cellular aerobic catabolism, are able to modulate cerebral perfusion.26 Therefore, multiple vasoactive factors are involved in linking neural activity to local cerebral perfusion.
Adding to the complexity of neurovascular coupling, the mechanisms involved vary depending on the segment of the cerebral vasculature. The role of astrocytes seems to be restricted to capillaries, and not upstream arterioles, whereas in arterioles, neurovascular coupling seems to be mediated by NMDA receptor-induced neuronal NO production.29 Microvascular endothelial cells may play a unique role in the retrograde propagation of activity-induced neurovascular signals.30 This retrograde propagation of vasodilation is necessary, as upstream vessels must also relax in order to adequately supply increased flow to the vascular segment of the activated regions downstream and prevent “flow steal” from neighboring interconnected vascular territories. The mechanisms of retrograde propagation in cerebral vessels have not been fully elucidated, but in peripheral vessels, it is known to be driven through a fast Ca2+-activated K+ (KCa) channel-mediated component, and a slow NO and prostanoid-mediated component.10,31 In the cerebral vasculature, the endothelial Kir channels have been implicated in mediating the fast component, instead of the KCa channel involved in the peripheral vasculature.32 Thus, neurovascular coupling results from a concerted effort involving various cell types and vasoactive modulation on different segments of the cerebrovascular tree to increase flow in the activated areas.
Cerebral perfusion and cognitive function
Adequate brain perfusion is critical for optimal brain health. Several cellular events occur with gradual reductions in CBF. First, protein synthesis is inhibited at CBF of 35–55 ml/100 g/min.33 Next, metabolism switches to anaerobic glycolysis at CBF at 20–35 ml/100 g/min,34 and overall glucose metabolism declines at CBF 20–25 ml/100 g/min.35 Finally, cellular ion homeostasis fails below 10–12 ml/100 g/min36 leading to irretrievable cell damage and death. Concerning the impact of hypoperfusion on cognition, reduction in CBF from 47 to 37 ml/100 g/min produced a transient deterioration of sustained attention. However, patients whose CBF dropped to 27 ml/100 g/min suffered from impaired sustained attention which persisted until the carotid obstruction was reversed.37 It is important to note that these are average CBF levels, and there was substantial variability within the groups, indicating that these are not hard-drawn lines identifying how much CBF needs to be reduced in order to lead to cognitive impairment.
Cognitive impairment is observed across a wide range of conditions associated with reduced cerebral perfusion. In patients with transient ischemic attack (TIA), acute perfusion deficits in the carotid artery territory leads to decreased cognitive performance after 90 days.38 The link between TIA and subsequent dementia is clinically relevant, since it suggests that TIA may lead to cognitive impairment independently of the increased risk for future strokes. Chronic carotid stenosis caused by atherosclerosis increases the risk of cognitive deterioration.39,40 Furthermore, in heart failure, low ejection fraction is associated with reduced verbal memory,41 as well as impaired cerebral autoregulation, vasomotor tone,42 and reduced CBF.43
Reduced CBF may precede the development of dementia.44 CBF and cerebrovascular reactivity are reduced in patients with mild cognitive impairment.45 In elderly adults, low CBF is associated with faster brain atrophy46 and cognitive deterioration.47 Most importantly, a recent prospective study linked cerebral hypoperfusion with accelerated cognitive decline and an increased risk of dementia in the general population.48 Although previous cross-sectional studies found an association with hypoperfusion in mild cognitive impairment and AD cohorts,49 this is the first study to extend these findings to pre-symptomatic individuals in the general population, thus providing further evidence of the importance of adequate cerebral perfusion for maintenance of brain health. These findings, collectively, highlight the dependence of cognitive function on cardiovascular and cerebrovascular health and on adequate cerebral perfusion.
HTN is a major cause of CBF dysfunction
The brain is a major target of end-organ damage in HTN. HTN has profound effects on the cerebral vasculature leading to both structural and functional alterations affecting the NVU at all levels of the cerebrovascular tree.27 These alterations may promote vascular insufficiency, leading to neuronal dysfunction and cognitive impairment.50 This section will discuss the effects of HTN on the cerebral vasculature. Selected papers describing the alterations in cerebrovascular function induced by HTN are present in Table 1.
Table 1.
Selected examples of cerebrovascular alterations in models of hypertension.
| Model | Cerebrovascular effects | Method | References | ||
|---|---|---|---|---|---|
| Genetic | SHR | Impaired autoregulation | Cranial window (LDF) | Toyoda et al.144 | |
| Impaired neurovascular coupling | Cranial window (LSF) Thin skull (2PLSM) | Calcinaghi et al.96 | |||
| Endothelium-independent responses | |||||
| Enhanced constriction (Ser) | Basilar artery rings | Winquist et al.145 | |||
| Reduced calcium sensitivity | |||||
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Pressurized MCA | Toth et al.146 | |||
| Cranial window (LSF) | Freitas et al.147 | ||||
| SHR-SP | Impaired autoregulation | Cranial window (LDF) | Smeda et al.148 | ||
| Endothelium-independent responses | |||||
| No effect (Adenosine) | Cranial window (pial a. Ø) | Mayhan et al.149 | |||
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Cranial window (pial or basilar a. Ø) | Mayhan et al.149 Kitazono et al.150 | |||
| Impaired dilatation (BK) | Cranial window (pial a. Ø) | Yang et al.151 | |||
| Impaired dilatation (A23187) | Yang et al.152 | ||||
| BPH | Impaired neurovascular coupling | Cranial window (LDF) | Faraco et al.88 | ||
| Endothelium-independent responses | |||||
| No effect (Adenosine) | Cranial window (LDF) | Faraco et al.88 | |||
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Cranial window (LDF) | Faraco et al.88 | |||
|
| |||||
| Salt-sensitive | Dahl SS | Impaired autoregulation | Cranial window (LDF) Pressurized MCA Ø | Fan et al.153 Smeda et al.154 | |
| DOCA-salt | Impaired neurovascular coupling | Cranial window (LDF) | Faraco et al.119 | ||
| Endothelium-independent responses | |||||
| Enhanced constriction (Ser) | Basilar artery strips | Soltis et al.155 | |||
| No effect (Adenosine) | Cranial window (LDF) | Faraco et al.119 | |||
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Cranial window (LDF) | Faraco et al.119 | |||
| Pressurized MCA/Pen. Art. Ø | Matin et al.156 | ||||
|
| |||||
| Ang II | Topical neocortex | Impaired neurovascular coupling | Cranial window (LDF) | Kazama et al.95 | |
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Cranial window (LDF) | Faraco et al.119 | |||
| Acute i.v. | Impaired neurovascular coupling | Cranial window (LDF) | Kazama et al.93 | ||
| Endothelium-independent responses | |||||
| No effect (Adenosine) | Cranial window (LDF) | Girouard et al.72 | |||
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Cranial window (LDF) | Girouard et al.72 | |||
| Carotid rings | Didion et al.157 | ||||
| Impaired dilatation (A23187) | Cranial window (LDF) | Capone et al.98 | |||
| Slow pressor | Impaired neurovascular coupling | Cranial window (LDF) | Kazama et al.95 | ||
| Endothelium-independent responses | |||||
| Enhanced constriction (Ser) | Carotid rings | Ryan et al.158 | |||
| No effect (Adenosine) | Cranial window (LDF) | Capone et al.94 | |||
| Endothelium-dependent responses | |||||
| Impaired dilatation (ACh) | Carotid rings | Schrader et al.77 | |||
| Pressurized basilar a. Ø | Chrissobolis et al.70 | ||||
| Cranial window (LDF) | Capone et al.94 | ||||
| Impaired dilatation (BK) | Cranial window (LDF) | Capone et al.94 | |||
| Impaired dilatation (A23187) | Cranial window (LDF) | Capone et al.94 | |||
| Pressurized basilar a. Ø | Johnson et al.159 | ||||
SHR: spontaneously hypertensive rat; SHR-SP: Stroke-prone SHR; SS: salt sensitive; LDF: laser Doppler flowmetry; LSF: laser speckle flowmetry; 2PLSM: 2-photon laser scanning microscopy; Ser: Serotonin; ACh: Acetylcholine; BK: Bradykinin; A23187: calcium-ionophore; Ø: diameter; MCA: middle cerebral artery.
