

Abstract
Neutrophil serine proteases (NSPs), such as neutrophil elastase (NE), are activated by dipeptidyl peptidase 1 (DPP1) during neutrophil maturation. High NSP levels can be detrimental, particularly in lung tissue, and inhibition of NSPs is therefore an interesting therapeutic opportunity in multiple lung diseases, including chronic obstructive pulmonary disease (COPD) and bronchiectasis. We conducted a randomized, placebo‐controlled, first‐in‐human study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple oral doses of the DPP1 inhibitor AZD7986 in healthy subjects. Pharmacokinetic and pharmacodynamic data were analyzed using nonlinear mixed effects modeling and showed that AZD7986 inhibits whole blood NE activity in an exposure‐dependent, indirect manner—consistent with in vitro and preclinical predictions. Several dose‐dependent, possibly DPP1‐related, nonserious skin findings were observed, but these were not considered to prevent further clinical development. Overall, the study results provided confidence to progress AZD7986 to phase II and supported selection of a clinically relevant dose.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Dipeptidyl peptidase 1 (DPP1) is critical to the activation of neutrophil serine proteases (NSPs) during neutrophil maturation. Pharmacological inhibition of DPP1 has been shown to reduce NSP activity in preclinical species, but no clear effect has been shown in man. A complete absence of DPP1 activity is associated with palmoplantar hyperkeratosis and periodontitis.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What is the safety and tolerability of the DPP1 inhibitor AZD7986 after dosing in healthy subjects, and is there an exposure‐dependent relationship between AZD7986 and whole blood activity of the NSP neutrophil elastase (NE)?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Daily dosing of AZD7986 led to an exposure‐related reduction in NE activity with a delayed onset of effect consistent with neutrophil maturation rates. AZD7986 was generally well tolerated. However, several nonserious, possibly DPP1‐related, adverse skin events were observed.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Inhibition of DPP1 remains a tractable target for disease modification in patients suffering from neutrophil‐driven inflammatory diseases, such as COPD and related lung diseases.
Dipeptidyl peptidase 1 (DPP1, also known as cathepsin C) is broadly expressed in human tissues, but particularly in cells of hematopoietic lineage such as neutrophils. In neutrophils, DPP1 controls the activation of the neutrophil serine proteases (NSPs): neutrophil elastase (NE), proteinase 3 (Pr3), and cathepsin G (CatG).1 Activation is achieved by removal of the N‐terminal dipeptide sequences of NSP zymogens, a process taking place during azurophilic granule assembly early in the cell maturation process in the bone marrow (Figure 1 a).1, 2, 3
Figure 1.
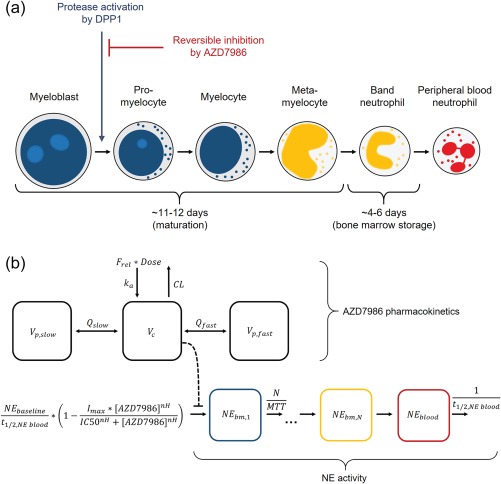
(a) An illustration of neutrophil maturation stages, time of NSP activation, and expected neutrophil maturation rates in healthy individuals.3, 22 (b) Outline of the final model used to describe AZD7986 PK and NE activity data. AZD7986 PK was modeled by a three‐compartment model (upper part) and whole blood NE activity by a transit compartment model (lower part). AZD7986 plasma concentrations were assumed to inhibit the amount of active NE entering the first transit bone marrow (bm) compartment.
Although considered protective under normal conditions,4 NSPs do not seem to be critical for overall neutrophil function. This hypothesis is supported by the fact that patients with Papillon‐Lefèvre syndrome (PLS)—a rare autosomal recessive disease characterized by mutations of the DPP1 gene and near‐complete loss of DPP1 function and NSP activity4, 5—do not suffer from major infections.6 The main symptoms of PLS instead include palmoplantar hyperkeratosis and severe periodontitis.7, 8, 9 However, it is currently unclear if these are a consequence of low NSP activity or linked to some other DPP1‐dependent mechanism.
In contrast to low NSP activity, high levels or overactivity of NSPs have been shown to have detrimental effects, particularly in lung tissue. For instance, increased lung levels of NSPs have been implicated in several of the key pathological features of chronic obstructive pulmonary disease (COPD), including inflammation, mucus hypersecretion, and emphysema.10, 11, 12 Furthermore, elevated sputum NE activity has been associated with exacerbations and lung function decline in patients with bronchiectasis.13 Targeting NSP activation is therefore an interesting therapeutic opportunity for the treatment of several potentially neutrophil‐driven lung diseases, including COPD and bronchiectasis, but also alpha1‐antitrypsin deficiency and cystic fibrosis.14, 15
AZD7986 is a small‐molecule, competitive, and reversible inhibitor of DPP1, with high potency and selectivity as measured in vitro.16 In human primary bone marrow‐derived CD34 + neutrophil progenitor cells, AZD7986 has been shown to completely inhibit activation of all three NSPs (NE, Pr3, and CatG) in a similar concentration‐dependent manner (IC50, ∼60–200 nM, Hill coefficient, ∼0.65). Furthermore, in vivo inhibition of NSP activity has been confirmed in rat blood, both with AZD7986 and structurally similar compounds.16, 17 Importantly, both the onset of inhibition and recovery of NSP activity in rat blood is delayed compared to initiation and cessation of dosing, consistent with the hypothesis that NSP activation is limited to a relatively short timeframe during early neutrophil maturation in the bone marrow.18 In rat blood, steady‐state inhibition of NSP activity was reached within 8 days of dosing. In man, however, the time to steady state is expected to be longer, considering the slower maturation process of human neutrophils (Figure 1 a).
