

Abstract
Background
The merozoite surface protein-1 (MSP-1) gene encodes for a leading malaria vaccine candidate antigen. However, its extensive polymorphic nature represents a major obstacle to the development of a protective vaccine. Previously, a pilot study was carried out to explore the sequence variation of the C-terminal 42 kDa fragment within P. knowlesi MSP-1 gene (PkMSP-142) based on 12 clinical samples; however, further study on an adequate sample size is vital in estimating the genetic diversity of the parasite population.
Methods
In the present study, we included a larger sample size of P. knowlesi (83 samples) covering eight states of Malaysia to determine the genetic polymorphism, natural selection and haplotype groups of the gene fragment coding PkMSP-142. The region flanking PkMSP-142 was amplified by PCR and directly sequenced. Genetic diversity, haplotype diversity, population genetic differentiation and natural selection were determined in order to study the polymorphic characteristic of PkMSP-142.
Results
A high level of genetic diversity (Hd = 0.970 ± 0.007; л = 0.01079 ± 0.00033) was observed among the 83 P. knowlesi samples, confirming the extensive genetic polymorphism exhibited among the P. knowlesi population found in Malaysia. A total of 18 distinct haplotypes with 17 amino acid changes were identified, whereby 15 were new haplotypes. High population differentiation values were observed within samples from Peninsular Malaysia and Malaysian Borneo. The 42 kDa fragments of P. knowlesi from Malaysian Borneo were found to be acting on balancing selection whilst purifying selection was suggested to act on isolates from Peninsular Malaysia. The separation of PkMSP-142 haplotypes into two main groups based on geographical separation has further supported the existence of two distinct P. knowlesi lineages.
Conclusions
A high level of genetic diversity was observed among PkMSP-142 in Malaysia, whereby most of the polymorphisms were found within the 33 kDa region. Taken together, these data will be useful in order to understand the nature of P. knowlesi population in Malaysia as well as the design and development of a MSP-142 based knowlesi malaria vaccine.
Electronic supplementary material
The online version of this article (10.1186/s13071-018-3234-5) contains supplementary material, which is available to authorized users.
Keywords: Plasmodium knowlesi, Malaysia, Merozoite surface protein, Genetic polymorphism, Natural selection
Background
The scale-up of malaria control interventions has resulted in a substantial decline in global malaria morbidity and mortality. Despite this achievement, malaria remains a serious global health burden, resulting in 216 million cases annually and nearly half of the world’s population are at risk of malaria. The annual malaria-associated mortality reached 44,500 cases, primarily in children under five [1].
Plasmodium knowlesi, a zoonotic malaria parasite that is commonly found in long-tailed, pig-tailed and banded leaf macaques [2, 3], has recently been recognized as the fifth malaria-causing species in humans [4, 5]. Malaysia has achieved great success in controlling malaria over recent decades, in particular with marked reductions in the incidences of Plasmodium falciparum and Plasmodium vivax [6]. However, with increasing number of reported human knowlesi infections, P. knowlesi is now the predominant species occurring in this country, particularly in the state of Sabah, comprising 62% of cases in 2013 [7, 8].
Although the global reported prevalence of human infection with P. knowlesi is considerably less than that caused by P. falciparum and P. vivax [8], the overall human burden of P. knowlesi infection may be substantially underestimated due to the use of routine microscopy which might lead to misidentification as other human malaria species [9]. Furthermore, the increasing overlap between macaque, human and vector habitats pose a major challenge for malaria control and elimination programmes [10]. Coupled with recent reports of severe and fatal consequences of knowlesi malaria in humans [11–14], this evidence further highlights the public health importance of this simian parasite, particularly in Malaysia.
Among the polymorphic loci of Plasmodium, merozoite surface protein-1 (MSP-1) has been widely used to study genetic diversity and is a prime vaccine candidate in clinical trials for many years [15]. Merozoite surface protein-1 (MSP-1) is a high molecular mass protein found on the surface of the blood stage of the parasite; it plays a key role during erythrocyte invasion [16, 17]. This 190 kDa precursor undergoes two steps of proteolytic processing during merozoite maturation. First, it is cleaved into four major fragments of 83, 30, 38 and 42 kDa, which remain on the merozoite surface as a glycosylphosphatidylinositol-anchored complex. Before erythrocyte invasion, the MSP-142 fragment undergoes a second cleavage, resulting in the generation of the 33 and 19 kDa (MSP-133 and MSP-119) fragments, where the latter remains on the surface as the merozoite, which enters the erythrocyte [18, 19].
This 42 kDa fragment of MSP-1 is a promising vaccine candidate due to its high immunogenicity [20]. Several studies have reported that antibodies directed against the 42 and 19 kDa fragments of MSP-1 (MSP-142 and MSP-119) can interrupt merozoite invasion in vitro [21, 22]. However, extensive genetic polymorphism has also been reported in MSP-142 of P. falciparum [23] and P. vivax [24, 25] among global isolates and this remains a major obstacle hampering the development of an effective malaria vaccine.
The genetic variation in the central repeat region of MSP-1 of P. falciparum [26–28] and P. vivax [24, 25] have been relatively well studied, but very little is known about the genetic diversity in this 42 kDa fragment of MSP-1 gene within P. knowlesi (PkMSP-142). A pilot study of the sequence variation in P. knowlesi MSP-142 has been previously described based on 12 clinical samples collected from hospitals in two states of Malaysia, i.e. Selangor and Sabah [29]. Nevertheless, small sample sizes can lead to significant errors in estimating the genetic diversity of the species. Therefore it is vital to include more samples in order to accurately characterize the genetic diversity, understand the parasite population history and, consequently, to assess the impact of elimination interventions [30].
The present study is an expansion from our previous work to further understand the genetic diversity, natural selection and haplotype groups of the gene fragment coding PkMSP-142, whereby a larger sample size of P. knowlesi isolates (83 isolates) covering more states of Malaysia (eight states) are being included. The level of genetic diversity of P. knowlesi population in Malaysia may provide insight into trends in parasite transmission and be useful for the design and development of an MSP-142 based knowlesi malaria vaccine.
Methods
Blood samples and geographical origin
A total of 645 blood samples, collected from different states of Malaysia between 2011 and 2014, were screened via nested PCR for human Plasmodium species (i.e. P. falciparum, P. vivax, P. malariae, P. ovale and P. knowlesi) as previously described [5, 31]. Of these, 83 samples were found positive with single infection of P. knowlesi (Table 1), consisting of 43 clinical samples of P. knowlesi collected from health clinics and hospitals located in Selangor and Sabah, while 40 were P. knowlesi community samples collected among indigenous populations of various sub-tribes from 10 villages of six other states of Malaysia. The locations of the collection sites are shown in Fig 1.
Table 1.
