

Abstract
Myeloid malignancies, including myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia, are characterized by abnormal proliferation and differentiation of hematopoietic stem and progenitor cells (HSPCs). Reports on analysis of bone marrow samples from patients have revealed a high incidence of mutations in splicing factors in early stem and progenitor cell clones, but the mechanisms underlying transformation of HSPCs harboring these mutations remain unknown. Using ex vivo cultures of primary human CD34+ cells as a model, we find that mutations in splicing factors SRSF2 and U2AF1 exert distinct effects on proliferation and differentiation of HSPCs. SRSF2 mutations cause a dramatic inhibition of proliferation via a G2‐M phase arrest and induction of apoptosis. U2AF1 mutations, conversely, do not significantly affect proliferation. Mutations in both SRSF2 and U2AF1 cause abnormal differentiation by skewing granulo‐monocytic differentiation toward monocytes but elicit diverse effects on megakaryo‐erythroid differentiation. The SRSF2 mutations skew differentiation toward megakaryocytes whereas U2AF1 mutations cause an increase in the erythroid cell populations. These distinct functional consequences indicate that SRSF2 and U2AF1 mutations have cell context‐specific effects and that the generation of myeloid disease phenotype by mutations in the genes coding these two proteins likely involves different intracellular mechanisms. stem cells 2018;36:1663–1675
Keywords: Acute myelogenous leukemia, Apoptosis, CD34+, Hematologic malignancies, Umbilical cord blood, Hematopoietic stem cells, Differentiation, Proliferation
Significance Statement.
Acquired somatic mutations in splicing factors SRSF2 and U2AF1 are associated with many myeloid diseases, including myelodysplastic syndromes, acute myeloid leukemia, myeloproliferative neoplasms, primary myelofibrosis, and chronic myelomonocytic leukemia. This study demonstrated that expression of U2AF1 and SRSF2 mutations have diverse effects on CD34+ hematopoietic stem and progenitor cells. SRSF2 mutations inhibit proliferation through induction of cell cycle arrest and apoptosis, whereas U2AF1 mutations do not significantly affect proliferation. Mutations of both SRSF2 and U2AF1 skew granulomonocytic differentiation towards monocytes. However, the effects on megakaryo‐erythroid differentiation are diverse. SRSF2 mutations skew megakaryo‐erythroid differentiation toward megakaryocytes, whereas U2AF1 mutations cause an increase in the erythroid cell populations. These results identify distinct functional consequences of SRSF2 and U2AF1 mutations in human hematopoietic stem and progenitor cells and provide a system for investigating the underlying molecular mechanisms.
Introduction
Somatic mutations of the pre‐mRNA splicing machinery are associated with a spectrum of myeloid diseases. In the genes encoding splicing factors SRSF2 and U2AF1, the frequency of mutations varies between ∼5% and 25% in myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), myeloproliferative neoplasms, and primary myelofibrosis (PMF), but in cases with chronic myelomonocytic leukemia (CMML), the incidence of mutations is higher in SRSF2 (∼45%) than in U2AF1 (∼5%) 1, 2, 3, 4, 5. Mutations of both SRSF2 and U2AF1 are associated with an increase in the risk of transformation to secondary AML and a decrease in overall survival 6. Clonal analysis of tumor samples from patients with MDS and CMML revealed that splicing gene mutations are present in early hematopoietic stem and progenitor cells (HSPCs) 7, 8. The recurrent nature of the mutations and the increased risk of transformation signify their role in disease development. Interestingly, mutations in both SRSF2 and U2AF1 are heterozygous and mutually exclusive of each other and other splicing gene mutations. Studies in human cells and mice have demonstrated that a copy of the wildtype (WT) allele is essential for survival of cells harboring a mutant allele 9, 10. These data indicate critical regulatory roles for the WT proteins and a gain of new function for the mutants. However, the mechanisms by which the acquisition of mutations in SRSF2, U2AF1, and other splicing genes leads to transformation of HSPCs are not known.
Myeloid disease mutations in SRSF2 and U2AF1 occur predominantly at hotspot residues 4, 6, 11. In SRSF2, missense mutations and insertions/deletions occur exclusively at or near the amino acid residue Pro95 in the linker between the RNA recognition motif (RRM) and the arginine–serine (RS)‐rich domain. Missense substitutions leading to a Pro95His/Leu/Arg (P95H/L/R) change are most predominant. In U2AF1, mutations are missense changes at the two highly conserved amino acid positions, S34 or Q157 that lie within the N‐ and C‐terminal Zn‐finger domains, respectively. Although a direct consequence of the mutations in SRSF2, U2AF1, and other splicing genes is alteration in mRNA splice patterns, few splicing alterations have been linked to impairment of hematopoietic differentiation 9, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23. The strongest evidence associating functional consequences of mutation‐induced splicing aberrations to disease phenotype has come from a report on the effects of U2AF1‐S34F mutation in human CD34+ cells, where it was demonstrated that the phenotype associated with the U2AF mutation could be recapitulated by expressing altered isoforms of genes encoding the H2A histone variant (H2AFY) and serine/threonine kinase receptor‐associated protein (STRAP) 19. Experiments carried out by Yip et al. also demonstrated that expression of H2AFY and STRAP isoforms associated with normal myelopoiesis could partially rescue the defects in erythroid differentiation of patient derived cells. Other processes reported to be affected by splicing factor mutations include 3′‐end cleavage‐polyadenylation and R‐loop resolution during transcription 24, 25, 26. U2AF1‐S34F induced altered usage of a distal cleavage and polyadenylation site in the autophagy related factor 7 (ATG7) pre‐mRNA and was found to be sufficient for transformation of immortalized mouse hematopoietic BaF3 cells 24. Mutations of both SRSF2 and U2AF1 were found to cause an increase in R‐loops, which can potentially cause genomic instability 25, 26.
Studies using knock‐in mouse models for SRSF2‐P95H and U2AF1‐S34F have significantly advanced the understanding of the effects of mutations on hematopoiesis. These studies reported that competitive repopulation capacity of the hematopoietic stem cells (HSCs) from mutant mice (SRSF2P95H/WT and U2AF1S34F/WT) was impaired and in transplantation experiments, the recipient mice exhibited MDS‐like symptoms, including leukopenia, anemia, and defective erythropoiesis 10, 16, 17, 23. However, the heterozygous mice themselves exhibited a milder phenotype. Other models using primary human CD34+ HSPCs have also successfully identified increased self‐renewal and abnormal differentiation as cellular phenotypes associated with leukemic chromosomal translocations and somatic mutations 19, 27, 28, 29, 30, 31, 32. In this study, we examined the impact of myeloid disease mutations in splicing factors SRSF2 and U2AF1 on ex vivo proliferation and myeloid differentiation of primary human CD34+ HSPCs. Our analyses revealed that while mutations of both U2AF1 and SRSF2 cause abnormal differentiation of the myeloid lineage cells, only SRSF2 mutations inhibit proliferation through induction of cell cycle arrest and apoptosis. These data indicate differential functional consequences of introducing mutations in SRSF2 and U2AF1 in human HSPCs and provide a system for investigating the underlying molecular mechanisms.
Materials and Methods
Cell Lines and Plasmids
The cell lines HEK‐293T, MS‐5, and K052 (a generous gift from Omar Abdel‐Wahab, SKMCC) were cultured in Dulbecco's Modified Eagle Media (DMEM) containing l‐glutamine and high glucose (ThermoFisher Scientific, Waltham, MA) and 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, CA) and 1% Penicillin–Streptomycin (Pen–Strep; Sigma‐Aldrich, St. Louis, MO). K562 and TF1a (American Type Culture Collection, Manassas, VA) cells were cultured in RPMI medium with l‐glutamine (ThermoFisher Scientific, Waltham, MA), 10% FBS, and 1% Pen–Strep.
DNA fragments for expression of a fusion protein consisting of green fluorescent protein (GFP), self‐cleaving 2A peptide, and FLAG epitope (DYKDDDDK) tagged splicing factor were generated by PCR amplification and cloned into the pCCL‐MNDU3c‐X2 plasmid (a generous gift from Don Kohn, University of California, Los Angeles). These plasmids expressed both GFP and the U2AF1 or SRSF2 proteins under the MNDU3c promoter.
Statistical Analysis
All immunophenotypic analyses were performed at least four times and data are represented as mean ± standard error (Figs. 1E and 3; Supporting Information Figs. S2 and S5–S7). For longitudinal comparison of expansion of GFP+ cells (Fig. 2) and lineage markers (Supporting Information Fig. S5) between cultures expressing mutations of SRSF2 and U2AF1 to the WT proteins, statistical analyses were performed with STATA version 14 using the linear mixed model. These analyses compared the observed differences in the mean fold change (relative to day 1) in the number of GFP+ cells using the WT as a reference (Fig. 2; Supporting Information Table S1) or the percentage of cells expressing lineage markers for the mutations with the WT as a reference (Supporting Information Fig. S5; Supporting Information Table S2). All expression measurement approximated a normal distribution following a log transformation. All p values were two‐sided and p < .05 was considered statistically significant. For comparisons of mature myeloid lineage populations (n = 4), apoptosis (n = 4), and cell cycle (n = 3), statistical analyses on the indicated days were performed using the Mann–Whitney test in GraphPad Prism v7. Data are represented as mean ± standard error (Figs. 3, 4, 5, 6). The p values for RT‐PCR analyses were calculated using the two‐tailed Student t‐test in Microsoft Excel (Fig. 6; Supporting Information Fig. S12).
