

Abstract
Background
Toll‐like receptor 7 (TLR7) is an endosomal TLR that is activated by single‐stranded RNA, including endogenous microRNAs (e.g., let‐7b). Increased hepatic expression of TLRs, microRNAs, and inflammatory mediators is linked to ethanol (EtOH) exposure and to alcoholic liver disease (ALD). ALD invovles chronic hepatic inflammation that can progress to alcoholic hepatitis (AH), a particularly severe form of ALD. This study aimed to investigate TLR7 expression in patients with different liver disease phenotypes and in mouse liver following alcohol exposure.
Methods
Hepatic mRNA expression was determined by RNA sequencing of liver tissue from patients with liver disease or normal liver tissue. Mice were exposed to subchronic EtOH followed by administration of the TLR7 agonist imiquimod. Primary human hepatocytes were exposed to EtOH or imiquimod in vitro.
Results
RNAseq analysis revealed that hepatic expression of TLR7 and let‐7b microRNA, an endogenous TLR7 ligand, was significantly increased in AH patients. Hepatic expression of TLR7 and let‐7b positively correlated with hepatic IL‐8 mRNA expression. In mice, EtOH increased hepatic TLR7 mRNA expression and enhanced imiquimod‐induced expression of the pro‐inflammatory mediators TNF α, MCP‐1, and iNOS. In vitro, EtOH significantly increased hepatocyte TLR7 mRNA and the TLR7 agonist, imiquimod, induced hepatocyte expression of TNF α and IL‐8 mRNA. EtOH also increased the release of let‐7b in microvesicles from hepatocytes, suggesting that EtOH can increase the expression of both the receptor and its endogenous ligand.
Conclusions
These studies suggest that increased TLR7 signaling caused by increased expression of TLR7 and its endogenous ligand let‐7b may contribute to the enhanced inflammatory response associated with AH.
Keywords: Liver Disease, microRNA, Pro‐Inflammatory Cytokines, Human, Hepatocyte
Alcoholic liver disease (ALD) is a leading cause of cirrhosis and acute‐on‐chronic liver failure. ALD is the main cause of mortality associated with organ damage in adults with chronic alcohol abuse (Stickel et al., 2017). ALD is a spectrum of disease states that progresses from mild, nonsymptomatic disease (e.g., alcoholic steatohepatitis [ASH]) to more severe chronic ALDs including fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). In addition, patients with underlying liver disease and excessive alcohol intake can develop episodes of alcoholic hepatitis (AH), a particularly severe form of acute‐on‐chronic liver injury associated with a short‐term mortality rate of up to 30 to 50% (Lanthier and Starkel, 2017). AH is characterized by hepatic inflammation, including increased IL‐8 levels, neutrophil infiltration, and hepatocyte damage. Although ethanol (EtOH) is known to enhance the hepatic pro‐inflammatory response, the mechanisms that trigger the acute onset of AH remain unclear. Furthermore, treatment with anti‐inflammatory corticosteroids, the current first‐line therapy for AH, is not effective in many patients (Thursz et al., 2015). Therefore, identification of disease‐specific mechanisms and novel therapeutic targets for AH represent major needs in clinical hepatology.
ALD progresses from early, subclinical disease to more advanced stages, including cirrhosis and AH, through poorly understood mechanisms that may represent multiple hepatic insults or “hits” (Day and James, 1998; Tsukamoto et al., 2009). In this paradigm of disease progression, lipid accumulation caused by EtOH metabolism serves as a first “hit” which sensitizes the liver to additional insults including enhanced pro‐inflammatory signaling, mitochondrial stress, and oxidative stress. The subsequent “hit” of enhanced pro‐inflammatory signaling is consistent with progressive increases in inflammation characterized by increased release of pro‐inflammatory cytokines from resident macrophages and sensitization of parenchymal cells to the pro‐inflammatory signals. Researchers have further demonstrated that enhanced inflammatory signaling during ALD is mediated, at least in part, via increased expression of Toll‐like receptors (TLRs). Many studies investigating the role of TLRs in ALD have focused on the role of TLR4 and its pathogen‐associated ligand, lipopolysaccharide (LPS), which is increased in the blood by alcohol consumption. Those studies have found that TLR4 contributes to hepatic inflammation and fibrosis in multiple murine models of alcohol‐induced liver injury (Hritz et al., 2008; Uesugi et al., 2001; Zhao et al., 2008). More recently, endogenous molecules that contain damage‐associated molecular patterns have also been shown to activate TLRs, suggesting that endogenous ligands may contribute to the progressive pro‐inflammatory response associated with the development of ALD. In contrast to TLR4 signaling, the roles of other TLRs in mediating EtOH induction of hepatic immune responses remain less clear. A study by Gustot and colleagues (2006) found increased hepatic expression of multiple TLR receptors (e.g., TLR1, TLR2, TLR6, TLR7, and TLR8) after chronic feeding of EtOH‐containing liquid diet. The up‐regulation of multiple liver TLRs by EtOH exposure was associated with enhanced hepatic TNFα levels and liver injury (Gustot et al., 2006) in response to exposure to TLR agonists. However, additional studies are required to investigate how multiple TLRs, including TLR7, impact the progression of human ALD and the onset of AH.
Previous studies report increased TLR7 expression both in experimental ASH caused by chronic alcohol exposure in mice (Gustot et al., 2006) and in humans with end‐stage ALD (Starkel et al., 2010). However, the functional effect of increased TLR7 expression in the setting of ALD remains to be determined. TLR7 is known to respond to viral single‐stranded RNA (ssRNA), and recent studies have found it can also respond to endogenous microRNAs (Coleman et al., 2017; Lehmann et al., 2012). Activation of TLR7 by ssRNAs or small molecule agonists (e.g., imiquimod) stimulates innate immune responses including induction of antiviral interferons and pro‐inflammatory cytokines (Novak et al., 2008; Ramirez‐Ortiz et al., 2015). In this study, we hypothesized that EtOH‐induced TLR7 expression would increase hepatic inflammation in response to the TLR7 agonist imiquimod. To test the effects of EtOH exposure on the TLR7‐mediated inflammatory responses, mice were preexposed to EtOH, and then, the response to the TLR7 agonist, imiquimod, was determined. Previous studies have demonstrated that hepatocyte‐like cell lines (e.g., HepG2, Huh7) express TLR7 and are activated by TLR7 agonists (Lee et al., 2006; Sarkar et al., 2015). Furthermore, as a primary site of alcohol metabolism, hepatocytes are a prime target for alcohol‐induced toxicity. Therefore, we determined the effects of EtOH and imiquimod on hepatocyte TLR7 expression and inflammatory signaling. Because there is currently no mouse model of EtOH exposure that adequately produces the clinical sequelae of AH, the TLR7 signaling pathway, including expression of the TLR7 receptor and the endogenous let‐7 miRNA agonists, was also determined in human liver samples from patients with liver disease, including AH. Here, we report that EtOH increased TLR7 mRNA expression in the liver of mice and in cultured primary human hepatocytes. We further found that EtOH preexposure increased TLR7 induction of pro‐inflammatory genes in mice. RNA sequencing of human liver tissue revealed that AH is associated with increased hepatic mRNA expression of the TLR7 receptor as well as let‐7 microRNAs which can endogenously activate TLR7 signaling. Importantly, hepatic expression of both the receptor and endogenous ligands correlate with hepatic mRNA expression of IL‐8, a known driver of AH. Together, these findings support the hypothesis that increased TLR7 signaling contributes to progressive increases in liver inflammation and injury caused by alcohol and may represent a “second hit” involved in the development of severe ALD and AH.
Materials and Methods
Animals and Treatment
Eight‐week‐old male C57BL/6J mice were exposed to EtOH (5 g/kg, intragastric [i.g.], 25% EtOH, w/v) or water (isovolumetric control, i.g.) daily for 10 consecutive days as previously described (Qin et al., 2008). Mice were administered the TLR7 agonist imiquimod (R837; InvivoGen, San Diego, CA; 2.5 mg/kg, intraperitoneal [i.p.]) or saline (control) 24 hours after the last dose of EtOH. Liver tissue was collected 2 hours after imiquimod (or saline) injection and was immediately snap‐frozen or formalin‐fixed for histology. All protocols were approved by the Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health regulations for the care and use of laboratory animals.
Cell Culture
Cryopreserved primary human hepatocytes (cPHH) were purchased from Triangle Research Laboratories (Research Triangle Park, NC) and maintained following the supplier's protocols. The cPHH were seeded on collagen‐coated 12‐well plates overlaid with Matrigel (Corning, Oneonta, New York) and allowed to attach overnight before exposure to EtOH (100 mM; 24 hours) or imiquimod (1 μg/ml; 2 hours). For TLR7 silencing studies, 15 pmol of negative scrambled control or TLR7 Silencer small interfering RNA (siRNA) (Ambion, Carlsbad, CA) was mixed with Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA) and added to cPHH for 24 hours. After removing the transfection media, cells were overlaid with Matrigel and allowed to grow for an additional 24 hours prior to stimulation with imiquimod (1 μg/ml). VL‐17A cells were a kind gift from the Zeisel Lab at UNC's Nutrition Research Institute and were maintained as previously described (Donohue et al., 2006). Immediately before use, mirVana let‐7 mimic (Applied Biosystems, Carlsbad, CA) was mixed with DOTAP Liposomal Transfection Reagent (Roche, Mannheim, Germany). The VL‐17A cells were exposed to the liposomal let‐7 mimic and to EtOH (100 mM) for 24 hours. RNA and media were collected at the termination of all cell culture experiments.
Microvesicle Isolation
Microvesicles (MVs) were isolated by sequential centrifugation as described previously (Coleman et al., 2017). The resulting MV‐containing pellet was immediately suspended in Qiazol (Qiagen, Valencia, CA) for total RNA (tRNA) extraction. This preparation results in MVs ranging between 100 nm and 1 mm in diameter.
RNA Isolation and qPCR
tRNA was isolated from mouse liver or cultured cells using TRIzol (Life Technologies, Carlsbad, CA) and phenol:chloroform extraction. One microgram of RNA was reverse transcribed into cDNA using a commercially available kit (Life Technologies). Real‐time qPCR was performed with primer sets based on previously published sequences (Table 1) and the SYBR Green PCR Master Mix (Life Technologies). qPCR products were run on the Bio‐Rad CFX system (Hercules, CA). Gene expression was normalized to 18S. tRNA for microRNA analysis was isolated using Qiazol (Life Technologies) and phenol:chloroform extraction. qPCR analysis of miR‐21 and let‐7b microRNA was performed using miRVana TaqMan assays (Life Technologies) following manufacturer's instructions. microRNA expression was normalized to RNU6B (U6). The comparative 2−ΔΔCT method was used to determine fold changes in mRNA and microRNA expression compared to control.
Table 1.
