

Abstract
Objective
The aim of this multi‐centre, randomised, double‐blind, placebo‐controlled trial was to compare the efficacy and safety of the fixed combination of 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, and 1.5 mg benzocaine (study drug marketed as Dorithricin®) in repeat dosing for 3 days to match placebo lozenges in the treatment of acute pharyngitis in adults.
Methods
Patients (pts, aged ≥18 years) with acute pharyngitis, ie, non‐streptococcal sore throat and moderate‐to‐severe pain (intensity NRS ≥ 7; VAS ≥ 50) were assigned to study drug (n = 160) or matching placebo (n = 161). Efficacy was assessed by investigator for 2 hours post initial dose (p.i.d.), and 3 days later (Visit 2). Primary efficacy endpoint was the complete resolution of throat pain and difficulty in swallowing at Visit 2 (3 days p.i.d.). Safety and local tolerability were also assessed.
Results
Seventy‐two hours (p.i.d.), complete resolution of throat pain and difficulty in swallowing were achieved by 44.6% patients on study drug compared with 27.2% patients on placebo (difference 17.4% (CI [5.8%; 29.7%]; 64% improvement [GEE, P = 0.0022]).
Until 2 hours p.i.d., reduction in symptoms was better with study drug (P < 0.005). Treatment satisfaction was higher with study drug (patients′/investigators′ assessment (78.9%/78.9% vs 55.0%/55.6% for placebo) and was well tolerated, overall safety profile was comparable to placebo.
Conclusion
The strength of this randomised controlled trial lies in the endpoint of complete remission after 3 days p.i.d., especially in the light of other trials addressing acute pharyngitis. The results of this study show a significant benefit of the study drug over placebo in the treatment of acute pharyngitis. Local treatment with the fixed combination (0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, and 1.5 mg benzocaine) provides a rapid analgesic effect and is effective in relieving both severe throat pain as well as difficulty in swallowing associated with acute pharyngitis leading to a 64% improved complete remission within 72 hours. The triple active combination is a suitable treatment option for patients in the self‐management of acute pharyngitis and sore throat. Clinical trial registration: ClinicalTrials.gov, NCT03323528.
What's known
Triple active lozenges contain tyrothricin, benzalkonium chloride, and benzocaine and thus, combine potent anaesthetic and local antimicrobial activity. The clinical efficacy has been studied in two randomized controlled trials (RCT) in the past 35, 36. The improvement of moderate‐to‐severe pain in acute pharyngitis was not addressed before.
What's new
In this randomized, double‐blind, placebo‐controlled, multi‐centre trial the fixed combination of 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, and 1.5 mg benzocaine demonstrates rapid and sustained relief of moderate‐to‐severe acute sore throat pain and difficulty in swallowing: upon single dosing, a significant reduction in pain and difficulty in swallowing was seen already 5 minutes after first initial dose lasting over 2 hours. Upon repeat dosing, a significant 64% improvement in complete remission at day 3 post initial dosing was achieved. Triple active lozenge provides a safety profile similar to placebo. The strength of this randomized controlled trial lies in the endpoint of complete remission after 3 days p.i.d., especially in light of other study designs addressing only the analgesic effects within the first two hours p.i.d. in patients with acute pharyngitis.
1. INTRODUCTION
Acute pharyngitis is one of the most common complaints encountered in clinical practice. Although such infections are self‐limited and typically last only for a few days, patients substantially suffer from associated symptoms. In particular, sore throat and dysphagia affect patients during their everyday life. In the majority of cases, the infection that causes pharyngitis is initially viral in nature1, 2 and may be superinfected by bacteria due to inadequate use of antibiotics and disturbed microflora3, 4, 5, 6 or diverse viral mechanisms which include disruption of the epithelial barrier, upregulation of adhesion proteins, production of viral factors, and dysfunction of immune system components.1, 2, 7, 8, 9, 10
Except for streptococcus infections occurring in approximately 15% of patients and haemorrhagic fever who clearly need systemic antibiotic regimes first line11 in non‐streptococcal, viral pharyngitis treatment is usually symptomatic addressing relief in pain: Lozenges and sprays are available with a variety of active ingredients for treating sore throat, but only one with a triple combination of synergistically active ingredients12: Dorithricin® lozenges contain: (a) Benzocaine, a local anaesthetic sodium channel blocker with analgesic activity which confers a fast and sustained pain relief13 (b) Benzalkonium chloride, a biocide with antimicrobial and antiviral activity14, 15, 16, 17 (c) Tyrothricin, a small, cationic, amphiphilic, antimicrobial peptide (AMP), a naturally occurring antimicrobial non‐resorbed agent with a broad spectrum. As part of the innate immune system of vertebrates, AMPs have direct antimicrobial function, acting as mediators of inflammation and their antimicrobial spectrum covers Gram‐positive and ‐negative bacteria, in case of superinfection, as well as fungi and certain viruses.18, 19, 20 Recent studies revealed antiviral activity was also in the fixed triple combination.21
This study was designed to investigate the efficacy and safety/tolerability of the triple combination after multiple dosing, randomly assigned and compared with a matching placebo lozenge in adult patients with acute non‐bacterial pharyngitis characterised by moderate‐ to‐ severe sore throat pain and difficulty in swallowing. The primary outcome was complete remission of symptoms after 3 days. Data were collected from January till June 2017.
2. PATIENTS AND METHODS
2.1. Patients
Male and female outpatients aged ≥18 years were eligible, given a recent onset of sore throat of ≤24 hours duration, diagnosed with acute pharyngitis defined by a Tonsillo‐Pharyngitis Assessment (TPA)‐score of ≥5 assessed by the investigator. For the TPA each of the following signs and symptoms are rated by points from 0 to 3 according to the severity of the symptoms: oral temperature, oropharyngeal colour, size of tonsils, number of oropharyngeal enanthems, largest size of anterior cervical lymph nodes, number of anterior cervical lymph nodes, and maximum tenderness of some anterior cervical lymph nodes (Table 1).18, 19, 20 In addition, patients were required to score their difficulty in swallowing ≥50 mm on the 0‐100 mm visual analogue scale (VAS) and pain intensity of ≥7 on an 11‐point numeric rating scale (NRS).
Table 1.
| Finding | 0 Points | 1 Point | 2 Points | 3 Points |
|---|---|---|---|---|
| Oral temperature | ≤37°C | 37.1‐37.2°C | 37.3‐37.7°C | ≥37.8°C |
| Oropharyngeal colour | Normal/pink | Slightly red | Red | Beefy red |
| Size of tonsils | Normal/absent | Slightly enlarged | Moderately enlarged | Much enlarged |
| Number of oropharyngeal enanthems (vesicles, petechiae or exudates) | None | Few | Several | Many |
| Largest size of anterior cervical lymph nodes | Normal | Slightly enlarged | Moderately enlarged | Much enlarged |
| Number of anterior cervical lymph nodes | Normal | Slightly increased | Moderately increased | Greatly increased |
| Maximum tenderness of some anterior cervical lymph nodes | Not tender | Slightly tender | Moderately tender | Very tender |
A positive rapid streptococcus A test (rapid antigen detection test) sensitive for the major bacterial pathogen responsible for sore throat or a strong suspicion (McIsaac score ≥3) or purulent tonsillitis implied the patient's non‐eligibility to avoid the need for antibiotic therapy.22 Other exclusion criteria consisted of potential confounding factors for assessment and results, such as the use of any systemic analgesics/local analgesics (NSAIDs) in the throat area within 36 hours prior to screening and during the study, the use of local anaesthetics for the treatment of sore throat within 2 days prior to screening and during the study, the use of any systemic anti‐inflammatory drug/local anti‐inflammatory drug in the throat area (eg, glucocorticoids) within 4 weeks prior to screening and during the study, and the use of any other “sore throat medication” or other “cold medication” (lozenges, drops, sprays) that could have interfered with the results of the study within 7 days prior to screening and during the study.
Between January 2017 and June 2017, 328 patients attending one of the 15 participating practices in Germany were screened for eligibility into the study. Study centres were run by registered doctors in private practices with a focus on general practice (GPs, 9 centres) or specialised in otorhinolaryngology (ENT, 6 centres). All patients gave their written informed consent according to national regulations. The trial was approved by the national regulatory authority, the Federal Institute for Drugs and Medical Devices (BfArM), positive ethics vote was granted by the responsible national lead independent Ethics Committee of Bavaria (Munich, DE). The study conformed to the ICH guidelines of Good Clinical Practice and was conducted in accordance with German drug law and the Declaration of Helsinki.
2.2. Study design
The primary objective of this study was to demonstrate that the effect of the fixed combination of 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, and 1.5 mg benzocaine is superior to placebo with no active substances in the treatment of acute pharyngitis. The study was designed as a prospective, randomised, parallel‐group, placebo‐controlled, double‐blind, multi‐centre, phase IV trial.
Patient data were collected by the investigator during two study visits using an electronic case report form (eCRF). Additionally, a paper‐based diary and questionnaires were used for the patient to document symptoms, drug administration, side effects, and smoking habits, and to answer consumer‐related questions from Visit 1 to Visit 2:
On day 0 (Visit 1), eligible patients were examined: The investigator performed the Tonsillo‐Pharyngitis Assessment (TPA ≥ 5) and McIsaac scoring (<3) and patients were assigned randomly by the investigator, according to their chronological order of arrival, either in the test product group or in the placebo group following a previously established randomisation list in a 1:1 ratio in a sequential order. To guarantee a satisfactory level of blinding, the investigational medicinal products used in this study did not contain mint oil as flavouring excipient. Randomisation list was performed by the sponsor's department for the production of clinical trial medication by using the software Rancode 3.6 professional (IDV Munich); this person also created the emergency envelopes. Treatment units were sequentially numbered using a computer‐generated randomisation list by the sponsor. Randomisation was stratified by centre with block size of 4.
