

SUMMARY
We define a homeostatic function for innate immune signaling within neurons. A genetic analysis of the innate immune signaling genes IMD, IKKβ, Tak1 and Relish demonstrates that each is essential for presynaptic homeostatic plasticity (PHP). Subsequent analyses define how the rapid induction of PHP (occurring in seconds) can be coordinated with the life-long maintenance of PHP, a time course that is conserved from invertebrates to mammals. We define a novel bifurcation of presynaptic innate immune signaling. Tak1 (Map3K) acts locally and is selective for rapid PHP induction. IMD, IKKβ and Relish are essential for long-term PHP maintenance. We then define how Tak1 controls vesicle release. Tak1 stabilizes the docked vesicle state, which is essential for the homeostatic expansion of the readily releasable vesicle pool. This represents a mechanism for the control of vesicle release, and an interface of innate immune signaling with the vesicle fusion apparatus and homeostatic plasticity.
ETOC
Harris et al., demonstrate that neuronal innate immune signaling coordinates the rapid induction and sustained expression of presynaptic homeostatic plasticity. Signaling includes the Map3K Tak1, which directly interfaces with the vesicle release machinery by stabilizing the docked synaptic vesicle state.
INTRODUCTION
Emerging models for innate immune signaling within the nervous system are consistent with an emergency surveillance system that is primarily mediated by astrocytes and microglia (Stevens et al., 2007; Schafer and Stevens, 2013). However, essential components of both the adaptive and innate immune signaling systems are also present within neurons. For example, with regard to adaptive immunity, the major histocompatibility complex class I (MHC-I) proteins are neuronally expressed, regulated by neural activity and participate in the anatomical refinement of neural circuitry during development (Corriveau et al., 1998; Huh et al., 2000; Lee et al., 2014). Likewise, in both Drosophila and mammals, the Rel/NF-κB family transcription factors, a downstream target of innate immune signaling receptors, have been shown to be neuronally expressed and participate in learning related and circadian plasticity (Ahn et al., 2008, Meffert et al., 2003; Chen et al., 2016). Although it is well established that neurons can respond to glial-derived cytokines (Stellwagen & Malenka, 2006), the action of innate immune signaling within neurons remains poorly understood. Another mystery concerns the function of neuronal innate immunity throughout the health-span of an organism. Does neuronal innate immune signaling represent a latent system, ready to engage injury and infection, or does it perform additional functions that are relevant to the day-to-day activity within the adult brain? A role for the cytokine TNF-alpha, secreted by astrocytes, has been established during the homeostatic regulation of glutamate receptor abundance (Stellwagen and Malenka, 2006). Could innate immune signaling act more generally within neurons as a mediator of other forms of homeostatic plasticity, thereby regulating the activity of individual neurons, synapses, circuitry and behavior throughout life?
Presynaptic homeostatic plasticity (PHP) is a highly evolutionarily conserved form of homeostatic signaling that is similarly expressed at the NMJ of Drosophila, mouse and human (Frank et al., 2006; Wang et al., 2016; Cull-Candy et al., 1980), and is expressed at central synapses in both Drosophila (Kazama and Wilson, 2008) and rodents (Kim and Ryan, 2010; Zhao et al., 2011; Mitra et al., 2011). Recently, in Drosophila, a highly conserved innate immune receptor, PGRP-LC, was shown to be essential for the rapid induction and sustained expression of PHP (Harris et al., 2015). However, the signaling system downstream of the PGRP-LC receptor has yet to be characterized in neurons in any organism. Furthermore, the mechanism by which this innate immune signaling pathway modulates the release of neurotransmitter at the presynaptic terminal remains entirely unknown. Indeed, most of what we understand about innate immune signaling in neurons revolves around either innate immune receptor signaling at the plasma membrane, or the transcriptional control of immune effector proteins such as antimicrobial factors and cytokines.
Here we explore the function of the canonical innate immune signaling system that acts downstream of the PGRP-LC receptor during PHP at the Drosophila NMJ. We define a novel organization of this innate immune signaling system within these neurons. In so doing, we also define how this signaling system interfaces with the presynaptic release mechanism. We provide multiple lines of evidence that the kinase Tak1, a downstream effector of the PGRP-LC receptor, functions to stabilize the docked synaptic vesicle state. This activity is essential for both baseline neurotransmission and the expression of PHP. Taken together, our data argue that innate immune signaling may encompass an emergency, feed-forward response in neurons that rapidly induces homeostatic plasticity at the presynaptic release site via Tak1, and a delayed transcriptional response that sets the stage for the prolonged maintenance of homeostatic plasticity, a process that can persist throughout the life of the organism.
RESULTS
The innate immune signaling cascade, termed the Immune Deficient (IMD) signaling cascade, is initiated by ligand induced dimerization of the PGRP-LC receptor and assembly of a protein complex (IKK complex) that includes the adaptor protein Imd (Immune deficiency), the serine threonine kinase Tak1 (TGF-β activated kinase 1, a Map3K) and the serine threonine kinase IKKβ (note: there is only a single IKK encoded in the Drosophila genome, termed IKKβ). Once initiated, IMD signaling drives nuclear translocation and activation of the NF-κB-type transcription factor Relish (Silverman et al., 2000; Choe et al., 2002; Myllymäki et al., 2014). While additional signaling complexity exists, the signaling constituents including Imd, IKKβ, Tak1 and Rel represent several principal stages of signal transduction from receptor activation at the plasma membrane to transcription in the nucleus (Figure 1A). Therefore, we assayed whether each of these genes is necessary for presynaptic homeostatic plasticity (PHP) (Figure 1).
Figure 1. Innate immune signaling during the rapid induction of PHP.
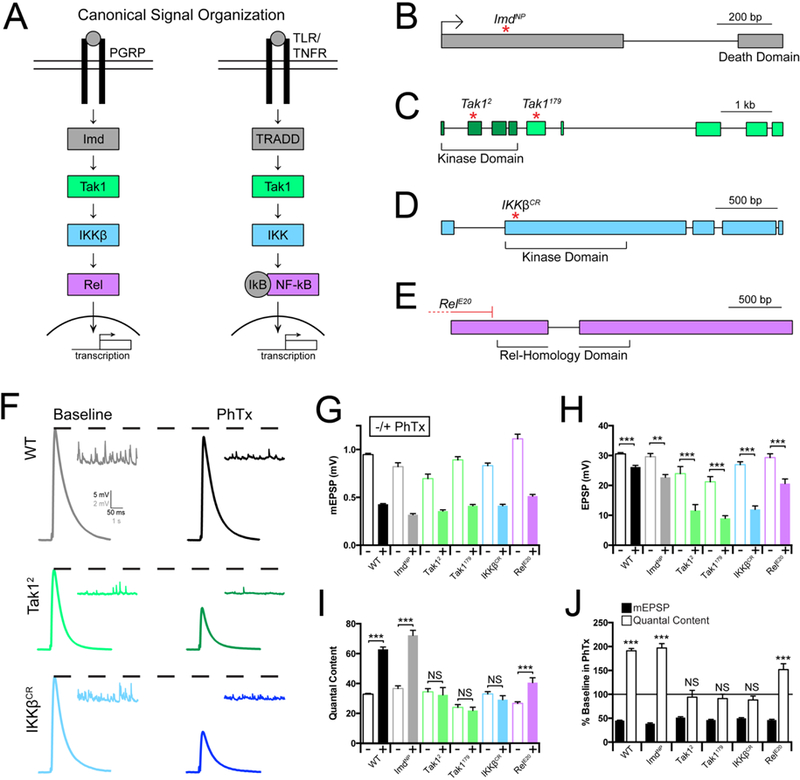
(A) Diagram of the Drosophila melanogaster Immune Deficient (IMD) pathway and homologous mammalian innate immune signaling. (B-E) Gene loci for Imd, Tak1, IKKβ, and Rel, with genetic reagents indicated. (F) Representative traces for EPSP (scale, 5 mV, 50 ms) and mEPSP (scale, 2 mV, 1 s) at baseline, and in the presence of philanthotoxin (PhTx) for the indicated genotypes. (G) Average mEPSP amplitude for each genotype in the absence (light bars) or presence (dark bars) of PhTx. (H) Average EPSP amplitude in the absence (light bars) or presence (dark bars) of PhTx. (I) Average quantal content in the absence (light bars) or presence (dark bars) of PhTx. (J) mEPSP amplitudes (filled bars) and quantal content (open bars) for each genotype in the presence of PhTx, normalized to baseline values in the absence of PhTx. Data are presented as average (+/− SEM) and statistical significance determined by Student’s t-test (unpaired, two-tailed). See also Supplemental Figures 1 and 3.
First, we assembled loss of function mutations that specifically disrupt the genes encoding Imd, IKKβ, Tak1 and Rel (Figure 1B). The ImdNP mutation is a transposon insertion residing within the first coding exon and is predicted to be a functional null. We acquired and validated two independently derived mutations in the Tak1 gene. The Tak1179 mutation was originally defined as a missense mutation in the kinase domain (Delaney et al., 2006). However, re-sequencing this line reveals that Tak1179 harbors a missense mutation (N384>T) in a conserved regulatory domain. The Tak12 mutation is an early stop codon prior to the catalytic sites within the kinase domain and considered to be a functional null mutation (Vidal et al., 2001). These classifications are supported by our subsequent phenotypic analyses (see below). We also acquired a deficiency that uncovers the Tak1 gene locus. We, then, generated a mutation in IKKβ (IKKβCR) using CRISPR/Cas9 technology, causing a frame shift that results in a stop codon early in the kinase domain, predicted to be a functional null mutation (see methods). Finally, the RelE20 mutation is a large deletion removing all possible transcription start sites and is a molecular null mutation (Hedengren et al., 1999).
Tak1 and IKKβ are necessary for the rapid induction of PHP
Presynaptic homeostatic plasticity (PHP) can be rapidly induced by application of the glutamate receptor antagonist Philanthotoxin-433 (PhTx; 10–20µM) (Frank et al., 2006). PhTx inhibits muscle specific glutamate receptors harboring the GluRIIA subunit, causing a decrease in postsynaptic mEPSP amplitudes. Within 10 minutes of PhTx application, retrograde signaling induces a potentiation of presynaptic neurotransmitter release (quantal content; see methods) that offsets the decrease in mEPSP amplitude and restores EPSP amplitudes toward baseline levels (Figure 1G-I, black traces and bars; Frank et al., 2006).
We began our pathway interrogation with Imd. The Imd gene encodes an adaptor protein that can directly bind the intracellular domain of PGRP-LC and is essential for the induction of the innate immune response (Lemaitre et al., 1995; Choe et al., 2005). Here we demonstrate that baseline neurotransmission is normal in the ImdNP mutant (Figure 1G-I). When PhTx is applied, mEPSP amplitudes are decreased by ~50%. In response, we observe a robust homeostatic increase in quantal content (Figure 1I, J) that restores EPSP amplitude to toward baseline levels (Figure 1H). Thus, Imd is not required for the rapid induction of PHP, despite the fact that the PGRP-LC receptor is essential (Harris et al., 2015).
We subsequently repeated our assays of baseline transmission in the Tak1 and IKKβ mutant backgrounds. First, we demonstrate that the Tak1 mutations have a defect in baseline transmission (0.3mM extracellular calcium – [Ca2+]e). Specifically, mEPSP amplitudes are significantly decreased in both Tak1 alleles and EPSP amplitudes are also diminished (Figure 1G, H). There is a significant decrease in baseline quantal content in Tak1179 (p<0.005), but no significant decrease in quantal content in Tak12 (p>0.05). When we assay the IKKβ mutant, we find it behaves in a similar manner, revealing mild decreases in both mEPSP and EPSP amplitudes and no change in quantal content relative to WT.