Primers Used for Real‐Time PCR Analysis
| Gene symbol | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| TLR7 | ATG TGG ACA CGG AAG AGA CAA | GGT AAG GGT AAG ATT GGT GGT G |
| TLR8 | GAA AAC ATG CCC CCT CAG TCA | CGT CAC AAG GAT AGC TTC TGG AA |
| TLR3 | GTG AGA TAC AAC GTA GCT GAC TG | TCC TGC ATC CAA GAT AGC AAG T |
| TLR4 | ATG GCA TGG CTT ACA CCA CC | GAG GCC AAT TTT GTC TCC ACA |
| TLR9 | CTC CAA CCG TAT CCA CCA CC | GAG AAG TGC AGG GGG CTA AG |
| TNFα | GAC CCT CAC ACT CAG ATC ATC TTC T | CCT CCA CTT GGT GGT TTG CT |
| MCP‐1 (CCL2) | ATT GGG ATC ATC TTG CTG GT | CCT GCT GTT CAC AGT TGC C |
| KC (CXCL1) | GCA CCC AAA CCG AAG TCA TAG C | CTT GGG GAC ACC TTT TAG CAT CT |
| MIP‐2 (CXCL2) | AAG TTT GCC TTG ACC CTG AAG | ATC AGG TAC GAT CCA GGC TTC |
| IFNα | AAT GAC CTG CAA GGC TGT CT | CAG GGG CTG TGT TTC TTC TC |
| IFNβ | CAC AGC CCT CTC CAT CAA CT | GCA TCT TCT CCG TCA TCT CC |
| IL‐1β | CTG GTG TGT GAC GTT CCC ATT A | CCG ACA GCA CGA GGC TTT |
| IL‐6 | GGC CTT CCC TAC TTC ACA AG | ATT TCC ACG ATT TCC CAG AG |
| IL‐10 | GCT CTT ACT GAC TGG CAT GAG | CGC AGC TCT AGG AGC ATG TG |
| HMGB1 | CCA TTG GTG ATG TTG CAA AG | CTT TTT CGC TGC ATC AGG TT |
| iNOS | GCT ATG GCC GCT TTG ATG TG | TCG AAC TCC AAT CTC GGT GC |
| gp91 | CAG GAG TTC CAA GAT GCC TG | GAT TGG CCT GAG ATT CAT CC |
| COX‐2 | CGA GGC CAC TGA TAC CTA TTG C | GCT GGC CTG GTA CTC AGT AGG TT |
| F4/80 | CTT TGG CTA TGG GCT TCC AGT C | GCA AGG AGG ACA GAG TTT ATC GTG |
| Cd11b | GAG GCC CCC AGG ACT TTA AC | CTT CTT GGT AGC GGG TTC T |
| CD317 | CAC AGG CAA ACT CCT GCA AC | TCC TGG TTC AGC TTC GTG AC |
| MARCO‐2 | GAA ACA AAG GGG ACA TGG G | TCC ACA CCT GCA ATC CCT G |
| Myd88 | TCA TGT TCT CCA TAC CCT TGG T | AAA CTG CGA GTG GGG TCA G |
| IRF7 | GGT GTG TCC CCA GGA TCA TTT | GCA TAG GGT TCC TCG TAA ACA |
| Neat1 lncRNA | ATT GTA GGA GCC AAC CTG CC | TAC CAG ACC GCT GAC ACA AC |
| HuR | ATG AAG ACC ACA TGG CCG AAG ACT | AGT TCA CAA AGC CAT AGC CCA AGC |
| TIMP‐1 | CCC ACC CAC AGA CAG CCT TC | CGG CCC GTG ATG AGA AAC TC |
| αSMA | TGC CAT CAT GCG TCT GGA CT | GCC GTG GCC ATC TCA TTT TC |
| TGFβ | TGC TAA TGG TGG ACC GCA AC | GGC GTA TCA GTG GGG GTC AG |
| Col1α1 | GTC TTC CTG GCC CCT CTG GT | AGC AGG GCC AGT CTC ACC AC |
| 18S | GGT AAC CCG TTG AAC CCC AT | CAA CGC AAG CTT ATG ACC CG |
Western Blot
Snap‐frozen liver tissue was homogenized in ice‐cold RIPA buffer (Sigma‐Aldrich, St. Louis, MO) supplemented with protease and phosphatase inhibitors (Sigma‐Aldrich). Lysates were centrifuged at 21,000×g to remove particulates. Protein concentrations were determined with a commercially available BCA assay (ThermoFisher, Grand Island, NY). Samples were diluted in DTT‐containing reducing running buffer (ThermoFisher) to a final protein concentration of 40 μg protein per well. Electrophoresis was performed using MiniProtean TGX gel (Bio‐Rad) and transferred onto nitrocellulose membranes using the Bio‐Rad Turbo Transfer system. Membranes were blocked with Odyssey blocking buffer (Li‐COR Biotechnology, Lincoln, NE) for 1 hour prior to overnight incubation at 4°C with primary antibody. Secondary antibody incubation was performed at room temperature for 1 hour in TBST. Membranes were visualized and bands quantified using the Li‐COR Odyssey imaging system™. Values for proteins of interest were normalized to β‐actin expression for each subject and are reported as fold of control.
RNA Sequencing of Human Liver Tissues
Hepatic mRNA expression was determined using a focused analysis of tRNA sequencing data from human liver tissues representing multiple etiologies of liver disease including AH, nonalcoholic steatohepatitis (NASH), and normal liver tissues (Argemi et al., 2018). Biopsies from patients with AH (n = 29) or NASH (n = 9) and fragments of normal liver tissue (n = 10) were obtained from the National Institute on Alcohol Abuse and Alcoholism–funded InTeam consortium biorepository core as described elsewhere (Argemi et al., 2018). All AH patients had a histological diagnosis of AH; inclusion criteria for AH have been described previously (Colmenero et al., 2007; Dominguez et al., 2008, 2009). Patients with NASH were diagnosed according to Kleiner's criteria (Kleiner et al., 2005). A laparoscopic liver biopsy was obtained from these patients during bariatric surgery. Fragments of normal liver tissue were obtained from patients undergoing hepatic resection of liver metastases as previously described (Affo et al., 2013).
For sequencing, tRNA was extracted from healthy, AH, or NASH liver tissues using TRIzol (Affo et al., 2013). Extracted tRNA was analyzed with the Agilent 2100 Bioanalyzer system (Agilent Biotechnologies, Palo Alto, CA). High‐quality tRNA was used for library construction using the Illumina TruSeq Stranded Total RNA Ribo‐Zero Gold kit (Illumina, San Diego, CA). Multiplexed samples were sequenced using Illumina HiSeq2000 platform using a read length of 2 × 100 bases. Short read alignment was performed using STAR alignment algorithm with default parameters (Dobin et al., 2013). Normalization of gene expression level across samples was computed as transcripts per million mapped reads (Wagner et al., 2012).
Validation of select RNAseq data was performed in separate, confirmatory cohorts of AH and control liver tissues using real‐time qPCR. The confirmatory cohort of AH tissues (n = 17) was collected at the University of North Carolina (UNC) at Chapel Hill Hospital as part of the human biorepository of the InTeam Consortium. These biopsies were collected by transjugular approach in patients diagnosed with AH by histological analysis. The confirmatory cohort of control tissues consisted of fragments of normal liver tissue (n = 7) from patients undergoing hepatic resection of liver metastases that were provided by the UNC at Chapel Hill Lineberger Cancer Center's Tissue Procurement Facility.
Clinical data for all patient cohorts are included in Table 2. All human studies conformed to the ethical guidelines of the 1875 Declaration of Helsinki and were approved by the local institutional review committee. Informed consent was obtained in writing from each patient prior to inclusion in the study.
Table 2.
Clinical Data for Human Subjects
| RNAseq | qPCR | ||||
|---|---|---|---|---|---|
| Control (n = 10) | NASH (n = 9) | AH (n = 29) | Control (n = 7) | AH (n = 16) | |
| Demographics | |||||
| Age median (IQR) | 32 (29–49) | 49.5 (43–53) | 46 (42.5–49.5) | 59 (54–68) | 50 (45–60) |
| Gender male n (%) | 7 (70) | 2 (25) | 18 (62) | 6 (54) | 26 (74) |
| Analytic parameters median (IQR) | |||||
| Hemoglobin (g/dl) | 14.6 (12.9–15.5) | 14.3 (12.5–14.9) | 11.6 (10.0–13.2) | 12.3 (11.1–14) | 11.1 (9.67–12.00) |
| Leukocyte count ×109/l | 5.73 (5.22–7.06) | 8.1 (6.9–10.2) | 8.3 (6.7–12.50) | 7.4 (6.05–8.15) | NA |
| Platelet count ×109/l | 237 (210–282) | 262 (221–361) | 124 (83–208)a | 236 (186–350.50) | 124 (79–159)a |
| AST (U/l) | 25 (16.75–31.25) | 30 (25–36.3) | 125 (100–226)a | 30 (22.5–46) | 132 (101.5–202.25)a |
| ALT (U/l) | 21.5 (19.25–25.75) | 40 (31–49.5) | 44 (31–59)a | 27 (25.5–42) | 44 (33–57.25)a |
| Albumin (g/dl) | 4.6 (4.43–4.6) | 4.5 (4.4–4.6) | 2.9 (2.3–3.2) | 3.8 (3.65–4.23) | 2.25 (1.88–2.90) |
| Creatinine (mg/dl) | 0.8 (0.74–0.9) | 1.05 (0.84–1.1) | 0.85 (0.65–1.10) | 0.89 (0.74–0.97) | 0.7 (0.55–1.11) |
| Bilirubin (mg/dl) | 0.6 (0.5–0.7) | 0.6 (0.4–0.9) | 12.0 (2.6–26.7)a | 0.6 (0.5–0.85) | 12.80 (7.31–20.38)a |
| INR | 1.03 (0.99–1.48) | NA | 1.56 (1.35–1.74) | 1.1 (1.05–1.2) | 1.8 (1.6–1.9) |
Human specimens, including liver tissue and serum, were collected in accordance with approved IRB protocols. Biopsies were collected from AH patients using a transjugular approach. Patients with nonalcoholic hepatitis (NASH, n = 9) were also included as diseased controls. Fragments of normal liver tissue (“healthy,” n = 10) were obtained from patients undergoing hepatic resection of liver metastases as previously described (Affo et al., 2013). Clinical chemistries were performed at the time of hospital admission.
IQR, interquartile range; AST, aspartate aminotransferase; ALT, alanine aminotransferase; INR, international normalized ratio; NASH, nonalcoholic steatohepatitis; AH, alcoholic hepatitis.
p < 0.05 compared to control.
Statistics
Results are reported as mean ± SEM. Analysis of variance with Bonferroni's post hoc test (for parametric data) or t‐test was used for the determination of statistical significance among treatment groups, as appropriate. Correlations between variables were evaluated using Pearson's r correlation. A p‐value less than 0.05 was selected before the study as the level of significance.