The effect of DPP1 inhibition in humans has previously been investigated in a phase I study with the irreversible DPP1 inhibitor GSK2793660.19 However, although the authors report sustained inhibition of DPP1 in whole blood during 21 days of dosing, little to no effect on NSP activity was demonstrated. Instead, the only clear downstream effect reported was epidermal desquamation on palmar and plantar surfaces, findings not unlike clinical manifestations of PLS.
Herein we present the pharmacokinetic, pharmacodynamic, and safety results of a single and multiple ascending dose study with AZD7986, showing—for the first time—significant downstream effects on NSP activity in man with a DPP1 inhibitor. Specifically, we present a comprehensive model‐based pharmacokinetic‐pharmacodynamic (PK‐PD) analysis of the relationship between AZD7986 PK and the inhibition and dynamics of whole blood NE activity, and its relation to neutrophil maturation and turnover. We also give a detailed report of observed adverse events associated with known symptoms of PLS, and discuss the importance of the study results to support further clinical development of AZD7986.
METHODS
Subjects and study design
The study was designed as a randomized, placebo‐controlled, dose‐escalation study (http://clinicaltrial.gov number NCT0230357), performed in accordance with the Declaration of Helsinki and the International Conference on Harmonisation/Good Clinical Practice. It was carried out at the Parexel Early Phase Clinical Unit (Harrow, London, UK) and was approved by the national regulatory authority (Medicines and Healthcare Products Regulatory Agency) and the Independent Ethics Committee (NRES London Brent). All subjects were healthy and provided written informed consent prior to initiation of any study‐specific procedures. The study consisted of two parts. In Part I, subjects received single ascending oral doses of AZD7986, and in Part II, subjects received multiple ascending oral doses of AZD7986. Doses, length of treatment (in Part II), and number of subjects were adjusted based on review of safety, PK, and PD data from previous cohorts.
Data collection and analysis
Safety assessments were performed at appropriate intervals to timely capture any signs of potential safety concerns. Furthermore, samples for PK and PD analysis were collected at frequent timepoints to allow for accurate characterization of PK and PD parameters. Safety monitoring and the sampling and analysis of PK and PD data is described thoroughly in the Supplementary Methods.
RESULTS
Subjects and study design
In total, 81 healthy male subjects were randomized to receive either a single (Part I) or multiple (Part II) oral doses of AZD7986 or placebo. Subject demographics can be found in Table S1. In Part I, five different doses (5, 15, 35, 50, or 65 mg) of AZD7986 were administered in cohorts of nine subjects (of which three subjects received placebo). In Part II, three different doses (10, 25, or 40 mg) were administered once daily in cohorts of 9, 11, and 16 subjects, respectively (of which three, three, and six subjects received placebo). Subjects in the 10 mg cohort were dosed for 21 days, while subjects in the 25 and 40 mg cohorts were dosed for 28 days. All subjects remained at the clinical unit for 1 week after cessation of dosing, and subjects in the 25 and 40 mg cohorts were also instructed to return for frequent follow‐up visits up to 1 month after leaving the unit. The decision to extend the dosing and follow‐up period in the 25 and 40 mg cohorts was made based on emerging NE activity data from the 10 mg cohort.
Pharmacokinetics
AZD7986 PK profiles from Parts I and II of the study are shown in Figure 2 a,b. Plasma concentrations increased in a dose‐dependent, supra‐proportional way, reaching maximum levels (Cmax) at 0.5–1.5 hours after dosing and thereafter declining in a multiphasic manner. Terminal half‐life after a single dose of AZD7986 was estimated to be 20–26 hours, and there was a trend towards an increased half‐life after repeated dosing (26–34 hours). Steady state was confirmed for all doses in Part II based on analysis of predose plasma concentrations on Days 18–20 and Days 21/28. Between‐subject variability in Cmax and area under the curve (AUC), as calculated using noncompartmental analysis (NCA) was generally low to moderate, with a coefficient of variation between 10–40% (Table S2).
Figure 2.
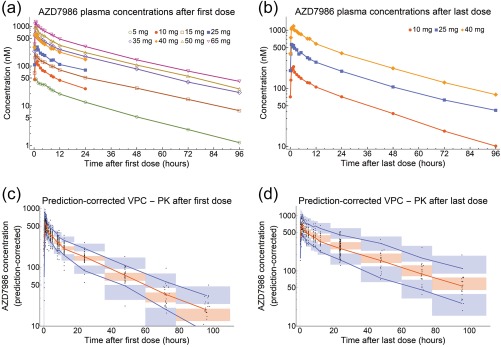
Observed geometric mean AZD7986 plasma concentrations after single/first dose (a) and after once‐daily dosing for 21 or 28 days (b). Data shown in (a) come from both Parts I and II, while data shown in (b) come from Part II only. In Part I, plasma concentrations were measured for up to 96 hours following a single dose of AZD7986. In Part II, plasma concentrations were measured for up to 24 hours following the first dose and 96 hours following the last dose of AZD7986. (c,d) Visual predictive checks (VPCs) of the final PK model. VPCs were prediction‐corrected and stratified on day (c = first dose, d = Day 21/28). Black dots show the individual observations. Blue lines show the 5th and 95th percentiles of the observed data, and red lines show the median of the observed data. The blue and red shaded areas show the 95% CIs of the model‐predicted 5th, median, and 95th percentiles based on simulated data from 1,000 simulations.