Next, we assayed PHP in the Tak1 and IKKβ mutants. When we applied PhTx to the Tak1 and IKKβ mutants, we found that PHP was completely blocked (Figure 1H, I). For both alleles of Tak1 as well as the IKKβ mutant, mEPSP amplitudes are decreased by ~50% in the presence of PhTx. However, there is no statistically significant increase in quantal content, diagnostic of a failure of PHP. As a consequence of failed PHP, EPSP amplitudes remain dramatically reduced compared to each mutant in the absence of PhTx. As a further genetic test, we placed the Tak12 allele in trans to a deficiency that uncovers the Tak1 gene locus (Tak12/Df), and placed the IKKβ mutant in trans to a deficiency that uncovers the IKKβ gene locus. In both instances, we replicate the findings described above, observing a complete block of PHP (Supplemental Figure 1A-D). We conclude that both Tak1 and IKKβ are essential for the rapid induction of PHP.
Finally, we repeated our assays for baseline transmission and PHP in the Rel mutant background. Note that the rapid induction of PHP is independent of both transcription and translation (Frank et al., 2006), suggesting that Rel should not be required. At baseline, the Rel mutant revealed a slight, but significant increase in mEPSP amplitudes, no change in EPSP amplitude and a significant decrease in quantal content compared to wild type (Figure 1G-I). When PhTx is applied to the Rel mutant, we find that PHP is robustly expressed, as predicted. We observe a large decrease in mEPSP amplitude and a statistically significant, homeostatic increase in presynaptic release. We note, however, that the magnitude of the increase in presynaptic release is significantly less than that observed in wild type, indicating a suppression of the homeostatic response (Figure 1I, J). From these data we conclude Rel is dispensable for the rapid initiation of PHP, but may contribute to the capacity of a motoneuron to fully express PHP, perhaps by participating in the steady state integrity of the IMD signaling cascade.
Imd, IKKβ, and Relish are necessary for the sustained expression of PHP.
PHP can also be induced by genetic deletion of the GluRIIA subunit of the postsynaptic glutamate receptors (Petersen et al., 1997). Since this perturbation is present throughout the four days of larval life, PHP in the GluRIIA mutation has been considered to reflect the long-term, sustained expression of PHP (Davis, 2013). We generated double mutant combinations of the GluRIIA mutation with the Imd, IKKβ, Tak1 and Rel mutations. Each double mutant combination was compared to the baseline functionality of the single Imd, IKKβ, Tak1 and Rel mutations alone, thereby accounting for the slight differences in baseline release in each individual mutant background. As shown previously, the GluRIIA mutation causes a decrease in mEPSP amplitude and a homeostatic increase in quantal content that returns EPSP amplitudes toward wild type levels, diagnostic of PHP (Figure 2A, B, C). When we examine the GluRIIA, ImdNP double mutant, we find a complete block of PHP (Figure 2A, E, F, G). We then demonstrate that PHP is also blocked in both the GluRIIA; IKKβ and the GluRIIA; RelE20 double mutants, arguing that the IMD signaling system is necessary for the long-term maintenance of PHP (Figure 2A, E, F, G). However, when we examined the Tak12; GluRIIA double mutants, we find that PHP is completely normal (Figure 2B, C; Supplemental Figure 1A-D). Indeed, this finding was repeated for a second allele of Tak1 (Tak1179) when placed in the GluRIIA mutant background (Figure 2B, C; Supplemental Figure 2A-D). Thus, Tak1 is selectively required for the rapid induction of PHP.
Figure 2. Innate immune signaling during the sustained expression of PHP.
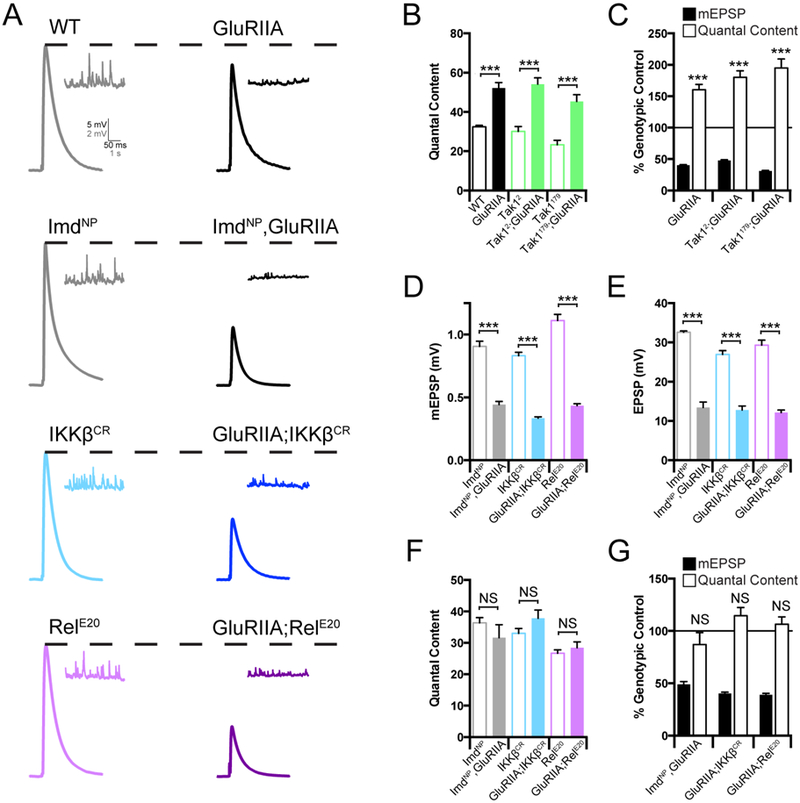
(A) Representative traces for EPSP (scale, 5 mV, 50 ms) and mEPSP (scale, 2 mV, 1 s) at baseline, and in the background of the GluRIIA mutation, for the indicated genotypes. (B) Average quantal content for each genotype in the absence (open bars) or presence (filled bars) of the GluRIIA mutation. (C) mEPSP amplitudes (filled bars) and quantal content (open bars) for each genotype in the GluRIIA background, normalized to the control genotype in the absence of GluRIIA. (D) Average mEPSP amplitude for each genotype in the absence (open bars) or presence (filled bars) of the GluRIIA mutation. (E) Average EPSP amplitude for each genotype in the absence (open bars) or presence (filled bars) of the GluRIIA mutation. (F) Average quantal content in the absence (open bars) or presence (filled bars) of the GluRIIA mutation. (G) Average mEPSP and quantal content normalized to genotypic controls as in C. Data are presented as average (+/−SEM) and statistical significance determined by Student’s t-test (unpaired, two-tailed). See also Supplemental Figure 2.
To this point, our data argue for a bifurcation in the IMD signaling cascade during PHP. The PGRP-LC receptor is necessary for both the rapid and long-term expression of PHP. Downstream of PGRP-LC, IKKβ and Tak1 are necessary for the rapid induction process, but Tak1 is dispensable for the long-term maintenance process. Conversely, Imd and Rel are selectively required for the long-term maintenance of PHP, consistent with the prior demonstration that the rapid induction of PHP is independent of transcription and translation (Frank et al., 2006). Only IKKβ is necessary for both the rapid and long-term form of PHP, suggesting that it represents a point of signaling bifurcation (see discussion).
Impaired PHP does not correlate with altered synapse morphology.
One formal possibility is that differences in synapse growth could account for the genotype-specific effects on homeostatic plasticity. We assessed synaptic growth by quantifying synaptic bouton number and the number of presynaptic active zones. Active zones were identified by immunostaining with anti-Bruchpilot, which labels the presynaptic T-bar that resides at the active zone center and is coincident with presynaptic calcium channels (Kittel et al., 2006). We find that the Tak12 mutation has a moderate reduction in bouton number and a corresponding increase in active zone density, compared to wild type (Supplemental Figure 3A-D). Conversely, the Tak1179 mutants have a small, but significant increase in bouton number. We also observe a small, but statistically significant reduction in active zone density in Imd as well as an increase in both active zone number and density in IKKβ. The difference in bouton number observed in Tak1179 compared to Tak12 is interesting and could be consistent with Tak12 being a more severe mutation (see also below). However, since both mutations completely block PHP, including when Tak12 is placed in trans to a deficiency, we conclude that the changes in morphology, while potentially interesting, are not the cause of impaired PHP. Indeed, throughout this data set, there is no consistent change in bouton number or active zone number that could account for altered synaptic transmission or PHP, considering both the short and long-term expression of PHP. Finally, it should be emphasized that, across all of mutations tested, anatomical changes are small, particularly compared to the nearly 20-fold growth of the NMJ that occurs during the four days of larval development (Schuster et al., 1996).
Tak1 is required presynaptically for the rapid induction of PHP.
Tak1 and IKKβ represent a branch of the IMD signaling cascade that is necessary for the rapid induction of PHP. As such, these proteins could define the first known functional interface of presynaptic innate immune signaling with the mechanisms of neurotransmitter release. Therefore, we chose to define how Tak1 mediates this interface by examining the role of Tak1 during baseline release and PHP in greater detail. First, we attained genetic evidence linking Tak1 to the function of PGRP-LC during PHP. A heterozygous deficiency that uncovers the Tak1 locus has no effect on the rapid induction of PHP (Figure 3A-E). We chose to use this reagent because it is, unquestionably, a heterozygous deletion of the Tak1 locus. As shown previously (Harris et al., 2015), a heterozygous mutation in PGRP-LC is also without effect (Figure 3A-E). However, when these two heterozygous mutations are placed in trans to each other, PHP is completely blocked. Taken together with biochemical and genetic evidence in both flies and mammals (Gottar et al., 2002; Sato et al., 2005; Shim et al., 2005), these data are consistent with Tak1 acting downstream of PGRP-LC during PHP (Figure 3A-E).
Figure 3. Tak1 Interacts genetically with PGRP-LC.
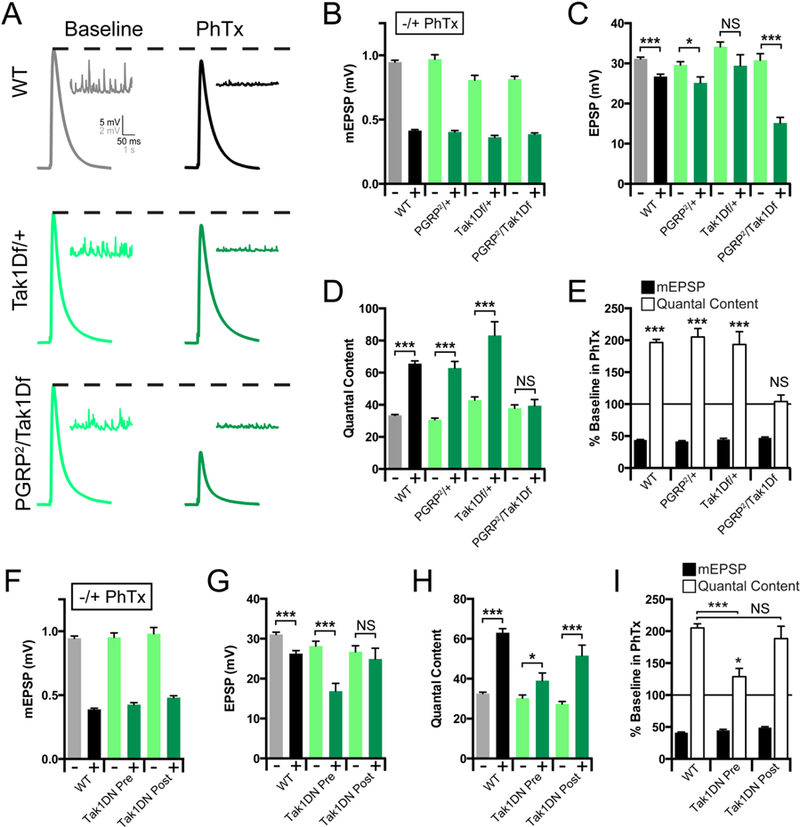
(A) Representative traces for EPSP (scale, 5 mV, 50 ms) and mEPSP (scale, 2 mV, 1 s) at baseline, and in the presence of PhTx, for the indicated genotypes. (B) Average mEPSP amplitude for each genotype in the absence (light bars) or presence (dark bars) of PhTx. (C) Average EPSP amplitude in the absence (light bars) or presence (dark bars) of PhTx. (D) Average quantal content in the absence (light bars) or presence (dark bars) of PhTx. (E) mEPSP amplitudes (filled bars) and quantal content (open bars) for each genotype in the presence of PhTx, normalized to baseline values in the absence of PhTx. (F) Average mEPSP amplitude for each genotype in the absence (light bars) or presence (dark bars) of PhTx. (G) Average EPSP amplitude in the absence (light bars) or presence (dark bars) of PhTx. (H) Average quantal content in the absence (light bars) or presence (dark bars) of PhTx. (I) mEPSP amplitudes (filled bars) and quantal content (open bars) for each genotype in the presence of PhTx, normalized to baseline values in the absence of PhTx. Data are presented as average (+/− SEM) and statistical significance determined by Student’s t-test (unpaired, two-tailed). See also Supplemental Figure 4.
