

A “tug-of-war” between kinases and phosphatases establishes the phosphorylation states of proteins. While serine and threonine phosphorylation can be catalyzed by more than 400 protein kinases, the majority of serine and threonine dephosphorylation is carried out by seven phosphoprotein phosphatases (PPPs). We have developed a chemical proteomic strategy for the systematic and quantitative interrogation of endogenous PPP catalytic subunits and their interacting proteins, including regulatory and scaffolding subunits (the “PPPome”) in human cancer cell lines, mouse tissues, and yeast species.
Keywords: Phosphorylation, Protein complex analysis, Protein Phosphatases, Protein-Protein Interactions, Affinity proteomics, Quantification
Graphical Abstract

Highlights
Chemical proteomics strategy for quantitative profiling of phosphoprotein phosphatases.
Compatible with quantitative multiplexing approaches.
Applicable to many samples types including tissues from human to yeast.
Abstract
A “tug-of-war” between kinases and phosphatases establishes the phosphorylation states of proteins. While serine and threonine phosphorylation can be catalyzed by more than 400 protein kinases, the majority of serine and threonine dephosphorylation is carried out by seven phosphoprotein phosphatases (PPPs). The PPP family consists of protein phosphatases 1 (PP1), 2A (PP2A), 2B (PP2B), 4 (PP4), 5 (PP5), 6 (PP6), and 7 (PP7). The imbalance in numbers between serine- and threonine-directed kinases and phosphatases led to the early belief that PPPs are unspecific and that kinases are the primary determinants of protein phosphorylation. However, it is now clear that PPPs achieve specificity through association with noncatalytic subunits to form multimeric holoenzymes, which expands the number of functionally distinct signaling entities to several hundred. Although there has been great progress in deciphering signaling by kinases, much less is known about phosphatases.
We have developed a chemical proteomic strategy for the systematic interrogation of endogenous PPP catalytic subunits and their interacting proteins, including regulatory and scaffolding subunits (the “PPPome”). PP1, PP2A, PP4, PP5, and PP6 were captured using an immobilized, specific but nonselective PPP inhibitor microcystin-LR (MCLR), followed by protein identification by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in a single analysis. Here, we combine this approach of phosphatase inhibitor bead profiling and mass spectrometry (PIB-MS) with label-free and tandem mass tag (TMT) quantification to map the PPPome in human cancer cell lines, mouse tissues, and yeast species, through which we identify cell- and tissue-type-specific PPP expression patterns and discover new PPP interacting proteins.
More than three-quarters of all proteins are phosphorylated at one or more sites in human cells (1). A “tug of war” between protein kinases and protein phosphatases establishes the phosphorylation states of proteins to control their function. Proper temporal and spatial coordination of these opposing activities is essential to maintain appropriate phosphorylation site occupancy and regulate cellular signaling. Deregulation of either kinase or phosphatase activities disrupts this balance and is commonly observed in human diseases, including cancer.
Serine/threonine phosphorylation is catalyzed by over 400 protein kinases (2). Intriguingly, the majority (> 90%) of serine/threonine dephosphorylation is carried out by the small family of phosphoprotein phosphatases (PPP)1, most prominently PP1 and PP2A (3–5). The PPP family also includes PP2B, PP4–7, PPT, and PPZ (supplemental Fig. 1). PPP catalytic subunits are among the proteins with the highest degree of sequence conservation from yeast to human: they share the same catalytic mechanism, and the residues required for catalysis are 100% conserved among PPP family members (6, 7). The early belief that PPPs are unspecific and that kinases are the primary determinants of protein phosphorylation derived from two observations: the imbalance in numbers of serine and threonine directed protein kinases and phosphatases and the fact that kinases, compared with free PPP catalytic subunits, exhibit greater site selectivity in vitro (7–9). However, it has recently become clear that PPP specificity and regulation is achieved when catalytic subunits associate with noncatalytic subunits to form multimeric holoenzymes (7). In mammals, the PPP family consists of seven enzymes (PP1, PP2A, PP2B, PP4, PP5, PP6, and PP7). PPP catalytic subunits interact with a diverse array of regulatory and scaffolding subunits (>200), endogenous protein inhibitors, and substrates to modulate their activity and establish substrate specificity (3). PP1 forms dimeric holoenzymes with more than 150 regulatory subunits (3), while PP2A, PP4, and PP6 mostly form trimeric holoenzymes with greater than 30, five, and six regulatory and scaffolding subunits, respectively (7).
While the combinatorial approach to PPP complexity closes the gap in numbers between phosphorylation and dephosphorylation signaling entities, it also creates an analytical challenge for investigating PPP signaling and its responses to external cues and stresses, drug treatment, and pathological rewiring. To do so, system-wide approaches are needed that capture PPP catalytic subunits and their interacting proteins, the PPPome, as a whole. Indeed, many PPP-associated proteins participate in more than one protein complex, and their associations are spatially and temporally regulated by a plethora of factors (10). Thus, to understand PPP signaling it is essential to investigate the interactions of the catalytic and noncatalytic subunits that constitute the PPP holoenzymes, rather than their protein abundances.
Great progress has been made in deciphering protein phosphorylation by kinases (11), in large part due to a chemical proteomic strategy that utilizes kinase inhibitors immobilized on beads and MS to enrich and quantify large swaths of the human kinome (12–19). By way of one example, pioneering work utilizing this technology enabled breakthrough discoveries in describing the expressed cancer kinome and its reprogramming upon kinase inhibition (16, 17). However, while kinome profiling provides global insights into one aspect of phosphorylation signaling in cancer, we lack this information for the opposing dephosphorylation reaction.
We have developed a chemical proteomic strategy that utilizes an immobilized nonselective PPP inhibitor MCLR combined with MS-based proteomics for the efficient capture, identification, and quantification of endogenously expressed PPPs, including PP1, PP2A, PP4, PP5, PP6, PPT, and PPZ and their interacting proteins in a single analysis (named PIB-MS) (supplemental Fig. 1).
EXPERIMENTAL PROCEDURES
Phylogenetic Analysis
PPP protein sequences from the UniProt database for human, mouse, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Neurospora crassa, and Candida albicans were analyzed by http://www.phylogeny.fr/simple_phylogeny.cgi in one-click mode using MUltiple Sequence Comparison by Log-Expectation (MUSCLE) alignment, Gblocks curation, PhyML phylogeny, and radial tree by TreeDyn tree rendering.
Cell Lines, Mice, Yeasts, and Antibodies
293FT, HeLa, MCF7, SF126, SF268, SF839, SKBR3, SW1088, T47D, and U87 were grown as adherent cultures in Dulbecco's modified Eagle's media (Cellgro Mediatech, Inc., Manassas, VA) with 10% fetal bovine serum (Hyclone, Logan, Utah) and penicillin—streptomycin (100U/ml and 100 μg/ml, respectively; Cellgro Mediatech, Inc.) at 37 °C in a humidified incubator with 5% CO2. 293FT cells were purchased from LifeTechnology (Carlsbad, CA). SF126, SF268, SF839, and U87 cells were a gift from Dr. Mark Israel (Dartmouth College). MCF7, SKBR3, and T47D cells were a gift from Dr. Todd Miller (Dartmouth College). All cell lines were regularly tested for mycoplasma contamination. Mouse tissues from six-week-old male C57BL/6J mice were a gift from Dr. Matthew Havrda (Dartmouth College). Yeast cells were harvested by centrifugation at 8,000 g at 4 °C, washed once with 200 ml of ice-cold PBS, and then centrifuged again. The pellet was resuspended in 1/3 w/v ice-cold PBS containing complete protease inhibitors (Roche, South San Francisco, CA) and 1 mm Phenylmethanesulfonyl fluoride (PMSF) and then frozen dropwise in liquid nitrogen. Frozen pellets were lysed by 2 min of grinding in a prechilled coffee bean grinder; lysis efficiency of the resulting powder was ∼80% as judged by microscopy. S. cerevisiae powder was a gift from Dr. Charles Cole (Dartmouth College). S. pombe powder was a gift from Dr. James Moseley (Dartmouth College). N. crassa powder was a gift from Dr. Jay Dunlap (Dartmouth College). C. albicans powder was a gift from Dr. Lawrence Myers (Dartmouth College). Antibodies against PP1, PP2A, PP4C, PP5C, and PP6C were purchased from Bethyl Laboratories (Montgomery, TX).
PIB Synthesis
PIBs were synthesized as described previously (20). Briefly, 1 mg/ml MCLR in ethanol (1 vol., Millipore, Burlington, MA) was reacted with water (1.5 vol.), DMSO (2 vol., SIGMA, St. Louis, MO), 5 N NaOH (0.67 vol.), and 1 g/ml cysteamine hydrochloride (1 vol., SIGMA). The reaction mixture was briefly purged with N2 gas and incubated at 50 °C for 1 h. The solution was then cooled and mixed with an equal volume of glacial acetic acid, diluted fivefold with 0.1% v/v TFA, followed by dropwise addition of 100% v/v TFA to reduce the pH to 1.5. The sample was applied to a C18 solid-phase extraction cartridge (ThermoFisher Scientific, Chelmsford, MA) equilibrated with 0.1% v/v TFA. The column was washed with 0.1% v/v TFA in 10% acetonitrile, followed by elution with 0.1% v/v TFA in acetonitrile. The aminoethanethiol-MC eluate was dried by vacuum centrifugation and dissolved in 0.02 ml of methanol. 1.5 g (per mg of MCLR used) of activated CNBr Sepharose beads (GE Healthcare, Marlborough, MA) were swelled in water for 15 min followed by washing with 1 mm cold HCl to make 4.5 ml of bead slurry. The aminoethanethiol-MC was coupled to the activated beads in coupling buffer (100 mm NaHCO3, pH 8.2) for 1.5 h at room temperature. The beads were blocked with 100 mm Tris-HCl, pH 8.0, for 1 h, followed by washing six times alternately with 50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, and 50 mm sodium acetate, pH 4.0, 0.5 m NaCl. The beads were stored in 25 mm Tris-HCl, pH 7.5, at 4 °C until use.