AZD7986 plasma exposure in Parts I and II was adequately described using a three‐compartment model (Figure 1 b) with first‐order absorption, dose‐dependent oral bioavailability, and a change in slowly equilibrating peripheral volume of distribution and bioavailability between single/first dose and steady state. Final model parameters are presented in Table 1. Dose‐dependent oral bioavailability was modeled using a power model and estimated to increase almost 2‐fold between 5 and 65 mg. Furthermore, slowly equilibrating peripheral volume of distribution and bioavailability were estimated to change by 34% and 24%, respectively, between Day 1 and steady state. Visual predictive checks20 of the model are presented in Figure 2 c,d, and a detailed summary of the model development process can be found in the Supplementary Methods.
Table 1.
Summary of PK and NE activity model parameter estimates
| Parameter | Description | Estimate | 90% CI |
|---|---|---|---|
| AZD7986 pharmacokinetics | |||
| Vc (L/kg) | Apparent volume per kg bodyweight of central plasma compartment | 0.368 | (0.257, 0.529) |
| Vp,fast (L/kg) | Apparent volume per kg bodyweight of rapidly equilibrating tissue compartment | 2.45 | (2.15, 2.79) |
| Vp,slow (L/kg) | Apparent volume per kg bodyweight of slowly equilibrating tissue compartment (single dose) | 1.97 | (1.70, 2.28) |
| Vp,slow,steadystate_change (%) | Percent change in volume of slowly equilibrating tissue compartment between first dose and steady state | 34.0 | (23.6, 45.2) |
| CL (L/h) | Apparent clearance from central compartment | 12.0 | (10.7, 13.4) |
| Qfast (L/h) | Apparent intercompartmental clearance between plasma and rapidly equilibrating tissues | 122 | (96.4, 154) |
| Qslow (L/h) | Apparent intercompartmental clearance between plasma and slowly equilibrating tissues | 14.7 | (11.2, 19.4) |
| Frel,5 mg | Relative bioavailability at 5 mg (single dose) | 1 (fixed) | — |
| Fslope | Slope parameter for bioavailability vs. dose power model | 0.228 | (0.180, 0.288) |
| Fsteadystate_change (%) | Percent change in bioavailability between first dose and steady state | 24.3 | (22.5, 26.2) |
| ka (h−1) | Absorption rate constant | 0.809 | (0.640, 1.02) |
| Alag (h) | Absorption lag time | 0.159 | (0.144, 0.176) |
| IIV Vc (%) | Interindividual variability in Vcentral | 53.6 | (23.8, 75.5) |
| IIV CL (%) | Interindividual variability in CL | 22.2 | (15.4, 27.5) |
| IIV Frel (%) | Interindividual variability in Frel | 18.2 | (13.4, 22.0) |
| IIV ka (%) | Interindividual variability in ka | 28.8 | (20.4, 35.5) |
| IIV Alag (%) | Interindividual variability in Alag | 21.3 | (0, 30.3) |
| εPK (%) | Additive error on log scale | 10.5 | (10.3, 10.7) |
| Neutrophil elastase activitya | |||
| N | Number of transit compartments in neutrophil maturation model | 27b | — |
| MTT (h) | Mean transit time from NE activation to neutrophil release into systemic circulation | 348 | (318, 381) |
| t1/2, NE blood (h) | Blood neutrophil half‐life | 7 (fixed)c | — |
| IC50 (nM) | Half‐maximum inhibitory concentration of AZD7986 | 394 | (319, 486) |
| Imax | Maximum achievable inhibition of NE activity | 1 (fixed)d | — |
| nH | Hill coefficient | 0.697 | (0.594, 0.819) |
| IIV MTT (%) | Interindividual variability in MTT | 10.6 | (0, 15.8) |
| IIV IC50 (%) | Interindividual variability in IC50 | 33.3 | (0, 53.5) |
| εNE (%) | Additive error on log scale | 14.9 | (14.2, 15.6) |
A complete list of NE activity parameter estimates can be found at the end of the Supplementary Methods.
See Figure S2 for objective function values using different numbers of compartments.
Fixed according to reported vascular neutrophil half‐life in healthy subjects.22
Fixed based on in vitro and preclinical data supporting complete NE inhibition at high AZD7986 concentrations.
Whole blood neutrophil elastase activity
The relative change from baseline in whole blood NE activity after repeated dosing of AZD7986 (Part II) is shown in Figure 3. A clear response to treatment—starting ∼12 days after treatment initiation—was observed when comparing the three active doses to placebo. Although the dosing and observation period used for the 10 mg cohort was too short to clearly see maximum effect, data indicated an increased inhibition with increasing dose, and NE activity was ultimately reduced by more than 50% on average in the 40 mg dose group. AZD7986 had no clinically significant effect on the number of neutrophils in blood (Figure S1), suggesting that DPP1 inhibition does not affect the maturation or release of neutrophils into the systemic circulation.
Figure 3.
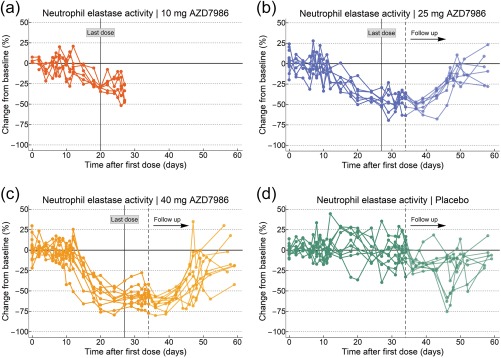
Observed percent change from baseline in whole blood NE activity after 21 or 28 days of repeated dosing with 10, 25, or 40 mg AZD7986 (a–c) or placebo (d). Each line represents data from one subject. For each subject, baseline NE activity was defined as the median of all observations up to Day 7 after initiation of dosing. Follow‐up NE activity data were not collected in the 10 mg cohort. Placebo data (d) have been pooled from all three cohorts.