The over-expression of Tak1 is cell lethal, consistent with a role in apoptosis (Takatsu et al., 2000), obviating genetic rescue experiments based on GAL4/UAS-dependent transgene overexpression. Therefore, we assessed the tissue specificity of Tak1 during PHP by generating a kinase dead transgene and expressing this protein pre- versus postsynaptically. We generated an epitope tagged, kinase dead transgene (UAS-Tak1-K46R-FLAG; referred to as Tak1-DN) based on a previously published kinase dead transgene (Takatsu et al., 2000). When this transgene is expressed presynaptically, PHP is strongly suppressed (Figure 3F-I; p<0.005). Next, we demonstrate that muscle-specific over-expression of UAS-Tak1-DN does not impair PHP, compared to wild type (Figure 3F-I). Finally, we demonstrate that the Tak1-DN protein traffics to the presynaptic terminal, based on immunolabeling for the FLAG epitope tag (Supplemental Figure 4). From these data, combined with evidence of a strong genetic interaction with the PGRP-LC receptor and the previously published evidence that PGRP-LC is present at the presynaptic terminal (Harris et al., 2015), we conclude that Tak1 is an essential presynaptic kinase that is required for the rapid induction of PHP.
Tak1 is not required for normal synaptic anatomy
To further characterize the effects of Tak1 on synapse organization, we pursued immunostaining of key synaptic proteins. We quantified the staining intensities of the presynaptic proteins Synaptotagmin 1 (Syt1), Cysteine String Protein (CSP), and Complexin (Cpx). The abundance and distribution of all three proteins were similar comparing wild type to Tak1 (Supplemental Figure 4B-G). We also examined immunolabeling for the microtubule associated protein Futsch (Map1b-like) and observe qualitatively normal organization and appearance of the presynaptic microtubule cytoskeleton (data not shown). Finally, we quantified the abundance of the post-synaptic glutamate receptor subunits GluRIIA and GluRIIC by immunolabeling. Glutamate receptor staining in Tak1179 is wild type. We observe a trend towards a reduction in GluRIIA staining in the Tak12 mutation, but this effect did not reach statistical significance (Supplemental Figure 4B-D). But, since PHP is blocked in both Tak1179 as well as Tak12, altered GluRIIA levels cannot account for impaired PHP. Furthermore, the long-term maintenance of PHP, induced by complete absence of GluRIIA, remains fully functional in Tak1 mutants. Once again, these data argue that a small (statistically not significant) allele-specific difference in the levels of GluRIIA protein cannot be causal for a block of PHP.
Loss of Tak1 strongly impairs evoked and spontaneous neurotransmitter release.
To investigate the role of Tak1 during baseline transmission and PHP, we measured release and PHP across a range of extracellular calcium concentrations using two-electrode voltage clamp to quantify synaptic currents. At 1.5mM external calcium ([Ca2+]e) there is a strong defect in baseline EPSC amplitude in Tak1 mutants compared to wild type (Figure 4A, C, E). Indeed, baseline EPSC amplitude in the Tak12 mutant is decreased by more than ~70% compared to wild type (Figure 4A, C, E). We observe a ~30% decrease in mEPSP amplitude that accounts for a fraction of the observed defect in average evoked release. The change in mEPSP amplitude at elevated [Ca2+]e (compare to Figure 1) could reflect small differences in postsynaptic receptor subunit composition that were not resolved in our immunostaining assays (Supplemental Figure 4). However, the defect in presynaptic release is pronounced. It was observed in both Tak1 alleles and it is consistent across a nearly 10-fold range of extracellular calcium (0.4 to 3mM [Ca2+]e) (Figure 4E), inclusive of [Ca2+]e where no change in mEPSP amplitude is observed for Tak1179 (Figure 1). Finally, we also confirm that PHP remains completely blocked at elevated (1.5mM) extracellular calcium (Figure 4A-D). Thus, Tak1 is essential for baseline synaptic transmission and for the rapid induction of PHP.
Figure 4. Tak1 is necessary for baseline vesicle fusion and PHP under physiological conditions.
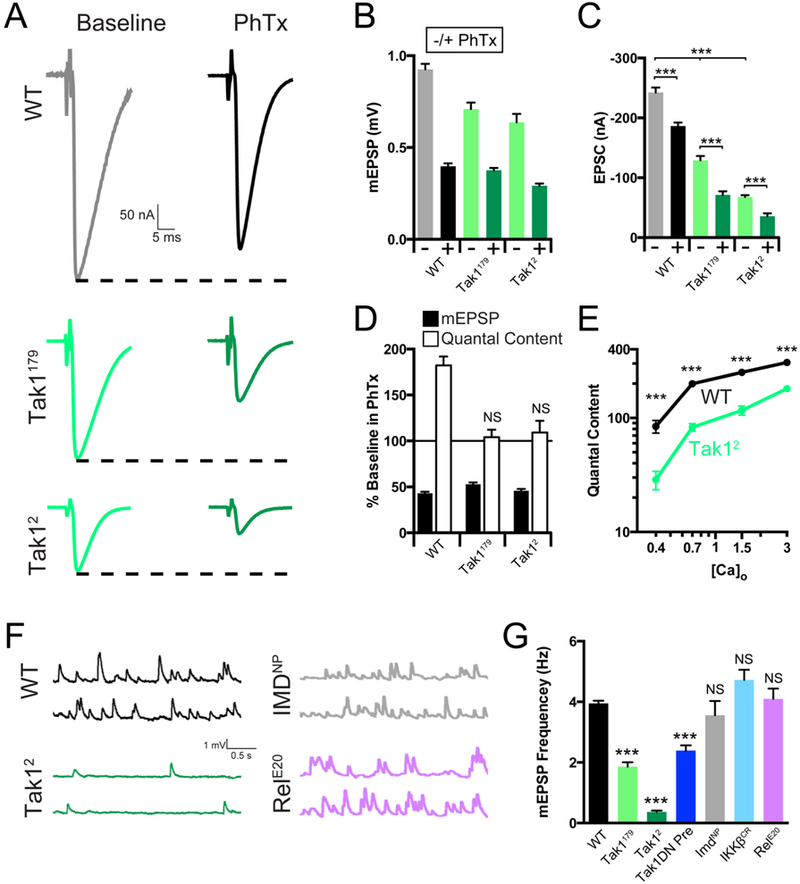
(A) Representative traces for EPSC (scale, 50 nA, 5 ms) at baseline, and in the presence of PhTx, for the indicated genotypes. (B) Average mEPSP amplitude for each genotype in the absence (light bars) or presence (dark bars) of PhTx. (C) Average EPSC amplitude in the absence (light bars) or presence (dark bars) of PhTx. (D) mEPSP amplitudes (filled bars) and quantal content (open bars) for each genotype in the presence of PhTx, normalized to baseline values in the absence of PhTx. (E) Calcium cooperativity curves for the indicated genotypes. Neurotransmitter release was measured at 0.4, 0.7, 1.5, and 3mM extracellular calcium concentration. Quantal content was calculated by dividing EPSC amplitudes by mEPSC amplitudes. (F) Representative traces for mEPSP (scale, 1 mV, 0.5 s) for the indicated genotypes. (G) Average mEPSP frequency for the indicated genotypes. Data are presented as average (+/− SEM) and statistical significance determined by Student’s t-test (unpaired, two-tailed) (C) or by one-way ANOVA with Tukey’s multiple comparisons test (G).
During our experiments, we noted that the rate of spontaneous miniature release is dramatically reduced in the Tak1 mutants compared to wild type (Figure 4F). This effect is observed in both Tak1 alleles. Spontaneous event frequency was reduced by ~50% in Tak1179 and by a dramatic ~90% in Tak12 (Figures 4F, G). This effect was also observed when the kinase dead Tak1 transgene is expressed presynaptically in a wild type background, confirming that this effect is due to impaired Tak1 function within the presynaptic motoneuron, and confirming that this is related to the kinase activity of Tak1 protein. It should be emphasized that the magnitude of this effect is similar to that observed in mutations that directly affect the vesicle fusion machinery, including unc-18 loss-of-function mutations and complexin overexpression (Wu et al., 1998; Weimer et al., 2003; Jorquera et al., 2012), highlighting a potentially profound action of presynaptic Tak1 on the vesicle release mechanism. The rate of spontaneous release at the Drosophila NMJ is largely independent of extracellular calcium (Davis and Goodman, 1998), emphasizing that this function for Tak1 may reflect a direct action at the presynaptic release site.
Given the effects of Tak1 on spontaneous synaptic vesicle fusion rates, we took care to rule out the possibility that a change in spontaneous fusion rate is the cause of impaired PHP expression. If mEPSPs are the physiological events that are monitored by the putative homeostatic sensor (Frank et al., 2006), then a large decrease in mEPSP frequency could prolong the time course necessary for full induction of PHP. PHP is normally induced with a 10-minute PhTx incubation (Frank et al., 2006; see methods). We doubled the incubation time for the Tak12 allele to 20 minutes and PHP remained blocked (see Methods). To further address this issue, we assessed the rate of spontaneous release in the other IMD signaling cascade mutations (Figure 4F, G). No significant difference was observed in the IKKβ, Imd or Rel mutants. From these data, it appears that Tak1 has a novel, constitutive, presynaptic function that is necessary for normal spontaneous vesicle fusion as well as normal evoked neurotransmitter release. In addition, we show that Tak1 is essential for PHP. Therefore, we hypothesize that these functions of Tak1 are mechanistically related.
Decreased release and normal calcium influx in Tak1 mutants
To further characterize the properties of presynaptic vesicle release at the Tak1 mutant synapse, we examined synaptic modulation during short trains of action potentials (APs) delivered at a frequency of 50 Hz. Strikingly, while wild type synapses show characteristic synaptic depression, the Tak1 mutant animals displayed profound short-term facilitation (Figure 5A, B). We also performed this experiment in animals expressing dominant negative Tak1 selectively in motoneurons. Again, we observe a significant baseline defect in neurotransmitter release accompanied by strong facilitation (Figure 5A, B).
Figure 5. Tak1 functions downstream of action-potential induced presynaptic calcium influx.