PIB Titration
Five microliters of packed PIB resin was incubated with increasing amounts of recombinant PP1 in PIB pulldown buffer (50 mm Tris-HCl, pH 7.5, (SIGMA), 500 mm NaCl (SIGMA), 5 mm beta-glycerophosphate (SIGMA), 0.5% Triton X-100 (SIGMA), 0.1 mm DTT (SIGMA), and one EDTA-free protease inhibitor tablet (Roche) per 10 ml of lysis buffer at 4 °C for 1 h while rotating. Afterward, PIBs were washed with PIB pulldown buffer and eluted with 1% sodium dodecyl sulfate (SDS) overnight at room temperature. Ten percent of elutes were analyzed by SDS-PAGE gel electrophoresis and visualized by Coomassie Brilliant Blue staining.
PPP Catalytic Subunit Purification and Activity Assay
For PP1β, PP2AB, PP4C, and PP6C enrichments, 293FT Freestyle cells were transiently transfected with p3XFlag-CMV10-PP1β, -PP2AB, -PP4C, or -PP6C, respectively. Forty-eight hours after transfection, cells were lysed in PIB pulldown buffer and the lysate was split in a 1:4 ratio to achieve a similar yield of PPP catalytic subunit in enrichments from total lysates versus lysates depleted with PIBs. PIBs were added to the depleted condition, and lysates were incubated for 45 min at 4 °C. PIBs were collected by centrifugation and supernatant was transferred to a new tube. PPP catalytic subunits were purified using anti-Flag M2 affinity gel (SIGMA) and eluted with 3xFLAG-peptide (SIGMA) (final concentration 150 ng/μl) as previously described (21). Purifications were performed in triplicates. From each preparation, 10% was removed for trichloroacetic acid (TCA) precipitation and interactome analysis. PPP catalytic subunit abundances in each sample were determined by label-free intensity-based absolute quantification (iBAQ) quantification and confirmed by Western blotting using anti-Flag antibody (SIGMA). Phosphatase activity assays were carried out according to Chattopadhyay et al. (22). Briefly, total and depleted PP6C were diluted to a final concentration of 60 nm in 6,8-Difluoro-4-Methylumbelliferyl Phosphate (DiFMUP) buffer (30 mm Hepes, pH 7, (SIGMA), 1 mm DTT, 0.01% Triton X-100 (SIGMA), 0.1 mg/ml BSA (SIGMA), 10 mm sodium ascorbate (SIGMA), 1 mm MnCl2). Reactions were started by the addition of DiFMUP to a final concentration of 125 μm DiFMUP. Assays were read every 45 s on for 45 min on a SpectraMax i3X (Molecular Devices, San Jose, CA) plate reader.
PIB Pulldowns
Cell, mouse tissues, or yeast powder were lysed in PIB pulldown buffer (50 mm Tris-HCl, pH 7.5, (SIGMA), 500 mm NaCl (SIGMA), 5 mm beta-glycerophosphate (SIGMA), 0.5% Triton X-100 (SIGMA), 0.1 mm DTT (SIGMA), and one EDTA-free protease inhibitor tablet (Roche) per 10 ml of lysis buffer) at a concentration of 1 mg/ml lysates were sonicated three times for 10 s with intermittent cooling on ice and clarified by centrifuging at 4 °C for 15 min at 21,000 × g. Lysates were transferred to a new tube, 10 μl of solid packed PIB resin were added, and incubated at 4 °C for 1 h while rotating. Afterward, PIBs were washed three times with pulldown buffer, and proteins were eluted with 1% SDS (SIGMA) overnight at room temperature. For competition with free MCLR, 1 μm MCLR (Millipore) was added to lysates after clarifying for 15 min before addition of PIBs. For competition with okadaic acid (OA), 100 nm OA (LC Labs, Woburn, MA) were added. For MCLR titration, increased amounts of MCLR were added to lysates after clarifying for 15 min before addition of PIBs. Proteins were precipitated by the addition of TCA to a final concentration of 20%, incubated on ice for 15 min and spun at 4 °C for 15 min at 21,000 × g, followed by two washes with 10% TCA and two washes with acetone (Burdick & Jackson, Muskegon, MI). Pelleted proteins were dried, resuspended in 25 μl of either 25 mm ammonium bicarbonate (SIGMA) for label-free analysis or 133 mm Hepes (SIGMA), pH 8.5, for TMT-labeling containing 50 ng trypsin (Promega, Madison, WI) and incubated at 37 °C overnight. For label-free analysis, digests were quenched with 25 μl 5% formic acid (Burdick & Jackson)/50% acetonitrile (Burdick & Jackson), and dried in a vacuum centrifuge. For TMT-labeling, acetonitrile to a final concentration of 20% was added and peptides were transferred to dried, individual TMT reagent (ThermoFisher Scientific), vortexed, and mixed with the reagent. After 1 h at room temperature, each digest was quenched with 3 μl of 500 mm ammonium bicarbonate solution for 10 min, mixed into the two sets of multiplexes, diluted threefold with 0.1% TFA in water, and desalted using C18 solid-phase extraction cartridges (ThermoFisher Scientific). The desalted multiplexes were dried by vacuum centrifugation.
MCLR Titration Data Analysis
For MCLR competitive binding assays in 293FT cell lysates, increasing amounts of MCLR (10 fM to 10 μm) were added to lysates and PIB pulldowns were performed. After TCA precipitation and digestion, each condition was labeled with an individual TMT channel and analyzed by LC-MS/MS as described above in technical triplicates. TMT intensities for PP1α, PP1β, PP1γ, PP2A, and PP4C-6C were normalized to the maximum measured amount of the respective protein. Curve fitting was performed in GraphPad Prism by least-squares nonlinear regression one site—Fit logIC50.
Experimental Design and Statistical Rationale
Cell Line Analyses
PIB pulldowns were performed in triplicates or quadruplets from 0.5 mg total protein in the presence or absence of 1 μm MCLR and analyzed by label-free LC-MS/MS and data analysis as described above. Common contaminants, including keratin, collagen, 40S and 60S ribosomal proteins, and heterogeneous nuclear ribonucleoprotein proteins, were removed from the analysis if they were not previously identified as PPP-binding proteins. For a protein to be considered for further analysis it had to be identified with total peptide count > 1 in control samples for known PPP subunits or an average total peptide count > 2 for unknowns, more than one quantification in control samples, and have a unique matches count > 0. Furthermore, the difference in iBAQ quantification in control or MCLR-treated samples had to be statistically significant (p value < 0.05; two-tailed Student's t test, unequal variance), or the protein must have been completed competed off by MCLR, and the difference in average iBAQ abundance between control and MCLR-treated samples has to be more than the smallest difference for any catalytic subunit. For comparison of OA and MCLR specificity, missing values were set to log2 of 13 in Perseus (23). To determine binding specificity, control, and MCLR-treated HeLa pulldowns were compared. To distinguish PP1 and PP5 from PP2A, PP4, and PP6 subunits, binding upon MCLR and OA treatment was compared by two-tailed Student's t test, unequal variance, and principal component analysis. For comparison of PPP catalytic subunit abundances between cell lines, the subunit had to be present in all samples. iBAQ abundances were averaged across all replicates on a per-cell-line basis. iBAQ abundances for all proteins present in all samples were summed and total abundances were normalized. Normalized iBAQ abundances were analyzed by hierarchical clustering of rows and columns using the Euclidean distance and average linkage in Perseus (23). For network analysis, proteins identified as specifically interacting with PPP catalytic subunits were compared with PP1α, PP1β, PP1γ, PP2A, and PP4C-6C interactors in the BioGRID database (24) and analyzed in Cytoscape (25, 26). Potentially new PPP-interacting proteins were connected to known interactors using the STRING database.
Mouse Tissue Analysis
PIB pulldowns were performed in triplicates from 0.5 mg total protein in the presence or absence of 1 μm MCLR, TMT-labeled, and analyzed by replicate injection by LC-MS/MS as described above. Common contaminants, including keratin, collagen, 40S and 60S ribosomal proteins, and heterogeneous nuclear ribonucleoprotein proteins, were removed from the analysis if they were not previously identified as PPP-binding proteins. For a protein to be considered for further analysis it had to be identified with a total peptide count > 1 for known PPP subunits or an average total peptide count > 2 for unknowns, in at least one of the replicate injections and have unique matches count > 0. Missing values were imputed in Perseus (23) from a normal distribution to enable visualization by Volcano plot. Statistical analysis of protein quantification was carried out in Perseus (23) by two-tailed Student's t test on a per-tissue-type basis. For comparison of PPP subunit abundances between different tissues, the same internal standard consisting of a PPP pulldown from a mixed lysate of all mouse tissues was included as a seventh channel. TMT intensities were normalized to the internal standard on a per-multiplex basis, followed by normalization to the median of the total amount of catalytic subunits in each channel across all multiplexes. Normalized TMT intensities were analyzed by hierarchical clustering of rows and columns using the Euclidean distance and average linkage in Perseus (23).