In the 25 and 40 mg cohorts, NE activity was assessed at multiple follow‐up visits to study the recovery of activity after cessation of dosing (Figure 3). Noticeably, there was a slight drop and increased variability in measured activity after the subjects left the clinical unit. These effects were most apparent in the placebo group, but seen also for the two AZD7986 doses (25 and 40 mg). Regardless, however, follow‐up data clearly showed that NE activity recovers once treatment is stopped, and that the recovery phase exhibits very similar dynamics compared to the onset of effect.
To model the dynamics of NE activity, a transit compartment model was used to represent the maturation process of neutrophils in the bone marrow from the time of NE activation to neutrophil release into the systemic circulation (Figure 1 b). The transit compartment model was linked to the AZD7986 PK model by assuming that AZD7986 plasma concentrations inhibit the amount of active NE entering the first transit compartment. The level of inhibition was described by a sigmoid Emax function and placebo effects modeled by a three‐parameter logistic function of time, multiplied with the NE activity in the last (blood) compartment (Supplementary Methods). Visual predictive checks of the model can be found in Figure 4 and show that the model adequately captures the response and variability in NE activity data.
Figure 4.
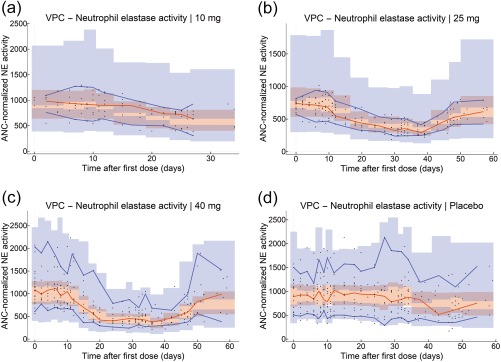
Visual predictive checks (VPCs) of the final NE activity model. VPCs were stratified on dose group (a = 10 mg, b = 25 mg, c = 40 mg, d = pooled placebo). Black dots show the individual observations of ANC‐normalized NE activity (see Supplementary Methods). Blue lines show the 5th and 95th percentiles of the observed data, and red lines show the median of the observed data. The blue and red shaded areas show the 95% CIs of the model‐predicted 5th, median, and 95th percentiles based on simulated data from 1,000 simulations.
Basically, the use of a transit compartment model assumes that the neutrophil lifespan from NE activation to systemic release is gamma distributed,21 with a mean and variance described by the mean transit time (MTT) and number of transit compartments (N). For the population of healthy subjects in this study, the neutrophil MTT was estimated to be ∼15 days (Table 1). Furthermore, the number of compartments found to best describe the data was 27 (Table 1, Figure S2), indicating that lifespan variance is low and that neutrophils spend a similar amount of time in the bone marrow before being released into the circulation. Together, the estimated MTT, N, and PK of AZD7986 imply an average time to steady state of whole blood NE activity—after initiation of dosing—of about 25 days.
The sigmoid relationship between AZD7986 exposure and inhibition of NE activity was best described by a Hill coefficient of 0.697 and a half‐maximal inhibitory AZD7986 concentration (IC50) of 394 nM (Table 1). The estimated steady‐state mean dose and concentration responses, together with 95% confidence intervals (CIs), are presented in Figure 5 a,b, showing that 10, 25, and 40 mg of once‐daily dosing of AZD7986 result in a significant mean steady‐state inhibition of NE activity of 30, 49, and 59%, respectively. Figure 5 c further highlights the model‐predicted dynamics of NE activity after prolonged dosing for 12 weeks with different doses of AZD7986, and Figure 5 d illustrates the inherent insensitivity of the system to missed doses, using the mid dose (25 mg) studied in Part II as an example.
Figure 5.
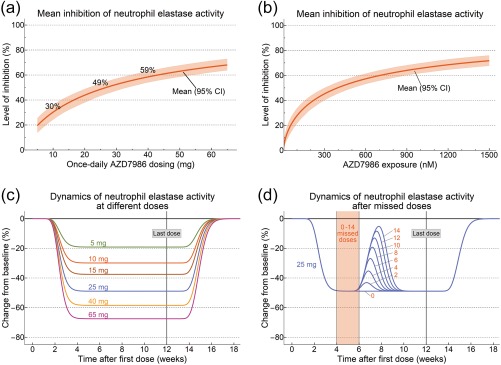
Estimated mean and 95% CIs for the steady‐state dose (a) and exposure (b) response of whole blood NE activity after once‐daily dosing with AZD7986. The mean level of inhibition and confidence intervals were derived using population typical PK and PD parameters and their estimated variance‐covariance. Percentages shown in (a) indicate the estimated mean inhibition for the three once‐daily doses (10, 25, and 40 mg) of AZD7986 studied in Part II. (c,d) Simulations with the final model highlighting the response of the system at different doses (c) and after a period of 0‐14 days of missed doses (d). Red numbers in (d) indicate number of missed doses.
General safety results
Overall, AZD7986 was well tolerated. No serious adverse events (SAEs) were reported and all other adverse events (AEs) were considered mild or moderate in intensity. No trends were observed in clinical laboratory results, 12‐lead ECGs, or telemetry results, and there was no signal of increased infection risk. One subject, receiving 25 mg AZD7986, discontinued treatment on Day 13 due to a skin AE of lichenoid drug eruption. On reevaluation of the medical history, it was discovered that the subject had been diagnosed with lichen planus previously. Nevertheless, the event was assessed to be possibly related to AZD7986 by the investigator.
