

Abstract
Rationale:
Hypophosphatasia is an inborn error of metabolism that can appear any time in life, mainly with bone manifestations due to low alkaline phosphatase activity. Asfotase alfa is a specific enzyme reposition treatment that has shown promising results in children; however, there are few reports about the outcomes in adult patients.
Patient concerns:
A 36-year-old male presented with an early history of craniosynostosis, short stature, and multiple fractures since the age of 13 years—which needed numerous surgical corrections. He was admitted with a previous diagnosis of osteogenesis imperfecta, taking alendronate, calcium carbonate, cholecalciferol, and calcitriol. Bone mineral density was low (lumbar spine Z-score = −3.0 SD), with impairment of all parameters of high-resolution peripheral quantitative computed tomography (HR-pQCT). Kidney impairment was also observed with reduced creatinine clearance, nephrolithiasis, and nephrocalcinosis.
Diagnosis:
Alkaline phosphatase was unexpectedly low (6 U/L, reference value: 30–120 U/L), with high serum vitamin B6 (260 mcg/L, reference value: 5.2–34.1). Genetic testing showed a homozygous missense mutation in ALPL gene c.443 C>T: p.Thr148Ile.
Intervention:
Asfotase alfa was requested due to important bone deterioration, ambulatory disability, and kidney impairment. It was given subcutaneously 2 mg/kg per dose, 3 times a week, for 12 months before reassessment.
Outcomes:
Bone mineral densities of the lumbar spine and whole body, besides almost all HR-pQCT microstructural parameters of the distal tibia, showed improvements and the patient was able to walk without assistant device. Kidney function did not further deteriorate.
Lessons:
Hypophosphatasia should be considered as a differential diagnosis in young patients with multiple fractures and kidney impairment, since the use of antiresorptive drugs, calcium and vitamin D, commonly used to treat fractures, worsen its symptoms and prognosis. A 12-month asfotase alfa treatment improved bone density and structural parameters even in an adult patient with late diagnosis.
Keywords: alkaline phosphatase, bone, bone remodeling, fractures, hypophosphatasia, nephrolithiasis
1. Introduction
Hypophosphatasia (HPP) is an inborn error of metabolism with a birth prevalence of roughly 1:100,000.[1] It is considered an inherited bone metabolism disorder with mainly bone manifestations that can appear any time in life.[2] There is no specific mutation that promotes this disease; however, mutant alleles have a dominant-negative effect or a loss of effect upon the alkaline phosphatase ALPL gene.[3] The direct consequence of a malfunctioning enzyme is elevated concentration of its substrates; in this case: inorganic pyrophosphate, pyridoxal phosphate, and phosphoethanolamine.[4,5]
HPP has 6 clinical subtypes,[4] but 4 of them are more relevant: perinatal, infantile, juvenile, and adult forms. It is important to highlight that when symptoms appear or remain during adulthood, the diagnosis is more challenging due to its many possible differentials, such as osteogenesis imperfecta, rickets, osteoporosis, chondrocalcinosis, nephrocalcinosis, and nephrolithiasis.
Laboratory findings such as unexplainably low serum alkaline phosphatase activity, elevation of serum pyridoxal-5′-phosphate (vitamin B6) or urinary phosphoethanolamine may support the clinical diagnosis.[6]ALPL gene mutations may also be investigated for confirmation; to date, more than 350 mutations causing hypophosphatasia have been described.[7]
Wrong or late diagnosis is noxious, since treating for differential diagnosis can exacerbate symptoms. Most bone metabolism disorders respond to calcium plus calcitriol supplementation; and bisphosphonates treat conditions like osteoporosis and osteogenesis imperfect; however, these same drugs worsen symptoms and prognosis in hypophosphatasia.[8]
A new specific treatment, asfotase alfa, a human recombinant tissue-nonspecific alkaline phosphatase with a bone-targeting domain, was approved in 2015 in the United States, Japan, and European Union for pediatric patients with hypophosphatasia, regardless of their current age during treatment.[9,10] Asfotase alfa has shown promising results in children,[11] however, there are few reports about the outcomes of the specific treatment using enzyme reposition in adults.[12] Hence, this report aims to describe a case of an adult patient with hypophosphatasia, presenting mainly with fractures, and the therapeutic outcomes 12 months after enzyme reposition.
