

Abstract
Ageing is characterized by the impairment of the acute innate immune response and the upregulation of low-grade inflammation, i.e. inflammaging. At the cellular level, telomeres are considered as a marker of biological ageing as their length is progressively eroded in the absence of repair mechanisms. However, the link between telomeres and inflammaging remains underexplored. We aimed to identify proteins that are differentially expressed between age classes in response to an acute inflammatory challenge. We challenged young (two months) and old (12 months) C57BL/6 mice using bacterial lipopolysaccharide (LPS) and measured telomere length and proteomic profiles in splenocytes. In total, 233 out of the 1966 proteins we quantified differed among experimental groups. A hierarchical clustering analysis revealed that nine of those 233 proteins were differently expressed among the experimental groups. Young mice responded to LPS by increasing the expression of proteins involved in the innate immune response, and interestingly, in telomere length maintenance. However, this regulation was impaired at older ages. These results are in agreement with the assumption that the strength of selection declines with age, potentially explaining the maintenance of costly, dysregulated, immune responses at old age. We suggest that the immune response is competing with the telomere maintenance process, highlighting how telomeres reflect the ageing trade-off even in a species where telomere length is not related to lifespan.
Keywords: immune challenge, ageing, proteomics, telomere, LPS
1. Introduction
The immune system is usually depicted as a two edged sword, conferring benefits but also generating costs. While the benefits of immune protection are obvious [1], the immune response has been shown to incur energy-dependent and -independent costs. Constitutive and induced immune responses draw host resources away from other physiological functions and this can generate trade-offs between immunity and life-history traits, such as growth or reproduction [2,3]. In addition to these resource-based trade-offs, immunity can incur costs if immune effectors target host tissues and organs [4]. These costs are referred to as immunopathology. Although immune-mediated diseases have been identified for quite a long time, the importance of immunopathology in the context of the ecology and evolution of host–parasite interactions has emerged as a new concept during the last two decades [5]. The risk of immune-induced damage is particularly strong during the process of acute and chronic inflammation. Broadly speaking, infection induces the activation of innate immunity effectors within minutes to hours [6]. Such rapid response and the associated local inflammation then favour mounting of the adaptive immune response (i.e. via lymphocyte recruitment [7]) within days and weeks. Innate and adaptive responses not only differ in terms of rapidity but also in terms of specificity. While adaptive response is highly specific, innate immunity is essentially based on the recognition of conserved pathogen molecular patterns such as structural components of the bacterial wall (lipopolysaccharide (LPS) in Gram-negative bacteria or peptidoglycan in Gram-positive bacteria), by pattern recognition receptors (such as Toll-like receptors) and the induction of cytotoxic effectors (oxidative burst) [7,8]. By their inherent function/nature, these effectors cannot discriminate invading pathogens from host cells and tissues and therefore can induce substantial damage if they get out of control [9].
Failure in the resolution of acute and chronic inflammation can induce life-threatening diseases [10,11]. Indeed, inflammation and activation of oxidative attacks on intruders may have many collateral negative impacts on the host itself, including ultimate costs on life-history traits [12]. Ageing has been shown to be associated with the upregulation of low-grade inflammation, i.e. inflammaging [13,14], which makes elderly more susceptible to many degenerative age-associated pathologies, such as cancer, diabetes, cardio-vascular diseases [15] and ultimately impairs lifespan. Old individuals have also been shown to pay disproportionately high costs of inflammatory activation when regulatory mechanisms are experimentally broken down, suggesting that ageing individuals are more prone to immunopathology [16]. In a companion study, we notably reported that LPS-induced oxidative stress in the liver induces immunopathology costs in old mice [17]. Accumulation of senescent cells in old organisms may be one of the mechanisms leading to chronic inflammation [18].
Telomeres are repeated DNA sequences that protect chromosome extremity. The length of these repeated motifs is supposed to reflect biological age, because cellular replication and stress erode them (although mechanisms that restore telomere caps exist) [19]. Telomere length is thus used as a marker of cell and organismal senescence [20,21–24]. Telomeres have been previously linked with the immune cell response to pathogens [25]. They are particularly susceptible to erosion during infection and the associated inflammatory response. This was shown for instance in a wild passerine, where chronic malaria infection induces an accelerated erosion of telomeres in red and white blood cells, resulting in a subsequent significant reduction of lifetime reproductive output and lifespan [26]. It is therefore possible that an upregulated chronic inflammation at old age (inflammaging) and the lower ability of old individuals to cope with an acute inflammatory response are accelerating factors promoting telomere attrition [27], leading to a vicious circle between telomere attrition, cellular senescence and inflammaging.
The aim of this study was twofold. First, we wished to explore if an acute inflammatory challenge induces age-dependent variation in protein expression in an immune organ, the spleen, using a quantitative proteomic approach. Second, we investigated whether telomere length differed with age and whether the proteomic profile activated by the immune challenge could reveal mechanisms involved in telomere attrition. In doing so, we wished to test if the cost of an immune challenge is mirrored in telomere malfunction even in a species were telomerase is active at adulthood.