Structural alterations
Cerebral blood vessels undergo several adaptive changes in response to the increase in arterial pressure in an attempt to protect microvessels from the increased pulsatile and mechanical stress.51 Vascular smooth muscle cells undergo a rearrangement of their organization leading to a decrease in the wall-to-lumen ratio without major changes in cross-sectional area (eutrophic remodeling). On the other hand, smooth muscle cells hypertrophy leads to a drastic increase in cell size in conjunction with the accumulation of extracellular matrix components, ultimately resulting in a decreased lumen size (hypertrophic remodeling). Deposition of collagen and fibronectin causes stiffening of the vessel walls,27 which is associated with clinically silent brain lesions in patients with HTN, and has been identified as a predictor of both stroke and cognitive decline.3 Additionally, HTN-induced vessel hypertrophy leads to increased vascular resistance as the lumen diameter is reduced, and is a potential risk factor for cerebrovascular disease in patients.52 These alterations have a profound effect on cerebral perfusion as they lead to increased vascular resistance and stiffening of arteries, but may also affect autoregulation (see next section). Furthermore, HTN promotes build-up of atherosclerotic plaques in cerebral blood vessels, which may compromise CBF by causing stenosis and nonlaminar flow.53 Atherosclerotic lesions are commonly found at sites of turbulent flow, including the carotid bifurcation,27 potentially due to a combination of free radicals and shear stress leading to vascular damage, inflammation, and immune cell accumulation.53
Small vessel disease (SVD) affecting deep white matter small arteries and arterioles is another major cerebrovascular consequence of HTN.27 SVD is characterized by arteriolosclerosis, marked by loss of smooth muscle cells, narrowing of the lumen, thickening of the vessel wall, and hyaline deposits (lipohyalinosis).54 These microvascular alterations are particularly damaging since: (a) the subcortical white matter is located at the border-zone between two arterial territories, i.e. descending terminal branches from the pial microcirculation and ascending branches arising from the first segment of the middle cerebral artery, and (b) the affected vessels are terminal arterioles with little or no potential for a compensatory collateral flow.27 Therefore, SVD often leads to white matter lesions (white matter disease), which remain the leading cause of cognitive impairment of vascular bases.3
Functional alterations
Cerebrovascular autoregulation
HTN has a detrimental effect on autoregulation, as it shifts the autoregulatory pressure-flow curve to the right. Although the autoregulation shift is intended to protect the microvessels from the mechanical impact of the elevated transmural pressure, this adaptive response may ultimately increase the risk for cerebral hypoperfusion if BP falls.27 The mechanisms underlying the detrimental effects of HTN on cerebrovascular autoregulation have not been fully elucidated, but they may involve both structural and functional factors. While the structural remodeling induced by HTN may impair cerebral autoregulation by altering the mechanical properties of cerebral blood vessels, functional alterations may also play a role. For example, the myogenic tone is increased in several models of HTN, which may results in vasoconstriction and contribute to the shift of the autoregulatory curve.27 The importance of cerebral autoregulation is highlighted by the fact that the magnitude of autoregulatory dysfunction correlates with the severity of periventricular white matter injury.55 Experimental data suggest that these alterations may be modulated by anti-hypertensive medication. Specifically, animal studies have shown that normalizing BP with ACE inhibitors normalizes the lower limit of the CBF autoregulation,56 suggesting that the dysfunction induced by HTN on autoregulation is not permanent and may be amenable to treatment.57
Endothelial function
Major cardiovascular risk factors (HTN, diabetes, hypercholesterolemia, etc.) have the common effect of disrupting endothelial function.58 Brain endothelial dysfunction has been extensively described in HTN. In the cerebral circulation, responses to endothelium-dependent vasodilators appear to be impaired in several models of HTN (see Table 1). One main mechanism is the impairment of NO signaling and bioavailability. Alterations of eNOS may underlie the impairment in various models, varying from decreased expression,59 mislocalization,60 altered phosphorylation61 and eNOS uncoupling.62 eNOS uncoupling refers to a dysfunctional state of the enzyme induced by a lack of cofactors that lead to the production of the free radical superoxide instead of NO.63 One of such cofactors, tetrahydrobiopterin (BH4) may facilitate L-arginine binding to eNOS, enabling electron transfer from the reductase to the oxidase domain, minimizing oxidative decay of the heme prosthetic group,64,65 and stabilizing eNOS dimerization.66 In the presence of ROS, BH4 is oxidized to dihydrobiopterin (BH2) so that the conversion of L-arginine to L-citrulline leads to production of O2ċ− instead of NO.63 Thus, reduced BH4 in HTN may lead to eNOS uncoupling resulting both in reduced NO production and exacerbation of vascular oxidative stress.
A major source of vascular ROS responsible for endothelial dysfunction is NADPH-oxidase.53 This enzyme usually requires assembly of cytosolic and membrane-bound subunits for its catalytic activity, i.e. reduction of molecular oxygen to superoxide.53 The catalytic subunit (Nox) includes five main isoforms (Nox1-5). Nox 1, 2, 4, and 5 have all been reported in brain tissue and blood vessels.67–70 However, single-cell RNA sequencing data indicate that only Nox2 is expressed in brain endothelial and myeloid cells, and, that although Nox1 and Nox4 are expressed in peripheral vessels, neither is detected in brain vascular smooth muscle or endothelial cells.71 In HTN induced by sustained administration of low doses of the octapeptide angiotensin II (Ang II; slow pressor HTN), ROS production occurs predominantly through Nox2, and superoxide is the main contributor to impaired cerebral endothelial function.72 However, Nox1 was found to play small role as well.72 Superoxide could impair NO-dependent responses by scavenging NO, leading to the production of peroxynitrite.73 Indeed, our previous studies have identified an important role for peroxynitrite. Slow pressor Ang II infusion induced 3-nitrityrosine (3-NT) immunoreactivity, a peroxynitrite marker, in cerebral blood vessels, and topical application of peroxynitrite scavengers or decomposition catalysts reduced 3-NT immunoreactivity and prevented the neurovascular dysfunction.74 The source of the superoxide involved in 3-NT production is likely to be Nox2, since mice lacking Nox2 did not show elevated ROS production, 3-NT immunoreactivity, or neurovascular impairment.74,75
Given that HTN is associated with a systemic immune response76 and endothelial cells are in direct contact with the blood circulation, it is conceivable that immune mediators may affect cerebrovascular function. For example, interleukin (IL)-6 is an important mediator of endothelial dysfunction in carotid arteries of mice with HTN induced by Ang II.77 On the other hand, the anti-inflammatory cytokine IL-10 protects endothelial function in carotid arteries following Ang II HTN.78 Recently, IL-17 has been implicated in both CNS and cardiovascular diseases, including Ang II HTN.79 Increased levels of IL-17 have been observed in Ang II HTN79 and the production of IL-17 is elevated in human CD4+ T cells of hypertensive patients.80 Chronic infusion of IL-17 in mice increased BP and caused endothelial dysfunction in aortic rings.81 Additionally, IL-17 knockout mice79 and germ-free mice lacking the IL-17 response to Ang II infusion82 are protected from endothelial dysfunction. However, in the mouse cerebral microcirculation, IL17 infusion induces endothelial dysfunction in the absence of HTN.9 Therefore, the role of IL-17 in the cerebral vasculature during HTN remains to be determined.
BBB
HTN has profound effects on BBB permeability. Several animal models of HTN are associated with disruption of the BBB.83,84 Disruption of the BBB has also been observed in patients with HTN85 and in white matter damage associated with SVD.86 The cellular and molecular mechanisms underlying the disruption of the BBB in HTN have not been elucidated. Early studies in which acute changes in BP were induced in laboratory animals suggested a role of mechanical effects of elevated pressure on cerebral blood vessels.87 However, more recent studies in models of sustained HTN produced by chronic Ang II administration have shown that the breakdown of the BBB is independent of the BP elevation.88 A loss of tight junction components, including occludin and claudin-5, has been described, but only in aged mice.89
Pericytes have been implicated in BBB alterations in models of AD,24 and may also be involved in HTN. Pericyte migration and contraction are stimulated by Ang II,90 suggesting that they respond to hypertensive stimuli. Human brain pericytes upregulate Nox4 expression in response to Ang II stimulation,91 suggesting that these cells may be a contributing cellular source of ROS in human HTN. One study indicated an increase in “granular” pericyte size and activity accompanied by a degeneration of “filamentous” pericytes during HTN in stroke-prone spontaneously hypertensive rats (SHR-SP).92 However, recent single-cell RNA sequencing studies have failed to molecularly identify pericyte subtypes in the brain microvasculature.71 Therefore, the functional implication of different pericyte morphology remains unclear. Of note, the RNAseq studies mentioned above were performed in normal mice and the possibility that HTN alters pericyte structure and function cannot be ruled out, and this presents a new opportunity to identify molecular and genetic changes in individual vascular cell types in HTN.