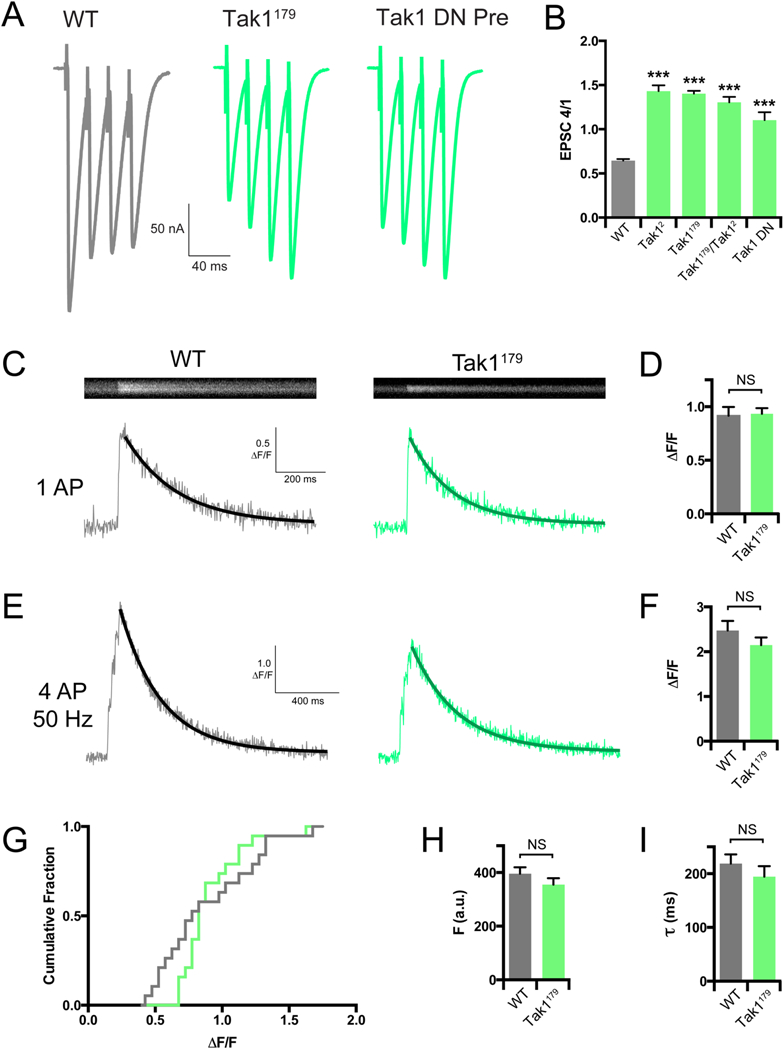
(A) Representative traces for EPSC trains (scale, 50 nA, 40 ms). Stimulation frequency was 50 Hz, and extracellular calcium concentration was 1.5 mM. UAS-Tak1DN was expressed presynaptically with OK371-GAL4. (B) Average ratio of EPSC #4 in the train divided by EPSC #1 for the indicated genotypes. (C) Representative line scans and calcium transients (scale, 0.5 ΔF/F, 200 ms) for WT (grey bar) and Tak1179 (green bar). (D) Average ΔF/F for the indicated genotypes. (E) Representative calcium transients (scale, 1 ΔF/F, 400 ms) for the indicated genotypes. Stimulation was identical to that used in F. (F) Average ΔF/F for the indicated genotypes. (G) Cumulative frequency distributions of ΔF/F for WT (grey line) and Tak1179 (green line). (H) Average baseline fluorescence as a measure of dye-loading for the indicated genotypes. (I) Average decay constants of the transients in H and I for the indicated genotypes. Data are presented as average (+/− SEM) and statistical significance determined by Student’s t-test (unpaired, two-tailed) (D-I) or by one-way ANOVA with Tukey’s multiple comparisons test (B).
A decrease in evoked release coupled to enhanced facilitation is indicative of a decreased release probability, which could result from reduced presynaptic calcium entry. To address this possibility, we imaged spatially averaged calcium transients within individual synaptic boutons using the fluorescent calcium indicator dye Oregon Green BAPTA-1 (OGB-1). We delivered single action potentials (AP) and trains of 4 APs at 50 Hz at 1.5 mM [Ca2+]e. Surprisingly, we find no difference in the amplitude of calcium transients (ΔF/F) in either stimulation protocol, comparing WT and Tak1179 (Figure 5C-G). Quantification of dye loading and the time constant of calcium transient decay were unchanged in Tak1179 compared to wild type (Figure 5H, I). We previously resolved a decrease in presynaptic calcium influx that was responsible for a ~30% decrease in presynaptic release, using identical calcium imaging methodology (Gaviño et al., 2015). The Tak1179 mutation decreases release by nearly 50% and, yet, we cannot resolve a change in calcium influx. From these data, we conclude that the effects of Tak1 on presynaptic release are independent of calcium influx.
Loss of Tak1 does not influence baseline RRP.
We next asked whether Tak1 influences the readily releasable synaptic vesicle pool (RRP) under baseline conditions and following the induction of PHP. We measured the size of the RRP in response to brief trains of high frequency stimulation as previously described (Müller et al., 2012; Harris et al., 2015 ). Again, we observed pronounced facilitation in the early phase of the stimulus train in Tak1 mutants, compared to synaptic depression in wild type animals (Figure 6A, B). As a consequence of the facilitation observed in Tak1 mutants, and despite having a smaller initial EPSC compared to wild type, the cumulative EPSC amplitude and RRP in Tak1179 are not significantly different compared to wild type (p = 0.061; data not shown. ) and are significantly increased in Tak12 (p<0.05). However, if we compare initial EPSC amplitude to the size of the RRP in Tak1 mutants and wild type animals, an estimate of presynaptic release probability can be obtained (Ptrain). There is a dramatic and consistent decrease in Ptrain in both Tak1 mutants compared to wild type (Figure 6D).
Figure 6. Tak1 controls the homeostatic modulation and dynamics of the readily releasable pool.
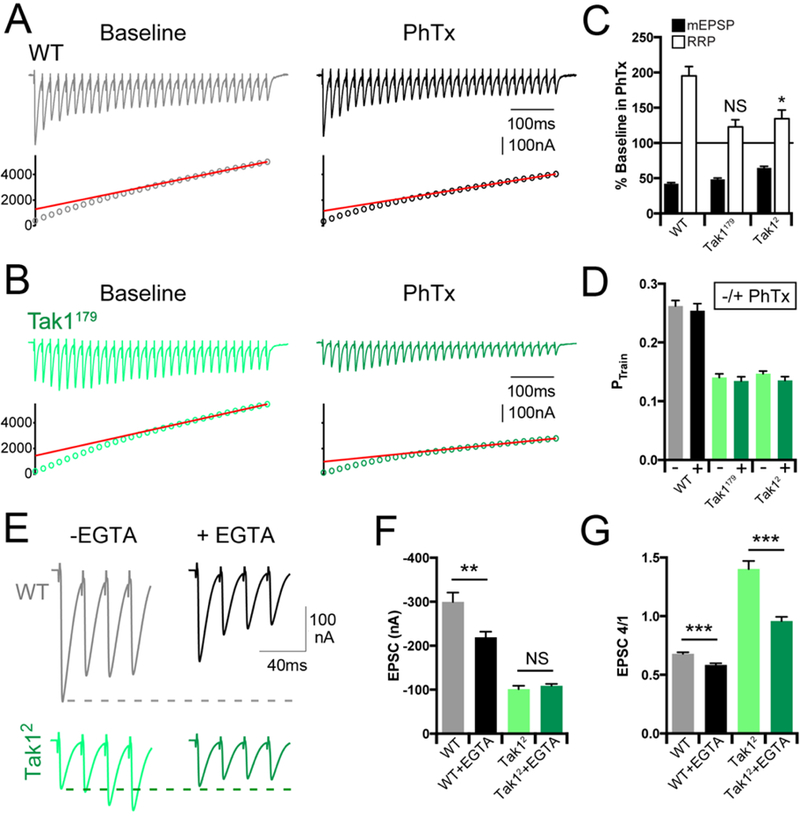
(A, B) Representative EPSC trains (scale, 100 nA, 100 ms) at baseline, and in the presence of PhTx, for the indicated genotypes. Trains are 30 stimuli delivered at 60 Hz. Extracellular calcium concentration is 3 mM. (C) Average data for mEPSP amplitudes (filled bars) and readily releasable pool size (open bars) in the presence of PhTx, normalized to baseline, for the indicated genotypes. (D) Average data for probability of release calculated as PTrain = amplitude of the first EPSC divided by the cumulative EPSC in the indicated genotypes. (E) Representative EPSC traces (scale, 100 nA, 40 ms) at baseline (light traces), and after incubation with EGTA-AM (50 µM) (dark offset traces) for the indicated genotypes. (F) Average data for EPSC amplitude at baseline (light bars), and after incubation with EGTA-AM (dark bars) for the indicated genotypes. (G) Average data for the amplitude of the 4th EPSC in a train divided by the 1st EPSC in a train. Data are presented as average (+/− SEM) and statistical significance determined by Student’s t-test (unpaired, two-tailed).
Next, we examined the effects of EGTA-AM, comparing wild type with Tak1. EGTA has a relatively slow calcium binding rate (Smith et al., 1984) and can be used to probe the functional coupling of vesicle to sites of presynaptic calcium influx in Drosophila, just as at mammalian central synapses (Schneggenburger and Neher, 2005; Wang et al., 2016). We demonstrate that application of EGTA-AM to the wild type synapse (50µM) causes a decrease in average EPSC amplitude in response to a single action potential and causes more pronounced synaptic depression during a short stimulus train (Figure 6E-G). This is consistent with the conclusion that wild type synapses harbor docked, primed vesicles with a range of calcium-coupling efficiencies, a portion of which are EGTA-sensitive. By contrast, Tak12 is insensitive to EGTA-AM following single action potential stimulation, although facilitation of EPSC amplitude during a stimulus train is blunted, as expected (Figure 6E-G). EGTA-insensitivity in response to a single stimulus argues that docked/primed vesicles in Tak12, at the resting state, are tightly coupled to sites of calcium entry.
When these data are taken together, we conclude that loss of Tak1 causes a primary defect in the initial EPSC amplitude of a stimulus train. During a stimulus train, elevated intra-terminal calcium compensates for the lack of Tak1 and drives facilitation of vesicle release. Finally, loss of Tak1 does not impair the total number of vesicles that are available for release (RRP) during repetitive stimulation and enhanced facilitation. One possible conclusion is that loss of Tak1 somehow limits the number of docked/primed synaptic vesicles that exists at a resting state, in the absence of stimulation. This steady state deficit then seems to be over-come by elevated intra-terminal calcium that occurs during repetitive stimulation. Such a steady state defect the in pool of docked/primed synaptic vesicles is consistent with the dramatic reduction in mEPSP frequency, no change in action potential-dependent calcium influx and the insensitivity of single-action potential release to the presence of EGTA-AM.
Loss of Tak1 prevents the homeostatic modulation of the RRP
The expression of PHP has been linked to a near doubling of the RRP and mutations that prevent the expansion of the RRP have been shown to block PHP, including mutations that delete the PGRP-LC innate immune receptor (Müller et al., 2012; Harris et al., 2015). Therefore, we repeated our measurements of RRP in the presence of PhTx to induce PHP. In wild type, as shown previously (Müller et al., 2012; Harris et al., 2015), we observe a significant expansion of the RRP (Figure 6A, C). However, in the Tak1179 mutants, the homeostatic expansion of the RRP is completely blocked, an observation that mirrors the effects of the PGRP-LC receptor mutation (Harris et al., 2015). In the Tak12 mutant there is a small, statistically significant, increase in the RRP. But, this effect is dramatically less that that observed in wild type supporting the conclusion that Tak1 is essential for the expansion of the RRP during PHP (Figure 6C). From these data, we conclude that Tak1 is essential for the homeostatic expansion of the RRP. If loss of Tak1 somehow limits the number of docked/primed synaptic vesicles, then the absence of Tak1 may prevent the expansion of this pool of vesicles during PHP, thereby blocking the expression mechanism. Thus, we sought experiments to directly test whether Tak1 regulates the pool of docked/primed synaptic vesicles.
Tak1 stabilizes the docked/primed pool of synaptic vesicles.
