

Abstract
Biotechnological strategies using renewable materials as starting substrates are a promising alternative to traditional oleochemical processes for the isolation of different fatty acids. Among them, long chain mono‐unsaturated fatty acids are especially interesting in industrial lipid modification, since they are precursors of several economically relevant products, including detergents, plastics and lubricants. Therefore, the aim of this study was to develop an enzymatic method in order to increase the percentage of long chain mono‐unsaturated fatty acids from Camelina and Crambe oil ethyl ester derivatives, by using selective lipases. Specifically, the focus was on the enrichment of gondoic (C20:1 cisΔ11) and erucic acid (C22:1 cisΔ13) from Camelina and Crambe oil derivatives, respectively. The pursuit of this goal entailed several steps, including: (i) the choice of a suitable lipase scaffold to serve as a protein engineering template (Candida antarctica lipase A); (ii) the identification of potential amino acid targets to disrupt the binding tunnel at the adequate location; (iii) the design, creation and high‐throughput screening of lipase mutant libraries; (iv) the study of the selectivity towards different chain length p‐nitrophenyl fatty acid esters of the best hits found, as well as the analysis of the contribution of each amino acid change and the outcome of combining several of the aforementioned residue alterations and, finally, (v) the selection and application of the most promising candidates for the fatty acid enrichment biocatalysis. As a result, enrichment of C22:1 from Crambe ethyl esters was achieved either, in the free fatty acid fraction (wt, 78%) or in the esterified fraction (variants V1, 77%; V9, 78% and V19, 74%). Concerning the enrichment of C20:1 when Camelina oil ethyl esters were used as substrate, the best variant was the single mutant V290W, which doubled its content in the esterified fraction from approximately 15% to 34%. A moderately lower increase was achieved by V9 and its two derived triple mutant variants V19 and V20 (27%).
Keywords: Biocatalysis, Candida antarctica lipase A, lipid modification, long chain mono-unsaturated fatty acid enrichment, semi-rational protein design
Introduction
Fatty acids (FA) and their alkyl‐derivatives are interesting building blocks for their application in oleochemistry.1 Particularly, long‐chain FA have been used in industry for the production of surfactants, erucamide as polymer building block, and lubricants.2 In order to obtain these valuable compounds, natural occurring oils can be utilized as an environmentally friendly alternative to fossil raw materials. In this context, plant oils are of great interest not only because they come from renewable resources, but also due to the possibility of modifying the oil composition to fit the desired necessities by new plant breeding and gene modification technologies.3 Regardless of the aforementioned strategies, enrichment of a desirable FA from oil can be further pursued by classical separation technologies, like distillation or crystallization. However, such classical techniques work with the implementation of harsh conditions like high pressure, high temperature and the usage of polluting agents like organic solvents.4 Moreover, the isolation in high yields and purities of a desired compound employing these classical separation technologies is often not sufficient for direct downstream processing. An attractive alternative is the application of enzymes for the enrichment of certain FA, as this represents an eco‐friendly and often more efficient alternative.5 In this sense some of the features that biocatalysts offer involve the possibility of working under mild reaction conditions or their high chemo‐, stereo‐ and regioselectivity when using both, natural as well as unnatural substrates.
Unsurprisingly, the most employed enzymes in lipid modification are lipases (EC 3.1.1.3). These proteins belong to the α/β‐hydrolase fold family and naturally catalyze the hydrolysis of triacylglycerides (TAG) into glycerol and free FA (FFA), displaying particular chemo‐, regio‐ and stereoselectivity.6 For that reason the use of lipases has been widely described for the enrichment of, for instance, long‐chain poly‐unsaturated FA from fish oil,7 erucic acid from Cruciferae plant oils,4,8 and trans‐FA from frying oils.9 Additionally, protein engineering has revealed itself as an important tool to overcome the limitations displayed by these enzymes, such as reduced enzymatic efficiency and stability under challenging industrial conditions, or to enhance the enzyme selectivity towards different substrates. In this sense, several examples of its implementation in the improvement of selectivity were reported for lipases from Rhizopus oryzae,10 Candida rugosa,11 Moesziomyces antarcticus (formerly: Pseudozyma/Candida antarctica).9b,12 Although both, directed evolution and rational design methodologies can be applied, some lipases display an a priori favorable feature from the (semi)rational design point of view: the unique architecture of their substrate binding site, which forms a tunnel‐like cavity that fits the acyl chain of the FA.13 Thus, amino acids surrounding this region are interesting targets for rational design approaches, since altering these residues could modify the tunnel's shape and, therefore, vary the lipase FA selectivity. This concept has been proven for lipase 1 from C. rugosa 11 and lipase A from M. antarcticus 9b,12a,14 for enhancing selectivity towards medium chain or trans fatty acids.
Consequently, the aim of the present study is the enrichment of mono‐unsaturated long chain fatty acids from plant oils by an improved lipase. More specifically, we focused on gondoic and erucic acids, which are present in high contents in Camelina sativa and Crambe abyssinica seed oils, respectively. Separation of these FA from the other FA present in the oil is challenging, due to the high content of C18‐FA of different saturation levels, which are highly similar in their physical properties to the mentioned mono‐unsaturated fatty acids (MUFAs). Within a screening of commercially available lipases and the knowledge about already described lipases in the literature, CAL‐A was chosen as best option to accomplish this task by engineering its tunnel‐type binding site,15 even though the wild type enzyme does not display hydrolytic preference for the targeted FA.16
Results and Discussion
In order to achieve the isolation of erucic and gondoic acids, a variant capable of efficiently discern and selectively remove FA depending on their chain length was required. This goal could be reached following two strategies: (i) by preferentially hydrolyzing FA longer than C18 and therefore causing their accumulation as FFA; or to the contrary, (ii) by hampering the ester bond cleavage of those FA, thus increasing their concentration in the ester‐bound fraction as the shorter FA are released (Scheme 1). Given the characteristics of both, the substrates and the structure of the enzyme, the second approach was chosen as it was deemed the most straightforward strategy.
Scheme 1.
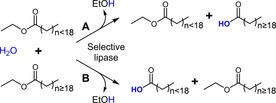
Possible approaches for enrichment of long chain FA either as FFA (A) or esters (B) catalyzed by a selective lipase. In this example fatty acid ethyl esters derived from the plant oils were used as substrates.
Semi‐rational Design
Selecting the Target Residues
The CAL‐A structure exhibits a classic α/β hydrolase fold, containing the catalytic triad (S184, D334 and H366), as well as a well‐defined lid domain, which takes part in forming a long (∼30 Å) substrate binding pocket.15 Both, the binding pocket and the catalytic site, are deeply buried in the enzyme and covered by a motile flap17 (Figure 1). Hence, to adopt the active state, CAL‐A has to undergo a conformational change, in which the flap domain moves away, giving the substrate access to the catalytic centre and the binding site.
Figure 1.
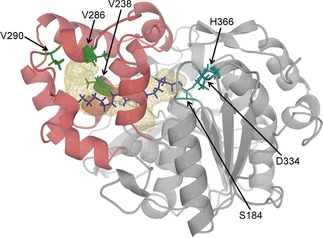
CAL‐A crystal structure (PDB: 3GUU) showing the lipase in its inactive state. The docked C18:1 fatty acid (purple) is used to illustrate its probable location in the CAL‐A wt binding tunnel (wireframe). The tunnel is mainly formed by the six consecutive helices of the cap domain (shown in dark red). Amino acid residues V238, V286 and V290 (green) are facing the tunnel and placed nearby the terminal carbon (Cω) of the FA. The active site residues are highlighted in blue.
In order to create CAL‐A variants able to discriminate FA longer than C18, the structural conformation of the acyl‐binding tunnel was analyzed. The aim was the identification of potential amino acid targets located at a position where the blocking of the binding tunnel would allow the hydrolysis of fatty acids with 18 carbon atoms or shorter, while preventing the hydrolysis of longer acyl groups.
Consequently, the crystal structure of CAL‐A (PDB: 3GUU) was used for docking studies with oleic acid (C18:1 cisΔ9), gondoic acid (C20:1 cisΔ11), and erucic acid (C22:1 cisΔ13). The following residues facing the tunnel in the surroundings of the oleic acid‘s terminal carbon (Cω) were identified as possible hotspots: V238, V286, and V290 (Figure 1). When in silico substitution of these residues by bulkier amino acids was performed, it did not result in a full blockage of the tunnel. Nonetheless, synergistic effects (which are hard to predict) at these positions appeared promising to accomplish this goal.
Mutagenesis Strategy and Library Generation
In order to test this hypothesis experimentally, saturation mutagenesis of each chosen position with all possible amino acids should be performed. Furthermore, to study the potential synergistic effects when altering different amino acids, more than one position must be saturated simultaneously in the created library. However, saturation mutagenesis at all three positions at the same time by using NNN degenerate primers would lead to a huge library of ∼800,000 clones to cover 95% of all possible amino acid combinations.18 A more reasonable alternative would be to reduce the number of generated clones by decreasing the number of codons introduced, thus creating a “small, but smart” mutagenesis library. However, even when choosing this approach the number of clones to be screened would be 24,000 or 32,000, depending on the strategy used (Table S1).19 Therefore, to reach a compromise between a library size experimentally feasible to be screened and exploring all possible amino acid combinations, the 22c‐trick19b was chosen to obtain three mutant libraries where only two positions were saturated at the same time. Hence, the created libraries were: library I (V238X and V286X), library II (V238X and V290X) and library III (V286X and V290X). This resulted in a drastic reduction of the necessary screening effort to 1,450 clones per library. Moreover, further combinations of the best amino acid substitutions observed could be subsequently explored for a better blockage of the binding pocket.
High‐Throughput Screening
The three pET22‐plasmid based mutant libraries created were transformed into E. coli BL21 (DE3) C41 and screened on the robotic platform LARA (laboratory automation robotic assistant).20 The process started by inoculating each well of a 96‐well microtiter plate with a single colony obtained after transforming the E. coli cells with the corresponding mutant library. Subsequently, protein variants were overexpressed in the microtiter plate wells, harvested and the selectivity assay was performed. Fortunately, when CAL‐A variants were overexpressed, their accumulation in the periplasm caused a self‐lysis of the cells resulting in the release of the lipase variant to the supernatant as it was previously described.9b,16 This fact allowed us to use this culture fraction instead of the cell free extract for the hydrolytic activity measurements. Consequently, the supernatant was used in three separate p‐nitrophenol (pNP) assays towards pNP‐myristate (pNP−C14:0) as control substrate, pNP‐oleate (pNP−C18:1) and pNP‐erucate (pNP−C22:1) to determine the chain length selectivity of each mutant. Activity of the different clones towards these substrates was calculated. The hydrolytic activity towards pNP−C18:1 and pNP−C22:1 was compared to the activity towards pNP−C14:0. Surprisingly, the results of the CAL‐A wt showed a two‐fold higher (∼80%) activity towards pNP−C22:1 while the average activity towards pNP−C18:1 was around 30–40% of the activity determined towards pNP−C14:0.