Neurovascular coupling
Neurovascular coupling was first shown to be attenuated in mice with acute or slow pressor Ang II HTN.93 Interestingly, the cerebrovascular dysfunction in slow-pressor Ang II HTN actually precedes the onset of HTN and persists following normalization of BP at the end of infusion.94 Attenuation of neurovascular coupling could also be induced by topical neocortical application of Ang II,95 indicating that it was not a result of the BP elevation. In support of this hypothesis, sustained elevation of BP with the alpha-adrenergic agonist phenylephrine did not induce a dysfunctional response.95 Similarly, preventing the BP elevation during Ang II infusion or administration of Ang II at subpressor doses significantly attenuated the neurovascular coupling response during whisker stimulation.94 Thus, in this model of HTN, the dysfunction can be attributed to Ang II signaling, rather than the BP elevation. However, as described in the previous section, chronic effects of BP elevation on arterial structure cannot be discounted. Neurovascular coupling to whisker stimulation is also impaired in genetic models of lifelong HTN, including blood pressure high (BPH) mice88 and spontaneously hypertensive rats (SHR),96 which also exhibit elevated levels of circulating Ang II. Importantly, neurovascular dysfunction has also been reported in human HTN. Hypertensive patients showed an attenuated increase in CBF in response to brain activation during memory tasks, compared to normotensive subjects.97
The mechanisms underlying the effects of Ang II HTN on neurovascular coupling are not completely clear. The response is not explained through direct effects of Ang II on neuronal activity, as Ang II does not affect the shape or amplitude of the field potentials induced by whisker stimulation when delivered either acutely95 or chronically.94 A central component to the neurovascular dysfunction has been shown by studies indicating that chronic Ang II infusion acts on the subfornical organ (SFO), one of the circumventricular organs, leading to activation of hypothalamic pathways releasing vasopressin and inducing expression of the potent vasoconstrictor endothelin-1 in cerebral arterioles.98 In this model, AT1R inhibition by losartan superfusion over the neocortex improves neurovascular dysfunction only partially, indicating that other mechanisms are also at play.98 Scavenging radicals in the SFO with CuZnSOD viral gene transfer prevents the neurovascular dysfunction, indicating the critical role of ROS in the SFO.98 Topical application of an ETA receptor blocker partially improves the neurovascular dysfunction and the residual deficit is completely rescued by losartan, implicating both local AT1R and ETA receptors in its mechanisms98 (Figure 2). Thus, unlike acute administration of pressor doses or neocortical application of Ang II, SFO activation of central pathways is necessary for the neurovascular dysfunction induced by slow-pressor Ang II HTN. In addition, a contribution of COX-1 mediated production of PGE2 acting on EP1 receptors has also been reported in this model.99
Figure 2.
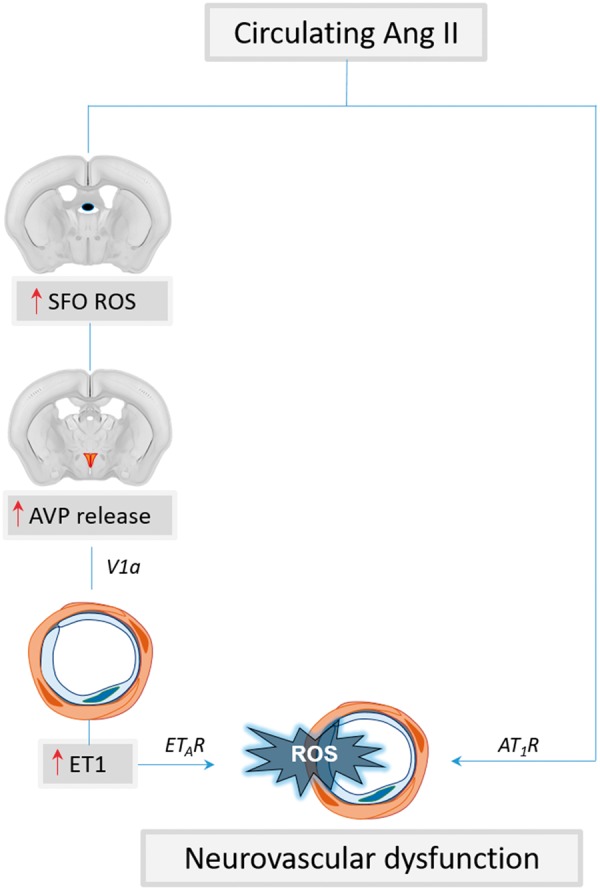
Mechanisms of slow-pressor Ang II HTN-induced neurovascular dysfunction. Circulating Ang II activates ROS production in the subfornical organ (SFO) leading to increased vasopressin (AVP) release from the hypothalamic paraventricular nucleus. AVP, in turn, leads to upregulation of endothelin-1 (ET1) in cerebral arterioles. ET1 and Ang II contribute to vascular oxidative stress and neurovascular dysfunction through ETA and AT1 receptors, respectively.
Perivascular macrophages
As discussed in the previous section, vascular oxidative stress is ultimately responsible for neurovascular dysfunction in models of Ang II HTN, but the cellular source(s) of the ROS remained unclear. Although capable of producing ROS,100 smooth muscle cells do not seem to be a major source of oxidative stress in the cerebral circulation.101 Furthermore, the ability of cerebral endothelial cells to produce toxic amounts of ROS is relatively limited compared to “professional” ROS producing cells such as macrophages.102
Recent data have revealed that perivascular macrophages (PVMs) are the major source of ROS mediating the neurovascular dysfunction. Brain PVM are innate immune cells closely apposed to cerebral arterioles and have emerged as critical constituents of the NVU.103 PVM and meningeal macrophages represent the bulk of macrophages in the normal brain. PVM reside in the Virchow-Robin space, delimited by the glia limitans and the vascular basement membrane, and are attached to the outer wall of intracerebral arteries and veins. They are distinct from other perivascular cells and microglia for their immune phenotype (CD45high, Iba1low, CD206+, CD163+) and propensity to phagocytosis. Their origin and function has been recently reviewed.103
In addition to their role in immune surveillance, recent data indicate that PVM are a major source of ROS with a negative impact on cerebrovascular function.88,103 Due to their myeloid origin, PVM express functional AT1R and have the potential to produce large amounts of ROS through Nox2.104 This is of interest, because, as discussed above, cerebrovascular dysfunction in slow-pressor Ang II HTN is mediated by vascular oxidative stress from a Nox2-containing NADPH-oxidase.93,98 Therefore, we recently tested the hypothesis that PVM are a source of ROS mediating the cerebrovascular and cognitive dysfunction induced by Ang II HTN. We found that in Ang II HTN, AT1 receptors on PVM mediate ROS production through Nox2-derived radicals leading to impairment in endothelium-dependent vasodilation and neurovascular coupling.88 This is in agreement with data showing that PVM depletion with clodronate in SHRSP improves the remodeling and endothelium-dependent vasodilation of middle cerebral artery.105 PVM depletion also improved cognitive function assessed in BPH mice, a model of lifelong HTN.88 These data indicate that PVM are a critical source of ROS responsible for the deleterious neurovascular and cognitive effects of HTN (Figure 3). Given that the cerebral endothelium, but not arteriolar smooth muscle, also express AT1R and Nox2,71,93 it is possible that these cells play a contributing role to the radical production during Ang II HTN. However, their contribution is assumed to be smaller because single-cell RNA sequencing data indicate that the expression of Nox2 in endothelial cells is limited compared to that in microglia and PVM.71 Furthermore, the observation that removal or genetic modification of PVM completely abolishes vascular ROS production,88 suggests that the endothelium and vascular smooth muscle cannot be the primary source of ROS. In contrast, in the HTN induced by acute Ang II administration, PVM do not participate in the neurovascular dysfunction9 implicating the endothelium in the ROS production (Figure 3).