By examining the recovery of synaptic vesicle release after strong stimulus-dependent depletion of the synaptic vesicle pool, we are able to gain insight into how Tak1 influences the behavior of the docked/primed pool of synaptic vesicles. We investigated recovery from strong synaptic depression at elevated extracellular calcium (3mM), consistent with previously published methods (Müller et al., 2015). The recovery from synaptic depression occurs with two time constants at the Drosophila NMJ, just as it does at mammalian central synapses (Müller et al, 2015; Sakaba & Neher, 2001). An initial phase of fast recovery is associated with the replenishment of a low-release probability vesicle pool that is thought to reside at a distance from sites of presynaptic calcium influx. A second, slower phase of recovery is associated with the replenishment of a high release probability vesicle pool, tightly coupled to sites of calcium entry (Müller et al., 2015; Sakaba & Neher, 2001). We delivered high frequency stimulus trains to deplete the RRP (30 APs at 60 Hz). We then delivered paired-pulse stimuli at varying intervals following cessation of the stimulus train, allowing us to quantify the rate of EPSC recovery (Figure 7A, C). First, we plot the rate of recovery as a percent of the initial EPSC amplitude (specific to each genotype since Tak1 reduces initial EPSC amplitudes). As expected, we observe two time constants of recovery in wild type, a fast time constant (τfast = 53ms) and a slow time constant (τslow = 6.25s). These values are quantitatively similar to previous measurements made in this system (Hallermann et al., 2010, Müller et al., 2015). By contrast, recovery i n the Tak1179 and Tak12 mutants is highly unusual, becoming tri-phasic and preventing estimation of recovery time constants. The initial phase of EPSC recovery in Tak1179 and Tak12 is significantly enhanced compared to wild type (Figure 7B). Indeed, EPSC amplitude is fully recovered within the first 50–100ms in Tak1179, a time point when wild type remains 50% depressed. However, the initial recovery of EPSC amplitude is not sustained in either of the Tak1 mutants. EPSC amplitude decays between 200ms and 5s in the post-train interval, before finally entering a new, slow phase of recovery to a steady state that is significantly smaller than that observed in wild type (Figure 7A, D). This unusual recovery profile has never been previously observed at a synapse, to our knowledge.
Figure 7. Tak1 stabilizes a high release probability synaptic vesicle pool.
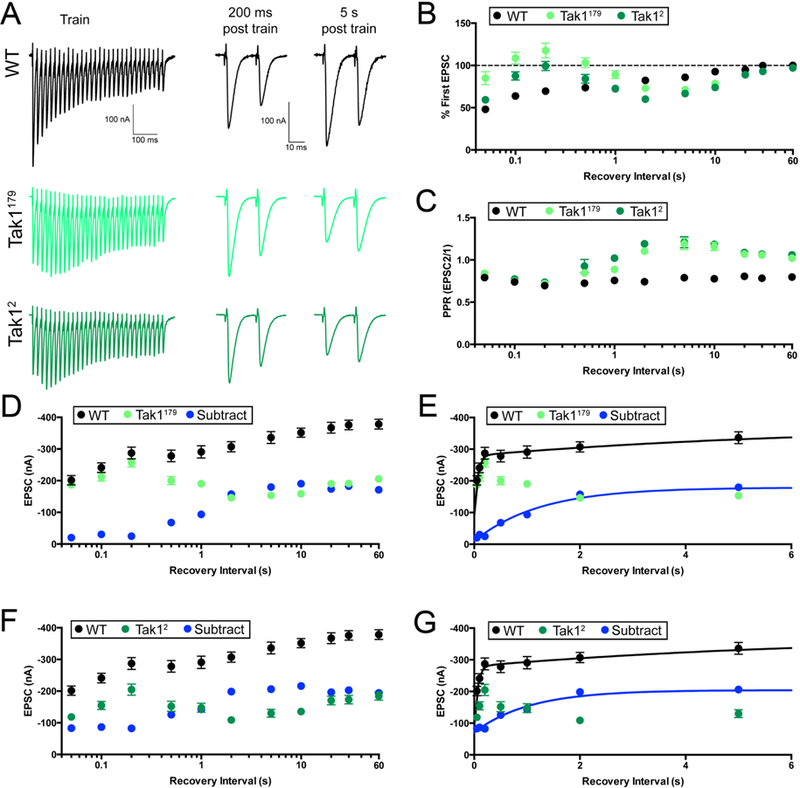
(A) Representative traces for trains used to deplete the vesicle pool (scale, 100 nA, 100 ms), and paired EPSC pulses at recovery intervals of 200 ms and 5 s after the train (scale, 100 nA, 10 ms), for the indicated genotypes. (B) Summary data showing average recovery time courses in the indicated genotypes. (C) Average paired-pulse ratio, calculated as the amplitude of EPSC #2 divided by that of EPSC #1, at each recovery interval, for the indicated genotypes. Inter stimulus interval for paired pulse is 20 ms. (D) Average data for recovery time courses of raw EPSC amplitudes in the indicated genotypes. The data labeled “subtract” (blue) are calculated by subtraction of the average amplitude in Tak1179 from that of WT, at each tested recovery interval. (E) View of the first 6 seconds of the data presented in D, now plotted on a linear x axis. WT data are fit with a double-exponential function (τfast=0.053 s, τslow=6.25 s). Subtraction data could not be fit with a double-exponential function and are fit with a single exponential function (τ=1.23 s). Fits were calculated using all the data out to 90 s, but the x axis is limited to 6 s for ease of visualization. (F) Average data for recovery time courses of raw EPSC amplitudes in the indicated genotypes. WT data are repeated from D. “Subtract” was generated as in D, subtracting Tak12 amplitudes from WT. (G) View of the first 6 seconds of the data in F. Subtraction data are again fit with a single exponential function (τ=1.04 s). WT data are repeated from E. Data are presented as average (+/− SEM).
Next, we assessed EPSC recovery with paired-pulse stimulation, quantifying the paired-pulse ratio (EPSC2/EPSC1) at each time point in the recovery phase. In wild type, PPR is unchanged throughout the recovery phase, always resembling the pre-stimulus PPR, showing synaptic depression (Figure 7C). By contrast, PPR in both Tak1 mutants changes dramatically during recovery from depression. Prior to the stimulus train, both Tak1 mutants show pronounced facilitation (Figure 7A). Immediately following the stimulus train, at the 50–200ms time point, the time at which EPSC amplitudes have fully recovered to baseline levels, we find that PPR in both Tak1 mutants converts from facilitation to depression, resembling wild type (Figure 7A,C). Then, during the 200ms-5s post-train interval, initial EPSC amplitude decays in both Tak1 mutants and PPR reverts to strong facilitation (exceeding that observed at baseline) (Figure 7A,C). Finally, in the last phase of recovery (20s-90s), PPR reverts to levels of facilitation observed prior to the stimulus train (Figure 7A,C). Thus, during an initial recovery phase, Tak1 mutants revert to ‘wild type’ release, both in terms of EPSC amplitude and paired pulse depression. However, this ‘wild type’ state cannotbe sustained in Tak1, and it rapidly decays (between 200ms and 5s after cessation of the stimulus train).
Finally, we re-analyzed the EPSC recovery kinetics without normalization to the initial EPSC amplitude. In this analysis, EPSC amplitudes prior to the stimulus train are significantly smaller in Tak1 mutants compared wild type. During the stimulus train, EPSC amplitudes depress to a similar amplitude in Tak1 mutants compared wild type. During the first 200ms of recovery, the EPSC in Tak1179 becomes identical to that observed in wild type (Figure 7A, D), and nearly so in the Tak12 mutant (Figure 7A, F). However, neither mutant is able to sustain this ‘wild type’ level of release, and EPSC amplitudes decay over the next 5s (Figure 7A,D,F). To further characterize this effect, we subtracted the Tak1 recovery curves from the wild type recovery curve, yielding a subtracted curve that represents the fraction of recovery that is dependent upon Tak1 (Figure 7D-G, blue points, lines). The subtracted curve (blue line) makes it clear that Tak1 mutants recover normally during the first 200 ms. Then, the recovery fails and Tak1 EPSC amplitudes not only fall below the wild type curve, they never fully reach wild type levels.
One interpretation of these data, taking into account all of our observations, is that the initial phase of recovery (50–200ms) is completely independent of Tak1 function. As such, wild type and Tak1 begin to recover from the same depressed EPSC amplitude, recover with initially similar kinetics, and PPR is identical during this time interval. The activity of the Tak1 kinase is revealed only after the first 200ms of recovery. During this time interval, Tak1 appears to be necessary to stabilize the newly recovered vesicle pool. Therefore, in the absence of Tak1, the rate of either de-priming or undocking exceeds the rate of vesicle replenishment causing a gradual decay in EPSC amplitude.
Ultrastructural evidence that Tak1 stabilizes the docked vesicle pool.
Taken together, our electrophysiological data argue that Tak1 normally functions to stabilize either the docked or primed vesicle state. While there is evidence that synaptic vesicle docking and priming are reversible reactions (Murthy and Stevens, 1999; He et al., 2017), the molecular mechanisms that specify the rates of undocking and de-priming remain largely unknown. Thus, we sought to pinpoint the effects of Tak1 at the presynaptic terminal. If Tak1 is required to stabilize the docked vesicle pool, then we should see a decrease in docked vesicles at the active zone by electron microscopy. By contrast, if Tak1 is specifically required to stabilize the primed vesicle state, then we may find normal numbers of docked vesicles despite a highly significant decrease in EPSC amplitude and mEPSP frequency.
First, we examined wild type and both Tak1 mutants ultrastructurally. Active zones were identified by standard criteria: 1) narrow synaptic cleft, 2) electron density within the synaptic cleft, 3) clustered presynaptic vesicles and 4) the presence of a characteristic presynaptic electron density referred to as a T-bar. For each active zone, we quantified several parameters: 1) the number synaptic vesicles that reside within 150nm and 400nm of the center of the pedestal of the T-bar, where presynaptic voltage gated calcium channels reside (Kittel et al., 2006), 2) the distance from each vesicle to its nearest neighboring vesicle (see Methods for details of sample acquisition and analysis) and 3) the number of docked synaptic vesicles. Docked vesicles were defined by first determining the center of each vesicle (see Methods). Any vesicle residing at distance equal to or less than a vesicle radius (15nm) from the plasma membrane was scored as a docked vesicle and was confirmed visually as touching the presynaptic plasma membrane. There is a striking, highly significant decrease in the number of synaptic vesicles at the release site (Figure 8A-D), whether this measurement is made within 150nm or 400nm of the T-bar. A similar change is also observed in the inter-vesicle distance (Figure 8D). Thus, there is a general defect in the packing density of synaptic vesicles at or near the presynaptic release site. We also observe a striking defect in the number of docked vesicles. We find that wild type active zones have, on average, 2 docked vesicles per active zone (1.9 ± 0.4, n = 16). By contrast, the Tak12 mutant has a dramatic reduction in the average number of docked vesicles (0.2 ± 0.1, n= 36), with many active zones being completely devoid of docked vesicles (Figure 8E, F). These data argue that Tak1 directly or indirectly stabilizes the docked vesicle state. The decrease in vesicle packing density could be a reflection of how Tak1 functions to stabilize docked vesicles, possibly indicative of an altered connection of vesicles to the presynaptic cytomatrix.
Figure 8. Impaired synaptic vesicle distribution and docking at the active zones of Tak1 mutants.