Adverse events of special interest
Due to the known symptoms of PLS, subjects' gingiva and skin were closely monitored throughout the study.
A few subjects reported mild gingival bleeding, although these events typically did not occur spontaneously, but due to probing of the gingiva. There was also no difference seen between AZD7986 and placebo. Six subjects (numbered 1–6 below), however, all participating in Part II of the study, had skin‐related AEs considered to be of special interest.
Subject 1, receiving placebo, reported mild skin exfoliation, unilaterally, on the right foot (Day 20) and hand (Day 23). In addition, fissures were observed at the base of the right foot. The symptoms improved spontaneously and were almost resolved on Day 44.
Subject 2, receiving 25 mg AZD7986, reported mild patches of exfoliated skin and mild hyperkeratosis in the palms on Day 23 (Figure 6 a). The subject remained on treatment until Day 28 as planned, and on Day 37 the findings had disappeared.
Figure 6.
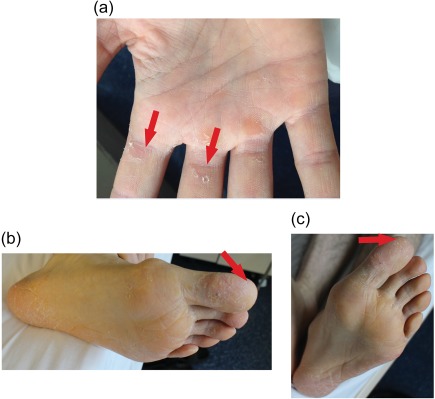
Skin findings after once‐daily repeated dosing with AZD7986. (a) Exfoliation in the right hand of Subject 2 after receiving 25 mg of AZD7986 for 23 days. (b) Scaling and erythema on the left sole and toe of Subject 3 after receiving 40 mg of AZD7986 for 23 days. (c) Spontaneous skin recovery and decreased scaling on the toe of Subject 3 after receiving 40 mg of AZD7986 for 26 days. Red arrows in (b,c) indicate area on the left toe to be compared between Day 23 and 26.
Subject 3, receiving 40 mg of AZD7986, experienced mild skin exfoliation in the heel bone area on both feet on Day 14. The exfoliation had later spread to the rest of the soles including the toes on Day 23, with addition of desquamation and moderate fissuring on the front foot pads and big toes (Figure 6 b). The scaling decreased spontaneously on Day 26 without discontinuation of treatment (Figure 6 c). Symptoms were almost resolved on Day 35 and reported completely resolved on Day 60.
Subject 4, receiving 40 mg of AZD7986, noted some rough skin on the knuckles of both hands and big toes on Day 5. On examination, dry and hyperkeratotic scaly patches were observed, especially in the dorsal part of the third metacarpophalangeal joints. In addition, mild skin hypertrophy and skin exfoliation were observed on the plantar surfaces of the big toes. The subject remained on treatment as planned and some symptoms improved spontaneously during the treatment period. On Day 35, skin hypertrophy was only present on the big toes and third metacarpophalangeal joint of the left hand. On Day 60, all symptoms apart from very mild skin hypertrophy had disappeared.
In addition, Subjects 5 and 6, receiving 40 mg AZD7986, also had skin events considered to be possibly related to DPP1 inhibition. Subject 5 had dry skin on the fingertips of both hands appearing on Day 21 and reported resolved on Day 32. Subject 6 noted some mild dryness on both arms and elbows on Day 13 that spontaneously resolved 2 days later. Both subjects stayed on treatment as planned.
In summary, five of the six skin findings of special interest were seen in subjects treated with AZD7986 and four were seen in the highest dose group. Subject 3, having the most pronounced symptoms, was also the one with highest average AZD7986 exposure (data not shown). Interestingly, in three of the subjects, symptoms appeared before any significant reduction of whole blood NE activity had been achieved, and most symptoms were also reported as resolved while NE activity was still low (Figure S3).
DISCUSSION
To our knowledge, this is the first study showing clear downstream effects of sustained DPP1 inhibition on whole blood NSP activity in man. Although we measured only the activity of NE, in vitro and preclinical data suggest that AZD7986 has similar effects on both Pr3 and CatG.16, 17 Using population PK‐PD modeling, we estimated the mean steady‐state inhibition of NE activity to 30, 49, and 59% for the three once‐daily doses (10, 25, and 40 mg) tested in Part II.
As expected from preclinical studies,17, 18 both the reduction and recovery of NE activity were delayed compared to initiation and cessation of AZD7986 dosing. Based on the hypothesis that this delay is caused by the maturation time of neutrophils in the bone marrow, we estimated the average neutrophil lifespan—from NE activation to neutrophil release into the circulation—to 15 days using a transit compartment model. This number is in good agreement with reported data on neutrophil maturation rates under healthy conditions (Figure 1 a).3, 22 Our model further implies that almost a month of treatment is required before steady‐state inhibition of whole blood NSP activity can be achieved (noting that this time may be shorter in subjects with increased neutrophil turnover, e.g., due to infection).22, 23 Since AZD7986 is targeted to treat chronic diseases, the delayed onset of effect is, however, not considered a limitation. Instead, AZD7986's mechanism of action can be considered beneficial, since NSP activity, both systemically and in tissues, is expected to stay low even if multiple doses of AZD7986 are missed. This is a clear advantage compared to direct NSP inhibitors, which are required in high local concentrations to continuously block NSP activity,24, 25 and may help overcome compliance challenges during chronic treatment.