Figure 3.

PVM mediate cerebrovascular dysfunction in slow-pressor Ang II HTN but not acute Ang II HTN. In slow-pressor Ang II HTN and BPH mice (left), circulating Ang II crosses the BBB and acts on AT1R on PVM to increase production of ROS by activating a Nox2-containing NADPH oxidase. Nox2-derived radicals, in turn, cause neurovascular dysfunction and cognitive deficits. ET1 also plays a role in the dysfunction (see Figure 2), but it remains unclear how this peptide contributes to ROS production in PVM. In contrast, in acute administration of pressor doses of Ang II (right), circulating Ang II activates AT1R and Nox2 on endothelial cells to increase ROS production and induce neurovascular dysfunction.
Dietary salt, cerebrovascular dysfunction and cognitive impairment
A diet rich in salt has been linked to increased incidence of cerebrovascular diseases, an effect first attributed to the elevation in BP observed with high-salt intake.106 However, subsequent studies revealed that high-salt intake had detrimental effects independent of BP.107 Thus, dietary salt is now recognized as an independent risk factor for stroke and dementia.7,8
Effects of salt on the cerebral vasculature
High dietary salt can result in arterial stiffness in animal models of HTN, an effect independent of the BP elevation.108 In peripheral vessels, this effect has been attributed to salt-induced increases in the pro-fibrotic effects of transforming growth factor beta (TGFβ).109,110 In agreement with these experimental data, arterial stiffness was found to be lower in patients on a low-salt diet versus normal-salt diet.111 Furthermore, reducing sodium consumption in HTN patients lowered stiffness in large peripheral arteries.112 Whether a similar reversal occurs in cerebral vessels remains to be established. High-salt diet also results in functional alterations in systemic vessels. For example, the mesenteric arteries of rats fed a high-salt diet show enhanced vasoconstriction to norepinephrine113 and effect attributed to an increase in the contractile properties of the vascular smooth muscle.114
Relatively little is known on the effect of dietary salt on the cerebral vasculature. Rats fed a high-salt diet have an impaired cerebral smooth muscle cell response to prostacyclin.115 Following short-term high-salt diet, rat pial arterioles exhibit impaired responses to ACh and to the prostaglandin I2 receptor agonist iloprost.116 A subsequent study in the same model found that the impaired ACh-induced vasodilation of MCA could be attributed to the suppression of peripheral Ang II and brain Cu/Zn SOD.117 Additionally, high-salt intake in rats abolished ADP-induced vasodilation,118 suggesting the involvement of eNOS-independent mechanisms. Neurovascular coupling during whisker stimulation and endothelium-dependent responses are impaired with a model of salt retention associated with HTN (DOCA-salt model),119 but the BBB is not affected.120 Similarly, chronic administration of a high-salt diet (8% NaCl) does not alter the BBB in normal mice,9 but it enhances BBB dysfunction in SHR-SP121 which already have BBB alterations at baseline.122 Therefore, the evidence suggest that dietary salt may induce endothelial dysfunction without affecting the BBB, unless there is pre-existing vascular damage, such as in SHR-SP.
Dietary salt, endothelial dysfunction and cognitive impairment
High-salt intake has long been associated with elevated BP, and the deleterious cardiovascular effects of salt-rich diets have traditionally been attributed to HTN.123 However, epidemiological data have unveiled a link between dietary salt, stroke, dementia and white matter damage independently of HTN.124 In a recent study, we examined the effect of dietary salt on cerebrovascular function and cognition. We found that high-salt diet (4–8% NaCl, 8–16 times the salt content of the normal mouse chow) for two to six months leads to cerebral endothelial dysfunction in the absence of HTN, without affecting neurovascular coupling.9 Uncertainties concerning basal salt requirements and consumption in humans notwithstanding,125 a 4–8% salt diet approaches the highest levels of estimated salt consumption in humans.126 Dietary salt-induced endothelial dysfunction is associated with a global reduction in CBF and cognitive impairment, attesting to the key role of the cerebrovascular endothelium in maintaining cognitive health. Therefore, this study provides evidence that endothelial dysfunction is sufficient to produce cognitive impairment in the absence of neurovascular coupling dysfunction. However, the relative contribution of neurovascular coupling and endothelial dysfunction to cognitive health in humans as in animal models is not clear.
We then explored the potential mechanisms of the cerebrovascular and cognitive effect of salt. In humans as in mice, a high-salt diet acts on the gut adaptive immune system leading to expansion of T-helper lymphocytes producing IL-17 (Th17).127–129 We found that this adaptive immune response induces the endothelial dysfunction and cognitive deficits. In particular, the Th17 response led to an increase in circulating IL-17, which, in turn, induced inhibitory eNOS phosphorylation (at Thr495) and reduced the bioavailability of endothelial NO. The inhibitory phosphorylation is mediated by Rho-kinase (ROCK), and ROCK inhibition prevented high-salt diet-induced Thr495 phosphorylation, cerebrovascular dysfunction, and cognitive impairment.9 The key role of IL-17 was confirmed by the observation that increasing circulating IL-17 to the same level reached with the high-salt diet reproduced the neurovascular and cognitive dysfunction induced by high-salt diet.9 Additionally, mice lacking IL-17 or treated with IL-17 neutralizing antibodies were protected from the detrimental effects of high dietary salt.9 Attesting to the cellular specificity of the neurovascular alterations induced by high salt, PVM, which are critical for the cerebrovascular effects of Ang II HTN, are not involved in the mechanisms of the endothelial dysfunction.9 These findings unveil a new gut-brain axis in which an adaptive immune response initiated in the gut by dietary salt acts on the cerebral endothelium to induce NO deficit and, consequently, cognitive impairment (Figure 4). The pathways downstream of endothelial NO leading to cognitive impairment remain to be established and are presently being investigated.
Figure 4.
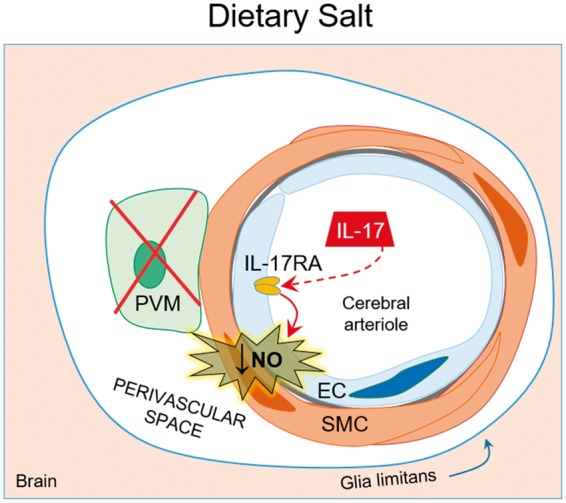
Dietary salt induces endothelial dysfunction and cognitive deficits. High dietary salt stimulates Th17 polarization in the gut leading to increased circulating IL-17. IL-17 acts on the cerebral endothelium to induce inhibitory phosphorylation of eNOS through Rho-kinase, thus reducing NO production and bioavailability. The resulting endothelial dysfunction in the cerebral vasculature is associated with cognitive impairment. Remarkably, neurovascular coupling is not affected. Of note, PVM do not play a role in the cerebrovascular dysfunction of dietary salt.