Results
EtOH Exposure Increases Hepatic TLR7 mRNA Expression and Pro‐Inflammatory Responses in Mice
The purpose of this experiment was to determine whether exposure to EtOH and/or the TLR7 agonist imiquimod altered hepatic mRNA expression of TLR7 and other immune signaling molecules. Previous studies have used multiple binge EtOH exposures to investigate EtOH sensitization to TLR4 and TLR3 responses (Beier et al., 2009; Massey and Arteel, 2012; Qin and Crews, 2012; Qin et al., 2008). For our study, we determined the effect of multiple EtOH binges on hepatic TLR expression. Accordingly, mice were exposed to EtOH (5 g/kg/d, i.g.) or water (control) by oral gavage once per day for 10 days. EtOH was allowed to clear before administration of the TLR7 agonist imiquimod (24 hours after the last dose of EtOH) since alcohol intoxication could confound interpretation of the TLR7 agonist response (Massey and Arteel, 2012). The imiquimod‐induced inflammatory response was assessed 2 hours after imiquimod administration.
The average blood alcohol concentration 1 hour after EtOH treatment was 279 ± 11 mg/dl. Histological analysis of liver tissue by hematoxylin and eosin (H&E) staining revealed no morphological changes in mice exposed to EtOH, imiquimod, or a combination of both at the time point studied (Fig. S1). Hepatic mRNA expression of Toll‐like receptors TLR3, TLR4, TLR7, TLR8, and TLR9 was determined by qPCR (Table 3). Hepatic mRNA expression of TLR7 and TLR4 was significantly increased by EtOH exposure by 256 and 323%, respectively (Fig. 1 A). Western blot analysis indicated that EtOH increased TLR4 protein levels by 126% compared to water controls (p = 0.028) (Fig. S2). EtOH also increased mRNA of pro‐inflammatory cytokines INFα (340%), IFNβ (372%), HMGB1 (179%) as well as the anti‐inflammatory mediator IL‐10 (983%), and the COX‐2 oxygenase (1,350%, Table 3). To determine whether EtOH induction of TLR7 expression increased TLR7 responses, the small molecule TLR7 agonist imiquimod was administered 24 hours after the last dose of EtOH and liver tissue was collected 2 hours later. Imiquimod alone significantly increased mRNA expression of TNFα (298%) and KC (4,405%), a mouse homolog of IL‐8 which is a known driver of AH, as well as mRNA expression of MCP‐1 (445%), IFNα (304%), IFNβ (295%), and MIP‐2 (263%). However, the TLR7 agonist did not increase TLR7 or TLR4 mRNA expression, whereas EtOH increased both. Interestingly, imiquimod did significantly increased hepatic levels of the lncRNA NEAT1 (448%) (Fig. 1 B), which has been implicated as a regulator of a unique subset of inflammatory cytokines, including human IL‐8. EtOH treatment did increase multiple imiquimod TLR7 responses in the liver (Table 3; Fig. 1 B). The combination of EtOH and imiquimod increased TLR9 mRNA (581%) (Fig. 1 A) and significantly enhanced expression of TNFα (701%) and iNOS (1,417%) (Fig. 1 B) as well as MCP‐1 (Table 3). The combination of EtOH and imiquimod also significantly increased MyD88 mRNA by 612% over control, consistent with enhancement of multiple TLR signaling pathways including TLR7. EtOH alone did not alter the mRNA expression of the fibrogenic mediators collagen 1α1, αSMA, TGFβ, or TIMP‐1 at the time point studied. In contrast, imiquimod significantly increased expression of collagen 1α1 by 200% (Table 3) and TIMP‐1, an inducible protease inhibitor associated with pro‐inflammatory responses, by 660% compared to control (Fig. 1 B). EtOH preexposure enhanced imiquimod‐induced TIMP‐1 expression to more than 920% of control. Among the immune signals assessed, EtOH robustly enhanced imiquimod induction of mRNA expression of pro‐inflammatory cytokines (TNFα, MCP‐1) and oxidases (iNOS, COX‐2) as well as the receptor MARCO‐2 and the immune receptor adaptor Myd88 in the liver. These findings suggest that EtOH induction of hepatic TLR7 levels and responses may also impact other immune signaling pathways. In summary, EtOH treatment of mice induces TLR7 mRNA as well as the hepatic response to the TLR7 agonist imiquimod.
Table 3.
Hepatic mRNA Expression of Toll‐Like Receptors, Inflammatory Signaling Molecules, Oxygenases, Cell Receptors, Intracellular Signaling Mediators, and Fibrogenic Markers in Mice Exposed to EtOH, Imiquimod, or the Combination (EtOH+Imiq)
| Gene symbol | Control | EtOH | Imiquimod | EtOH + Imiq. |
|---|---|---|---|---|
| Toll‐like receptors | ||||
| TLR7 | 1.00 ± 0.09 | 2.56 ± 0.20a | 1.26 ± 0.12 | 2.54 ± 0.51a |
| TLR8 | 1.00 ± 0.13 | 1.81 ± 0.28 | 1.41 ± 0.20 | 1.81 ± 0.27 |
| TLR3 | 1.17 ± 0.27 | 0.742 ± 0.22 | 1.24 ± 0.25 | 1.79 ± 0.40 |
| TLR4 | 1.00 ± 0.36 | 3.23 ± 0.33a | 1.46 ± 0.16 | 2.20 ± 0.35a |
| TLR9 | 1.00 ± 0.43 | 2.93 ± 1.28 | 2.96 ± 0.93 | 5.81 ± 1.31a |
| Inflammatory signaling molecules | ||||
| TNFα | 1.00 ± 0.23 | 2.49 ± 0.97 | 2.98 ± 0.72a | 7.01 ± 0.83a , b , c |
| MCP‐1 (CCL2) | 1.00 ± 0.18 | 2.30 ± 1.62 | 4.45 ± 1.01a | 10.04 ± 2.78a , b , c |
| KC (CXCL1) | 1.00 ± 0.18 | 6.72 ± 4.75 | 44.05 ± 9.46a , b | 55.76 ± 7.98a , b |
| MIP‐2 (CXCL2) | 1.00 ± 0.27 | 0.72 ± 0.51 | 2.63 ± 0.40a , b | 2.87 ± 0.61a , b |
| IFNα | 1.00 ± 0.34 | 3.40 ± 0.84a | 3.04 ± 0.47a | 4.88 ± 1.02a |
| IFNβ | 1.00 ± 0.45 | 3.72 ± 0.96a | 2.95 ± 0.42a | 4.78 ± 1.06a |
| IL‐1β | 1.00 ± 0.22 | 1.02 ± 0.40 | 1.71 ± 0.50 | 1.83 ± 0.39 |
| IL‐6 | 1.00 ± 0.39 | 0.70 ± 0.17 | 1.42 ± 0.37 | 1.47 ± 0.18 |
| IL‐10 | 1.00 ± 0.28 | 9.83 ± 3.39a | 5.06 ± 2.47 | 5.37 ± 3.54 |
| HMGB1 | 1.00 ± 0.13 | 1.79 ± 0.36a | 1.11 ± 0.17 | 1.53 ± 0.19a |
| Oxygenases | ||||
| iNOS | 1.00 ± 0.17 | 0.77 ± 0.06 | 3.56 ± 1.03a | 14.17 ± 7.43a , b , c |
| gp91 | 1.00 ± 0.13 | 1.14 ± 0.025 | 1.36 ± 0.48 | 1.19 ± 0.19 |
| COX‐2 | 1.00 ± 0.26 | 13.48 ± 7.07a | 2.189 ± 0.78 | 18.47 ± 5.08a |
| Inflammatory cell receptors | ||||
| F4/80 | 1.00 ± 0.26 | 0.87 ± 0.11 | 0.87 ± 0.21 | 0.94 ± 0.17 |
| Cd11b | 1.00 ± 0.13 | 2.24 ± 0.43a | 1.59 ± 0.23a | 1.88 ± 0.24a |
| CD317 | 1.00 ± 0.22 | 0.86 ± 0.26 | 0.66 ± 0.12 | 0.79 ± 0.30 |
| MARCO‐2 | 1.00 ± 0.27 | 3.14 ± 0.52a | 3.33 ± 1.04a | 10.48 ± 1.96a , b , c |
| Intracellular signaling | ||||
| Myd88 | 1.00 ± 0.08 | 1.96 ± 0.40a | 3.78 ± 0.73a | 6.121 ± 1.7a , b |
| IRF7 | 1.00 ± 0.17 | 1.17 ± 0.32 | 1.29 ± 0.16 | 0.84 ± 0.19 |
| Neat1 lncRNA | 1.00 ± 0.13 | 0.73 ± 0.24 | 4.48 ± 1.72a | 4.74 ± 1.43a |
| HuR | 1.00 ± 0.12 | 2.66 ± 0.58a | 1.19 ± 0.10 | 1.73 ± 0.12a |
| Fibrogenesis | ||||
| TIMP‐1 | 1.00 ± 0.14 | 1.59 ± 0.38 | 6.66 ± 1.58a | 9.20 ± 2.53a , b |
| αSMA | 1.00 ± 0.06 | 0.67 ± 0.20 | 1.08 ± 0.20 | 0.66 ± 0.15 |
| TGFβ | 1.00 ± 0.24 | 1.75 ± 0.45 | 2.10 ± 0.59 | 1.30 ± 0.33 |
| Col1α1 | 1.00 ± 0.15 | 1.25 ± 0.21 | 2.09 ± 0.31a | 2.20 ± 0.35a |
Data are shown as mean ± SE.
p < 0.05 compared to control.
p < 0.05 compared to EtOH alone.
p < 0.05 compared to imiquimod alone.
Figure 1.
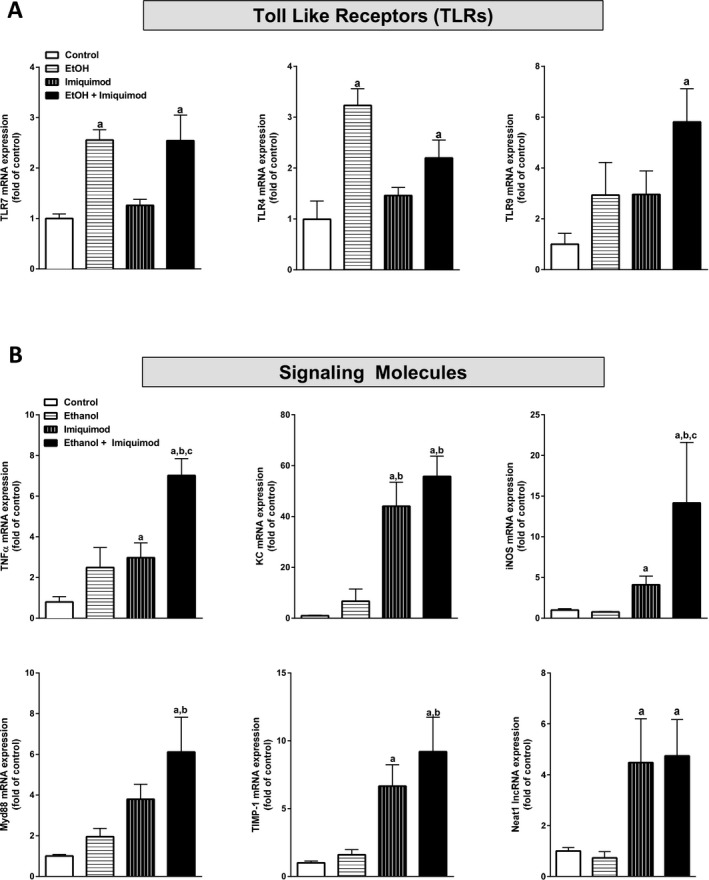
EtOH treatment (10 days) significantly increases hepatic expression of TLR mRNA and increases the inflammatory response induced by the TLR7 agonist imiquimod in mice. Male C57BL/6J mice were exposed to EtOH (5 g/kg, i.g.) for 10 days. Twenty‐four hours after the last EtOH dose, some mice were exposed to the TLR7 agonist imiquimod (2.5 mg/kg, i.p.) as described in Materials and Methods. Hepatic mRNA expression of (A) the Toll‐like receptors 7, 4, and 9 and (B) the signaling molecules TNF α, KC, iNOS, MyD88, TIMP‐1, and NEAT1 lncRNA were determined by qPCR. Data are shown as mean ± SEM (n = 5). a, p < 0.05 compared to control; b, p < 0.05 compared to EtOH alone; c, p < 0.05 compared to imiquimod alone.