The study plan consisted of a stationary single‐dose phase up to 2 hours after first dosing, then an ambulatory multiple‐dose phase up to Day 3 (Visit 2), ie, 72 (−1/+2) hours after the start of treatment.
Intensity of throat pain was assessed using an 11‐point numeric rating scale (11‐point NRS) with 0 representing one pain extreme (no pain) and 10 representing the other pain extreme (severe pain). The patient was instructed to evaluate the severity of throat pain at that moment. Patients had to have a baseline NRS score ≥7, at screening.
Difficulty in swallowing was assessed using a VAS, 100 mm in length, and (100‐mm VAS) anchored by two verbal descriptors, one for each extreme symptom (0 mm = not difficult, 100 mm = very difficult). The patient was instructed to swallow and to point on the scale how difficult it was to swallow at that moment. Patients had to have a baseline VAS score ≥50 mm, at screening (inclusion criterion).
During the stationary single‐dose phase in the centre, patients were instructed to suck the initial dose (two lozenges simultaneously) until it had dissolved, and were not allowed to eat, drink, smoke or take any concomitant medication. The patient assessed the symptoms' pain intensity and difficulty in swallowing over a period of 1 or 2 hours depending on patient′s availability on site at Visit 1: before the initial dose (t0) and 5 (±1), 10 (±1), 15 (±1), 20 (±1), 30 (±3), 45 (±3), 60 (±3), 75 (±3), 90 (±3), 105 (±3), and 120 (±6) minutes after the initial dose.
During the ambulatory multiple‐dose phase, patients were asked to keep a diary from Day 0 to Day 3 for monitoring throat pain and difficulty in swallowing (Days 0‐2), for recording the number of lozenges taken per day (Days 0‐3), and any further symptoms or side effects, and for recording smoking habits and the number of cigarettes, if applicable (Days 0‐3). Additionally (only on Day 3), the patient recorded in the diary if he/she would recommend the study drug to others and was willing to use the medication in the future.
On Days 0, 1, and 2, the patient assessed throat pain (11‐point NRS) and difficulty in swallowing (100‐mm VAS) in the evening before the administration of the last lozenge (documentation in the diary). If the two symptoms were not present any more at this point in time, the patient also recorded the approximate time of last throat pain and difficulty in swallowing on this day or the day before, if currently no symptoms were present.
On Day 3 (Visit 2), after 72 hrs treatment, patients returned to the centre to be interviewed and have a general and local examination by the investigator performing TPA. The patient assessed his/her throat pain and difficulty in swallowing in a patient questionnaire. Both, the patient and the investigator were asked to assess study medication with regard to tolerability and level of satisfaction and patients were asked to assess their willingness for recommendation using a 5‐point verbal rating scale (VRS).
Also, the consumption of investigational study drug (lozenges) was evaluated: Patients were provided 40 lozenges at the treatment start and reported in their diary about their lozenge consumption, which was reviewed by the investigator at the study end Visit based on the number of lozenges returned by the patient.
2.3. Efficacy assessments and derived endpoints, safety assessments
2.3.1. Primary efficacy endpoint
The primary endpoint variable was defined as the percentage of total responders assessed at Visit 2 (Approx. 72 hours after first application of treatment).
A patient was defined as total responder in case of a complete resolution of throat pain and difficulty in swallowing at Visit 2. This was documented as complete disappearance of both pharyngitis symptoms, ie, no throat pain (score = 0 on the 11‐point NRS scale) and no difficulties in swallowing (0 mm on the 100‐mm VAS scale) based on the questionnaire completed at the study site (Visit 2).
2.3.2. Secondary efficacy endpoints
As several secondary endpoint parameters were analysed such as:
the percentage of patients with complete resolution of throat pain 72 hours post initial dose (p.i.d.);
the percentage of patients with complete resolution of difficulty in swallowing 72 hours p.i.d.
the percentage of Early Responders (48 hours p.i.d.) and symptom‐free until study end;
And additionally, secondary endpoints such as
the baseline difference in throat pain at Visit 2 (average change in NRS score from t0 to 72 hours p.i.d.);
the baseline difference in difficulty to swallow at Visit 2 (average change in mmVAS from baseline to 72 hours p.i.d.);
the percentage of patients with complete resolution of throat pain 48 hours p.i.d. and symptom‐free until end of study (up to 72 hours p.i.d);
the percentage of patients with complete resolution of difficulty in swallowing 48 hours p.i.d. and symptom‐free until end of the study (up to 72 hours p.i.d);
the time to free of symptom(s) of throat pain and difficulty in swallowing, and separately for throat pain and difficulty in swallowing;
-
symptom relief after administration of the initial dose (two lozenges):
-
○
intensity of symptoms analysed by mixed model for repeated measures (MMRM) using centre as random effect, treatment as fixed effect, an indicator variable which states the documented assessment at 2 hours p.i.d. and 1 hour p.i.d. as fixed effect, baseline as covariate and baseline difference in symptom intensity as dependent variable repeated in time, separately for throat pain and difficulty in swallowing
-
o
time to symptom reduction were analysed by the Log‐rank test, separately for throat pain: time to reduction by at least 1 NRS score point and difficulty in swallowing: time to reduction by at least 10 mm on VAS
-
o
the percentage of patients with reduction in baseline symptom intensity by at least 50% 1 hour and 2 hours p.i.d. was analysed using GEE (analogous to primary endpoint analysis), separately for throat pain: at least 50% reduction of baseline NRS score, difficulty in swallowing: at least 50% reduction of baseline mm VAS
-
○
Finally, an analysis of prognostic factors: The primary endpoint variables were descriptively investigated by logistic regression with respect to prognostic factors (baseline scores, treatment compliance, gender, age, centre, smoking, and single TPA assessments at baseline). The level of significance for the detection of prognostic factors was defined as P < 0.1.
2.3.3. Safety endpoints
Safety and tolerability were assessed by analysis of treatment‐emergent adverse events (AEs). All AEs reported spontaneously by the patient or in response to non‐leading questioning or clinical exam by the investigator were recorded throughout the stationary phase and at study end. The seriousness, severity, management, outcome, and relationship with study drug of the event were recorded. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA, version 19.1). The examination of the oropharynx was made at baseline and at study end.
Further safety endpoints were: the tolerability of study medication assessed by patient and investigator; the percentage of patients requiring further medication for treatment of acute pharyngitis after end of study; the percentage of patients with an increase in throat pain intensity [NRS score points] or difficulty in swallowing [mm VAS] at Visit 2 compared to baseline (Visit 1, t0) requiring further medication.
2.3.4. Additional endpoints addressing treatment satisfaction
Additionally, the following parameters were assessed: the change in the TPA score and TPA single symptom scores from Visit 1 to Visit 2; patients' and investigators' satisfaction with study medication (efficacy); the recommendation of study drug to others and willingness to use the medication in the future.
2.4. Statistics, statistical methods
The study was planned to show superiority of the fixed combination of 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, and 1.5 mg benzocaine compared to placebo in the primary endpoint, defined as the percentage of patients with complete resolution of throat pain and difficulty in swallowing at Visit 2 (Day 3).
A centre had to randomise and to treat at least eight patients to be a standalone centre in the analysis (centres enrolling less than eight patients were pooled to one virtual centre).
Assuming a response rate of 15% higher for the test product compared to placebo (test product: 44.1% placebo: 29.0%) and a statistical power of 80% and a type I‐error rate of 2.5% (one‐sided) revealed 160 patients per treatment group (320 patients in total). Sample size was calculated using program PASS 11. No interim analysis was performed.
The analyses of the primary and secondary endpoints were performed using the full analysis set (FAS), ie, all patients were randomised with at least one documented application of trial medication and post‐baseline efficacy data for the primary endpoint (Visit 2). For the primary endpoint, this analysis was confirmatory. The analysis of per protocol (PP) set was performed additionally as a sensitivity analysis to determine the effects of the patients excluded from the PP (patients with major protocol deviations were excluded).
The analysis of the primary endpoint was performed applying a generalised estimation equation (GEE) model using logit as link function (SAS proc genmod) for binary response and treatment as factor. Study centre was included as confounding factor into the model.
Binary‐secondary efficacy endpoints were tested statistically analogously to the primary endpoint model. Baseline changes of endpoints in NRS score or VAS will be calculated by a linear mixed model using centre as random effect, treatment as fixed effect and the baseline difference of the respective endpoint as dependent variable. Subgroup analysis of the PP set was performed additionally as a sensitivity analysis to determine the effects of the patients excluded from the PP.
Statistical tests were performed two‐sided using an α‐level of 5% (type I error rate). The number of AEs and the number and percentage of patients with at least one AE were tabulated for each treatment group by system organ class (SOC) and preferred term (PT) using the Medical Dictionary for Regulatory Activities (MedDRA, Version 19.1). The number of patients with at least one drug‐related AE (ADR) was compared between treatment groups using Fisher's exact test. The log rank test was used to compare the time with first ADR between treatment groups.
3. RESULTS
3.1. Patients
A total of 328 patients were screened, 321 were randomised and analysed 160 (49.8%) to triple active study drug and 161 (50.2%) to placebo; all received the study treatment (FAS population), and 312 patients (97.2%) completed the study (Figure 1). Nine patients prematurely withdrew from the study. At baseline, treatment groups were well matched for age (mean: 35.1 years), sex (male:female ratio: 1:1.6), and baseline sore throat characteristics (Table 2).
Figure 1.