Figure 1.
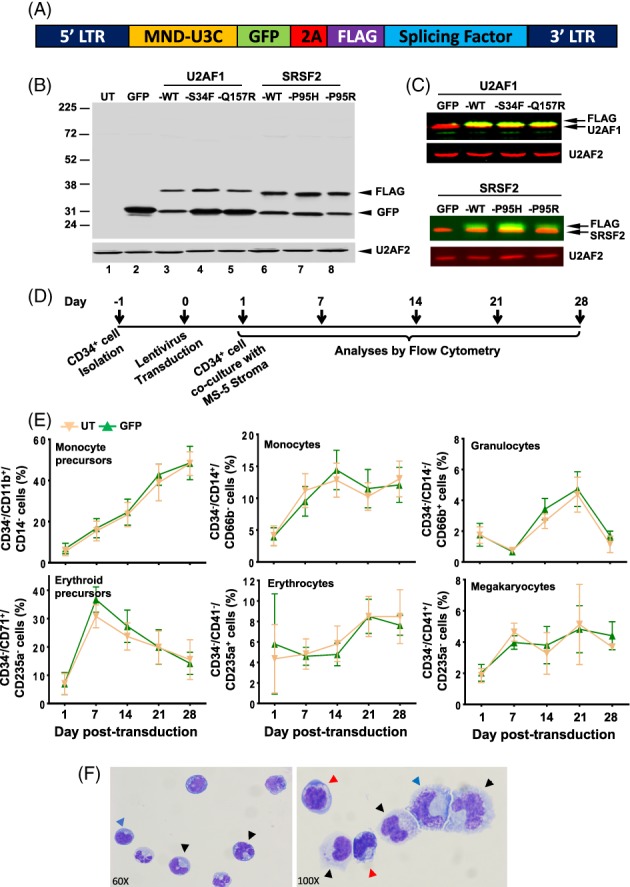
Transduction and differentiation of the CD34+ cells. (A): Schematic of plasmid construct used to make lentiviruses. (B): Western blot of K562 cell lysates showing expression of GFP and FLAG‐tagged splicing factors. U2AF2 was used as a loading control. (C): Western blot of K562 lysates showing expression of FLAG‐tagged splicing factors with endogenous U2AF1 (upper panel) and SRSF2 (lower panel). (D): Scheme for transduction and differentiation of CD34+ cells. (E): Immunophenotypic detection of lineage cells by flow cytometry. Percentages of CD34− population that were monocyte precursors (CD34−/CD11b+/CD14−), monocytes (CD34−/CD14+/CD66b−), granulocytes (CD34−/CD14−/CD66b+), erythroid precursors (CD34−/CD71+/CD235a−), erythrocytes (CD34−/CD41a−/CD235a+), and megakaryocytes (CD34−/CD41a+/CD235a−) are shown for cultures that were untransduced (UT) or expressing GFP alone. Representative scatter plots that were used to calculate the positive populations are shown in Supporting Information Figure S3. (F): Wright–Giemsa staining of untreated cultured cells depicting morphologic characteristic of erythroid cells (red arrowhead), granulocytes (black arrowhead), and monocytes (blue arrowhead).
Figure 3.
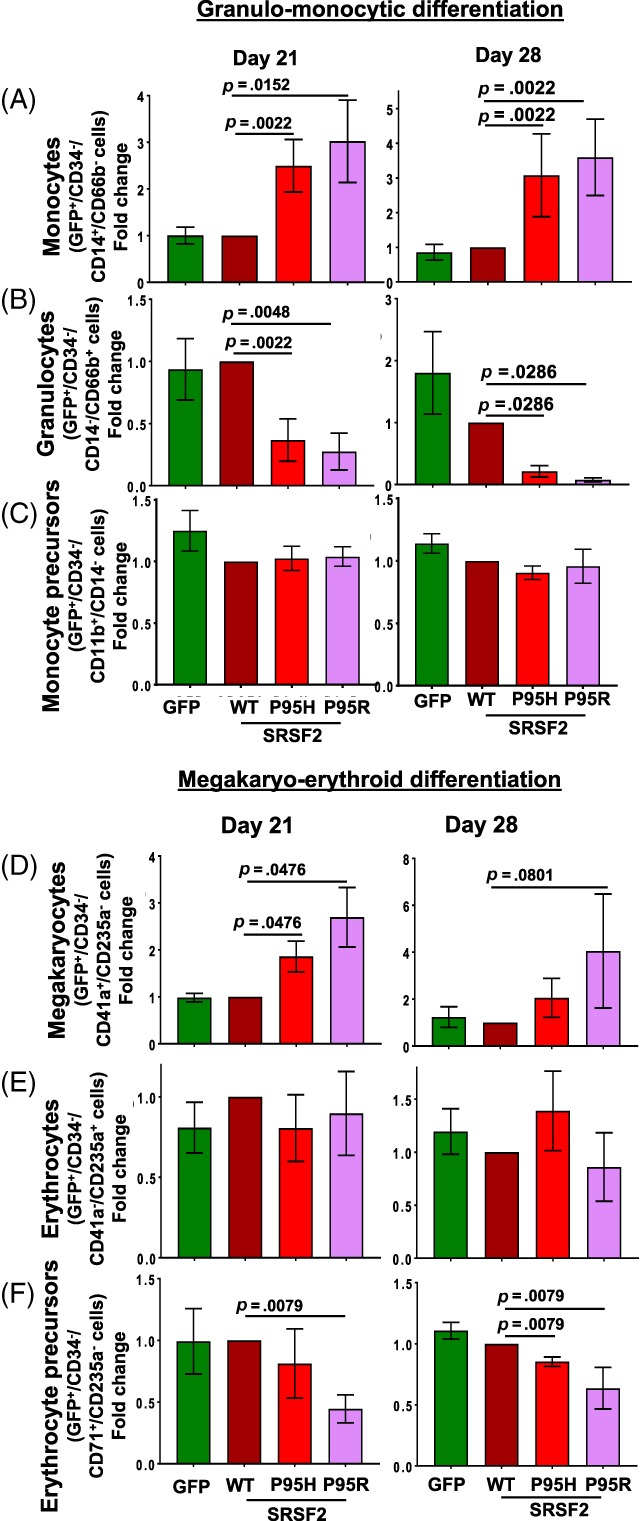
Mutations of SRSF2 skew myeloid differentiation. Immunophenotypic detection of lineage cells expressing the WT and mutant proteins was carried out by flow cytometry for granulo‐monocyte differentiation and megakaryo‐erythroid differentiation. Fold change in the percentages of (A) monocytes (CD34−/GFP+/CD14+/CD66b−), (B) granulocytes (CD34−/GFP+/CD14−/CD66b+), (C) monocyte precursors (CD34−/GFP+/CD11b+/CD14−), (D) megakaryocytes (CD34−/GFP+/CD41a+/CD235a−), (E) erythrocytes (CD34−/GFP+/CD41a−/CD235a+), and (F) erythroid precursors (CD34−/GFP+/CD71+/CD235a−) were calculated relative to the wildtype for days 21 and 28. Percentages of positive populations are shown in Supporting Information Figure S6B, S6C, respectively. All data are represented as mean ± standard error of four independent experiments. The p values were calculated using the Mann–Whitney test in GraphPad Prism v7. The p values ≤.05 were considered significant and are shown. Abbreviation: WT, wildtype.
Figure 2.