Distribution of P. knowlesi positive samples according to state in Malaysia (N = 83)
| State | District | Village/ Hospital/ Health clinic | n |
|---|---|---|---|
| Peninsular Malaysia | |||
| Perak | Slim River | Kg Sungai Bil | 15 |
| Tapah | Kg Batu 7 1/2 | 4 | |
| Kg Batu 8 | 7 | ||
| Selangor | Hulu Selangor | Hospital Kuala Kubu Bharu | 15 |
| Gombak | Hospital Selayang | 4 | |
| Petaling | Hospital Sungai Buloh | 1 | |
| Hulu Langat | Hospital Kajang | 1 | |
| Pahang | Pekan | Kg Chini | 1 |
| Lanchang | Kg Kuala Gandah | 1 | |
| Melaka | Alor Gajah | Kg Bukit Sebang | 1 |
| Negeri Sembilan | Jelebu | Kg Ulu Kelaka | 2 |
| Kelantan | Gua Musang | Kg Kuala Lah | 3 |
| Kg Aring 5 | 3 | ||
| Malaysian Borneo | |||
| Sabah | Kota Kinabalu | Hospital Queen Elizabeth II | 12 |
| Beluran | Telupid Health Clinic | 10 | |
| Sarawak | Sarikei | Pakan | 3 |
Fig. 1.

Geographical origin of samples used in this study
DNA extraction, PCR amplification and sequencing of PkMSP-142
Whole genomic DNA was extracted from 200 μl of collected blood samples using a commercially available QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions and stored at -20 °C until further use. PCR amplification of PkMSP-142 was performed in a 50 μl reaction volume consisting of 5 μl of DNA template, 1× PCR buffer (Promega, Madison, USA), 0.3 mM dNTPs (Promega, Madison, USA), 3 mM MgCl2 (Promega, Madison, USA), 2.0 U of Taq DNA polymerase (Promega, Madison, USA) and 0.4 μM forward and reverse primers (PkMSP1-42F: 5'-TCT ACC CCT GTT CGG CAA TG-3' and PkMSP1-42R: 5'-CCA TGG AAA GGA AGA AAA TCA ACA-3') based on the previously described region [20]. Thermal condition was as follows: an initial cycle of activation at 95 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 59 °C for 1 min and extension at 72 °C for 1 min, with a final extension of 72 °C for 10 min. The amplified products were examined in agarose gels at 1.5 % and then subjected to direct, automated sequencing (BigDye Terminator v.3.1 chemistry, Applied Biosystems, Foster City, USA). The quality of electropherograms with the forward and reverse nucleotide sequences was verified manually using Geneious v.9.0.4 software [32]. Sequence electropherograms were also carefully inspected for the presence of multiple sequence types.
Sequence and phylogenetic analysis
Nucleotide and deduced amino acid sequences were aligned and analyzed, using Geneious v.9.0.4 software [32] with the reference sequence encoded by the H strain of P. knowlesi (GenBank: XM_002258546). Phylogenetic trees were constructed using the neighbor-joining and maximum likelihood methods as described in MEGA7 [33]. Bootstrap replicates of 1000 were used to test the robustness of the trees. All newly generated sequences were deposited in the GenBank database (MH796675-MH796757) (Additional file 1: Table S1).
DNA sequence polymorphism analysis
DNA sequence polymorphism analysis was performed on the 83 PkMSP-142 sequences. DnaSP v.5.10.01 [34] was used to calculate the numbers of segregating sites (S) and haplotypes (H) as well as haplotype diversity (Hd), nucleotide diversity (л) and average number of pairwise nucleotide differences within the population (K). The numbers of synonymous nucleotide substitutions per synonymous site (dS), the number of non-synonymous substitutions per non-synonymous site (dN), and the difference between the non-synonymous and synonymous substitutions (dN-dS) were estimated using Nei & Gojobori’s method [35] with the Jukes and Cantor (JC) correction to detect evidence of natural selection in MEGA7. In brief, an excess of dN relative to dS is a clear signal of positive selection. Conversely, a lack of dN relative to dS suggests a negative or purifying selection. Tajima’s D [36] and Fu and Li’s D and F test [37] were further applied using DnaSP version 5.10.01 to evaluate the neutral model of molecular evolution. A significantly positive value of Tajima’s D indicates balancing selection, whereas a negative value suggests negative selection or population size expansion after a recent bottleneck. Wright’s fixation index (FST) was used to measure genetic differentiation between the PkMSP-142 fragments of Peninsular Malaysia and Malaysian Borneo isolates [38]. FST values between populations were calculated using DnaSP v.5.10.01 based on the differences in allele frequencies. They are interpreted as no or low (0–0.05), moderate (0.05–0.15) and high (0.15–0.25) genetic differentiation.
Results
Genetic polymorphism and amino acid changes of PkMSP-142
In this study, the region corresponding to PkMSP-142 was successfully amplified from all 83 P. knowlesi Malaysia samples (58 from Peninsular Malaysia, 25 from Malaysian Borneo). This fragment of 993 bp in size contained a region coding a protein sequence of 331 amino acids. Using the P. knowlesi H strain sequence published in GenBank (XM_002258546) as a reference, 61 segregating sites were identified. Singleton sites were found to be lower in frequency (12/61) than the parsimony-informative sites (having a minimum of two nucleotides each present at least twice) (49/61). Of these polymorphic sites, 47 were dimorphic and two were trimophic changes (Additional file 2: Figure S1).
Amino acid changes at 17 positions were identified as compared to the reference H strain sequence. Of these, 16 were dimorphic mutations with a change into two amino acid type, while one showed trimorphic mutations with change in three amino acid types (F1789S/Y) (Fig. 2). Eighteen haplotypes were deduced from the amino acid sequences with haplotype H18 having the highest frequency (26/83, 31.3%). The distribution of PkMSP-142 amino acid haplotypes in Peninsular Malaysia and Malaysian Borneo is shown in Table 2.
Fig. 2.

Amino acid sequence polymorphism in PkMSP-142. Dimorphic and trimorphic amino acid changes are shaded in red and yellow, respectively. The total numbers of sequences for each haplotype are listed in the right panel
Table 2.