Yeast Tissue Analysis
PIB pulldowns were performed in triplicates from 1 mg total protein in the presence or absence of 1 μm MCLR, TMT-labeled, and analyzed by replicate injection by LC-MS/MS as described above. Statistical significance of the difference in binding in the presence and absence of MCLR (p value < 0.05) was determined by two-tailed Student's t test, unequal variance). Protein–protein interactions of specifically identified proteins were extracted from the STRING database and analyzed in Cytoscape (25, 26).
Short Linear Motif Analysis
The human, mouse, S. cerevisiae, S. pombe, N. crassa, and C. albicans proteomes were investigated for the presence of short linear interaction motifs (SLiM) in protein sequences using SLiMSearch4 (27). [RK].{0,1}[VIL].[FW], [SG][I][L][KR], and [R]..[Q][VIL][KR].[YW] for PP1 and [LMFY]..[IVL].E, [LCVMIF][ST][P][ILVM].[E] for PP2A were entered into SLiMSearch4 and all results per organism were retrieved and compared with the list of proteins we identified as specifically bound to PIBs.
Label-free LC-MS/MS Analysis
PIB pulldowns were analyzed on a Q-Exactive Plus quadrupole Orbitrap mass spectrometer (ThermoScientific) equipped with an Easy-nLC 1000 (ThermoScientific) and nanospray source (ThermoScientific). Peptides were resuspended in 5% methanol/1.5% formic acid and loaded on to a trap column (1-cm length, 100-μm inner diameter, ReproSil, C18 AQ 5 μm 120 Å pore (Dr. Maisch, Ammerbuch, Germany)) vented to waste via a microtee and eluted across a fritless analytical resolving column (35-cm length, 100-μm inner diameter, ReproSil, C18 AQ 3 μm 120 Å pore) pulled in-house (Sutter P-2000, Sutter Instruments, San Francisco, CA) with a 60-min gradient of 5–30% LC-MS buffer B (LC-MS buffer A: 0.0625% formic acid, 3% acetonitrile; LC-MS buffer B: 0.0625% formic acid, 95% acetonitrile). The Q-Exactive Plus was set to perform an Orbitrap MS1 scan (r = 70K; automated gain control [AGC] target = 3e6) from 350–1500 Thomson, followed by higher energy collisional dissociation (HCD) MS2 spectra on the 10 most abundant precursor ions detected by Orbitrap scanning (r = 17.5K; AGC target = 1e5; max ion time = 75ms) before repeating the cycle. Precursor ions were isolated for HCD by quadrupole isolation at width = 0.8 Thomson and HCD fragmentation at 26 normalized collision energy. Charge state 2, 3, and 4 ions were selected for MS2. Precursor ions were added to a dynamic exclusion list ± 20 ppm for 20 s. Raw data were searched using COMET (release version 2014.01) in high-resolution mode (28) against a target–decoy (reversed) (29) version of the human (UniProt; downloaded 2/2013, 40,482 entries of forward and reverse protein sequences), or mouse (UniProt; downloaded 2/2013, 33,180 entries of forward and reverse protein sequences), or S. cerevisiae (UniProt; downloaded 2/2008, 13,242 entries of forward and reverse protein sequences), or S. pombe (UniProt; downloaded 2/2008, 10,178 entries of forward and reverse protein sequences), or N. crassa (UniProt; downloaded 3/2006, 20,516 entries of forward and reverse protein sequences), or C. albicans (UniProt; downloaded 3/2017, 14,008 entries of forward and reverse protein sequences) proteome sequence database with a precursor mass tolerance of ± 1 Da and a fragment ion mass tolerance of 0.02 Da, and requiring fully tryptic peptides (K, R; not preceding P) with up to three miscleavages. Static modifications included carbamidomethyl cysteine and variable modifications included oxidized methionine. Searches were filtered using orthogonal measures including mass measurement accuracy (± 3ppm), Xcorr for charges from +2 through +4, and dCn targeting a <1% false discovery rate at the peptide level. Quantification of LC-MS/MS spectra was performed using MassChroQ (30) and the iBAQ method (31).
TMT-based Quantitative Data Analysis
TMT-labeled PIB pulldowns were analyzed on an Orbitrap Fusion (32) mass spectrometer (ThermoScientific) equipped with an Easy-nLC 1000 (ThermoScientific). Peptides were resuspended in 8% methanol/1% formic acid across a column (45-cm length, 100-μm inner diameter, ReproSil, C18 AQ 1.8 μm 120 Å pore) pulled in-house across a 2-h gradient from 8% acetonitrile/0.0625% formic acid to 37% acetonitrile/0.0625% formic acid. The Orbitrap Fusion was operated in data-dependent, SPS-MS3 quantification mode (33, 34) wherein an Orbitrap MS1 scan was taken (scan range = 350–1,500 m/z, r = 120K, AGC target = 2.5e5, max ion injection time = 100 ms), followed by ion trap MS2 scans on the most abundant precursors for 4 s (max speed mode, quadrupole isolation = 0.6 m/z, AGC target = 4e3, scan rate = rapid, max ion injection time = 60 ms, minimum MS1 scan signal = 5e5 normalized units, charge states = 2, 3, and 4 included, collision-induced dissociation (CID) energy = 33%) and Orbitrap MS3 scans for quantification (r = 15K, AGC target = 2e4, max ion injection time = 125 ms, HCD collision energy = 48%, scan range = 120–140 m/z, synchronous precursors selected = 10). The raw data files were searched using COMET with a static mass of 229.162932 on peptide N termini and lysines and 57.02146 Da on cysteines and a variable mass of 15.99491 Da on methionines against the target–decoy version of the respective FASTA database (UniProt; www.uniprot.org) and filtered to a <1% false discovery rate at the peptide level. Quantification of LC-MS/MS spectra was performed using in-house developed software. For protein abundance measurements, TMT peptides intensities were summed to protein intensities.
RESULTS
Synthesis and Assessment of PIBs
MCLR is a naturally occurring small molecule inhibitor of PP1, PP2A, PP4–6, PPT1, and PPZ1 that inhibits them at nanomolar concentration (31). We began by immobilizing MCLR on activated Sepharose beads (35) to generate PIBs. To assess the capacity of PIBs, we incubated them with varying amounts of recombinant PP1 (supplemental Fig. 2A), washed the beads, eluted PP1, and analyzed the eluates by SDS-PAGE and Coomassie staining (supplemental Fig. 2B), which established the binding capacity of PIBs to be up to 3 mg/ml (supplemental Fig. 2B). To test the performance of PIBs in removing PPPs from lysates, we performed pulldown experiments from HeLa cells (Fig. 1). Cells were lysed, lysates were split in half, and either control treated or treated with MCLR for 15 min to distinguish specific from unspecific binders. Equal amounts of PIBs were incubated with each lysate, followed by washing, elution, tryptic digestion of the eluates, and analysis by LC-MS/MS, database searching, and iBAQ (supplemental Table I). To distinguish specific from unspecific binders, iBAQ areas of proteins identified in control- and MCLR-treated lysates were compared. Using this approach, we readily detected all MCLR-sensitive catalytic PPP subunits (PP1α, PP1β, PP1γ, PP2A, PP4C, PP5C, and PP6C) as well as associated subunits and interacting proteins (supplemental Table I). In the absence of MCLR, the log2 iBAQ areas of proteins in replicate analyses were comparable (r = 0.92) (Fig. 2A). However, upon preincubation of lysates with free MCLR, binding of catalytic and associated subunits and PPP interacting proteins was strongly reduced or abolished (Fig. 2B). Statistical analysis of triplicate log2 ratios of control/MCLR iBAQ areas showed a reproducible reduction in PIB binding upon MCLR addition (supplemental Table I). By comparing PIB binding in the presence and absence of MCLR, we identified 71 known PPP subunits, as well as seven proteins previously not known to be PPP interactors as specifically captured by PIB-MS in HeLa cells (supplemental Table I). In addition, we identified on average 335 proteins that were unspecifically bound to PIBs, even upon addition of MCLR (Fig. 2C). Interestingly, the summed iBAQ area of specific PIB binders was on average 8.8-times higher compared with unspecific binders and was on average 200-fold reduced upon addition of MCLR (Fig. 2D). We also confirmed our label-free MS results by Western blotting (supplemental Fig. 3).
Fig. 1.

PIB-MS workflow. Workflow for PIB-MS. HeLa cells were lysed, split in half, and control- or MCLR-treated before PIB pulldown. Washed pulldowns were eluted, TCA precipitated, trypsin-digested and analyzed by LC-MS/MS. Statistical analysis of triplicates was performed to distinguish specific (green dots) from unspecific (black dots) binders.
Fig. 2.