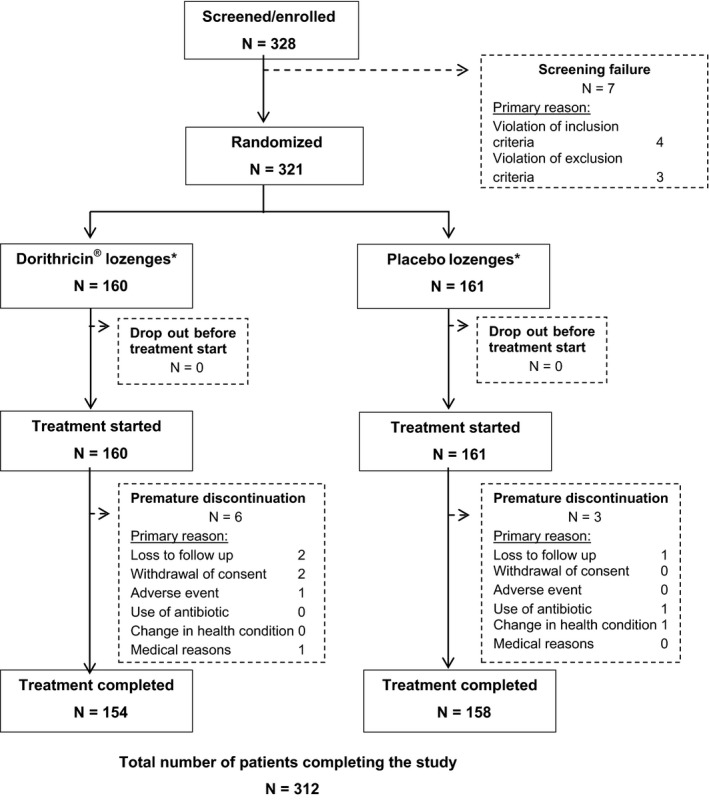
Patient disposition chart with all patients enrolled and total number of patients completing the study (N = 312)
Table 2.
Patient population: baseline characteristics of target disease (acute pharyngitis)
| Dorithricin® | Placebo | Total | |
|---|---|---|---|
| Number of patients randomised and receiving investigational treatment (SES) | 160 (100%) | 161 (100%) | 321 (100%) |
| Mean age ± SD (y) | 37.4 (14.0) | 35.5 (13.8) | 36.5 (13.9) |
| Gender (% male) | 39.4 | 44.1 | 41.7 |
| Mean TPA ± SD | 10.4 (9.0) | 10.8 (8.7) | 10.6 (8.9) |
| Tonsillo‐pharyngitis assessment (TPA) ≥5 (% pts) | 100 | 100 | 100 |
| Throat pain intensity (11‐point NRS ≥7) (% pts) | 20 | 19 | 20 |
| Throat pain intensity (11‐point NRS ≥8) (% pts) | 80 | 81 | 80 |
| Difficulty in swallowing (0‐100 mm VAS ≥ 5) | 100 | 100 | 100 |
| McIsaac‐Score (<3 in all pts) (mean [SD]) | 1.39 (0.49) | 1.41 (0.5) | 1.40 (0.5) |
| McIsaac‐Score −1 (% pts) | 3.1 | 4.3 | 3.7 |
| McIsaac‐Score 0 (% pts) | 8.8 | 5.6 | 7.2 |
| McIsaac‐Score 1 (% pts) | 36.3 | 32.3 | 34.3 |
| McIsaac‐Score 2 (% pts) | 51.9 | 57.8 | 54.8 |
| Fever in anamnesis or temperature ≤38°C (% pts) | 98.8 | 99.4 | 99.1 |
| Presence of cough (% pts) | 47.5 | 51.6 | 49.5 |
| Painful anterior lymph nodes (% pts) | 61.9 | 59.0 | 60.4 |
| Swollen tonsils or tonsil exudates (% pts) | 55.0 | 60.2 | 57.6 |
| Age > 45 y (% pts) | 33.8 | 24.8 | 29.3 |
| Treatment compliance (8 ± 2 lozenges/day) (% pts (SD)) | 98.9 (10.8) | 98.9 (11.0) | 98.9 (10.8) |
| Number of pts in full analysis set (FAS) | 156 (97.5%) | 160 (99.4%) | 316 (98.4%) |
| Number of pts in per protocol population (PP) | 140 (87.5%) | 146 (90.7%) | 286 (89.1%) |
3.2. Efficacy
3.2.1. Primary efficacy endpoint
Three days treatment with verum and placebo lozenges had a clinically relevant analgesic effect on sore throat and achieved clinically relevant improvement in swallowing in adult patients with acute pharyngitis.
Complete resolution of throat pain and difficulty in swallowing 72 hours post initial dose (p.i.d.) was achieved by 44.6% of 156 patients in the verum group compared with 27.2% of 160 patients in the placebo group (Figure 2). The difference in total responder rates of 17.4% (CI [5.8%; 29.7%]) was statistically significant in favour of study drug (P = 0.0022; FAS) corresponding to a 64% improvement in the test product. The sensitivity analysis in the PP population confirmed the results (P = 0.0019).
Figure 2.

Primary endpoint (FAS): responder free of both symptoms (throat pain and difficulty in swallowing) at 72 h (n = 316); related secondary endpoints: responders free of throat pain or difficulty in swallowing at 48 and 72 h (n = 316)
3.2.2. Secondary endpoints related to primary endpoint
Figure 2 also displays complete resolution of throat pain and difficulty in swallowing 48 hours p.i.d. and symptom free until the end of the study achieved by 11.3% of 156 patients in the verum group compared with 3.4% of 160 patients in the placebo group (Figure 2). The treatment group difference obtained from the GEE model was calculated as 7.9% (CI [1.1%; 22.5%]) and was statistically significant in favour of the test product (P = 0.0115; FAS). The sensitivity analysis in the PP population confirmed the results (P = 0.0093).
The difference in responder rates for single symptoms of acute pharyngitis (ie, throat pain and difficulty in swallowing) were statistically significant in favour of study drug regarding complete resolution of throat pain 72 hours p.i.d. and complete resolution of difficulty in swallowing 72 hours p.i.d. or 48 hours p.i.d. and symptom‐free until study end (all P‐values <0.05, Table 2). The difference in the responder rates regarding complete resolution of throat pain 48 hours p.i.d. and symptom‐free until study was close to statistical significance in the FAS (P = 0.0528), but reached statistical significance in favour of study drug in the PP analysis (P = 0.0485).
3.2.3. Further evaluations
The baseline differences in difficulty to swallow and throat pain at Visit 2 showed a better improvement in throat pain of 0.5 NRS score points (FAS, CI [−0.1; 1.0] points) and improvement in difficulty to swallow of 3.3 mm VAS (CI [−1.9; 8.50] with verum, thus, in the PP analysis, the baseline difference in throat pain at Visit 2 was statistically significant in favour of the test product (P = 0.0323).
Symptom relief within 1 and 2 hours after the initial dose of two lozenges (Visit 1/ Day 0)
The current analysis (sum of pain intensity differences, SPID) focused on the values at 60 minutes, being the mandatory time point at which 100% of patients had completed the questionnaire (mandatory assessment time p.i.d.). The sum of differences over 120 minutes p.i.d. was analysed additionally for the subgroup of patients who completed the questionnaire within 2 hours.
In both the treatment groups, the mean throat pain intensity and the mean intensity of difficulty in swallowing significantly decreased within 2 hours after administration of the initial dose of two lozenges (P < 0.0001).
The mean values of the SPID in throat pain (score points*min) and difficulty in swallowing (mm*min) 1 and 2 hours after the initial dose were higher in the verum group indicating greater reduction in pain intensity (Figure 3) and swallowing difficulty (Figure 4) with study drug compared with placebo at both the timepoints. The group differences were all statistically significant in favour of study drug (P‐values <0.005).
Figure 3.

Course of the mean intensity of throat pain over 2 h after initial dose ± 95% CL (FAS) measured on a 11‐point numeric rating scale (NRS) ranging from 0 (no pain) to 10 (severe pain): SPID 1 h −108.9 vs −78.3 points*min and SPID 2 h −241.9 vs −182.2 points*min; significant differences at each time point (5‐120 min) (P < 0.005; P < 0.0068 at 20 min)
Figure 4.

Course of mean intensity of difficulty in swallowing over 2 h after initial dose ± 95% CL (FAS) measured on a 100‐mmVAS scale ranging from 0 mm (not difficult) to 100 mm (very difficult): SPID 1 h −876.5 vs −582.8 mm*min and SPID 2 h −2068.3 vs −1404.2 mm*min; significant differences at each time point (5‐120 min) (P < 0.005)
The median time to symptom relief (ie, reduction in throat pain by at least 1 score point on the 11‐point NRS / reduction in swallowing difficulty by at least 10 mm on 100‐mm VAS) was shorter in the verum group compared with the placebo group for both pain relief (5 vs 15 minutes p.i.d.) and for relief in swallowing difficulty (10 vs 30 minutes p.i.d.). The differences between groups were statistically significant in favour of study drug (Log rank test: all P‐values <0.005).
The percentage of patients with at least 50% symptom reduction from baseline was higher in the verum group compared with the placebo group both for throat pain (23.1% vs 13.8% of patients with at least 50% NRS score reduction within 1 hour and 28.1% vs 22.6% of patients within 2 hours) and difficulty in swallowing (14.7% vs 8.1% of patients with at least 50% mmVAS reduction within 1 hour and 24.2% vs 15.8% of patients within 2 hours). All differences between groups were statistically significant (P < 0.05) in favour of study drug.
Tonsillo‐pharyngitis assessment
Changes in the presence and severity of signs and symptoms of acute pharyngitis calculated as TPA score different between the verum group and the placebo group with regard to the percentage of patients who had an improvement (93.6% vs 98.1%, respectively), a worsening (1.3% vs 3.9%, respectively), or no change (2.6% vs 0.6%) compared with baseline (GEE, P = 0.0014).