Taking these results into account, in a first step to define a variant as a positive hit, those whose activity towards pNP−C22:1 was equal or inferior to their activity towards pNP−C18:1 were chosen. Then, the pNP−C22:1/pNP−C18:1 activity ratio was calculated for all of them, resulting in a CAL‐A wt pNP−C22:1/pNP−C18:1 ratio of approximately 2.6. To narrow down the number of positive clones, only those displaying a ratio lower than 0.8 were considered selective. The outcome was a pre‐selection of 107 clones from library I, 257 from library II and 173 from library III. Confirmation of their selectivity was achieved by repeating the screening experiment only with the pre‐selected clones by quadruplicate measurements. This re‐screening allowed restricting the number of chosen clones further and only those mutants displaying pNP−C22:1/pNP−C18:1 activity ratio lower than 0.5 (28 from Lib I, 59 from Lib II and 33 from Lib III) were sequenced. Non‐surprisingly, mutations on these clones encoded mostly for large, uncharged amino acids that could block the tunnel.
CAL‐A Variants Chain Length Selectivity Towards pNP Derivatives
Selectivity of the Variants Chosen from the Mutant Libraries
From the sequenced clones, the 15 most selective variants were chosen for further characterization (Table S2). Therefore, the variants were overexpressed in a small‐scale culture (50 ml) and purified based on the His‐tag present using immobilized metal ion affinity chromatography (IMAC). The activity of the purified variants was determined spectrophotometrically towards a range of different chain length pNP‐FA esters, including pNP−C14:0, −C18:1, −C18:2 (−linoleate), −C18:3 (−linolenoate), −C20:1, and −C22:1 (Figure 2).
Figure 2.
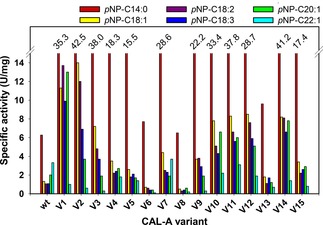
Chain length selectivity profile displayed by each CAL‐A variant towards pNP−C14:0 (▪), −C18:1(▪), −C18:2 (▪), −C18:3 (▪), −C20:1 (▪) and −C22:1 (▪).
The chain length profile of CAL‐A wt showed that this lipase does not possess a pronounced chain length specificity, i. e., CAL‐A wt did not sharply differentiate between pNP‐FA esters with chain length ranging from C14 to C22 (Figure 2). These results match those previously published by our group.12a Interestingly, when compared to the wild type enzyme, almost all CAL‐A variants maintained or increased their activity towards the pNP‐FA esters shorter than C18. In particular, CAL‐A variants V2, V3 and V14 presented up to six times increased activity for the hydrolysis of pNP−C14:0 when compared to CAL‐A wt. The exceptions to this observation were CAL‐A variants V6 and V8 that – although they exhibited a similar hydrolytic activity towards pNP−C14:0− had notably decreased activity towards longer chain pNP‐FA derivatives (Figure 2).
Regarding to the effect of the amino acid substitutions, the selectivity of the variants towards the hydrolysis of pNP‐FA esters shorter than C20 was increased in all cases. This shift in the substrate preference was reflected in both activity ratios calculated: pNP−C20:1/pNP−C18:1 and pNP−C22:1/pNP−C18:1 (Figure 3). The values obtained were especially promising for the erucic acid derivative, where the most reduced hydrolytic activities were determined. It is noteworthy that variants belonging to each one of the three created libraries were present among the 15 most selective clones. Moreover, as it is subsequently described, when the number of variants was further narrowed, this fact continued being true, indicating the impact of all three chosen positions on the FA selectivity.
Figure 3.
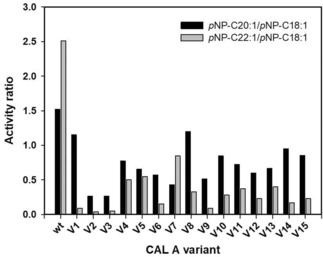
Activity ratios calculated for CAL‐A variants: pNP−C20:1/pNP−C18:1 (▪), pNP−C22:1/pNP−C18:1 (▪).
As the outcome of this selectivity investigation, four variants were identified with a C22:1‐over‐C18:1 value lower than 0.1 (more than twenty‐fold lower than CAL‐A wt: ∼2.5): V1 (CAL‐A V238L, V290L), V2 (CAL‐A V238Y, V290N), V3 (CAL‐A V238I, V290M), and V9 (CAL‐A V286Q, V290W).
Regarding to the variants discriminating between C20:1−over−C18:1, as mentioned above also all of them had a better selectivity than CAL‐A wt (pNP−C20:1/pNP−C18:1 ratio 1.52). In this case, the ratios achieved ranged between 1.15 and 0.26, standing out CAL‐A V2 (CAL‐A V238Y, V290N), V3 (CAL‐A V238I, V290M), V6 (CAL‐A V238L, V286H, V290M) and V12 (CAL‐A V238L, V290N) with pNP−C20:1/pNP−C18:1 ratio below 0.6. Taking into account these results, variants V1, V2, V3, V9 and V12 were selected to further study their behavior towards Crambe and Camelina oil derivatives. CAL‐A V6 was discarded as a candidate due to its low activity compared to the other variants and its low overexpression levels.
Prior to perform the oil enrichment biocatalysis, a better expression system was required. The successful expression of CAL‐A in Pichia pastoris was described in previous studies.9b Hence, mutations present in the variants′ corresponding gene were transferred to a methanol inducible P. pastoris overexpression vector, thus creating the matching pPICZα‐CALA‐His mutant plasmids. The resulting constructs were used to transform P. pastoris GS115 cells. Up to 10 colonies from each transformation were then seeded in three different plates: YPDS containing either, 100 μg/ml or 500 μg/ml of Zeocin, and YPMS‐tributyrin containing 100 μg/ml of Zeocin. Among them, from each transformation the colony displaying the bigger halo and better growth was selected, meaning higher protein overexpression level and Zeocin resistance, in the tributyrin and YPDS‐Zeocin (500 μg/ml) plates (which are usually indicators of multiple gene insertions), respectively. CAL‐A expression experiments were performed on a small scale in shake flasks (50 ml). The variants were secreted to the supernatant due to the α‐factor signal peptide. Subsequently, supernatants were desalted and used to confirm their hydrolytic activities towards different chain length pNP‐FA esters (Figure S1). As expected, the variants obtained displayed similar ratios for pNP−C20:1/pNP−C18:1 or pNP−C22:1/pNP−C18:1 as their E. coli overexpressed counterparts. Additionally, after desalting an aliquot from each supernatant, protein concentration was measured, ranging between 1.6 and 2.1 mg/ml, (Table S3) and secreted proteins were analyzed via SDS‐PAGE (Figure S2).
Moreover, modelling of variants V1, V2, V3, V9 and V12 containing an oleic acid molecule inside the binding pocket was performed. The analysis of the structures indicated the possible changes that the binding site could undergo in each variant due to the corresponding amino acid substitutions (Figure 4). As it was intended, in all five resulting models, the tunnel was either cut or narrowed, explaining the newly achieved selectivity.
Figure 4.
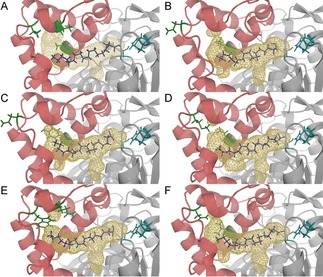
Binding pockets of CAL‐A wt (A) and CAL‐A variants V1 (B), V2 (C), V3 (D), V9 (E) and V12 (F). The docked C18:1 (purple) is used to illustrate the probable location of the FA in CAL‐A binding tunnel (wireframe). The tunnel is mainly formed by the six consecutive helices of the cap domain shown in red. Amino acid residues V238, V286 and V290 (green) are facing the tunnel and placed nearby to the Cω. The active site residues are highlighted in blue. Note: Helix αD1 is removed for a better view onto the binding tunnel.
Analysis of Single Mutations in the Most Selective Variants
To investigate the contribution of each amino acid change present in the double mutants variants created in the combinatorial approach, the corresponding single mutants were generated and overexpressed in P. pastoris in a similar manner as described before. Dialyzed supernatants containing the corresponding overexpressed variant were used to study their pNP‐FA chain length profile (Figure 5A). All single mutants, with the exception of V238Y, showed decreased activity ratios of pNP−C20:1/pNP−C18:1 and pNP−C22:1/pNP−C18:1 in comparison to the wildtype ranging between 0.6–2.0 and 0.1–0.8, respectively (Figure 5B). However, these decreased ratios of the single mutants did not reach to the level of their corresponding double mutants, with the exception of single mutant V290W. From this fact it can be assumed that the distinct selectivity of the remaining four double mutant variants (V1, V2, V3, and V12) resulted from the synergistic effects derived from the simultaneous change of both amino acids. It is noteworthy that only mutant V290W reached activity ratios comparable to the ones of its related double mutant V9, suggesting its major contribution for the selectivity created in this variant. While the pNP−C22:1/pNP−C18:1 ratios of both, V9 and V290W, have been reported as 0.1, the pNP−C20:1/pNP−C18:1 ratio of V290W was with 0.4 slightly worse in comparison to V9 with 0.3. As a consequence, a priori synergistic effects of mutation V286Q with V290 W could improve the selectivity towards C20:1 further.
Figure 5.
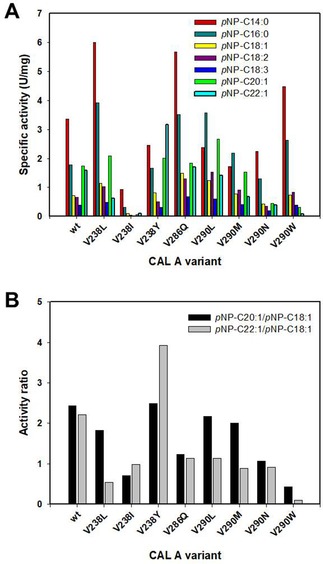
(A) Chain length profile displayed by the selected CAL‐A variants (wt, V238L, V238I, V238Y, V286Q, V290L, V290M, V290N and V290W) overexpressed in P. pastoris towards pNP−C14:0 (▪),−C16:0 (▪), −C18:1 (▪), −C18:2 (▪), −C18:3 (▪), −C20:1 (▪) and −C22:1 (▪). (B) Activity ratios calculated for the aforementioned CAL‐A variants: pNP−C20:1/pNP−C18:1 (▪), pNP−C22:1/pNP−C18:1 (▪).