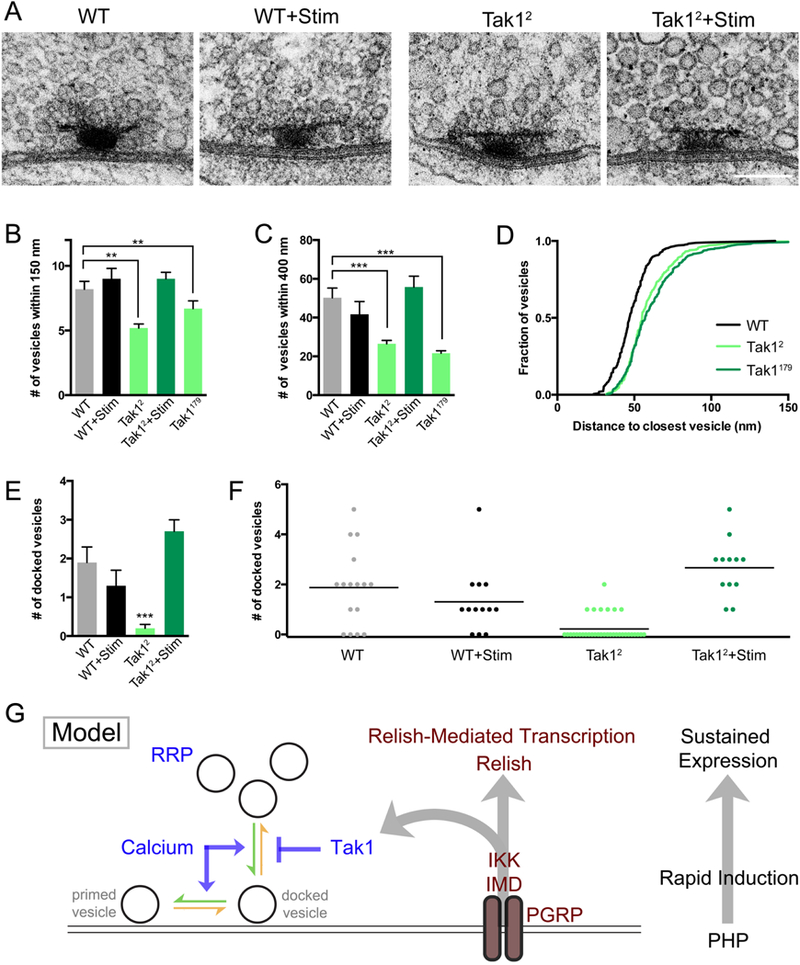
(A) Representative electron micrographs of presynaptic active zones in the indicated genotypes. Scale bar represents 100 nm. (B) Average number of vesicles within 150 nm of the base of the T-bar for the indicated genotypes. (C) Average number of vesicles within 400 nm of the base of the T-bar for the indicated genotypes. (D) Cumulative frequency distribution of the distance between each vesicle and its nearest neighboring vesicle for the indicated genotypes. (E) Average number of docked vesicles per active zone in the indicated genotypes. Only Tak12 is statistically different from wild type. Data are presented as average (+/− SEM) and statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test. (F) Individual data points for number of docked vesicles in the indicated genotypes, re-plotted from (E) where statistical significance is indicated. Each dot represents one active zone. Line indicates the mean. (G) Model. At right: the IMD signaling cascade couples the mechanisms of rapid induction with the long-term expression of PHP. Center: diagram of the IMD signaling cascade initiated by the PGRP receptor, which catalyzes the assembly of the IMD complex inclusive of the proteins IMD, IKK and Tak1. Left: Tak1 inhibits the rate of vesicle un-docking (orange arrow) at an individual presynaptic release site. The opposing rate of vesicle docking is potentiated by intracellular calcium, among other factors. The readily releasable vesicle pool (RRP) is indicated.
Our electrophysiological data demonstrate that presynaptic release is temporarily rescued following a stimulus train (Figure 7). We hypothesize that this rescue is caused by the effects of elevated presynaptic calcium, which could potentiate the rate of vesicle docking and temporarily overcome the defect caused by loss of Tak1. This hypothesis predicts that we should see restoration of the number of docked vesicles immediately following a stimulus train. We delivered a stimulus train (10 seconds 100 Hz) and immediately (within ~1s) immersed the NMJ with fixative at 4˚C. Under these conditions, wild type active zones are normal, with no change in the number of docked synaptic vesicles (1.3 ± 0.4, n = 13). By contrast, the Tak1 mutant synapse shows a complete restoration of the population of docked synaptic vesicles, with every active zone having at least one docked vesicle and having on average 2 docked vesicles, just as observed in wild type (2.7 ± 0.3, n = 12) (Figure 8E, F). This represents a complete ultrastructural rescue that correlates with the complete rescue of synaptic transmission immediately following a stimulus train. Thus, we propose that elevated intra-terminal calcium that occurs during a stimulus train transiently overcomes the loss of Tak1 by driving the rate of synaptic vesicle docking and priming (Figure 8G). Then, as intra-terminal calcium levels drop following the stimulus train, docked vesicles are no longer stabilized at the active zone and EPSC amplitude gradually reverts to the impaired levels observed at rest in the Tak1 mutant background. The resting state in Tak1 represent a new steady state that reflects a normal rate of vesicle docking and priming and an enhanced rate of vesicle undocking due to the lack of Tak1.
In conclusion, our data argue that Tak1 is a molecular interface of presynaptic innate immune signaling with the release mechanism. Tak1 functions at the presynaptic terminal to stabilize docked synaptic vesicles. Tak1 is also required for the rapid induction of presynaptic homeostatic plasticity. As such, activation of Tak1 would be expected to potentiate the stability of the docked vesicle pool, providing a molecular mechanism for the rapid, homeostatic potentiation of the readily releasable vesicle pool, a well established expression mechanism of presynaptic homeostatic plasticity at the Drosophila NMJ that is conserved at the mammalian NMJ and at mammalian central synapses (Cull-Candy et al., 1980; Kim and Ryan, 2010; Zhao et al., 2011; Mitra et al., 2011).
DISCUSSION
We have tested the core signaling components of an innate immune signaling pathway (IMD signaling) for a role during presynaptic homeostatic plasticity. Our data support a model in which IMD signaling bifurcates downstream of the presynaptic innate immune receptor PGRP-LC to achieve immediate, local modulation of the presynaptic release apparatus via Tak1, and prolonged maintenance of the homeostatic response via the transcription factor Relish (Figure 8G). This model allows the innate immune signaling system to rapidly alter presynaptic release (seconds to minutes) and simultaneously initiate a Rel-dependent consolidation of PHP. It is noteworthy that the consolidation of PHP can persist for months in insects (Mahoney et al., 2014) and decades in humans (Cull-Candy et al., 1980). It has become clear that the molecular mechanisms responsible for the rapid induction and sustained expression are genetically separable (Davis, 2013). Our findings provide an explanation for how the rapid induction and sustained expression of PHP are mechanistically coordinated.
The canonical function of innate immunity is to recognize invading pathogens or non-normal molecular patterns and induce a rapid, inflammatory reaction that is sustained for as long as the invasion persists. During an innate immune response, Tak1-dependent signaling turns on more rapidly, and turns off more rapidly, than Rel-mediated transcription (Boutros et al., 2002; Park et al., 2004). As such, Tak1 signaling can be considered as a feed-forward activator of the cellular immune response. Based on our data, we propose that innate immune signaling in the nervous system is transfigured to detect non-normal neurophysiology, although the molecular event that reports altered neural (synaptic) function and is subsequently detected by presynaptic PGRP-LC remains a mystery. In our study, we place Tak1 at the presynaptic release site where homeostatic plasticity is rapidly induced. Thus, just as in canonical innate immunity, Tak1 is ideally situated to act as a feed-forward potentiometer that controls vesicle fusion, thereby achieving a rapid compensatory change in presynaptic release following postsynaptic glutamate receptor inhibition. Subsequent activation of Rel-mediated transcription, provides a sustained response that can be maintained for the duration of the perturbation.
Tak1 inhibits the rate of vesicle un-docking at baseline and during PHP
We provide evidence that Tak1 has a potent, constitutive function to stabilize the docked vesicle pool, defined both electrophysiologically and at the EM level (Figure 7, 8). This can explain the dramatic defect in spontaneous miniature release frequency, which occurs without a change in the total number of presynaptic release sites (Figure 4). Fewer docked vesicles can explain the defect in EPSC amplitude despite normal action-potential induced calcium influx. We also demonstrate that vesicles can be mobilized to the release site during a stimulus train, temporarily achieving wild type release rates before declining back to a reduced baseline (Figure 7). This effect reflects a stimulus dependent re-population of the docked vesicle pool, as demonstrated by our EM analysis directly following a train of action potentials. The stimulus, and presumably calcium-dependent, re-population of the docked vesicle pool explains why there is pronounced short-term synaptic facilitation in the Tak1 mutants compared to short-term depression in wild type. Essentially, the number of docked vesicles is increased in a stimulus dependent manner, something that does not occur in wild type, based on our EM analysis (Figure 8). Finally, our analysis of recovery from vesicle depletion pinpoints the time at which Tak1 activity becomes essential. The recovery of synaptic transmission is completely normal in Tak1 for the first 500ms following a stimulus train. It is only after this time point that recovery becomes defective.
As a working model (Figure 8G), we propose that Tak1 functions to inhibit the rate of vesicle undocking (Figure 8G, orange arrows) and, thereby, persistently stabilizes the docked/primed vesicle pool. Immediately following a stimulus train, calcium-dependent mechanisms actively mobilize vesicles to the active zone and drive calcium-dependent vesicle docking and priming (Sakaba and Neher, 2001; Figure 8G, green arrows). Thus, calcium-dependent potentiation of the forward rate of docking/priming transiently overcomes the enhanced un-docking rate caused by loss of Tak1 and the number of docked vesicles is restored to wild type levels. As intra-terminal calcium levels drop following the stimulus train, the rates of docking and un-docking re-equilibrate in the Tak1 mutant, reaching a steady state with many fewer docked vesicles compared to wild type.
This model can also explain the failure of PHP in a Tak1 mutant. The rapid induction of PHP occurs following inhibition of postsynaptic glutamate receptors and is expressed in response to a single action potential. This requires an expansion of the resting pool of docked/primed vesicles and this cannot be achieved in the absence of Tak1. Accordingly, enhanced Tak1 activity, following activation of the innate immune receptor PGRP, would inhibit the rate of vesicle un-docking and potentiate the pool of docked/primed vesicles, necessary for the rapid induction of PHP. Thus, Tak1 functions as a molecular potentiometer controlling the docked/primed vesicle pool during PHP. A question for future experimentation concerns how baseline Tak1 activity is established to achieve highly reproducible, baseline synaptic transmission in wild type.
Organization of innate immune signaling in Drosophila motoneurons
Most of our knowledge regarding the organization of innate immune signaling pathways are based on assays that detect the nuclear translocation of Rel/NF-κB, or quantify Rel/NF-κB -mediated transcription (Sato et al., 2005; Delaney et al., 2006; Rothwarf & Karin, 1999; Choe et al., 2002). Thus, the logic and spatial organization of innate immune signaling in neurons has yet to be clearly defined. Our data argue for several novel features of signaling organization in a neuron, beyond the local synaptic action of Tak1. In canonical innate immune signaling receptor activation catalyzes the assembly of an intracellular complex that includes Imd, Tak1 and IKKβ. Based upon our evidence in Drosophila motoneurons, and consistent with published models of IMD signaling, we propose that activation of the PGRP receptor drives assembly of the IMD signaling complex that includes a poly-ubiquitin chain as well as the three proteins IMD, Tak1 and IKKβ (Zhou et al., 2005). Then, we suggest that IKKβ is necessary for this signaling complex to either assemble correctly, or signal efficiently. Thus, IKKβ is necessary for both the short and long-term forms of PHP, just as the PGRP receptor. Finally, once this complex is assembled and activated, two independent signal transduction cascades are initiated. One requires Tak1 and acts locally within the presynaptic terminal to achieve the rapid induction of PHP. The second pathway requires the function of IMD and signals via Rel-mediated transcription, necessary for the long-term maintenance of PHP (Figure 8G). We note, however, that other models for IKKβ activation should also be considered (Polley et al., 2013; Häcker & Karin, 2016). Finally, our data raise an important question regarding how signaling is conveyed from synaptic IKKβ to nuclear Rel, a topic of future studies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Graeme W. Davis (Graeme.davis@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila melanogaster was the species used in this study. Flies were raised on standard cornmeal media. Both male and female flies were used. All experiments were conducted with animals at the third instar larval stage.