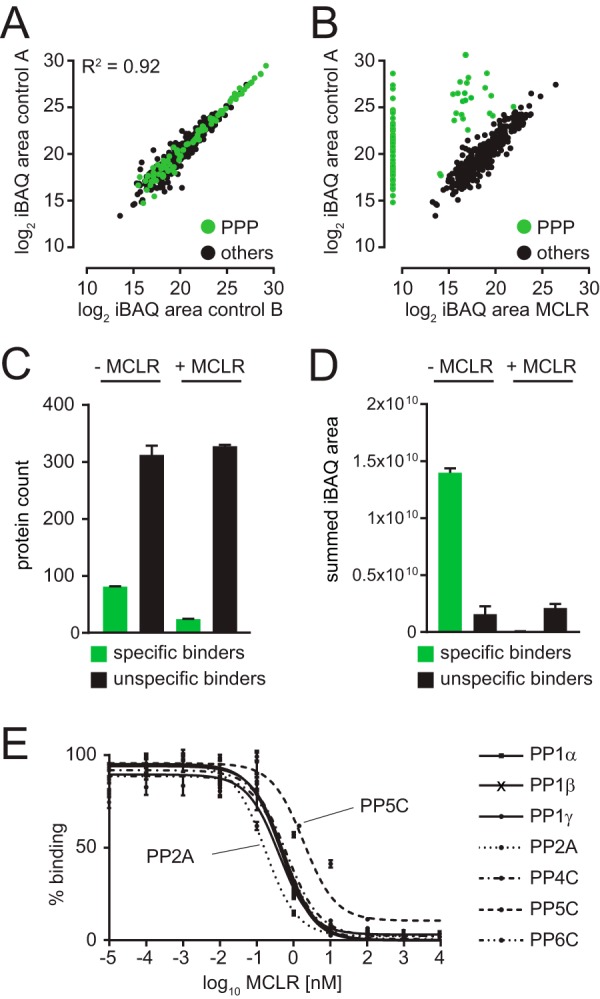
Identification of specifically bound proteins by MCLR competition. (A) Scatter plot of log2 iBAQ area of biological replicates of PIB pulldowns from control HeLa cell lysates. (B) Scatter plot of log2 iBAQ area of PIB pulldowns from control versus MCLR-treated HeLa cell lysates. (C) Bar graph depicting the number of proteins identified as specific or unspecific bound to PIB in the presence and absence of MCLR. (D) Bar graph depicting summed iBAQ areas of specific or unspecific bound to PIB in the presence and absence of MCLR. (E) MCLR competitive binding assays in 293FT cell lysates with increasing amounts of MCLR (10 fM to 10 μm). Overlay of titration curves for PP1α, PP1β, PP1γ, PP2A, PP4, PP5, and PP6. Error bars indicate the standard deviation of triplicate analyses. Note that PP2A is the most sensitive and PP5 is the least sensitive to MCLR.
Next, we performed quantitative profiling of PPP binding to PIBs to determine the dose–response of PPP catalytic subunit recovery to free MCLR from 10 fM to 10 μm. Consistent with relative IC50 values determined in vitro (36), we found that PP1α, PP1β, PP1γ, PP4C, and PP6C (IC50 0.41–0.69 nm) showed comparable dose–responses to MCLR (supplemental Figs. 4A-4E). PP2A was the most (IC50 0.17 nm), and PP5C was the least (IC50 1.94 nm) sensitive to MCLR in competitive binding assays in 293FT cell lysates (supplemental Figs. 4F and 4G, Fig. 2E, supplemental Table II).
Compared with MCLR, OA displays a 100-fold difference in inhibition of PP1 and PP5C compared with PP2A and PP4C in vitro (36). To determine if we can detect these differences by PIB-MS, we treated HeLa cell lysates with DMSO (control), 100 nm OA or 1 μm MCLR, and subsequently performed PIB-MS on these lysates. Indeed, we found that while all PPP subunits were lost upon addition of 1 μm MCLR (Fig. 3A), the addition of 100 nm OA selectively disrupted PIB binding to PP2A, PP4C, and PP6C as well as their associated subunits, while PP1 and PP5C were still bound (Fig. 3B). PCA of iBAQ areas of PPP subunits in control, OA-, and MCLR-treated samples clearly distinguished the three groups (Fig. 3C). Thus, through the use of PPP inhibitors with differential PPP inhibition selectivity, it is possible to distinguish PP1 and PP5 and from PP2A-like (PP2A, PP4, PP6) PPP catalytic and associated subunits by PIB-MS.
Fig. 3.
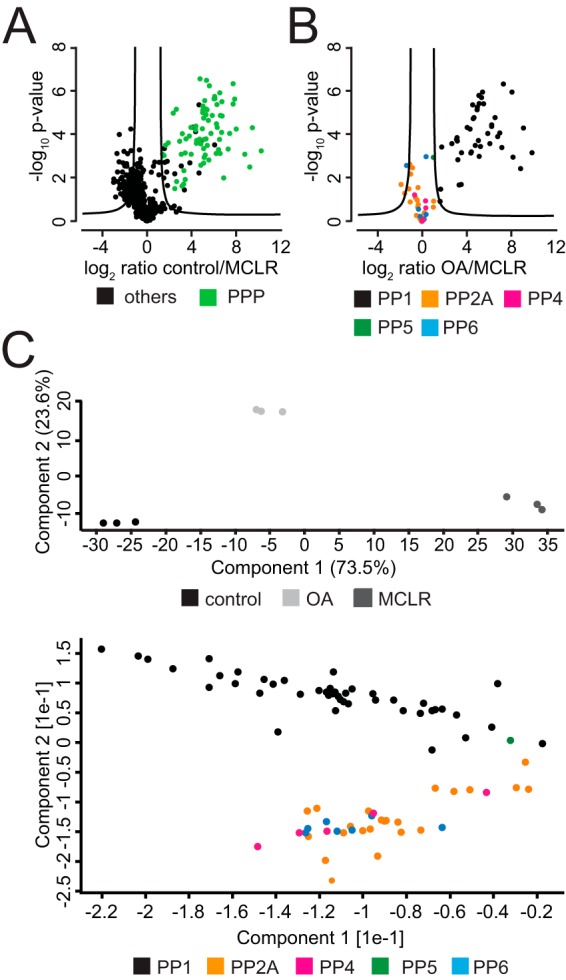
Comparison of PPP binding upon MCLR and OA treatment. (A) Volcano plot of PIB-MS analyses of control and MCLR-treated HeLa lysates. (B) Volcano plot of PIB-MS analyses of known PPP subunits of OA- and MCLR-treated HeLa lysates. (C) Principal component analysis of PIB-MS analyses of known PPP subunits of OA- and MCLR-treated HeLa lysates.
PPPome Abundance Analysis in Glioblastoma and Breast Cancer Cell Lines
PPPs are often functionally inactivated in many cancers, including breast, colorectal, uterine, lung, and melanoma, through somatic mutations, decreased subunit expression, and changes in posttranslational modifications that regulate phosphatase activity (37–44). PP2A-like phosphatases are considered tumor suppressors based on the tumor-promoting activities of their naturally occurring small molecule inhibitors (45). Furthermore, PP2A is the target of oncogenic viruses, which alter PP2A enzymatic activity and substrate specificity (46–48). Mutations in PP2A and PP6 subunits can reduce or alter regulatory and scaffolding subunit binding, shifting the balance in phosphorylation signaling in cells (41, 43, 44, 49, 50).
To evaluate the performance of PIB-MS in detecting these phenomena, we performed PIB pulldowns from lysates of three human breast cancer (SKBR3, MCF7, T47D) and five human glioblastoma (SF126, SF268, SF539, SW1088, U87) cell lines (supplemental Table IV). For each cell line, we performed PIB-MS on at least three biological replicates in the presence and absence of MCLR to distinguish specific from unspecific binders. Next, we performed agglomerative hierarchical clustering on averaged iBAQ areas identified for each PPP catalytic subunit and specific binder identified in each cell line (Fig. 4A). Interestingly, this analysis revealed that cell lines clustered according to their cancer type of origin, suggesting that breast cancer and glioblastoma cell lines have specific PPP expression patterns indicative of their origin.
Fig. 4.

PPP profiling in human cancer cell lines. (A) Hierarchical clustering of rows and columns using the Euclidean distance and average linkage of proteins specifically identified across all human breast cancer and human glioblastoma cell lines. (B) Network analysis of all proteins specifically identified in any breast cancer or glioblastoma cell line. (C) Validation of potentially new PPP interactors.
The majority of proteins identified by PIB-MS were known PPP subunits. We identified all MCLR-sensitive PPP human catalytic subunits (PP1α, PP1β, PP1γ, PP2AA, PP2AB, PP4C, PP5C, and PP6C). While PP1 forms heterodimeric holoenzymes with regulatory subunits that can also be substrates, PP2A, PP4, and PP6 form heterotrimeric enzymes with regulatory and scaffolding subunits which in turn recruit substrates. This difference is reflected in the number of regulatory subunits. PP1 interacts with >150 regulatory subunits in a highly regulated and context-specific manner (3). In contrast, PP2A, PP4, and PP6 holoenzymes are formed in a more constitutive manner and the regulation occurs between regulatory subunits and substrates (51, 52). In the PIB-MS analyses, we identified 82 known PPP-associated subunits or interacting proteins, the majority of which (59%) were PP1 specific (Fig. 4B). Furthermore, we detected all known PP4 and PP6 and the majority of PP2A regulatory and scaffolding subunits. We compared the results of the PIB-MS analyses with PP2A interactome datasets (51, 53), which revealed that PIB-MS identifies both PP2A scaffolding (2AAA and 2AAB) subunits as well as members of the four families of regulatory B subunits (B55, B56, PR72, and striatins) but not proteins interacting with B subunits (supplemental Table IV). This is consistent with observations made in kinome profiling, where for the most part only the kinase itself or the kinase catalytic subunit in complex with regulatory subunits is detected and quantified but not substrates (15, 16).