Surprisingly, the single mutation of V238Y showed an increased activity ratio of pNP−C22:1/pNP−C18:1 of 3.9, being nearly two‐fold higher than the wildtype. This opposite effect in selectivity was reversed again by the synergistic effects in the double mutant V2 (V238Y, V290N) to a ratio of 0.4.
Creation of Triple Mutants
As aforementioned, the main aim behind the creation of three different libraries bearing two saturated positions each was to explore the possible synergistic effects of substituting more than one amino acid simultaneously. The study of the selectivity of the single mutant and double mutant variants confirmed the significance of the influence of these effects for the reshaping of the tunnel architecture appropriately.
In the previous experiments it was clearly observed how, with the exception of CAL‐A V290W, the discrimination between FA esters of a chain length longer than C18 was increased when a pair of residues was altered, compared to the single residue modifications. Therefore, several combinations of the amino acid substitutions from the best double variants were investigated (Table 1). Ideally, these new variants would lead to a better blockage of the binding pocket, resulting in a sharper FA discrimination. Overexpression of lipase variants V16–V20 was confirmed by SDS‐PAGE (Figure S2) and protein concentration was determined (Table S3), prior to the analysis of the hydrolytic activity of the supernatants (Figure 6).
Table 1.
Amino acid changes present in the triple mutant CAL‐A variants.
| Triple Mutants | Amino acid | ||
|---|---|---|---|
| V238 | V286 | V290 | |
| V16 | L | Q | L |
| (V1+V286Q) | |||
| V17 | Y | Q | N |
| (V2+V286Q) | |||
| V18 | I | Q | M |
| (V3+V286Q) | |||
| V19 | L | Q | W |
| (V9+V238L) | |||
| V20 | I | Q | W |
| (V9+V238I) | |||
Figure 6.
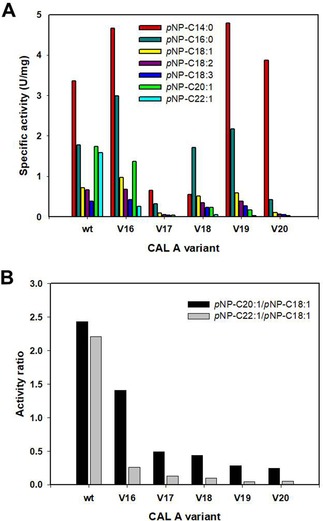
(A) Chain length profiles displayed by the selected CAL‐A variants (wt, V16, V17, V18, V19 and V20) overexpressed in P. pastoris towards pNP−C14:0 (▪),−C16:0 (▪), −C18:1 (▪), −C18:2 (▪), −C18:3 (▪), −C20:1 (▪) and −C22:1 (▪). (B) Activity ratios calculated for the aforementioned CAL‐A variants: pNP−C20:1/pNP−C18:1 (▪), pNP−C22:1/pNP−C18:1 (▪).
Comparing the generated triple mutants to the already investigated variants, their tendency to hydrolyze the longer FA was further decreased. Unfortunately, with the exception of V16, also the activity towards C18 FA was partially affected, leading to lower specific activities towards these substrates. Especially acute was the aftermath in V20, which displayed a sharp decrease in activity with substrates longer than pNP−C14:0. These results indicate that an accumulation of bulky residues at those positions in the binding tunnel may cause a larger steric hindrance, hence hampering as well the hydrolysis of FA shorter than C20. Despite this fact, the pNP−C22:1/pNP−C18:1 activity ratio calculated for the triple mutant variants was further diminished when compared to their parental double mutant variants. In this case the obtained values ranged from 0.25 to 0.04, while those obtained for the variants with two altered amino acids varied between 0.4 and 0.06. Similarly, the values of the pNP−C20:1/pNP−C18:1 activity ratio of the new variants were lowered (0.5–0.25) regarding to their precursors (1.0–0.5), excluding V16 that remained alike (∼1.4). Consequently, these variants were selected as good candidates for further application in gondoic and erucic FA enrichment. Only V17 was excluded due to both, low overexpression yields and low specific activity.
High Cell Density Expression of CAL‐A Variants
One of the main advantages of using P. pastoris as host is that it can grow to very high cell densities, thus, expressing and secreting recombinant proteins accordingly, while only very few proteins from the host cell are present in the fermentation broth.21 Taking advantage of this fact, large‐scale production of CAL‐A wt and the most promising variants, i. e. V290W, V1, V2, V3, V9, V16, V18, V19 and V20, was performed to obtain sufficient amounts of protein for the biocatalysis experiments.
Fermentation of the clones was a fed‐batch process where cells were inoculated and grown on glycerol, followed by a fed‐batch with glycerol (50%) step and the subsequent induction phase involving fed‐batch with methanol. After 162 h the culture's supernatant was used for FA enrichment. During this process the cultures growth and protein overexpression was monitored by measuring the absorbance at 600 nm, the wet cell weight, and the lipase activity towards pNP−C14:0. Additionally, final protein concentration in the supernatant was determined. Absorbance at 600 nm increased along time from 1.2–2.7 (t0) to 270–370 (t162), depending on the variant overexpressed. In a similar way, the final wet cell weight achieved was approximately 0.2 g/ml of culture for all clones. In contrast, both, lipase concentration and hydrolytic activity strongly depended on the variant studied. In this sense, enzyme concentration in the dialyzed supernatant ranged between 1.1 and 2.5 mg/ml, while the activity towards pNP−C14:0 varied more than ten‐fold (120.1 U/mg from V20 to 1,672.4 U/mg from V1) (Table S4).
In addition, to verify that the amino acid changes introduced did not affect the fermented variants stability, their melting temperatures were analyzed and compared to the wt (Table 2). As a result it could be confirmed that, although the melting temperature decreased up to 6.5 °C for V2 and V19, in all cases it remained above 82 °C, indicating a high stability towards thermal unfolding.
Table 2.
Melting temperatures obtained for the CAL‐A wt and fermented variants.
| CAL‐A variant | Melting temperature (°C) |
|---|---|
| wt | 89.0 |
| V290W | 87.9 |
| V1 | 89.0 |
| V2 | 82.5 |
| V3 | 87.8 |
| V9 | 83.6 |
| V16 | 83.1 |
| V18 | 83.5 |
| V19 | 82.5 |
| V20 | 84.2 |
Lipase Specificity on Transesterified Oil Substrates
Besides discriminating towards different FA chain lengths, lipases can display several types of selectivities when acting on TAG. For instance, lipase regioselectivity, i. e. the ability to distinguish between primary (sn‐1,3) and secondary (sn‐2) ester functionalities in a triacylglycerol molecule, may affect the hydrolysis pattern of these enzymes when acting on determined oil.22 Formerly, CAL‐A was described as a sn‐2 selective lipase,23 this information was proven to be an artifact due to acyl migration in the products of this regio‐unspecific lipase.24 Nonetheless, lipase regioselectivity is an important factor to take into account when performing FA selective enrichment on an oil substrate. For instance, C22:1 in Crambe oil is mostly located at the sn‐1,3 positions of the glycerol and the remaining FA in this oil are prone to be located at the sn‐2 position.25 Interestingly, although some studies showed that C22:1 enrichment could be achieved utilizing sn‐1,3 specific lipases, this strategy was less successful than using lipases with natural selectivity towards long‐chain fatty acids like C. rugosa or G. candidum lipase.4,8b In order to analyze the enrichment capacity of the newly created CAL‐A variants depending exclusively on the different chain length of the FA present in the oils, FA ethyl ester (EE) derivatives obtained from Crambe and Camelina oils were used as substrates. Regarding their composition, Crambe oil and, thus, its EE derivatives are mainly composed of C22:1 (58.7%) and unsaturated FA of C18 (31.2%) (Figure S3). On the other hand, Camelina oil and its derived EE contain primarily C18:3 (38.0%) and similar amounts of C18:1, C18:2, and C20:1 (15.5% of each) (Figure S4).
Biocatalysis Using Crambe Ethyl Esters as Substrate
When performing the enrichment biocatalysis with the selected variants on the Crambe esterified FA (EFA) fraction, CAL‐A wt was also included as control to verify the created selectivities. Depending on their behavior the used biocatalysts could be differentiated in two groups: (i) those which enriched the desired C22:1 in EFA fraction as it was planned; and (ii) those which caused an accumulation of the FA in the FFA (Figure 7).
Figure 7.
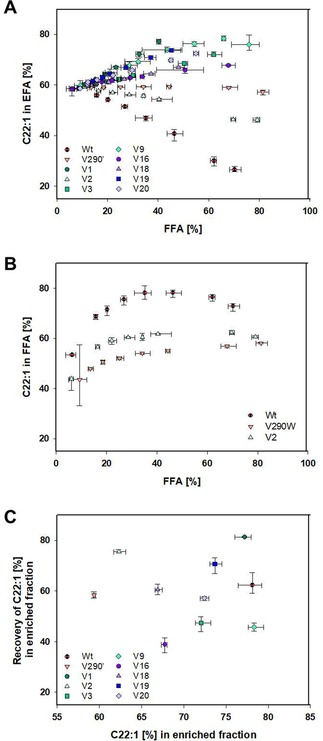
Content of C22:1 in the Crambe EFA (A) or FFA (B) fraction over content of FFA as mean averages of triplicates displayed with their errors, as well as their theoretical recovery of C22:1 (C), which is defined as the amount of C22:1 present in the enriched fraction (either FFA or EFA) in comparison to the total amount of C22:1 in the sample (in both FFA and EFA), over the C22:1 content in the most enriched fraction.