AZD7986 inhibited NE activity in an exposure‐dependent manner, with an estimated plasma IC50 (394 nM) marginally higher (∼4‐fold) than predicted using human primary bone marrow‐derived CD34 + neutrophil progenitor cells.16 Interestingly, the estimated Hill coefficient of 0.697 is also consistent with neutrophil progenitor cell data, although less than the expected Hill coefficient of 1 seen with isolated DPP1 and a fixed substrate concentration. A possible explanation for the shallower shape of the cellular and in vivo concentration–responses could be continuous accumulation of NSP zymogens inside the cells when activation by DPP1 is inhibited. Although NSP zymogens are believed to be efficiently degraded if not activated during neutrophil maturation,18 a certain degree of intracellular zymogen accumulation may still occur during the early stages when the zymogens are produced. Since the inhibition of NSP activation by AZD7986 is dependent on the concentration of competing substrate,26 accumulating zymogens could create a complex relationship between AZD7986 and overall activation of NSPs,27, 28 where the concentration–response becomes shallow at high concentrations.
The pharmacokinetic evaluation of AZD7986 showed a half‐life and Cmax/Cmin ratio supporting sustained DPP1 coverage using a once‐daily dosing regimen. This had been a major focus of the preclinical discovery program and demonstrates the benefits of robust PK property optimization prior to candidate drug selection.16 PK data, however, also showed minor signs of supra‐proportionality and time dependency, which warrant further research. The model‐based PK analysis points to nonlinearity and time‐dependency in bioavailability and volume of distribution as possible reasons. This hypothesis is supported by in‐house data indicating that AZD7986 may be a substrate for efflux transporters. Saturation of such transporters in the gut can lead to an increase in bioavailability, while efflux saturation in kidneys or other tissues can lead to an increased volume of distribution.29, 30, 31 In‐house data does not indicate saturable metabolic clearance routes or plasma protein binding, and so is consistent with clearance linearity in the PK model.
AZD7986 was generally well tolerated, with no signs of increased infections, consistent with the hypothesis that DPP1 inhibition does not affect the overall function or recruitment of neutrophils.6, 18 However, five subjects experienced nonserious skin events possibly related to treatment. Of these five subjects, four were seen in the highest dose group, and two of the subjects had bilateral findings as typically observed in PLS patients.9
The pathophysiology of palmoplantar hyperkeratosis in PLS patients has not been confirmed. It has been hypothesized that NSPs play a crucial role in epidermal barrier function, and that imbalance in proteolytic activity may cause various skin disorders such as redness and scaling.32 However, as suggested by Miller et al.,19 an alternative explanation for the skin events may be that DPP1 also plays an important role in maintaining the structural integrity of plantar and palmar epidermal surfaces, by contributing to the processing of keratins.33 This hypothesis is supported by the observations in this study. First, AZD7986 caused dose‐dependent skin events, but the timing of events did not correlate with the dynamics of NE activity. Second, our skin findings appear less prominent than the seven cases of skin desquamation reported for the irreversible DPP1 inhibitor GSK2793660, even though GSK2793660 showed no clear effect on NSP activity.19 In fact, contrary to the findings in our study, the frequency and intensity of skin events caused by GSK2793660 led to premature study termination.
The idea that different pathways are responsible for the desirable (reduced NSP activity) and undesirable (skin symptoms) effects of DPP1 inhibition, could possibly also explain the discrepancy in NSP inhibition and frequency of skin events between GSK2793660 and AZD7986. Although GSK2793660 was reported to achieve sustained, near‐complete DPP1 inhibition in whole blood,19 this does not necessarily imply a continuous high level of DPP1 inhibition in all cells and tissues. In fact, GSK2793660 was administered once daily, but in contrast to AZD7986, exhibited a very short half‐life (1.5 hours). This means that during a significant part of the dosing interval, newly formed neutrophil precursor cells in the bone marrow, expressing DPP1, may have enough time to activate a considerable amount of NSPs. Such a time window could be of less importance for the DPP1‐dependent processes in skin.
We did not directly measure the level of DPP1 inhibition in the present study, but our NE and safety data strongly suggest that AZD7986 inhibits DPP1 to a significant extent. Both NSP inhibition and the development of skin symptoms appear to require high inhibition of DPP1.7, 26, 34, 35, 36 Unaffected PLS family members (heterozygotes), with no apparent skin symptoms, have been described to have DPP1 activity ranging from 13–47% compared to controls,7, 36 suggesting >80–90% inhibition is required before symptoms develop. Even if high DPP1 inhibition is needed for both desirable and undesirable effects, however, we do see opportunities for a therapeutic window within the studied dose range of AZD7986.
Because AZD7986 is a first‐in‐class molecule, the level of NSP inhibition needed to achieve clinical efficacy in relevant diseases is unknown. However, data from preclinical models show that a 40% reduction in NE activity attenuates tissue destruction in a mouse smoking model.37 This is consistent with in‐house data of elastin degradation showing that a 50% reduction in NSPs result in a marked increase in bovine elastin protection. Furthermore, peripheral blood NE activity has been reported to be 2‐fold higher in early‐onset COPD patients vs. healthy nonsmoker subjects,38 suggesting that 50% inhibition of NE may be therapeutically relevant in patients. Supported by this evidence and the results presented herein, a once‐daily dose of 25 mg AZD7986 should be able to achieve a clinically relevant reduction in NSP activity, with low risk of potentially limiting skin effects.
In conclusion, this study shows that inhibition of DPP1 remains a tractable target for disease modification in patients suffering from neutrophil‐driven inflammatory diseases, such as COPD, bronchiectasis, and related lung diseases. Importantly, based on 1) in vitro and preclinical studies providing good predictions of AZD7986 PK, potency, and required duration of treatment; 2) a flexible study design allowing for changes in dosing, sampling, and follow‐up visits; 3) careful monitoring of subjects' skin based on known symptoms of PLS; and 4) the use of modeling to provide sound, mechanistic interpretations and predictions of NE data, we could provide confidence to progress clinical development of AZD7986. As reported by Chalmers et al.,13 NE may play an important role in bronchiectasis, and the hypothesis of 25 mg AZD7986 as a clinically relevant dose is currently being studied in bronchiectasis patients (http://clinicaltrials.gov number NCT03218917).