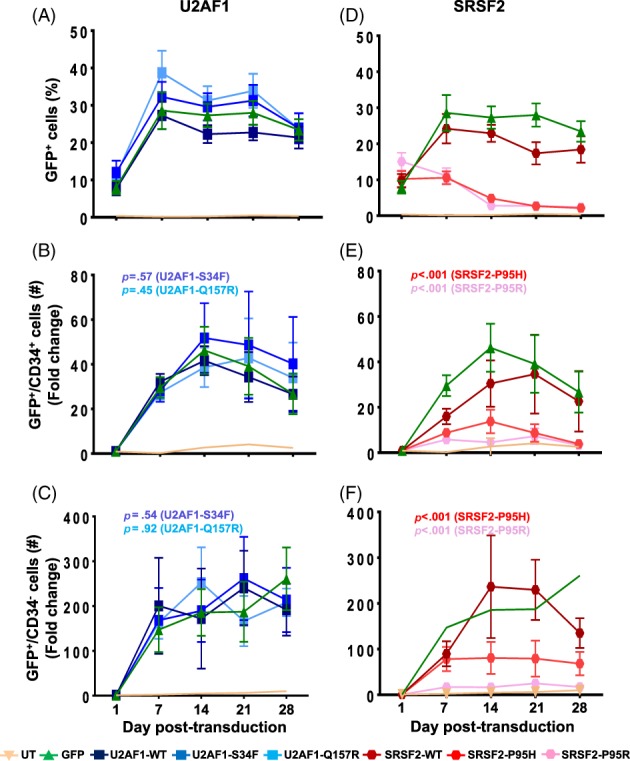
SRSF2 mutations inhibit expansion of the CD34+ cells. GFP and CD34 expression were determined by flow cytometry. Percentages of GFP+ cells in cultures expressing GFP alone and U2AF1 wildtype (WT) and mutants (A) and SRSF2 WT and mutants (D) are shown. Fold change in number of GFP+/CD34+ and GFP+/CD34− cells were calculated relative to day 1 for cultures expressing GFP alone, the WT and mutant proteins of U2AF1 (B, C) and SRSF2 (E, F). Representative scatter plots for analysis of GFP+ and CD34+ cells are shown in Supporting Information Figure S4. All data are represented as mean ± standard error of eight independent experiments. The p values were calculated using the linear mixed effect model in STATA. The p values ≤.05 were considered significant and are shown here. All statistical data are shown in Supporting Information Table S1.
Figure 4.
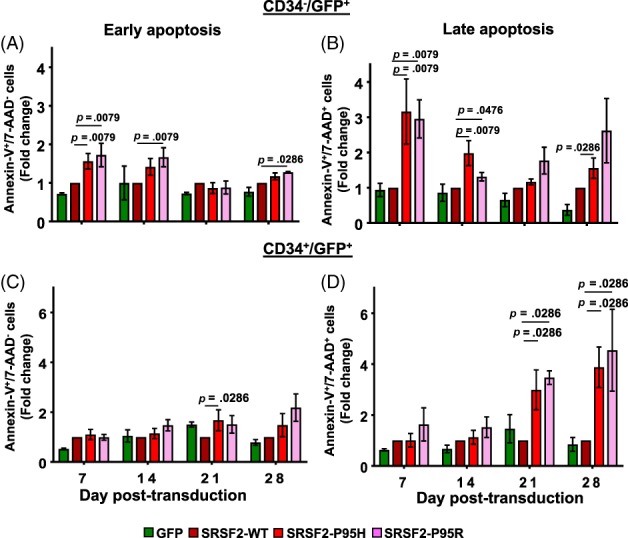
SRSF2 mutations induce apoptosis. Fraction of cells undergoing apoptosis was determined by flow cytometry analysis after Annexin‐V and 7‐AAD staining. Fold change in fractions of early apoptotic (Annexin‐V+/7‐AAD−) and late apoptotic (Annexin‐V+/7‐AAD+) cells in the GFP+/CD34− (A, B) and the GFP+/CD34+ (C, D) populations expressing SRSF2 mutations were calculated relative to the wildtype protein. All data are represented as mean ± standard error of four independent experiments. The p values were calculated using the Mann–Whitney test in GraphPad Prism v7. The p values ≤.05 were considered significant and are shown. Representative scatter plots that were used to calculate positive populations and graphs depicting percentages of positive population are shown in Supporting Information Figure S8A.
Figure 5.
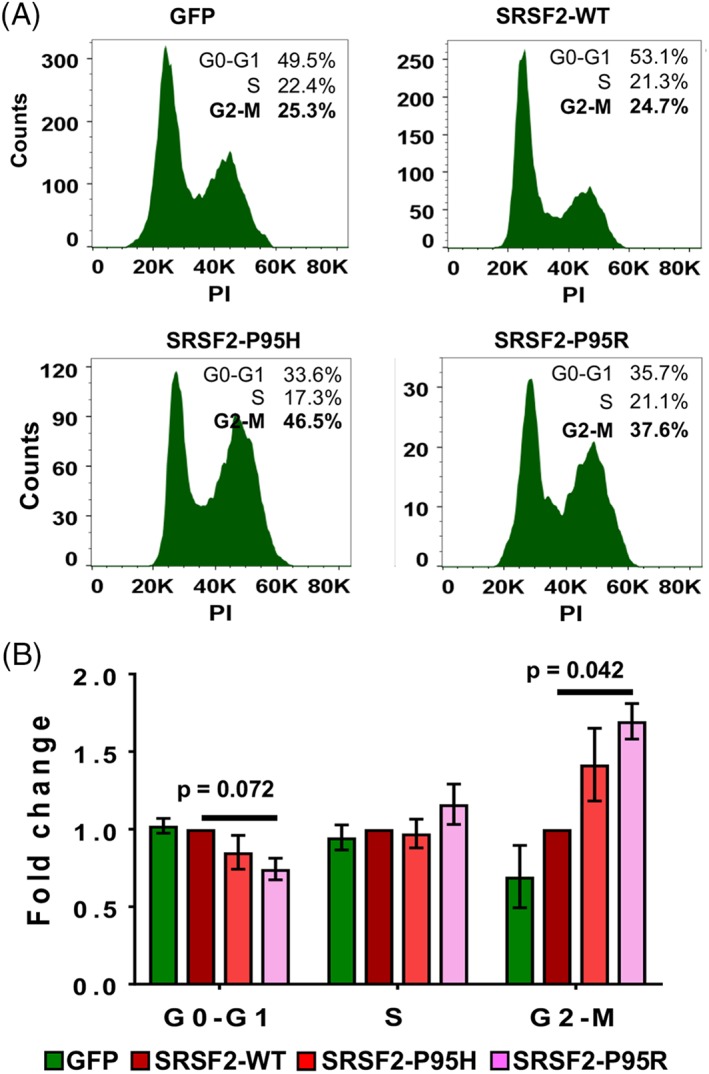
SRSF2 mutations cause a G2‐M phase arrest in the CD34+ cells. Cell cycle phase distribution of CD34+/GFP+ cells was determined by DNA content measurements after propidium iodide staining on day 14 post‐transduction. (A): Histograms for cells expressing GFP alone, SRSF2‐WT, SRSF2‐P95H, and SRSF2‐P95R from a representative experiment are shown. (B): Fold change in percentages of the G0‐G1, S, and G2‐M phases were calculated relative to wildtype. All data are represented as mean ± standard error of three independent experiments. The p values were calculated using the Mann–Whitney test in GraphPad Prism v7. The p values ≤.05 were considered significant and are shown.
Figure 6.
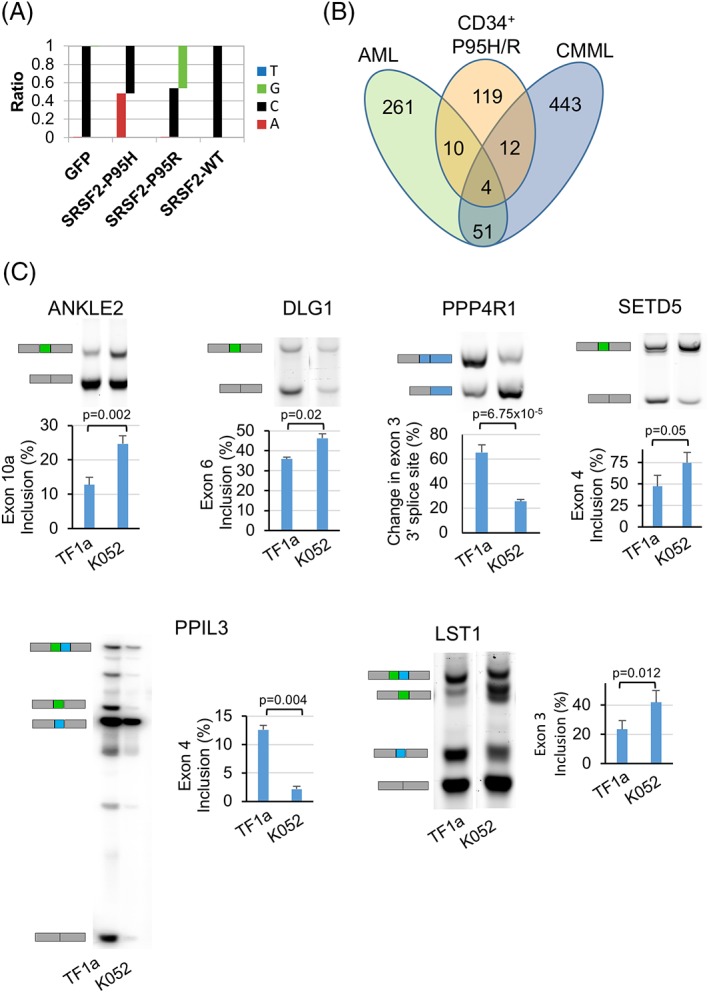
SRSF2 mutations alter splicing profiles of CD34+ cells. (A): Frequency of occurrence of nucleotides G, C, A, and T showing C → A and C → G change in the reads spanning exon 2 from RNA‐Seq libraries from cells expressing SRSF2‐P95H and SRSF2‐P95R, respectively. (B): Venn diagrams showing the overlap among the genes that were aberrantly spliced upon expression of SRSF2 mutations in CD34+ cells, patients with acute myeloid leukemia and chronic myelomonocytic leukemia carrying SRSF2 mutations (Kim et al. 17). (C): RT‐PCR analysis showing validation of targets of SRSF2 mutations in TF1a and K562 cells. Gel images showing mRNA isoforms that include and skip cassette exons 10a, 6, and 4 in ANKLE1, DLG1, and SETD5, respectively. Isoforms for mutually exclusive exons 3 and 4 in LST1 and exons 4 and 5 in PPIL3 and change in use of alternative 3′ splice site in PPP4R1 are also shown. Percentages of exon inclusion are represented as mean ± standard error of three independent experiments. The p values were calculated using the Student t‐test in Microsoft Excel. The p values ≤.05 were considered significant and are shown.