Number of isolates for each of the PkMSP-142 amino acid haplotypes in the two main study areas of Malaysia (N = 83)
| Amino acid haplotypes | Study area | Total | |
|---|---|---|---|
| Peninsular Malaysia | Malaysian Borneo | ||
| H1 | 1 | 1 | |
| H2 | 14 | 14 | |
| H3 | 3 | 3 | |
| H4 | 5 | 5 | |
| H5 | 2 | 2 | |
| H6 | 4 | 4 | |
| H7 | 3 | 3 | |
| H8 | 6 | 6 | |
| H9 | 6 | 6 | |
| H10 | 2 | 2 | |
| H11 | 1 | 1 | 2 |
| H12 | 2 | 2 | |
| H13 | 1 | 1 | |
| H14 | 2 | 2 | |
| H15 | 1 | 1 | |
| H16 | 1 | 1 | 2 |
| H17 | 1 | 1 | |
| H18 | 26 | 26 | |
| Total | 58 | 25 | 83 |
Nucleotide diversity and natural selection
DNA sequence polymorphisms analyses were performed to determine the nucleotide diversity and genetic differentiation at PkMSP-142 region as well as for its 33 and 19 kDa fragments among the Malaysian P. knowlesi samples. The average number of pairwise nucleotide differences (K) for entire MSP-142 region of all 83 samples was found to be 10.7338, while the overall haplotype diversity (Hd) and nucleotide diversity (л) were 0.970 ± 0.007 and 0.01079 ± 0.00033, respectively. The Malaysian Borneo PkMSP-142 was noted to have slightly higher diversity (π = 0.01024 ± 0.00061) than the Peninsular Malaysia PkMSP-142 (π = 0.009119 ± 0.00031). Analysis of the genetic diversity of the 33 and 19 kDa fragments of the samples from the entire population, revealed that PkMSP-133 is more divergent as compared to the PkMSP-119 fragment, suggesting that the nucleotide diversity was predominantly concentrated in PkMSP-133 (Table 3). The overall haplotype diversity (Hd) and nucleotide diversity (π) for PkMSP-133 was 0.963 ± 0.008 and 0.01010 ± 0.00044, respectively.
Table 3.
Estimates of nucleotide diversity, natural selection and neutrality tests of PkMSP1-42 (N = 83)
| Domain | S | л (SD) | K | Hd (SD) | dS | dN | dN-dS (SE) | Z-test (P-value) | Tajima’s D test | Fu & Li’s test | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dN = dS | dN > dS | dN < dS | D | F | |||||||||
| 42 kDa | |||||||||||||
| Entire (n = 83) | 61 | 0.01079 (0.00033) |
10.7338 | 0.970 (0.007) |
0.030 | 0.004 | -0.026 (0.006) |
<0.0001 | 1.000 | <0.0001 | -0.49252 | -0.03809 | -0.26440 |
| Peninsular Malaysia (n = 58) | 51 | 0.009119 (0.00031) |
9.14156 | 0.956 (0.014) |
0.029 | 0.002 | -0.027 (0.006) |
<0.0001 | 1.000 | <0.0001 | -0.68468 | -1.11074 | -1.13645 |
| Malaysian Borneo (n = 25) | 31 | 0.01024 (0.00061) |
10.18667 | 0.910 (0.030) |
0.035 | 0.004 | -0.031 (0.008) |
<0.0001 | 1.000 | <0.0001 | 0.75884 | 0.74035 | 0.87508 |
| 33 kDa | |||||||||||||
| Entire (n = 83) | 46 | 0.01010 (0.00044) |
7.89891 | 0.963 (0.008) |
0.026 | 0.004 | -0.022 (0.006) |
<0.0001 | 1.000 | <0.0001 | -0.52113 | 0.55872 | 0.15928 |
| Peninsular Malaysia (n = 58) | 37 | 0.00758 (0.00037) |
5.92922 | 0.946 (0.015) |
0.032 | 0.002 | -0.030 (0.008) |
<0.0001 | 1.000 | <0.0001 | -0.92293 | -1.07888 | -1.21884 |
| Malaysian Borneo (n = 25) | 25 | 0.01082 (0.00062) |
8.46000 | 0.893 (0.034) |
0.034 | 0.005 | -0.029 (0.009) |
0.001 | 1.000 | 0.001 | 0.84595 | 0.96956 | 1.09115 |
| 19 kDa | |||||||||||||
| Entire (n = 83) | 15 | 0.00779 (0.00064) |
2.83485 | 0.916 (0.011) |
0.046 | 0.002 | -0.044 (0.016) |
0.001 | 1.000 | 0.004 | -0.33245 | -1.37274 | -1.18780 |
| Peninsular Malaysia (n = 58) | 14 | 0.00708 (0.00067) |
3.21234 | 0.906 (0.019) |
0.068 | 0.002 | -0.066 (0.024) |
0.003 | 1.000 | 0.001 | -0.02596 | -0.81452 | -0.64299 |
| Malaysian Borneo (n = 25) | 6 | 0.00811 (0.00117) |
1.72667 | 0.773 (0.048) |
0.038 | 0.000 | -0.038 (0.018) |
0.001 | 1.000 | <0.0001 | -0.25913 | -0.25602 | -0.12303 |
Abbreviations: N number of isolates, S number of segregating sites, K average number of pairwise nucleotide differences, Hd haplotype diversity, л observed average pairwise nucleotide diversity, dS nucleotide diversity of synonymous mutation per synonymous site, dN nucleotide diversity of non-synonymous mutation per non-synonymous site; dN, dS the difference of dN and dS with their standard deviation estimated by bootstrap with 1000 pseudoreplicates, SD standard deviation
The genetic differentiation of PkMSP-142 as well as PkMSP-133 and PkMSP-119 fragments among the samples from Peninsular Malaysia and Malaysian Borneo were compared using Wright’s fixation index (FST). The findings showed high genetic differentiation within the P. knowlesi populations originating from Peninsular Malaysia and Malaysian Borneo in both PkMSP-142 and PkMSP-133 fragments, respectively (FST: 0.23677, P < 0.001; 0.28257, P < 0.001). However, moderate genetic differentiation was observed in PkMSP-119 fragments between Peninsular Malaysia and Malaysian Borneo (FST: 0.05729, P < 0.001) (Table 4).
Table 4.
FST indices for PkMSP-142, PkMSP-133 and PkMSP-119 from Peninsular Malaysia and Malaysian Borneo
| PkMSP-142 | PkMSP-133 | PkMSP-119 | |
|---|---|---|---|
| Malaysian Borneo | |||
| Peninsular Malaysia | 0.23677* | 0.28257* | 0.05729* |
*P < 0.001
In order to examine whether natural selection contributed to the diversity observed in PkMSP-142, we further analyzed the average difference of dN-dS for all PkMSP-142 sequences. The significant excess of synonymous substitutions (dN-dS = -0.026, P < 0.05) of PkMSP-142 for all 83 samples as well as the samples from Peninsular Malaysia (dN-dS = -0.027, P < 0.05) and Malaysian Borneo (dN-dS = -0.031, P < 0.05), respectively, suggested a purifying selection for MSP-142 fragment of P. knowlesi populations from both Peninsular Malaysia and Malaysian Borneo areas. The negative values of Tajima’s D test (-0.68468) as well as Fu and Li’s D (-1.11074) and F (-1.13645) further supported the proposal for a purifying selection pressure of the PkMSP-142 region for samples from Peninsular Malaysia. On the other hand, the high positive values of Tajima’s D (0.75884), Fu and Li’s D (0.74035) and F (0.87508) found among PkMSP-142 of Malaysian Borneo samples instead suggested balancing selection acts on the fragment in the samples collected from this region (Table 3). When analysis was done for 33 and 19 kDa fragments separately, similar findings were observed in the PkMSP-133 fragment whereby negative purifying selection was suggested to be acting among Peninsular Malaysia samples. High positive values of Tajima’s D and Fu and Li’s D and F among Malaysian Borneo samples suggesting that PkMSP-133 fragments from this region may be under balancing selection pressure. On the other hand, purifying selection is most likely to be acting at the fragment of PkMSP-119 among both Peninsular Malaysia and Malaysian Borneo samples (Table 3).