Clinical considerations
HTN is by far the most important modifiable risk factor for cerebrovascular disease leading to stroke and dementia.50 Mounting evidence has also linked HTN to the risk of AD, the leading cause of dementia in the elderly.130 An important consideration is that approximately 50% of clinically diagnosed AD feature vascular pathology (Figure 1), thus targeting vascular risk factors as a preventive measure for AD is increasingly important. Midlife HTN increases the risk of clinical diagnosis of AD later in life131 and at autopsy the brains of hypertensive patients have more amyloid plaques and neurofibrillary tangles, pathological hallmarks of AD, than normotensive subjects.132 Recent imaging studies using markers of amyloid or tau and positron emission tomography have shown that HTN increases amyloid and tau deposition.133–135 Furthermore, arterial stiffness, a correlate of HTN, is also associated with greater accumulation of amyloid markers.136 These findings have raised the possibility that HTN promotes AD pathology, but the mechanisms by which HTN and other vascular risk factors may exert this effect remain to be established. Experimental studies have shown that slow-pressor Ang II HTN promotes amyloid-β deposition in mouse models of amyloid accumulation, an effect due to enhancement of amyloid-β cleavage from the amyloid precursor protein through the enzyme β-secretase.119 Similarly, experimental HTN has been shown to induce tau phosphorylation,137,138 but the mechanism of the effects is not entirely clear. Furthermore, the role of AD pathology in the cognitive deficits of HTN in animal models as in humans remains to be established. Highlighting the complexity of the link between HTN and AD is the clinical observation that the association between these conditions is age dependent, such that later in life HTN no longer correlates with AD risk.139 This is probably due to the fact that, as dementia develops, reduced activity, nutritional factors and possibly central autonomic dysfunction lead to BP lowering.
Given that many of cardiovascular risk factors have recently also been linked to cognitive impairment, cardiovascular health is now recognized as a key requirement for optimal brain health.140 Indeed, the recommendations for promoting and maintaining optimal brain health are similar to those proposed for cardiovascular health, and include a combination of physical exercise, healthy diet (with reduced sodium consumption), and control of vascular risk factors (such as HTN) as a strategy.140 Physical exercise reduces the risk of cardiovascular disease and improves endothelial function, thus potentially reducing the risk for dementia.58 Importantly, reducing sodium consumption remains an important recommendation for BP and cardiovascular risk management.123
Although anti-hypertensive medications remain the cornerstone of therapy, definitive evidence that HTN treatment reduces the risk of dementia is lacking.50 Several observations studies have suggested that treatment of HTN reduced the risk of dementia,50 but only one double-blinded, randomized and placebo controlled clinical trial has thus far shown a clear benefit of treatment of HTN on subsequent cognitive impairment,141 while other trials have not.50 A major problem has been that midlife HTN increases the risk of late-life dementia decades later, and studies to date have lacked the length of follow up or the proper cognitive assessment to assess the impact of HTN treatment on cognitive outcomes. Another challenge has been that it has been difficult to differentiate between direct effects of BP lowering on the development of dementia, and indirect effects resulting from the associated reduction in the risk of stroke, in itself a significant cause of cognitive impairment (post-stroke dementia).50
A related question is whether certain classes of anti-hypertensive agents are superior in terms of dementia prevention. In animal models, angiotensin receptor blockers have been found to be neuroprotective.142 However, strong evidence is lacking in patients with HTN. Although some studies have suggested certain drug classes superiority,143 their findings are weakened by underpowered design and lack of equivalent cognitive end points.50 Therefore, additional double-blinded clinical trials with extended follow up and assessment of cognitive outcomes are needed to address these critical questions. In this regard, results from the SPRINT-MIND trial are eagerly awaited in the hope that they may provide guidance on the use of antihypertensive medications to prevent that deleterious effects of HTN on cognition.
Conclusions
We have examined the damaging effects of HTN and dietary salt on the brain, focusing on microvascular dysfunction and its negative impact on cognition. The evidence suggests that HTN and dietary salt alter cerebrovascular structure and function profoundly, resulting in vascular insufficiency and cognitive impairment. Vascular oxidative stress has long been known to be a major culprit, especially in HTN, but the sources and targets of ROS remain to be clearly identified. While PVM have emerged as a previously unappreciated cellular source of ROS in the vasculopathy of Ang II HTN, the potential contribution of other vascular cells remains unclear. How radicals interact with cerebrovascular cells to induce vascular and neuronal dysfunction also remains to be fully established. Reduced NO bioavailability may play a role, but considering the multitude of neurovascular mediators interacting with the cerebral vasculature, other agents are also likely to be involved.
A remarkable specificity in the cellular bases of vascular dysfunction has also emerged. For example, PVM are critical for the neurovascular and cognitive dysfunction in HTN, but are not involved in the cerebrovascular effects of dietary salt, which is exclusively mediated by endothelial cell dysfunction (Figures 3 and 4). Single cells RNAseq studies of cerebrovascular cells in health and disease would provide important clues to unveil the earliest disease-linked molecular change in vascular cells that may yield novel diagnostic and therapeutic insights.
The downstream events linking the vascular dysfunction to the synaptic dysfunction underlying cognitive impairment remain to be defined. How does alteration of specific vascular cells drive neuronal dysfunction in brain regions critical for cognition? Is a mismatch between energy supply and demands due to hemodynamic insufficiency the sole factor? Or, are there yet-undiscovered neurovascular links through which vascular cells can directly influence neuronal function? The role of vascular growth factors in neuronal development and survival is well described, but are there also disease specific processes at play?
These are some of the outstanding questions that remain to be addressed. Rapid advances in neurovascular biology and a host of new methodological approaches and disease models promise to expand our understanding of these damaging processes and to identify predictive biomarkers and viable therapeutic targets. These preclinical studies are essential for subsequent clinical efforts to better define the natural history of the pathogenic impact of HTN and dietary salt on cognitive function and develop disease-modifying treatments for age-related dementia, one of the greatest public health challenges of our times.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the following grants: 17POST33370064 (MMS), R01 NS37853, R37 NS89323, and R01 NS100447 (CI). Support from the Feil Family Foundation is gratefully acknowledged.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Gale SA, Acar D, Daffner KR. Dementia. Am J Med Epub ahead of print 6 February 2018. DOI: 10.1016/j.amjmed.2018.01.022. [DOI] [PubMed] [Google Scholar]
- 2.WHO. Global action plan on the public health response to dementia 2017 – 2025. [ISBN: 978-92-4-151348-7], Geneva: World Health Organization, 2017. [Google Scholar]
- 3.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011; 42: 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azarpazhooh MR, Avan A, Cipriano LE, et al. Concomitant vascular and neurodegenerative pathologies double the risk of dementia. Alzheimers Dement 2018; 14: 148–156. [DOI] [PubMed] [Google Scholar]
- 5.Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 2010; 120: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakris G, Sorrentino M. Redefining hypertension – assessing the new blood-pressure guidelines. N Engl J Med 2018; 378: 497–499. [DOI] [PubMed] [Google Scholar]
- 7.Heye AK, Thrippleton MJ, Chappell FM, et al. Blood pressure and sodium: association with MRI markers in cerebral small vessel disease. J Cereb Blood Flow Metab 2016; 36: 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strazzullo P, D'Elia L, Kandala NB, et al. Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ 2009; 339: b4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faraco G, Brea D, Garcia-Bonilla L, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci 2018; 21: 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 2017; 96: 17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cipolla MJ. The cerebral circulation, 2nd ed San Rafael (CA): Morgan & Claypool Life Sciences, 2016. [PubMed] [Google Scholar]