2. Material and methods
(a). Experimental design
The experiment was conducted on 26 C57BL/6 J male mice from our own colony and originated from Janvier Labs. 12 mice were two months old (hereafter young), and 14 mice were 12 months old (hereafter old), reared in age-separated cages of six to seven animals under constant temperature (24 ± 2°C), humidity (50–55%) and photoperiod (13 L : 11 D cycle) and fed ad libitum on a standard diet (SAFE A03). Mice in each age class were randomly allocated to one of the two experimental groups: six young and eight old mice received an intraperitoneal injection of 25 µg kg−1 body mass of LPS (LPS; Escherichia coli, Sigma Aldrich, a non-lethal dose based on a previous study [17]), and the other mice were kept as controls and injected with the same volume of sterile phosphate buffered solution (PBS). LPS is a TLR4 ligand that stimulates neutrophils and macrophages, induces the release of pro-inflammatory cytokines (IL-6, IFN-γ, TNF-α, IL-1β), cytotoxic reactive oxygen and nitrogen species (ROS, RNS) (e.g. [28]). LPS also induces a series of physiological and behavioural changes collectively called sickness behaviour that includes loss of appetite and reduced activity. LPS-induced sickness behaviour usually lasts for 24–48 h, depending on the injected dose, after which animals resume their normal behaviour [17] One old mouse died in the LPS group, reducing the sample size of this group to seven.
Twenty-four hours post-challenge, all mice were euthanized by cervical dislocation, the spleen was removed and separated into two aliquots that were immediately snap-frozen in liquid nitrogen before storage at −80°C, until telomere length measurements by quantitative polymerase chain reaction, proteomic and western blot analyses (methodological details are available in the electronic supplementary material, S1). Body mass (±0.1 g) was also measured just before the intraperitoneal injection and 24 h post-challenge (electronic supplementary material, S3). We chose this short-term measurement (and age classes) based on previous LPS injection experiments conducted on the same mouse line [17] showing that body mass loss and oxidative costs were maximal at that time (i.e. the putative cost being easier to measure at the telomere level) and will not be compensated by the triggering of telomerase activity during recovery.
(b). Statistical analyses
We selected a three-step analysis for the proteomic and molecular data obtained on spleen tissues. As we did in previous studies [17,29], we first sorted the significant effects of age (young and old) and treatment (PBS and LPS) as fixed factors using ANOVA for each of the 1966 quantified proteins (electronic supplementary material, table S2). We thus selected the 233 proteins for which (i) ANOVA p-values were below 0.05, and (ii) the post hoc Tukey test p-value (with Bonferroni correction) was below 0.05 and the absolute fold change was >1.5 for at least one pairwise comparison. In a second step, we used this subsample to run a hierarchical clustering on principal component analysis (PCA) to obtain a dendrogram that assembles proteins based on similarities of variation according to experimental groups [30]. This second step was done to further reduce the number of variables to avoid multicollinearity among a too large number of variables. We chose to set up the hierarchical clustering analysis with four clusters, based on inertia gain choice (four clusters explained a total of 82% of the total variance while three clusters explained 74%), and because two of these clusters (3 and 4) explained a large fraction of the total variance in both axes (81.1% and 6.1%, respectively, figure 1). These clusters (3 and 4) encompassed nine proteins. The final step of analysis consisted of merging the proteomic variables selected so far (nine proteins) and the physiological and molecular data (body mass and telomere length) to run a PCA with varimax rotation (determinant of 0.001). PCA resulted in three axes (PCA1, PCA2 and PCA3) explaining 36.2%, 24.4% and 13.9% of the variance, respectively (total 74.5%). Kaiser–Meyer–Olkin value was of 0.66 and Bartlett's test of sphericity confirmed an adequate correlation among variables for PCA (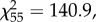 p < 0.001). Given the number of variables, only components of the axis with an eigenvalue over 0.4 were retained (Kaiser's criterion). PCA scores of each individual were then used as the dependent variable in a generalized linear model (GLM) with age (young versus old), immune challenge (LPS versus PBS) and their interaction. To confirm the significance of the relationship between individual telomere length and the protein signals, we chose to run an additional analysis on the raw values (not on the PCA values). To do so, we investigated the association between telomere length and the nine proteins (among which some were previously known to be involved in telomere dynamics, e.g. chitinase-like protein 3) using a model selection after testing for the absence of multicollinearity using variance inflation factor (minimum tolerance index greater than 0.19). For western blot analysis of shelterin proteins, we performed one-way ANOVA with Tukey post hoc tests. Adjustment of p-values was done according to Bonferroni−Holm (p < 0.05), after checking for normality distribution (Shapiro−Wilk test) and homoscedasticity (Bartlett test). All statistics were run on the program R using RStudio (v. 1.1.414). Hierarchical clustering and PCA were conducted using FactoMineR R package [31]. We used the dredge function of the MuMIn package (v. 1.15.1) to run model selection. Normality of the residuals was checked using quantile-quantile (QQ) plots and Kolmogorow–Smirnov tests. Significance threshold used is p ≤ 0.05.
p < 0.001). Given the number of variables, only components of the axis with an eigenvalue over 0.4 were retained (Kaiser's criterion). PCA scores of each individual were then used as the dependent variable in a generalized linear model (GLM) with age (young versus old), immune challenge (LPS versus PBS) and their interaction. To confirm the significance of the relationship between individual telomere length and the protein signals, we chose to run an additional analysis on the raw values (not on the PCA values). To do so, we investigated the association between telomere length and the nine proteins (among which some were previously known to be involved in telomere dynamics, e.g. chitinase-like protein 3) using a model selection after testing for the absence of multicollinearity using variance inflation factor (minimum tolerance index greater than 0.19). For western blot analysis of shelterin proteins, we performed one-way ANOVA with Tukey post hoc tests. Adjustment of p-values was done according to Bonferroni−Holm (p < 0.05), after checking for normality distribution (Shapiro−Wilk test) and homoscedasticity (Bartlett test). All statistics were run on the program R using RStudio (v. 1.1.414). Hierarchical clustering and PCA were conducted using FactoMineR R package [31]. We used the dredge function of the MuMIn package (v. 1.15.1) to run model selection. Normality of the residuals was checked using quantile-quantile (QQ) plots and Kolmogorow–Smirnov tests. Significance threshold used is p ≤ 0.05.