2. Case report
CC, a 36-year-old male, presented with an early history of craniosynostosis, requiring craniotomy at 12 and 24 months of age. He had always had short stature, with a final height of 130.5 cm. During childhood, there was no history of premature tooth loss; nonetheless, a history of multiple fractures began 23 years prior to admission, after falling from his own height, which led to a left femur neck fracture that required osteosynthesis. Afterward, he fractured the same site twice, needing correction of bone nonunion. Two years later, when he was 15 years old, a knee arthroscopy meant to investigate meniscal damage led to a distal right femur fracture. Twelve years after the last episode, at age 27, a fall from bed caused fractures of the right humerus, right pelvis, and right femur neck, requiring internal fixation of humerus and hip arthroplasty. Since then, his mobility reduced, and he became unable to independently dress himself, go to work and even move around the house. This brief history is depicted in a simplified timeline (Fig. 1).
Figure 1.
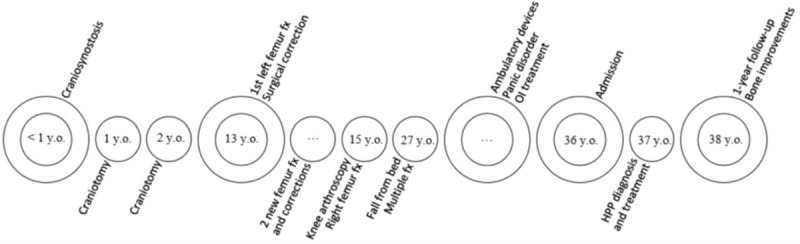
Patient timeline since birth. fx = fracture, HPP = hypophosphatasia, OI = osteogenesis imperfecta, Y.O. = years old.
At the time of admission, the patient had a diagnosis of osteogenesis imperfecta, treating with calcium carbonate, cholecalciferol, calcitriol, and taking psychotropic drugs for panic disorder. He had a history of using alendronate 70 mg weekly for eight years until one year prior to admission.
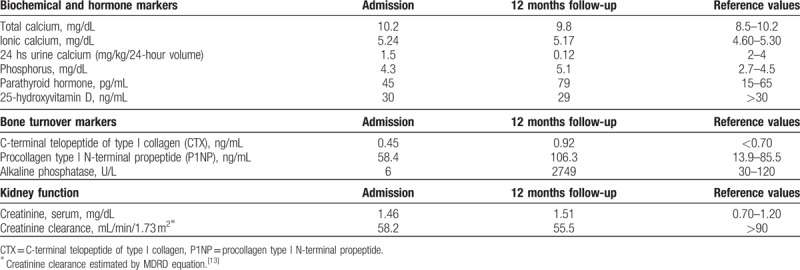
In order to reassess and further evaluate his bone metabolism, a laboratory workup was performed. No abnormalities in serum biochemical and hormone markers were seen. Bone turnover markers were also within normal range, but alkaline phosphatase was unexpectedly low. Additionally, renal function was altered: creatinine clearance estimated by Modification of Diet in Renal Disease (MDRD) equation[13] showed a stage III chronic kidney disease (Table 1).
Table 1.
Initial and follow-up laboratory evaluation.
Common radiographs were performed to evaluate the multiple fractures and orthopedic procedures: evident kyphoscoliosis and vestiges of craniotomy were observed (Fig. 2).
Figure 2.