RNA‐Seq Library Preparation and Sequencing
On day 7 post‐transduction, CD34+/GFP+ cells were isolated from cultures expressing GFP alone, SRSF2‐WT, SRSF2‐P95H, or SRSF2‐P95R using BD FACS Aria II. Total RNA was isolated using the Total RNA Purification Plus Micro Kit (Norgen Biotek, Thorold, ON). rRNA depleted RNA‐Seq libraries were generated using the Stranded RNA‐Seq Kit with RiboErase (KAPA Biosystems, Wilmington, MA). Three replicate libraries, each derived from CD34+ cells obtained from a different donor, were generated for each set of samples. The libraries were subjected to 100 nt paired‐end sequencing to a depth of 40 million reads on Illumina Hi‐Seq 4000.
Bioinformatics Analysis
Reads from the CD34+/GFP+ cells were mapped to the current human genome (GRCh38) using Hisat 2 33. rMATS version 3.08 was used to carry out differential splicing analysis of cassette exons, and alternative 3′ and 5′ splice sites in SRSF2‐WT, SRSF2‐P95H, and SRSF2‐P95R samples 34. Differential intron retention was analyzed using DexSeq 35, 36. Differences in gene expression between the SRSF2 WT and mutant samples were determined using EdgeR after assigning reads to annotated transcripts using Rsubread 37, 38, 39. Alterations in polyadenylation were examined using the DaPars algorithm 40.
Results
Transduction, Culture, and Differentiation of Cord Blood Derived CD34+ Cells
We used lentiviruses to co‐express GFP and either WT or mutant splicing factors in human umbilical cord blood (UCB) derived CD34+ cells. The lentiviral vectors were bi‐cistronic and expressed a GFP‐splicing factor fusion protein that is linked by a 2A self‐cleaving peptide bridge 41 (Fig. 1A). Vectors expressed FLAG‐tagged versions of WT splicing factor SRSF2 (SRSF2‐WT) and its two mutations (SRSF2‐P95H and SRSF2‐P95R), and of WT U2AF1 (U2AF1‐WT) and its two mutations (U2AF1‐S34F and U2AF1‐Q157R). We used K562 myeloid leukemia cells to test the expression levels of the proteins from the lentiviral constructs. Western analysis of lysates from K562 cells that were transduced with the vectors and then sorted for GFP+ cells displayed efficient cleavage of the 2A peptide bridge and robust expression of GFP (Fig. 1B, lanes 2–8), the U2AF1‐WT and mutant proteins (lanes 3–5) and the SRSF2‐WT and mutant proteins (lanes 6–8). The ratio of FLAG‐tagged proteins to endogenous was calculated from blots that were probed with anti‐FLAG and anti‐U2AF1 or anti‐FLAG and anti‐SRSF2 antibodies (Fig. 1C). The fractions of tagged proteins relative to endogenous for U2AF1‐WT, U2AF1‐S34F, and U2AF1‐Q157R were estimated to be 0.45, 0.42, and 0.36, respectively, and for SRSF2‐WT, SRSF2‐P95H, and SRSF2‐P95R were estimated to be 0.49, 0.56, and 0.43, respectively. Therefore, this indicates that in the transduced cells, the proportion of the mutant proteins was equivalent to that of the endogenous WT proteins.
The onset of myeloid lineage commitment is marked by the generation of common myeloid progenitors (CMPs) from HSCs 42, 43, 44 (Supporting Information Fig. S1). Differentiation of CMPs involves further commitment to one of two more lineage‐restricted pathways through either the megakaryocyte erythroid progenitor (MEP) or granulocyte–monocyte progenitor (GMP) stages. MEPs give rise to megakaryocytes and erythrocytes, and GMPs give rise to granulocytes and monocytes. Lineage commitment during differentiation is accompanied by loss of CD34 and gain of lineage‐specific cell surface markers.
CD34+ cells were isolated from human UCB, transduced, and then co‐cultured with MS5 stromal cells for 4 weeks in medium containing cytokines to support proliferation and myeloid differentiation 45 (Fig. 1D). Cells were harvested weekly and immunophenotyped for markers of mature erythrocytes (CD235a/glycophorin A), megakaryocytes (CD41a), granulocytes (CD66b), and monocytes (CD14). The level of CD11b that is expressed in both granulocyte and monocyte precursors and of CD71 (transferrin receptor) that is present on the erythroid precursors was also analyzed 46, 47, 48. A comparison of untransduced and GFP expressing cells showed that there was a rapid and sustained increase in the fraction of cells expressing CD11b and CD14 (Supporting Information Fig. S2). An initial increase in CD71 was followed by a decline in its expression, presumably due to maturation of the precursor cells. The onset of CD66b, CD41a, and CD235a expression began later, day 7–14, and reached maximum by day 21. Presence of CD34−/CD11b+/CD14− and CD34−/CD71+/CD235− cells confirmed the generation of monocyte and erythroid precursors, respectively (Fig. 1E). The maturation of the precursors to lineage cells was established by the presence of CD34−/CD14+/CD66b− (monocyte), CD34−/CD14−/CD66b+ (granulocyte), CD34−/CD41+/CD235a− (megakaryocyte), and CD34−/CD41−/CD235a+ (erythrocyte) cell populations. Representative scatter plots that were used to calculate positive populations are depicted in Supporting Information Figure S3. Morphological examination after Wright‐Giemsa staining confirmed the phenotype of the lineage cells includes mature monocytes, granulocytes, and erythroblasts in the cultures (Fig. 1F). Thus, under the culture conditions used in this study, the UCB CD34+ cells differentiated to myeloid lineage cells and the expression of GFP did not have any significant effects on the ex vivo cultures.
SRSF2 Mutations Inhibit Expansion of the CD34+ Cells
To analyze the effects of mutations, we compared the fraction of GFP+ cells post‐transduction. In cultures transduced with GFP alone, U2AF1‐WT, or SRSF2‐WT, maximal GFP expression was achieved by day 7 (Fig. 2). The cultures for U2AF1 mutations S34F and Q157R also exhibited fractions of GFP+ cells similar to GFP alone and U2AF1‐WT (Fig. 2A). However, in the cases of SRSF2 mutations P95H and P95R, growth of the GFP+ cells was impaired (Fig. 2D).
We analyzed the impact of mutations on CD34+ and CD34− populations by the linear mixed effect model and longitudinally compared the mean difference in fold change in the number of GFP+ cells using the WT protein as a reference. Representative scatter plots that were used to calculate the GFP+/CD34+ and GFP+/CD34− populations are shown in Supporting Information Figure S4. Analysis for fold change in cell numbers revealed that rates of increase in the number of GFP+/CD34+ cells expressing U2AF1 mutations S34F and Q157R were not significantly different from those of cells expressing the WT protein (Fig. 2B and Table S1, p > .05, n = 8). Relative to day 1 post‐transduction, there was a ∼40‐ to 50‐fold increase in GFP+/CD34+ cells in cultures generated by the control GFP vector, as well as by U2AF1‐WT and mutants. The rates of conversion of CD34+ to CD34− phenotype were also not affected by expression of the U2AF1 mutations (Fig. 2C, p > .05, n = 8). Conversely, in comparison to SRSF2‐WT, SRSF2‐P95H, or SRSF2‐P95R mutations significantly inhibited expansion of the CD34+ cells (Fig. 2E, p < .001, n = 8) and also the conversion of these stem and progenitor populations to CD34− cells (Fig. 2F, p < .001, n = 8). In comparison to SRSF2‐WT, in cultures expressing SRSF2‐P95H and SRSF2‐P95R, the fraction of GFP+/CD34− cells was significantly suppressed to ∼3%–5%, likely indicating reduced differentiation. In both the CD34+ and CD34− cell populations, the effects of SRSF2‐P95R appeared to be greater than those of SRSF2‐P95H.
