

Abstract
Objective
We aimed to evaluate racial differences in the clinical features of neuromyelitis optica spectrum disorder.
Methods
This retrospective review included 603 patients (304 Asian, 207 Caucasian, and 92 Afro-American/Afro-European), who were seropositive for anti–aquaporin-4 antibody, from 6 centers in Denmark, Germany, South Korea, United Kingdom, United States, and Thailand.
Results
Median disease duration at last follow-up was 8 years (range 0.3–38.4 years). Asian and Afro-American/Afro-European patients had a younger onset age than Caucasian patients (mean 36, 33, and 44 years, respectively; p < 0.001). During the disease course, Caucasian patients (23%) had a lower incidence of brain/brainstem involvement than Asian (42%) and Afro-American/Afro-European patients (38%) (p < 0.001). Severe attacks (visual acuity ≤0.1 in at least one eye or Expanded Disability Status Scale score ≥6.0 at nadir) at onset occurred more frequently in Afro-American/Afro-European (58%) than in Asian (46%) and Caucasian (38%) patients (p = 0.005). In the multivariable analysis, older age at onset, higher number of attacks before and after immunosuppressive treatment, but not race, were independent predictors of severe motor disabilities at last follow-up.
Conclusion
A review of a large international cohort revealed that race affected the clinical phenotype, age at onset, and severity of attacks, but the overall outcome was most dependent on early and effective immunosuppressive treatment.
Neuromyelitis optica spectrum disorder (NMOSD) is a rare, severe, inflammatory, CNS disorder characterized by optic neuritis (ON), longitudinally extensive transverse myelitis, and area postrema syndrome (APS).1 Recent epidemiologic studies have revealed that NMOSD prevalence varies by race, with a higher prevalence in nonwhite than in white populations.2,3 A study conducted in Australia and New Zealand showed that NMOSD was 3 times more prevalent in individuals with Asian ancestry than in the remaining population with predominantly European ancestry.4 The NMOSD/multiple sclerosis ratio is reported to be higher in Asia than in Western countries.5 Therefore, comparative studies across different races are needed to determine whether the epidemiologic differences observed reflect true racial differences in the NMOSD phenotype and prognosis.
Studies investigating the clinical characteristics of NMOSD have been conducted in several countries, including China,6 Denmark,7 France,8 Germany,9 Iran,10 Japan,11,12 Korea,13 Spain,14 Thailand,15 Turkey,16 and United Kingdom.11 However, it is difficult to compare the clinical features between races using the results of these studies because of variability in inclusion criteria, patient selection methods/criteria, and characteristics investigated, and mixed racial groups. A few studies have reported some differences among racial groups in the clinical manifestations and outcome of NMOSD: nonwhite patients had a younger onset age11 and more attacks involving the brain or brainstem11,17 and tended to have worse outcomes (severe motor deficit or visual disability).11,14,18 However, very few studies have been conducted, and they are limited by the relatively small sample sizes, lack of data from multiple country cohorts, and lack of multivariable analysis to investigate true associations among races and outcomes after adjustment for confounding factors. Therefore, the aim of this study was to improve our understanding of the variations of the clinical manifestations and outcomes in NMOSD across different racial groups, categorized as Asian, Afro-American/Afro-European, and Caucasian using data from multiple countries/continents.
Methods
Patients
We included 610 consecutively identified patients with anti–aquaporin-4 (AQP4) antibody–seropositive NMOSD during 2006–2017 and followed at 6 departments of neurology worldwide: University of Southern Denmark (Odense), Charité–University Medicine (Berlin, Germany), Heinrich Heine University (Düsseldorf, Germany), Research Institute and Hospital of National Cancer Center (Goyang, Korea), University of Oxford (UK), The Johns Hopkins University School of Medicine (Baltimore, MD), and Siriraj Hospital, Mahidol University (Bangkok, Thailand) (figure 1). Case ascertainment and data entry were performed between March 2017 and August 2017 by neurologists at each center. All patients were tested for anti-AQP4 antibodies using cell-based or enzyme-linked immunosorbent assays. Seven Hispanic or other/mixed race patients were excluded because of low sample sizes. Finally, 603 patients were included in the study.
Figure 1. Flowchart of patient inclusion in the study.

AQP4 = aquaporin-4; NMOSD = neuromyelitis optica spectrum disorder; UK = United Kingdom; USA = United States.