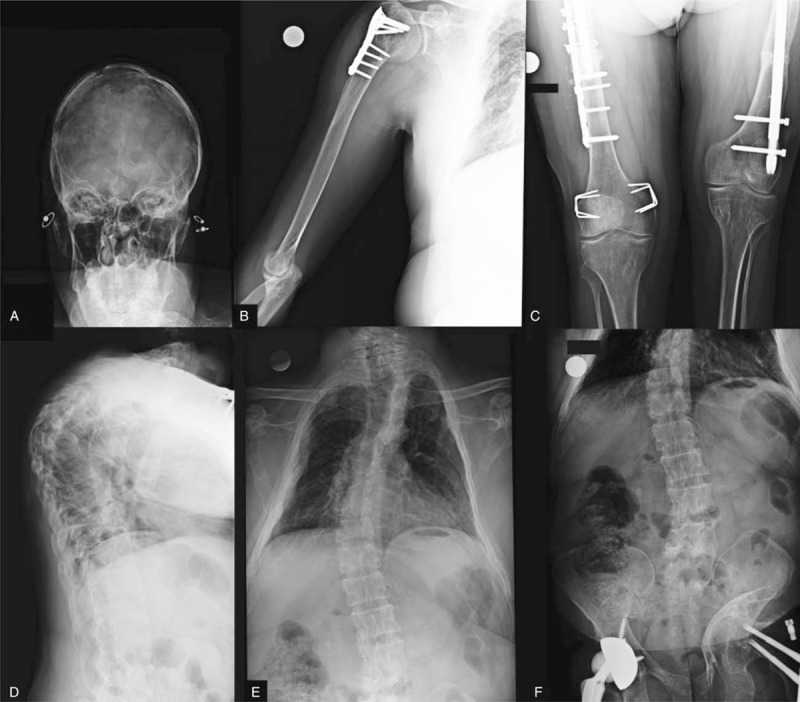
Conventional radiographs. (A) Skull, with signs of craniotomy; (B) right humerus, with proximal screws and plates fixation; (C) femur and knees: bone malunion/nonunion bilaterally despite screws and plates fixations; (D, E) thoracolombar spine [(D) lateral; (E and F): anterior–posterior scans], showing kyphoscoliosis and right total hip arthroplasty.
Bone densitometry was determined by dual energy x-ray absorptiometry (DXA, Hologic QDR 4500A, Bedford, MA) and showed low bone mineral density (BMD) for chronological age, with lumbar spine Z-score of −3.0 SD and a 1/3 distal left radius Z-score of −4.4 SD. Hip and whole body BMD were analyzed, but not initially considered for diagnostic purposes due to the presence of bilateral hip prosthesis, and internal fixations in right knee and humerus.
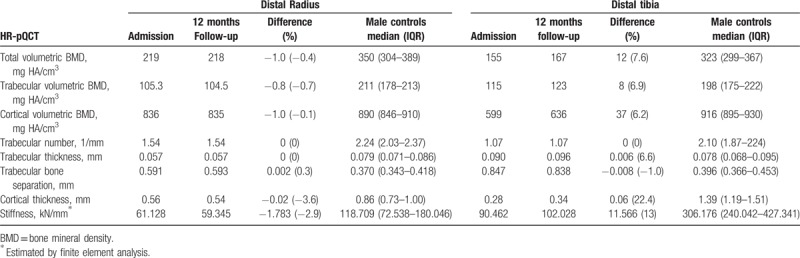
High-resolution peripheral quantitative computed tomography (HR-pQCT, XtremeCT, SCANCO Medical AG, Brüttisellen, Switzerland) was performed to analyze cortical and trabecular bone compartments, including volumetric density, structure parameters, and biomechanical bone strength, and it showed an impairment of them all (Table 2).
Table 2.
Volumetric bone density, structure and strength parameters obtained by high-resolution peripheral quantitative computed tomography (HR-pQCT), compared at admission, 12 months follow-up, and with healthy male controls 30–39 years old.
Kidney ultrasound showed chronic parenchymatous nephropathy with nephocalcinosis, bilateral nonobstructive nephrolithiasis with parietal calcifications, and stones ranging from 0.6 to 0.8 cm, and cysts on the right kidney.
Continuous evaluation proceeded: serum vitamin B6 260 mcg/L (reference value: 5.2–34.1), alkaline phosphatase persisted low: 12 U/L (reference value: 30–120). Genetic testing (Centro de Genomas, Sao Paulo, SP, Brazil) showed a homozygous missense mutation in ALPL gene c.443 C>T: p.Thr148Ile, which has already been identified in other patients with hypophosphatasia.[14] A probably benign heterozygous mutation was also found in the ALPL gene: c.1190-266_1190-251del16 (rs 145806416). The only available relative who accepted to be tested was his mother, who presented the same missense mutation in ALPL gene c.443 C>T: p.Thr148Ile, in heterozygosity.