EtOH and TLR7 Agonist Imiquimod Responses in Culture cPHH
Hepatocytes are the main site of alcohol metabolism and, accordingly, are particularly susceptible to the effects of alcohol. Additionally, previous studies have shown that hepatocytes express TLR7 and are sensitive to TLR7 activation (Lee et al., 2006; Sarkar et al., 2015) and, furthermore, that hepatocytes can contribute to hepatic innate immune response through multiple signaling pathways (Crispe, 2016). Therefore, we investigated the effect of EtOH or imiquimod on the mRNA expression of TLR7, TLR signaling molecules, and innate immune genes in cPHH. EtOH exposure (100 mM, 24 hours) significantly increased TLR7 mRNA expression by nearly 400% in the hepatocytes (Fig. 2 A). Furthermore, EtOH exposure increased the mRNA expression of the TLR signaling pathway including the TLR adaptor protein MyD88 (450%) and the NFκB subunit NFκB‐p65 (550%). EtOH also significantly increased mRNA expression of the inflammatory mediators IL‐8 (300%) and TNFα (270%). In contrast to EtOH exposure, the TLR7 agonist imiquimod did not affect the expression of TLR7 mRNA at the concentration (1 μg/ml) and time point (2 hours) studied (Fig. 2 B). However, imiquimod stimulation did robustly increase mRNA expression of MyD88 (800%), NFκB‐p65 (1,200%), IL‐8 (1,250%), and TNFα (3,800%) and also increased levels of the lncRNA NEAT1 (170%). To confirm that imiquimod was stimulating the hepatocyte inflammatory response through the TLR7 receptor, cultured hepatocytes were transfected with control siRNA or TLR7 siRNA 24 hours before exposure to imiquimod. Transfection with TLR7 siRNA decreased TLR7 mRNA expression by 60% ± 12% (Fig. 2 C) and blunted imiquimod‐induced TNFα mRNA expression by 52% ± 15% (Fig. 2 D). Thus, EtOH increased hepatocyte TLR7 mRNA expression and imiquimod induced pro‐inflammatory cytokine (e.g., TNFα) expression via activation of TLR7 receptor in primary human hepatocytes.
Figure 2.
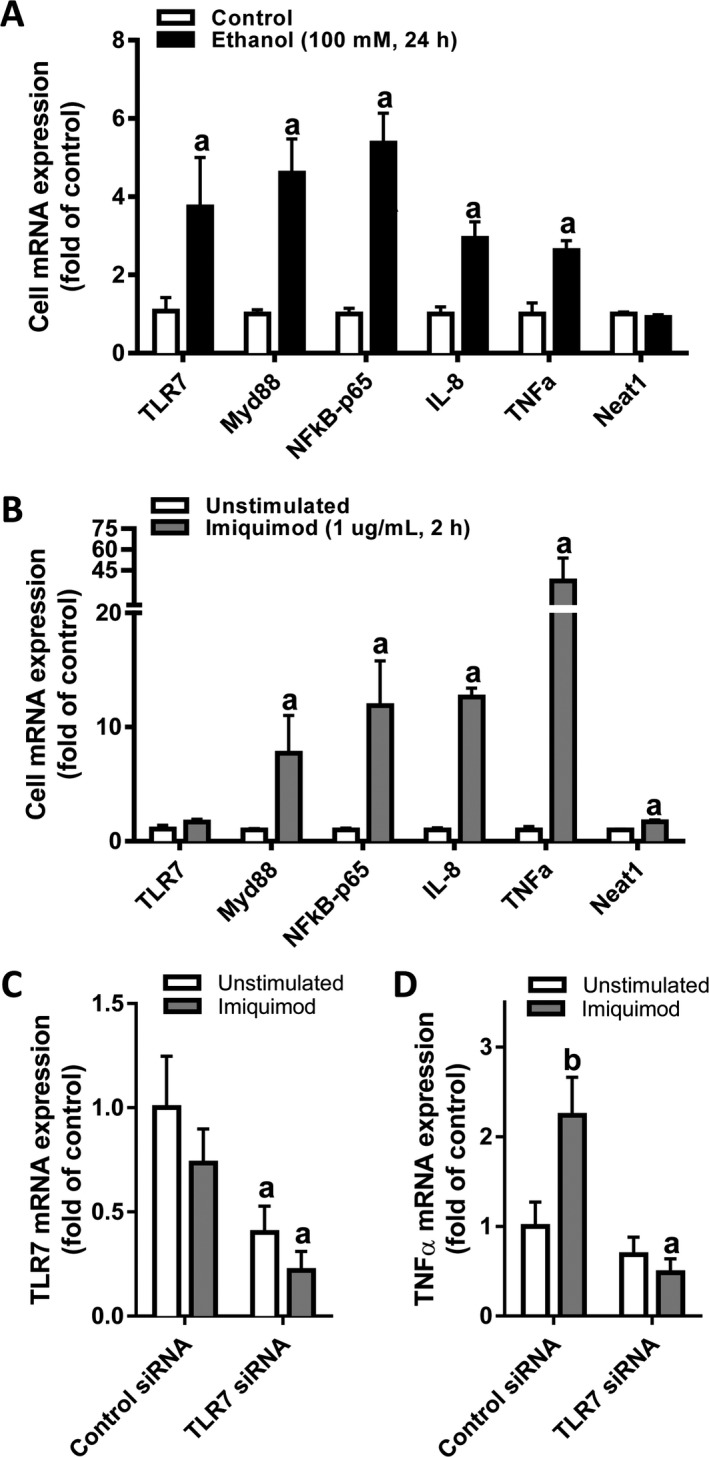
EtOH or imiquimod exposure increases mRNA expression of pro‐inflammatory mediators in cultured primary human hepatocytes. Primary human hepatocytes were exposed to (A) EtOH (100 mM, 24 hours) or (B) imiquimod (1 μg/ml, 2 hours). For siRNA studies (C,D), primary human hepatocytes were exposed to control or TLR7‐targeted siRNA as described in Materials and Methods. Cellular mRNA was collected at the termination of the experiments. Intracellular mRNA expression of TLR7, MyD88, NF κB, IL‐8, TNF α, and NEAT1 was determined by qPCR. Data are shown as mean ± SEM (n = 4). a, p < 0.05 compared to control siRNA; b, p < 0.05 compared to unstimulated control.
EtOH and Imiquimod Increase MV Levels of the TLR7 Agonist Let‐7b in Cultured Primary Human Hepatocytes
EtOH exposure increases vesicle release from cultured hepatocytes (Momen‐Heravi et al., 2015), and other studies find that EtOH increases let‐7 microRNAs released in MVs (Coleman et al., 2017). Therefore, we measured the level of total MV‐associated RNA and of MV‐associated TLR7‐activating microRNAs let‐7b and miR‐21 in the conditioned media of primary human hepatocytes exposed to EtOH or the TLR7 agonist imiquimod. As expected, EtOH significantly increased the release of MV‐associated RNA in the hepatocyte media by 175% and let‐7b mRNA by 200% (Fig. 3 A), but did not affect the release of MV‐associated miR‐21. Imiquimod increased the release of MV‐containing let‐7b mRNA about 200%, similar to EtOH exposure (Fig. 3 B). Thus, both EtOH and imiquimod increased human hepatocyte release of let‐7b in MVs.
Figure 3.
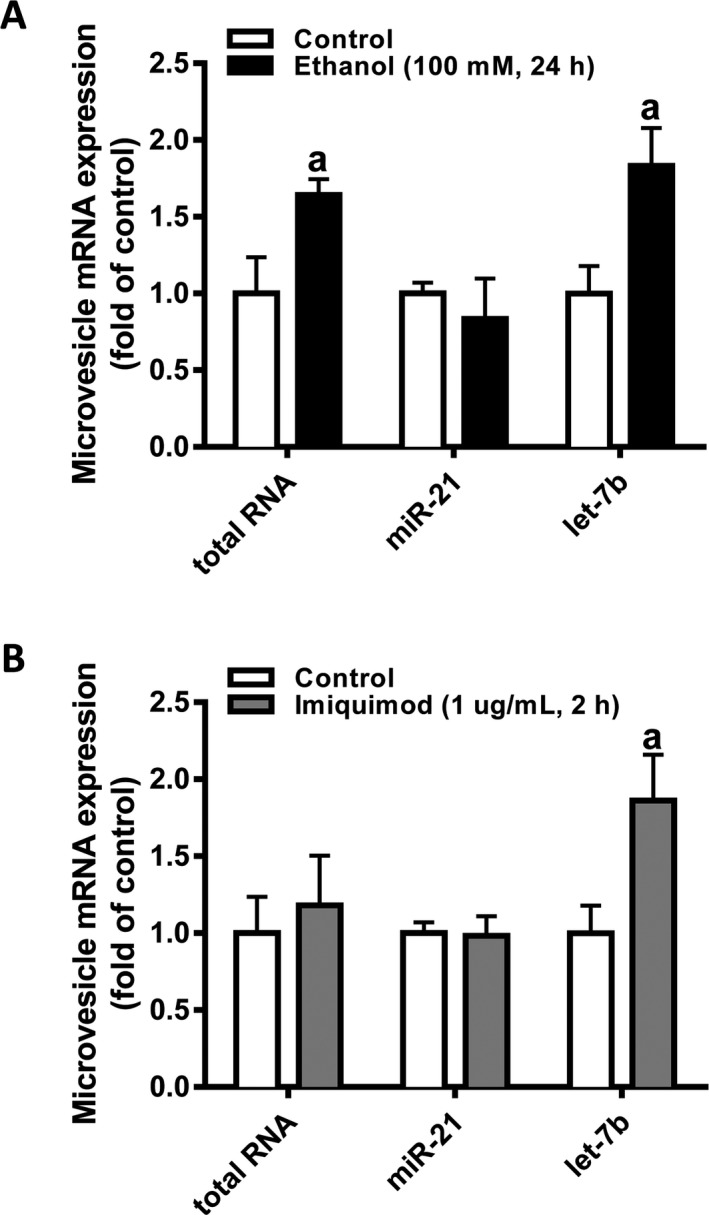
EtOH or imiquimod exposure increases the level of let‐7b in microvesicles released from primary human hepatocytes. tRNA was isolated from hepatocyte‐derived MVs as described in Materials and Methods. 5 ng of MV‐associated tRNA was used for qPCR for the TLR7‐binding microRNAs miR‐21 and let‐7b in MVs released from hepatocytes exposed to EtOH (A) or imiquimod (B). Data are shown as mean ± SEM (n = 4). a, p < 0.05 compared to control.