We evaluated the following factors: age; sex; ethnicity; type of syndrome (6 core clinical characteristics according to the 2015 criteria1) at onset, at second attack, and at third attack; number of attacks before and after immunosuppressive treatment; severity of the first attack; presence of longitudinally extensive transverse myelitis on MRI at onset (lesion extending more than 3 vertebral segments); brain MRI abnormalities (when an NMOSD-typical brain lesion pattern was observed according to the 2015 criteria1); presence of symptomatic brain attack; time to treatment; treatment duration; type of treatment; concurrent systemic or localized autoimmune disease; and CSF-specific oligoclonal bands. Race was classified as self-identified (University of Southern Denmark; University of Oxford, UK; The Johns Hopkins University School of Medicine, Baltimore, MD) or subjectively assigned by the hospital administration/physician (Charité–University Medicine, Germany; Heinrich Heine University, Germany; Research Institute and Hospital of National Cancer Center, Korea; Siriraj Hospital, Thailand). A relapse was defined as a new or worsening acute neurologic symptom lasting ≥24 hours and not explained by fever, infection, or metabolic condition. A severe relapse was defined as an Expanded Disability Status Scale (EDSS) score of ≥6.0 at the nadir of the attack, or an increase of ≥0.5 points if the patient had a baseline EDSS score of ≥6.0.19 In ON cases, a severe relapse was defined as a new worsening of visual acuity ≤0.1 at the nadir of the attack. If baseline vision was light perception, hand motion, or counting fingers, any decrease accompanied with MRI evidence of ON was regarded as a severe relapse.19 The outcome reached at last follow-up was evaluated by the EDSS score. Severe motor disability was defined as a sustained EDSS score ≥6.0, and severe visual disability was defined as sustained converted visual function scale score (cVFSS) ≥3. Patients were classified into 1 of 3 groups according to age at onset: pediatric onset (age at onset <18 years), young adult onset (age at onset 18–49 years), and late adult onset (age at onset >49 years).
Statistical analysis
Demographic and clinical information was compared across the 3 racial groups. For categorical variables, we used the Pearson χ2 test, and Fisher exact test was used when the expected values in any of the cells of a contingency table were below 5. For continuous variables following a normal distribution, we performed 1-way analysis of variance and then independent t test was used as post hoc analysis. If a continuous variable did not follow a normal distribution, we applied the Kruskal-Wallis and Mann-Whitney U tests. The Kaplan-Meier technique was used to estimate the median time from disease onset to severe motor disability (EDSS score ≥6.0). We examined the effect of race on the manifestations of NMOSD using a binary logistic regression model, adjusting for age at onset (pediatric onset, young adult onset, and late adult onset) and sex. To investigate the relationship between race and severe disabilities, we used a binary logistic regression model with an EDSS score ≥6.0 and a cVFSS ≥3 as a binary outcome, while controlling for covariates such as age at onset (as a continuous variable), sex (male vs female), disease duration, number of attacks before and after immunosuppressive treatment, type of syndrome at onset, and country. Backward variable selection method with an elimination criterion of p > 0.05 was applied. To determine whether race affects time from disease onset to the occurrence of severe irreversible disabilities, we analyzed Cox proportional hazard regression model. The models were adjusted for age at onset (as a continuous variable), sex (male vs female), type of syndrome at onset, and whether patients initiated immunosuppressive treatment before attaining severe disabilities. Occurrence of severe irreversible motor (EDSS score ≥6.0) or visual disability (cVFSS ≥3) was the event of interest. Patients who had not yet reached severe disabilities, but were followed for a known period, were right censored. A p value <0.05 was considered statistically significant. All results were analyzed using SAS software, version 9.4 (SAS Institute Inc., Cary, NC) and Stata Statistical software, version 14.2 (StataCorp LP, College Station, TX).
Standard protocol approvals, registrations, and patient consents
The data used were anonymized and were shared subject to the individual center's local ethical requirements. The informed consent requirement was waived.
Data availability
Individual deidentified patient data will not be available in a publicly accessible repository to protect the interests of the patients and investigators.