METHOD DETAILS
For all experiments except electron microscopy, the w1118 strain was used as the wild type control. For electron microscopy the w−3605 strain was used as the wild type control. Animals were raised at 22–25°C. ImdNP1182, RelE20, Tak12, Tak1179, and the Tak1 deficiency (Df(1)BSC645) were obtained from the Bloomington Drosophila Stock Center. PGRP-LC2 was provided by Kathryn Anderson. The GluRIIASP16 mutation (Petersen et al., 1997) and the OK371-GAL4 (Mahr and Aberle, 2006) driver have been previously described.
The IKKβCR mutation was generated following the protocol of (Kondo and Ueda, 2013). IKKβ gRNA was selected using the CRISPR optimal target finder website (http://tools.flycrispr.molbio.wisc.edu/targetFinder). The gRNA sequence AAGTCAAGCTCAGCGAGCGT was cloned into the pCDF3-dU6:3gRNA vector (Addgene plasmid #49410, Simon Bullock). Flies expressing the UAS-gRNA were crossed with flies expressing UAS-Cas9 in the germline (nos-GAL4VP14, UAS-cas9). Male offspring were used to create unique stable lines after removing the UAS-cas9 and removing in the next generation the UAS-gRNA. Putative IKKβ mutants were sequenced to identify the nature of the Cas9 mediated mutation using the primers CTTATAGCCTTTCGGCTGAGTTAG and CCACCGTTACAGTATTCCAGC. The resulting mutation, is a deletion of the 162nd nucleotide in IKKβ cDNA, creating an early stop codon shortly thereafter.
Generation of UAS-Tak1DN-FLAG
The UAS-Tak1DN-FLAG construct was generated by amplifying the full-length Tak1 open reading frame from the BDGP DGC cDNA clone LD42274 and cloning into pENTR/D-TOPO (Invitrogen). Duplex PCR was used to simultaneously amplify the Tak1 open reading frame and generate the dominant negative mutation. Specifically, the following three primer pairs were used: 1) caccATGGCCACAGCATCGCTGG and GAAGAACTCCcTGACGGCAA, 2) TTGCCGTCAgGGAGTTCTTC and CGCATTGTGATGCGGTGGG, 3) caccATGGCCACAGCATCGCTGG and CGCATTGTGATGCGGTGGG. Amino acid 46 was changed from K to R. The Tak1 cDNA was then directly cloned into the UAS-C-terminal FLAG (3x FLAG) vector, pTWF (T. Murphy, Drosophila Genomics Resource Center) using the Gateway recombination cloning system (Invitrogen). Transgenic flies were generated by standard injection methods (BestGene).
Electrophysiology
Sharp-electrode recordings were performed as previously described (Davis and Goodman, 1998; Müller et al., 2012). Two-electrode voltage-clamp recordings were performed with an Axoclamp 2B amplifier. The extracellular HL3 saline contained the following (in mM): 70 NaCl, 5 KCl, 10 MgCl2, 10 NaHCO3, 115 sucrose, 4.2 trehalose, 5 HEPES, and 0.3 CaCl2 (unless otherwise specified). For acute pharmacological homeostatic challenge, partially dissected larvae were incubated in Philanthotoxin-433 (PhTx; 10–20 µM; Sigma-Aldrich) for 10 min according to published methods (Frank et al., 2006). Quantal content was estimated by calculating the ratio of EPSP amplitude/average mEPSP amplitude and then averaging recordings across all NMJs for a given genotype. Selection criteria: intracellular recordings in current clamp configuration with a resting membrane potential more depolarized than minus 60 millivolts or with an input resistance of less than 4MΩ were excluded from analysis, being diagnostic of poor muscle impalement with sharp electrodes. EPSC data were analyzed identically. The RRP was estimated by cumulative EPSC analysis (Müller et al., 2012). Muscles were clamped at −65 mV, and EPS C amplitudes during a stimulus train (60 Hz, 30 stimuli, 3 mM extracellular calcium) were calculated as the difference between peak and baseline before stimulus onset of a given EPSC. For recovery of the vesicle pool, stimulus trains (60 Hz, 30 stimuli, 3 mM extracellular calcium) were first given to deplete the pool to steady-state. Recovery was then interrogated with paired pulses (2 pulses, 50 Hz) at varying recovery intervals post-train. Electrophysiology data were analyzed with custom-written routines in Igor Pro 6.37 (Wavemetrics) and MATLAB 8.5 (Mathworks) (Gaviño et al., 2015). Additional methodological considerations are as follows. Replication: All sample sizes, for all experiments are either stated directly in the text of the manuscript or in Supplemental Table 1. All experiments were performed in at least two independent genetic crosses. Ramdomization: wild type recordings were routinely incorporated in all electrophysiological tests of experimental genotypes. Key experiments regarding the identification and characterization of innate immune signaling pathway mutations were independently replicated by two individuals. Necessary sample size was computed using a power analysis. Given experimentally determined variance of baseline synaptic transmission and homeostatic plasticity as well as an effect size in the range of 75–200%, it is estimated that sample sizes of 5 are sufficient. However, in most experiments, sample sizes greatly exceed this level, as evidenced in Supplemental Table 1.
Calcium Imaging
Ca2+ imaging experiments were done as described in (Müller et al., 2012). Third instar larvae were dissected and incubated in ice-cold HL3, 0.3 mM calcium, containing 5 mM Oregon green 488 BAPTA-1 (OGB-1; hexapotassium salt, Invitrogen) and 1 mM Alexa 568 (Invitrogen). After incubation for 10 min, the preparation was washed with ice-cold HL3 for 10–15 min. Single-action-potential-evoked spatially averaged Ca2+ transients were measured at an extracellular [Ca2+] of 1.5 mM using a confocal laser scanning system (Ultima, Prairie Technologies) at room temperature. Fluorescence changes were quantified as ∆F/F = (F(t) – F baseline)/(Fbaseline – Fbackground), where F(t) is the fluorescence in a region of interest (ROI), Fbaseline is the mean fluorescence from a 50-ms period preceding the stimulus, and Fbackground is the background fluorescence from an adjacent ROI without any indicator-containing cellular structures.
Anatomical Analyses
Third-instar larvae were dissected in 0 Ca2+ HL3 and fixed for 1–2 min in Bouin’s fixative (100%; Sigma-Aldrich), for 15 min in PFA (4% in PBS), or for 7 minutes in ice-cold ethanol, and incubated overnight at 4°C with primar y antibodies. The following primary antibodies were used at the indicated dilutions: mouse anti-Bruchpilot (Brp), 1:100 (nc82; Kittel et al., 2006); mouse anti-CSP, 1:250; mouse anti-GluRIIA, 1:100; rabbit anti-synaptogamin, 1:1000; rabbit anti-complexin, 1:500; mouse anti-FLAG, 1:50; rabbit anti-GluRIIC, 1:1000; and rabbit anti-Dlg, 1:10,000. Cy5-conjugated or Cy3-conjugated anti-HRP was used at 1:50, (Invitrogen), and applied for 1 h at room temperature. Larval preparations were mounted in Vectashield (Vector Laboratories). An Axiovert 200 inverted microscope (Carl Zeiss), a 63x or 100x (1.4 NA) Plan Apochromat objective (Carl Zeiss), a cooled CCD camera (CoolSNAP HQ; Roper Scientific), and Slidebook 5.0 (3i) were used for deconvolution microscopy. Synaptic boutons were identified by anti-Dlg. Active zones were identified by anti-Brp and quantified using automated functionality in ImageJ and a watershed plug-in (http://bigwww.epfl.ch/sage/soft/watershed/). Parameters were: radius=1, 8-connected, Min/Max=0,170–210. For staining intensity quantification, a mask was made on the CSP signal (for CSP and Synaptotagmin 1), the GluRIIA signal (for GluRIIA and GluRIIC), or the Complexin signal (for Complexin). The average pixel intensity across the mask is reported. For Supplemental Figure 3A, a Nikon Ti microscope, a 60x Plan Apochromat objective, a Photometrics Evolve Delta EMCCD camera, and a Yokogawa CSU-22 spinning disk confocal were used, in the UCSF Nikon Imaging Center.