Mutations in SRSF2 and U2AF1 Skew Differentiation
To determine the impact of mutations on myeloid differentiation, we examined the expression of lineage markers on the GFP+ cells. Longitudinal analysis of the precursor and mature lineage markers on the total GFP+ population indicated that the fraction of cells expressing CD11b, CD14, CD41a, CD71, CD235a, and CD66b were similar in cultures transduced with GFP alone, U2AF1‐WT, and SRSF2‐WT vectors (Supporting Information Fig. S5). Comparison of the percentages of cells expressing the individual markers showed that upon expression of U2AF1‐S34F or U2AF1‐Q157R, there was a statistically significant increase in the fraction of cells expressing CD11b, CD14, CD66b, CD41a, and CD235a (p ≤ .001, n = 4), but not CD71 (p > .05, n = 4) (Supporting Information Fig. S5; Supporting Information Table S2). In contrast, expression of SRSF2‐P95H or SRSF2‐P95R caused a significant reduction in the fraction of GFP+ cells expressing all lineage markers examined (p ≤ .05, n = 4) except CD66b (p > .05, n = 4), relative to SRSF2‐WT and GFP controls, thereby indicating that SRSF2 mutations reduced the percentage of cells expressing lineage markers, whereas U2AF1 mutations enhanced the fraction of cells expressing lineage markers.
We further compared lineage populations in the transduced cultures. We compared fold change in the fraction of precursor and mature lineage cells in cultures expressing mutant proteins to WT on days 7, 14, 21, and 28. However, on day 7 the fractions of GFP+ cells expressing the lineage markers were very low (Supporting Information Fig. S5). The percentages of GFP+/CD34− cells expressing the precursor and mature lineage markers for granulo‐monocyte and megakaryo‐erythroid differentiation for days 14, 21, and 28 are illustrated in Supporting Information Figure S6A–S6C, respectively. The trends in fold change on all three days were similar, but the differences between WT and mutations were found not to be significant on day 14 (Fig. S6D). The analyses for days 21 and 28 revealed that mutations in both SRSF2 and U2AF1 skewed differentiation (Fig. 3; Supporting Information Fig. S7). During granulo‐monocyte differentiation, in comparison to SRSF2‐WT, SRSF2‐P95H, or SRSF2‐P95R elicited a significant increase in the fraction of monocytes (GFP+/CD34−/CD14+/CD66b−) with a concomitant decrease in granulocytes (GFP+/CD34−/CD14−/CD66b+) (Fig. 3A, 3B; p < .05, n = 4). Expression of SRSF2 mutations did not affect the monocyte precursors (GFP+/CD34−/CD11b+/CD14−) (Fig. 3C). In megakaryo‐erythroid differentiation, in comparison to the WT protein, SRSF2‐P95H and SRSF2‐P95R caused a significant increase in the fraction of megakaryocytes (GFP+/CD34−/CD41a+/CD235a−) (Fig. 3D; p < .05, n = 4). This change was accompanied by a concomitant decrease in the erythroid precursors (GFP+/CD34−/CD71+/CD235a−) (Fig. 3E; p < .05, n = 4), but not the mature erythroid population (GFP+/CD34−/CD41a−/CD235a+) (Fig. 3F).
The effects of U2AF1 mutations on granulo‐monocyte differentiation were similar to the SRSF2 mutations (Supporting Information Fig. S7). There was an increase in monocytes with a concomitant decrease in granulocytes and no change in monocyte precursors upon expression of U2AF1‐S34F and U2AF1‐Q157R (Supporting Information Fig. S7A–S7C). In the case of megakaryo‐erythroid differentiation, the effects of U2AF1 mutations were opposite. Expression of U2AF1‐S34F and U2AF1‐Q157R led to a decrease in the megakaryocyte fraction (Supporting Information Fig. S7D), which was accompanied by an increase in the erythroid precursors, but not the mature erythroid cells (Supporting Information Fig. S7E, S7F). Notably, the effects of U2AF1 mutations were less dramatic than those of SRSF2 mutations. Although the direction of the observed change was consistent on both days, the extent of change was small and the statistical significance was variable. It is likely that in CD34+ cells, appearance of more pronounced effects of U2AF1 mutation on the cellular phenotype are delayed or require a secondary event.
Thus, the immunophenotypic analyses of differentiation revealed that mutations of SRSF2 and U2AF1 cause abnormal differentiation, but their qualitative and quantitative effects are variable. Mutations of both proteins skew granulo‐monocytic differentiation toward monocytes but have different effects on megakaryo‐erythroid differentiation. The SRSF2 mutations skew differentiation toward megakaryocytes whereas U2AF1 mutations cause a decrease in the generation of megakaryocytes with a compensatory increase in the erythroid populations.
SRSF2 Mutations Induce Apoptosis and G2‐M Arrest
To determine the underlying reason for lack of increase in the CD34− and CD34+ cell numbers, we determined (via weekly staining with Annexin‐V and 7‐AAD) whether expression of SRSF2 mutations was inducing apoptosis. This analysis revealed that for CD34− cells, in comparison to SRSF2‐WT, expression of SRSF2 mutation P95H or P95R caused a significant increase in both early (Annexin‐V+/7‐AAD−) (Fig. 4A; p < .05, n = 4) and late (Annexin‐V+/7‐AAD+) apoptosis (Fig. 4B; p < .05, n = 4). For the CD34+ cells, expression of SRSF2‐P95H or SRSF2‐P95R did not increase early apoptosis (Fig. 4C; p > .05, n = 4) but caused a marked increase in the fraction of late apoptotic cells on days 21 and 28, which did not occur at earlier time points (Fig. 4D; p < .05, n = 4). In comparison to U2AF1‐WT, the U2AF‐S34F and U2AF1‐Q157R mutations did not significantly induce either early or late apoptosis (Supporting Information Fig. S8B). This delayed induction of apoptosis upon expression of SRSF2‐P95H and SRSF2‐P95R explains the loss of GFP+/CD34+ cells on days 21 and 28, but it does not account for the lack of expansion of the CD34+ cells expressing mutations at earlier time points (Fig. 2B).
To further elucidate the effects of SRSF2 mutations, we examined cell cycle profiles of the CD34+ and CD34− populations by measuring DNA content using propidium iodide (PI) staining. On day 7, the distribution of the GFP+/CD34+ cells in different phases of cell cycle was similar in cells expressing GFP, SRSF2‐WT, and the mutations P95H or P95R (data not shown). However, by day 14, in comparison to cells expressing SRSF2‐WT, there was a ∼twofold increase in the fraction of GFP+/CD34+ cells in the G2‐M phase upon expression of the SRSF2 mutation P95H or P95R (Fig. 5A, 5B; p ≤ .05, n = 3). This increase in G2‐M cells was accompanied by a decrease in the fraction of G0‐G1 cells. The percentage of cells in S phase was found to be similar in the cells expressing SRSF2‐WT and mutations thereof. To further examine the effect of mutations on the S phase, we performed the BrdU incorporation assay on day 14. In cultures expressing GFP alone, SRSF2‐WT and mutations thereof exhibited similar percentages of GFP+/CD34+/BrdU+ cells, thereby confirming the absence of an effect on the S phase upon expression of SRSF2 mutations in comparison to WT (Supporting Information Fig. S9A). As the CD34− population is postmitotic, the GFP+/CD34− cells were found not to be cycling. They were in the G0‐G1 phase and expression of mutations did not have any effect in comparison to the WT protein (Supporting Information Fig. S9B). Thus, the data show that upon expression of the SRSF2‐P95H and SRSF2‐P95R mutations in the CD34+ cells, a G2‐M phase arrest occurs first and is followed by massive cell death at later time points. There is loss of CD34− cells because of apoptosis, which likely leads to the reduced percentages of differentiated cells upon expression of SRSF2 mutations.
SRSF2 Mutations Do Not Adversely Affect K562 Cells
We also examined the effects of U2AF1 and SRSF2 mutations on proliferation and phorbol myristic acid (PMA) induced differentiation of K562 cells. These analyses did not reveal significant differences in the rate of proliferation of K562 cells expressing the U2AF1 and SRSF2 WT and mutant proteins (Supporting Information Fig. S10A). In comparison to the WT proteins, expression of the mutants caused neither an increase in early (Annexin‐V+/7‐AAD−) nor late (Annexin‐V+/7‐AAD+) apoptotic cells (Supporting Information Fig. S10B). We also determined the influence of mutations on PMA induced differentiation of K562 cells stably expressing GFP and the U2AF1 and SRSF2 WT and mutant proteins. Post PMA treatment, flow cytometry analysis for CD11b (monocyte) and CD41a (megakaryocyte) did not yield significant differences in the levels of either marker on cells expressing the U2AF1 or SRSF2 mutations in comparison to those expressing the WT proteins (Supporting Information Fig. S10C, S10D).