After confirming the diagnosis, specific treatment with enzyme reposition (asfotase alfa—2 mg/kg/dose, 3 times a week) was requested to slow disease progression,[15] aiming for an improvement especially in bone and motor functions.[16] Twelve months after treatment, a new bone mineral densitometry was performed and showed significant improvements in lumbar spine and whole body BMDs (Fig. 3), since the least significant change in our laboratory is 0.023 g/cm2 at lumbar spine and 0.022 g/cm2 for the whole body scan. A bone microarchitecture evaluation was also repeated (Table 2). There was an improvement in almost every parameter in the distal tibia, with exception to trabeculae number and trabecular bone separation. No significant changes were noted in the distal radius parameters, considering the coefficient of variation (CV) of HR-pQCT from our laboratory (distal tibia: density CV = 0.25%–1.16%, structure CV = 0.78%–6.35%; distal radius: density CV = 0.93%–1.41%, structure CV = 0.78%–6.35%) (Table 2).[17]
Figure 3.
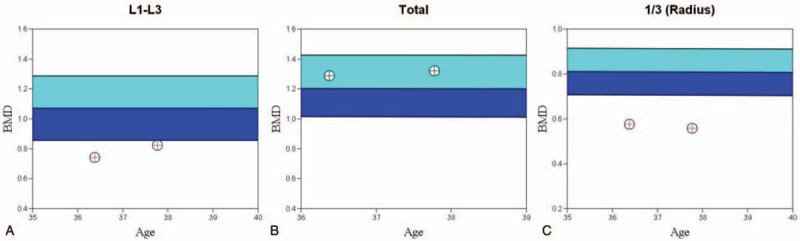
Graphs showing temporal evolution of bone mineral density. (A) Lumbar spine BMD (L1–L3): 0.742 to 0.822 g/cm2 (0.08 g/cm2, or 10.8% increase). (B) Whole body BMD: 1.287 to 1.320 g/cm2 (0.033 g/cm2, or 2.5% increase). (C) Left proximal third distal radius BMD: 0.575 to 0.559 g/cm2 (−0.017 g/cm2 or 2.9% decrease). BMD = bone mineral density.
3. Discussion
It is important to draw attention to hypophosphatasia as a differential diagnosis of multiple fractures as the main clinical manifestation in an adult patient since childhood. Osteogenesis imperfecta, which is another relevant differential diagnosis, was ruled out because the patient also presented with craniosynostosis and kidney impairment. Moreover, the remarkably low levels of alkaline phosphatase observed pointed toward hypophosphatasia. However, in this patient, the low levels of this enzyme could be due to bisphosphonate chronic use, since this drug inhibits bone turnover.[18] The fact that other bone turnover markers, as C-terminal telopeptide of type I collagen (CTX) and procollagen type I N-terminal propeptide (P1NP), were at normal ranges in addition to the finding of elevated vitamin B6 strongly suggested hypophosphatasia as the main diagnosis,[2,6,19,20] which was confirmed by genetic testing.
Hence, bisphosphonates, calcium, and vitamin D supplementation were discontinued, since these drugs worsen HPP: without proper alkaline phosphatase function, there can be no calcium and phosphorus mineralization. Therefore, providing more calcium and vitamin D leads to its build up specially in the kidneys, worsening nephrocalcinosis and nephrolithiasis.[4] Vitamin D supplementation may also lead to hypercalcemia and increase calciuria.[21] Regarding bisphosphonates, they worsen bone mineralization, which is already compromised, leading to even more severe bone fragility.[22,23] Furthermore, tissue nonspecific alkaline phosphatase can be inhibited by N-containing bisphosphonates[24] and may increase atypical fractures even in carriers of heterozygous mutations.[25] Indeed, hypophosphatasia should be considered as a differential diagnosis of atypical fractures caused by bone mineralization deterioration due to bisphosphonate chronic use.