Phylogenetic analysis
Phylogenetic trees of the 18 PkMSP-142 amino acid haplotypes were constructed using both neighbor-joining and maximum likelihood methods. Both methods produced phylogenetic trees of similar topology and analysis revealed that the Malaysian PkMSP-142 haplotypes were clustered into two main clades. Most of the samples collected from Malaysian Borneo formed one cluster while P. knowlesi samples from Peninsular Malaysia and two shared haplotypes formed another cluster with a laboratory line, the H strain, which also originated from Peninsular Malaysia (Figs. 3, 4). Another neighbor-joining tree was constructed for both PkMSP-133 and PkMSP-119 fragments to determine if they contributed to the haplotype clustering. We found that the phylogenetic tree of PkMSP-133 displayed a similar clustering pattern to the tree constructed using PkMSP-142, suggesting the 33 kDa fragment is playing the role of haplotype clustering as compared to PkMSP-119 (Fig. 5).
Fig. 3.
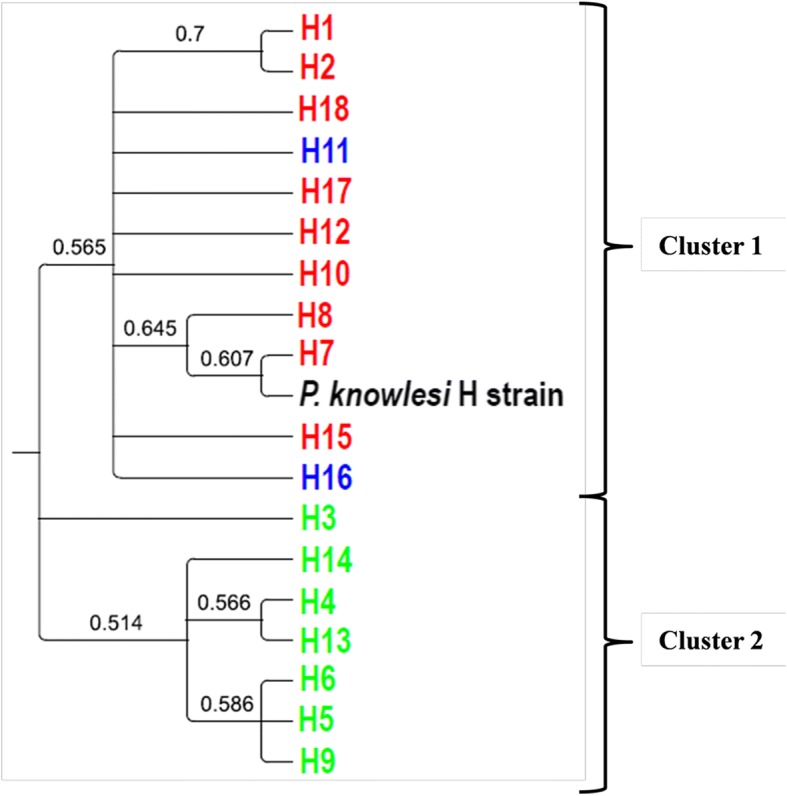
Phylogenetic tree of PkMSP-142 haplotypes. The neighbor-joining method was used to construct the tree. Numbers at nodes indicate percentage support of 1000 bootstrap replicates; only bootstrap values above 50% are shown. Haplotypes from Peninsular Malaysia and Malaysian Borneo are indicated in red and light green, respectively, while the shared haplotypes are indicated in blue
Fig. 4.
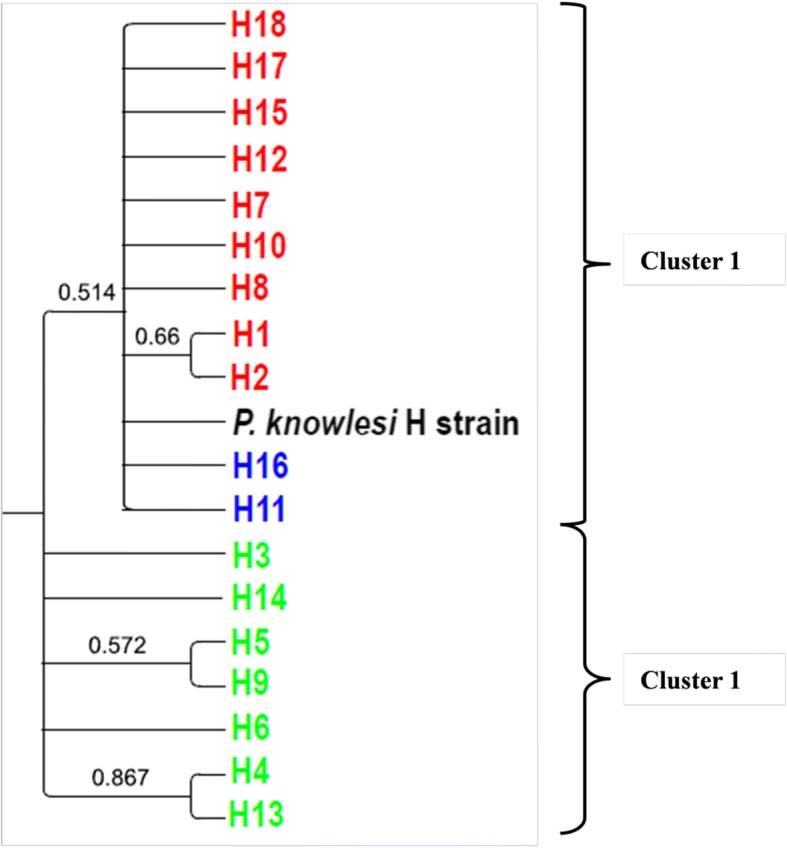
Phylogenetic tree of PkMSP-142 haplotypes. The maximum likelihood method was used to construct the tree. Numbers at nodes indicate percentage support of 1000 bootstrap replicates; only bootstrap values above 50% are shown. Haplotypes from Peninsular Malaysia and Malaysian Borneo are indicated in red and light green, respectively, while the shared haplotypes are indicated in blue
Fig. 5.