In addition to known PPP subunits, we also identified 26 proteins that are potentially new PPP-associated subunits or interacting proteins (Fig. 4B). These potentially new PPP-associated proteins included the catalytic subunit of casein kinase II alpha (CSK21), which is implicated in the regulation of many cellular processes, including DNA damage response (54), mitotic progression (55, 56), and circadian rhythm (57, 58). Others such as GCC1 and SNX1 function in transport through the trans-Golgi network (59–61), while RAB6A and RAB14 contribute to the transport from Golgi to endoplasmic reticulum (ER) and endosomes (62, 63). To see if these interactions were previously observed in large scale protein–protein interaction analyses, we compared our dataset with the BioGRID (24) and STRING protein interaction databases (Fig. 4B). Indeed, some of the proteins were found in the BioGRID or STRING databases to interact with PPP subunits, including KLC1, IDH3A, CSK2, NSA2, and COX41 (Fig. 4B). To identify if these potentially new PPP-associated proteins are PP1 or PP2A interactors, we investigated their primary amino acid sequences for conserved short linear motifs (SLiM), such as RVxF, SILK, or MyPHoNE, which are required for interaction of PP1 regulatory and catalytic subunits (64), or LxxIxE indicative of PP2A-B56 binding (51, 52) (supplemental Table IV). Based on these predictions, we selected three potential PP1 interactors: KLC1, SMD1, and IDH3A, and one potential PP2A interactor GCC1 for follow-up analyses. The four genes were cloned into a Flag-tag containing vector and individually co-expressed with either myc-tagged PP1γ or -PP2AB in 293T cells. To determine if these proteins interacted with the predicted PPP, we immunoprecipitated PP1γ or PP2AB using anti-myc antibody and probed the eluates with antibodies against the myc or Flag epitope by Western blotting (Fig. 4C). In each immunoprecipitation, we detected either myc-PP1γ or myc-PP2AB. Importantly, we also readily detected the respective PPP interacting, Flag-tagged protein, confirming that these proteins do interact in PPP holoenzymes.
Endogenous Inhibitors of PPPs are Absent from PIB-MS Analyses
Importantly, we did not identify known endogenous inhibitors of PPP activity such as α4, SET, IPP1/2, or CP1–17 (3, 7, 65) as specifically bound in any of our PIB-MS analyses. This was not due to their low expression, weak interaction, or problems with their MS-based detection since they are readily detectable in affinity purifications of catalytic PPP subunits (21). Endogenous PPP inhibitors function in part by displacing regulatory and scaffolding subunits, blocking or distorting the active site of the catalytic subunit, or altering the ability of the PPP holoenzyme to bind to substrates (3, 7, 65, 66). For instance, PP2A, PP4, and PP6 catalytic subunits all bind to α4 (also called IGBP1 in humans, TAP42 in yeast, and Tap46 in plants) (67–69). During holoenzyme biogenesis, α4 binds to nascent, partially folded PP2A, PP4, and PP6 catalytic subunits, inhibits their activities and prevents aggregation and polyubiquitination (65, 70–73). In addition, upon cellular stress (e.g. DNA damage and heat shock), PP2A holoenzymes, and potentially PP4 and PP6 holoenzymes as well, are disassembled and the catalytic subunits are bound by α4 (70). Thus, we hypothesized that this inactive subset of PPP complexes might not be accessible for PIB capture. To test this, we compared the relative abundances of interacting proteins in Flag-PP1β, PP2AB, PP4C, or PP6C affinity purifications from 293FT cell lysates (total) and 293FT cell lysates depleted with PIBs (depleted) (Fig. 5A). Under normal growth conditions, only a small portion of PPP catalytic subunits are associated with inhibitors. Thus, to compensate for this we split the lysate from Flag-PPP catalytic subunit transfected 293FT cells in a 1:4 ratio and depleted PPPs from the larger portions of the lysate using PIBs (Fig. 5A). After depletion, Flag-PPP catalytic subunits that did not bind to PIBs were immunoprecipitated from the PIB-depleted lysates. Separately, Flag-PPP catalytic subunits were immunoprecipitated from the remaining nondepleted, total lysate. We then compared the phosphatase activities of equal amounts of PP1, PP2AB, PP4C, and PP6C in enzyme activity assays before and after PIB depletion. Strikingly, phosphatase activity was reduced in the samples after PIB depletion by two- to sixfold (Fig. 5B), supporting the notion that the PPP catalytic subunits not captured by PIBs are likely in inhibitory complexes that block access to the active site and also make them inaccessible to PIB capture. To further investigate this, we compared the interactomes of PPP catalytic subunits from total and PIB-depleted lysates (Fig. 5C). In these analyses, we found that the interactome from PP1β catalytic subunits after PIB depletion were enriched in Inhibitor 2 (IPP2), while the interactomes of PP2AB, PP4C, and PP6C were enriched in α4/IGBP1 after PIB depletion. Conversely, the PP4C and PP6C interactomes after PIB depletion were significantly reduced in all regulatory subunit binding. For PP2AB, B55, PR72, and striatin regulatory subunits were decreased, while surprisingly the scaffold 2AAA and B56 subunits were increased. In contrast to IPP2 and α4, we do detect 2AAA and B56 subunits by PIB-MS. These observations suggested to us that there might be a population of PP2A-2AAA-B56 complexes for which access for MCLR-Sepharose to the active site is hindered. To further explore this possibility, we compared the structures of PP2A-2AAA-B55, PP2A-2AAA-B56, and PP2A-α4 (65, 74, 75). Closer inspection of the PP2A-2AAA-B56 structure showed that the amino acids 92 to 118 are part of an extended loop that is in close contact with the PP2A active site. A similar interaction does not exist in the PP2A-2AAA-B55 structure. Based on these observations, we surmise that under certain conditions, the 92–118 loop in B56 might block access to the active site, which would result in PIB inaccessible PP2A-2AAA-B56 complexes, although future experiments are required to formally test this hypothesis. We could not determine the subunit composition of PIB-bound PPP catalytic complexes because elution of PIBs requires denaturing conditions that result in the disassembly of PPP holoenzymes. Taken together, these results suggest that PIB-MS profiles the catalytically active, MCLR-accessible portion of the PPPome that is not bound to endogenous inhibitors.
Fig. 5.

PIBs enrich active PPP holoenzyme complexes. (A) Workflow for purification of PPP catalytic subunits before and after PIB depletion. (B) Western blot analysis of normalized PPP catalytic subunit inputs from purification from total and PIB depleted 293FT cells. Quantification of end-point measurement of PPP catalytic subunit activity purified from total and PIB depleted 293FT cell lysates. (C) Relative quantification of iBAQ abundances of regulatory/scaffolding subunits and endogenous inhibitors (IPP2 and α4/IGBP1) from catalytic subunit purifications from total and PIB depleted 293FT lysates. * p value less than 0.05, ** p value less than 0.01, and *** p value less than 0.005.
Quantitative Profiling of the PPPome in Tissue Samples
A major advantage of PIB-MS is the ability to profile endogenous PPP catalytic and interacting proteins without the need to introduce tags into endogenous PPP loci or overexpress PPPs. To evaluate the performance of PIB pulldown in tissues, we performed PIB pulldowns on six mouse organs (brain, heart, kidney, liver, spleen, and testis). To increase throughput, we combined PIB-MS with isobaric labeling (Fig. 6A). Organs were resected from mice, lysed, and control-treated or treated with MCLR. To compare PPP subunit abundances between different tissues, we included an internal standard composed of a mixed lysate of all mouse tissues. PIB eluates were digested with trypsin per normal, followed by labeling with TMT reagents. The resulting labeled peptides were combined and analyzed by single-shot LC-MS/MS; proteins were identified and quantified based on the intensity of the TMT-reporter ion. Specificity of binding was determined by comparing summed TMT-reporter ion intensities for each protein in biological triplicates treated or untreated with MCLR for each organ type (supplemental Fig. 5, supplemental Table VI). Using this approach, we were able to identify and quantify MCLR-sensitive PPP catalytic subunits, 76 known associated PPP subunits, and 16 proteins that specifically bound to PIBs but were not previously identified as PPP interacting proteins (Fig. 6B, supplemental Table VI). We observed significant tissue-type-specific expression of PPP subunits and interacting proteins. Taken together, these analyses demonstrated that PIB-MS can easily be performed in tissues and combined with TMT-labeling to achieve quantitative analyses of endogenous PPP expression.
Fig. 6.