Considering the patient's important ambulatory disability due to multiple fractures, which could worsen with disease progression, treatment with enzyme reposition was recommended based on literature.[6,12] Enzyme replacement is achieved with asfotase alfa, a human recombinant tissue-nonspecific alkaline phosphatase. Its modifications from the original molecule include an IgG Fc domain, and a repetitive C-terminal extension of 10 Asp residues, which was designed to have a high affinity for hydroxyapatite crystals (e.g., a bone-targeting domain). Once in the bone, asfotase alfa assembles into an enzymatic active tetramer capable of hydrolysing inorganic pyrophosphate, the same role human tissue-nonspecific alkaline phosphatase plays in the bone. It is hypothesized that asfotase alfa, as alkaline phosphatase, regulates bone mineralization by regulating inorganic pyrophosphate concentrations, which are both a substrate for hydroxyapatite crystal formation and an inhibitor of its nucleation and growth.[26]
An improvement of areal/volumetric bone mineral density, microarchitecture parameters and biomechanical proprieties at distal tibia and stabilization of these parameters at distal radius were observed after 12 months of treatment (Table 2 and Fig. 4), which was also associated with a better quality of life, since the patient was able to walk without assistant devices.
Figure 4.
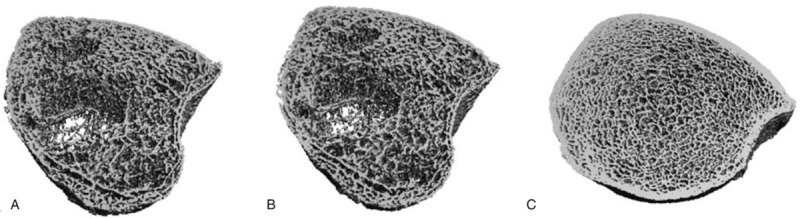
High-resolution peripheral quantitative computed tomography images of distal tibia. (B) patient at admission. (B) patient after treatment for 12 months (B) age- and gender-matched control.
In conclusion, unexpectedly or persistently low values of alkaline phosphatase with bone and systemic manifestations should draw attention to hypophosphatasia as an important diagnosis. This disease has specific treatment and the late diagnosis and wrong treatment, mainly with antiresorptive drugs and calcium plus vitamin D supplementation, commonly used to treat many of HPP's differentials diagnoses, could increase morbidity and lead to mortality. Additionally, considering its difficult diagnosis and complex management, patients with HPP should be referred to tertiary or academic medical centers, which can provide specialists in metabolic bone disorders and easier access to newer treatments.[20]
Recently, asfotase alfa treatment showed promising results in children, and this case report, as other researches,[12,27] boost evidence that it may be successful also in adult patients with hypophosphatasia.
4. Patient consent statement
Informed written consent was obtained from the patient for publication of this case report and accompanying images.
Author contributions
Conceptualization: Rosa Maria Rodrigues Pereira.
Funding acquisition: Rosa Maria Rodrigues Pereira.
Investigation: Thiago Quadrante Freitas, Andre Silva Franco, Rosa Maria Rodrigues Pereira.
Project administration: Rosa Maria Rodrigues Pereira.
Supervision: Rosa Maria Rodrigues Pereira.
Writing – original draft: Thiago Quadrante Freitas, Andre Silva Franco, Rosa Maria Rodrigues Pereira.
Writing – review & editing: Thiago Quadrante Freitas, Andre Silva Franco, Rosa Maria Rodrigues Pereira.
Footnotes
Abbreviations: ALPL = alkaline phosphatase, liver/bone/kidney, BMD = bone mineral density, CTX = C-terminal telopeptide of type I collagen, CV = coefficient of variation, HPP = hypophosphatasia, HR-pQCT = high-resolution peripheral quantitative computed tomography, MDRD = modification of diet in renal disease, P1NP = procollagen type I N-terminal propeptide, SD = standard deviation.
Funding information: This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico # 301805/2013-0, #305556/2017-7 to RMRP and Federico Foundation (RMRP).
The authors have no conflicts of interest to disclose.