FUNDING
The study was funded by AstraZeneca.
CONFLICT OF INTEREST
At the time this work was conducted, R.P., J.M., A.J., B.L., J.MO., M.R., J.R., S.P., K.S., and P.G. were employees of AstraZeneca, and L.C. and P.F. were employees of Parexel. T.E. was contracted by AstraZeneca to provide input on study results.
AUTHOR CONTRIBUTIONS
R.P., Ju.M., B.L., and P.G. wrote the article; R.P., Ju.M., A.J., B.L., Jo.M., M.R., S.P., L.C., P.F., K.S., and P.G. designed the research; R.P., M.R., and P.F. performed the research; R.P., Ju.M., A.J., B.L., Jo.M., M.R., J.R., L.C., P.F., T.E., K.S., and P.G. analyzed the data.
Supporting information
Figure S1. Geometric mean absolute neutrophil counts for the different doses tested in Part II, indicating no effect of treatment. Since samples were not always taken on the same day for every subject, data were binned based on an interval of 5 days (at baseline, 1‐5 days, 6‐10 days, etc.) before calculating the geometric means.
Figure S2. Change in objective function value when increasing the number of transit compartments in the NE activity model. Minimum value obtained with 27 compartments.
Figure S3. Appearance/resolution of skin symptoms versus change from baseline in NE activity for Subject 2‐6 (A‐E) with skin findings possibly related to DPP1 inhibition. Filled red arrows indicate appearance or worsening of symptoms. Open green arrows indicate improvement or resolution of symptoms.
SUPPLEMENTARY MATERIAL is linked to the online version of the article at http://www.wileyonlinelibrary.com.cpt
Supplementary Methods. Document describing details related to (1) monitoring of adverse events, (2) sampling and measuring of AZD7986 PK and whole blood neutrophil elastase (NE) activity, and (3) the development of the AZD7986 pharmacokinetic (PK) and NE activity non‐linear mixed effects models.
Table S1. Subject demographics.
Table S2. Summary of AZD7986 AUC and Cmax parameters from non‐compartmental analysis.
ACKNOWLEDGMENTS
The first two authors contributed equally to this work.
References
- 1. Pham, C.T. , Ivanovich, J.L. , Raptis, S.Z. , Zehnbauer, B. & Ley, T.J. Papillon‐Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J. Immunol. 173, 7277–7281 (2004). [DOI] [PubMed] [Google Scholar]
- 2. Fouret, P. , du Bois, R.M. , Bernaudin, J.F. , Takahashi, H. , Ferrans, V.J. & Crystal, R.G. Expression of the neutrophil elastase gene during human bone marrow cell differentiation. J. Exp. Med. 169, 833–845 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cowland, J.B. & Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 273, 11–28 (2016). [DOI] [PubMed] [Google Scholar]
- 4. Korkmaz, B. , Horwitz, M.S. , Jenne, D.E. & Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 62, 726–759 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dalgic, B. , Bukulmez, A. & Sari, S. Eponym: Papillon‐Lefevre syndrome. Eur. J. Pediatr. 170, 689–691 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Sørensen, O.E. et al Papillon‐Lefevre syndrome patient reveals species‐dependent requirements for neutrophil defenses. J. Clin. Invest. 124, 4539–4548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Toomes, C. et al Loss‐of‐function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat. Genet. 23, 421–424 (1999). [DOI] [PubMed] [Google Scholar]
- 8. Haneke, E. The Papillon‐Lefevre syndrome: Keratosis palmoplantaris with periodontopathy. Report of a case and review of the cases in the literature. Hum. Genet. 51, 1–35 (1979). [DOI] [PubMed] [Google Scholar]
- 9. Moss, T.A. , Spillane, A.P. , Almquist, S.F. , McCleskey, P.E. & Wisco, O.J. Palmoplantar keratoderma with progressive gingivitis and recurrent pyodermas. Cutis 93, 193–198 (2014). [PubMed] [Google Scholar]
- 10. Hiemstra, P.S. , van Wetering, S. & Stolk, J. Neutrophil serine proteinases and defensins in chronic obstructive pulmonary disease: effects on pulmonary epithelium. Eur. Respir. J. 12, 1200–1208 (1998). [DOI] [PubMed] [Google Scholar]
- 11. Park, J.A. , He, F. , Martin, L.D. , Li, Y. , Chorley, B.N. & Adler, K.B. Human neutrophil elastase induces hypersecretion of mucin from well‐differentiated human bronchial epithelial cells in vitro via a protein kinase C{delta}‐mediated mechanism. Am. J. Pathol. 167, 651–661 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandhaus, R.A. & Turino, G. Neutrophil elastase‐mediated lung disease. COPD 10 Suppl 1, 60–63 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Chalmers, J.D. et al Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am. J. Respir. Crit. Care Med. 195, 1384–1393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Polverino, E. , Rosales‐Mayor, E. , Dale, G.E. , Dembowsky, K. & Torres, A. The role of neutrophil elastase inhibitors in lung diseases. Chest 152, 249–262 (2017). [DOI] [PubMed] [Google Scholar]
- 15. Stockley, R.A. Alpha1‐antitrypsin review. Clin. Chest Med. 35, 39–50 (2014). [DOI] [PubMed] [Google Scholar]
- 16. Doyle, K. et al Discovery of second generation reversible covalent DPP1 inhibitors leading to an oxazepane amidoacetonitrile based clinical candidate (AZD7986). J. Med. Chem. 59, 9457–9472 (2016). [DOI] [PubMed] [Google Scholar]