Figure 1.

(a) Hierarchical dendogram built on the 233 proteins detected in mouse spleen (n = 24). The number of protein clusters selected (k = 4, indicated by the arrow in dots) was chosen because it increased the percentage of total variance explained by the classification by 7.4% and then thereafter only by 4%. Among the four clusters thereby selected, two were explaining more of the total variation of axes 1 and 2 (cluster 3 and 4 indicated in shadow), which gathered nine proteins (cluster 3, ID 2156, 1548, 277, 2242, 656; cluster 4, ID 928, 471, 132 546). Numbers refer to identification (ID) no. in electronic supplementary material, table S2. Height represents the numbers of mice present in each cluster. (b) Hierarchical dendrogram built using mouse spleen proteomic profiles (based on the 233 proteins quantified), showing mouse distribution (n = 24) among clusters (k = 3, separating old mice, and young PBS (dark shadow) and young LPS (grey shadow) groups. Setting the number of clusters to four or more did not show any sharp increase in the total variance explained (9% for four clusters, and 8% for five clusters). Clusters of mice correspond to the protein clusters represented in figure 1a. Height represents the numbers of proteins present in each cluster.
3. Results
The proteomics analysis yielded the robust identification of 2788 proteins, of which 1966 fulfilled the criteria for quantification (electronic supplementary material, S2, and table S2). A total of 285 proteins were differentially expressed as they exhibited ANOVA p-values below 0.05, and this number dropped to 233 when considering Tukey p-values below 0.05 and absolute fold changes above 1.5 for at least one pairwise comparison. Hierarchical clustering pointed to nine of these proteins as major contributors belonging to two different clusters (3 and 4) that explained most of the variation on both axes of the dendrogram (figure 1a), characterizing young PBS and young LPS groups (figure 1b). To test how these proteomic data, once added to biological variables, vary with age and treatment, we used these nine proteins, changes in body mass during the 24 h post-challenge and telomere length to run a PCA. The contribution of the different variables to the three PCs (PCA1, 2 and 3) are reported in table 1. PCA1 (36.2% of variance explained) positively loaded with protein S100-A9, lysozyme C-2 and annexin A1; PCA2 (24.4% of variance explained) had positive loads with coronin 1A and RAS-C3, and negative loads with endoplasmin HSP90b1, GRP-78, carbonic-anhydrase 1 and changes in body mass; finally PCA3 (13.9% of variance explained) positively loaded with endoplasmin HSP90b1, GRP-78 and chitinase-like protein 3, and negatively with telomere length. Based on gene ontology annotations and literature analysis, the three PCs may be attributed to the following biological functions: immune response and inflammation (PCA1), response to stress and ageing (PCA2) and stress and telomere function/length regulation (PCA3).
Table 1.
Nature of the variables forming the main axes (PCA1, PCA2 and PCA3) of the PCA conducted on proteomic and physiological variables describing spleen cellular responses to an immune challenge (LPS) among old and young mice. (Only eigenvalues over 0.4 were taken into account and are indicated (see text for details). Total variance explained by the model: 74.5%. ID no. refers to ‘identification no.’ of proteins that are listed in the electronic supplementary material, table S2. Numbers in italics indicate the proportion of total variance explained by each principal component axes.)
| ID no. | PCA1 | PCA2 | PCA3 | |
|---|---|---|---|---|
| proteomic signals | ||||
| protein S100-A9 | 2156 | 0.922 | ||
| lysozyme C-2 | 1548 | 0.819 | ||
| annexin A1 | 277 | 0.882 | ||
| endoplasmin Hsp90b1 | 928 | −0.441 | 0.742 | |
| GRP-78 kDa | 132 | −0.463 | 0.588 | |
| carbonic-anhydrase 1 | 471 | −0.853 | ||
| coronin 1A | 656 | 0.865 | ||
| RAS-C3 | 2242 | 0.817 | ||
| chitinase-like protein 3 | 546 | 0.818 | ||
| physiological signals | ||||
| body mass loss | −0.535 | |||
| telomere length | −0.892 | |||
| % variance explained | 36.2 | 24.4 | 13.9 | |
We then used the individual PCA values on each of the three axes as the dependent variables in a general linear model to investigate how these values varied as a function of age and immune treatment. The results showed that PCA1 was significantly affected by the interaction age × immune treatment. In particular, young LPS-treated mice had higher PCA1 values compared with the other experimental groups, suggesting an upregulated expression of protein S100, annexin A1 and lysozyme (table 2). Old mice had similar PCA1 values, whatever their immune treatment (LPS versus PBS; estimates 0.267 ± 0.317, NS).
Table 2.
Generalized linear models (GLMs) explaining the variability in the components axes (PCA1, PCA2, PCA3) of proteomic and physiological signals in old and young mice challenged with PBS or LPS (n = 6 in each group). (GLM analysis separated by treatment (a) was used to assess interaction age × immune treatment differences among groups (indicated in italics). Estimates and mean values are given ±s.e. and only significant differences among age × treatment interactions are indicated (using Bonferroni correction). Residuals of each models followed a normal distribution (checked using Kolmogorov–Smirnov test and QQ plot).)