The first group included almost all newly generated variants: V1, V3, V9, V16, V18, V19 and V20 (Figure 7A). These lipases showed their preference for the hydrolysis of FA shorter C20, thus achieving enrichments of C22:1 in the EFA fraction ranging from 67.7 to 78.3%. Especially high enrichment levels were achieved by V1 (77.2%), V9 (78.3%), V19 (73.7%), and V20 (72.4%). On the other hand, an increase of the C22:1 concentration in the FFA fraction was possible by using CAL‐A wt. This result was surprising since, although a higher activity for the hydrolysis of C22:1 when compared to the hydrolysis of C18:1 was previously observed while testing the pNP‐derivatives, leading to a pNP−C22:1/pNP−C18:1 ratio of ∼2.5, it was not anticipated to have such a large impact when using the complex FAEE mixture (Figures 3, 5, 6 and S1). Nevertheless, an enrichment of C22:1 in the FFA fraction could be observed accomplished by the wt enzyme up to a concentration of 78.1% (Figure 7B). It is noteworthy to mention that these results demonstrate that it was possible to engineer the complete inversion of the CAL‐A wt selectivity.
Additionally, even though variants V290W and V2 displayed pNP−C22:1/pNP−C18:1 ratios quite low (0.1 and 0.4, respectively), these lipases also showed some enrichment of C22:1 in the FFA fraction, but only up to a concentration of 62.3%.
Regarding to the hydrolysis yields accomplished, even though the amount of enzyme present to reach a sufficient degree of substrate hydrolysis (10–40%) was estimated (Figure S5), maximal FFA contents obtained significantly varied after 48 h, ranging from 40.4% for V1 to 81.4% for V290W. Especially, when examining V9 and its corresponding triple mutants V19 and V20, a similar slope can be observed leading to the assumption that similar maximal values of enriching C22:1 can be attained by reaching a higher degree of hydrolysis. However, the steepest slope was observed for V1 as well as the highest theoretical recovery of 81.3% (Figure 7C). This value is defined as the amount of C22:1 present in the enriched fraction (either FFA or EFA) in comparison to the total amount of C22:1 in the sample (in both FFA and EFA). In comparison, the theoretical recovery for the wt only reached 62.3% and for V9 as far as 45.7%.
The results achieved in this work are comparable to those described in previous studies using C. rugosa and G. candidum lipases as biocatalysts.4 However, it must be taken into account that, to the best of our knowledge, the proteins used in the aforementioned examples were commercial samples composed of a mixture of several isoenzymes in different proportions. Since modifications of the fermentation conditions lead to different lipase amounts and (iso)enzymatic profiles, reproducibility of the results could hence be challenging.26 On the other hand, CAL‐A wt is a well‐studied lipase with great potential in biocatalysis due to its high level of overexpression, stability and the possibility of immobilization.21,27 Therefore, the described CAL‐A variants represent a very attractive alternative for C22:1 enrichment from plant oils.
Biocatalysis Using Camelina Ethyl Esters as Substrate
Interestingly, when analyzing the results for the reactions with Camelina EFA, the same long chain FA enrichment behavior for the enzyme variants could not be observed. In this case, the best results for the enrichment of C20:1 in the EFA fraction were obtained with the variants derived from CAL‐A V290W. The double, V9, and triple mutants, V19 and V20, enriched C20:1 from a concentration of 15.4% to approx. 27% (Figure 8A). Interestingly, the highest enrichment, 34.1%, was obtained with the single mutant V290W. However, the theoretical recovery determined for the best four variants was moderate reaching 52–59% (Figure 8B). It is certainly intriguing that variant V290W was not able to enrich C22:1 in the Crambe EFA, but it enriched C20:1 in the Camelina EFA. As it is explained below, a concrete structural reason for this reduced activity towards C20:1 but not towards C22:1 could not be identified by molecular modeling studies (Figure S6). Regarding V1 and its derivative V16 the data analysis detected a similar behavior in enrichment, producing a slight decrease of gondoic acid in the EFA fraction. This fact indicated that for these two variants the chain length selectivity cut‐off was generated between C20 and C22. These results are endorsed by the results obtained in the pNP‐FA selectivity experiment where both variants showed pNP−C20:1/pNP−C18:1 ratios decreased in comparison to CAL‐A wt but were close to 1, which means similar rates in hydrolysis for both substrates (Figure 3 and 6). Concerning V2, V3 and V18, only a minor enrichment was produced in the EFA, not representing more than 5% on any of these cases. Finally, despite the pNP−C22:1‐ and pNP−C20:1/pNP−C18:1 ratios obtained for CAL‐A wt were quite similar (2.2 and 2.4, respectively), no significant C20:1 enrichment was observed for CAL‐A wt in the FFA fraction.
Figure 8.
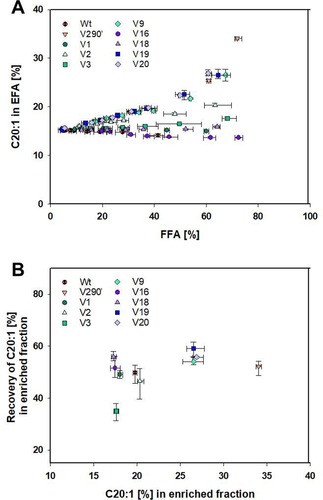
Content of C20:1‐EE in the Camelina EFA (A) fraction over content of FFA as mean averages of triplicates displayed with their errors, as well as their theoretical recovery of C20:1 (B), which is defined as the amount of C20:1 present in the enriched fraction (either FFA or EFA) in comparison to the total amount of C20:1 in the sample (in both FFA and EFA), over the C20:1 content in the most enriched fraction.
The different performance of the variants can be explained due to two non‐exclusive reasons: (i) although Camelina and Crambe samples are comparable concerning the FA types present, there is a great difference in their concentrations. Thus, while in both oils FA chain lengths range between C14 to C24, the concentration of the desired FA is dissimilar, representing 15.4% (C20:1) and 58.7% (C22:1), respectively. This may be especially significant for CAL‐A wt, as crowding effects could play a role in the conversion of the long‐chain EFA by this enzyme. Hence, in the Crambe EFA it is more likely that the enzyme meets a molecule of C22:1‐EE in comparison to contacting a molecule of C20:1‐EE using Camelina EFA as substrate. Secondly, (ii) in the robotic screening the assay was directed towards the identification of clones with lower pNP−C22:1/pNP−C18:1 ratio. As a consequence, variants discerning shorter FA than erucic and not gondoic acid were favored (“You get what you screen for”).28 Consequently, V1 was identified with a ratio of 0.4 and V16 was derived from it with an improved ratio of 0.3, while they are not able to discriminate towards C20:1.
Bioinformatic Analysis of Changes in the Tunnel Structure
When analyzing the main FA binding tunnel of CAL‐A there are striking features to mention. The tunnel is located in the cap domain of the enzyme15 and can reach a length of up to 36.1 Å (Figure 9). The beginning of the tunnel is buried inside the protein scaffold at the active site, which is connected to smaller tunnels important as solvent or small ion channels (Figure 9A). The main part of the tunnel (33.7 Å) has a radius ≥1.5 Å, being large enough to fit the cross section of a FA molecule, i. e. a carboxylic acid with a long aliphatic chain containing as cross section units C2H4 with a radius of approx. 1.44 Å. In close proximity of the active site a minor appendix leaves the main acyl‐binding tunnel. This groove was investigated by Brundiek et al. as an alternative binding pocket able to bind medium chain FA.12a The major binding tunnel displays a linear section of 15 Å in the beginning and takes then several turns until it narrows down and is exposed to the surface in the lid region.
Figure 9.
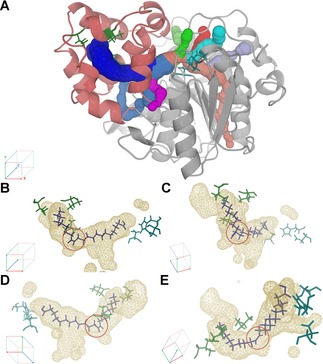
Elucidation of the CAL‐A FA binding tunnel structure with (A) CAVER tunnel computation and (B–E) the results of the molecular dynamics studies of C22:1 binding to CAL‐A wt. The main tunnel is shown from different angles. Highlighted in the red circle is the cisΔ13 double bond of the C22:1 molecule. The FA (purple) was previously docked in the CAL‐A crystal structure (3GUU) displaying the binding tunnel (wireframe), the cap domain (red), the catalytic residues (blue) and the hotspots for protein engineering (green).
In the previous molecular modeling experiments it was observed that the conformation of the tunnel underlies a certain degree of freedom as well as the binding of the FA molecule. However, the FA selectivity towards different chain length of MUFAs of the wt enzyme was striking. CAL‐A wt preferred the hydrolysis of C20:1 and C22:1 over C18:1. An explanation for this was found in the tunnel architecture, which allows the cis double bond to adopt an angle closer to its natural conformation (110°). While the cis double bond of C18:1 (cisΔ9) is located in the linear part of the FA binding tunnel (Table S5), the cis double bonds of C20:1 (cisΔ11) and C22:1 (cisΔ13) suit with the first turn of the tunnel (Figure 9B–E).
When analyzing changes introduced to the FA binding tunnel of CAL‐A by its mutants, it can be said that each variant displayed an at least by 10 Å shortened tunnel (Table 3; Figure S6). However, convincing structural reasons for the distinct selectivities were not found neither for the distinct chain‐length selectivity of V1 and V16 between C20 and C22 FA, nor the decreased activity of V290 W towards C20:1, but not C22:1.
Table 3.
Summary of the distance from the catalytic active site to the first bottleneck determined for CAL‐A wt, the single mutant V290W, the double mutants V1–3, 9, and 12, and the triple mutants V16–20. More specifically, the tunnel graphs are displayed for the FA binding tunnel calculated from the catalytic Ser185 with a minimal probe radius of 0.6–0.7. Path lengths were calculated with a probe radius of 1.50 Å using Caver Analyst 1.0.
| Variant | Length [Å] | Variant | Length [Å] | Variant | Length [Å] |
|---|---|---|---|---|---|
| Wt | 33.65 | V1 | 20.85 | V16 | 22.24 |
| V290W | 19.33 | V2 | 21.65 | V17 | 22.23 |
| – | – | V3 | 19.09 | V18 | 20.42 |
| – | – | V9 | 22.26 | V19 | 20.31 |
| – | – | V12 | 22.26 | V20 | 21.91 |
Conclusions
The work presented herein describes the semi‐rational protein design of CAL‐A selectivity to enhance the enrichment of mono‐unsaturated long chain FA from Camelina and Crambe seed oil ethyl ester derivatives.