Results
Clinical and demographic data of the 603 patients with a median disease duration of 8 (range: 0.3–38.4) years are summarized in table 1. Of the 603 patients, 304 were Asian, 207 were Caucasian, and 92 were Afro-American/Afro-European. Compared with the Caucasian patients, Afro-American/Afro-European and Asian patients were younger at disease onset (table 1) (p < 0.001). Late adult onset patients (>49 years of age) were more frequently Caucasian (40%) than Asian (17%) and Afro-American/Afro-European (13%; p < 0.001; figure 2). The disease course was recurrent in 511 patients (85%), and 92 patients (15%) experienced only a single attack. Disease duration was longer in patients with recurrent attacks than in patients with a single attack (median 9.4 vs median 2.5 years, p < 0.001). When we excluded the 149 patients who started immunosuppressive therapy before the second attack, 98% of patients (438/448) revealed a relapsing disease course. At disease onset, a severe attack at nadir (EDSS score ≥6.0 or visual acuity ≤0.1 in at least one eye) occurred in 266 (45%) of 587 patients. Severe attacks at disease onset included ON (39%), myelitis (38%), multifocal attack (14%), and brain/brainstem attack (9%). At last follow-up, 26% of patients (155/596) had an irreversible EDSS score ≥6.0, and 35% of patients (203/576) had an irreversible cVFSS ≥3. When we excluded the 8 patients with a disease duration of less than 6 months, the median interval to irreversible severe motor disability was 24 years (95% confidence interval [CI] 18.718–29.282) (figure 3). Of the 598 patients with known history of treatment, 583 patients (98%) received immunosuppressive therapy; the median time from onset to treatment was 1.4 years (range 0–29.7 years). The classic presentation of simultaneous ON and myelitis at onset was found in only 21 patients (4%). Among the 244 patients who presented with myelitis and had initial spinal cord MRI, 190 (78%) had longitudinally extensive transverse myelitis lesions. During the disease course, brain attack occurred in 212 patients (35%).
Table 1.
Comparison of demographic and clinical features between the 3 racial groups

Figure 2. Distribution of patients by age at disease onset and race.
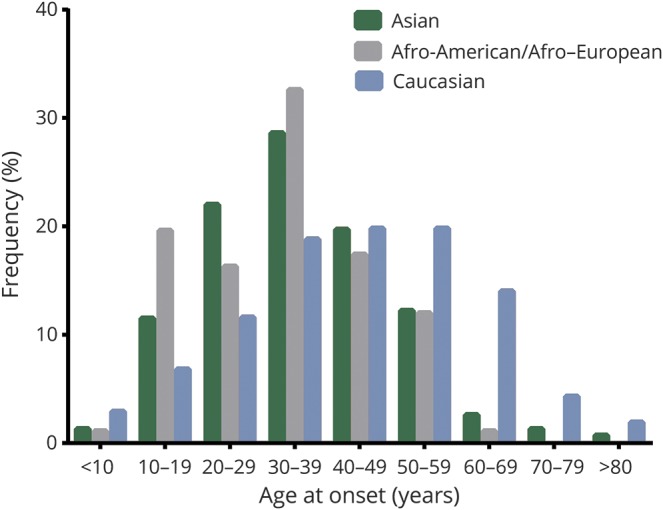
Each bar represents one age group in each ethnic group.
Figure 3. Kaplan-Meier curve for the probability to reach an score of ≥6.0.
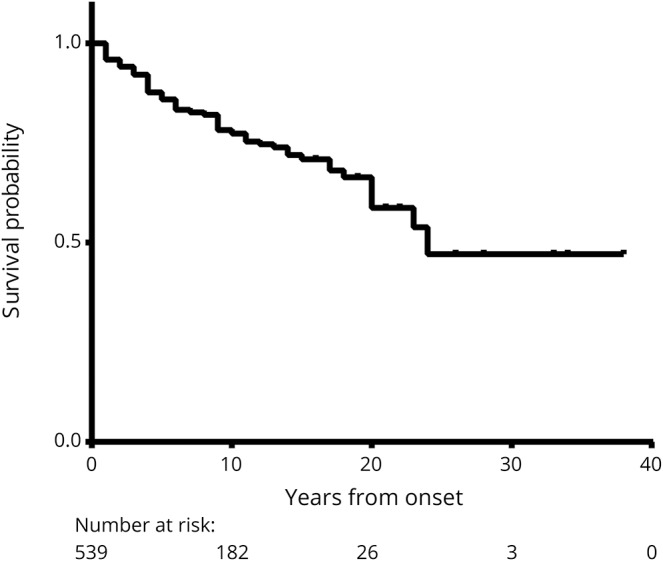
Expanded Disability Status Scale
Comparison of clinical characteristics and outcomes among racial groups
Caucasian patients (54%) were more likely than Asian patients (43%) to have myelitis at the initial presentation (p = 0.04), but they had a lower frequency of brain/brainstem involvement at onset (16%) than Asian (22%) and Afro-American/Afro-European patients (27%, p = 0.043) (table 1). Afro-American/Afro-European patients (58%) had a severe attack at onset more frequently than Asian (46%) and Caucasian (38%, p = 0.005) patients. The proportion of patients with each of the 6 core clinical features of the 2015 NMOSD criteria (ON, myelitis, APS, brainstem syndrome, diencephalic syndrome, symptomatic cerebral syndrome)1 during the disease course, excluding cerebral syndrome, did not differ according to race; cerebral syndrome was most frequently observed in Asian (21%) followed by Afro-American/Afro-European (11%) and Caucasian (3%, p < 0.001) patients. When we assumed an unknown anti-AQP4 antibody status, Afro-American/Afro-European patients (23%) were more likely to fulfill the 2015 criteria than Asian (7%) and Caucasian (12%) patients at onset (p < 0.001), because attacks that involve multiple locations simultaneously at onset were more common in Afro-American/Afro-European patients than in other racial groups. Caucasian patients (61%) were less likely to fulfill the 2015 criteria than Asian (72%) and Afro-American/Afro-European (76%) patients after a third attack (p = 0.032), because having a subsequent event in the same location was more common in Caucasian patients than in other racial groups after a third attack. CSF-specific oligoclonal bands were found more frequently in Caucasian (26%) and Afro-American/Afro-European (26%) patients than in Asian patients (13%, p = 0.011).