| variables: physiological and proteomic signals | estimates | d.f. | F | p-value |
|---|---|---|---|---|
| (a) PCA1 | ||||
| age (old versus young) | 0.319 ± 0.317 | 1, 23 | 7.208 | 0.007 |
| immune treatment (LPS versus PBS) | 2.110 ± 0.317 | 1, 23 | 28.047 | <0.001 |
| age × immune treatment | 1, 23 | 16.867 | <0.001 | |
| young LPS versus old LPS | 1.524 ± 0.317 | <0.001 | ||
| versus old PBS | 1.791 ± 0.408 | <0.001 | ||
| versus young PBS | 2.110 ± 0.419 | <0.001 | ||
| (b) PCA2 | ||||
| age (old versus young) | −1.172 ± 0.466 | 1, 23 | 10.846 | 0.001 |
| immune treatment (LPS versus PBS) | −0.276 ± 0.386 | 1, 23 | 0.332 | 0.565 |
| age × immune treatment | 1, 23 | 0.068 | 0.794 | |
| (c) PCA3 | ||||
| age (old versus young) | 1.900 ± 0.368 | 1, 23 | 27.656 | <0.001 |
| immune treatment (LPS versus PBS) | 0.780 ± 0.517 | 1, 23 | 0.907 | 0.341 |
| age × immune treatment | 1, 23 | 4.217 | 0.040 | |
| young LPS versus old PBS | −1.119 ± 0.368 | 0.014 | ||
| versus young PBS | 0.781 ± 0.370 | 0.034 | ||
| young PBS versus old PBS | −1.614 ± 0.408 | <0.001 | ||
| versus old LPS | −1.190 ± 0.419 | <0.001 | ||
The PCA2 axis was only affected by age (table 2), with old mice having lower values than young mice. This suggests an age-dependent upregulation of carbonic-anhydrase 1, GRP 70 kDa and Hsp 90, and a downregulation of Coronin 1A and RAS-C3.
As for PCA1, the PCA3 axis was also affected by the interaction between age and the immune treatment (table 2): while young mice significantly increased their PCA3 values in response to LPS, old mice presented unchanged PCA3 values after the immune treatment (and, overall, had higher mean values throughout the study). This suggests an upregulated expression of GRP 70 kDa, Hsp 90 and chitinase-like protein 3, and shorter telomeres in old compared to young mice.
Given that chitinase-like protein 3 and Hsp 90 (over-expressed in old mice), and the protein S100-A9 (over-expressed in young mice in the LPS group) have been shown to have a function in the regulation of telomere length, we further investigate the relationship between telomere length and the proteomic profile at the individual level. Model selection indicates that chitinase-like protein 3 and Hsp 90 proteins were the best predictors of telomere length independently of the treatment or age (table 3). In addition, we found that chitinase-like protein 3 was negatively correlated with telomere length, both when the regression was run using the entire dataset or when separated by treatments (PBS and LPS; figure 2). The regression was not significant when the dataset was split according to age (young mice only, r = 0.373, r² = 0.139, F1,11 = 1.619, p = 0.232; old mice only r = 0.530, r² = 0.281, F1,11 = 3.903, p = 0.076).
Table 3.
Model of the relationship between (i) the spleen proteomic expression levels of nine proteins and (ii) the spleen telomere length (log-transformed). (The estimates and p-values for the fixed effects are averaged over the seven best general linear models for which the Δ Akaike information criterion (AIC) less than 2 in comparison to the best model (AIC = −47.6, which used annexin A1 and chitinase-like 3 as fixed factors (indicated in bold)). Fixed factors used in those seven best models are indicated in italics.)
| fixed effect | estimates | s.e. | z-value | p-value |
|---|---|---|---|---|
| intercept | 0.105 | 0.089 | 1.130 | 0.259 |
| chitinase-like protein 3 | −7.926−10 | 2.535−10 | 2.967 | 0.003 |
| annexin A1 | 7.587−10 | 5.000−10 | 1.439 | 0.150 |
| lysozyme C-2 | −1.238−9 | 8.560−10 | 1.380 | 0.168 |
| endoplasmin Hsp90b1 | −1.087−9 | 8.788−10 | 1.194 | 0.232 |
| protein S100-A9 | 1.051−9 | 9.059−10 | 1.160 | 0.250 |
| carbonic-anhydrase 1 | 3.030−10 | 3.610−10 | 0.915 | 0.360 |
| RAS-C3 | −3.124−10 | 7.753−10 | 0.403 | 0.687 |
| GRP-78 kDa | 3.365−10 | 6.524−10 | 0.490 | 0.624 |
| coronin 1A | −1.344−10 | 6.473−10 | 0.196 | 0.845 |
Figure 2.

Regression checking for linear relationship between individual telomere length and proteomic levels of expression of the chitinase-like 3 protein. Lines represent 95% confidence interval. Log10 (T/S ratio) = 0.10–7.95 × 10−10 (chitinase-like 3 protein), r = 0.636, r² = 0.404, F1,23 = 14.934, p = 0.001. Dispersion of the experimental groups is shown by different colours, filled circles indicate young PBS mice, open circles indicate young LPS mice, dark grey circles indicate old PBS mice and light grey circles indicate old LPS mice. Regressions tested by treatment are also both significant (PBS: r = 0.635, r² = 0.403, F1,11 = 6.763, p = 0.026, LPS: r = 0.623, r² = 0.389, F1,11 = 6.357, p = 0.030). (Online version in colour.)
Finally, among the different shelterin proteins involved in telomere length regulation, western blot analysis highlighted that only TRF1-interacting nuclear factor 2 (TIN2) expression levels exhibited significant differences among experimental groups: there was a significantly higher level of expression for LPS-injected younger mice compared to old ones (figure 3; see the electronic supplementary material, S4 for statistics and electronic supplementary material, S5 for additional representative blots).
Figure 3.