Hepatic TLR7 Expression Is Increased in AH
In the absence of an adequate animal model of AH, we studied the association of TLR7 with human AH using RNA sequencing data of human liver tissue from patients with liver disease of different etiologies including NASH and AH. TLR7 mRNA expression was increased by more than 300% in patients with AH, but was not changed in liver of patients with NASH (Fig. 4 A). Importantly, TLR7 mRNA expression was increased by nearly 30‐fold in AH liver tissue compared to control liver tissue when confirmed by qPCR in a separate cohort of patients (Fig. 4 B). TLR7 mRNA expression correlated with serum aspartate aminotransferase (AST) levels (r = 0.5994, p = 0.001) and with hepatic IL‐8 mRNA levels (r = 0.6384, p = 0.0002) in AH patients (Fig. 4 C,D). In agreement with these results, TLR7 mRNA expression also correlated with serum AST levels (r = 0.7699, p < 0.001) and IL‐8 mRNA (r = 0.734, p = 0.0004) in a confirmatory cohort of AH patients (Table 4). Thus, TLR7 mRNA is increased in AH, but not NASH, and correlates with serum AST, a marker of liver damage, and increased hepatic mRNA expression of IL‐8, a known driver of AH (Hill et al., 1993; Liu et al., 2015).
Figure 4.
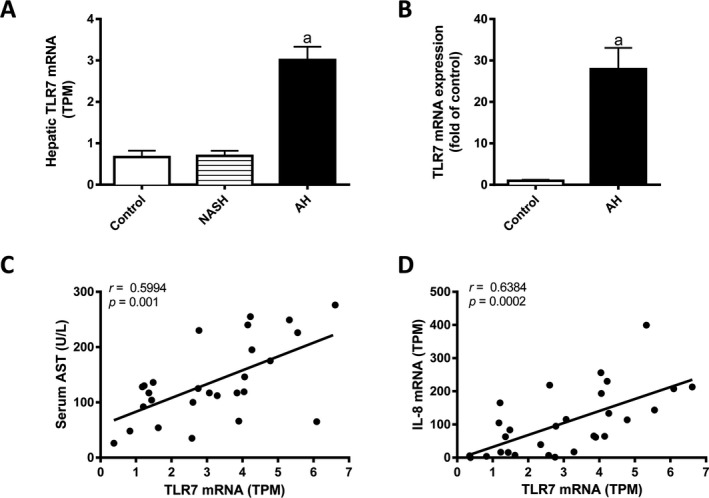
Hepatic TLR7 mRNA expression is increased in patients with AH and correlates with liver injury and IL‐8 mRNA levels in humans. (A) RNAseq was performed on liver tissue from alcoholic hepatitis (AH; n = 29), nonalcoholic steatohepatitis (NASH; n = 9), or normal fragments of tissue (control, n = 10). TLR7 mRNA levels are shown as transcripts per kilobase million (TPM). (B) TLR7 mRNA expression was validated in a separate cohort of control (n = 7) and AH (n = 17) liver tissues by qPCR. Correlations between hepatic TLR7 and (C) serum AST or (D) hepatic IL‐8 were determined by Pearson's coefficient. mRNA expression data are shown as mean ± SEM. a, p < 0.05 compared to control.
Table 4.
Summary of Clinical Correlations
| R‐value | p‐Value | |
|---|---|---|
| Cohort 1 (RNAseq) | ||
| TLR7 versus AST | 0.5994 | 0.0010 |
| TLR7 versus IL‐8 | 0.6384 | 0.0002 |
| TR7 versus let‐7b | 0.3351 | 0.0433 |
| TLR7 versus let‐7d | 0.2240 | NS |
| TLR7 versus let‐7a1 | 0.3775 | 0.0213 |
| TLR7 versus let‐7f1 | 0.4957 | 0.0025 |
| TLR7 versus let‐7f2 | 0.2608 | NS |
| TLR7 versus let‐7g | 0.2329 | NS |
| TLR7 versus NEAT1 | 0.2874 | NS |
| Let‐7b versus IL‐8 | 0.5181 | 0.0010 |
| Let‐7d versus IL‐8 | 0.5255 | 0.0008 |
| Let‐7a1 versus IL‐8 | 0.5534 | 0.0004 |
| Let‐7f1 versus IL‐8 | 0.5798 | 0.0003 |
| Let‐7f2 versus IL‐8 | 0.0781 | NS |
| Let‐7g versus IL‐8 | 0.2013 | NS |
| NEAT1 versus IL‐8 | 0.3470 | 0.0328 |
| et‐7b versus AST | 0.3521 | 0.0481 |
| Let‐7d versus AST | 0.1792 | NS |
| Let‐7a1 versus AST | 0.2361 | NS |
| Let‐7f1 versus AST | 0.5654 | 0.0009 |
| Let‐7f2 versus AST | 0.0264 | NS |
| Let‐7g versus AST | 0.2124 | NS |
| Let‐7b versus NEAT1 | 0.7010 | <0.0001 |
| Let‐7d versus NEAT1 | 0.6923 | <0.0001 |
| Let‐7a1 versus NEAT1 | 0.6151 | <0.0001 |
| Let‐7f1 versus NEAT1 | 0.5285 | 0.0013 |
| Let‐7f2 versus NEAT1 | 0.5905 | 0.0001 |
| Let‐7g versus NEAT1 | 0.6883 | <0.0001 |
| Cohort 2 (qPCR) | ||
| TLR7 versus AST | 0.7699 | <0.0001 |
| TLR7 versus IL‐8 | 0.7340 | 0.0004 |
| TLR7 versus let‐7b | 0.8992 | <0.0001 |
| Let‐7b versus AST | 0.5590 | 0.0045 |
| Let‐7b versus IL‐8 | 0.4875 | 0.0292 |
Correlations were performed using Pearson's coefficient as described in Materials in Methods.
NS, nonsignificant; AST, aspartate aminotransferase.
Expression of Let‐7 microRNAs, the Endogenous Ligands of TLR7, Is Increased in AH
TLR7 responds to ssRNA, viruses, and endogenous microRNAs as well as small molecule drugs, such as imiquimod. Because the abundant let‐7 microRNAs have been identified as endogenous TLR7 agonists (Coleman et al., 2017), the relative levels of let‐7 microRNAs in hepatic tissue of patients with AH and NASH were compared to levels in fragments of normal liver tissue. RNA sequencing revealed that 6 let‐7 microRNAs were increased in patients with AH including let‐7b (246%), let‐7a1 (500%), let‐7d (220%), let‐7f1 (400%), let‐7f2 (180%), and let‐7g (250%) (Fig. 5). Increased expression of let‐7b was confirmed by qPCR in a separate cohort of AH patients (Fig. 5 I). As expected, IL‐8 expression was significantly increased in the AH patient cohort compared to controls (Fig. 6 A). Furthermore, expression of IL‐8 correlated with liver expression of let‐7b (r = 0.5181, p = 0.001), let‐7d (r = 0.5255, p = 0.0008), let‐7a1 (r = 0.5534, p = 0.0004) (Fig. 6 B–D), and let‐7f1 (r = 0.5798, p = 0.003; Table 4). NEAT1 is a lncRNA that has recently been shown to regulate IL‐8 expression and additionally has been shown to be activated downstream of TLR stimulation (West et al., 2014). Interestingly, NEAT1 lncRNA expression was also increased in the liver tissue of AH patients (Fig. 6 E) and correlated with hepatic IL‐8 mRNA (r = 0.3470, p = 0.0328; Table 4), and let‐7 miRNA expression (Fig. 6 F–H). Serum AST levels were also positively correlated with hepatic expression of let‐7b (r = 0.3521, p = 0.0481) and let‐7f1 (r = 0.5654, p = 0.0009) (Table 4). The positive correlation of hepatic IL‐8 mRNA (r = 0.4875, p = 0.0292) and serum AST levels (r = 0.559, p = 0.0045) with hepatic let‐7b expression was confirmed in the confirmatory cohort of AH patients (Table 4).
Figure 5.
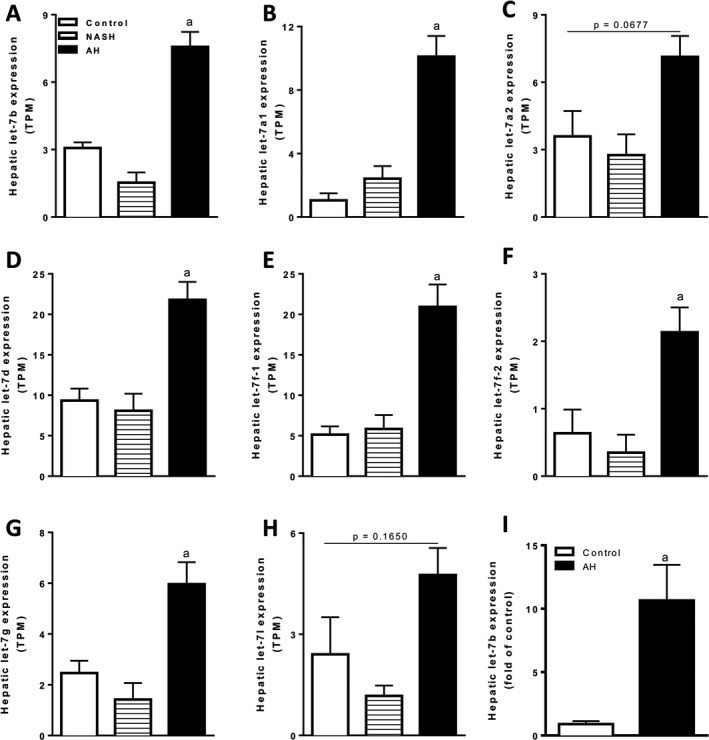
Let‐7b microRNA expression is significantly increased in the liver of patients with AH. (A–H) Let‐7b microRNA expression was determined by RNAseq in liver tissue from alcoholic hepatitis (AH; n = 29), nonalcoholic steatohepatitis (NASH; n = 9), or fragments of normal tissue (control, n = 10). Sequencing data are shown as transcripts per kilobase million (TPM). (I) Let‐7b microRNA levels were validated by qPCR in a confirmatory cohort of control (n = 7) and AH (n = 17) patients. qPCR data are shown as fold of control. a, p < 0.05 compared to control.
Figure 6.