There was no difference between the racial groups in the proportion of patients who received immunosuppressive treatment. Time from onset to initiation of immunosuppressive treatment was shorter in Afro-American/Afro-European patients (median 1 year) than in Asian patients (median 2.2 years, p = 0.024; table 1). Regarding type of treatment, fewer Caucasian patients (13%) were treated with mycophenolate mofetilas first-line treatment compared to Asian (23%) and Afro-American/Afro-European (27%) patients (p = 0.005; table 2). The type of current treatment also differed in rate of azathioprine use: Caucasian patients (34%) used azathioprine more frequently than Afro-American/Afro-European patients (20%, p = 0.023). All 3 ethnic groups showed reduction in median annualized relapse rate after immunosuppressive treatment (table 2). Caucasian patients had higher EDSS scores at last follow-up than Asian patients (median 4.0 vs median 3.0, p = 0.039) and higher frequency of EDSS score of 6.0 (32% vs 22%, p = 0.037).
Table 2.
Comparison of treatment outcomes between racial groups

Comparison of clinical characteristics and outcomes by age at disease onset
Data available from Dryad (table e-1, doi.org/10.5061/dryad.hs87bk0) show the demographics, clinical characteristics, and outcomes of the 3 groups of patients according to age at onset. The disease duration was shorter in late adult onset patients (p < 0.001) than in pediatric onset and young adult onset patients. Late adult onset patients showed more frequent myelitis (p < 0.001), less frequent ON (p = 0.008) and APS (p = 0.017) at disease onset (p = 0.017), higher proportion with severe motor disability from the first attack (p < 0.001) and at last follow-up (p < 0.001) than pediatric onset and young adult onset patients. These findings were also consistently obtained when we investigated the frequency of these features according to age (in decades) at onset. See data available from Dryad (appendix, doi.org/10.5061/dryad.hs87bk0).
Multivariable analysis of predictors for clinical manifestations and severe disability
To investigate whether the different clinical manifestations between races were more affected by differences in age at onset than by differences in race, we conducted multivariable analysis, the results of which are shown in table 3. After adjustment for age at onset, the proportion of patients with symptomatic brain attack was higher for Asian than for Caucasian patients (odds ratio [OR] 2.165, 95% CI 1.433–3.270). Afro-American/Afro-European (OR 2.713, 95% CI 1.601–4.596) and Asian (OR 1.589, 95% CI 1.082–2.332) patients experienced more severe attacks at onset than did Caucasian patients. The proportion of patients who experienced a severe attack at least once was higher in Afro-American/Afro-European than in Caucasian patients (OR 1.885, 95% CI 1.035–3.431).
Table 3.
Demographic factors associated with occurrence of neuromyelitis optica spectrum disorder symptoms

When all of the examined prognostic factors were included in the multivariable analysis on severe motor disability (EDSS score ≥6.0) at last follow-up, the effect of age was reinforced: older patients appeared to exhibit a higher risk of obtaining an EDSS score of 6.0 (per year) (OR 1.05, 95% CI 1.034–1.067) (table 4). Higher number of attacks before (OR 1.133, 95% CI 1.075–1.193) and after (OR 1.185, 95% CI 1.076–1.306) immunosuppressive therapy was associated with attaining severe motor disability. Disease duration (per year longer) (OR 1.05, 95% CI 1.011–1.09), higher number of attacks before therapy (OR 1.125, 95% CI 1.059–1.194), and ON presentation at onset (OR 5.61, 95% CI 3.746–8.394) were independently associated with severe residual visual disability at last follow-up (table 4). However, race was not an independent predictor of severe motor or visual disabilities at last follow-up. Cox regression analysis revealed no differences on time from disease onset to severe motor or visual disability among the racial groups (data available from Dryad, appendix and figures e-1 and e-2, doi.org/10.5061/dryad.hs87bk0).