Initially, molecular modelling on the CAL‐A structure allowed the identification of three possible hotspots for site‐directed saturation mutagenesis: V238, V286, and V290. This led to the creation of three different “small, but smart” libraries bearing two saturated positions simultaneously: library I (V238X and V286X), library II (V238X and V290X) and library III (V286X and V290X). High‐throughput screening in LARA, and the subsequent characterization of the pNP‐FA ester hydrolysis preference, allowed the identification of five CAL‐A variants, i. e. V1 (V238L, V290L), V2 (V238Y, V290N), V3 (V238I, V290M), V9 (V286Q, V290W) and V12 (V238L, V290N), displaying significantly lower pNP−C20:1/pNP−C18:1, pNP−C22:1/pNP−C18:1 ratios (between 0.6–1.3 and 0.4–0.06, respectively), in comparison to CAL‐A wt (2.4 and 2.2, respectively). Consequently, the capability of these variants of accepting FA longer than C18 was dramatically decreased. The fact that amino acid changes present in all three positions influenced the catalytic specificity confirmed the suitability of the chosen site‐directed mutagenesis targets. Furthermore, analysis of single and triple mutants derived from the aforementioned variants highlighted the relevance of the synergistic effects achieved by altering more than one residue at the same time. In general, when compared to their parental double mutant variants, single mutant variants displayed higher pNP−C20:1/pNP−C18:1, pNP−C22:1/pNP−C18:1 activity ratios (between 0.4–2.5 and 0.1–3.9, respectively), while in the triple mutants these values were decreased (between 0.25–1.4 and 0.04–0.25, respectively).
Regarding to the FA enrichment process with Crambe ethyl ester derivatives, it is noteworthy that when the biocatalysis was mediated by CAL‐A wt, up to 78% enrichment of C22:1 was observed in the FFA fraction. On the other hand, a complete inversion of this selectivity was achieved when the best variants of the above were tested in the same procedure. Hence, C22:1 was enriched in the EFA fraction to 77% with V1, 78% with V9, and 74% with V19. Within these variants the best theoretical yield of C22:1 was determined for V1 with 81%. In the case of using Camelina oil ethyl esters as substrate, the lipases behaved differently. CAL‐A wt could not enrich C20:1 in neither, the EFA nor in the FFA fraction, while the best variant was the single mutant V290 W, which more than doubled the content of C20:1 in the EFA fraction to 34%. The next best catalysts comprised V9 and its two derived triple mutant variants V19 and V20, which were able to increase the percentage of C20:1 up to 27%.
Therefore, the generated variants have demonstrated their capability to enrich long chain mono‐unsaturated FA, utilizing as starting substrate complex mixtures of FA ester derivatives obtained from renewable sources. In addition, different lipase variants could be chosen in a practical situation to best fit the process requirements, depending on the substrates used, the required purity and the desired recovery of FA. Moreover, further optimization of the biocatalysis reaction conditions would probably lead to both, better recovery yields and higher level of enrichment. Thus, they represent promising candidates for their application in industrial lipid modification as a more environmentally friendly alternative to traditional oleochemical processes.
Experimental Section
Materials
Unless stated otherwise all chemicals were purchased from Fluka (Buchs, Switzerland), Sigma (Steinheim, Germany), Merck (Darmstadt, Germany), VWR (Hannover, Germany), or Carl Roth (Karlsruhe, Germany). Primers were synthesized by Sigma (Steinheim, Germany) and Thermo Fisher Scientific (Waltham, MA, USA). Sequencing was performed by Eurofins MWG Operon (Luxembourg, Luxembourg). E. coli TOP10 strain was obtained from Invitrogen (Carlsbad, CA, USA) and E. coli BL21 (DE3) C41 strain was purchased from Lucigen (Middelton, USA). Plasmids pGAPZ and pPICZα were purchased from Invitrogen (Carlsbad, USA).
CAL‐A Cloning and Overexpression in E. coli
The plasmid pET22b bearing the gene encoding CAL‐A from Moesziomyces antarcticus (Candida antarctica) was previously cloned in our group.12a E. coli C41(DE3) cells were transformed with pET22b(+)_ppro_CALA_His or the corresponding derivatives by electroporation (MicroPulser™ electroporator). Similarly, in order to preserve them, E. coli Top10 cells were transformed with pPICZα or pGAPZα vector bearing the desired CAL‐A wt/mutant gene.
Overexpression of CAL‐A wt and the different variants in E. coli C41 was achieved by growing a colony bearing the plasmid in a 5 ml LB O/N culture containing ampicillin (0.1 mg/ml) at 37 °C and 180 rpm. Depending on the experimental setup, 50 ml of ZYM‐5052 medium29 was inoculated with 1–2% volume of the E. coli O/N culture. After inoculation, cultures were incubated at 37 °C and 180 rpm for 6 h and, then, cultivated at 30 °C for 22 h. The cultures were harvested at 3900 g for 60 min at 4 °C. The supernatant (SN) was discarded and the cell pellet was resuspended in 10 ml sodium phosphate buffer (50 mM; pH 7.5) containing sodium chloride (1 mM). Cell lysis was performed by ultrasonication (Sonoplus HD2070 UW2070 KE76, Bandelin electronic, Berlin, Germany) at 30% power and 50 cycles for 5 to 20 min on ice. The suspension was centrifuged at 3900 g for 30 min at 4 °C. Subsequently, the supernatant, containing the cell free extract (CFE), was separated from the cell pellet and snap frozen with liquid N2. The frozen samples were stored at −80 °C until further usage.
CAL‐A Cloning and Overexpression in P. pastoris
Prior to the electroporation of P. pastoris GS115 competent cells, the pPICZα (wt, V290W, double mutants and triple mutants) or pGAPZα (wt and single mutants) vector bearing the desired CAL‐A wt/mutant gene was purified from a 20 ml (LB‐low salt containing 25 μg/ml Zeocin) O/N culture of the corresponding E. coli Top10 strain and digested using PmeI or AvrII restriction enzyme to obtain the pure linearized plasmid. After electroporation of the GS115 competent cells with the linear plasmid (5–10 μg), 1 ml of ice cold sorbitol (1 M) was added. The mixture was incubated at 30 °C during 1 h without shaking. Subsequently, 250 μl were seeded in YPDS plates containing 1% yeast extract, 2% peptone, 2% dextrose, 1 M sorbitol, 2% agar and 100 μg/ml Zeocin. After 72 h incubation at 30 °C the transformed colonies grew.
Selection of the best P. pastoris clones: once obtained the transformants, screening of the best clones was performed by plating 10 μl from a 0.5 ml YPD (1% yeast extract, 2% peptone, 2% dextrose and 100 μg/ml Zeocin) O/N culture in three different plates:(i) an indicator plate supplemented with tributyrin30 and YPDS plates containing either (ii) 100 or (iii) 500 μg/ml of Zeocin. These plates were incubated during 96 h at 30 °C. One clone from each transformation was chosen based on the size of the halo and the resistance towards Zeocin. The selected clones were then used for lipase overexpression, as well as stored at −80 °C in YPD medium containing 30% of glycerol for conservation and further use.
P. pastoris overexpression of CAL‐A variants in small scale: a freshly plated P. pastoris GS115 colony transformed with the desired CAL‐A gene was used to inoculate 5 ml BMGY medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6.0), 1.34% YNB, 4×10−5% biotin and 1% glycerol) with Zeocin (0.1 mg/ml) in a 250 ml Erlenmeyer baffled flask. The culture was shaken overnight at 30 °C and 180 rpm. Using 5 ml of overnight culture 45 ml of BMGY media were inoculated and kept at 30 °C and 180 rpm.
When P. pastoris clones were overexpressing the desired protein under the control of the inducible promoter (double and triple mutants), after 24 h the BMGY cultures were centrifuged at 4500 g for 15 min and washed with 5 ml sterilized 50 mM sodium phosphate buffer with 1 mM NaCl at pH 7.5. The washed cells were centrifuged again and the cell pellet was resuspended with 50 ml BMMY (1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6.0), 1.34% YNB, 4×10−5% biotin and 0.5% methanol) containing Zeocin (0.1 mg/ml). Subsequently, the culture mixture was incubated at 30 °C and 180 rpm for 96 h in a 250 ml Erlenmeyer baffled flask. Every 24 h 250 μl of sterilized methanol (0.5% final culture volume) was added to maintain the protein overexpression. After 96 h of induction the culture was harvested by centrifugation (20 min, 4500 rpm, 4 °C). The supernatant (∼50 ml) was collected, its hydrolytic activity towards pNP−C14:0 measured and a SDS‐PAGE run as described below. Then, the corresponding supernatant was divided into aliquots and frozen at −80 °C.
When overexpressing variants under the control of the constitutive promoter a similar protocol was followed. However, since the constitutive promoter needs no additional feeding with methanol for induction, it was used YPD media instead of the BMGY and BMMY media.
Fed‐batch P. pastoris fermentation for the overexpression of CAL‐A variants V290W, V1, V2, V3, V9, V16, V18, V19 andV20: a 250 ml Erlenmeyer baffled flask with 50 ml of BMGY medium was inoculated with a freshly plated P. pastoris colony per variant. The flask was incubated for ∼48 h at 30 °C on an orbital shaker at 180 rpm. Once the cell density reached OD600 ∼50, 0.6 l of Basal Salts Medium (BSM was made as described by Invitrogen in Pichia Fermentation Process Guidelines) were seeded with 30 ml of the shake flask culture in a 1 l fermenter. In the glycerol batch phase the culture was grown until the glycerol was completely consumed (∼24 h). Then, the glycerol fed‐batch phase was initiated by adding glycerol 50% containing 1.2% PTM1 salts at 5 ml/min/l during 4 h to increase the cell biomass. After that time, the feeding was stopped and the MeOH induction was not started until all the remaining glycerol was consumed (∼3 h). During the MeOH fed‐batch phase the methanol was introduced slowly to avoid cell death. Therefore, MeOH flow started on 1.5 ml/min/l and was increased up to 3 ml/min/l when the culture was fully adapted to methanol utilization and methanol limitation occurred. The induction phase continued during 96 h. The temperature was kept constant at 30 °C and the pH was maintained between 5.0–6.0 using NH4OH (30%) and H3PO4 (50%). The airflow and the stirrer speed were adjusted to maintain the dissolved oxygen (DO) above 20%. When the fermentation finished the culture was centrifuged at 4500 rpm during 1 h and the supernatant was collected and frozen at −80 °C for later lyophilization. The fermentation process was followed by taking 10 ml samples at least once a day. Samples were analyzed for cell growth (OD600 and wet cell weight) and hydrolytic activity of the SN towards pNP−C14:0.