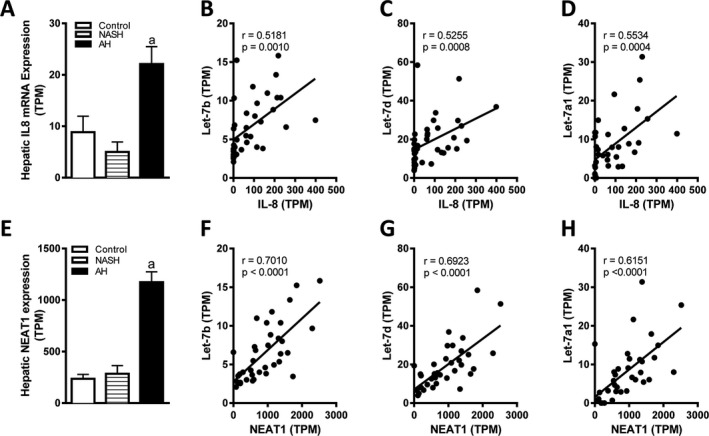
Hepatic expression of let‐7b, let‐7d, and let‐7a1 correlates with hepatic IL‐8 and Neat1 expression in patients with AH. (A) Hepatic expression of IL‐8 was determined by RNAseq in liver tissue from AH (n = 29), nonalcoholic steatohepatitis (NASH; n = 9), or fragments of normal liver tissue (control, n = 10). Correlations between hepatic IL‐8 mRNA expression and (B) let‐7b, (C) let‐7d, and (D) let‐7a1 expression were determined by Pearson's coefficient. (E) NEAT1 lncRNA expression was analyzed in the RNA sequencing data of AH, NASH, and control liver tissue. Correlations between hepatic NEAT1 expression and (F) let‐7b, (G) let‐7d, and (H) let‐7a1 were determined by Pearson's coefficient. mRNA expression data are shown as transcripts per kilobase million (TPM). a, p < 0.05 compared to control.
Effect of a Let‐7b Mimic and EtOH Induction of TLR7 in VL‐17A Cells
We investigated let‐7 agonist actions on TLR7 and immune responses using a let‐7b mimic prepared in liposomes and VL‐17A human HCC‐derived cells that express EtOH‐metabolizing enzymes. Consistent with results in cultured primary human hepatocytes, EtOH increased mRNA expression of TLR7 (200%) and TNFα (160%) in the VL‐17A hepatocytes (Fig. 7). Addition of 5 nmol of liposomal let‐7b mimic did not affect TLR7 or TNFα mRNA expression in controls. Similar to imiquimod, let‐7b mimic alone increased TNFα mRNA expression by 150% compared to scrambled control and EtOH exposure further increased TNFα mRNA expression caused by the let‐7b mimic to more than 250%. Therefore, a let‐7b mimic, when encapsulated in liposomal vesicles, increases TNFα pro‐inflammatory cytokine expression that is further increased with EtOH preexposure. Taken together with our findings in human liver tissue, these data suggest that EtOH induction of let‐7 microRNAs, the let‐7‐responsive TLR7 receptor, and subsequent acute phase pro‐inflammatory responses may be a key component in the transition to acute AH.
Figure 7.
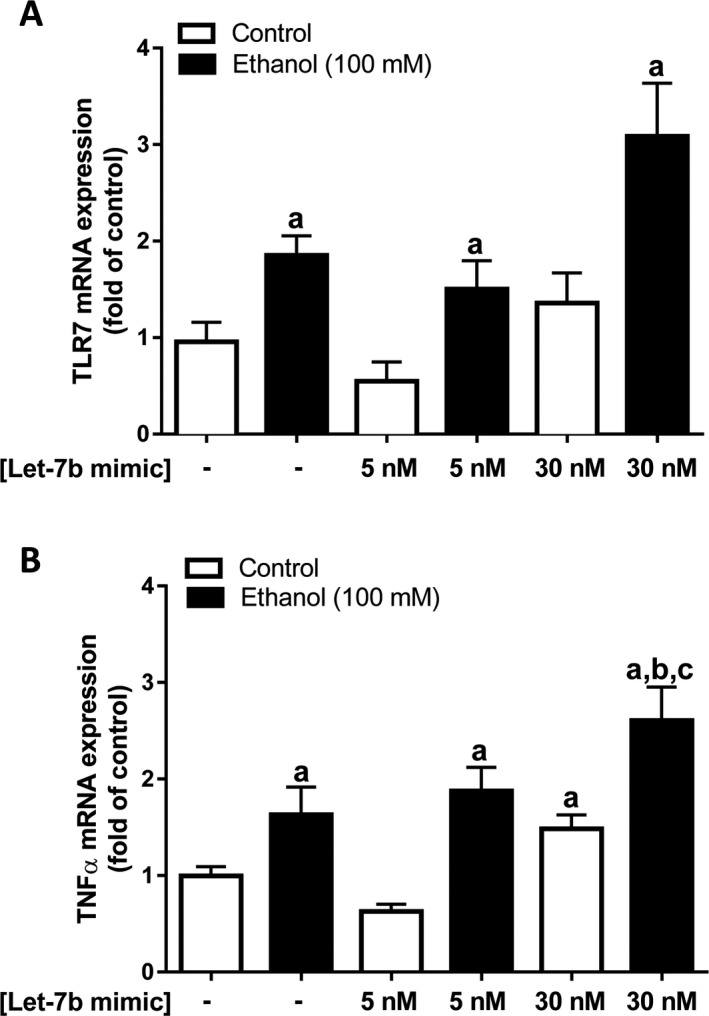
EtOH exposure increases TLR7 mRNA and let‐7‐induced TNF α and mRNA expression in cultured VL‐17A cells, a recombinant human HCC cell line which express alcohol‐metabolizing enzymes. VL‐17A cells were exposed to EtOH (100 mM) for 24 hours followed by exposure to liposomal let‐7 mimic (5 nM or 30 nM), a TLR7 agonist, for 24 hours. TLR7 (A) and TNF α (B) mRNA expressions were determined by qPCR. Data are shown as mean ± SEM (n = 3 to 4). a, p < 0.05 compared to NT; b, p < 0.5 compared to 5 nM let‐7 mimic; c, significant effect of EtOH.
Discussion
We report here that EtOH exposure increases TLR7 mRNA expression in mouse liver tissue, in cultured primary human hepatocytes, and in a human hepatocyte cell line. We find that EtOH and TLR7 agonists increase expression of overlapping, but unique, immune signaling molecules and that the combination of EtOH exposure followed by TLR7 activation enhances pro‐inflammatory mediator expression in mice. Additionally, TLR7 activation by imiquimod in human hepatocytes induces IL‐8, a unique human cytokine known to contribute to AH (Hill et al., 1993; Liu et al., 2015). Furthermore, we demonstrate that both EtOH and imiquimod increase expression of MyD88 and NFκB‐p65, common elements in TLR signaling, and additionally induce the release of an endogenous TLR7 ligand, let‐7b, in hepatocyte‐derived MVs. For the first time, we report that a let‐7 mimic induces TNFα in a human hepatocyte cell line. Taken together, these findings suggest that EtOH can increase TLR7 signaling through both induction of the TLR7 receptor and TLR7 signaling components (e.g., MyD88, NFκB) and through increased release of the endogenous TLR7 ligand, let‐7. For the first time to our knowledge, we also report that both TLR7 expression and let‐7b expression are increased in patients with AH and positively correlate with expression of IL‐8, a known driver of AH; NEAT1 lncRNA, a positive regulator of IL‐8 expression (West et al., 2014); and with serum AST levels, an index of liver injury. These results from patients with liver disease are consistent with our in vivo and in vitro results as well as our hypothesis that increased let‐7 and TLR7 signaling may be a key component contributing to the transition to acute AH from less severe stages of ALD.
Our study finds that EtOH exposure induces liver TLR4 expression in mice, which is in agreement with previous studies supporting the role of TLR4 in ALD. LPS, a gut‐derived endotoxin known to be increased in blood by EtOH consumption (Bode et al., 1987), increases liver inflammation and steatohepatitis (Gao and Tsukamoto, 2016) via TLR4 and may contribute to increased alcohol drinking (Blednov et al., 2011). Although TLR4 is closely linked to steatohepatitis, the factors that trigger progression from earlier stages of ALD to severe forms of ALD (e.g., HCC and AH) are poorly understood. For this study, we were particularly interested in studying the role of TLR7 in AH. TLR7 is an endosomal TLR which is distinct from TLR4. Previous studies have reported altered hepatocyte TLR7 expression in patients with liver disease (Firdaus et al., 2014; Leake, 2014; Lee et al., 2006; Lin et al., 2013), including end‐stage alcoholic cirrhosis (Starkel et al., 2010), and have found TLR7 responses in hepatocyte cell lines (e.g., HepG2, Huh‐7) (Lee et al., 2006; Sarkar et al., 2015). Interestingly, unsaturated fatty acids reduce TLR7 expression, increase insulin‐like growth factor 1 (IGF1), and increase oxidative stress in mouse hepatocytes and genetic ablation of TLR7 led to fatty liver, reactive free radicals, and IGF1 up‐regulation in TLR7 knockout mice, consistent with the involvement of TLR7 in hepatic fat accumulation (Kim et al., 2016). TLR7 responds to ssRNA, including viral ssRNA such as hepatitis C virus (HCV) (Negash et al., 2013), linking TLR7 to chronic viral hepatitis‐associated cirrhosis HCC. Indeed, hepatocytes may be uniquely involved in hepatic TLR7 signaling due to their susceptibility to HCV infection, a ssRNA virus that activates hepatocyte TLR7 and induces inflammatory responses (e.g., TNFα induction) in hepatocytes. In mouse hepatocytes, TLR7 can induce autophagy responses through IGF1 that are Myd88 dependent and reduced by fat accumulation (Kim et al., 2016). In this study, EtOH induced TLR7 mRNA expression in both mice and in cultured hepatocytes, whereas imiquimod, the TLR7 agonist, did not increase the receptor's mRNA expression at the time points studied. Also, we report for the first time that IL‐8 is induced by both EtOH and TLR7 agonists in cultured primary human hepatocytes. TLR7‐mediated induction of innate immune genes in hepatocytes is consistent with previous studies linking hepatocytes to the hepatic immune response (Crispe, 2016). Increases in MyD88 and pNFκB induced by either EtOH or imiquimod expression could support the enhancement of multiple TLR signaling pathways which may amplify autocrine and paracrine induction of innate immune genes. These findings suggest that induction of hepatocyte TLR7 signaling may play an important role in innate immune induction in the context of alcohol exposure.
In a mouse model of alcohol exposure followed by TLR7 stimulation, we find that EtOH induces expression of TLR7 and that the combination of EtOH pretreatment followed by TLR7 activation in mice resulted in a unique pro‐inflammatory profile which consisted of simultaneous induction of both TLR7‐responsive genes (e.g., KC, MIP‐2, Neat1) and EtOH‐responsive genes (e.g., TLR4, TLR7, COX2, HMGB1) as well as increased induction of TNFα, MCP‐1, iNOS, MARCO‐2, and TIMP‐1 compared to EtOH or imiquimod exposure alone. Importantly, combined exposure also blunted EtOH induction of IL‐10, an immunoregulatory cytokine important for maintaining inflammatory tone that is decreased in AH. Increased induction of a distinct subset of innate immune genes and concomitant loss of IL‐10 induction in the co‐exposed mice is consistent with EtOH induction of TLR7 contributing to a hyperacute phase response like that which is associated with AH (Fig. 8).