Table 4.
Univariable and multivariable logistic regression analysis of patients with severe motor disability or severe visual disability at last follow-up

Discussion
This is the largest published study comparing clinical manifestations and outcome of NMOSD among 3 major racial groups. Our results confirm that ethnicity influences age at onset and NMOSD phenotype. Asian and Afro-American/Afro-European patients had a younger mean onset age than Caucasian patients, a higher prevalence of a brain attack at onset as well as during the disease course, and more frequent brain abnormalities on MRI. Afro-American/Afro-European patients were more likely than Asian and Caucasian patients to have a severe attack at onset. Our findings are consistent with the results of studies showing that nonwhite patients tend to have younger onset age and more brain symptoms or MRI abnormalities than white patients.11,17,18 The ethnic disparity in the prevalence of brain attacks during the disease course and severe attacks were not attenuated after adjustment for age at onset and sex.
This study also described several clinical features of NMOSD. While myelitis and ON are predominant manifestations in NMOSD, brain/brainstem attacks occurred in 35% of patients throughout the disease course. Consistent with previous studies,20,21 APS occurred mainly at the onset of disease (10% of patients) and in 15% of patients throughout the disease course. However, simultaneous ON and myelitis phenotype at disease onset, which is known as Devic syndrome, was found in only 4% of patients. Recently, anti-myelin oligodendrocyte glycoprotein antibodies have been detected in a subgroup of patients with NMOSD phenotype, and anti-myelin oligodendrocyte glycoprotein antibody–associated inflammatory CNS diseases may account for some cases of Devic syndrome.22–25 Despite the notion that NMOSD is characterized by severe attacks leading to visual loss or mobility difficulties, more than half of our patients (55%) did not initially present with a severe attack (visual acuity ≤0.1 in at least one eye or EDSS score ≥6.0 at nadir). Irreversible severe motor and visual disability from the first attack occurred in only 6% and 10% of patients, respectively. This suggests that early initiation of immunosuppressive therapy is likely to prevent irreversible disability in a majority of patients. Among patients who presented with myelitis at onset, 22% revealed spinal cord lesions extending fewer than 3 vertebral segments on the spinal cord MRI, which is slightly higher than the 14% with an initial myelitis attack reported in previous studies.26,27 Thus, clinicians should be careful not to exclude the possibility of NMOSD in patients who present with short transverse myelitis. Consistent with previous reports,8,11,28,29 clinical features and outcomes differed according to age at onset. Late adult onset patients seemed to more frequently have myelitis at onset, irreversible severe motor disability from the first attack, and severe motor disability at last follow-up despite shorter disease duration, but less frequent ON at onset than other groups.
Heterogeneity of NMOSD and the differences in clinical features may be mainly genetic in origin. Human leukocyte antigen (HLA) predisposition has been suggested as a genetic factor associated with NMOSD. In Japanese and Chinese patients, NMOSD susceptibility is associated with the HLA-DPB1*0501 allele,30,31 whereas in Caucasian, Afro-Caribbean, and Indian patients, it is associated with the HLA-DPB1*03 allele,32–34 with no association between the HLA-DPB1*0501 allele and NMOSD in Caucasian patients.35 However, more genetic studies are required to identify specific genes related to NMOSD pathogenesis and to understand how these genes may influence variability in phenotype and disease activity among racial groups.