References
- [1].Fraser D. Hypophosphatasia. Am J Med 1957;22:730–46. [DOI] [PubMed] [Google Scholar]
- [2].Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev 2013;10suppl 2:380–8. [PubMed] [Google Scholar]
- [3].Mornet E. Molecular genetics of hypophosphatasia and phenotype–genotype correlations. Subcell Biochem 2015;76:25–43. [DOI] [PubMed] [Google Scholar]
- [4].Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep 2016;14:95–105. [DOI] [PubMed] [Google Scholar]
- [5].Buchet R, Millán JL, Magne D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol 2013;1053:27–51. [DOI] [PubMed] [Google Scholar]
- [6].Shapiro JR, Lewiecki EM. Hypophosphatasia in adults: clinical assessment and treatment considerations. J Bone Miner Res 2017;32:1977–80. [DOI] [PubMed] [Google Scholar]
- [7].Mornet E. The Tissue Nonspecific Alkaline Phosphatase Gene Mutations Database. Available at: http://www.sesep.uvsq.fr/03_hypo_mutations.php Accessed March, 2018. [Google Scholar]
- [8].Whyte MP. Hypophosphatasia—aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2016;12:233–46. [DOI] [PubMed] [Google Scholar]
- [9].Pharmaceuticals A. StrensiqTM (asfotase alfa) injection, for subcutaneous use. US prescribing information. Available at: http://alxn.com/Documents/strensiq_pi-10-2015.aspx Published 2015. Accessed April 4, 2018. [Google Scholar]
- [10].Scott LJ. Asfotase alfa in perinatal/infantile-onset and juvenile-onset hypophosphatasia: a guide to its use in the USA. BioDrugs 2016;30:41–8. [DOI] [PubMed] [Google Scholar]
- [11].Scott LJ. Asfotase alfa: a review in paediatric-onset hypophosphatasia. Drugs 2016;76:255–62. [DOI] [PubMed] [Google Scholar]
- [12].Remde H, Cooper MS, Quinkler M. Successful asfotase alfa treatment in an adult dialysis patient with childhood-onset hypophosphatasia. J Endocr Soc 2017;1:1188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levey AS, Coresh J, Greene T, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med 2006;145:247–54. [DOI] [PubMed] [Google Scholar]
- [14].Spentchian M, Merrien Y, Herasse M, et al. Severe hypophosphatasia: characterization of fifteen novel mutations in the ALPL gene. Hum Mutat 2003;22:105–6. [DOI] [PubMed] [Google Scholar]
- [15].Orimo H. Pathophysiology of hypophosphatasia and the potential role of asfotase alfa. Ther Clin Risk Manag 2016;12:777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Whyte MP. Hypophosphatasia: an overview For 2017. Bone 2017;102:15–25. [DOI] [PubMed] [Google Scholar]
- [17].Lima GL, Paupitz JA, Aikawa NE, et al. A randomized double-blind placebo-controlled trial of vitamin D supplementation in juvenile-onset systemic lupus erythematosus: positive effect on trabecular microarchitecture using HR-pQCT. Osteoporos Int 2017;29:587–94. [DOI] [PubMed] [Google Scholar]
- [18].Glendenning P, Paul Chubb SA, Vasikaran S. Clinical utility of bone turnover markers in the management of common metabolic bone diseases in adult. Clin Chim Acta 2018;481:161–70. [DOI] [PubMed] [Google Scholar]
- [19].Millán JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int 2016;98:398–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Whyte MP. Hypophosphatasia: enzyme replacement therapy brings new opportunities and new challenges. J Bone Miner Res 2017;32:667–75. [DOI] [PubMed] [Google Scholar]
- [21].Cranney A, Horsley T, O’Donnell S, et al. Effectiveness and safety of vitamin D in relation to bone health. Evid Rep Technol Assess (Full Rep) 2007;158:1–235. [PMC free article] [PubMed] [Google Scholar]
- [22].2017;Marini F, Brandi ML. Atypical femur fractures: a distinctive tract of adult hypophosphatasia. 14:324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].2018;Allen MR, Allen MR. Recent advances in understanding bisphosphonate effects on bone mechanical properties. 16:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vaisman DN, McCarthy AD, Cortizo AM. Bone-specific alkaline phosphatase activity is inhibited by bisphosphonates: role of divalent cations. Biol Trace Elem Res 2005;104:131–40. [DOI] [PubMed] [Google Scholar]
- [25].Sutton RAL, Mumm S, Coburn SP, et al. “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res 2012;27:987–94. [DOI] [PubMed] [Google Scholar]
- [26].Millán JL, Narisawa S, Lemire I, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res 2008;23:777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kitaoka T, Tajima T, Nagasaki K, et al. Safety and efficacy of treatment with asfotase alfa in patients with hypophosphatasia: Results from a Japanese clinical trial. Clin Endocrinol (Oxf) 2017;87:10–9. [DOI] [PubMed] [Google Scholar]