SRSF2 Mutations Alter Splicing in the CD34+ Cells
To determine the effects of SRSF2 mutations on pre‐mRNA splicing in the CD34+ cells, we carried out RNA‐Seq analysis. On day 7 post‐transduction, the GFP+/CD34+ cells were sorted for RNA isolation and strand‐specific libraries were prepared from total RNA from cells expressing GFP alone, SRSF2‐WT, SRSF2‐P95H, or SRSF2‐P95R. RNA‐Seq reads mapped to SRSF2 exon 2 demonstrated C → A and C → G substitutions in cells transduced with vectors expressing SRSF2‐P95H and SRSF2‐P95R, respectively. In these samples, the fractions of mutant transcripts determined by the number of reads covering the mutation site were 0.41, 0.24, and 0.48 for the SRSF2‐P95H replicates and 0.47, 0.56, and 0.46 for the SRSF2‐P95R replicates (Fig. 6A depicts data for one representative experiment). These values are in agreement with the protein expression levels that were estimated by Western analysis and confirm that expression of the mutants was at a proportion equivalent to SRSF2‐WT (Fig. 1C). In principal component analysis on the basis of gene expression, the samples clustered together by the donor (Supporting Information Fig. S11A). In contrast to previous studies in human cell lines and mouse models, comparison of the WT and mutant samples did not identify any significant changes in gene expression in the CD34+ cells. This could be because of large differences in background gene expression of the individual CD34+ cell donors and likely reflects high genetic variance. In addition, examination of alternative polyadenylation sites by the dynamic analysis of alternative polyadenylation from RNA‐Seq (DaPars) algorithm did not identify significant changes that were common by comparing both SRSF2 mutations P95H and P95R with GFP and SRSF2‐WT (Supporting Information File S1) 40.
Splicing events were quantified to identify cassette exons, 5′ and 3′ splice sites, mutually exclusive exons, and retained introns that were altered upon expression of SRSF2‐P95H and SRSF2‐P95R in comparison to SRSF2‐WT and also to GFP alone (Supporting Information Files [Link], [Link]) 34. Using a cutoff of >10% for change in splicing and a false discovery rate (FDR) below 0.01, we found that a relatively small fraction (<1%) of splicing events of any class was affected by the SRSF2 mutations and that the affected events were both enhanced and repressed by the mutations (Table S3). We further filtered the cassette exons and 5′ and 3′ splice site events for changes that occur in the same direction for both mutations, for FDR below 0.01, and a splicing change of >10% for at least one of the mutations (Supporting Information File S6). This identified 228 altered splicing events (122 cassette exons, 39 altered 5′ splice sites, and 67 altered 3′ splice sites) via comparison of the mutations to SRSF2‐WT (Supporting Information File S6). Similarly, comparison with the GFP alone transduced cells identified 182 altered splicing events (98 cassette exons, 39 altered 5′ splice sites, and 45 altered 3′ splice sites) upon expression of the mutations. Further comparison of the two target sets (WT vs. P95H/R and GFP vs. P95H/R) identified 71 overlapping genes with splicing alterations (Supporting Information File S6). Previous studies have reported enrichment of CCNG motif and depletion of GGNG motif in the exons that are altered in response to SRSF2 mutations 17. However, the number of overlapping exons in our dataset was too small for performing statistical analysis. The list of cassette exons that contained either CCNG or GGNG motifs is provided in Supporting Information File S7. Examination for retained introns did not identify any significant events that were altered in response to both mutations. In unsupervised hierarchical clustering based on the inclusion levels of the altered cassette exons, the samples clustered by the donor (Supporting Information Fig. S11B). As with gene expression, this could be due to large differences in splice patterns in CD34+ cells from individual donors.
We next compared the differentially spliced genes in CD34+ cells with those reported in other studies to be altered in response to SRSF2 mutations in hematopoietic cells. Comparisons with CMML and AML tumor samples identified relatively few intersecting genes in association with SRSF2 mutations P95H and P95R (Fig. 6B; Supporting Information Table S4) 17. A comparison with SRSF2‐P95H targets reported to be differentially spliced in K562 cells identified five genes (Supporting Information Table S4) 20. Similarly, comparisons with SRSF2‐P95H targets in mice also identified very few overlapping genes (Supporting Information Tables S5 and S6) 17, 23. Some of the previously reported splicing alterations in mice, including CDC25C, EZH2, and BCL6 corepressor (BCOR), were not identified as targets of SRSF2 mutations in the CD34+ cells 17, 49. These differences could be because of variations in the RNA‐Seq analysis pipeline and/or filtering cutoff values as well as the cell type and model system analyzed.
Using overlap with AML and/or CMML samples and conservation across mammals as criteria, we selected six differentially spliced target genes for confirmation. These overlapping targets have known functions in cell cycle regulation (Ankyrin repeat and LEM Domain‐Containing Protein 2; ANKEL2), pre‐mRNA splicing (Peptidylprolyl Isomerase Like 3; PPIL3), signal transduction (Leukocyte specific transcript 1; LST1), protein dephosphorylation (Protein Phosphatase 4 regulatory subunit 1; PPP4R1), and tumor suppression (Discs Large MAGUK Scaffold Protein 1; DLG1) 50, 51, 52, 53, 54. We also included a histone modifier SETD5, which was found to be a target in mouse KSL and myeloid progenitors, but not in K562 cells, AML or CMML tumor samples (Supporting Information Tables S4–S6) 55. Differences in exon inclusion levels that were predicted by RNA‐Seq were validated by RT‐PCR using RNA from leukemia cell lines TF1a and K052 that are WT and P95H for SRSF2, respectively 10. In comparison with TF1a cells, there is an increased inclusion of exon 10a of ANKLE2, exon 6 of DLG1, and exon 4 of SETD5 in K052 (Fig. 6C). Although, the altered exons in LST1 and PPIL3 were identified as cassette exons in rMATS analysis, these were found to be mutually exclusive. In both LST1 and PPIL3, the individual exons change as predicted (Fig. 6C). In LST1, increased inclusion of exon 3 and decreased inclusion of exon 4 is observed in K052 cells compared to TF1a. In PPIL3, decreased inclusion of exon 4 is accompanied with increased inclusion of exon 5. Lastly, there is an increased usage of the distal 3′ splice site of exon 3 in PPP4R1 in K052 cells. To determine if exogenous expression of SRSF2 mutations mimics the splicing alterations in TF1a cells, we transduced them with vectors to express GFP alone, SRSF2‐WT or SRSF2‐P95R and then sorted them for GFP+ cells. RT‐PCR analysis using RNA from the transduced cells showed that expression of SRSF2‐P95R recapitulated the effects on splicing of ANKLE 2, PPP4R1, and LST1 transcripts in TF1a cells (Supporting Information Fig. S12). Relative to GFP alone and SRSF2‐WT controls, TF1a cells expressing SRSF2‐P95R exhibited increased inclusion of exon 10a in ANKLE2 and exon 3 in LST1, and an increased usage of the distal 3′ splice site of exon 3 in PPP4R1. The data indicate that exogenous expression of SRSF2‐P95R mutation is sufficient to induce these splicing changes.
Discussion
The longitudinal analysis in this study demonstrated that under the ex vivo differentiation conditions used, the responses of CD34+ HSPCs to mutations of the splicing factors SRSF2 and U2AF1 are diverse and affect multiple lineages (Fig. 7). Effects of SRSF2 mutations, P95H and P95R are dramatically antiproliferative and are associated with a G2‐M phase arrest of the CD34+ cells and induction of apoptosis in the CD34− cells (Fig. 7A). These data indicate that in cells expressing the SRSF2 mutations, initiation of differentiation induces apoptosis, which leads to reduced differentiation. SRSF2 mutations also cause abnormal differentiation by skewing granulo‐monocytic differentiation toward monocytes, and megakaryo‐erythroid differentiation toward megakaryocytes. Conversely, U2AF1 mutations S34F and Q157R do not affect proliferation and their effects on differentiation are not as dramatic (Supporting Information Fig. S13). Like SRSF2 mutations, U2AF1 mutations also skew granulo‐monocytic differentiation toward monocytes but have the opposite effect on megakaryo‐erythroid differentiation. They cause a decrease in generation of megakaryocytes, which is accompanied by an accumulation of the erythroid population.
Figure 7.
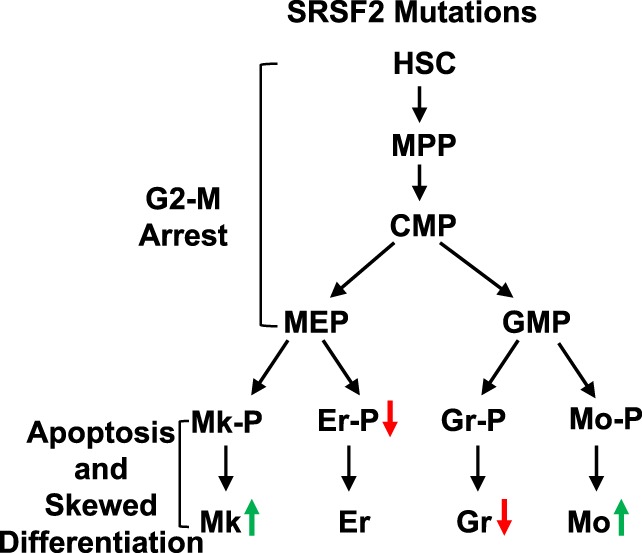
Mutations in SRSF2 cause abnormal myeloid differentiation. Schematic representation of defects in proliferation and differentiation that occur due to expression of SRSF2 mutations in human CD34+ hematopoietic stem and progenitor cells. The red and green arrows indicate a decrease and an increase in the fraction of the indicated population, respectively. Abbreviations: CMP, common myeloid progenitor; Er, erythrocyte; Er‐P, erythroid precursor; GMP, granulocyte–monocyte progenitor; Gr, granulocyte. Gr‐P, granulocyte precursor; HSC, hematopoietic stem cell; MEP, megakaryocyte‐erythrocyte progenitor; Mk, megakaryocyte; Mk‐P, megakaryocyte precursor; Mo, monocyte; Mo‐P, monocyte precursor; MPP, multipotent progenitor.