CAL‐A Purification
The presence of the C‐terminal His‐tag enabled a one‐step purification procedure of CAL‐A by IMAC. The fraction containing the soluble protein (25 ml) was applied to 5 ml of Co+2−IDA agarose column (Roti®garose‐His/Co Beads; Roth, Karlsruhe, Germany) pre‐equilibrated with 50 mM sodium phosphate buffer containing 300 mM NaCl and 30 mM imidazole, pH 7.0. The column was washed with equilibrating buffer (50 ml) in order to remove the unbound material. The desired protein was eluted with 10 ml of the same buffer containing 300 mM imidazole. Subsequently, in order to desalt and concentrated the resulting protein solution two methods could be used: (i) concentration of the eluted fraction by centrifugal filter units, Vivaspin 20 (10 kDa cut off, Satorius AG, Göttingen, Germany), to 1 ml. This volume was subsequently washed five times with washing buffer (5 mM sodium phosphate buffer, pH 7.5). Alternatively, (ii) dialysis with ZelluTrans dialysis membranes (10 kDa cut off, Roth, Karlsruhe, Germany) was performed, followed by lyophilisation.
Determination of Protein Concentration
Protein concentration was determined by the bicinchoninic acid (BCA) assay (Pierce™ BCA Protein Assay Kit, ThermoFisher Scientific, Waltham, USA) with bovine serum albumin as a standard. Absorbance of all samples was measured in triplicates (λ=562 nm) (Tecan Infinite M2000 Pro, Tecan Group Ltd., Männedorf, Switzerland).
SDS‐PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) was performed using a 12.5% and 5% acrylamide in the resolving and stacking gels, respectively. Gels were run on a Mini‐PROTEAN® Tetra System apparatus (Bio‐Rad) at 200 V for 40 min. Molecular weight marker was purchased from either Carl‐Roth (Roti®‐Mark Standard) or from Thermofisher (Unstained Protein MW Marker). Proteins were stained with Coomassie Brilliant Blue G‐250 (Bradford).
Determination of Protein Melting Temperature
Measurement of the melting temperature for the CAL‐A variants overexpressed in high cell density cultures were performed using a Prometheus NT.48 equipment (NanoTemper Inc., Germany). Approximately 4 μl of each sample containing 1 mgprot/ml was aspirated into standard‐treated glass capillaries (NanoTemper Inc., Germany). Capillaries were loaded into a 48‐capillary sample holder and thermostated inside the instrument at 20 °C for ∼5 min before measurements. Excitation power was set at 10%. Intrinsic fluorescence was measured at 330 and 350 nm in presence of a heat gradient ranging from 20 to 95 °C (ΔT=0.1 °C/min).
Hydrolysis of pNP‐Esters of Different Chain Length
CAL‐A hydrolytic activity was determined by following the initial hydrolysis rate of pNP‐esters spectrophotometrically during 5 min (TECAN Infinite M2000 Pro) at λ=410 nm (ϵ=10,998 M−1 cm−1) in a 96‐well microtiter plate. The assays were performed in sodium phosphate buffer (50 mM; pH=7.5) containing NaCl (1 mM), isopropanol (10% v/v), Triton X‐100 (0.5% v/v), the fatty acid derivative (1 mM) and the enzyme. Reactions were initiated by the addition of the substrate and the Triton X‐100 dissolved in isopropanol. As the standard substrate for measuring enzyme activity pNP−C14:0 was chosen. One unit of lipase activity was defined as the amount of enzyme releasing 1 μmol pNP per minute. All measurements were performed at least in triplicates.
Chain length selectivity was investigated by measuring the hydrolytic activity towards several p‐nitrophenyl esters, including pNP‐myristate (C14:0), ‐palmitate (C16:0), ‐oleate (C18:1), ‐linoleate (C18:2), ‐linolenate (C18:3), ‐gondoate (C20:1), and ‐erucate (C22:1).
Chemical Synthesis of pNP‐Fatty Acid Esters
pNP−C14:0 and pNP−C16:0 were purchased from Fluka, while the FA esters derived from C18:1 (cisΔ9), C18:2 (cisΔ9, cis‐Δ12), C18:3 (cisΔ9, cisΔ12, cisΔ15), C20:1 (cisΔ11) and C22:1 (cisΔ13) acids were synthesized in one‐gram scale using the corresponding acid choride derivatives (NuCheck, USA) as described by Brundiek et al. (2012).31 To a 250 ml round‐bottom flask, with magnetic stirring, under an inert atmosphere (N2), anhydrous ZnCl2 (0.54 g, 3.96 mmol), anhydrous dichloromethane (30 ml), and pNP (1.0 g) were added. The corresponding acid chloride (1.0 g) was added dropwise by a syringe, and the reaction mixture was refluxed until complete conversion was observed by thin layer chromatography (∼1 h). The reaction mixture was then cooled to room temperature in an ice bath. Afterwards, 30 ml of water were added to the reaction mixture to, subsequently, decant the organic layer. The aqueous phase was extracted with diethyl ether (Et2O, 2×30 ml). The combined organic layers were further washed with NaHCO3 (2×30 ml) and, then, dried with anhydrous Na2SO4. After filtration and evaporation in vacuo this yielded a dark brown product. Purification was performed by flash chromatography (hexane/Et2O, 10:1) to afford the pure pNP‐FA derivative as a clear, beige oil. The identity of all compounds synthesized was confirmed by 1H NMR spectroscopy (300 MHz; dilute solution in CDCl3) (Figure S7).
pNP−C18:1 (0.83 g, 62% yield): 1H‐NMR (300 MHz; dilute solution in CDCl3): δ=8.27 (2H, d, J=9.25 Hz, 2×Ar−H), 7.27 (2H, d, J=9.25 Hz, 2×Ar−H), 5.43–5.30 (2H, m, −HC=CH−), 2.60 (2H, t, J=7.55 Hz, α‐CH2), 2.11–1.91 (4H, m, H2C−HC=CH−CH2), 1.83–1.69 (2H, m, β‐CH2), 1.49–1.19 (20H, m), and 0.88 (t, J=6.61 Hz, CH3).
pNP−C18:2 (1.14 g, 85% yield): 1H‐NMR (300 MHz; dilute solution in CDCl3): δ=8.26 (dd, J=9.25 Hz, 2H, 2×Ar−H), 7.27 (dd, J=9.25 Hz, 2H, 2×Ar−H), 5.45–5.27 (m, 4H, 2×−HC=CH−), 2.78 (t, J=6.0 Hz, 2H =CH−CH2−HC=), 2.59 (t, J=7.45 Hz, 2H, α‐CH2), 2.11–1.99 (m, 4H, H2C−HC=CH−CH2), 1.82–1.70 (m, 2H, β‐CH2), 1.40–1.24 (m, 14H), and 0.89 (t, J=6.61 Hz, 3H, CH3).
pNP−C18:3 (0.98 g, 74% yield): 1H‐NMR (300 MHz; dilute solution in CDCl3): δ=8.26 (dd, J=9.25 Hz, 2H, 2×Ar−H), 7.27 (dd, J=9.25 Hz, 2H, 2×Ar−H), 5.54–5.19 (m, 6H, 3×−HC=CH−), 2.81 (t, J=5.71 Hz, 2H =CH−CH2−HC=), 2.60 (t, J=7.42 Hz, 2H, α‐CH2), 2.14–1.97 (m, 4H, H2C−HC=CH−CH2), 1.82–1.68 (m, 2H, β‐CH2), 1.41–1.24 (m, 13H), and 0.98 (t, J=7.4 Hz, 3H, CH3).
pNP−C20:1 (1.17 g, 89% yield): 1H‐NMR (300 MHz; dilute solution in CDCl3): δ=8.27 (dd, J=9.20 Hz, 2H, 2×Ar−H), 7.27 (dd, J=9.20 Hz, 2H, 2×Ar−H), 5.37–5.32 (m, 2H, −HCOCH−), 2.60 (t, J=7.45 Hz, 2H, α‐CH2), 2.06–1.96 (m, 4H, H2C−HC=CH−CH2), 1.80–1.65 (m, 2H, β‐CH2), 1.37–1.25 (m, 24H), and 0.88 (t, J=6.60 Hz, 3H, CH3).
pNP‐C22:1 (1.04 g, 81% yield): 1H‐NMR (300 MHz; dilute solution in CDCl3): δ=8.25 (dd, J=9.30 Hz, 2H, 2×Ar−H), 7.27 (dd, J=9.30 Hz, 2H, 2×Ar−H), 5.37–5.32 (m, 2H, −HC=CH−), 2.60 (t, J=7.45 Hz, 2H, α‐CH2), 2.06–1.96 (m, 4H, H2C−HC=CH−CH2), 1.81–1.70 (m, 2H, β‐CH2), 1.45–1.21 (m, 28H), and 0.88 (t, J=6.60 Hz, 3H, CH3).
Bioinformatic Tools
Molecular modelling was performed using YASARA (www.yasara.org) based on the 2.1 Å‐resolution X‐ray structure of molecule A of CAL‐A (PDB: 3GUU). Structural refinement was carried out by a molecular dynamics simulation in water. Docking experiments with the refined structure were performed using oleic, gondoic, and erucic acids as ligands. The simulation cell was arranged including the catalytic residues and the whole binding tunnel of CAL‐A. For the docking experiment the dockrunensemble macro from YASARA was used. The binding of the docked FA was further investigated for CAL‐A wt and V290W by molecular dynamic simulations with an Amber14 forcefield utilizing the mdrun macro of YASARA under physiological conditions and a frame rate of 250 ps. Snapshots were evaluated concerning the FA binding of the tunnel and the tunnel architecture.
Further characterization of the tunnel was performed with the software CAVER Analyst 1.032 (www.caver.cz). Tunnels were computed starting from the catalytic Ser184 with a maximal distance of 5 Å and a solvent radius of 0.7 Å.