TIN2 protein levels in mice spleen. Representative western blots for protein levels of TERF1-interacting nuclear factor 2 (TIN2) in the spleen of mice are shown in (a). Fold changes in young mice injected with LPS and old mice injected with PBS or LPS relative to the mean value in young mice injected with PBS (which was arbitrarily set to 1) are shown in (b). Values that do not share the same superscript letter are significantly different (see the electronic supplementary material, S4 and S5).
4. Discussion
Our results show that the induction of an acute inflammatory response produced age-dependent changes in the proteomic profile of mouse splenocytes. LPS challenge induced an upregulated expression of protein S100-A9, lysozyme C-2 and annexin A1 in young mice compared to the other groups. Old mice had a significantly higher level of expression of proteins associated with immune response and oxidative stress, as well as shorter telomeres compared with young individuals.
Analysis of the PCA1 axis (which summarizes the variation of protein S100-A9, lysozyme and annexin A1) showed higher values in LPS-treated young mice compared with control mice. These proteins play key roles as anti-bacterial effectors and in the regulation of the inflammatory response. For instance, lysozyme degrades the membrane of Gram-positive bacteria [32], and protein S100-A9, released by apoptotic neutrophils, favours the resolution of the inflammatory response by promoting bacterial clearance by macrophages and downregulates pro-inflammatory signals [33]. Similarly, annexin A1, a protein expressed in innate immune cells, has an anti-inflammatory effect [34] and mice that do not express annexin A1 (individuals with a knocked out gene) suffer from fatal septic shock following an LPS injection [35]. Therefore, young mice challenged with LPS had a proteomic signature in splenocytes, indicating both the upregulation of anti-bacterial effectors and proteins involved in the fine-tuned control of the acute inflammatory response.
In a companion study, we showed that the liver proteome of C57BL/6 old mice, exposed to a 24 h LPS challenge, was characterized by the upregulation of proteins involved in the oxidative balance compared to young mice; in the same study, the expression of proteins with specific anti-bacterial functions was not affected by the immune challenge [17]. Using spleen tissue, we found a comparable pattern of variation for PCA2, old mice being similar in their protein profiles before and after injection, but having an upregulated expression of HSP90, HSP70 (GRP-78 kDa) and carbonicanhydrase 1 compared to young mice. Carbonic-anhydrase 1 has been previously reported to be over-expressed in old mice [36]. Of particular interest is the upregulated expression of HSP90 and HSP70 in old mice, because these proteins have pleiotropic functions, with both protective [37] and potentially damaging properties [38–40]. Compared with young mice, old mice also had a lower expression of RAS3 and coronin 1A. These proteins are involved in the regulation of the stress response [41] and immunodeficiency [42].
(a). Telomere maintenance as a cost of innate immunity
Mice have highly active telomerase [43,44], and as a consequence, it is generally assumed that telomere length does not decrease with age in laboratory mice [45]. However, this does not mean that mice are not exposed to telomere erosion when facing stressful events, i.e. when telomerase activity is potentially inhibited, for instance, by increased corticosterone levels [46]. Marginal telomere shortening with age in some mice tissue has been previously reported (see [43]), and telomere shortening over generations in telomerase knock-out mice does negatively affect organ functioning and lifespan [47]. This particularly affects highly proliferative tissues which are lined by the haematopoietic cells [48]. In agreement with this, our finding that old mice had shorter telomeres compared to young mice (see the electronic supplementary material, table S3) could be explained by a transient inhibition of telomerase activity owing to the trade-off between telomere maintenance and the immune response. This is in agreement with the recent idea that telomere maintenance per se could be energetically costly [49]. Telomere shortening in old mice was concomitant with over-expression of HSP90, HSP70 and chitinase-like protein 3. Interestingly, HSP90 binds to the catalytic subunit of the telomerase, allowing the assembly of the whole enzyme and maintenance of its activity [50]. Immune cells are quite diverse in function, from haematopoietic stem cells to peripheral blood cells, all which have different proliferative capacities and telomerase activity [51]. The dynamics of telomere length in white blood cells during infection and particularly after mitogen and LPS injections was previously studied [52]. Subsequent results provided evidence for an increased telomerase activity in response to infection, mostly restricted to lymphocyte cell lines. However, mice lacking the telomerase catalytic subunit TERC−/− also showed higher inflammatory responses [53] and we detected the chitinase-like protein 3 in mice spleen, which is highly expressed in activated macrophages [54]. This suggests that: (i) macrophages may indeed be largely present in our sampled cell population, and (ii) in old mice, HSP90 expression may be linked to an (unsuccessful) response to preserve telomeres via telomerase activity. Chitinase-like protein 3 has a dual key role for the immune system: (i) by inducing macrophage activation via the degradation of bacterial chitin, which triggers pro-inflammatory cytokine secretion, and (ii) by insuring a negative feedback loop on the inflammation process necessary to the remodelling of injured tissues [55]. The finding that chitinase-like protein 3 is highly expressed in old mice as well as in young mice after LPS injection is in agreement with a lower capacity to control inflammation at old ages. In addition, chitinase-like protein 3 expression and telomere ends and telomerase dysfunction have been previously linked [56,57], suggesting that chitinase-like protein 3 is a relevant ageing biomarker. Again, our results are in agreement with a direct or indirect implication of chitinase-like protein 3 in telomere maintenance, because we found that chitinase-like protein 3 expression levels are negatively linked with telomere lengths irrespectively of age but in relation to the LPS treatment (figure 3). As a consequence, chitinase-like protein 3 may be an element explaining the immune trade-off faced by young mice injected with LPS, independently of previously suggested pathways (e.g. collateral direct oxidative stress). Chitinase-like protein 3 may be one of the links between inflammatory response and telomere shortening. Although the nature of such a mechanism remains to be established, macrophage functioning is likely to be involved. Another pathway that may be involved concerns the shelterin TIN2 expression, which clearly tended to be upregulated in young LPS mice. TIN2 is regulating the access of telomerase to telomere ends and is necessary for telomere maintenance and telomere end adequate functioning [58,59]. In addition, TIN2 has been shown to insure cell viability independently of any telomere length regulation [60], underlying that its expression may also participate in the mitigation of negative side effects of inflammation.