- 12.Koller A, Toth P. Contribution of flow-dependent vasomotor mechanisms to the autoregulation of cerebral blood flow. J Vasc Res 2012; 49: 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol 1902; 28: 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson EC, Wang Z, Britz G. Regulation of cerebral blood flow. Int J Vasc Med 2011; 2011: 823525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brayden JE, Earley S, Nelson MT, et al. Transient receptor potential (TRP) channels, vascular tone and autoregulation of cerebral blood flow. Clin Exp Pharmacol Physiol 2008; 35: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubanyi GM, Freay AD, Kauser K, et al. Mechanoreception by the endothelium: mediators and mechanisms of pressure- and flow-induced vascular responses. Blood Vessels 1990; 27: 246–257. [DOI] [PubMed] [Google Scholar]
- 17.Andresen J, Shafi NI, Bryan RM., Jr Endothelial influences on cerebrovascular tone. J Appl Physiol 2006; 100: 318–327. [DOI] [PubMed] [Google Scholar]
- 18.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 2006; 26: 1215–1225. [DOI] [PubMed] [Google Scholar]
- 19.Kraehling JR, Sessa WC. Contemporary approaches to modulating the nitric oxide-cGMP pathway in cardiovascular disease. Circ Res 2017; 120: 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvajal JA, Germain AM, Huidobro-Toro JP, et al. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell 2000; 184: 409–420. [DOI] [PubMed] [Google Scholar]
- 21.Harrison DG, Widder J, Grumbach I, et al. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J Intern Med 2006; 259: 351–363. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Z, Nelson AR, Betsholtz C, et al. Establishment and dysfunction of the blood-brain barrier. Cell 2015; 163: 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Zvi A, Lacoste B, Kur E, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 2014; 509: 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci 2016; 19: 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallmann R, Mayer DN, Berg EL, et al. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn 1995; 202: 325–332. [DOI] [PubMed] [Google Scholar]
- 26.Attwell D, Buchan AM, Charpak S, et al. Glial and neuronal control of brain blood flow. Nature 2010; 468: 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension 2013; 62: 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lecrux C, Hamel E. Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philos Trans R Soc Lond B Biol Sci 2016; 371: pii: 20150350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mishra A, Reynolds JP, Chen Y, et al. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci 2016; 19: 1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen BR, Kozberg MG, Bouchard MB, et al. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc 2014; 3: e000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tallini YN, Brekke JF, Shui B, et al. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res 2007; 101: 1300–1309. [DOI] [PubMed] [Google Scholar]
- 32.Longden TA, Dabertrand F, Koide M, et al. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 2017; 20: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mies G, Ishimaru S, Xie Y, Seo K, et al. Ischemic thresholds of cerebral protein synthesis and energy state following middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab 1991; 11: 753–761. [DOI] [PubMed] [Google Scholar]
- 34.Hossmann KA, Fischer M, Bockhorst K, et al. NMR imaging of the apparent diffusion coefficient (ADC) for the evaluation of metabolic suppression and recovery after prolonged cerebral ischemia. J Cereb Blood Flow Metab 1994; 14: 723–731. [DOI] [PubMed] [Google Scholar]
- 35.Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part I: pathophysiology. J Neurosurg 1992; 77: 169–184. [DOI] [PubMed] [Google Scholar]
- 36.Harris RJ, Symon L, Branston NM, et al. Changes in extracellular calcium activity in cerebral ischaemia. J Cereb Blood Flow Metab 1981; 1: 203–209. [DOI] [PubMed] [Google Scholar]
- 37.Marshall RS, Lazar RM, Pile-Spellman J, et al. Recovery of brain function during induced cerebral hypoperfusion. Brain 2001; 124(Pt 6): 1208–1217. [DOI] [PubMed] [Google Scholar]
- 38.Bivard A, Lillicrap T, Marechal B, et al. Transient ischemic attack results in delayed brain atrophy and cognitive decline. Stroke 2018; 49: 384–390. [DOI] [PubMed] [Google Scholar]
- 39.Balestrini S, Perozzi C, Altamura C, et al. Severe carotid stenosis and impaired cerebral hemodynamics can influence cognitive deterioration. Neurology 2013; 80: 2145–2150. [DOI] [PubMed] [Google Scholar]
- 40.Gardener H, Caunca MR, Dong C, et al. Ultrasound markers of carotid atherosclerosis and cognition: the Northern Manhattan study. Stroke 2017; 48: 1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Festa JR, Jia X, Cheung K, et al. Association of low ejection fraction with impaired verbal memory in older patients with heart failure. Arch Neurol 2011; 68: 1021–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erkelens CD, van der Wal HH, de Jong BM, et al. Dynamics of cerebral blood flow in patients with mild non-ischaemic heart failure. Eur J Heart Fail 2017; 19: 261–268. [DOI] [PubMed] [Google Scholar]
- 43.Roy B, Woo MA, Wang DJJ, et al. Reduced regional cerebral blood flow in patients with heart failure. Eur J Heart Fail 2017; 19: 1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hays CC, Zlatar ZZ, Wierenga CE. The utility of cerebral blood flow as a biomarker of preclinical Alzheimer's Disease. Cell Mol Neurobiol 2016; 36: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beishon L, Haunton VJ, Panerai RB, et al. Cerebral hemodynamics in mild cognitive impairment: a systematic review. J Alzheimers Dis 2017; 59: 369–385. [DOI] [PubMed] [Google Scholar]
- 46.Zonneveld HI, Loehrer EA, Hofman A, et al. The bidirectional association between reduced cerebral blood flow and brain atrophy in the general population. J Cereb Blood Flow Metab 2015; 35: 1882–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benedictus MR, Leeuwis AE, Binnewijzend MA, et al. Lower cerebral blood flow is associated with faster cognitive decline in Alzheimer's disease. Eur Radiol 2017; 27: 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolters FJ, Zonneveld HI, Hofman A, et al. Cerebral perfusion and the risk of dementia: a population-based study. Circulation 2017; 136: 719–728. [DOI] [PubMed] [Google Scholar]
- 49.Johnson NA, Jahng GH, Weiner MW, et al. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 2005; 234: 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iadecola C, Yaffe K, Biller J, et al. Impact of hypertension on cognitive function: a scientific statement from the American Heart Association. Hypertension 2016; 68: e67–e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Avolio A, Kim MO, Adji A, et al. Cerebral haemodynamics: effects of systemic arterial pulsatile function and hypertension. Curr Hypertens Rep 2018; 20: 20. [DOI] [PubMed] [Google Scholar]
- 52.Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol 2011; 300: H1566–H1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res 2006; 71: 247–258. [DOI] [PubMed] [Google Scholar]
- 54.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- 55.Matsushita K, Kuriyama Y, Nagatsuka K, et al. Periventricular white matter lucency and cerebral blood flow autoregulation in hypertensive patients. Hypertension 1994; 23: 565–568. [DOI] [PubMed] [Google Scholar]
- 56.Vorstrup S, Barry DI, Jarden JO, et al. Chronic antihypertensive treatment in the rat reverses hypertension-induced changes in cerebral blood flow autoregulation. Stroke 1984; 15: 312–318. [DOI] [PubMed] [Google Scholar]
- 57.Strandgaard S, Paulson OB. Cerebral blood flow in untreated and treated hypertension. Neth J Med 1995; 47: 180–184. [DOI] [PubMed] [Google Scholar]
- 58.Trigiani LJ, Hamel E. An endothelial link between the benefits of physical exercise in dementia. J Cereb Blood Flow Metab 2017; 37: 2649–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamakawa H, Jezova M, Ando H, et al. Normalization of endothelial and inducible nitric oxide synthase expression in brain microvessels of spontaneously hypertensive rats by angiotensin II AT1 receptor inhibition. J Cereb Blood Flow Metab 2003; 23: 371–380. [DOI] [PubMed] [Google Scholar]
- 60.Gerzanich V, Ivanova S, Zhou H, et al. Mislocalization of eNOS and upregulation of cerebral vascular Ca2+ channel activity in angiotensin-hypertension. Hypertension 2003; 41: 1124–1130. [DOI] [PubMed] [Google Scholar]
- 61.Kang KT, Sullivan JC, Spradley FT, et al. Antihypertensive therapy increases tetrahydrobiopterin levels and NO/cGMP signaling in small arteries of angiotensin II-infused hypertensive rats. Am J Physiol Heart Circ Physiol 2011; 300: H718–H724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong YF, Kataoka K, Tokutomi Y, et al. Beneficial effects of combination of valsartan and amlodipine on salt-induced brain injury in hypertensive rats. J Pharmacol Exp Ther 2011; 339: 358–366. [DOI] [PubMed] [Google Scholar]