PPP profiling in mouse tissues. (A) Scheme of TMT workflow. Triplicate PIB pulldowns were performed from mouse organ lysates derived from three six-week-old male C57BL/6J mice in the presence and absence of MCLR. For comparison of PPP subunit abundances between different tissues, an internal standard consisting of a PPP pulldown from a mixed lysate of all mouse tissues was included as a seventh channel (shown in black). After pulldown, samples and internal standard were TMT-labeled, mixed, and analyzed by single-shot LC-MS/MS. Green dots indicate specifically bound proteins in control versus MCLR-treated lysates. (B) Hierarchical clustering of rows and columns using the Euclidean distance and average linkage of PPP subunits specifically identified by PIB-MS from mouse tissues.
PPP catalytic subunits are highly conserved from yeast to human (7). In addition, fungi express PPT1 and PPZ1, which have similarity with PP5C (76) and PP1 (77) in higher eukaryotes, respectively (supplemental Fig. 1). To extend PPP profiling beyond mammals, we performed PIB-MS experiments in four different yeast species (S. cerevisiae, S. pombe, C. albicans, and N. crassa). Indeed, we were able to capture the homologous catalytic subunits of PP1, PP2A, PP4C, and PP6C, as well as the yeast-specific subunits PPT1 and PPZ1 (Fig. 7, supplemental Table VII). Furthermore, we identified known and new PPP interacting proteins (Fig. 7) supporting our assertion of PIB-MS as a general strategy for PPP profiling across eukaryotes.
Fig. 7.

PPP profiling of four yeast species. Network analysis of proteins identified as specifically PIB-bound in each of the four yeast species.
DISCUSSION
In this work, we present a chemical proteomics strategy we call PIB-MS for quantitative profiling of endogenous PPP catalytic and associated noncatalytic subunits from cells and tissues of diverse eukaryotes in a single analysis. Using this approach, we are able to identify and quantify PP1, PP2A, PP4, PP5, and PP6 subunits as well as the yeast-specific PPP family members PPT and PPZ, but not PP2B and PP7 (supplemental Fig. 1). PIB-MS is easily implemented, compatible with quantitative multiplexing approaches such as isobaric labeling, and widely applicable to investigations into phosphorylation signaling by phosphoprotein phosphatases from yeast to humans. In the future, it would be exciting to extend this approach to PP2B and PP7 to profile the whole PPP family using a multiplexed inhibitor bead-based approach, as has been done for human kinases (15, 16).
PPPs achieve substrate specificity through the formation of multimeric holoenzyme complexes of the catalytic subunit with other noncatalytic scaffolding and regulatory subunits. While this strategy greatly expands the number of unique PPP signaling entities, it requires mapping of protein–protein interactions to investigate PPP signaling and its rewiring in cells and tissues upon drug treatments and cellular stresses. Proteome-wide protein expression analyses do not discriminate among the total amount of PPP associated subunits from those in complex with catalytic subunits, missing crucial information essential for deciphering PPP signaling.
PIB-MS bridges this gap by allowing for the facile identification and quantification of endogenous, MCLR-sensitive PPP holoenzymes. Using this approach, we can reproducibly identify and quantify PP1, PP2A, PP4, PP5, and PP6 catalytic subunits and on average 80 known associated subunits from 0.5 mg of total protein lysate (Figs. 1, 4, and 5). A major advantage of this strategy is that it does not require the introduction of tags to PPP catalytic subunits, exogenous expression of tagged PPPs, or the use of PPP-specific antibodies. In-depth analyses of phosphatase protein–protein interactions in protein-centric or proteome-wide analyses have comprehensively annotated associated proteins (3, 7, 40, 78–81), allowing us to link proteins identified by PIB-MS to their respective PPP catalytic subunits. PIB-MS can also be performed on many samples types, including tissues and tumors. In addition, combining PIB-MS with postdigestion labeling approaches such as multiplexes isobaric labeling allows for the quantitative comparison of PPP expression across many samples types.
As observed with other affinity enrichment strategies that utilize chemical compounds immobilized to Sepharose resin, we detected a large number of unspecifically bound proteins (supplemental Fig. 2). However, pretreatment of lysates with MCLR results in a 200-fold reduction in PIB binding for specific interactors, allowing us to effectively distinguish them from unspecific contaminants. Furthermore, the summed iBAQ area of specific binders is 8.8-times higher compared with unspecific ones. Thus, statistical analysis of replicate analyses readily identifies specifically bound proteins that are potentially new PPP interactors. In addition, we found that comparison of protein binding behavior to PIBs in the presence of MCLR or OA allows us to distinguish PP1 and PP5 from PP2A, PP4, and PP6 subunits (Fig. 3).
Intriguingly, in our analysis of different cell lines, we failed to specifically observe endogenous inhibitors such as α4, SET, IPP1/2, CP1–17 (3, 7, 65) by PIB-MS (Fig. 5). We hypothesized that this was likely due to the ability of these inhibitory proteins to block the PPP catalytic subunit active site. Consistent with this hypothesis, we found that PPP catalytic subunits not bound by PIBs displayed reduced activity in phosphatase activity assays. Furthermore, the interactomes of catalytic subunits not bound by PIBs showed increased binding to endogenous inhibitors (Fig. 5). This suggests that PIB-MS profiles a subproteome of PPP holoenzymes that are actively engaged in phosphorylation signaling.
PPP catalytic subunits are among the most conserved proteins from yeast to human (7). We demonstrate that PIB-MS is not restricted to human cells but can be employed to investigate PPP signaling in mouse and yeast tissues. Thus, PIB-MS is a general strategy for PPP profiling across the eukaryotic kingdom.
To understand how protein phosphorylation regulates complex biological processes, we must investigate both forward and reverse reactions. Although there has been great progress in deciphering signaling by kinases, much less is known about phosphatases. Approaches similar to PIB-MS for kinases have greatly expanded our understanding of the kinome and its rewiring in cancer (13–19). We anticipate that PIB-MS will yield similar information and greatly complement kinome profiling by providing the to-date missing insights into the dephosphorylation reaction and its regulation and dysregulation in basic biology and disease.
DATA AVAILABILITY
Raw MS data for the experiments performed in this study are available at MassIVE (MSV000081226) and PRIDE accession (PXD006902).
Supplementary Material
Acknowledgments
We would like to thank Mark Israel and Todd Miller for cell lines; Matthew Havrda for mouse tissues; Charles Cole, James Moseley, Jay Dunlap, and Lawrence Myers for yeast samples. We would like to thank Gus Lienhard and Bill Wickner for reading the manuscript and critical discussion of the data. We would like to thank members of Kettenbach lab, Jakob Nilsson, and Scott Gerber for critical discussions of the data.
Footnotes
* This work was supported by grants from NIH/NIGMS (R35GM119455 and P20GM113132) and The V Foundation for Cancer Research (V2016-022) to A.N.K. and the NCCC Cancer Center Support Grant P30CA023108. The Orbitrap Fusion Tribrid mass spectrometer was acquired with support from NIH (S10-OD016212).
 This article contains supplemental material Tables I, II, IV, VI, and VII and Figs. 1–5.
This article contains supplemental material Tables I, II, IV, VI, and VII and Figs. 1–5.
1 The abbreviations used are:
- PPP
- phosphoprotein phosphatases
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MCLR
- microcystin -LR
- PIB
- phosphatase inhibitor beads
- PIB-MS
- phosphatase inhibitor beads and mass spectrometry.