Previous studies suggested that telomere length is involved in the capacity of individuals to respond to infection [61,62]. Based on our data, we hypothesize here, using a proteomic exploratory approach, that the age-related costs of the innate response to LPS imply at least partly a telomere and/or telomerase-related pathway. In mice, even if telomere length is not related to lifespan [63], transient telomere shortening may be the outcome of life-history trade-offs via the direct competition for the energy of telomere maintenance pathways or owing to the indirect effect of stress hormones.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank Dr Hélène Gachot-Neveu, Aurélie Kranitsky and Marie-Laure Rizzi for their work in the animal husbandry.
Ethics
The present experiment has been conducted under the permit no. AL/05/34/12/12 delivered by the ‘Comité Régional d'Ethique en Matière d'Expérimentation Animale de Strasbourg’. Animals were maintained in captivity within the IPHC-DEPE animal husbandry (French Department of Veterinary Services no. E67-482-18) and under standardized rearing conditions previously indicated in [17]. F.C. is the holder of an animal experimentation licence granted by French authorities (no. 67-78).
Data accessibility
Raw data are given in the supplementary material files (electronic supplementary material, S2).
Authors' contributions
F.C. conceived the study and carried out the experimental work. F.B. and M.B.-D. conducted proteomic and western blot analyses while S.Z. conducted telomere length measurements. F.C. ran the statistical analyses and F.C., G.S., F.B. and B.F. drafted the manuscript. All authors gave final approval for publication.
Competing interests
We declare no competing interests.
Funding
This work was financially supported by the CNRS and Strasbourg University (H2E project; IdEx Unistra), the French Proteomic Infrastructure (ProFI; ANR-10-INSB-08-03), and a CNRS ‘Projet Exploratoire Premier Soutien’ (PEPS INEE, 2012). During the tenure of this study, M.B.-D. was the recipient of a grant from the Strasbourg University (IdEx Unistra).
References
- 1.Sorci G, Lippens C, Léchenault C, Faivre B. 2017. Benefits of immune protection versus immunopathology costs: a synthesis from cytokine KO models. Infect. Genet. Evol. 54, 491–495. ( 10.1016/j.meegid.2017.08.014) [DOI] [PubMed] [Google Scholar]
- 2.Bonneaud C, Mazuc J, Gonzalez G, Haussy C, Chastel O, Faivre B, Sorci G. 2003. Assessing the cost of mounting an immune response. Am. Nat. 161, 367–379. ( 10.1086/346134) [DOI] [PubMed] [Google Scholar]
- 3.Klasing KC, Laurin DE, Peng RK, Fry DM. 1987. Immunologically mediated growth depression in chicks: influence of feed intake, corticosterone and interleukin-1. J. Nutr. 117, 1629–1637. ( 10.1093/jn/117.9.1629) [DOI] [PubMed] [Google Scholar]
- 4.Apanius V. 1998. Ontogeny of immune function. In Avian growth and development. Evolution within the altricialprecocial spectrum (eds Starck JM, Ricklefs RE), pp. 203–222. Oxford, UK: Oxford University Press. [Google Scholar]
- 5.Graham AL, Allen JE, Read AF. 2005. Evolutionary causes and consequences of immunopathology. Ann. Rev. Ecol. Evol. Syst. 36, 373–397. ( 10.1146/annurev.ecolsys.36.102003.152622) [DOI] [Google Scholar]
- 6.Martin LB. 2009. Stress and immunity in wild vertebrates: timing is everything. Gen. Comp. Endocrinol. 163, 70–76. ( 10.1016/j.ygcen.2009.03.008) [DOI] [PubMed] [Google Scholar]
- 7.Playfair J, Bancroft G. 2006. Infection and immunity. Oxford, UK: Oxford University Press. [Google Scholar]
- 8.Emre Y, Hurtaud C, Nübel T, Criscuolo F, Ricquier D, Cassard-Doulcier A-M. 2007. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem. J. 402, 271 ( 10.1042/bj20061430) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorci G, Faivre B. 2009. Inflammation and oxidative stress in vertebrate host-parasite systems. Phil. Trans. R. Soc. B 364, 71–83. ( 10.1098/rstb.2008.0151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coussens LM, Werb Z. 2002. Inflammation and cancer. Nature 420, 860–867. ( 10.1038/nature01322) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. 2008. Oxidative damage: induced inflammation initiates age-related macular degeneration. Nat. Med. 14, 194–198. ( 10.1038/nm1709) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasselquist D, Nilsson J.-Å. 2012. Physiological mechanisms mediating costs of immune responses: what can we learn from studies of birds? Anim. Behav. 83, 1303–1312. ( 10.1016/j.anbehav.2012.03.025) [DOI] [Google Scholar]
- 13.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. 2000. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. NY Acad. Sci. 908, 244–254. ( 10.1111/j.1749-6632.2000.tb06651.x) [DOI] [PubMed] [Google Scholar]
- 14.Frasca D, Blomberg BB. 2015. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 17, 7–19. ( 10.1007/s10522-015-9578-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Licastro F, Candore G, Lio D, Porcellini E, Colonna-Romano G, Franceschi C, Caruso C. 2005. Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immunity Ageing 2, 8 ( 10.1186/1742-4933-2-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belloni V, Faivre B, Guerreiro R, Arnoux E, Bellenger J, Sorci G. 2010. Suppressing an anti-inflammatory cytokine reveals a strong age-dependent survival cost in mice. PLoS ONE 5, e12940 ( 10.1371/journal.pone.0012940) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plumel MI, et al. 2016. Differential proteomics reveals age-dependent liver oxidative costs of innate immune activation in mice. J. Proteom. 135, 181–190. ( 10.1016/j.jprot.2015.09.008) [DOI] [PubMed] [Google Scholar]