- 63.Yang YM, Huang A, Kaley G, et al. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol 2009; 297: H1829–H1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A 1998; 95: 9220–9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem 2001; 276: 14533–14536. [DOI] [PubMed] [Google Scholar]
- 66.Cai S, Khoo J, Mussa S, et al. Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia 2005; 48: 1933–1940. [DOI] [PubMed] [Google Scholar]
- 67.Miller AA, Drummond GR, Schmidt HH, et al. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res 2005; 97: 1055–1062. [DOI] [PubMed] [Google Scholar]
- 68.Ago T, Kitazono T, Kuroda J, et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke 2005; 36: 1040–1046. [DOI] [PubMed] [Google Scholar]
- 69.Hashad AM, Sancho M, Brett SE, et al. Reactive oxygen species mediate the suppression of arterial smooth muscle T-type Ca(2+) channels by angiotensin II. Sci Rep 2018; 8: 3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jha JC, Watson AMD, Mathew G, et al. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin Sci 2017; 131: 981–990. [DOI] [PubMed] [Google Scholar]
- 71.Vanlandewijck M, He L, Mae MA, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018; 554: 475–480. [DOI] [PubMed] [Google Scholar]
- 72.Chrissobolis S, Banfi B, Sobey CG, et al. Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol 2012; 113: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Silva TM, Faraci FM. Effects of angiotensin II on the cerebral circulation: role of oxidative stress. Front Physiol 2012; 3: 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Girouard H, Park L, Anrather J, et al. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol 2007; 27: 303–309. [DOI] [PubMed] [Google Scholar]
- 75.Girouard H, Park L, Anrather J, et al. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol 2006; 26: 826–832. [DOI] [PubMed] [Google Scholar]
- 76.Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med 2018; 215: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schrader LI, Kinzenbaw DA, Johnson AW, et al. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol 2007; 27: 2576–2581. [DOI] [PubMed] [Google Scholar]
- 78.Didion SP, Kinzenbaw DA, Schrader LI, et al. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension 2009; 54: 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Madhur MS, Lob HE, McCann LA, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010; 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Itani HA, McMaster WG, Jr., Saleh MA, et al. Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension 2016; 68: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nguyen H, Chiasson VL, Chatterjee P, et al. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 2013; 97: 696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karbach SH, Schonfelder T, Brandao I, et al. Gut microbiota promote angiotensin ii-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 2016; 5: pii: e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rosenberg GA. Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab 2012; 32: 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huisa BN, Caprihan A, Thompson J, et al. Long-term blood-brain barrier permeability changes in binswanger disease. Stroke 2015; 46: 2413–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dankbaar JW, Hom J, Schneider T, et al. Age- and anatomy-related values of blood-brain barrier permeability measured by perfusion-CT in non-stroke patients. J Neuroradiol 2009; 36: 219–227. [DOI] [PubMed] [Google Scholar]
- 86.Rosenberg GA. Binswanger's disease: biomarkers in the inflammatory form of vascular cognitive impairment and dementia. J Neurochem 2018; 144: 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mayhan WG, Faraci FM, Heistad DD. Disruption of the blood-brain barrier in cerebrum and brain stem during acute hypertension. Am J Physiol 1986; 251(6 Pt 2): H1171–1175. [DOI] [PubMed] [Google Scholar]
- 88.Faraco G, Sugiyama Y, Lane D, et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest 2016; 126: 4674–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Toth P, Tucsek Z, Sosnowska D, et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab 2013; 33: 1732–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matsugi T, Chen Q, Anderson DR. Contractile responses of cultured bovine retinal pericytes to angiotensin II. Arch Ophthalmol 1997; 115: 1281–1285. [DOI] [PubMed] [Google Scholar]
- 91.Kuroda J, Ago T, Nishimura A, et al. Nox4 is a major source of superoxide production in human brain pericytes. J Vasc Res 2014; 51: 429–438. [DOI] [PubMed] [Google Scholar]
- 92.Tagami M, Nara Y, Kubota A, et al. Ultrastructural changes in cerebral pericytes and astrocytes of stroke-prone spontaneously hypertensive rats. Stroke 1990; 21: 1064–1071. [DOI] [PubMed] [Google Scholar]
- 93.Kazama K, Anrather J, Zhou P, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res 2004; 95: 1019–1026. [DOI] [PubMed] [Google Scholar]
- 94.Capone C, Faraco G, Park L, et al. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol Heart Circ Physiol 2011; 300: H397–H407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kazama K, Wang G, Frys K, et al. Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol 2003; 285: H1890–H1899. [DOI] [PubMed] [Google Scholar]
- 96.Calcinaghi N, Wyss MT, Jolivet R, et al. Multimodal imaging in rats reveals impaired neurovascular coupling in sustained hypertension. Stroke 2013; 44: 1957–1964. [DOI] [PubMed] [Google Scholar]
- 97.Jennings JR, Muldoon MF, Ryan C, et al. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology 2005; 64: 1358–1365. [DOI] [PubMed] [Google Scholar]
- 98.Capone C, Faraco G, Peterson JR, et al. Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci 2012; 32: 4878–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Capone C, Faraco G, Anrather J, et al. Cyclooxygenase 1-derived prostaglandin E2 and EP1 receptors are required for the cerebrovascular dysfunction induced by angiotensin II. Hypertension 2010; 55: 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res 2006; 71: 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park L, Anrather J, Forster C, et al. Abeta-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab 2004; 24: 334–342. [DOI] [PubMed] [Google Scholar]
- 102.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007; 87: 245–313. [DOI] [PubMed] [Google Scholar]
- 103.Faraco G, Park L, Anrather J, et al. Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med 2017; 95: 1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wenzel P, Knorr M, Kossmann S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011; 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- 105.Pires PW, Girgla SS, McClain JL, Kaminski NE, van Rooijen N, Dorrance AM. Improvement in middle cerebral artery structure and endothelial function in stroke-prone spontaneously hypertensive rats after macrophage depletion. Microcirculation 2013; 20: 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zemel MB, Sowers JR. Salt sensitivity and systemic hypertension in the elderly. Am J Cardiol 1988; 61: 7h–12h. [DOI] [PubMed] [Google Scholar]
- 107.Farquhar WB, Edwards DG, Jurkovitz CT, et al. Dietary sodium and health: more than just blood pressure. J Am Coll Cardiol 2015; 65: 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Safar ME, Temmar M, Kakou A, et al. Sodium intake and vascular stiffness in hypertension. Hypertension 2009; 54: 203–209. [DOI] [PubMed] [Google Scholar]
- 109.Ying WZ, Sanders PW. Dietary salt increases endothelial nitric oxide synthase and TGF-beta1 in rat aortic endothelium. Am J Physiol 1999; 277(4 Pt 2): H1293–H1298. [DOI] [PubMed] [Google Scholar]
- 110.Sanders PW. Vascular consequences of dietary salt intake. Am J Physiol Renal Physiol 2009; 297: F237–F243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Avolio AP, Clyde KM, Beard TC, et al. Improved arterial distensibility in normotensive subjects on a low salt diet. Arteriosclerosis 1986; 6: 166–169. [DOI] [PubMed] [Google Scholar]
- 112.Gates PE, Tanaka H, Hiatt WR, et al. Dietary sodium restriction rapidly improves large elastic artery compliance in older adults with systolic hypertension. Hypertension 2004; 44: 35–41. [DOI] [PubMed] [Google Scholar]
- 113.Sofola OA, Knill A, Hainsworth R, et al. Change in endothelial function in mesenteric arteries of Sprague-Dawley rats fed a high salt diet. J Physiol 2002; 543(Pt 1): 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adegunloye BJ, Sofola OA. Effect of dietary salt loading and high-calcium diet on vascular smooth muscle responses and endothelium function in rats. Clin Exp Pharmacol Physiol 1997; 24: 814–818. [DOI] [PubMed] [Google Scholar]
- 115.Zhu J, Yu M, Friesema J, et al. Salt-induced ANG II suppression impairs the response of cerebral artery smooth muscle cells to prostacyclin. Am J Physiol Heart Circ Physiol 2005; 288: H908–H913. [DOI] [PubMed] [Google Scholar]