REFERENCES
- 1. Sharma K., D'Souza R. C. J., Tyanova S., Schaab C., Wisniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Reports 8, 1583–1594 [DOI] [PubMed] [Google Scholar]
- 2. Manning G., Whyte D. B., Martinez R., Hunter T., and Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 3. Bollen M., Peti W., Ragusa M. J., and Beullens M. (2010) The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moorhead G. B., Trinkle-Mulcahy L., and Ulke-Lemée A. (2007) Emerging roles of nuclear protein phosphatases. Nature Rev. Mol. Cell Biol. 8, 234–244 [DOI] [PubMed] [Google Scholar]
- 5. Virshup D. M., and Shenolikar S. (2009) From promiscuity to precision: Protein phosphatases get a makeover. Mol. Cell 33, 537–545 [DOI] [PubMed] [Google Scholar]
- 6. Choy M. S., Swingle M., D'Arcy B., Abney K., Rusin S. F., Kettenbach A. N., Page R., Honkanen R. E., and Peti W. (2017) PP1: Tautomycetin complex reveals a path toward the development of PP1-specific inhibitors. J. Amer. Chem. Soc. 139, 17703–17706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brautigan D. L. (2013) Protein Ser/Thr phosphatases—The ugly ducklings of cell signalling. FEBS J. 280, 324–345 [DOI] [PubMed] [Google Scholar]
- 8. Miller M. L., Jensen L. J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S. A., Bordeaux J., Sicheritz-Ponchen T., Olhovsky M., Pasculescu A., Alexander J., Knapp S., Blom N., Bork P., Li S., Cesareni G., Pawson T., Turk B. E., Yaffe M. B., Brunek S., and Linding R. (2008) Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kettenbach A. N., Wang T., Faherty B. K., Madden D. R., Knapp S., Bailey-Kellogg C., and Gerber S. A. (2012) Rapid determination of multiple linear kinase substrate motifs by mass spectrometry. Chem. Biol. 19, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruepp A., Brauner B., Dunger-Kaltenbach I., Frishman G., Montrone C., Stransky M., Waegele B., Schmidt T., Doubieu O. N., Stümpflen V., and Mewes H. W. (2008) CORUM: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 36, D646–D650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pawson T., and Scott J. D. (2005) Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 30, 286–290 [DOI] [PubMed] [Google Scholar]
- 12. Klaeger S., Heinzimeir S., Wilhelm M., Polzer H., Vick B., Koenig P.-A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., Zecha J., Reiter K., Qiao H., Helm D., Koch H., Schoof M., Canevari G., Casale, Re Depaolini S., Feuchtinger A., Wu Z., Schmidt T., Rueckert L., Becker W., Huenges J., Garz A.-K., Gohlke B.-O., Zolg D. P., Kayser G., Vooder T., Preissner R., Hahne H., Tönisson N., Kramer K., Götze K., Bassermann F., Schlegl, Ehrlich H.-C., Aiche S., Walch A., Greif P. A., Schneider S., Felder E. R., Ruland J., Médard G., Jeremias I., Spiekermann K., and Kuster B. (2017) The target landscape of clinical kinase drugs. Science 358, eean4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sutton C. W. (2012) The role of targeted chemical proteomics in pharmacology. British J. Pharmacol. 166, 457–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Godl K., Wissing J., Kurtenbach A., Habenberger P., Blencke S., Gutbrod H., Salassidis K., Stein-Gerlach M., Missio A., Cotten M., and Daub H., (2013) An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl. Acad. Sci. USA 100, 15434–15439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C., Reader V., Sweetman G., Bauer A., Bouwmeester T., Hopf C., Kruse U., Neubauer G., Ramsden N., Rick J., Kuster B., and Drewes G. (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature Biotechnol. 25, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 16. Duncan J. S., Whittle M. C., Nakamura K., Abell A. N., Midland A. A., Zawistowski J. S., Johnson N. L., Granger D. A., Jordan D. V., Darr D. B., Usary J., Kuan P.-F., Smalley D. M., Major B., He X., Hoadley K. A., Zhou B., Sharpless N. E., Perou C. M., Kim W. Y., Gomez S. M., Chen X., Jin J., Frye S. V., Earp H. S., Graves L. M., and Johnson G. L. (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Midland A. A., Whittle M. C., Duncan J. S., Abell A. N., Nakamura K., Zawistowski J. S., Carey L. A., Earp H. S. III, Graves L. M. Gomez S. M., and Johnson G. L. (2012) Defining the expressed breast cancer kinome. Cell Res. 22, 620–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson G. L., Stuhlmiller T. J., Angus S. P., Zawistowski J. S., and Graves L. M. (2014) Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 20, 2516–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stuhlmiller T. J., Miller S. M., Zawistowski J. S., Nakamura K., Beltran A. S., Duncan J. S., Angus S. P., Collins K. A. L., Granger D. A., Reuther R. A., Graves L. M., Gomez S. M., Kuan P.-F., Parker J. S., Chen X., Sciaky N., Carey L. A., Earp H. S., Jin J., and Johnson G. L. (2015) Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting family Bromodomains BET. Cell Reports 11, 390–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moorhead G., MacKintosh R. W., Morrice N., Gallagher T., and MacKintosh C. (1994) Purification of type 1 protein (serine/threonine) phosphatases by microcystin-Sepharose affinity chromatography. FEBS Lett. 356, 46–50 [DOI] [PubMed] [Google Scholar]
- 21. Rusin S. F., Schlosser K. A., Adamo M. E., and Kettenbach A. N. (2015) Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Science Signal. 8, rs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chattopadhyay D., Swingle M. R., Salter E. A., Wood E., D'Arcy B., Zivanov C., Abney K., Musiyenko A., Rusin S. F., Kettenbach A., Yet L., Schroeder C. E., Golden J. E., Dunham W. H., Gingras A.-C., Banerjee S., Forbes D., Wierzbicki A., and Honkanen R. E. (2016) Crystal structures and mutagenesis of PPP-family ser/thr protein phosphatases elucidate the selectivity of cantharidin and novel norcantharidin-based inhibitors of PP5C. Biochem. Pharmacol. 109, 14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 24. Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N. K., O'Donnell L., Oster S., Theesfeld S., Sellam A., Stark C., Breitkreutz B.-J., Dolinski K., and Tyers M. (2017) The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369–D379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saito R., Smoot M. E., Ono K., Ruscheinski J., Wang P.-L., Lotia S., Pico A. R., Bader G. D., and Ideker T. (2012) A travel guide to Cytoscape plugins. Nat. Methods 9, 1069–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krystkowiak I., and Davey N. E. (2017) SLiMSearch: A framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 45, W464–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eng J. K., Jahan T. A., and Hoopmann M. R. (2013) Comet: An open-source MS/MS sequence database search tool. Proteomics 13, 22–24 [DOI] [PubMed] [Google Scholar]
- 29. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 30. Valot B., Langella O., Nano E., and Zivy M. (2011) MassChroQ: A versatile tool for mass spectrometry quantification. Proteomics 11, 3572–3577 [DOI] [PubMed] [Google Scholar]
- 31. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 32. Senko M. W., Remes P. M., Canterbury J. D., Mathur R., Song Q., Eliuk S. M., Mullen C., Earley L., Hardman M., Blethrow J. D., Bui H., Specht A., Lange O., Denisov E., Makarov A., Horning S., and Zabrouskov V. (2013) Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 85, 11710–11714 [DOI] [PubMed] [Google Scholar]
- 33. Ting L., Rad R., Gygi S. P., and Haas W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McAlister G. C., Nusinow D. P., Jedrychowski M. P., Wühr M., Huttlin E. L., Erickson B. K., Rad R., Haas W., and Gygi S. P. (2014) MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moorhead G. B., Haystead T. A., and MacKintosh C. (2007) Synthesis and use of the protein phosphatase affinity matrices microcystin-Sepharose and microcystin-biotin-Sepharose. Meth. Mol. Biol. 365, 39–45 [DOI] [PubMed] [Google Scholar]
- 36. Swingle M., Ni L., and Honkanen R. E. (2007) Small-molecule inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Meth. Mol. Biol. 365, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sangodkar J., Farrington C. C., McClinch K., Galsky M. D., Kastrinsky D. B., and Narla G. (2016) All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 283, 1004–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sents W., Ivanova E., Lambrecht C., Haesen D., and Janssens V. (2013) The biogenesis of active protein phosphatase 2A holoenzymes: A tightly regulated process creating phosphatase specificity. FEBS J. 280, 644–661 [DOI] [PubMed] [Google Scholar]
- 39. Grech G., Baldacchino S., Saliba C., Grixti M. P., Gauci R., Petroni V., Fenech A. G., and Scerri C. (2016) Deregulation of the protein phosphatase 2A, PP2A in cancer: Complexity and therapeutic options. Tumour Biol. 37, 11691–11700 [DOI] [PubMed] [Google Scholar]
- 40. Eichhorn P. J., Creyghton M. P., and Bernards R. (2009) Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 1795, 1–15 [DOI] [PubMed] [Google Scholar]
- 41. Hodis E., Watson I. R., Kryukov, Arold S. T., Imielinski M., Theurillat J.-P., Nickerson E., Auclair D., Li L., Place C., DiCara D., Ramos A. H., Lawrence M. S., Cibulskis K., Sivachenko A., Voet D., Saksena G., Stransky N., Onofrio R. C., Winckler W., Ardlie K., Wagle N., Wargo J., Chong K., Morton D. L., Stemke-Hale K., Chen G., Noble M., Meyerson M., Ladbury J. E., Davies M. A., Gershenwald J. E., Wagner S. N., Hoon D. S. B., Schadendorf D., Lander E. S., Gabriel S. B., Getz G., Garraway L. A., and Chin L. (2012) A landscape of driver mutations in melanoma. Cell 150, 251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krauthammer M., Kong Y., Ha B. H., Evans P., Bacchiocchi A., McCusker J. P., Cheng E., Davis M. J., Goh G., Choi M., Ariyan S., Narayan D., Dutton-Regester K., Capatana A., Holman E. C., Bosenberg M., Sznol M., Kluger H. M., Brash D. E., Stern D. F., Materin M. A., Lo R. S., Mane S., Ma S., Kidd K. K., Hayward H. K., Lifton R. P., Schlessinger J., Boggon T. J., and Halaban R. (2012) Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haesen D., Asbagh A. A., Derua R., Schrauwen S., Hoorne Y., Amant F., Waelkins E., Sablina A., and Janssens V., (2016) Recurrent PPP2R1A mutations in uterine cancer act through a dominant-negative mechanism to promote malignant cell growth. Cancer Res. 76, 5719–5731 [DOI] [PubMed] [Google Scholar]