Patients' and investigators' satisfaction with study medication
Treatment satisfaction (ratings of “satisfied” and “very satisfied” combined) was higher for the 156 (100.0%) patients treated with study medication than for the 160 (100.0%) patients receiving placebo lozenges as shown by the assessments of patients (78.9% vs 55.0%, P < 0.0015) and the investigators' assessments in FAS (78.9% vs 55.6%, P < 0.001; Figure 5).
Figure 5.

Treatment satisfaction: Patients' and investigators' ratings of “satisfied” and “very satisfied” with treatment (SES) and patients' recommendation to others and willingness to use medication in the future (SES)
3.3. Investigational drug consumption and willingness to use study medication in the future
The mean adherence to treatment (compliance) was 98.9% (SD 10.8%) corresponding to 8 ± 2 lozenges per day with no difference between the two treatment groups (Table 2). Patients' willingness to use the study medication in the future and to recommend the study medication to others (Figure 5) were higher in the verum group compared with the placebo group (75.0% vs 47.8% and 76.9% vs 50.9%, respectively) (Table 3); the differences between treatment groups were statistically significant (P < 0.0001).
Table 3.
Responder rates—primary and secondary endpoint analyses (FAS)
| Dorithricin® responder | Placebo responder | P‐value (GEE)e | |||
|---|---|---|---|---|---|
| N | % | N | % | ||
| Primary endpoint | |||||
| Total responders (complete resolution of throat pain + difficulty in swallowing 72 h p.i.d.)a + c | 72/156 | 46.2 | 49/160 | 30.6 | 0.0022 |
| Secondary endpoints | |||||
| Early responders (complete resolution of throat pain + difficulty in swallowing 48 h p.i.d.) and remaining symptom‐freea + c | 17/156 | 10.9 | 6/160 | 3.8 | 0.0115 |
| Complete resolution of throat pain | |||||
| 72 h p.i.d.a | 77/156 | 49.4 | 63/160 | 39.4 | 0.0459 |
| 48 h p.i.d. and remaining symptom freeb | 21/156 | 13.5 | 12/160 | 7.5 | 0.0528 |
| Complete resolution of difficulty in swallowing | |||||
| 72 h p.i.d.c | 83/156 | 53.2 | 59/160 | 36.9 | 0.0017 |
| 48 h p.i.d. and remaining symptom freed | 22/156 | 14.1 | 11/160 | 6.9 | 0.0237 |
GEE, generalised estimation equation; IMPs, investigational medicinal products; NRS, numeric rating scale (0‐10 score points); VAS, visual analogue scale (0‐100 mm).
Complete resolution of throat pain 72 h p.i.d. was defined as NRS score = 0 documented in the patient questionnaire on Day 3/Visit 2, approximately 72 h after initial dosing of IMPs.
Complete resolution of throat pain 48 h p.i.d. and symptom‐free until the end of the study was defined as NRS score = 0 and yes/no question answered with “no throat pain” both in the diary on Day 2 (48 h p.i.d.) and the patient questionnaire at Visit 2 (72 h p.i.d.).
Complete resolution of difficulty in swallowing 72 h p.i.d. was defined as VAS = 0 mm documented in the patient questionnaire on Day 3/Visit 2, approximately 72 h after initial dosing of IMPs.
Complete resolution of difficulty in swallowing 48 h p.i.d. and symptom‐free until the end of the study was defined as VAS = 0 mm and yes/no question answered with “no difficulty in swallowing” documented both in the diary on Day 2 (48 h p.i.d.) and the patient questionnaire at Visit 2 (72 h p.i.d.).
Two‐sided test (GEE model using logit as link function for binary response and treatment).
3.4. Safety
3.4.1. Treatment‐emergent adverse events
Study drug was well tolerated and the overall safety profile was comparable with placebo. Overall, 42 out of 321 treated patients (13.1%) reported a total of 68 treatment‐emergent adverse events (TEAEs). The incidence of TEAEs was higher in the verum group (26/160 patients, 16.3%, reporting 43 TEAEs) compared with the placebo group (16/161 patients, 9.9%, reporting 25 TEAEs; Table 4).
Table 4.
Adverse events summary; most frequent treatment‐emergent adverse events; drug‐related treatment‐emergent adverse events (FAS population)
| Dorithricin® (N = 160, 100%) | Placebo (N = 161, 100%) | |
|---|---|---|
| TEAEs classified as bacterial infections | ||
| Pharyngitis bacterial | 1 (0.6%) | 2 (1.2%) |
| Nasopharyngitis | 0 | 1 (0.6%) |
| Otitis media | 0 | 1 (0.6%) |
| Tonsillitis | 0 | 2 (1.2%) |
| Sinusitis | 0 | 1 (0.6%) |
| Patients with ≥1 SAE | 1 (0.6%) pneumonia | 1 (0.6%) tonsillitis |
| Patients with ≥1 TEAE | 26 (16.3%) | 16 (9.9%) |
| Patients with ≥1 TEAE leading to premature treatment discontinuation | 3 (1.9%) | 2 (1.2%) |
| TEAEs not related to trial medication | ||
| Palpitations | 1 (0.6%) | 0 |
| Abdominal pain upper | 2 (1.3%) | 0 |
| Hypoaesthesia oral | 2 (1.3%) | 0 |
| Nausea | 3 (1.9%) | 3 (1.9%) |
| Chills | 0 | 2 (1.2%) |
| Influenza like illness | 1 (0.6%) | 0 |
| Pyrexia | 2 (1.3%) | 1 (0.6%) |
| Headache | 6 (3.8%) | 6 (3.7%) |
| Cough | 3 (1.9%) | 2 (1.2%) |
| Drug‐related TEAEs | ||
| Patients with ≥1 drug‐related TEAE | 10 (6.3%) | 3 (1.9%) |
| Nausea | 1 (0.6%) | 0 |
| Abdominal pain upper | 1 (0.6%) | |
| Breath odour | 1 (0.6%) | |
| Enteritis | 1 (0.6%) | |
| Hypoaesthesia oral | 2 (1.2%) | |
| Nausea | 1 (0.6%) | 1 (0.6%) |
| Cough | 1 (0.6%) | 1 (0.6%) |
| Dyspnoea | 1 (0.6%) | |
| Oropharyngeal pain | 1 (0.6%) | |
SAE, serious adverse event; TEAE, treatment emergent adverse event. Drug‐related TEAEs, TEAEs for which relationship with study drug was suspected.
Thirteen out of 321 patients (4.1%) experienced at least 1 TEAE with a possible, probable or certain causal relationship to the study drug (drug‐related TEAE), 10 out of 160 patients (6.3%) treated with study drug and 3 out of 161 patients (1.9%) receiving placebo.
The majority of TEAEs was mild‐ to‐ moderate in intensity. Two out of 321 patients (0.6%) experienced a severe TEAE, ie, pneumonia with hospital admission 1 day after Visit 1 in 1 of 160 patients (0.6%) assigned to but considered unrelated to the test product and one case of tonsillitis in 1 of 161 patients (0.6%) receiving placebo. Deaths did not occur. All TEAEs had resolved by the end of the study. The three TEAEs in 160 patients (1.9%) leading to premature termination of study drug were mild influenza like illness, mild cough, and mild febrile infection (each experienced by 1 patient, 0.6% each). The TEAEs leading to premature termination of placebo were chills and pyrexia both of moderate intensity experienced by the same patient (0.6%). Drug‐related TEAEs (MedDRA PT) experienced in both treatment groups (verum vs placebo) were nausea (1.9% vs 0.6%) and cough (0.6% vs 0.6%); drug‐related TEAEs that were only reported in the placebo group were pharyngitis bacterial, and in the verum group were oral hypoaesthesia (1.3%), and abdominal pain upper, enteritis, dyspnoea, and oropharyngeal pain (each event 0.6%). The median time to onset of the first drug‐related TEAE after treatment start tended to be longer in the verum group compared with the placebo group (8.5 vs 3.2 hours; P = 0.4513).
Bacterial infections such as pharyngitis, nasopharyngitis, otitis media, tonsillitis, sinusitis were reported, seven cases plus one severe case in the placebo group (8/161 patients; 5.2%) compared with one case of nasopharyngitis in the verum group (1/160 patients; 0.6%; P = 0.0186); by including the case of pneumonia with hospital admission at day 2 (P = 0.0505); without both severe TEAEs (P = 0.0327; Table 4).
3.4.2. Vital signs
Measurements in the practice by investigator at baseline (Visit 1) and study end (Visit 2) did not show any clinically relevant changes in average blood pressure, pulse or body temperature in any treatment group (SES).
3.4.3. Global judgement of tolerability
The frequency of “good” and “excellent” ratings for tolerability of investigational treatment was comparable between the verum group (51.9% and 40.65, respectively, by patients / 53.1 and 40.0% by investigators) and the placebo group (49.7% or 47.8% by patients /(51.6% or 46.6% by investigators) (P = 0.3378 and P = 0.1650, respectively, SES).
3.4.4. Need of further treatment for acute pharyngitis after end of study
The percentage of patients requiring further medication for treatment of acute pharyngitis after study end was a little higher in the verum group compared with the placebo group (8.8% vs 5.6%, P = 0.2886). The difference between both the groups was also not significant regarding the percentage of patients requiring further medication due to an increase in throat pain intensity and/or difficulty in swallowing compared to baseline (2.5% vs 1.2%; P = 0.4480; SES).