In 2015, new NMOSD criteria were proposed, allowing an NMOSD diagnosis in patients without anti-AQP4 antibody serostatus1 on the basis of the presence of at least 2 core clinical features, at least one of which should be ON, myelitis, or APS. However, these criteria do not consider race and its potential influence on NMOSD manifestation. In the current study, we found that 5 core clinical features, except symptomatic cerebral syndrome, did not differ across racial groups: cerebral syndrome was most frequently observed in Asian (21%) followed by Afro-American/Afro-Caribbean (11%) and Caucasian (3%) patients. These findings may explain why cerebral lesions on MRI have been reported occasionally in Asian patients with NMOSD,36–39 but rarely in Western case series. When we compared the proportion of patients who satisfied the 2015 criteria among the races, assuming an unknown anti-AQP4 antibody status, results revealed that the proportion of Afro-American/Afro-European patients was higher than that of the other racial groups at disease onset because of more frequent attacks involving multiple sites. However, when we compared the proportion of patients who satisfied the 2015 criteria after the third attack, the proportion of Caucasians (61%) was less frequent than that of Asians (72%) and Afro-American/Afro-Europeans (77%) because a higher proportion of Caucasian patients experienced a subsequent event in the same location. Of note, without a positive anti-AQP4 antibody test, about 30% of the total cohort did not satisfy the 2015 criteria even after the third attack. Therefore, an anti-AQP4 antibody test is required to diagnose NMOSD early in some patients, particularly more so in Caucasian than in Asian and Afro-American/Afro-European patients.
NMOSD may be a more aggressive disease in Afro-American/Afro-European patients than in Caucasian and Asian patients, and thus, Afro-American/Afro-European patients have a greater risk of severe disability during the early phase of the disease. Despite these differences, the risk of attaining severe motor or visual disabilities at last follow-up in Afro-American/Afro-European patients was not different from that in Asian and Caucasian patients in the present study. These results are inconsistent with previous studies that reported worse outcome in Afro-Caribbean than in Caucasian patients.11,14,18 These discrepancies may be attributable to treatment factors. Earlier initiation of immunosuppressive therapy in Afro-American/Afro-European than in Asian patients and the higher proportion of patients who were treated with mycophenolate mofetil as first-line therapy in Afro-American/Afro-European than in Caucasian patients might have influenced the final outcome. In the multivariable analysis, a higher number of attacks before and after immunosuppressive therapy but not race was a strong independent predictor of severe motor disabilities at last follow-up.
Because our cohort was not population-based, there are inherent limitations in terms of site participation, patient selection, and reporting biases. Race was not classified consistently in this study, and classification of race based on phenotypical traits and impressions of probable geographic ancestry and cultural identity was not reflected at the genetic level. In addition, we cannot rule out the influence of other factors, such as timing and type of acute treatment and heterogeneity in immunosuppressive treatment regimens on final outcomes, and other potential sources of ethnic disparities derived from health care professional-related factors. Limitations notwithstanding, this study contributes to the limited literature on the differences among racial groups. The major strengths of the present study included the large sample size and multicenter cohort from different countries, which allowed us to compare clinically important variables by race and to adjust for potential confounders of the relationship between race and residual disabilities.
Race contributes to the heterogeneity of NMOSD clinical manifestations. Considering that cerebral attacks and brain MRI abnormalities develop more frequently in Asian and Afro-American/Afro-European patients, clinicians should give more attention to distinguishing NMOSD from multiple sclerosis in these non-Caucasian patients. Because severe attacks were more frequent in Afro-American/Afro-European than in Asian and Caucasian patients, these patients may need more potent immunosuppressive therapy to prevent worsening of disability. Race is important for determining disease manifestation and activity, but early and effective treatment is more important for determining long-term outcomes. Better understanding of the racial differences in NMOSD may yield important insight into NMOSD pathogenesis, improve early diagnosis, and help in identifying the best treatment strategy, tailored to the needs of individual target populations.
Glossary
- APS
area postrema syndrome
- AQP4
aquaporin-4
- CI
confidence interval
- cVFSS
converted visual function scale score
- EDSS
Expanded Disability Status Scale
- HLA
human leukocyte antigen
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
- OR
odds ratio
Author contributions
Su-Hyun Kim: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis, study supervision. Maureen A. Mealy: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval, acquisition of data. Michael Levy: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Felix Schmidt: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Klemens Ruprecht: data acquisition, accepts responsibility for conduct of research and will give final approval, study supervision. Friedemann Paul: drafting/revising the manuscript, data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval, acquisition of data. Marius Ringelstein: data acquisition, accepts responsibility for conduct of research and will give final approval, acquisition of data. Orhan Aktas: data acquisition, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Hans-Peter Hartung: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Nasrin Asgari: drafting/revising the manuscript, data acquisition, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Jessica Li Tsz-Ching: data acquisition, accepts responsibility for conduct of research and will give final approval, acquisition of data. Sasitorn Siritho: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Naraporn Prayoonwiwat: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Hyun-June Shin: data acquisition, accepts responsibility for conduct of research and will give final approval. Jae-Won Hyun: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision. Mira Han: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. Maria Isabel S. Leite: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval, acquisition of data. Jacqueline Palace: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Ho Jin Kim: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision.