- 18.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153, 1194–1217. ( 10.1016/j.cell.2013.05.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monaghan P, Haussmann M. 2006. Do telomere dynamics link lifestyle and lifespan? Trends Ecol. Evol. 21, 47–53. ( 10.1016/j.tree.2005.11.007) [DOI] [PubMed] [Google Scholar]
- 20.Blackburn EH. 2000. Telomere states and cell fates. Nature 408, 53–56. ( 10.1038/35040500) [DOI] [PubMed] [Google Scholar]
- 21.Angelier F, Vleck CM, Holberton RL, Marra PP, Blount J. 2013. Telomere length, non-breeding habitat and return rate in male American redstarts. Funct. Ecol. 27, 342–350. ( 10.1111/1365-2435.12041) [DOI] [Google Scholar]
- 22.Bize P, Criscuolo F, Metcalfe NB, Nasir L, Monaghan P. 2009. Telomere dynamics rather than age predict life expectancy in the wild. Proc. R. Soc. B 276, 1679–1683. ( 10.1098/rspb.2008.1817) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heidinger BJ, Blount JD, Boner W, Griffiths K, Metcalfe NB, Monaghan P. 2012. Telomere length in early life predicts lifespan. Proc. Natl Acad. Sci. USA 109, 1742–1748. ( 10.1073/pnas.1113306109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cawthon R, Smith K, Obrien E, Sivatchenko A, Kerber R. 2003. Association between telomere length in blood and mortality in people aged 60 years or older. The Lancet 361, 393–395. ( 10.1016/s0140-6736(03)12384-7) [DOI] [PubMed] [Google Scholar]
- 25.De Punder K, Heim C, Przesdzing I, Wadhwa PD, Entringer S. 2018. Characterization in humans of in vitro leucocyte maximal telomerase activity capacity and association with stress. Phil. Trans. R. Soc. B 373, 20160441 ( 10.1098/rstb.2016.044110.6084/m9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asghar M, Hasselquist D, Hansson B, Zehtindjiev P, Westerdahl H, Bensch S. 2015. Hidden costs of infection: chronic malaria accelerates telomere degradation and senescence in wild birds. Science 347, 436–438. ( 10.1126/science.1261121) [DOI] [PubMed] [Google Scholar]
- 27.Blasco MA. 2002. Immunosenescence phenotypes in the telomerase knockout mouse. Springer Seminars Immunopathology 24, 75–85. ( 10.1007/s00281-001-0096-1) [DOI] [PubMed] [Google Scholar]
- 28.Meng F, Lowell CA. 1997. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 185, 1661–1670. ( 10.1084/jem.185.9.1661) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plumel MI, Stier A, Thiersé D., van Dorsselaer A, Criscuolo F, Bertile F. 2014. Litter size manipulation in laboratory mice: an example of how proteomic analysis can uncover new mechanisms underlying the cost of reproduction. Front. Zool. 11, 41 ( 10.1186/1742-9994-11-41) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James FC, McCulloch CE. 1990. Multivariate analysis in ecology and systematics panacea or Pandora's box? Ann. Rev. Ecol. Evol. Syst. 21, 129–166. ( 10.1146/annurev.es.21.110190.001021) [DOI] [Google Scholar]
- 31.Lê S, Josse J, Husson F. 2008. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25, 2–18. [Google Scholar]
- 32.Elsbach P, Weiss J, Levy O. 1992. Oxygen-independent antimicrobial systems of phagocytes. Inflammation 3, 603–636. [Google Scholar]
- 33.De Lorenzo BHP, Godoy LC, Novaes e Brito RR, Pagano RL, Amorim-Dias MA, Grosso DM, Lopes JD, Mariano M. 2010. Macrophage suppression following phagocytosis of apoptotic neutrophils is mediated by the S100A9 calcium-binding protein. Immunobiology 215, 341–347. ( 10.1016/j.imbio.2009.05.013) [DOI] [PubMed] [Google Scholar]
- 34.Perretti M, D'Asquisto F. 2009. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70. ( 10.1038/nri2470) [DOI] [PubMed] [Google Scholar]
- 35.Damazo AS, Yona S, D'Acquisto F, Flower RJ, Oliani SM, Perretti M. 2005. Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am. J. Pathol. 166, 1607–1617. ( 10.1016/s0002-9440(10)62471-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staunton L, Zweyer M, Swandulla D, Ohlendieck KAY. 2012. Mass spectrometry-based proteomic analysis of middle-aged vs. aged vastus lateralis reveals increased levels of carbonic anhydrase isoform 3 in senescent human skeletal muscle. Int. J. Mol. Med. 30, 723–733. ( 10.3892/ijmm.2012.1056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi AD, et al. 2013. Hsp90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am. J. Respir. Cell Mol. Biol. 50, 170–179. ( 10.1165/rcmb.2012-0496OC) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Triantafilou K, Triantafilou M, Dedrick RL. 2001. A CD14-independent LPS receptor cluster. Nat. Immunol. 2, 338–345. ( 10.1038/86342) [DOI] [PubMed] [Google Scholar]