In the case of SRSF2 mutations, our observations on differentiation defects are in agreement with studies of knock‐in mouse models. In transplantation experiments, mice injected with SRSF2‐P95H mutant HSCs exhibited impaired erythropoiesis compared to mice carrying the WT HSCs 23. These recipient mice also exhibited increase in bone marrow megakaryocytes. However, the U2AF1 mutations induced defects in the lineage populations are not as impactful and appear to be distinct from those reported in other studies using knock‐in mice and human CD34+ model system 16, 19. These variations could be caused by the differences in culture conditions including the presence of serum, stroma, and combinations of cytokines. The study by Yip et al. used separate cultures for granulo‐monocytic and erythroid differentiation 19. In their study, U2AF1‐S34F was found to skew differentiation toward granulocytes in granulo‐monocytic cultures and cause a block in differentiation in erythroid cultures. However, in our analyses, cells were exposed to a cocktail of cytokines that promotes differentiation along all myeloid lineages. The critical role of hematopoietic cytokines in lineage commitment during hematopoiesis is well characterized 56, 57. Studies using other leukemia models have shown that variations in cytokine composition can impact phenotypic outcome 58. Therefore, further investigations of the role of cytokine milieu and culture conditions such as hypoxia and serum will be necessary to delineate the correlation between microenvironment and lineage outcomes upon expression of splicing factor mutations.
In CD34+ cells, SRSF2 mutation‐induced alterations were observed in pre‐mRNA splicing, but not in the use of polyadenylation sites in 3′‐UTRs. Interestingly, the SRSF2 mutations also did not induce significant changes in the gene expression profile of the CD34+ cells. This likely reflects large background variations in the transcriptome of the donor CD34+ cells. In accordance with previous reports on SRSF2 mutation‐induced splicing changes, the splicing alterations we have observed in CD34+ cells are few and their magnitude of change is small. Several of the splicing alterations identified in CD34+ cells overlap with the changes seen in individuals with AML and CMML. These overlapping splicing alterations also persist in the K052 acute myeloblastic leukemia cells that harbor the SRSF2‐P95H mutation. Thus, these leukemia cells lines may be useful for delineating the pathways downstream of SRSF2 mutation‐induced splicing alterations. It has been proposed that the phenotypic manifestations in disease may be due to cumulative effects of several small changes in splicing 20. Studies examining modulation of U2AF1‐S34F induced splicing alterations in human CD34+ cells support this hypothesis. It was demonstrated that expression of the normal myelopoiesis associated isoforms of H2AFY and STRAP could partially reverse abnormalities in erythroid differentiation 19, thereby, supporting the idea that multiple splicing aberrations may be involved in the development of the disease phenotype. Notably, alterations in splicing of H2AFY and STRAP mRNAs were not detected in response to SRSF2 mutations. Thus, this supports the conclusion that phenotypic effects of mutations of SRSF2 and U2AF1 are mediated via different mechanisms. This lack of a set of commonly altered splice patterns in response to different splicing gene mutations is fueling another theory that leukemogenesis might involve non‐splicing functions of the mutant proteins 59. These non‐splicing roles may be in 3′‐end processing of mRNAs or in resolution of R‐loops during transcription 24, 25. SRSF2 and U2AF1 mutation‐induced defects in resolution of R‐loops in conjunction with a G2‐M arrest have been reported to occur in HEK293T cells and have been proposed as a “unifying” mechanism in myelodysplasia 25. It is not clear if R‐loops accumulate in human HSPCs expressing these mutations, as U2AF1 mutations were not found to affect proliferation. The role of the R‐loop mechanism, if any, in induction of the phenotypic effects observed here, clearly requires further investigation.
The dramatic induction of G2‐M arrest elicited by SRSF2 mutations appears to occur specifically in the human CD34+ HSPCs. Although, we did not find any adverse effects, SRSF2‐P95H induced inhibition of proliferation and G2‐M arrest in HEK293T cells was reported by others 25. These deleterious effects did not occur in mouse models 17, 23. This G2‐M arrest may be key to survival and adaptation of the HSPCs harboring SRSF2 mutations in leukemia and in preleukemic condition such as clonal hematopoiesis of indeterminate potential that has been identified as a strong risk factor for hematological malignancies 60. Delineating the triggers or conditions that allow reentry of the G2‐M arrested cells into the cell cycle may help to delineate the pathways that are involved in transformation of the CD34+ HSPCs.
SRSF2 mutation‐induced apoptosis of CD34− cells is highly reminiscent of increased apoptosis seen in MDS cells 61. In mouse knock‐in experiments, analysis of the influence of SRSF2 mutations on apoptosis has yielded conflicting results. In one study that used Mx1‐cre SRSF2 WT and P95H mutant mice, the SRSF2‐P95H mutation was reported to evoke an increase in apoptotic LSK cells in mutant mice over that in WT littermates 17. Yet a separate investigation in which the Vav‐cre system was used did not report any changes in the frequency of apoptotic LT‐HSCs or LSK cells in the mutant mice in comparison to WT 23. The differences in SRSF2 mutation‐induced apoptotic responses of CD34+ and CD34− cells and lack of such effects in K562 erythroleukemia cells suggests that the mutations have cell context‐specific effects. Therefore, the mutations in SRSF2 evaluated herein differentially affect the HSPCs and more committed CD34− precursor populations. Understanding the effects of such mutations on cell population hierarchies will be important in characterizing the molecular changes and intracellular pathways that are specifically involved in these compartments.
Summary
In summary, this study demonstrates that introduction of myeloid disease mutations of splicing factor SRSF2 have adverse effects on human myelopoiesis. SRSF2 mutations inhibit expansion of HSPCs by causing a G2‐M arrest and induce apoptosis upon initiation of differentiation. These mutations also skew myeloid differentiation. The RNA‐Seq analysis has identified several SRSF2‐mutation‐induced alterations in splicing of genes that overlap with targets that have been reported to be altered in patients with AML and CMML. Defining the role of these splicing alterations in proliferation and differentiation of HSPCs will provide insight into mechanisms of pathogenesis and lead to development and characterization of therapeutic targets.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supporting Information Figures
Supporting Information Tables
Supporting Information Supporting File S1
Supporting Information Supporting File S2
Supporting Information Supporting File S3
Supporting Information Supporting File S4
Supporting Information Supporting File S5
Supporting Information Supporting File S6
Supporting Information Supporting File S7
Acknowledgments
S.S. would like to thank Drs. Mark Haussler, Carol Haussler, Daruka Mahadevan, and Aparna Sertil for scientific comments. This work was made possible by support from the Flow Cytometry Core Facilities at the University of Arizona College of Medicine—Phoenix (directed by Dr. Mrinalini Kala) and Broad Stem Cell Research Center at University of California, Los Angeles (UCLA), and by the Arizona Cord Blood Program and the UCLA Center for AIDS Research Virology Core Lab (grant 5P30 AI028697). This work was supported in part by funds to S.S. from the National Institutes of Health/National Cancer Institute (R21CA170786), the American Cancer Society (the Institutional Research Grant 74‐001‐34‐IRG), and Arizona Area Health Education Centers (AzAHEC) program, funds to P.S. from Department of Defense Breast Cancer Research program (BC142286), and to C.S. from National Institutes of Health/National Center for Advancing Translational Science (KL2TR001882) and UCLA Broad Stem Cell Research Center clinical fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the AzAHEC.
Author Contributions
A.B., N.K., W.M., P.K., C.S., J.A.R., P.S., and S.S performed experiments and analyzed the data. C.H. and G.M.C. provided study material and contributed to discussions. A.B. and S.S. prepared the manuscript. S.S. conceived and supervised the study. All authors discussed results, edited the manuscript, and gave final approval of the manuscript.