Phylogenetic tree of PkMSP-133 and PkMSP-119 haplotypes. The neighbor-joining method was used to construct the tree. Numbers at nodes indicating percentage support of 1000 bootstrap replicates; only bootstrap values above 50% are shown. Haplotypes from Peninsular Malaysia and Malaysian Borneo are indicated in red and light green, respectively, while the shared haplotypes are indicated in blue
Discussion
The MSP-142 is one of the most outstanding malarial vaccine antigens, which is currently at an advanced stage of clinical evaluation [39–41]. However, its extensive polymorphic nature suggests that continuous survey of the genetic polymorphism from a wide range of field isolates is necessary. To date, a considerable amount of studies on MSP-142 have been carried out on P. falciparum [42, 43] and P. vivax [24, 25, 44] but there is a paucity of information on the structure, function or genetic variation in MSP-1 in P. knowlesi. Recent studies by Cheong et al. [20, 45] demonstrated the high immunogenicity of MSP-142 and its ability to elicit protective immunity in P. knowlesi. This suggests that PkMSP-142 may serve as a candidate for malaria vaccine design. However, further evaluation is needed to validate its potential and limitations.
Previously, we conducted a pilot study with sample size of 12 P. knowlesi clinical isolates to determine the genetic diversity of PkMSP-142 [29]. However, a larger sample size is important for precise estimation of genetic diversity parameters in order to characterize the parasite population [30]. Hence, we expanded our work using the same approach, but with a larger sample size covering more states of Malaysia in order to study the genetic polymorphism and natural selection of MSP-142 in Malaysian P. knowlesi samples at a broader scale. All 83 sequences of single P. knowlesi infections were classified into 18 distinct haplotypes with amino acid changes at 17 positions as compared to the reference H strain sequence. Most of the identified haplotypes were novel and have not been reported previously, except for haplotypes H2, H7 and H18 in our previous study [29]. Most of the amino acid substitutions were found concentrated in the PkMSP-133 region whereby only two dimorphic changes (N1762I, S1801Y) and one trimophic change (F1789S/Y) were seen in PkMSP-119. It is known that MSP-119 is highly conserved, in all Plasmodium species including PkMSP-119 of clinical isolates from Malaysia which has recently been reported [46].
The high level of genetic polymorphism observed in the present study (л = 0.0179) was in concordance with the result reported previously (л = 0.0132) [29], further confirming the extensiveness of genetic polymorphism among P. knowlesi population found in Malaysia. This finding is also in agreement with a previous analysis on MSP-142 fragments reported in P. knowlesi-infected orangutan samples from Kalimantan, Indonesia (л = 0.013) [47] as well as other P. knowlesi functional genes, such as Pk-DBPαII (π = 0.013 ± 0.002) [48] and Pk-RAP-1 (π = 0.01298 ± 0.00091) [49]. However, this diversity was lower than the MSP-142 fragment reported previously for both P. falciparum [23, 26, 28] and P. vivax [24, 25, 44]. With regards to non-human primate malaria, PkMSP-142 was also found to be less diverse than MSP-142 of P. cynomolgi and P. inui [47, 50]. The lower genetic diversity of P. knowlesi human infection as compared to other Plasmodium species may be related to the limited intensity of malaria transmission in which to date, human-to-human transmission has not been reported.
The rate of non-synonymous and synonymous mutations (dN-dS) is often used to evaluate the effect of natural selection on gene sequences. Most of the malaria surface antigens with relatively high polymorphism have been reported to be under positive-diversifying selection due to the accumulation of amino acid replacements that may hamper the ability of the host’s immune system to recognize the parasite [51]. However, in the present study, the significant negative values of dN-dS (-0.026) and Tajima’s D statistic (-0.49252) in the entire population of PkMSP-142 sequences suggest that the MSP-142 fragment in our P. knowlesi samples is under the influence of negative natural selection. This finding is in accordance with our previous study on 12 PkMSP-142 human P. knowlesi sequences from Malaysia [29] as well as P. knowlesi from orangutan samples from Kalimantan, Indonesia [47].
When the PkMSP-142 sequences of Peninsular Malaysia and Malaysian Borneo were further analysed as two different populations, some differences were observed whereby the Malaysian Borneo PkMSP-142 was found to have slightly higher diversity (π = 0.01024 ± 0.00061) as compared to those from Peninsular Malaysia (π = 0.009119 ± 0.00031). Although significant negative values of dN-dS were seen on PkMSP-142 as well as PkMSP-133 and PkMSP-119 of both Peninsular Malaysia and Malaysian Borneo populations, negative values of Tajima’s D postulated purifying selection were acting on 42 and 33 kDa fragments of P. knowlesi from Peninsular Malaysia, whereas positive values of Tajima’s D in PkMSP-142 and PkMSP-133 of Malaysian Borneo populations suggested they are under balancing selection pressure. This suggests that a bottleneck event may be happening among P. knowlesi in Peninsular Malaysia during the transition period of parasite transmission from macaque host to humans that drives population expansion or growth. On the other hand, the balancing selection pressure acting on P. knowlesi in Malaysian Borneo population suggests that the active expression of different alleles of the gene allow P. knowlesi to escape the human immune response, thus maintaining a high infection rate. Given that most of the human knowlesi infections were reported from Sabah and Sarawak, Malaysian Borneo [52], this might also be one of the reasons why the selection pressure acting on P. knowlesi sampled from respective Peninsular Malaysia and Malaysian Borneo populations in our study was different. Nevertheless, the low level of genetic polymorphism and negative purifying selection found among PkMSP-119 in both Peninsular Malaysia and Malaysian Borneo populations are in agreement with previous studies [29, 46].
Furthermore, the high genetic differentiation using Wright’s fixation index (FST) was seen in both PkMSP-142 and PkMSP-133 within Peninsular Malaysia and Malaysian Borneo P. knowlesi populations. This might be due to the geographical separation of Peninsular Malaysia and Malaysian Borneo by the South China Sea. These results are similar to previous findings at the genomic level as well as functional genes [46, 53, 54]. On the other hand, the moderate genetic differentiation of PkMSP-119 seen within Peninsular Malaysia and Malaysian Borneo populations further confirmed the theory of the 19 kDa fragment being a highly conserved region which is evolving towards a fixation stage.