Generating CAL‐A Mutant Libraries
Three different CAL‐A mutant libraries were generated to perform saturation mutagenesis simultaneously at two positions with a “small, but smart” focused library.19b Saturation positions for each library were: V238X and V286X for library I; V238X and V290X for library II; and V286X and V290X for library III. CAL‐A libraries I and II were generated via MegaWhop with specific primers for each variant to produce megaprimers (MP1 and MP2)33 (Table S6). The plasmid containing the CAL‐A gene (100 ng) was mixed with 10× Buffer B (5 μl), dNTPs (2 μl), 25 mM MgSO4 (1 μl), 0.5 μl OptiTaq, forward primer 16.21 (1 μl, Lib I+II), the corresponding reverse primer 16.19 (Lib I) or 16.22 (Lib II) (1 μl), and distilled water (38.5 μl). The PCR program included the following temperature steps: 95 °C for 2 min; subsequently 30 cycles of the following: 95 °C for 15 s, 55 °C for 30 s, 72 °C for 42 s; and finally a holding step at 72 °C for 10 min. The PCR product was purified (Nucleo Spin® Gel and PCR Clean‐up, Macherey & Nagel, Düren, Germany) and used in the optimized protocol for the MegaWhop as megaprimer. The plasmid containing the CAL‐A gene (100 ng) was mixed with 10× Pfu buffer (5 μl), dNTPs (1 μl), purified megaprimer (5 μl), undiluted PfuPlus polymerase (0.25 μl), and distilled water (37.75 μl). The following program was used: hold 94 °C for 2 min, 25 cycles of the following three steps: 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 437 s. Finally hold at 72 °C for 10 min. The template DNA was digested with 1 μl of DpnI (10 U/μl solution) for 3 h at 37 °C and 20 min at 80 °C.
The CAL‐A library III was generated via QuikChange™ with specific primers34 (Table S6). The plasmid containing the CAL‐A gene (100 ng) was mixed with 10× Buffer B (5 μl), dNTPs (2 μl), 25 mM MgSO4 (1 μl), 0.5 μl OptiTaq, forward primer 16.20 (1 μl), reverse primer 16.19 (1 μl), and distilled water (38.5 μl). The PCR program included the following temperature steps: hold at 95 °C for 2 min, afterwards 10 cycles of the following: hold at 95 °C for 15 s, 55 °C for 30 s, 72 °C for 437 s. Subsequent cycles (25) are prolonged by their elongation time by 20 s per cycle. Finally hold at 72 °C for 10 min. The template DNA was digested with 1 μl of DpnI (10 U/μl solution) for 3 h at 37 °C and 20 min at 80 °C.
The PCR‐products were first transformed into electrocompetent E. coli TOP10 cells and, after plasmid purification, transformed into electrocompetent E. coli C41 (DE3) cells, following the protocol previously described.
Screening of CAL‐A Variants using the Robotic Platform LARA
High‐throughput screening of the CAL‐A variant libraries was performed in the “laboratory automation robotic assisant” (LARA).20 For this purpose, ∼5,000 colonies transformed with each of the mutant libraries previously obtained were used to inoculate master MTP (M‐MTP) containing 220 μl per well of LB with ampicillin (0.1 mg/ml). Utilizing the QPix 420 Colony Picking System (Molecular Devices, USA) each well was inoculated with only one colony. The so prepared M‐MTPs were incubated at 37 °C and 700 rpm during 5 h and, subsequently, stored at 4 °C O/N until later usage. These plates served as a backup for the different libraries.
Overexpression of the CAL‐A library variants was achieved in microtiter plates (OE‐MTP) containing 230 μl/well of auto‐induction ZYM‐5052 medium29 per well and inoculated using the corresponding grown master MTP. Before inoculation M‐MTPs were revitalized by incubation for 2 h at 37 °C. After inoculation they were incubated at 37 °C and 700 rpm for 6 h. For better folding at the protein expression stage, the temperature was the temperature was changed to 30 °C for another 22 h. OD600nm was monitored during the process to verify the growth of the cells. The supernatants were transferred to a fresh plate and stored at 4 °C until performing the hydrolytic activity measurements after centrifugation of the OE‐MTPs at 4 °C and 4500 rpm.
Hydrolytic activity reaction conditions in LARA were similar to the conditions usually employed for hydrolytic activity measurements outside the robot. Initial hydrolysis rate of p‐nitrophenyl esters was followed spectrophotometrically for 5 min at λ=410 nm (ϵ=10.84 mM−1 cm−1) at 37 °C in a 96‐well microtiter plate. The assay was performed in sodium phosphate buffer (50 mM; pH=7.5) containing NaCl (1 mM), isopropanol (10% v/v), Triton X‐100 (0.5% v/v), the fatty acid derivative (1 mM, pNP−C14:0, pNP−C18:1 or pNP−C22:1) and 45 μl of SN sample for each reaction. Reactions were initiated by the addition of the substrate and the Triton X‐100 dissolved in isopropanol. The final reaction volume was 200 μl. One unit of lipase activity was defined as the amount of enzyme releasing 1 μmol p‐nitrophenol per minute.
Creation of pPICZα‐CALA‐His Vectors Containing the Desired Mutations
In order to overexpress in P. pastoris the enzyme CAL‐A wt and its most interesting variants, QuikChange reactions were performed using as template pPICZα‐CALA‐His/pGAPZα‐CALA‐His vector or the corresponding derivative. For this purpose, specific primers were designed to insert the desired mutation (Table S7 and S8). The reaction mixtures included the CAL‐A plasmid template (100 ng, ∼1 μl), 10× Buffer B (5 μl), dNTPs (2 μl), 25 mM MgSO4 (1 μl), 0.5 μl OptiTaq, forward primer (1 μl), reverse primer (1 μl), and distilled water (q.s. 50 μl). The PCR program included the following temperature steps: a denaturation step at 95 °C during 2 min; then 20 cycles of amplification including denaturation at 95 °C for 1 min, annealing at 50 °C for 1.5 min, and elongation at 72 °C for 2 min, and, finally, temperature was hold at 72 °C for 10 min. The template DNA was then digested with 1 μl of DpnI (10 U/μl solution) for 3 h at 37 °C and 20 min at 80 °C. The PCR‐products were first transformed into electrocompetent E. coli TOP10 cells as described above. When the presence of the desired mutation in the CAL‐A gene was confirmed by sequencing, a 30% glycerol stock of the strain was frozen at −80 °C and 20 ml of LB‐ls containing 25 μg/ml Zeocin were inoculated for plasmid purification and subsequent P. pastoris transformation (see Transformation of P. pastoris section).
Fatty Acid Enrichment Reactions
First, the right amount of lipase for the EFA enrichment reactions was tested by checking the rate of hydrolysis with 1, 5, 10, or 50 U of enzyme after 4 h reaction. The reaction solution contained 5 g Crambe ethyl esters or Camelina ethyl esters (Solutex, Zaragoza, Spain), 2 g gum arabic, and 100 ml buffer 50 mM sodium phosphate with 1 mM NaCl at pH 7.5. The emulsification was achieved by shearmixing at 24,000 rpm for 2 min (Ultra Turrax T25 basic, IKA Labortechnik, Staufen, Germany). Subsequently, 250 μl of the emulsion were dispensed in 1.5 ml polypropylene tubes for each time sample. The reaction was started by adding either buffer, or the corresponding Us of enzyme per reaction vessel.
Enzyme solutions were prepared using the lyophilised supernatants of the fermentation of P. pastoris GS115 strains overexpressing CAL‐A wild type and variants V290W, 1, 2, 3, 9, 16, 18, 19 and 20. Prior their use, enzyme solutions were dialyzed towards distilled, deionized water and their activity was determined towards pNP‐myristate as previously described. Reaction vessels were shaken at 1400 rpm and 37 °C. Reactions were stopped by adding 25 μl 4 N HCl. Samples were then vortexed and stored at −20 °C until extraction. After extraction of the samples, as described underneath, and subsequent evaporation of the solvent, samples were dissolved in 500 μl n‐hexane and 2 μl were loaded in TLC, as described later, to check for a sufficient rate of hydrolysis (10–40%).
Long chain fatty acid enrichment biocatalysis reactions were prepared in the same way as described above. Reactions were started by adding the corresponding amount of enzyme (5 or 10 U) and taking time samples after 0; 0.5; 1; 2; 4; 8; 24; and 48 h. All reactions were carried out in triplicates.
Thin‐Layer Chromatography
Thin‐layer chromatography (TLC) was used to preliminarily check the rate of hydrolysis of the EFA. For this purpose, 4 h time samples were extracted twice with 500 μl diethylether by vortexing for 1 min and centrifuging at 16200 g during 1 min. The upper organic layer was collected. The joined organic phases were dried with anhydrous Na2SO4 by vortexing for 1 min and centrifuging at 16200 g for 5 min. The organic solvent was then evaporated under a stream of nitrogen. Subsequently, the sample was dissolved in 500 μl n‐hexane and 2 μl of this mixture was spotted on a silica TLC plate (Silica gel on TLC Al plate, Sigma‐Aldrich). This plate was developed with a solvent composed of n‐hexane:diethyl ether:glacial acetic acid (79:20:1 v/v/v).35 For visualization, the TLC plate was stained with potassium permanganate solution (0.1 g/L KMnO4, 0.2 g/L CH3COOH, 0.5 g/L NaHCO3).
Fatty Acid Quantification of Different Oil Fractions
Reaction samples were extracted with 250 μl diethylether containing 2 mM tridecanoate ethyl ester and 2 mM tripentadecanoate (serving as internal standards), by vortexing for 1 min and centrifuging at 16 200 g during 1 min. The upper organic layer was collected. Each sample was extracted two more times with 500 μl pure diethylether. The joined organic phases were dried with anhydrous Na2SO4 by vortexing for 1 min and centrifuging at 16 200 g for 5 min. The organic solvent was then evaporated under a stream of nitrogen.
The residual oil was resuspended in 1 ml anhydrous n‐hexane and split in two aliquots for direct analysis of the FAEE and treatment with the methanolic hydrogensulfate method for esterification of the total FA.36 For the methanolic hydrogensulfate method, the 500 μl samples were treated with 50 μl 2,2‐dimethoxypropane and freshly prepared anhydrous 500 μl 2% methanolic hydrogen sulfate. The samples were shaken at 14000 rpm in a glass vial at 60 °C for 4 h. After the reaction, samples were extracted twice with 300 μl n‐hexane and the collected organic phases were dried over anhydrous Na2SO4. The organic layer was analyzed via GC (GC‐2010, Shimadzu, Duisburg, Germany).
For GC‐analysis of fatty acid methyl and ethyl esters (FAME and FAEE) a BPX‐70 column (SGE 0.25 mm diameter, 0.25 μm film thickness, 60 m length; SGE Analytical Science, Victoria, Australia) was used. Analysis of 1 μl of sample was carried out with a flow rate of 0.52 ml/min and a temperature program starting at (i) 180 °C for 33 min, followed by heating step (ii) to 230 °C at 10 °C/min and a final holding step for 5 min. Injector and detector temperatures were set at 240 °C and 290 °C, respectively.