References
- 1. Papaemmanuil E, Gerstung M, Bullinger L et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Malcovati L, Papaemmanuil E, Ambaglio I et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood 2014;124:1513–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartels S, Lehmann U, Busche G et al. SRSF2 and U2AF1 mutations in primary myelofibrosis are associated with JAK2 and MPL but not calreticulin mutation and may independently reoccur after allogeneic stem cell transplantation. Leukemia 2015;29:253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshida K, Sanada M, Shiraishi Y et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478:64–69. [DOI] [PubMed] [Google Scholar]
- 5. Patnaik MM, Lasho TL, Finke CM et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol 2013;88:201–206. [DOI] [PubMed] [Google Scholar]
- 6. Thol F, Kade S, Schlarmann C et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012;119:3578–3584. [DOI] [PubMed] [Google Scholar]
- 7. Itzykson R, Kosmider O, Renneville A et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121:2186–2198. [DOI] [PubMed] [Google Scholar]
- 8. Papaemmanuil E, Gerstung M, Malcovati L et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013;122:3616–3627. quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fei DL, Motowski H, Chatrikhi R et al. Wild‐type U2AF1 antagonizes the splicing program characteristic of U2AF1‐mutant tumors and is required for cell survival. PLoS Genet 2016;12:e1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee SC, Dvinge H, Kim E et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 2016;22:672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meggendorfer M, Roller A, Haferlach T et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012;120:3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alsafadi S, Houy A, Battistella A et al. Cancer‐associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 2016;7:10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brooks AN, Choi PS, de Waal L et al. A pan‐cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 2014;9:e87361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ilagan JO, Ramakrishnan A, Hayes B et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 2015;25:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Madan V, Kanojia D, Li J et al. Aberrant splicing of U12‐type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 2015;6:6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shirai CL, Ley JN, White BS et al. Mutant U2AF1 expression alters hematopoiesis and pre‐mRNA splicing in vivo. Cancer Cell 2015;27:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim E, Ilagan JO, Liang Y et al. SRSF2 mutations contribute to myelodysplasia by mutant‐specific effects on exon recognition. Cancer Cell 2015;27:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kesarwani AK, Ramirez O, Gupta AK et al. Cancer‐associated SF3B1 mutants recognize otherwise inaccessible cryptic 3′ splice sites within RNA secondary structures. Oncogene 2017;36:1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yip BH, Steeples V, Repapi E et al. The U2AF1S34F mutation induces lineage‐specific splicing alterations in myelodysplastic syndromes. J Clin Invest 2017;127:2206–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J, Lieu YK, Ali AM et al. Disease‐associated mutation in SRSF2 misregulates splicing by altering RNA‐binding affinities. Proc Natl Acad Sci USA 2015;112:E4726–E4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okeyo‐Owuor T, White BS, Chatrikhi R et al. U2AF1 mutations alter sequence specificity of pre‐mRNA binding and splicing. Leukemia 2015;29:909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Przychodzen B, Jerez A, Guinta K et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 2013;122:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kon A, Yamazaki S, Nannya Y et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 2017;131:621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park SM, Ou J, Chamberlain L et al. U2AF35(S34F) promotes transformation by directing aberrant ATG7 pre‐mRNA 3′ end formation. Mol Cell 2016;62:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L, Chen JY, Huang YJ et al. The augmented R‐loop is a unifying mechanism for myelodysplastic syndromes induced by high‐risk splicing factor mutations. Mol Cell 2018;69:412–425. e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen HD, Yadav T, Giri S et al. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol Cell 2017;65:832–847. e834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mazumdar C, Shen Y, Xavy S et al. Leukemia‐associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 2015;17:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kennedy JA, Barabe F. Investigating human leukemogenesis: From cell lines to in vivo models of human leukemia. Leukemia 2008;22:2029–2040. [DOI] [PubMed] [Google Scholar]
- 29. Qin H, Malek S, Cowell JK et al. Transformation of human CD34+ hematopoietic progenitor cells with DEK‐NUP214 induces AML in an immunocompromised mouse model. Oncogene 2016;35:5686–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chung KY, Morrone G, Schuringa JJ et al. Enforced expression of an Flt3 internal tandem duplication in human CD34+ cells confers properties of self‐renewal and enhanced erythropoiesis. Blood 2005;105:77–84. [DOI] [PubMed] [Google Scholar]
- 31. Lin S, Ptasinska A, Chen X et al. A FOXO1‐induced oncogenic network defines the AML1‐ETO preleukemic program. Blood 2017;130:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen SW, Dolnikov A, Passioura T et al. Mutant N‐ras preferentially drives human CD34+ hematopoietic progenitor cells into myeloid differentiation and proliferation both in vitro and in the NOD/SCID mouse. Exp Hematol 2004;32:852–860. [DOI] [PubMed] [Google Scholar]
- 33. Kim D, Langmead B, Salzberg SL. HISAT: A fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen S, Park JW, Lu ZX et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA‐Seq data. Proc Natl Acad Sci USA 2014;111:E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA‐seq data. Genome Res 2012;22:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reyes A, Anders S, Weatheritt RJ et al. Drift and conservation of differential exon usage across tissues in primate species. Proc Natl Acad Sci USA 2013;110:15377–15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 2012;40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liao Y, Smyth GK, Shi W. The subread aligner: Fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res 2013;41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia Z, Donehower LA, Cooper TA et al. Dynamic analyses of alternative polyadenylation from RNA‐seq reveal a 3′‐UTR landscape across seven tumour types. Nat Commun 2014;5:5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szymczak AL, Workman CJ, Wang Y et al. Correction of multi‐gene deficiency in vivo using a single 'self‐cleaving' 2A peptide‐based retroviral vector. Nat Biotechnol 2004;22:589–594. [DOI] [PubMed] [Google Scholar]
- 42. Hao QL, Zhu J, Price MA et al. Identification of a novel, human multilymphoid progenitor in cord blood. Blood 2001;97:3683–3690. [DOI] [PubMed] [Google Scholar]
- 43. Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997;91:661–672. [DOI] [PubMed] [Google Scholar]
- 44. Manz MG, Miyamoto T, Akashi K et al. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA 2002;99:11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parekh C, Sahaghian A, Kim W et al. Novel pathways to erythropoiesis induced by dimerization of intracellular C‐Mpl in human hematopoietic progenitors. Stem Cells 2012;30:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marsee DK, Pinkus GS, Yu H. CD71 (transferrin receptor): An effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol 2010;134:429–435. [DOI] [PubMed] [Google Scholar]
- 47. Dong HY, Wilkes S, Yang H. CD71 is selectively and ubiquitously expressed at high levels in erythroid precursors of all maturation stages: A comparative immunochemical study with glycophorin A and hemoglobin A. Am J Surg Pathol 2011;35:723–732. [DOI] [PubMed] [Google Scholar]
- 48. Lakschevitz FS, Hassanpour S, Rubin A et al. Identification of neutrophil surface marker changes in health and inflammation using high‐throughput screening flow cytometry. Exp Cell Res 2016;342:200–209. [DOI] [PubMed] [Google Scholar]
- 49. Skrdlant L, Stark JM, Lin RJ. Myelodysplasia‐associated mutations in serine/arginine‐rich splicing factor SRSF2 lead to alternative splicing of CDC25C. BMC Mol Biol 2016;17:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Draber P, Stepanek O, Hrdinka M et al. LST1/A is a myeloid leukocyte‐specific transmembrane adaptor protein recruiting protein tyrosine phosphatases SHP‐1 and SHP‐2 to the plasma membrane. J Biol Chem 2012;287:22812–22821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Asencio C, Davidson IF, Santarella‐Mellwig R et al. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell 2012;150:122–135. [DOI] [PubMed] [Google Scholar]
- 52. Chen J, Chen S, Wang J et al. Cyclophilin J is a novel peptidyl‐prolyl isomerase and target for repressing the growth of hepatocellular carcinoma. PLoS One 2015;10:e0127668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Ree JH, Nam HJ, Jeganathan KB et al. Pten regulates spindle pole movement through Dlg1‐mediated recruitment of Eg5 to centrosomes. Nat Cell Biol 2016;18:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu G, Ma Z, Qian J et al. PP4R1 accelerates cell growth and proliferation in HepG2 hepatocellular carcinoma. Onco Targets Ther 2015;8:2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Osipovich AB, Gangula R, Vianna PG et al. Setd5 is essential for mammalian development and the co‐transcriptional regulation of histone acetylation. Development 2016;143:4595–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Metcalf D. Hematopoietic cytokines. Blood 2008;111:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhu J, Emerson SG. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene 2002;21:3295–3313. [DOI] [PubMed] [Google Scholar]
- 58. Wei J, Wunderlich M, Fox C et al. Microenvironment determines lineage fate in a human model of MLL‐AF9 leukemia. Cancer Cell 2008;13:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Joshi P, Halene S, Abdel‐Wahab O. How do messenger RNA splicing alterations drive myelodysplasia? Blood 2017;129:2465–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Genovese G, Kahler AK, Handsaker RE et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kerbauy DB, Deeg HJ. Apoptosis and antiapoptotic mechanisms in the progression of myelodysplastic syndrome. Exp Hematol 2007;35:1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figures
Supporting Information Tables
Supporting Information Supporting File S1
Supporting Information Supporting File S2
Supporting Information Supporting File S3
Supporting Information Supporting File S4
Supporting Information Supporting File S5
Supporting Information Supporting File S6
Supporting Information Supporting File S7