Unlike a previous study in which no evidence of geographical clustering was seen among the PkMSP-142 [29], the phylogenetic tree analyses of the present study involving a larger sample size revealed that P. knowlesi MSP-142 haplotypes were clustered into two main groups: one in which samples from Peninsular Malaysia clustered with the laboratory H strain, and the other comprising most of the samples typed from Malaysian Borneo. This is in agreement with previous reports on other P. knowlesi functional proteins such as the Duffy binding protein (PkDBPαII) [48], Pknbpxa [55], PkAMA-1 domain [56] and PkMSP-3 [57] which reported bifurcation of haplotypes, indicating dimorphism of the genes. Recent microsatellite genotyping of P. knowlesi found in humans and macaques have also highlighted the presence of two divergent P. knowlesi populations which have been associated with two natural macaque reservoir host species, the long tailed (Macaca fascicularis) and the pig tailed macaque (Macaca nemestrina) [58].
It is important to characterize genetic diversity in order to understand parasite biology and disease pathogenesis, to evaluate the direct effects of diversity on clinical disease, as well as to develop an effective malaria vaccine [59]. Several studies have associated the genetic diversity of P. falciparum with the clinical outcome of a malaria infection comparing different genotypic determinants in mild and severe cases [60–62]. However, the limitation of our study is that we could not obtain the clinical status of all our 83 P. knowlesi-infected samples; therefore, we were not able to determine whether the severity of malaria episode was associated with a particular genotype of PkMSP-142.
Conclusions
The present study provides an in-depth analysis of the genetic diversity and natural selection of the PkMSP-142 gene among Malaysian samples. PkMSP-142 showed polymorphic characteristics that resulted in 18 distinct haplotypes. Synonymous and non-synonymous mutation analysis indicated negative purifying selection of the gene on the overall P. knowlesi Malaysia population. The separation of PkMSP-142 haplotypes into two main groups has further supported the existence of two distinct P. knowlesi lineages. Most polymorphisms were found in the 33 kDa fragment while the 19 kDa fragment was relatively conserved, highlighting that future studies should focus more in PkMSP-133 as this region appears to be more informative for the development of a knowlesi malaria vaccine.
Additional files
Table S1. Study samples and origin. (DOCX 20 kb)
Figure S1. Nucleotide variants of 38 haplotypes of P. knowlesi MSP1-42. Identical nucleotides are marked as dots, while polymorphic sites are shaded in light green (two variants) and light blue (three variants), respectively. (ZIP 699 kb)
Acknowledgements
We thank the staff from the Department of Orang Asli Development (JAKOA) for assisting the collection of community blood samples.
Funding
The research was supported by funding from University of Malaya Student Grant (PG056-2013A for NJY) and UM/MoHE High Impact Research Grant (H-20001-00-E000061 for YALL). BPH acknowledges The Ministry of Higher Education Malaysia under Fundamental Research Grant Scheme [Grant code: 600-RMI/FRGS 5/3 (136/2014) and FRGS/1/2015/ST03/UCSI/01/1] for supporting the community blood samples collection activities.
Availability of data and materials
All data supporting the conclusions of this study are included within the article and its additional files. The nucleotide sequences of PkMSP-142 generated in the present study were submitted to GenBank under accession numbers MH796675-MH796757.
Abbreviations
- dNTPs
Deoxynucleotide triphosphates
- PCR
Polymerase chain reaction
Authors’ contributions
YALL and IV designed the study and supervised the study process. BPH, XTG, AM, RN, YR and SHC collected the community blood samples. TW and TSY provided the clinical blood samples. NJY performed sequence and phylogenetic analyses. BPH helped in data analyses. NJY, YALL and IV wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study protocol was approved by the Medical Research Ethics Committee of the University of Malaya Medical Centre (MEC ref. no. 824.11). Participants’ informed consent was obtained prior to blood sample collection. For all child participants, written and signed, or thumb-printed informed consent was obtained from the parents or guardians on behalf of their children.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Nan Jiun Yap, Email: ynj1203@gmail.com.
Indra Vythilingam, Email: indrav@um.edu.my.
Boon Peng Hoh, Email: hoh.boonpeng@gmail.com.
Xiang Ting Goh, Email: gohxt88@gmail.com.
Azdayanti Muslim, Email: azdayantimuslim@gmail.com.
Romano Ngui, Email: romano@um.edu.my.
Yamuna Rajoo, Email: yamuna.rajoo@rmit.edu.au.
Seow Huey Choy, Email: seow33huey@gmail.com.
Timothy William, Email: tim7008@gmail.com.
Tsin Wen Yeo, Email: Tsin.Yeo@menzies.edu.au.
Yvonne Ai-Lian Lim, Email: limailian@um.edu.my.
References
- 1.World Health Organization. World Malaria Report 2017. http://apps.who.int/iris/bitstream/10665/259492/1/9789241565523-eng.pdf?ua=1. Accessed 16 June 2018.
- 2.Garnham PCC. Malaria Parasites and Other Haemosporidia. Oxford: Blackwell Scientific Publications; 1966. [Google Scholar]
- 3.Knowles R, Das Gupta BM. A study of monkey-malaria and its experimental transmission to man. Ind Med Gaz. 1932;67:301–321. [PMC free article] [PubMed] [Google Scholar]
- 4.Cox-Singh J, Davis TME, Lee KS, Shamsul SSG, Matusop A, Ratnam S, et al. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin Infect Dis. 2008;46:165–171. doi: 10.1086/524888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh B, Lee KS, Matusop A, Radhakrishnan A, Shamsul SSG, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–1024. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- 6.Ramasamy R. Zoonotic malaria - global overview and research and policy needs. Front Public Health. 2014;2:123. doi: 10.3389/fpubh.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.William T, Rahman HA, Jelip J, Ibrahim MY, Menon J, Grigg MJ, et al. Increasing incidence of Plasmodium knowlesi malaria following control of P. falciparum and P. vivax malaria in Sabah, Malaysia. PLoS Negl Trop Dis. 2013;7:e2026. doi: 10.1371/journal.pntd.0002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.William T, Jelip J, Menon J, Anderios F, Mohammad R, Mohammad TAA, et al. Changing epidemiology of malaria in Sabah, Malaysia: increasing incidence of Plasmodium knowlesi. Malar J. 2014;13:390. doi: 10.1186/1475-2875-13-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moyes CL, Henry AJ, Golding N, Huang Z, Singh B, Baird JK, et al. Defining the geographical range of the Plasmodium knowlesi reservoir. PLoS Negl Trop Dis. 2014;8:e2780. doi: 10.1371/journal.pntd.0002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vythilingam I. Plasmodium knowlesi in humans: a review on the role of its vectors in Malaysia. Trop Biomed. 2010;27:1–12. [PubMed] [Google Scholar]