- 39.Asea A. 2005. Stress proteins and initiation of immune response: chaperokine activity of Hsp72. Exerc. Immunol. Rev. 11, 34–45. [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Yang L, Chen K-L, Zhou B, Yan H, Zhou Z.-G, Li Y. 2014. Knockdown of GRP78 promotes apoptosis in pancreatic acinar cells and attenuates the severity of cerulein and LPS induced pancreatic inflammation. PLoS ONE 9, e92389 ( 10.1371/journal.pone.0092389.g001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bopp A, Wartlick F, Henninger C, Kaina B, Fritz G. 2013. Rac1 modulates acute and subacute genotoxin-induced hepatic stress responses, fibrosis and liver aging. Cell Death Dis. 4, e558 ( 10.1038/cddis.2013.57) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerber RA, O'Brien E, Cawthon RM. 2009. Gene expression profiles associated with aging and mortality in humans. Aging Cell 8, 239–250. ( 10.1111/j.1474-9726.2009.00467.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomes NMV, Shay JW, Wright WE. 2010. Telomere biology in metazoa. FEBS Lett. 584, 3741–3751. ( 10.1016/j.febslet.2010.07.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorbunova V, Bozzella MJ, Seluanov A. 2008. Rodents for comparative aging studies: from mice to beavers. Age 30, 111–119. ( 10.1007/s11357-008-9053-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kipling D, Cooke HJ. 1990. Hyervariable ultra-long telomeres in mice. Nature 347, 400–402. ( 10.1038/347400a0) [DOI] [PubMed] [Google Scholar]
- 46.Choi J, Fauce SR, Effros RB. 2008. Reduced telomerase activity in human T lymphocytes exposed to cortisol. Brain Behav. Immun. 22, 600–605. ( 10.1016/j.bbi.2007.12.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blasco MA, Lee H-W, Hande MP, Samper E, Lansdorp PM, De Pinho RA, Greider CW. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34. ( 10.1016/S0092-8674(01)80006-4) [DOI] [PubMed] [Google Scholar]
- 48.Lee H-W, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. 1998. Essential role of mouse telomerase in highly proliferative organs. Nature 392, 569–574. ( 10.1038/33345) [DOI] [PubMed] [Google Scholar]
- 49.Young AJ. 2018. The role of telomeres in the mechanisms and evolution of life-history trade-offs and ageing. Phil. Trans. R. Soc. B 373, 20160452 ( 10.1098/rstb.2016.0452) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holt SE, et al. 1999. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 13, 817–826. ( 10.1101/gad.13.7.817) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aubert G, Landsdorp PM. 2008. Telomeres and aging. Physiol. Rev. 88, 557–579. ( 10.1152/physrev.00026.2007) [DOI] [PubMed] [Google Scholar]
- 52.Yamada O, Motoji T, Mizoguchi H. 1996. Up-regulation of telomerase activity in human lymphocytes. Biochim. Biophys. Acta 1314, 260–266. ( 10.1016/S0167-4889(96)00104-8) [DOI] [PubMed] [Google Scholar]
- 53.Bhattacharjee RN, Banerjee B, Akira S, Hande MP. 2010. Telomere-mediated chromosomal instability triggers TLR4 induced inflammation and death in mice. PLoS ONE 5, e11873 ( 10.1371/journal.pone.0011873.g001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maresz K, Ponomarev ED, Barteneva N, Tan Y, Mann MK, Dittel BN. 2008. IL-13 induces the expression of the alternative activation marker Ym1 in a subset of testicular macrophages. J. Reprod. Immunol. 78, 140–148. ( 10.1016/j.jri.2008.01.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang M-J, He C-H, Takyar S, Elias JA. 2011. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 73, 479–501. ( 10.1146/annurev-physiol-012110-142250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Figura G, Hartmann D, Song Z, Rudolph KL. 2009. Role of telomere dysfunction in aging and its detection by biomarkers. J. Mol. Med. 87, 1165–1171. ( 10.1007/s00109-009-0509-5) [DOI] [PubMed] [Google Scholar]
- 57.Jiang H, et al. 2008. Proteins induced by telomere dysfunction and DNA damage represent biomarkers of human aging and disease. Proc. Natl Acad. Sci. USA 105, 11 299–11 304. ( 10.1073/pnas.0801457105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. 2011. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol. Cell 44, 647–659. ( 10.1016/j.molcel.2017.05.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Houghtaling BR, Canudas S, Smith S. 2012. A role for sister telomere cohesion in telomere elongation by telomerase. Cell Cycle 11, 19–25. ( 10.4161/cc.11.1.18633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiang YJ, Kim SH, Tessarollo L, Campisi J, Hodes RJ. 2004. Telomere-associated protein TIN2 is essential for early embryonic development through a telomerase-independent pathway. Mol. Cell. Biol. 24, 6631–6634. ( 10.1128/mcb.24.15.6631-6634.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stauffer J, Bruneaux M, Panda B, Visse M, Vasemägi A, Ilmonen P. 2017. Telomere length and antioxidant defense associate with parasite-induced retarded growth in wild brown trout. Oecologia 185, 365–374. ( 10.1007/s00442-017-3953-x) [DOI] [PubMed] [Google Scholar]
- 62.Ilmonen P, Kotrschal A, Penn DJ. 2008. Telomere attrition due to infection. PLoS ONE 3, e2143 ( 10.1371/journal.pone.0002143.t001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hemann MT, Greider CW. 2000. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 28, 4474–4478. ( 10.1093/nar/28.22.4474) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are given in the supplementary material files (electronic supplementary material, S2).