- 116.Liu Y, Rusch NJ, Lombard JH. Loss of endothelium and receptor-mediated dilation in pial arterioles of rats fed a short-term high salt diet. Hypertension 1999; 33: 686–688. [DOI] [PubMed] [Google Scholar]
- 117.Durand MJ, Lombard JH. Low-dose angiotensin II infusion restores vascular function in cerebral arteries of high salt-fed rats by increasing copper/zinc superoxide dimutase expression. Am J Hypertens 2013; 26: 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Foulquier S, Dupuis F, Perrin-Sarrado C, et al. High salt intake abolishes AT(2)-mediated vasodilation of pial arterioles in rats. J Hypertens 2011; 29: 1392–1399. [DOI] [PubMed] [Google Scholar]
- 119.Faraco G, Park L, Zhou P, et al. Hypertension enhances Abeta-induced neurovascular dysfunction, promotes beta-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab 2016; 36: 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rodrigues SF, Granger DN. Cerebral microvascular inflammation in DOCA salt-induced hypertension: role of angiotensin II and mitochondrial superoxide. J Cereb Blood Flow Metab 2012; 32: 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakagawa T, Hasegawa Y, Uekawa K, et al. Renal denervation prevents stroke and brain injury via attenuation of oxidative stress in hypertensive rats. J Am Heart Assoc 2013; 2: e000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ueno M, Sakamoto H, Tomimoto H, et al. Blood-brain barrier is impaired in the hippocampus of young adult spontaneously hypertensive rats. Acta Neuropathol 2004; 107: 532–538. [DOI] [PubMed] [Google Scholar]
- 123.Elijovich F, Weinberger MH, Anderson CA, et al. Salt sensitivity of blood pressure: a scientific statement from the American Heart Association. Hypertension 2016; 68: e7–e46. [DOI] [PubMed] [Google Scholar]
- 124.Makin SDJ, Mubki GF, Doubal FN, et al. Small vessel disease and dietary salt intake: cross-sectional study and systematic review. J Stroke Cerebrovasc Dis 2017; 26: 3020–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Titze J. Estimating salt intake in humans: not so easy!. Am J Clin Nutr 2017; 105: 1253–1254. [DOI] [PubMed] [Google Scholar]
- 126.Powles J, Fahimi S, Micha R, et al. Global, regional and national sodium intakes in 1990 and 2010: a systematic analysis of 24 h urinary sodium excretion and dietary surveys worldwide. BMJ Open 2013; 3: e003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kleinewietfeld M, Manzel A, Titze J, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013; 496: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wilck N, Matus MG, Kearney SM, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017; 551: 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013; 496: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Iadecola C, Gottesman R. Cerebrovascular alterations in Alzheimer's disease: incidental or pathogenic? Circ Res 2018; 123: 406–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gottesman RF, Schneider AL, Albert M, et al. Midlife hypertension and 20-year cognitive change: the atherosclerosis risk in communities neurocognitive study. JAMA Neurol 2014; 71: 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Petrovitch H, White LR, Izmirilian G, et al. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: the HAAS. Honolulu-Asia aging Study. Neurobiol Aging 2000; 21: 57–62. [DOI] [PubMed] [Google Scholar]
- 133.Gottesman RF, Schneider AL, Zhou Y, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 2017; 317: 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rodrigue KM, Rieck JR, Kennedy KM, et al. Risk factors for beta-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol 2013; 70: 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim HJ, Park S, Cho H, et al. Assessment of extent and role of tau in subcortical vascular cognitive impairment using 18F-AV1451 positron emission tomography imaging. JAMA Neurol 2018; 75: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hughes TM, Wagenknecht LE, Craft S, et al. Arterial stiffness and dementia pathology: atherosclerosis risk in communities (ARIC)-PET study. Neurology 2018; 90: e1248–e1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Diaz-Ruiz C, Wang J, Ksiezak-Reding H, et al. Role of hypertension in aggravating abeta neuropathology of AD Type and tau-mediated motor impairment. Cardiovasc Psychiatry Neurol 2009; 2009: 107286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shih YH, Wu SY, Yu M, et al. Hypertension accelerates Alzheimer's disease-related pathologies in pigs and 3xTg mice. Front Aging Neurosci 2018; 10: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de Bruijn RF, Ikram MA. Cardiovascular risk factors and future risk of Alzheimer's disease. BMC Med 2014; 12: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gorelick PB, Furie KL, Iadecola C, et al. Defining optimal brain health in adults: a presidential advisory from the American Heart Association/American Stroke Association. Stroke 2017; 48: e284–e303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Forette F, Seux ML, Staessen JA, et al. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med 2002; 162: 2046–2052. [DOI] [PubMed] [Google Scholar]
- 142.Saavedra JM. Beneficial effects of angiotensin II receptor blockers in brain disorders. Pharmacol Res 2017; 125(Pt A): 91–103. [DOI] [PubMed] [Google Scholar]
- 143.Staessen JA, Thijs L, Richart T, et al. Placebo-controlled trials of blood pressure-lowering therapies for primary prevention of dementia. Hypertension 2011; 57: e6–e7. [DOI] [PubMed] [Google Scholar]
- 144.Toyoda K, Fujii K, Ibayashi S, et al. Attenuation and recovery of brain stem autoregulation in spontaneously hypertensive rats. J Cereb Blood Flow Metab 1998; 18: 305–310. [DOI] [PubMed] [Google Scholar]
- 145.Winquist RJ, Bohr DF. Structural and functional changes in cerebral arteries from spontaneously hypertensive rats. Hypertension 1983; 5: 292–297. [DOI] [PubMed] [Google Scholar]
- 146.Toth P, Csiszar A, Sosnowska D, et al. Treatment with the cytochrome P450 omega-hydroxylase inhibitor HET0016 attenuates cerebrovascular inflammation, oxidative stress and improves vasomotor function in spontaneously hypertensive rats. Br J Pharmacol 2013; 168: 1878–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Freitas F, Estato V, Reis P, et al. Acute simvastatin treatment restores cerebral functional capillary density and attenuates angiotensin II-induced microcirculatory changes in a model of primary hypertension. Microcirculation 2017; 24: pii: e12416. [DOI] [PubMed] [Google Scholar]
- 148.Smeda JS, VanVliet BN, King SR. Stroke-prone spontaneously hypertensive rats lose their ability to auto-regulate cerebral blood flow prior to stroke. J Hypertens 1999; 17(12 Pt 1): 1697–1705. [DOI] [PubMed] [Google Scholar]
- 149.Mayhan WG, Faraci FM, Heistad DD. Impairment of endothelium-dependent responses of cerebral arterioles in chronic hypertension. Am J Physiol 1987; 253(6 Pt 2): H1435–H1440. [DOI] [PubMed] [Google Scholar]
- 150.Kitazono T, Faraci FM, Heistad DD. L-arginine restores dilator responses of the basilar artery to acetylcholine during chronic hypertension. Hypertension 1996; 27: 893–896. [DOI] [PubMed] [Google Scholar]
- 151.Yang ST, Mayhan WG, Faraci FM, et al. Endothelium-dependent responses of cerebral blood vessels during chronic hypertension. Hypertension 1991; 17: 612–618. [DOI] [PubMed] [Google Scholar]
- 152.Yang ST, Faraci FM, Heistad DD. Effects of cilazapril on cerebral vasodilatation in hypertensive rats. Hypertension 1993; 22: 150–155. [DOI] [PubMed] [Google Scholar]
- 153.Fan F, Geurts AM, Murphy SR, et al. Impaired myogenic response and autoregulation of cerebral blood flow is rescued in CYP4A1 transgenic Dahl salt-sensitive rat. Am J Physiol Regul Integr Comp Physiol 2015; 308: R379–R390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Smeda JS, Payne GW. Alterations in autoregulatory and myogenic function in the cerebrovasculature of Dahl salt-sensitive rats. Stroke 2003; 34: 1484–1490. [DOI] [PubMed] [Google Scholar]
- 155.Soltis EE, Bohr DF. Cerebral vascular responsiveness in deoxycorticosterone acetate-salt hypertensive rats. Am J Physiol 1987; 252(1 Pt 2): H198–H203. [DOI] [PubMed] [Google Scholar]
- 156.Matin N, Pires PW, Garver H, et al. DOCA-salt hypertension impairs artery function in rat middle cerebral artery and parenchymal arterioles. Microcirculation 2016; 23: 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Didion SP, Kinzenbaw DA, Faraci FM. Critical role for CuZn-superoxide dismutase in preventing angiotensin II-induced endothelial dysfunction. Hypertension 2005; 46: 1147–1153. [DOI] [PubMed] [Google Scholar]
- 158.Ryan MJ, Didion SP, Mathur S, et al. Angiotensin II-induced vascular dysfunction is mediated by the AT1A receptor in mice. Hypertension 2004; 43: 1074–1079. [DOI] [PubMed] [Google Scholar]
- 159.Johnson AW, Kinzenbaw DA, Modrick ML, et al. Small-molecule inhibitors of signal transducer and activator of transcription 3 protect against angiotensin II-induced vascular dysfunction and hypertension. Hypertension 2013; 61: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]