- 17. Gardiner, P. , Wikell, C. , Clifton, S. , Shearer, J. , Benjamin, A. & Peters, S.A. Neutrophil maturation rate determines the effects of dipeptidyl peptidase 1 inhibition on neutrophil serine protease activity. Br. J. Pharmacol. 173, 2390–2401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guarino, C. et al Prolonged pharmacological inhibition of cathepsin C results in elimination of neutrophil serine proteases. Biochem. Pharmacol. 131, 52–67 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Miller, B.E. et al Epithelial desquamation observed in a phase I study of an oral cathepsin C inhibitor (GSK2793660). Br. J. Clin. Pharmacol. 83, 2813–2820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krzyzanski, W. Interpretation of transit compartments pharmacodynamic models as lifespan based indirect response models. J. Pharmacokinet. Pharmacodyn. 38, 179–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bekkering, S. & Torensma, R. Another look at the life of a neutrophil. World J. Hematol. 2, 44–58 (2013). [Google Scholar]
- 23. Summers, C. , Rankin, S.M. , Condliffe, A.M. , Singh, N. , Peters, A.M. & Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 31, 318–324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gunawardena, K.A. , Gullstrand, H. & Perrett, J. Pharmacokinetics and safety of AZD9668, an oral neutrophil elastase inhibitor, in healthy volunteers and patients with COPD. Int. J. Clin. Pharmacol. Ther. 51, 288–304 (2013). [DOI] [PubMed] [Google Scholar]
- 25. Vogelmeier, C. , Aquino, T.O. , O'Brien, C.D. , Perrett, J. & Gunawardena, K.A. A randomised, placebo‐controlled, dose‐finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 9, 111–120 (2012). [DOI] [PubMed] [Google Scholar]
- 26. Methot, N. et al Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro‐enzyme processing. J. Biol Chem. 282, 20836–20846 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Christopherson, R.I. & Duggleby, R.G. Metabolic resistance: The protection of enzymes against drugs which are tight‐binding inhibitors by the accumulation of substrate. Eur. J. Biochem. 134, 331–335 (1983). [DOI] [PubMed] [Google Scholar]
- 28. Westley, A.M. & Westley, J. Enzyme inhibition in open systems. Superiority of uncompetitive agents. J. Biol Chem. 271, 5347–5352 (1996). [DOI] [PubMed] [Google Scholar]
- 29. Grover, A. & Benet, L.Z. Effects of drug transporters on volume of distribution. AAPS J. 11, 250–261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan, L.M. , Lowes, S. & Hirst, B.H. The ABCs of drug transport in intestine and liver: Efflux proteins limiting drug absorption and bioavailability. Eur. J. Pharm. Sci. 21, 25–51 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Lin, J.H. & Yamazaki, M. Role of P‐glycoprotein in pharmacokinetics: clinical implications. Clin. Pharmacokinet. 42, 59–98 (2003). [DOI] [PubMed] [Google Scholar]
- 32. de Veer, S.J. , Furio, L. , Harris, J.M. & Hovnanian, A. Proteases and proteomics: cutting to the core of human skin pathologies. Proteomics Clin. Appl. 8, 389–402 (2014). [DOI] [PubMed] [Google Scholar]
- 33. Nuckolls, G.H. & Slavkin, H.C. Paths of glorious proteases. Nat. Genet. 23, 378–380 (1999). [DOI] [PubMed] [Google Scholar]
- 34. Methot, N. et al In vivo inhibition of serine protease processing requires a high fractional inhibition of cathepsin C. Mol. Pharmacol. 73, 1857–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 35. Hamon, Y. et al Neutrophilic cathepsin C is maturated by a multistep proteolytic process and secreted by activated cells during inflammatory lung diseases. J. Biol Chem. 291, 8486–8499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hewitt, C. et al The role of cathepsin C in Papillon‐Lefevre syndrome, prepubertal periodontitis, and aggressive periodontitis. Hum. Mutat. 23, 222–228 (2004). [DOI] [PubMed] [Google Scholar]
- 37. Podolin, P.L.B. et al Cathepsin C inhibition: a novel approach for the treatment of chronic respiratory diseases. In ATS Conference San Francisco (2016).
- 38. Milara, J. , Juan, G. , Peiro, T. , Serrano, A. & Cortijo, J. Neutrophil activation in severe, early‐onset COPD patients versus healthy non‐smoker subjects in vitro: effects of antioxidant therapy. Respiration 83, 147–158 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Geometric mean absolute neutrophil counts for the different doses tested in Part II, indicating no effect of treatment. Since samples were not always taken on the same day for every subject, data were binned based on an interval of 5 days (at baseline, 1‐5 days, 6‐10 days, etc.) before calculating the geometric means.
Figure S2. Change in objective function value when increasing the number of transit compartments in the NE activity model. Minimum value obtained with 27 compartments.
Figure S3. Appearance/resolution of skin symptoms versus change from baseline in NE activity for Subject 2‐6 (A‐E) with skin findings possibly related to DPP1 inhibition. Filled red arrows indicate appearance or worsening of symptoms. Open green arrows indicate improvement or resolution of symptoms.
SUPPLEMENTARY MATERIAL is linked to the online version of the article at http://www.wileyonlinelibrary.com.cpt
Supplementary Methods. Document describing details related to (1) monitoring of adverse events, (2) sampling and measuring of AZD7986 PK and whole blood neutrophil elastase (NE) activity, and (3) the development of the AZD7986 pharmacokinetic (PK) and NE activity non‐linear mixed effects models.
Table S1. Subject demographics.
Table S2. Summary of AZD7986 AUC and Cmax parameters from non‐compartmental analysis.