- 11.Barber BE, William T, Grigg MJ, Menon J, Auburn S, Marfurt J, et al. A prospective comparative study of knowlesi, falciparum and vivax malaria in Sabah, Malaysia: high proportion with severe disease from Plasmodium knowlesi and P. vivax but no mortality with early referral and artesunate therapy. Clin Infect Dis. 2012;56:383–397. doi: 10.1093/cid/cis902. [DOI] [PubMed] [Google Scholar]
- 12.Cox-Singh J, Hiu J, Lucas SB, Divis PC, Zulkarnean M, Chandran P, et al. Severe malaria - a case of fatal Plasmodium knowlesi infection with post-morterm findings: a case report. Malar J. 2010;9:10. doi: 10.1186/1475-2875-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daneshvar C, Davis TME, Cox-Singh J, Rafa’ee MZ, Zakaria SK, Divis PC, et al. Clinical and laboratory features of human Plasmodium knowlesi infection. Clin Infect Dis. 2009;49:852–860. doi: 10.1086/605439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kantele A, Jokiranta TS. Review cases with the emerging fifth human malaria parasite Plasmodium knowlesi. Clin Infect Dis. 2010;52:1356–1362. doi: 10.1093/cid/cir180. [DOI] [PubMed] [Google Scholar]
- 15.Takala SL, Plowe CV. Genetic diversity and malaria vaccine design, testing and efficacy: preventing and overcoming “vaccine resistant malaria”. Parasite Immunol. 2009;31:560–573. doi: 10.1111/j.1365-3024.2009.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holder AA, Freeman RR. Protective antigens of rodent and human bloodstage malaria. Philos Trans R Soc Lond B Biol Sci. 1984;307:171–177. doi: 10.1098/rstb.1984.0117. [DOI] [PubMed] [Google Scholar]
- 17.Holder AA, Blackman MJ, Burghaus PA, Chappel JA, Ling IT, McCallum-Deighton N, et al. A malaria merozoite surface protein (MSP1)-structure, processing and function. Mem Inst Oswaldo Cruz. 1992;87:37–42. doi: 10.1590/S0074-02761992000700004. [DOI] [PubMed] [Google Scholar]
- 18.Blackman MJ, Ling IT, Nicholls SC, Holder AA. Proteolytic processing of the Plasmodium falciparum merozoite surface protein-1 produces a membrane-bound fragment containing two epidermal growth factor-like domains. Mol Biochem Parasitol. 1991;49:29–33. doi: 10.1016/0166-6851(91)90127-R. [DOI] [PubMed] [Google Scholar]
- 19.Blackman MJ, Dennis ED, Hirst EM, Kocken CH, Scott-Finnigan TJ, Thomas AW. Plasmodium knowlesi: secondary processing of the malaria merozoite surface protein-1. Exp Parasitol. 1996;83:229–239. doi: 10.1006/expr.1996.0069. [DOI] [PubMed] [Google Scholar]
- 20.Cheong FW, Fong MY, Lau YL, Mahmud R. Immunogenecity of bacterial expressed recombinant Plasmodium knowlesi merozoite surface protein-142 (MSP-142) Malar J. 2013;12:454. doi: 10.1186/1475-2875-12-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nwuba RI, Sodeinde O, Anumudu CI, Omosun YO, Odaibo AB, Holder AA, et al. The human immune response to Plasmodium falciparum includes both antibodies that inhibit merozoite surface protein 1 secondary processing and blocking antibodies. Infect Immun. 2002;70:5328–5331. doi: 10.1128/IAI.70.9.5328-5331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patino JAG, Holder AA, McBride JS, Blackman MJ. Antibodies that inhibit malaria merozoite surface protein-1 processing and erythrocyte invasion are blocked by naturally acquired human antibodies. J Exp Med. 1997;186:1689–1699. doi: 10.1084/jem.186.10.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamani Z, Razavi MR, Sadeghi S, Naddaf S, Pourfallah F, Mirkhani F, et al. Sequence diversity of the C-terminal region of Plasmodium falciparum merozoite surface protein 1 in southern Iran. Southeast Asian J Trop Med Public Health. 2009;40:1–9. [PubMed] [Google Scholar]
- 24.Dias S, Longacre S, Escalante AA, Udagama-Randeniya PV. Genetic diversity and recombination at the C-terminal fragment of the merozoite surface protein-1 of Plasmodium vivax (PvMSP-1) in Sri Lanka. Infect Genet Evol. 2011;11:145–156. doi: 10.1016/j.meegid.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Kang JM, Ju HL, Kang YM, Lee DH, Moon SU, Sohn WM. Genetic polymorphism and natural selection in the C-terminal 42 kDa region of merozoite surface protein-1 among Plasmodium vivax Korean isolates. Malar J. 2012;11:206. doi: 10.1186/1475-2875-11-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehrizi AA, Zakeri S, Salmanian AH, Sanati MH, Djadid ND. Plasmodium falciparum: sequence analysis of the gene encoding the C-terminus region of the merozoite surface protein-1, a potential malaria vaccine antigen, in Iranian clinical isolates. Exp Parasitol. 2008;118:378–385. doi: 10.1016/j.exppara.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Pan D, Hu J, Ma Q, Pan W, Li M. Diversity and prevalence of the C-terminal region of Plasmodium falciparum merozoite surface protein 1 in China. Acta Trop. 2010;116:200–205. doi: 10.1016/j.actatropica.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Viputtigul K, Tungpukdee N, Ruangareerate T, Luplertlop N, Wilairatana P, Gaywee J, et al. C-terminal polymorphism of Plasmodium falciparum merozoite surface protein-1 (MSP-1) from Tak Province, Thailand. Southeast Asian J Trop Med Public Health. 2013;44:1–12. [PubMed] [Google Scholar]
- 29.Yap NJ, Goh XT, Koehler AV, William T, Yeo TW, Vythilingam I, et al. Genetic diversity in the C-terminus of merozoite surface protein 1 among Plasmodium knowlesi isolates from Selangor and Sabah Borneo, Malaysia. Infect Genet Evol. 2017;54:39–46. doi: 10.1016/j.meegid.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 30.Bashalkhanov S, Pandey M, Rajora OP. A simple method for estimating genetic diversity in large populations from finite sample sizes. BMC Genet. 2009;10:84. doi: 10.1186/1471-2156-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh B, Bobogare A, Cox-Singh J, Snounou G, Abdullah MS, Rahman H. A genus and species specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am J Trop Med Hyg. 1999;60:687–692. doi: 10.4269/ajtmh.1999.60.687. [DOI] [PubMed] [Google Scholar]
- 32.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 35.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 36.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wright S. The genetical structure of populations. Ann Eugenics. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 39.Ellis RD, Sagara I, Doumbo O, Wu Y. Blood stage vaccines for Plasmodium falciparum: current status and the way forward. Hum Vaccin. 2010;6:627–634. doi: 10.4161/hv.6.8.11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huaman MC, Martin LB, Malkin E, Narum DL, Miller LH, Mahanty S, et al. Ex vivo cytokine and memory T cell responses to the 42-kDa fragment of Plasmodium falciparum merozoite surface protein-1 in vaccinated volunteers. J Immunol. 2008;180:1451–1461. doi: 10.4049/jimmunol.180.3.1451. [DOI] [PubMed] [Google Scholar]