Study funding
This work was supported by the Bio & Medical Technology Development Program (M3A9B6069339) through the Ministry of Science & ICT, Republic of Korea, and by the Deutsche Forschungsgemeinschaft (DFG Exc 257 to F.P.).
Disclosure
S. Kim and M. Mealy report no disclosures relevant to the manuscript. M. Levy currently receives research support from the NIH, Maryland Technology Development Corporation, Sanofi, Genzyme, Alexion, Alnylam, Shire, Acorda, and ApoPharma. He also received personal compensation for consultation with Alexion, Acorda, and Genzyme, and he serves on the scientific advisory boards for Alexion, Acorda, and Quest Diagnostics. F. Schmidt reports no disclosures relevant to the manuscript. K. Ruprecht has received research grants from the German Ministry of Education and Research (BMBF/KKNMS, Competence Network Multiple Sclerosis), Novartis, Merck Serono, and the Charité Research Fund; honoraria for consultancy or speaking and travel reimbursement from Novartis, Bayer Healthcare, Biogen Idec, Merck Serono, Sanofi-Aventis/Genzyme, Teva Pharmaceuticals, and the Guthy-Jackson Charitable Foundation. F. Paul reports no disclosures relevant to the manuscript. M. Ringelstein received consulting and speaker honoraria as well as travel reimbursements from Bayer, Biogen, Genzyme, Teva, Merz, Ipsen, and Novartis. O. Aktas, H. Hartung, N. Asgari, J. Tsz-Ching, S. Siritho, N. Prayoonwiwat, H. Shin, J. Hyun, and M. Han report no disclosures relevant to the manuscript. M. Leite receives support from NHS National Specialized Commissioning Group for Neuromyelitis Optica UK and NIHR Oxford Biomedical Research Centre; received travel funding and speaker honoraria from Biogen Idec; and received a travel grant from Novartis. J. Palace reports no disclosures relevant to the manuscript. H. Kim has lectured and consulted for, and received honoraria from, Bayer Schering Pharma, Biogen, Celltrion, Eisai, Genzyme, HanAll BioPharma, MedImmune, Merck Serono, Novartis, Teva-Handok, and UCB; received a grant from the Ministry of Science & ICT; accepted research funding from Genzyme, Kael-GemVax, Merck Serono, Teva-Handok, and UCB; serves on a steering committee for MedImmune; is a coeditor for the Multiple Sclerosis Journal—Experimental, Translational and Clinical and an associated editor for the Journal of Clinical Neurology. Go to Neurology.org/N for full disclosures.
Publication history
Received by Neurology February 18, 2018. Accepted in final form August 14, 2018.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flanagan EP, Cabre P, Weinshenker BG, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol 2016;79:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol 2012;69:1176–1180. [DOI] [PubMed] [Google Scholar]
- 4.Bukhari W, Prain KM, Waters P, et al. Incidence and prevalence of NMOSD in Australia and New Zealand. J Neurol Neurosurg Psychiatry 2017;88:632–638. [DOI] [PubMed] [Google Scholar]
- 5.Kim SH, Kim HJ. Central nervous system neuroinflammatory disorders in Asian/Pacific regions. Curr Opin Neurol 2016;29:372–380. [DOI] [PubMed] [Google Scholar]
- 6.ZhangBao J, Zhou L, Li X, et al. The clinical characteristics of AQP4 antibody positive NMO/SD in a large cohort of Chinese Han patients. J Neuroimmunol 2017;302:49–55. [DOI] [PubMed] [Google Scholar]
- 7.Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology 2011;76:1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collongues N, Marignier R, Zephir H, et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 2010;74:736–742. [DOI] [PubMed] [Google Scholar]
- 9.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashtari F, Safaei A, Shaygannejad V, Najafi MA, Vesal S. Neuromyelitis optica spectrum disease characteristics in Isfahan, Iran: a cross-sectional study. J Res Med Sci 2017;22:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012;135:1834–1849. [DOI] [PubMed] [Google Scholar]
- 12.Nagaishi A, Takagi M, Umemura A, et al. Clinical features of neuromyelitis optica in a large Japanese cohort: comparison between phenotypes. J Neurol Neurosurg Psychiatry 2011;82:1360–1364. [DOI] [PubMed] [Google Scholar]
- 13.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Clinical spectrum of CNS aquaporin-4 autoimmunity. Neurology 2012;78:1179–1185. [DOI] [PubMed] [Google Scholar]