- 44. Ruediger R., Pham H. T., and Walter G. (2001) Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene 20, 10–15 [DOI] [PubMed] [Google Scholar]
- 45. Bialojan C., and Takai A. (1988) Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 256, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mumby M. (2007) PP2A: Unveiling a reluctant tumor suppressor. Cell 130, 21–24 [DOI] [PubMed] [Google Scholar]
- 47. Shtrichman R., Sharf R., Barr H., Dobner T., and Kleinberger T. (1999) Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 96, 10080–10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pallas D. C., Shahrik L. K., Martin B. L., Jaspers S., Miller T. B., Brautigan D. L., and Roberts T. M. (1990) Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60, 167–176 [DOI] [PubMed] [Google Scholar]
- 49. Ruediger R., Ruiz J., and Walter G. (2011) Human cancer-associated mutations in the Aalpha subunit of protein phosphatase 2A increase lung cancer incidence in Aalpha knock-in and knockout mice. Mol. Cell. Biol. 31, 3832–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen W., Arroyo J. D., Timmons J. C., Possemato R., and Hahn W. C. (2005) Cancer-associated PP2A alpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 65, 8183–8192 [DOI] [PubMed] [Google Scholar]
- 51. Hertz E. P. T., Kruse T., Davey N. E., López-Mendez B., Sigurösson J. O., Montoya G., Olsen J. V., and Nilsson J. (2016) A conserved motif provides binding specificity to the PP2A-B56 phosphatase. Mol. Cell 63, 686–695 [DOI] [PubMed] [Google Scholar]
- 52. Wang X., Bajaj R., Bollen M., Peti W., and Page R. (2016) Expanding the PP2A interactome by defining a B56-specific SLiM. Structure 24, 2174–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F.-U., Ban N., Malmström L., and Aebersold R. (2012) Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352 [DOI] [PubMed] [Google Scholar]
- 54. Keller D. M., Zeng X., Wang Y., Zhang Q. H., Kapoor M., Shu H., Goodman R., Lozano G., Zhao Y., and Lu H. (2001) A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol. Cell 7, 283–292 [DOI] [PubMed] [Google Scholar]
- 55. Sayed M., Pelech S., Wong C., Marotta A., and Salh B. (2001) Protein kinase CK2 is involved in G2 arrest and apoptosis following spindle damage in epithelial cells. Oncogene 20, 6994–7005 [DOI] [PubMed] [Google Scholar]
- 56. St-Denis N. A., Derksen D. R., and Litchfield D. W. (2009) Evidence for regulation of mitotic progression through temporal phosphorylation and dephosphorylation of CK2alpha. Mol. Cell. Biol. 29, 2068–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tamaru T., Hirayama J., Isojima Y., Nagai K., Norioka S., Takamatsu K., and Sassone-Corsi P. (2009) CK2alpha phosphorylates BMAL1 to regulate the mammalian clock. Nature Struct. Mol. Biol. 16, 446–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tamaru T., Hattori M., Honda K., Nakahata Y., Sassone-Corsi P., van der Horst G. T. J., Ozawa T., and Takamatsu K. (2015) CRYDrives cyclic CK2-mediated BMAL1 phosphorylation to control the mammalian circadian clock. PLoS Biol. 13, e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lieu Z. Z., Derby M. C., Teasdale R. D., Hart C., Gunn P., and Gleeson P. A. (2007) The golgin GCC88 is required for efficient retrograde transport of cargo from the early endosomes to the trans-Golgi network. Mol. Biol. Cell 18, 4979–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mari M., Bujni M. V., Zeuschner D., Geerts W. J. C., Griffith J., Petersen C. M., Cullen P. J., Klumperman J., and Geuze H. J. (2008) SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic 9, 380–393 [DOI] [PubMed] [Google Scholar]
- 61. Rojas R., Kametaka S., Haft C. R., and Bonifacino J. S. (2007) Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol. Cell. Biol. 27, 1112–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Matsuto M., Kano F., and Murata M. (2015) Reconstitution of the targeting of Rab6A to the Golgi apparatus in semi-intact HeLa cells: A role of BICD2 in stabilizing Rab6A on Golgi membranes and a concerted role of Rab6A/BICD2 interactions in Golgi-to-ER retrograde transport. Biochim. Biophsy. Acta 1853, 2592–2609 [DOI] [PubMed] [Google Scholar]
- 63. Linford A., Yoshimura S., Bastos R. N., Langemeyer L., Gerondopoulos A., Rigden D. J., and Barr F. A. (2012) Rab14 and its exchange factor FAM116 link endocytic recycling and adherens junction stability in migrating cells. Developmental cell 22, 952–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heroes E., Lesage B., Görnemann J., Beullens M., Van Meervelt L., and Bollen M. (2013) The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 280, 584–595 [DOI] [PubMed] [Google Scholar]
- 65. Jiang L., Stanevich V., Satyshur K. A., Kong M., Watkins G. R., Wadzinski B. E., Sengupta R., and Xing Y. (2013) Structural basis of protein phosphatase 2A stable latency. Nature Commun. 4, 1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dancheck B., Ragusa M. J., Allaire M., Nairn A. C., Page R., and Peti W. (2011) Molecular investigations of the structure and function of the protein phosphatase 1-spinophilin-inhibitor 2 heterotrimeric complex. Biochemistry 50, 1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ahn C. S., Han J. A., Lee H. S., Lee S., and Pai H. S. (2011) The PP2A regulatory subunit Tap46, a component of the TOR signaling pathway, modulates growth and metabolism in plants. Plant Cell 23, 185–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen J., Peterson R. T., and Schreiber S. L. (1998) Alpha 4 associates with protein phosphatases 2A, 4, and 6. Biochem. Biophys. Res. Commun. 247, 827–832 [DOI] [PubMed] [Google Scholar]
- 69. Nanahoshi M., Nishiuma T., Tsujishita Y., Hara K., Inui S., Sakaguchi N., and Yonezawa K. (1998) Regulation of protein phosphatase 2A catalytic activity by alpha4 protein and its yeast homolog Tap42. Biochemical and biophysical research communications 251, 520–526 [DOI] [PubMed] [Google Scholar]
- 70. Kong M., Ditsworth D., Lindsten T., and Thompson C. B. (2009) Alpha4 is an essential regulator of PP2A phosphatase activity. Mol. Cell. 36, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Santhanam A., Hartley A., Duvel K., Broach J. R., and Garrett S. (2004) PP2A phosphatase activity is required for stress and Tor kinase regulation of yeast stress response factor Msn2p. Eukaryotic Cell 3, 1261–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu J., Prickett T. D., Elliott E., Meroni G., and Brautigan D. L. (2001) Phosphorylation and microtubule association of the Opitz syndrome protein mid-1 is regulated by protein phosphatase 2A via binding to the regulatory subunit alpha 4. Proc. Natl. Acad. Sci. USA 98, 6650–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trockenbacher A., Suckow V., Foerster J., Winter J., Krauss S., Ropers H. H., Schneider R., and Schweiger S. (2001) MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 29, 287–294 [DOI] [PubMed] [Google Scholar]
- 74. Xu Y., Xing Y., Chen Y., Chao Y., Lin Z., Fan E., Yu J. W., Strack S., Jeffrey P. D., and Shi Y. (2006) Structure of the protein phosphatase 2A holoenzyme. Cell 127, 1239–1251 [DOI] [PubMed] [Google Scholar]
- 75. Xu Y., Chen Y., Zhang P., Jeffrey P. D., and Shi Y. (2008) Structure of a protein phosphatase 2A holoenzyme: Insights into B55-mediated Tau dephosphorylation. Mol. Cell 31, 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen M. X., McPartlin A. E., Brown L., Chen Y. H., Barker H. M., and Cohen P. T. (1994) A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 13, 4278–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen E., Choy M. S., Petrényi K., Kónya Z., Erdödi F., Dombrádi V., Peti W., and Page R. (2016) Molecular insights into the fungus-specific serine/threonine protein phosphatase Z1 in Candida albicans. mBio 7, e002872–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Youn J. Y., Dunham W. H., Hong S. J., Knight J. D. R., Bashkurov M., Ghen, Ginny I., Bagci H., Rathod B., MacLeod G., Eng S. W. M., Angers S., Morris Q., Fabian M., Côté J.-F., and Gingras A.-C. (2018) High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532 [DOI] [PubMed] [Google Scholar]
- 79. St-Denis N., Gupta G. D., Lin Z. Y., Gonzalez-Badillo B., Veri A. O., Knight J. D. R., Rajendran D., Couzens A. L., Currie K. W., Tkach J. M., Cheung S. W. T., Pelletier L, and Gingras A.-C. (2016) Phenotypic and interaction profiling of the human phosphatases identifies diverse mitotic regulators. Cell Reports 17, 2488–2501 [DOI] [PubMed] [Google Scholar]
- 80. Yadav L., Tamene F., Göös H., van Drogen A., Aebersold R., Gstaiger M., and Varjosalo M. (2017) Systematic analysis of human protein phosphatase interactions and dynamics. Cell Systems 4, 430–444 [DOI] [PubMed] [Google Scholar]
- 81. Huttlin E. L., Ting L., Bruckner R. J., Gebreab F., Gygi M. P., Szypt J., Tam S., Zarraga G., Colby G., Baltier K., Dong R., Guarani V., Vaites L. P., Ordureau A., Rad R., Erickson B. K., Wühr M., Chick J., Zhai B., Kolippakkam D., Mintseris J., Obar R. A., Harris T., Artavanis-Tsakonas S., Sowa M. E., De Camilli P., Paulo J. A., Harper J. W., and Gygi H. P. (2015) The BioPlex network: A systematic exploration of the human interactome. Cell 162, 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw MS data for the experiments performed in this study are available at MassIVE (MSV000081226) and PRIDE accession (PXD006902).