Electron Microscopy
Wandering third instar larvae were pinned and filleted in cold physiological saline (HL3) on a Sylgard pad in a small Petri dish. Once filleted, the saline was replaced with cold 2% glutaraldehyde in 0.1 M Na-cacodylate buffer, pH 7.4. After 10 minutes the fillets were de-pinned and transferred to 20 ml scintillation vials with fresh fixative and subsequently processed at room temperature with agitation on a tissue rotator. Glutaraldehyde fixation was continued for a total of one hour, followed by 6 × 10 min rinses with 0.1 M Na-cacodylate buffer, post-fixation with 1% osmium tetroxide in 0.1 M Na-cacodylate buffer, 6 × 10 min rinses with deionized water, and one hour en bloc staining with 1% aqueous uranyl acetate. Following en bloc staining, samples were rinsed 6 × 10 min with deionized water, dehydrated in an ethanol series to 100% in 10% steps for 10 min each, exchanged with 3 × 10 rinses with propylene oxide, and infiltrated with Eponate 12 resin (1:1 propylene oxide:resin for 30 min followed by 3 changes of 100% resin, the last one overnight). Samples were placed in silicone molds with fresh resin and the resin polymerized at 65 °C for 48 hou rs. Larvae imaged after stimulation were prepared as described for electrophysiological recording, and immediately (within 1 second) following cessation of stimulation, the physiological saline was replaced with icecold fixative and processed as described above. Serial sections of larval abdominal segment 2 were made with a Leica UCT ultramicrotome using a Diatome diamond knife (Diatome USA) and picked up on Synaptek slot grids (Ted Pella, Inc) with Pioloform films and a 2 nm layer of evaporated carbon. Sections were stained with 1% aqueous uranyl acetate followed by Sato’s lead. Sections were examined and imaged with an FEI T12 TEM (FEI, Hillsboro, OR) equipped with a Gatan U895 4k x 4k CCD camera. During imaging, care was taken to align the membranes at the T-bar/AZ with the optical axis of the electron microscope to provide the optimum orientation to detect docked vesicles. Images were analyzed using the ImageJ ROI manager. ROIs for all vesicles within 400 nm of an active zone (from the base of the T-bar) were acquired, and their XY coordinates were compared to the XY coordinates of the active zone, pre-synaptic membrane, and all vesicle ROIs. Thus, a list of distances was created and used to calculate the following parameters: number of vesicles within 400 and 150 nm of the active zone, vesicle diameter, distance to nearest neighboring vesicle, and if a vesicle was ‘docked’ to the pre-synaptic membrane. Vesicleswere considered ‘docked’ if the distance between the presynaptic membrane and the center of the vesicle was equal to or less than the radius of the vesicle.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details can be found in the Results, Figures, and Supplemental Table. Unpaired Student’s t-test was used for comparison of two groups (typically comparison of a genotype in the presence vs absence of a manipulation). Where three or more groups were compared, one-way ANOVA was used to determine significance, and Tukey’s test was used to compare individual groups. N represents number of synapses recorded from. All bar graphs display the mean plus standard error. Significance was defined as p<0.05 (one asterisk), with two asterisks indicating p<0.01 and three indicating p<0.005. A prior power analysis indicates samples size of 5 is sufficient given that our effect size is often 50–100% and synapse to synapse variance is low. None-the-less, our data sets generally greatly exceed this number.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Bruchpilot | Developmental studies hybridoma bank |
DSHB Cat# nc82 RRID:AB_2314867 |
| Mouse monoclonal anti-cysteine string protein | Developmental studies hybridoma bank |
DSHB Cat# DCSP-1 (ab49) RRID:0041B_2307345 |
| Mouse monoclonal anti-GluRIIA | Corey Goodman | DSHB Cat# 8B4D2 (MH2B) RRID:AB_528269 |
| Rabbit polyclonal anti-Complexin | Huntwork and Littleton, 2007 |
PMID:17873870 |
| Rabbit polyclonal anti-Synaptotagmin 1 | Littleton et al., 1993 | PMID:8269841 |
| Rabbit polyclonal anti-GluRIIC | Aaron DiAntonio | PMID:15182665 |
| Rabbit polyclonal anti-Dlg | Corey Goodman | DSHB Cat# 4F3 anti-discs large RRID:AB_528203 |
| Mouse monoclonal anti-FLAG | Sigma Aldrich | F1804 SIGMA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Philanthotoxin-433 | Santa Cruz Biotechnology |
sc-255421 |
| Experimental Models: Organisms/Strains | ||
| W | Bloomington Drosophila Stock Center |
#3605 |
| w; imdNP1182 | Kyoto Stock Center | #112555 |
| w, Tak12 | Bloomington Drosophila Stock Center |
#26272 |
| w, Tak1179 | Bloomington Drosophila Stock Center |
#26275 |
| w; IKKβCR | This work | N/A |
| w; RelE20 | Bloomington Drosophila Stock Center |
#9457 |
| w; GluRIIASP16 | Petersen et al., 1997 | PMID:9427247 |
| PGRP2 |
Choe et al., 2002 (ird72) |
PMID:11872802 |
| w, Df(1)BSC645 | Bloomington Drosophila Stock Center |
#25735 |
| w; Df(3R)BSC728 | Bloomington Drosophila Stock Center |
#26580 |
| w; Ok371-Gal4 | Mahr and Aberle, 2006 |
PMID:16378756 |
| BG57-Gal4 | Budnik et al., 1996 | PMID:8893021 |
| w; UAS-Tak1K46R-FLAG | This work | N/A |
| w−3605 | Yuh Nung Jan Lab | N/A |
| w; nos-Gal4VP14,UAS-cas9 | Kondo and Ueda, 2013 |
PMID:24002648 |
| Oligonucleotides | ||
| gRNA targeting IKKβ AAGTCAAGCTCAGCGAGCGT |
This paper | N/A |
| FWD primer for IKKβ target: CTTATAGCCTTTCGGCTGAGTTAG |
This paper | N/A |
| REV primer for IKKβ target: CCACCGTTACAGTATTCCAGC |
This paper | N/A |
| Tak1DN Duplex PCR primer a: caccATGGCCACAGCATCGCTGG |
This paper | N/A |
| Tak1DN Duplex PCR primer b: GAAGAACTCCcTGACGGCAA |
This paper | N/A |
| Tak1DN Duplex PCR primer c: TTGCCGTCAgGGAGTTCTTC |
This paper | N/A |
| Tak1DN Duplex PCR primer d: CGCATTGTGATGCGGTGGG |
This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pCDF3-dU6:3gRNA | Simon Bullock | Addgene plamid #49410 |
| cDNA LD42274 | Drosophila Genomics Resource Center |
DGRC: 2729, Flybase ID: FBcl0166830 |
| pENTR/D-TOPO | Invitrogen | K240020 |
| pTWF (UAS-C-terminal FLAG) | Drosophila Genomics Resource Center |
DGRC barcode: 1116 |
| Software and Algorithms | ||
| Igor Pro 6.37 | Wavemetrics | N/A |
| MATLAB R2015a | Mathworks | N/A |
Highlights:
Neuronal innate immune signaling controls presynaptic homeostatic plasticity.
Innate immune signaling coordinates rapid and long-term homeostatic plasticity.
Tak1 (Map3K) selectively controls the rapid, presynaptic induction of PHP
Tak1 (Map3K) controls the stability of the docked vesicle state.
Acknowledgements:
Supported by NIH grant number R35NS097212 to GWD. We thank Davis lab members, particularly Özgür Genç, B rian Orr, and Tingting Wang for assistance with data interpretation and analysis. Dedicated to the memory of Amy Tong.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: None
REFERENCES
- Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou H-C, and Sweatt JD (2008). c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn. Mem 15, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Agaisse H, and Perrimon N (2002). Sequential Activation of Signaling Pathways during Innate Immune Responses in Drosophila. Developmental Cell 3, 711– 722. [DOI] [PubMed] [Google Scholar]
- Chen X, Rahman R, Guo F, and Rosbash M (2016). Genome-wide identification of neuronal activity-regulated genes in Drosophila. eLife 5, e19942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe K-M, Lee H, and Anderson KV (2005). Drosophila peptidoglycan recognition protein LC (PGRP-LC) acts as a signal-transducing innate immune receptor. Proc. Natl. Acad. Sci. U.S.A 102, 1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe K-M, Werner T, Stöven S, Hultmark D, and Anderson KV (2002). Requirement for a Peptidoglycan Recognition Protein (PGRP) in Relish Activation and Antibacterial Immune Responses in Drosophila. Science 296, 359–362. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, and Shatz CJ (1998). Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21, 505–520. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, and Uchitel OD (1980). On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. The Journal of Physiology 299, 621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, and Goodman CS (1998). Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature 392, 82–86. [DOI] [PubMed] [Google Scholar]
- Delaney JR, Stöven S, Uvell H, Anderson KV, Engström Y, and Mlodzik M (2006). Cooperative control of Drosophila immune responses by the JNK and NF-kappaB signaling pathways. EMBO J 25, 3068–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, and Davis GW (2006). Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52, 663–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaviño MA, Ford KJ, Archila S, and Davis GW (2015). Homeostatic synaptic depression is achieved through a regulated decrease in presynaptic calcium channel abundance. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottar M, Gobert V, Michel T, Belvin M, Duyk G, Hoffmann JA, Ferrandon D, and Royet J (2002). The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 416, 640–644. [DOI] [PubMed] [Google Scholar]
- Häcker H, and Karin M (2006). Regulation and Function of IKK and IKK-Related Kinases. Sci. STKE 2006, re13–re13. [DOI] [PubMed]
- Hallermann S, Heckmann M, and Kittel RJ (2010). Mechanisms of short-term plasticity at neuromuscular active zones of Drosophila. HFSP J 4, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris N, Braiser DJ, Dickman DK, Fetter RD, Tong A, and Davis GW (2015). The Innate Immune Receptor PGRP-LC Controls Presynaptic Homeostatic Plasticity. Neuron 88, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He E, Wierda K, van Westen, R., Broeke JH, Toonen RF, Cornelisse LN, and Verhage M (2017). Munc13–1 and Munc18–1 together prevent NSF-dependent de-priming of synaptic vesicles. Nature Communications 8, 15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedengren M, Asling B, Dushay MS, Ando I, Ekengren S, Wihlborg M, and Hultmark D (1999). Relish, a central factor in the control of humoral but not cellular immunity in Drosophila. Mol. Cell 4, 827–837. [DOI] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, and Shatz CJ (2000). Functional requirement for class I MHC in CNS development and plasticity. Science 290, 2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, and Littleton JT (2012). Complexin Controls Spontaneous and Evoked Neurotransmitter Release by Regulating the Timing and Properties of Synaptotagmin Activity. J. Neurosci 32, 18234–18245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazama H, and Wilson RI (2008). Homeostatic matching and nonlinear amplification at identified central synapses. Neuron 58, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, and Ryan TA (2010). CDK5 serves as a major control point in neurotransmitter release. Neuron 67, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, et al. (2006). Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 312, 1051–1054. [DOI] [PubMed] [Google Scholar]
- Kondo S, and Ueda R (2013). Highly Improved Gene Targeting by Germline-Specific Cas9 Expression in Drosophila. Genetics 195, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, Datwani A, and Shatz CJ (2014). Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 509, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, Reichhart JM, and Hoffmann JA (1995). A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proc. Natl. Acad. Sci. U.S.A 92, 9465–9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney RE, Rawson JM, and Eaton BA (2014). An age-dependent change in the set point of synaptic homeostasis. J. Neurosci 34, 2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahr A, and Aberle H (2006). The expression pattern of the Drosophila vesicular glutamate transporter: a marker protein for motoneurons and glutamatergic centers in the brain. Gene Expr. Patterns 6, 299–309. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, and Baltimore D (2003). NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci 6, 1072–1078. [DOI] [PubMed] [Google Scholar]
- Mitra A, Mitra SS, and Tsien RW (2011). Heterogeneous reallocation of presynaptic efficacy in recurrent excitatory circuits adapting to inactivity. Nat. Neurosci 15, 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Liu KSY, Sigrist SJ, and Davis GW (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci 32, 16574–16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Genç Ö, and Davis GW (2015). RIM-b inding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron 85, 1056–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, and Stevens CF (1999). Reversal of synaptic vesicle docking at central synapses. Nature Neuroscience 2, 503–507. [DOI] [PubMed] [Google Scholar]
- Myllymäki H, Valanne S, and Rämet M (2014).heT Drosophila imd signaling pathway. J. Immunol 192, 3455–3462. [DOI] [PubMed] [Google Scholar]
- Park JM, Brady H, Ruocco MG, Sun H, Williams D, Lee SJ, Kato T, Richards N, Chan K, Mercurio F, et al. (2004). Targeting of TAK1 by the NF-κB protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev 18, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, and DiAntonio A (1997). Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19, 1237–1248. [DOI] [PubMed] [Google Scholar]
- Polley S, Huang D-B, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schröfelbauer B, Kim Y, Hoffmann A, Verma IM, et al. (2013). A Structural Basis for IκB Kinase 2 Activation Via Oligomerization-Dependent Trans Auto-Phosphorylation. PLOS Biology 11, e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwarf DM, and Karin M (1999). The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci. STKE 1999, RE1. [DOI] [PubMed]
- Sakaba T, and Neher E (2001). Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, and Akira S (2005). Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol 6, 1087–1095. [DOI] [PubMed] [Google Scholar]
- Schafer DP, and Stevens B (2013). Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Current Opinion in Neurobiology 23, 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, and Neher E (2005). Presynaptic calcium and control of vesicle fusion. Curr. Opin. Neurobiol 15, 266–274. [DOI] [PubMed] [Google Scholar]
- Shim J-H, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee K-Y, Bussey C, Steckel M, Tanaka N, et al. (2005). TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 19, 2668–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Stöven S, Pandey N, Hultmark D, and Maniatis T (2000). A Drosophila IkappaB kinase complex required for Relish cleavage and antibacterial immunity. Genes Dev 14, 2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Liesegang GW, Berger RL, Czerlinski G, and Podolsky RJ (1984). A stopped-flow investigation of calcium ion binding by ethylene glycol bis(beta-aminoethyl ether)-N,N’-tetraacetic acid. Anal. Biochem 143, 188–195. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, and Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Takatsu Y, Nakamura M, Stapleton M, Danos MC, Matsumoto K, O’Connor MB, Shibuya H, and Ueno N (2000). TAK1 participates in c-Jun N-terminal kinase signaling during Drosophila development. Mol. Cell. Biol 20, 3015–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, and Lemaitre B (2001). Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes Dev 15, 1900–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jones RT, Whippen JM, and Davis GW (2016). α2δ−3 Is Required for Rapid Transsynaptic Homeostatic Signaling. Cell Rep 16, 2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, and Jorgensen EM (2003). Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci 6, 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Littleton JT, Bhat MA, Prokop A, and Bellen HJ (1998). ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage‐dependent manner. The EMBO Journal 17, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Dreosti E, and Lagnado L (2011). Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J. Neurosci 31, 7492–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Silverman N, Hong M, Liao DS, Chung Y, Chen ZJ, and Maniatis T (2005). The role of ubiquitination in Drosophila innate immunity. J. Biol. Chem 280, 34048–34055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.