- 41.Malkin E, Long CA, Stowers AW, Zou L, Singh S, MacDonald NJ, et al. Phase 1 study of two merozoite surface protein 1 (MSP 142) vaccines for Plasmodium falciparum malaria. PLoS Clin Trials. 2007;2:e12. doi: 10.1371/journal.pctr.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gueye NSG, Ntoumi F, Vouvoungui C, Kobawila SC, NKombo M, Mouanga AM, et al. Plasmodium falciparum merozoite protein-1 genetic diversity and multiplicity of infection in isolates from Congolese children consulting in a pediatric hospital in Brazzaville. Acta Trop. 2018;183:78–83. doi: 10.1016/j.actatropica.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 43.Somé AF, Bazié T, Zongo I, Yerbanga RS, Nikiéma F, Neya C, et al. Plasmodium falciparum msp1 and msp2 genetic diversity and allele frequencies in parasites isolated from symptomatic malaria patients in Bobo-Dioulasso, Burkina Faso. Parasit Vectors. 2018;11:323. doi: 10.1186/s13071-018-2895-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou X, Tambo E, Su J, Fang Q, Ruan W, Chen JH, et al. Genetic diversity and natural selection in 42 kDa region of Plasmodium vivax merozoite surface protein-1 from China-Myanmar endemic border. Korean J Parasitol. 2017;55:473–480. doi: 10.3347/kjp.2017.55.5.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheong FW, Fong MY, Lau YL. Identification and characterization of epitopes on Plasmodium knowlesi merozoite surface protein-142 (MSP-142) using synthetic peptide library and phage display library. Acta Trop. 2016;154:89–94. doi: 10.1016/j.actatropica.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 46.Ahmed MA, Fauzi M, Han ET. Genetic diversity and natural selection of Plasmodium knowlesi merozoite surface protein 1 paralog gene in Malaysia. Malar J. 2018;17:115. doi: 10.1186/s12936-018-2256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacheco MA, Reid MJ, Schillaci MA, Lowenberger CA, Galdikas BM, Jones-Engel L, et al. The origin of malarial parasites in orangutans. PLoS One. 2012;7:e34990. doi: 10.1371/journal.pone.0034990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fong MY, Lau YL, Chang PY, Anthony CN. Genetic diversity, haplotypes and allele groups of Duffy binding protein (PkDBPαII) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Parasit Vectors. 2014;7:161. doi: 10.1186/1756-3305-7-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rawa MS, Fong MY, Lau YL. Genetic diversity and natural selection in the rhoptry-associated protein 1 (RAP-1) of recent Plasmodium knowlesi clinical isolates from Malaysia. Malar J. 2016;15:62. doi: 10.1186/s12936-016-1127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pacheco MA, Poe AC, Collins WE, Lal AA, Tanabe K, Kariuki SK, et al. A comparative study of the genetic diversity of the 42 kDa fragment of the merozoite surface protein 1 in Plasmodium falciparum and P. vivax. Infect Genet Evol. 2007;7:180–187. doi: 10.1016/j.meegid.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Escalante AA, Cornejo OE, Rojas A, Udhayakumar V, Lal AA. Assessing the effect of natural selection in malaria parasites. Trends Parasitol. 2004;20:388–395. doi: 10.1016/j.pt.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev. 2013;26:165–184. doi: 10.1128/CMR.00079-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed MA, Fong MY, Lau YL, Yusof R. Clustering and genetic differentiation of the normocyte binding protein (nbpxa) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia and Malaysia Borneo. Malar J. 2016;15:241. doi: 10.1186/s12936-016-1294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Assefa S, Lim C, Preston MD, Duffy CW, Nair MB, Adroub SA, et al. Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci USA. 2015;112:13027–13032. doi: 10.1073/pnas.1509534112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinheiro MM, Ahmed MA, Millar SB, Sanderson T, Otto TD, Lu WC, et al. Plasmodium knowlesi genome sequences from clinical isolates reveal extensive genomic dimorphism. PLoS One. 2015;10:e0121303. doi: 10.1371/journal.pone.0121303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fong MY, Wong SS, Silva JR, Lau YL. Genetic polymorphism in domain I of the apical membrane antigen-1 among Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Acta Trop. 2015;152:145–150. doi: 10.1016/j.actatropica.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 57.De Silva JR, Lau YL, Fong MY. Genetic clustering and polymorphism of the merozoite surface protein-3 of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Parasit Vectors. 2017;10:2. doi: 10.1186/s13071-016-1935-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Divis PC, Singh B, Anderios F, Hisam S, Matusop A, Kocken CH, et al. Admixture in humans of two divergent Plasmodium knowlesi populations associated with different macaque host species. PLoS Pathog. 2015;11:e1004888. doi: 10.1371/journal.ppat.1004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neafsey DE, Galinsky K, Jiang RHY, Young L, Skykes SM, Saif S, et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012;44:1046–1050. doi: 10.1038/ng.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ariey F, Hommel D, Scanf CL, Duchemin JB, Peneau C, Hulin A, et al. Association of severe malaria with a specific Plasmodium falciparum genotype in French Guiana. J Infect Dis. 2001;184:237–241. doi: 10.1086/322012. [DOI] [PubMed] [Google Scholar]
- 61.Kiwuwa MS, Ribacke U, Moll K, Byarugaba J, Lundblom K, Färnert A, et al. Genetic diversity of Plasmodium falciparum infections in mild and severe malaria of children from Kampala, Uganda. Parasitol Res. 2013;112:1691–1700. doi: 10.1007/s00436-013-3325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nicastri E, Paglia MG, Severini C, Ghirga P, Bevilacqua N, Narciso P. Plasmodium falciparum multiple infections, disease severity and host characteristics in malaria affected travellers returning from Africa. Travel Med Infect Dis. 2008;6:205–209. doi: 10.1016/j.tmaid.2008.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Study samples and origin. (DOCX 20 kb)
Figure S1. Nucleotide variants of 38 haplotypes of P. knowlesi MSP1-42. Identical nucleotides are marked as dots, while polymorphic sites are shaded in light green (two variants) and light blue (three variants), respectively. (ZIP 699 kb)
Data Availability Statement
All data supporting the conclusions of this study are included within the article and its additional files. The nucleotide sequences of PkMSP-142 generated in the present study were submitted to GenBank under accession numbers MH796675-MH796757.