Figure 8.
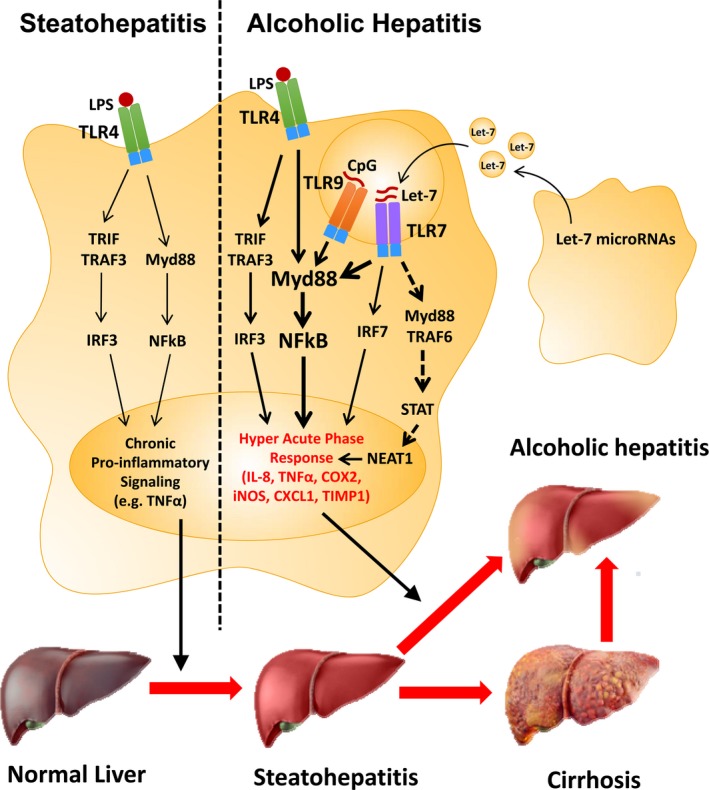
Activation of the TLR7 receptor by let‐7 microRNAs as a novel mechanism contributing to the onset of alcoholic hepatitis (AH). Increased systemic lipopolysaccharide (LPS) caused by EtOH has been shown to activate hepatic TLR4, which contributes to pro‐inflammatory signaling (e.g., TNF α) during alcoholic liver injury. However, the mechanisms that contribute to progression to acute AH remain unclear. In mouse hepatocytes, fat accumulation was associated with decreased TLR7 expression (Kim et al., 2016); however, in our human liver biopsy studies, NASH did not show decreased TLR7 mRNA expression. Our research indicates that both the TLR7 receptor and let‐7 microRNAs, endogenous ligands of TLR7, are increased in patients with AH. Therefore, we propose that increased hepatic TLR7 signaling via increased expression of the TLR7 receptor, the endogenous ligand (let‐7b), and TLR7 signaling components (e.g., MyD88, NF κB‐p65) may contribute to a hyperacute innate immune response which triggers progression to acute AH. Activation of the lncRNA NEAT1 via TLR7 may also contribute to activation of a unique subset of inflammatory genes, including IL‐8.
Although our findings in a mouse of alcohol exposure support enhancement of TLR7 pro‐inflammatory signaling by EtOH, the mouse model used is not a model of AH. AH is difficult to model in rodents in vivo and even the most injurious models of alcohol‐induced liver damage do not produce the clinical sequelae of AH. Therefore, we also analyzed hepatic TLR7 mRNA expression in patients with chronic liver disease, including AH. Our human studies find that TLR7 mRNA expression is uniquely increased in patients with AH, but not in NASH. AH has previously been linked to increased expression of both IL‐8 and its receptor CXCR2 (Liu et al., 2015). IL‐8 is a human cytokine with no direct homolog in rodents. Here, we report that hepatic TLR7 expression positively correlates with hepatic IL‐8 mRNA levels as well as with serum AST levels. Our discovery that AH has increased TLR7 expression compared to controls or NASH patients suggests that sensitization to TLR7 may contribute to severe ALD including AH. The correlations of TLR7 mRNA with serum AST and IL‐8 mRNA in both AH cohorts link TLR7 induction levels to markers of AH pathology.
Emerging evidence indicates that endogenous microRNAs including siRNAs and microRNAs can contribute to immune modulation via TLR7 signaling. Initial studies suggested that GU‐rich microRNAs are TLR7 agonists (Heil et al., 2004; Hornung et al., 2005) and more recent findings indicate that the endogenous let‐7 microRNA family can contribute to pathogenesis via TLR7 activation (Lehmann et al., 2012). The let‐7 family of microRNAs consists of 13 closely related genes that contain variations of the GU‐rich motif proposed to stimulate TLR7 (Roush and Slack, 2008). Let‐7b has specifically been implicated in TLR7 activation and immune signaling. For example, activation of neuronal TLR7 by let‐7b has been shown to contribute to neurodegeneration (Lehmann et al., 2012) and pain (Winkler et al., 2014). Additionally, Coleman and colleagues (2017) recently found that MV let‐7 released from microglia contributes to TLR7‐mediated neurotoxicity. Importantly, Coleman and colleagues (2017) also reported that EtOH induced the release of let‐7b from cultured microglia and that both TLR7 and let‐7b were increased in alcoholic brain. All of these findings suggest that TLR7‐let‐7 signaling can mediate innate immune activation in the absence of viral infection and, furthermore, that its activation is up‐regulated by EtOH exposure. Here, we report that hepatic let‐7 microRNA expression is increased in patients with AH. Indeed, 6 of the 8 let‐7 microRNAs detected by RNA sequencing are uniquely increased in the liver of patients with AH. Furthermore, hepatic expression of let‐7b, let‐7d, let‐7a1, and let‐7f1 levels positively correlates with hepatic IL‐8 mRNA and with the lncRNA NEAT1, a positive regulator of IL‐8 transcription. These are the first studies to our knowledge to implicate increased TLR7‐let‐7 signaling as a mechanism of innate immune activation in the context of AH. Increased expression of both the TLR7 receptor and endogenous TLR7 ligands (e.g., let‐7) in AH and correlation with expression of IL‐8, a known driver of AH, is consistent with TLR7 signaling being a “second hit” that contributes to progression to AH (Fig. 8).
Compartmentalization of TLR7 to the endosome allows for recognition of viral ssRNAs and microRNAs that are endocytosed while preventing constitutive activation by cytosolic ssRNAs. Accordingly, ssRNAs, which are encapsulated in liposomes or MVs, have greater TLR7‐stimulating effects than free ssRNA (Jackson and Linsley, 2010). Additionally, previous studies have shown that EtOH alters vesicle release from hepatocyte cell lines (Momen‐Heravi et al., 2015). Therefore, we determined the effect of EtOH on the release of MV‐associated let‐7b from cultured hepatocytes. We found that EtOH exposure increased both let‐7b and tRNA levels in MVs released from cultured primary human hepatocytes. To test the effects of encapsulated let‐7b on hepatocyte innate immune induction, we exposed VL‐17A cells to a liposome‐encapsulated let‐7b mimic (or negative control) in the presence or absence of EtOH. We found that that liposomal let‐7b mimic induces TNFα mRNA expression in VL‐17A cells (Fig. 7). Together, our finding that EtOH induces the release of MV let‐7b and that liposomal let‐7b can activate hepatocyte innate immune responses is consistent with EtOH increasing both TLR7 signaling via induction of both the TLR7 receptor and its endogenous ligand, let‐7.
In summary, we report that TLR7 signaling is activated by EtOH exposure through up‐regulation of both the TLR7 receptor and of its endogenous ligand let‐7b. Indeed, we found that increased TLR7 signaling, including induction of the receptor and endogenous microRNA ligands (e.g., let‐7b), is associated with AH and correlates with liver IL‐8 levels and serum AST. In an in vivo model of alcohol exposure, we demonstrate that EtOH induction of hepatic TLR7 expression sensitizes the liver to innate immune activation caused by the TLR7 agonist, imiquimod, including increased expression of TNFα, MCP‐1, iNOS, MyD88, and MARCO‐2. In vitro, we demonstrate that EtOH increases hepatocyte TLR7 expression and that imiquimod induces innate immune gene expression (e.g., TNFα, IL‐8) through the TLR7 receptor. Additionally, we demonstrate that both EtOH and imiquimod stimulate the release of MV let‐7b from hepatocytes and that a let‐7b mimic induces TNFα in a hepatocyte cell line. Taken together, our findings suggest that increased TLR7 signaling caused by EtOH is multifactorial and includes up‐regulation of the TLR7 receptor, induction of TLR7 signaling components (e.g., MyD88, NFκB), and a concomitant increase in the expression and release of endogenous microRNA ligands of TLR7 (e.g., let‐7b). The highly translational approach of this study, including in vitro studies, a mouse model of alcohol exposure, and analysis of human liver diseases, was fundamental to connecting TLR7 activation to IL‐8 induction since mice do not express IL‐8. Indeed, our data suggest that IL‐8 induction via TLR7 may mediate the hyperacute innate immune response critical for the onset of AH. Since TLR7 antagonists are currently in the drug development pipeline, future studies should determine whether disruption of TLR7 activation could prevent systemic inflammatory responses in vivo. Our findings are consistent with EtOH induction of both the TLR7 receptor and endogenous ligand let‐7b in contributing to the induction of acute phase innate immune genes, including IL‐8, which contribute to the onset of AH.
Funding
This study has been supported by the National Institute on Alcohol Abuse and Alcoholism (AA011605, AA007573, AA018051, AA020023, AA020024, AA019767, 1U01AA021908‐01) and the UNC Bowles Center for Alcohol Studies.
Conflict of interest
Authors have nothing to disclose.
Author Contributions
Expeirmental design: VLM, LQ, FTC. Data collection: VLM, LQ, Ju.C, PS‐B. Data analysis and interpretation: VLM, Jo.C, FTC, RB. Writing and revision of the manuscript: VLM, RB, FTC.
Supporting information
Fig. S1 Effect of EtOH and imiquimodexposure on liver morphology in mice. Male C57BL/6J mice were exposed to EtOH (5 g/kg, i.g.) for 10 days. Twenty‐four hours after the last EtOH dose, some mice were exposed to the TLR7 agonist imiquimod (2.5 mg/kg, i.p.) as described in Materials and Methods. Liver tissue was collected 2 hours after imiquimod (or saline) administration. Formalin‐fixed, paraffin embedded sections were cut at 5 μm and mounted on glass slides. Sections were deparaffinizedand stained with H&E. Representative photomicrographs (original magnification, 200×) are shown.