- 14.Sepulveda M, Armangue T, Sola-Valls N, et al. Neuromyelitis optica spectrum disorders: comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm 2016;3:e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siritho S, Nakashima I, Takahashi T, Fujihara K, Prayoonwiwat N. AQP4 antibody-positive Thai cases: clinical features and diagnostic problems. Neurology 2011;77:827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altintas A, Karabudak R, Balci BP, et al. Neuromyelitis optica and neuromyelitis optica spectrum disorder patients in Turkish cohort: demographic, clinical, and laboratory features. Neurologist 2015;20:61–66. [DOI] [PubMed] [Google Scholar]
- 17.Kremer L, Mealy M, Jacob A, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler 2014;20:843–847. [DOI] [PubMed] [Google Scholar]
- 18.Cabrera-Gomez JA, Kurtzke JF, Gonzalez-Quevedo A, Lara-Rodriguez R. An epidemiological study of neuromyelitis optica in Cuba. J Neurol 2009;256:35–44. [DOI] [PubMed] [Google Scholar]
- 19.Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler 2016;22:329–339. [DOI] [PubMed] [Google Scholar]
- 20.Iorio R, Lucchinetti CF, Lennon VA, et al. Intractable nausea and vomiting from autoantibodies against a brain water channel. Clin Gastroenterol Hepatol 2013;11:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apiwattanakul M, Popescu BF, Matiello M, et al. Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 2010;68:757–761. [DOI] [PubMed] [Google Scholar]
- 22.Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014;71:276–283. [DOI] [PubMed] [Google Scholar]
- 23.Probstel AK, Rudolf G, Dornmair K, et al. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflammation 2015;12:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm 2015;2:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation 2016;13:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flanagan EP, Weinshenker BG, Krecke KN, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015;72:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huh SY, Kim SH, Hyun JW, et al. Short segment myelitis as a first manifestation of neuromyelitis optica spectrum disorders. Mult Scler 2017;23:413–419. [DOI] [PubMed] [Google Scholar]
- 28.Seok JM, Cho HJ, Ahn SW, et al. Clinical characteristics of late-onset neuromyelitis optica spectrum disorder: a multicenter retrospective study in Korea. Mult Scler 2017;23:1748–1756. [DOI] [PubMed] [Google Scholar]
- 29.Long Y, Liang J, Wu L, et al. Different phenotypes at onset in neuromyelitis optica spectrum disorder patients with aquaporin-4 autoimmunity. Front Neurol 2017;8:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushita T, Matsuoka T, Isobe N, et al. Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009;73:171–176. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Dai Y, Qiu W, et al. HLA-DPB1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern Han Chinese. J Neuroimmunol 2011;233:181–184. [DOI] [PubMed] [Google Scholar]
- 32.Blanco Y, Ercilla-Gonzalez G, Llufriu S, et al. HLA-DRB1 typing in Caucasians patients with neuromyelitis optica [in Spanish]. Rev Neurol 2011;53:146–152. [PubMed] [Google Scholar]
- 33.Pandit L, Malli C, D'Cunha A, Mustafa S. Human leukocyte antigen association with neuromyelitis optica in a south Indian population. Mult Scler 2015;21:1217–1218. [DOI] [PubMed] [Google Scholar]
- 34.Deschamps R, Paturel L, Jeannin S, et al. Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to NMO and multiple sclerosis among the French Afro-Caribbean population. Mult Scler 2011;17:24–31. [DOI] [PubMed] [Google Scholar]
- 35.Zephir H, Fajardy I, Outteryck O, et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult Scler 2009;15:571–579. [DOI] [PubMed] [Google Scholar]
- 36.Ito S, Mori M, Makino T, Hayakawa S, Kuwabara S. “Cloud-like enhancement” is a magnetic resonance imaging abnormality specific to neuromyelitis optica. Ann Neurol 2009;66:425–428. [DOI] [PubMed] [Google Scholar]
- 37.Kim W, Park MS, Lee SH, et al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult Scler 2010;16:1229–1236. [DOI] [PubMed] [Google Scholar]
- 38.Cheng C, Jiang Y, Chen X, et al. Clinical, radiographic characteristics and immunomodulating changes in neuromyelitis optica with extensive brain lesions. BMC Neurol 2013;13:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saiki S, Ueno Y, Moritani T, et al. Extensive hemispheric lesions with radiological evidence of blood-brain barrier integrity in a patient with neuromyelitis optica. J Neurol Sci 2009;284:217–219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Individual deidentified patient data will not be available in a publicly accessible repository to protect the interests of the patients and investigators.