4. DISCUSSION
In this multi‐centre, double‐blind, randomised, placebo‐controlled, parallel‐group phase IV clinical trial, safety and efficacy of 3‐day oral treatment with the fixed combination of 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, and 1.5 mg benzocaine and matching placebo lozenges were compared in adult patients with acute pharyngitis recruited in Germany in 2017. Patients randomised are considered to be representative for the population affected by the target disease as patients were screened in private practices of specialists in otorhinolaryngology or of general practitioners which are more likely to be consulted by patients suffering from acute pharyngitis.
The study was designed to show superiority of study drug over placebo lozenges. The strength of the study design is its primary efficacy endpoint, defined as the percentage of patients with complete resolution of throat pain and difficulty in swallowing 72 hours after the first application of treatment (total responders) which is remarkable as most other trials for treating acute pharyngitis address only pain reduction and improved swallowing within the first 2 hours p.i.d.: In reviewing placebo‐controlled efficacy trials of lozenges in patients with acute pharyngitis based on a PubMed literature search for “lozenge AND pharyngitis AND randomized controlled trial” (Table 5), we conclude non‐steroidal anti‐inflammatory drugs (NSAIDs) and topical analgesics can provide short‐term pain relief within 1‐2 hours in comparison with placebo. However, these trials frequently did not address complete remission or failed in detecting superiority over placebo over the course of 3‐4 days ‐ often explained by the natural improvement in the disease in a few days.23, 24, 25
Table 5.
Placebo‐controlled trials of lozenges in patients with acute non‐streptococcal sore throat and moderate‐to‐severe pain according to PubMed search for “lozenge in pharyngitis” and “randomized controlled trial”
| Reference (Author, year) | Single (S) Multi‐centre (M) | Active treatment | Throat pain (onset, VAS score) | Number of pts (active/placebo) | Acute effect (% peak decrease in pain; relative effect to control) | Sustained effect (decrease of pts with symptoms; relative effect to control) |
|---|---|---|---|---|---|---|
| Schachtel, 198855 | S | Paracetamol 1000 mg | <4 d, VAS > 5 | 40/41 | 50% 3 h p.i.d.** | n.a. |
| 20% 5 h p.i.d.** | ||||||
| Bertin, 199156 | S | Paracetamol 10 mg/kg tid | <2 d | 78/76 (children 6‐12 y) | n.a. | 34% decrease of pts with symptoms 2 d p.i.d.** |
| Schachtel, 199357 | S | Paracetamol 15 mg/kg | <5 d, VAS > 5 | 38/39 (children 4‐12 y) | 30% 2 h p.i.d.* | n.a. |
| 7% 6 h p.i.d. | ||||||
| Schachtel, 199357 | S | Ibuprofen 10 mg/kg tid | <5 d, VAS > 5 | 38/39 (children 4‐12 y) | 25% 2 h p.i.d.* | n.a. |
| 22% 6 h p.i.d. | ||||||
| Schachtel, 198855 | S | Ibuprofen 400 mg | <4 d, VAS > 5 | 39/41 | 80% 3 h p.i.d.** | n.a. |
| 70% 5 h p.i.d.** | ||||||
| Bertin, 199156 | S | Ibuprofen 10 mg/kg tid | <2 d | 77/76 (children 6‐12 y) | n.a. | 56% decrease of pts with symptoms 2 d p.i.d.** |
| Schachtel, 199458 | S |
Ibuprofen 200 mg / Ibuprofen 400 mg |
<5 d, VAS > 5 | 10/10 | 32% 2 h p.i.d.* | n.a. |
| 47% 2 h p.i.d.** | ||||||
| Bouroubi,. 201632 | M | Ibuprofen 25 mg | <3 d, VAS > 6 | 194/191 | 13% 2 h p.i.d. and 24 h p.i.d. (TOTPAR, STRS)* | No difference to placebo from day 2 to day 4 |
| Schachtel, 199159 | S | Acetyl salicylic acid 800 mg | <4 d, VAS > 5 | 68/69 | 55% 1 h p.i.d.** | n.a. |
| Schachtel, 199159 | S | Acetyl salicylic acid 800 mg + caffeine 64 mg | <4 d, VAS > 5 | 7/69 | 75% 1 h p.i.d.** | n.a. |
| Sauvage, 199060 | S | Niflumic acid 1000 mg | <2 d | 118/113 | n.a. |
17% 2 d p.i.d.*
33% 4 d p.i.d.* |
| Schachtel, 201423 | S | Flurbiprofen 8.25 mg | <3 d, VAS > 5 | 101/97 | 59% greater reduction in throat pain, 45% less difficulty in swallowing, 44% less throat swelling 24 h p.i.d.** | No difference to placebo from day 2‐7 p.i.d. (38% greater relief of sore throat pain (P = 0.07), 36% greater improve‐ment in difficulty in swallowing (P = 0.09) |
| Aspley, 201630 | S | Flurbiprofen 8.25 mg | ≤4 d, throat pain VAS ≥ 6, difficulty in swallowing NRS ≥5 | 59/65 | 79.8% greater reduction in throat pain 24 h p.i.d.** | n.a. |
| Whiteside, 198261 | S | Benzydamine HCL 0.15% rinse 5x/d | <2 d | 25/26 | 42% decrease in mean pain score 3 d p.i.d. | Symptom‐free 88% pts vs 38% pts 7 d p.i.d.* |
| Schutz, 200262 | S | Ambroxol hydrochloride 20 mg | <2 d, VAS > 5 | 107/108 | 28% greater reduction in throat pain 3 h p.i.d.** | 20% greater reduction in throat pain 24 and 72 h p.i.d.* |
| Fischer, 200263 | S | Ambroxol hydrochloride 20 and 30 mg | <2 d, VAS > 5 | 160/158 | 31% and 37% greater reduction in throat pain 3 h p.i.d.** | n.a. |
| McNally, 201224 | S | Amylmetacresol/2,4‐dichlorobenzyl alcohol (AMC/DCBA) plus lidocaine lozenge or a hexylresorcinol lozenge or placebo | <4 d, throat pain VAS ≥ 6, difficulty in swallowing NRS ≥ 5 | 94/93/93 | 24%, 30% and 14% pain reduction with AMC/DCBA + lidocaine or placebo 3 h p.i.d.**, respectively | n.a. |
| Wade, 201125 | M | Amylmetacresol/2,4‐dichlorobenzyl alcohol (AMC 0.6 mg/DCBA 1.2 mg) vs unflavored placebo | ≤4 d, throat pain VAS ≥6, TPA ≥ 3 | 151/74 | AUC (0‐2 h): 43% pain reduction 2 h p.i.d.** | n.a. |
| Wonnemann, 200764 | S | Lidocaine 8 mg | ≤2 d, throat pain VAS ≥ 6 | 122/125 | AUC (0‐2 h): pain reduction 2 and 48 h p.i.d.** | n.a. |
| Busch, 201065 | Benzocain 8 mg | <2 d, VAS > 5 | 123/122 | 25% reduction in throat pain within 20 min, 2 h p.i.d.** | n.a. | |
| Chrubasik, 201131 | S | Benzocaine 8 mg | ≤2 d, throat pain VAS ≥ 6 | 83/82 | SPID 2 h p.i.d. −12 vs −5*** | n.a. |
| Eberhardt, 200435 | M | 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, 1.5 mg benzocaine | ≤1 d, throat pain VAS ≥ 3 | 59/59 | n.a. | Total responders free of throat pain and difficulty in swallowing: 75% vs 58% 3 d p.i.d.,** |
| Scholten, 200536 | M | 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, 1.5 mg benzocaine | ≤3 d, throat pain VAS ≥ 3 | 63/65 | n.a. | Total responders free of throat pain and difficulty in swallowing: 30% vs 10% 48 h p.i.d.,* |
| Palm, 2018 | M | 0.5 mg tyrothricin, 1.0 mg benzalkonium chloride, 1.5 mg benzocaine | ≤1 d, throat pain VAS ≥ 7 | 160/161 | SPID 2 h p.i.d. −20 vs −10*** | Total responders free of throat pain and difficulty in swallowing: 44.6% vs 27.2%*** 72 h p.i.d., 64% improved response rate*** |
p.i.d.: post initial dose; tid: three times daily; n.a.: not assessed.
*P‐value < 0.05.
**P‐value < 0.01.
***p‐value < 0.005.
The results of the present study showed a significant benefit of study drug over placebo in the treatment of acute pharyngitis. The primary endpoint (total responders 72 hours p.i.d.) revealed a 64% improvement (GEE, P < 0.0002) and most of the secondary endpoints also showed statistically significant improvements in throat pain and swallowing difficulty on study drug compared with placebo. Overall, patients and investigators were very satisfied with the treatment effect and the tolerability of study drug. This is supported by the fact that 77% of patients in the verum group were willing to use the study medication in the future and to recommend it to others compared with 48% of patients in the placebo group.
4.1. Efficacy: acute and sustained
A rapid onset and sustainability of pain relief are key features when assessing sore throat remedies.26 Onset of pain relief can be measured via several different outcome measures. Methods used have included summed pain intensity difference (SPID) and time‐weighted TOTal Pain Relief (TOTPAR) over a defined time period (eg, 15 or 120 minutes) after first dosing; TOTPAR is calculated as the area under the plot of relief scores over this interval, that is, the sum of the Sore Throat Relief Scale (STRS) scores obtained during this interval multiplied by the interval (expressed in hours) between successive ratings.27
The increased pain relief provided by the study drug lozenge vs. the matched placebo lozenge was rapid as demonstrated by a statistically significantly higher weighted TOTPAR than that provided by the placebo as early as within the first 5 minutes post initial dose. This significant effect of study drug was maintained up to each subsequent assessment timepoint during the 120 minutes p.i.d. and demonstrated superiority over placebo in relieving sore throat pain over 120 minutes after first dosing (P = 0.005), a result confirmed by supportive and sensitive analyses.
The sustainability of pain relief is often assessed by TOTPAR, but only over 120 minutes after first dosing. In contrast, we consider complete responders free of throat pain and difficulties in swallowing as a more relevant outcome parameter characterising sustainable treatment effect. Indeed, Singla showed that SPID and TOTPAR are very sensitive parameters to detect treatment differences early in clinical trials on acute pain.28 However, as clinically more meaningful, the authors of the meta‐analysis recommend Day 3 as the preferred assessment time for predefined primary endpoint in pharyngitis interventional trials.29
The ambulatory multiple dose phase of the study revealed significantly better results for patients receiving triple active lozenge on Day 3: They rated higher pain relief, improvement in difficulties in swallowing and combined a higher responder rate than those on placebo, based on both STRS and global efficacy assessments.
This is a remarkable finding as other intervention studies (Table 5) with marketed single compound products (NSAIDS, ibuprofen, flurbiprofen, benzocaine, lidocaine),23, 30, 31, 32, 33 failed in showing superiority beyond day 1 or 2: the investigators explained their non‐significant results by “the favourable natural progression of sore throat (which heals spontaneously in most patients within a few days),” “the natural decrease in sore throat pain intensity with time,” and with the “known placebo effect of lozenges.”32 Also, for marketed flurbiprofen, including the approved microgranules, no statistically significant pain relief effect beyond day 1 evening has been shown.33
The known placebo effect explained by the effect of sucking lozenges and the consequent stimulation of salivation induced by sucking34 was also seen in this study—despite the same qualifications such as natural progression of sore throat and natural decrease with time, ie, patients in the placebo group also experienced pain relief albeit less. Which discrepancies may explain the beneficial sustainable effect for study drug after 3 days? The design of the trial is comparable to others (placebo controlled, 1:1 balanced allocation to intervention), alike patient population with inclusion criteria of non‐bacterial cause of pharyngitis of acute to moderate throat pain. However, we included patients with an onset of symptoms within 24 hours and not up to 2 or 4 days like others did (Table 5) and we included patients only with pain intensity >7 in order to exclude milder courses. Clearly, the main difference among trials is the active intervention as others used single compound products, such as NSAIDs or benzocaine, or a double combination of amylmetacresol and 2,4‐dichlorobenzyl alcohol (AMC/DCBA).24 The triple combination used in this trial not only offers pain relief due to the sodium channel blocking agent benzocaine, but also antiviral and antimicrobial activity due to benzalkonium chloride and tyrothricin. The antiviral activity for benzalkonium chloride, as well as the antibacterial and antiviral property of tyrothricin, is known.18, 19, 20 Antiviral properties for the combination of all three compounds in Dorithricin® have been recently confirmed in an in vitro model.21 Thus, we can conclude the composite of three active ingredients with antibacterial, antiviral as well as anti‐inflammatory, analgesic activity has its value in blocking the inflammatory process seen in pharyngitis and its viral etiology and potential superinfection ‐ all supporting faster remission free of throat pain and difficulty in swallowing in acute pharyngitis.
4.2. Safety and tolerability
The occurrence of TEAEs considered related to the test product (ADRs) was low (10/160 patients, 6.3%). All ADRs were mild or moderate in intensity and had resolved by the end of the study. Hypersensitivity reactions including those of the skin were not reported for any patient treated with the test product. In the study, no relevant side effects were noted ‐ all mild and comparable to other studies on lozenges in patients with pharyngitis (Table 4 and 5). That is, acute side effects were not reported. Also, in previous studies, no relevant adverse drug effects were observed in the clinical studies conducted with Dorithricin® lozenges.35, 36
Benzocaine has been used as a local anaesthetic for more than a century. Its safety profile is well known.37 Methaemoglobinaemia is one of the most severe adverse effects known, but is usually associated with the administration of higher concentrations (eg, benzocaine 20% spray) applied in endoscopy, intubation, bronchoscopy, or similar invasive procedures.38 The authors of a non‐clinical in vivo study revealed a single oral dose of 1.6‐4.9 mg benzocaine per kg bodyweight would not induce methaemoglobin in humans.39 Considering these numbers it appears unlikely that the usage of benzocaine‐containing throat lozenges may lead to methaemoglobinaemia.
Benzalkonium chloride (N‐alkyl‐N‐benzyl‐N,N‐dimethyl ammonium chloride) is a quaternary ammonium compound with antimicrobial and antiviral activity that is also used as a preservative agent in topical medications such as eye and nose drops. With the usage of such nose drops, anaphylactic reactions to benzalkonium chloride have been described.40 Medical application of decongestant nose drops should also take into consideration the rare, but possible, allergic reaction to benzalkonium chloride. One single dose of one lozenge corresponds to only 0.01 mg/kg bodyweight; assuming that one lozenge is dissolved in 15 minutes and that on average 30 mL saliva is produced during that time, a maximum concentration of 0.0333% benzalkonium chloride would be theoretically achieved.41
The treatment of acute pharyngitis has been controversial for decades, with most of the debate addressing the immediate, delayed or no use of antibiotics: benefits of systemic antibiotics are modest by shortening the symptoms of the illness (by ca. 16 hours),42 by protecting against acute rheumatic fever or secondary bacterial infections (eg, acute otitis media).42, 43, 44 Risks are known adverse reactions (eg, nausea, rash, vaginitis, headache, gastrointestinal side effects), disturbance of the beneficial microbial community, especially in the gut microbiome with decreased microbial diversity,44, 45, 46 but also reported in throat microflora45 and their widespread use leads to bacterial resistance especially with broad‐spectrum antibiotics.43, 44, 46, 47, 48, 49 Penicillin resistance in Haemophilus influenzae is mainly due to the production of beta‐lactamases TEM‐1 and ROB‐1. Strep. pneumoniae resistance is due to changes in penicillin‐binding proteins. Resistance to tetracyclines, macrolides, trimethoprim‐sulphamethoxazole and fluoroquinolones depends on changes in target, active efflux and modifying enzymes involved.47, 48
One first hypothesis assumes that the usage of antibiotics predisposes to bacterial superinfection as prophylactic usage could imbalance the natural microbial flora and facilitate colonisation of bacterial pathogens or pathobionts.5, 6 The human upper respiratory tract is a reservoir of a diverse community of commensals and potential pathogens (pathobionts) including Streptococcus pneumoniae, H. influenzae, Moraxella catarrhalis and Staphylococcus aureus, which occasionally turn into pathogens.1
The other second hypothesis claims that virus infections of the respiratory tract imbalance the respiratory microbial community7, 8 and thus predispose to bacterial superinfections2, 8, 9, 10: Longitudinal studies have revealed a clear and positive association between viral and bacterial infections of the airway as acute otitis media, sinusitis, purulent nasopharyngitis, acute bronchitis, tonsillitis and pneumonia all often occur after local viral infection.2, 5, 7 Interactions between viruses and bacteria in the pathogenesis of respiratory infections have been investigated by others applying techniques used in microbiology and molecular biology1, 2, 7, 8, 9: The mechanisms by which viruses influence bacterial colonisation and invasion are diverse and include disruption of the epithelium barrier, upregulation of adhesion proteins, production of viral factors and dysfunction of immune system components.1, 2, 7, 9
In analogy with this controversy it is justified to question if AMPs such as tyrothricin might also disturb the physiological bacterial flora predisposing to bacterial superinfection and if AMPs could lead to bacterial resistance.
Clearly, our trial was not designed for addressing changes in microbiotic flora or induced resistance in the topical usage of antimicrobial tyrothricin.
In our study there were several cases of TEAEs classifiable as bacterial superinfection: bacterial pharyngitis, nasopharyngitis, otitis media, tonsillitis, sinusitis with seven cases in the placebo group (7/161 pts; 4.5%) plus one severe case of tonsillitis (5.2%) versus only one case (ie, nasopharyngitis) among the 160 patients (0.6%) assigned to the verum group (plus one case of pneumonia hospitalised at day 2) supporting the hypothesis that study medication is not enhancing the risk for potential superinfections (first hypothesis) but possibly the opposite, ie, supporting the second hypothesis. Our clinical findings with a significant difference in bacterial infections underline the antimicrobial mode of action. As published by American Pharm Association, Tyrothricin has an effect on oral microorganism and it was shown that total numbers of pathobionts are diminished for at least one‐half hour after dissolution of a single lozenge.50 Tyrothricin is an AMP and AMPs are an intrinsic part of the human innate immune system. Tyrothricin acts only topically, is not absorbed and represents a mixture of two different substances with 80% tyrocidins and 20% gramicidins.18 With their interplay, these peptides offer a broad antimicrobial spectrum counteracting Gram‐negative and Gram‐positive bacteria, especially streptococcus and staphylococcus frequently seen with bacterial infection in inflammatory pharyngitis.50
The notion of bacterial resistance needs to differentiate systemic antibiotics, especially broad‐spectrum antibiotics, from tyrothricin: The mixture of peptides in tyrothricin with different sequences and secondary structures is prevents the induction of bacterial resistance as this would require microorganisms developing different mechanism for resistance simultaneously which is practically not seen. Tyrothricin as AMP acts on the membrane of bacteria leading to lysis of bacteria cells within minutes.51 One should consider that the double lipid membrane layer of bacteria is highly conserved over evolution and seems not to be a suitable target for modification as resistance strategy without major consequences. Even more, tyrocidine and gramicidin interact with the membrane on different targets which would imply multiple and differing modifications of the membrane if resistance was achieved—on a theoretical level.52 Finally, the mode of action implies that tyrothricin does not have to enter the bacterial cell for its antibacterial potential, and in consequence, the theoretical strategy for developing resistance by expressing efflux pump systems is not valid. The fast mode of action counteracts the evolution of building resistance within bacterial populations. And tyrothricin acts only locally and thus prevents the cross building of resistance with systemically active antibiotics.18
These microbiological aspects are underlined by clinical evidence and for addressing the question of induced resistance by the topical usage of tyrothricin, we revert to trials designed for: Many studies have investigated the question of induced resistance.53 Locally applied tyrothricin has not led to resistance as shown in multiple studies, the latest by Strauss‐Grabo,54 excluding resistance against tyrothricin despite decades of tyrothricin usage. The authors tested Gram‐positive bacteria and yeast strains which all turned out to be highly susceptible to tyrothricin (MICs ≤ 4 mg/L), despite decade‐long use of the substance. The authors concluded, “No acquired resistance of the tested strains was determined.”54 Also, the topical usage on mucous membranes reviewed by Korting19 concluded, “in contrast to other antibiotics, AMPs (such as tyrothricin)” do not seem to propagate the development of antibiotic‐resistant micro‐organisms.”19
In consequence, an effective reduction in the germ count with locally acting mouth and throat preparations may not only reduce the risk of disease aggravation via, eg, bacterial superinfection, but also the unnecessary use of systemic antibiotics, mainly penicillin. The widespread overuse of systemic penicillin and other systemic antibiotics may expose patients unnecessarily to potential AEs and cause a dramatic increase in resistances.49
5. CONCLUSION
The results of this clinical study show that 3‐day oral treatment with Dorithricin® at the recommended dose of 8 lozenges per day is effective in relieving severe throat pain and difficulty in swallowing associated with acute pharyngitis within 48 and 72 hours.
Local treatment with Dorithricin® is safe, well tolerated and comparable to placebo treatment regarding the safety profile. Triple active throat lozenges provide rapid analgesic effects that last 2 hours, securing ongoing relief long after the lozenge has dissolved. The superior analgesic effects and improvements in functional impairment observed with triple active throat lozenges translate into clinically meaningful responder rates and are thus a suitable OTC treatment option for patients in the self‐management of acute sore throat.
DISCLOSURES
The authors have no conflict of interest. Medice Arzneimittel Pütter GmbH & Co.KG, 58638 Iserlohn, Germany, is the sponsor of the study and the manufacturer of the clinical trial material manufactured in 58638 Iserlohn, Germany.
AUTHOR CONTRIBUTIONS
All authors contributed towards data analysis, drafting and critically revising the paper, and agree to be accountable for all aspects of the work.
ACKNOWLEDGEMENTS
DoriPha investigators were Balck Katharina (38527 Meine), Berger‐Roscher Jürgen (medicoKit, 47574 Goch), Eckermann Tamara (80330 München), Heimlich Florian (ENT, 69126 Heidelberg), Horn Andreas (ENT, 69120 Heidelberg), Jung Thomas (94550 Künzing), Palm Jürgen (ENT, doc‐hno, 90552 Röthenbach), Samer Holger (83527 Haag), Schaale‐Maas Jürgen (ENT, 53359 Rheinbach), Schaefer Axel (Medizentrum, 53359 Essen), Stahl Hans‐Detlef (AmBeNet, 04107 Leipzig), Thieme Uta (ENT, 47051 Duisburg), Thinesse‐Mallwitz Manuela (80339 München), Walter Ulrike (90402 Nürnberg), Wienhold Karin (42103 Wuppertal), Yarin Yury (ENT, 01139 Dresden). Medical writing assistance was provided by Martin Carmen at MartinMed Ltd, and the study including clinical trial material was sponsored by Medice Arzneimittel Pütter GmbH & Co.KG, 58638 Iserlohn, Germany.
Palm J, Fuchs K, Stammer H, Schumacher‐Stimpfl A, Milde J for the DoriPha investigators . Efficacy and safety of a triple active sore throat lozenge in the treatment of patients with acute pharyngitis: Results of a multi‐centre, randomised, placebo‐controlled, double‐blind, parallel‐group trial (DoriPha). Int J Clin Pract. 2018;72:e13272 10.1111/ijcp.13272
REFERENCES
- 1. Bosch AA, Biesbroek G, Trzcinski K, Sanders E, Bogaert D. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 2013;9(1):e1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sajjan US, Jia Y, Newcomb DC, Bentley JK, Lukacs NW, et al. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM‐1 and TLR3 expression. FASEB J. 2006;20(12):2121‐2123. [DOI] [PubMed] [Google Scholar]
- 3. Panda S, El Khader I, Casellas F, et al. Short‐term effect of antibiotics on human gut microbiota. PLoS ONE. 2014;9:e95476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Willing BP, Russell SL, Finlay BB. Shifting the balance: antibiotic effects on host‐microbiota mutualism. Nat Rev Microbiol. 2011;9:233‐243. [DOI] [PubMed] [Google Scholar]
- 5. Planelles J. The pathogenic mechanism of superinfections in antibiotic therapy of bacterial infections. Z Arztl Fortbild (Jena). 1963;15(57):702‐707. [PubMed] [Google Scholar]
- 6. Alvarez C, Ramos JM, San Juan R, Lumbreras C, Aguado JM. Risk of superinfection related to antibiotic use. Are all antibiotics the same? Rev Esp Quimioter. 2005;18(1):39‐44. [PubMed] [Google Scholar]
- 7. Bakaletz LO. Viral potentiation of bacterial superinfection of the respiratory tract. Trends Microbiol. 1995;3(3):110‐114. [DOI] [PubMed] [Google Scholar]
- 8. Bisno AL. Acute pharyngitis. N Engl J Med. 2001;344(3):205‐211. [DOI] [PubMed] [Google Scholar]
- 9. Moore HCBS, Jacoby P, Taylor A, Harnett G, Bowman J, et al. The interaction between respiratory viruses and pathogenic bacteria in the upper respiratory tract of asymptomatic aboriginal and nonaboriginal children. Pediatr Infect Dis J. 2010;29(6):540‐545. [DOI] [PubMed] [Google Scholar]
- 10. Bisno AL. Pharyngitis In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia: Elsevier Churchill Livingstone; 2005:752‐758. [Google Scholar]
- 11. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279‐1282. [DOI] [PubMed] [Google Scholar]
- 12. Farrer F. Sprays and lozenges for sore throat. S Afr Pharm J. 2011;78:26‐31. [Google Scholar]
- 13. Kumar M, Chawla R, Goyal M. Topical anesthesia. J Anaesthesiol Clin Pharmacol. 2015;31(4):450‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weibel MA, Cortat M, Lebek G, LeCotonnec JY, Kitler ME, Barcherini G. An approach of the in vivo antibacterial activity of benzaconium chloride and comparison with other buccopharyngeal disinfectants. Arzneimittelforschung. 1987;37(4):467‐471. [PubMed] [Google Scholar]
- 15. Iwasawa A, Niwano Y, Kohno M, Ayaki M. Virucidal activity of alcohol‐based hand rub disinfectants. Biocontrol Sci. 2012;17(1):45‐49. [DOI] [PubMed] [Google Scholar]
- 16. Yamasaki H, Tsujimoto K, Ikeda K, Suzuki Y, Arakawa T, Koyama AH. Antiviral and virucidal activities of nα‐cocoyl‐L‐arginine ethyl ester in comparison to benzalkonium chloride. Adv Virol. 2011;11(28):e572868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wood A, Payne D. The action of three antiseptics/disinfectants against enveloped and non‐enveloped viruses. J Hosp Infect. 1998;38(4):283‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lang C, Staiger C. Tyrothricin‐An underrated agent for the treatment of bacterial skin infections and superficial wounds? Pharmazie. 2016;71(6):299‐305. [PubMed] [Google Scholar]
- 19. Korting HC, Schöllmann C, Stauss‐Grabo M, Schäfer‐Korting M. Antimicrobial peptides and skin: a paradigm of translational medicine. Skin Pharmacol Physiol. 2012;25(6):323‐334. [DOI] [PubMed] [Google Scholar]
- 20. Bals R. Antimicrobial peptides and peptide antibiotics. Med Klin. 2000;95(9):496‐502. [DOI] [PubMed] [Google Scholar]
- 21. Schmidbauer M. Dorithricin wirkt antiviral. Results of in‐vitro investigations. Pharmazeutische Zeitung. 2015;160(38):48‐52. [Google Scholar]
- 22. DEGAM. Halsschmerzen. Deutsche Gesellschaft für Allgemeinmedizin (DEGAM)‐Leitlinie Nr 14. 2009. https://www.degam.de/degam-leitlinien-379.html. Accessed 10th January 2018.
- 23. Schachtel B, Aspley S, Shephard A, Shea T, Smith G, Schachtel E. Utility of the sore throat pain model in a multiple‐dose assessment of the acute analgesic flurbiprofen: a randomized controlled study. Trials. 2014;15:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McNally D, Shephard A, Field E. Randomised, double‐blind, placebo‐controlled study of a single dose of an amylmetacresol/ 2,4‐dichlorobenzyl alcohol plus lidocaine lozenge or a hexylresorcinol lozenge for the treatment of acute sore throat due to upper respiratory tract infection. J Pharm Pharm Sci. 2012;15(2):281‐294. [DOI] [PubMed] [Google Scholar]
- 25. Wade AG, Morris C, Shephard A, Crawford G, Goulder G. A multicentre, randomised, double‐blind, single‐dose study assessing the efficacy of AMC/DCBA Warm lozenge or AMC/DCBA Cool lozenge in the relief of acute sore throat. BMC Fam Pract. 2011;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oxford JS, Leuwer M. Acute sore throat revisited: clinical and experimental evidence for the efficacy of over‐the‐ Counter AMC/DCBA throat lozenges. Int J Clin Pract. 2011;65:524‐530. [DOI] [PubMed] [Google Scholar]
- 27. Dworkin RH, Nagasago EM, Hetzel RD, Farrar JT. Assessment of pain and pain‐related quality of life in clinical trials In: Turk DC, Melzack R, eds. Handbook of Pain Assessment, chapter 34. New York: Guilford Press; 2001:659‐692. [Google Scholar]
- 28. Singla N, Hunsinger M, Chang PD, et al. Assay sensitivity of pain intensity versus pain relief in acute pain clinical trials: ACTTION systematic review and meta‐analysis. J Pain. 2015;16:683‐691. [DOI] [PubMed] [Google Scholar]
- 29. Thomas M, Del Mar C, Glasziou P. How effective are treatments other than antibiotics for acute sore throat. Br J Gen Pract. 2000;50:817‐820. [PMC free article] [PubMed] [Google Scholar]
- 30. Aspley S, Schachtel B. Efficacy of flurbiprofen 8.75 mg lozenge in patients with a swollen and inflamed sore throat. Curr Med Res Opin. 2016;32(9):1529‐1538. [DOI] [PubMed] [Google Scholar]
- 31. Chrubasik S, Beime B, Magora F. Efficacy of a benzocaine lozenge in the treatment of uncomplicated sore throat. Eur Arch Otorhinolaryngol. 2012;269(2):571‐577. [DOI] [PubMed] [Google Scholar]
- 32. Bouroubi A, Donazzolo Y, Donath F, et al. Pain relief of sore throat with a new anti‐inflammatory throat lozenge, ibuprofen 25 mg: a randomised, double‐blind, placebo‐controlled, international phase III study. Int J Clin Pract. 2017;71(9):e12961. [DOI] [PubMed] [Google Scholar]
- 33. Russo M, Bloch M, de Looze F, Morris C, Shephard A. Flurbiprofen microgranules for relief of sore throat: a randomised, double‐blind trial. Br J Gen Pract. 2013;63:149‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nathan A. Colds and sore throat. Pharm J. 1996;256:24‐27. [Google Scholar]
- 35. Eberhardt R, Maier‐Bosse I, Schlauch W. Halsschmerzen bei akuter Pharyngitis. Deutsche Apotheker Zeitung. 2004;144(7):753‐754. [Google Scholar]
- 36. Scholten T, Pautler M, Kober G. Dorithricin bei akuter Pharyngitis. Deutsche Apotheker Zeitung. 2005;145(1):81‐82. [Google Scholar]
- 37. Adriani J, Taylor M. Benzocaine's properties and uses as a topical anesthetic. Part I. Physicochemical and pharmacological description. Anesthesiol Rev. 1990; XVII(1):27‐33. [Google Scholar]
- 38. Moore TJ, Walsh CS, Cohen MR. Reported adverse event cases of methemoglobinemia associated with benzocaine products. Arch Intern Med. 2004;164(11):1192‐1196. [DOI] [PubMed] [Google Scholar]
- 39. Tungeln LS, Zhou T, Woodling KA, Doerge DR, Greenlees KJ, Beland FA. Benzocaine‐induced methemoglobinemia in an acute‐exposure rat model. Food Chem Toxicol. 2011;49:2530‐2535. [DOI] [PubMed] [Google Scholar]
- 40. Mezger E, Wendler O, Mayr S, Bozzato A. Anaphylactic reaction following administration of nose drops containing benzalkonium chloride. Head Face Med. 2012; Oct 18; 8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bundesinstitut für Risikobewertung . Health Assessment of Benzalkonium Chloride Residues in Food. BfR opinion No 032/2012. 2012. https://www.bfr.bund.de/cm/343/gesundheitliche-bewertung-der-rueckstaende-vonbenzalkoniumchlorid-in-lebensmitteln.pdf. Accessed 1 May 2018.
- 42. Spinks A, Glasziou PP, Del Mar CB. Antibiotics for sore throat. Cochrane Database Syst Rev. 2013;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kenealy T. Sore throat. BMJ Clin Evid. 2014;4:1509. [PMC free article] [PubMed] [Google Scholar]
- 44. Spurling GK, Del Mar CB, Dooley L, Foxlee R, Farley R. Delayed antibiotic prescriptions for respiratory infections. Cochrane Database Syst Rev. 2017;9:CD004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jakobsson HE, Jernberg C, Andersson AF, Sjölund‐Karlsson M, Jansson JK, Engstrand L. Short‐term antibiotic treatment has differing long‐term impacts on the human throat and gut microbiome. PLoS ONE. 2010;5:e9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Llor C, Bjerrum L. Antimicrobial resistance: risk associated with antibiotic overuse and initiatives to reduce the problem. Ther Adv Drug Saf. 2014;5(6):229‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steinman MA, Landefeld C, Gonzales R. Predictors of Broad‐Spectrum Antibiotic Prescribing for Acute Respiratory Tract Infections in Adult Primary Care. JAMA. 2003;289(6):719‐725. [DOI] [PubMed] [Google Scholar]
- 48. Mlynarczyk G, Mlynarczyk A, Jeljaszewicz J. Epidemiological aspects of antibiotic resistance in respiratory pathogens. Int J Antimicrob Agents. 2001;18(6):497‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith RA, Mikanatha NM, Read AF. Antibiotic resistance: a primer and call to action. Health Commun. 2015;30(3):309‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Florestano HJ, Bahler ME, Richards AB. Effect of actamer and tyrothricin on oral microorganisms. J Am Pharm Assoc Am Pharm Assoc. 1956;45(1):13‐16. [DOI] [PubMed] [Google Scholar]
- 51. Ruckdeschel G, Beaufort F, Nahler G, Belzer O. In vitro antibacterial activity of gramicidin and tyrothricin. Arzneimittelforschung. 1983;33(12):1620‐1622. [PubMed] [Google Scholar]
- 52. Prenner EJ, Lewis RN, McElhaney RN. The interaction of the antimicrobial peptide gramicidin S with lipid bilayer model and biological membranes. Biochim Biophys Acta. 1999;1462(1–2):201‐221. [DOI] [PubMed] [Google Scholar]
- 53. Lind HE, Swanton EM. The effect of prolonged use of a tyrothricin tooth paste on the tyrothricin resistance of oral microorganisms and their cross resistance to other antibiotics: Antibiot . Chemother. 1983;4(11):1161‐1166. [PubMed] [Google Scholar]
- 54. Stauss‐Gabo M, Atiye S, Le T, Kretschmar M. Decade‐long use of the antimicrobial peptide combination tyrothricin does not pose a major risk of acquired resistance with gram‐positive bacteria and Candida spp. Pharmazie. 2014;69(11):838‐841. [PubMed] [Google Scholar]
- 55. Schachtel BP, Fillingim JM, Thoden WR, Lane AC, Baybutt RI. Sore throat pain in the evaluation of mild analgesics. Clin Pharmacol Ther. 1988;44:704‐711. [DOI] [PubMed] [Google Scholar]
- 56. Bertin L, Pons G, d'Athis P, et al. Randomized, double‐blind, multicenter, controlled trial of ibuprofen versus acetaminophen (paracetamol) and placebo for treatment of symptoms of tonsillitis and pharyngitis in children. J Pediatr. 1991;119:811‐814. [DOI] [PubMed] [Google Scholar]
- 57. Schachtel BP, Thoden WR. A placebo‐controlled model for assaying systemic analgesics in children. Clin Pharmacol Ther. 1993;53:593‐601. [DOI] [PubMed] [Google Scholar]
- 58. Schachtel BP, Cleves GS, Konerman JP, Brown AT, Markham AO. A placebo‐controlled model to assay the onset of action of nonprescription‐strength analgesic drugs. Clin Pharmacol Ther. 1994;55:464‐470. [DOI] [PubMed] [Google Scholar]
- 59. Schachtel BP, Fillingim JM, Lane AC, Thoden WR, Baybutt RI. Caffeine as an analgesic adjuvant. A double‐blind study comparing aspirin with caffeine to aspirin and placebo in patients with sore throat. Arch Intern Med. 1991;151:733‐737. [DOI] [PubMed] [Google Scholar]
- 60. Sauvage JP, Ditisheim A, Bessede JP, David N. Double‐blind, placebo‐controlled, multi‐centre trial of the efficacy and tolerance of niflumic acid (Nifluril) capsules in the treatment of tonsillitis in adults. Curr Med Res Opin. 1990;11:631‐637. [DOI] [PubMed] [Google Scholar]
- 61. Whiteside MW. A controlled study of benzydamine oral rinse (Difflam) in general practice. Curr Med Res Opin. 1982;8:188‐190. [DOI] [PubMed] [Google Scholar]
- 62. Schutz A, Gund HJ, Pschorn U, et al. Local anaesthetic properties of ambroxol hydrochloride lozenges in view of sore throat. Clinical proof of concept. Arzneimittelforschung. 2002;52(3):194‐199. [DOI] [PubMed] [Google Scholar]
- 63. Fischer J, Pschorn U, Vix JM, et al. Efficacy and tolerability of ambroxol hydrochloride lozenges in sore throat. Randomised, double‐blind, placebo‐controlled trials regarding the local anaesthetic properties. Arzneimittelforschung. 2002;52(4):256‐263. [DOI] [PubMed] [Google Scholar]
- 64. Wonnemann M, Helm I, Stauss‐Grabo M, et al. Lidocaine 8 mg sore throat lozenges in the treatment of acute pharyngitis. A new therapeutic option investigated in comparison to placebo treatment. Arzneimittelforschung. 2007;57(11):689‐697. [DOI] [PubMed] [Google Scholar]
- 65. Busch R, Graubaum HJ, Grünwald J, Schmidt M. Double‐blind comparison of two types of benzocaine lozenges for the treatment of acute pharyngitis. Arzneimittelforschung. 2010;60(5):245‐248. [DOI] [PubMed] [Google Scholar]