Fig. S2. Effect of EtOH and imiquimodexposure on liver TLR4 protein expression in mice. Male C57BL/6J mice were exposed to EtOH (5 g/kg, i.g.) for 10 days. Twenty‐four hours after the last EtOH dose, some mice were exposed to the TLR7 agonist imiquimod (2.5 mg/kg, i.p.) as described in Materials and Methods. Liver tissue was collected 2 hours after imiquimod (or saline) administration. Western blot was performed as described in Materials and Methods. Quantitative data are shown as fold of control. *p < 0.05 compared to control.
References
- Affo S, Dominguez M, Lozano JJ, Sancho‐Bru P, Rodrigo‐Torres D, Morales‐Ibanez O, Moreno M, Millan C, Loaeza‐Del‐castillo A, Altamirano J, Garcia‐Pagan JC, Arroyo V, Gines P, Caballeria J, Schwabe RF, Bataller R (2013) Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut 62:452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argemi JLMU, Atkinson SR, Blokhin I, Massey VL, Gue JP, Cabezas J, Lozano JJ, van Booven D, Cao S, Vernetti LA, Arab JP, Ventura‐Cots M, Fondevila C, Starkel P, Dubuquoy L, Louvet A, Odena G, Altamirano J, Caballería J, Taylor DL, Berasain C, Wahlestedt C, Mong SP, Sancho‐Bru P, Mathurin P, Lackner K, Rusyn I, Shah V, Thursz MR, Mann J, Avila MA, Bataller R (2018) Defective HNF4a‐dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Hepatology 66:93A. [Google Scholar]
- Beier JI, Luyendyk JP, Guo L, von Montfort C, Staunton DE, Arteel GE (2009) Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide‐induced liver injury caused by ethanol in mice. Hepatology 49:1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA (2011) Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun 25(Suppl 1):S92–S105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, Kugler V, Bode JC (1987) Endotoxemia in patients with alcoholic and non‐alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 4:8–14. [DOI] [PubMed] [Google Scholar]
- Coleman LG Jr, Zou J, Crews FT (2017) Microglial‐derived miRNA let‐7 and HMGB1 contribute to ethanol‐induced neurotoxicity via TLR7. J Neuroinflammation 14:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenero J, Bataller R, Sancho‐Bru P, Bellot P, Miquel R, Moreno M, Jares P, Bosch J, Arroyo V, Caballeria J, Gines P (2007) Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology 132:687–697. [DOI] [PubMed] [Google Scholar]
- Crispe IN (2016) Hepatocytes as immunological agents. J Immunol 196:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, James OF (1998) Steatohepatitis: a tale of two “hits”? Gastroenterology 114:842–845. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez M, Miquel R, Colmenero J, Moreno M, Garcia‐Pagan JC, Bosch J, Arroyo V, Gines P, Caballeria J, Bataller R (2009) Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 136:1639–1650. [DOI] [PubMed] [Google Scholar]
- Dominguez M, Rincon D, Abraldes JG, Miquel R, Colmenero J, Bellot P, Garcia‐Pagan JC, Fernandez R, Moreno M, Banares R, Arroyo V, Caballeria J, Gines P, Bataller R (2008) A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol 103:2747–2756. [DOI] [PubMed] [Google Scholar]
- Donohue TM, Osna NA, Clemens DL (2006) Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol‐elicited cytotoxicity. Int J Biochem Cell Biol 38:92–101. [DOI] [PubMed] [Google Scholar]
- Firdaus R, Biswas A, Saha K, Mukherjee A, Pal F, Chaudhuri S, Chandra A, Konar A, Sadhukhan PC (2014) Modulation of TLR 3, 7 and 8 expressions in HCV genotype 3 infected individuals: potential correlations of pathogenesis and spontaneous clearance. Biomed Res Int 2014:491064 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4122007/. Accessed August 27, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Tsukamoto H (2016) Inflammation in alcoholic and nonalcoholic fatty liver disease: friend or foe? Gastroenterology 150:1704–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Deviere J, le Moine O (2006) Differential liver sensitization to toll‐like receptor pathways in mice with alcoholic fatty liver. Hepatology 43:989–1000. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S (2004) Species‐specific recognition of single‐stranded RNA via toll‐like receptor 7 and 8. Science 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- Hill DB, Marsano LS, McClain CJ (1993) Increased plasma interleukin‐8 concentrations in alcoholic hepatitis. Hepatology 18:576–580. [PubMed] [Google Scholar]
- Hornung V, Guenthner‐Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G (2005) Sequence‐specific potent induction of IFN‐alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11:263–270. [DOI] [PubMed] [Google Scholar]
- Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt‐Jones E, Szabo G (2008) The critical role of toll‐like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48:1224–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS (2010) Recognizing and avoiding siRNA off‐target effects for target identification and therapeutic application. Nat Rev Drug Discov 9:57–67. [DOI] [PubMed] [Google Scholar]
- Kim S, Park S, Kim B, Kwon J (2016) Toll‐like receptor 7 affects the pathogenesis of non‐alcoholic fatty liver disease. Sci Rep 6:27849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp‐Arida A, Yeh M, McCullough AJ, Sanyal AJ (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41:1313–1321. [DOI] [PubMed] [Google Scholar]
- Lanthier N, Starkel P (2017) Treatment of severe alcoholic hepatitis: past, present and future. Eur J Clin Invest 47:531–539. [DOI] [PubMed] [Google Scholar]
- Leake I (2014) Hepatocellular carcinoma. Treatment potential of targeting Toll‐like receptors in HCC. Nat Rev Gastroenterol Hepatol 11:518. [DOI] [PubMed] [Google Scholar]
- Lee J, Wu CC, Lee KJ, Chuang TH, Katakura K, Liu YT, Chan M, Tawatao R, Chung M, Shen C, Cottam HB, Lai MM, Raz E, Carson DA (2006) Activation of anti‐hepatitis C virus responses via Toll‐like receptor 7. Proc Natl Acad Sci USA 103:1828–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S (2012) An unconventional role for miRNA: let‐7 activates Toll‐like receptor 7 and causes neurodegeneration. Nat Neurosci 15:827–835. [DOI] [PubMed] [Google Scholar]
- Lin KJ, Lin TM, Wang CH, Liu HC, Lin YL, Eng HL (2013) Down‐regulation of Toll‐like receptor 7 expression in hepatitis‐virus‐related human hepatocellular carcinoma. Hum Pathol 44:534–541. [DOI] [PubMed] [Google Scholar]
- Liu H, French BA, Nelson TJ, Li J, Tillman B, French SW (2015) IL‐8 signaling is up‐regulated in alcoholic hepatitis and DDC fed mice with Mallory Denk Bodies (MDBs) present. Exp Mol Pathol 99:320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey VL, Arteel GE (2012) Acute alcohol‐induced liver injury. Front Physiol 3:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momen‐Heravi F, Bala S, Kodys K, Szabo G (2015) Exosomes derived from alcohol‐treated hepatocytes horizontally transfer liver specific miRNA‐122 and sensitize monocytes to LPS. Sci Rep 5:9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, Delker DA, Jo J, Bertoletti A, Hagedorn CH, Gale M Jr (2013) IL‐1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog 9:e1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak N, Yu CF, Bieber T, Allam JP (2008) Toll‐like receptor 7 agonists and skin. Drug News Perspect 21:158–165. [PubMed] [Google Scholar]
- Qin L, Crews FT (2012) Chronic ethanol increases systemic TLR3 agonist‐induced neuroinflammation and neurodegeneration. J Neuroinflammation 9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT (2008) Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez‐Ortiz ZG, Prasad A, Griffith JW, Pendergraft WF III, Cowley GS, Root DE, Tai M, Luster AD, El Khoury J, Hacohen N, Means TK (2015) The receptor TREML4 amplifies TLR7‐mediated signaling during antiviral responses and autoimmunity. Nat Immunol 16:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roush S, Slack FJ (2008) The let‐7 family of microRNAs. Trends Cell Biol 18:505–516. [DOI] [PubMed] [Google Scholar]
- Sarkar N, Panigrahi R, Pal A, Biswas A, Singh SP, Kar SK, Bandopadhyay M, Das D, Saha D, Kanda T, Sugiyama M, Chakrabarti S, Banerjee A, Chakravarty R (2015) Expression of microRNA‐155 correlates positively with the expression of Toll‐like receptor 7 and modulates hepatitis B virus via C/EBP‐beta in hepatocytes. J Viral Hepat 22:817–827. [DOI] [PubMed] [Google Scholar]
- Starkel P, de Saeger C, Strain AJ, Leclercq I, Horsmans Y (2010) NFkappaB, cytokines, TLR 3 and 7 expression in human end‐stage HCV and alcoholic liver disease. Eur J Clin Invest 40:575–584. [DOI] [PubMed] [Google Scholar]
- Stickel F, Datz C, Hampe J, Bataller R (2017) Pathophysiology and management of alcoholic liver disease: update 2016. Gut Liv 11:173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, Downs N, Gleeson D, Macgilchrist A, Grant A, Hood S, Masson S, McCune A, Mellor J, O'Grady J, Patch D, Ratcliffe I, Roderick P, Stanton L, Vergis N, Wright M, Ryder S, Forrest EH (2015) Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 372:1619–1628. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H (2009) “Second hit” models of alcoholic liver disease. Semin Liver Dis 29:178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG (2001) Toll‐like receptor 4 is involved in the mechanism of early alcohol‐induced liver injury in mice. Hepatology 34:101–108. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Lynch VJ (2012) Measurement of mRNA abundance using RNA‐seq data: RPKM measure is inconsistent among samples. Theory Biosci 131:281–285. [DOI] [PubMed] [Google Scholar]
- West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, Kingston RE (2014) The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell 55:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler CW, Taylor KG, Peterson KE (2014) Location is everything: let‐7b microRNA and TLR7 signaling results in a painful TRP. Sci Signal 7:pe14. [DOI] [PubMed] [Google Scholar]
- Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, Kolls JK (2008) TRIF and IRF‐3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 181:3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Effect of EtOH and imiquimodexposure on liver morphology in mice. Male C57BL/6J mice were exposed to EtOH (5 g/kg, i.g.) for 10 days. Twenty‐four hours after the last EtOH dose, some mice were exposed to the TLR7 agonist imiquimod (2.5 mg/kg, i.p.) as described in Materials and Methods. Liver tissue was collected 2 hours after imiquimod (or saline) administration. Formalin‐fixed, paraffin embedded sections were cut at 5 μm and mounted on glass slides. Sections were deparaffinizedand stained with H&E. Representative photomicrographs (original magnification, 200×) are shown.
Fig. S2. Effect of EtOH and imiquimodexposure on liver TLR4 protein expression in mice. Male C57BL/6J mice were exposed to EtOH (5 g/kg, i.g.) for 10 days. Twenty‐four hours after the last EtOH dose, some mice were exposed to the TLR7 agonist imiquimod (2.5 mg/kg, i.p.) as described in Materials and Methods. Liver tissue was collected 2 hours after imiquimod (or saline) administration. Western blot was performed as described in Materials and Methods. Quantitative data are shown as fold of control. *p < 0.05 compared to control.