Firstly, the samples were analyzed by peak identification of the different FAME′s to a standard FAME‐Mix (Sigma CRM47885, Steinheim, Germany) or to single FAEE standards, respectively (Figures S3 and S4). The peaks were normalized to their standards ethyl‐tridecanoate (FAEE) or methyl‐tridecanoate (H2SO4/MeOH method) and only the FAME peaks were treated with their theoretical response correction factors (Eq. 1) (RCF, Table 4).37
| (1) |
Table 4.
RCF for flame ionization detectors to convert to weight percent methyl ester.37
| FA | RCF | FA | RCF | FA | RCF |
|---|---|---|---|---|---|
| C13:0 | 1.06 | C18:2 | 0.99 | C20:3 | 0.97 |
| C15:0 | 1.03 | C18:3 | 0.98 | C22:0 | 0.97 |
| C16:0 | 1.02 | C20:0 | 0.98 | C22:1 | 0.97 |
| C18:0 | 1.00 | C20:1 | 0.98 | C24:0 | 0.96 |
| C18:1 | 0.99 | C20:2 | 0.97 | C24:1 | 0.96 |
The EFA or FFA amount of the samples was calculated by comparing the EFA amount determined by the EFA analysis towards the whole FA amount determined by the H2SO4/MeOH method (Eq. (2).
| (2) |
The theoretical yield of enriched FA was determined by comparing the total amount of this FA in the sample to its amount present in its enriched fraction (Either FFA or EFA) (Eq. (3).
| (3) |
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The COSMOS project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 635405. We are also grateful to Novozymes (Bagsvaerd, Denmark) for providing samples of CAL‐A.
K. Zorn, I. Oroz-Guinea, H. Brundiek, M. Dörr, U. T. Bornscheuer, Adv. Synth. Catal. 2018, 360, 4115.
Contributor Information
Henrike Brundiek, Phone: (+49) 3834 420 4367, FAX: (+49) 3834 420 744367.
Uwe T. Bornscheuer, Email: uwe.bornscheuer@uni-greifswald.de, Phone: (+49) 3834 420 4367, FAX: (+49) 3834 420 744367
References
- 1.
- 1a. Biermann U., Bornscheuer U., Meier M. A., Metzger J. O., Schäfer H. J., Angew. Chem. Int. Ed. 2011, 50, 3854–3871; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3938–3956; [Google Scholar]
- 1b. Schörken U., Kempers P., Eur. J. Lipid Sci. Technol. 2009, 111, 627–645; [Google Scholar]
- 1c. Borrelli G. M., Trono D., Int. J. Mol. Sci. 2015, 16, 20774–20840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leonard E. C., Ind. Crops Prod. 1993, 1, 119–123. [Google Scholar]
- 3.
- 3a. Lu C., Napier J. A., Clemente T. E., Cahoon E. B., Curr. Opin. Biotechnol. 2011, 22, 252–259; [DOI] [PubMed] [Google Scholar]
- 3b. Napier J. A., Graham I. A., Curr. Opin. Plant Biol. 2010, 13, 330–337. [DOI] [PubMed] [Google Scholar]
- 4. Tao C., He B. B., T. ASAE 2005, 48, 1471–1479. [Google Scholar]
- 5. Zorn K., Oroz-Guinea I., Brundiek H., Bornscheuer U. T., Prog. Lipid Res. 2016, 63, 153–164. [DOI] [PubMed] [Google Scholar]
- 6. Casas-Godoy L., Duquesne S., Bordes F., Sandoval G., Marty A., Methods Mol. Biol. 2012, 861, 3–30. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Kralovec J. A., Zhang S., Zhang W., Barrow C. J., Food Chem. 2012, 131, 639–644; [Google Scholar]
- 7b. McNeill G. P., Ackman R. G., Moore S. R., J. Am. Chem. Soc. 1996, 73, 1403–1407; [Google Scholar]
- 7c. Hoshino T., Yamane T., Shimizu S., Agric. Biol. Chem. 1990, 54, 1459–1467. [Google Scholar]
- 8.
- 8a. McNeill G. P., Sonnet P. E., J. Am. Chem. Soc. 1995, 72, 213–218; [Google Scholar]
- 8b. Volkova N., Li X., Zhu L.-H., Adlercreutz P., Sustain. Chem. Proc. 2016, 4; [Google Scholar]
- 8c. Kaimal T. N. B., Prasad R. B. N., Rao T. Chandrasekhara, Biotechnol. Lett. 1993, 15, 353–356; [Google Scholar]
- 8d. Sonnet P. E., Fogila T. A., Feairheller S. H., J. Am. Chem. Soc. 1993, 70, 387–391. [Google Scholar]
- 9.
- 9a. Borgdorf R., Warwel S., Appl. Microbiol. Biotechnol. 1999, 51, 480–485; [DOI] [PubMed] [Google Scholar]
- 9b. Brundiek H. B., Evitt A. S., Kourist R., Bornscheuer U. T., Angew. Chem. Int. Ed. 2012, 51, 412–414; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 425–428. [Google Scholar]
- 10.
- 10a. Joerger R. D., Haas M. J., Lipids 1994, 29, 377–384; [DOI] [PubMed] [Google Scholar]
- 10b. Klein R. R., King G., Moreau R. A., Haas M. J., Lipids 1997, 32, 123–130. [DOI] [PubMed] [Google Scholar]
- 11. Schmitt J., Brocca S., Schmid R. D., Pleiss J., Protein Eng. 2002, 15, 595–601. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Brundiek H. B., Padhi S. K., Kourist R., Evitt A., Bornscheuer U. T., Eur. J. Lipid Sci. Technol. 2012, 114, 1148–1153; [Google Scholar]
- 12b. Magnusson A. O., Rotticci-Mulder J. C., Santagostino A., Hult K., ChemBioChem 2005, 6, 1051–1056. [DOI] [PubMed] [Google Scholar]
- 13. Pleiss J., Fischer M., Schmid R. D., Chem. Phys. Lipids 1998, 93, 67–80. [DOI] [PubMed] [Google Scholar]
- 14.A. Svendsen, J. Vind, S. A. Patkar, K. Borch, Patent WO2008040738A1, 2008.
- 15. Ericsson D. J., Kasrayan A., Johansson P., Bergfors T., Sandstrom A. G., Backvall J. E., Mowbray S. L., J. Mol. Biol. 2008, 376, 109–119. [DOI] [PubMed] [Google Scholar]
- 16. Kirk O., Christensen M. W., Org. Process Res. Dev. 2002, 6, 446–451. [Google Scholar]
- 17. Widmann M., Juhl P. B., Pleiss J., BMC Genomics 2010, 11, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reetz M. T., Kahakeaw D., Lohmer R., ChemBioChem 2008, 9, 1797–1804. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Tang L., Gao H., Zhu X., Wang X., Zhou M., Jiang R., BioTechniques 2012, 52, 149–158; [DOI] [PubMed] [Google Scholar]
- 19b. Kille S., Acevedo-Rocha C. G., Parra L. P., Zhang Z. G., Opperman D. J., Reetz M. T., Acevedo J. P., ACS Synth. Biol. 2013, 2, 83–92. [DOI] [PubMed] [Google Scholar]
- 20. Dörr M., Fibinger M. P. C., Last D., Schmidt S., Santos-Aberturas J., Böttcher D., Hummel A., Vickers C., Voss M., Bornscheuer U. T., Biotechnol. Bioeng. 2016, 113, 1421–1432. [DOI] [PubMed] [Google Scholar]
- 21. Pfeffer J., Richter S., Nieveler J., Hansen C. E., Rhlid R. B., Schmid R. D., Rusnak M., Appl. Microbiol. Biotechnol. 2006, 72, 931–938. [DOI] [PubMed] [Google Scholar]
- 22. Muralidhar R. V., Chirumamilla R. R., Marchant R., Ramachandran V. N., Ward O. P., Nigam P., World J. Microbiol. Biotechnol. 2002, 18, 81–97. [Google Scholar]
- 23. Rogolska E., Cudrey C., Ferrato F., Verger R., Chirality 1993, 5, 24–30. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a.H. P. Heldt-Hansen, M. Ishii, S. A. Patkar, T. T. Hansen, P. Eigtved, Vol. 389, 1989, pp. 158–172;
- 24b. Briand D., Dubreucq E., Galzy P., Eur. J. Biochem. 1995, 228, 169–175. [DOI] [PubMed] [Google Scholar]
- 25. Grynberg H., Szczepanska H., J. Am. Chem. Soc. 1966, 43, 151–152. [Google Scholar]
- 26. María P. Domínguez de, Sánchez-Montero J. M., Sinisterra J. V., Alcántara A. R., Biotechnol. Adv. 2006, 24, 180–196. [DOI] [PubMed] [Google Scholar]
- 27. Müller J., Sowa M. A., Fredrich B., Brundiek H., Bornscheuer U. T., ChemBioChem 2015, 16, 1791–1796. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. You L., Arnold F. H., Protein Eng. 1996, 9, 77–83; [DOI] [PubMed] [Google Scholar]
- 28b. Schmidt-Dannert C., Arnold F. H., Trends Biotechnol. 1999, 17, 135–136. [DOI] [PubMed] [Google Scholar]
- 29. Studier F. W., Protein Expression Purif. 2005, 41, 207–234. [DOI] [PubMed] [Google Scholar]
- 30. Kouker G., Jaeger K.-E., Appl. Environ. Microbiol. 1987, 53, 211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brundiek H., Sass S., Evitt A., Kourist R., Bornscheuer U. T., Appl. Microbiol. Biotechnol. 2012, 94, 141–150. [DOI] [PubMed] [Google Scholar]
- 32. Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P., Biedermannova L., Sochor J., Damborsky J., PLoS Comput. Biol. 2012, 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyazaki K., in Methods in Enzymology, Vol. Volume 498 (Ed.: V. Christopher), Academic Press, 2011, pp. 399–406. [DOI] [PubMed] [Google Scholar]
- 34. Zheng L., Baumann U., Reymond J. L., Nucleic Acids Res. 2004, 32, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bertram M., Manschot-Lawrence C., Flöter E., Bornscheuer U. T., Eur. J. Lipid Sci. Technol. 2007, 109, 180–185. [Google Scholar]
- 36.
- 36a. Strelkov S., Elstermann M. von, Schomburg D., Biol. Chem. 2004, 385, 853–861; [DOI] [PubMed] [Google Scholar]
- 36b. Tai M., Stephanopoulos G., Metab. Eng. 2013, 15, 1–9. [DOI] [PubMed] [Google Scholar]
- 37. Christie W. W., Lipid analysis: Isolation, seperation, identification and structural analysis of lipids, Vol. 15, Third ed., The oil press lipid library, Shepperton (Middlesex, UK), 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary