

Abstract
Sphingosine kinase 1 (SphK1), the enzyme that produces the bioactive sphingolipid metabolite, sphin-gosine-1-phosphate, is a promising new molecular target for therapeutic intervention in cancer and inflammatory diseases. In view of its importance, the main objective of this work was to find new and more potent inhibitors for this enzyme possessing different structural scaffolds than those of the known inhibitors. Our theoretical and experimental study has allowed us to identify two new structural scaffolds (three new compounds), which could be used as starting structures for the design and then the development of new inhibitors of SphK1. Our study was carried out in different steps: virtual screening, synthesis, bioassays and molecular modelling. From our results, we propose a new dihydrobenzo[b] pyrimido[5,4-f]azepine and two alkyl{3-/4-[1-hydroxy-2-(4-arylpiperazin-1-yl)ethyl]phenyl}carbamates as initial structures for the development of new inhibitors. In addition, our molecular modelling study using QTAIM calculations, allowed us to describe in detail the molecular interactions that stabilize the different Ligand-Receptor complexes. Such analyses indicate that the cationic head of the different compounds must be refined in order to obtain an increase in the binding affinity of these ligands.
Keywords: Sphingosine kinase 1 inhibitors, Virtual screening, Synthesis, Bioassays, Molecular modelling
1. Introduction
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid metabolite that regulates many physiological functions [1–3]; however, it also has a pathological role in autoimmune dysfunction, inflammation, cancer and many other diseases [4–6]. S1P is generated intracellularly by the action of two sphingosine kinases (SphKs) named SphK1 and SphK2, which catalyze the ATP-dependent phosphorylation of sphingosine on its primary hydroxyl group.
It has been demonstrated that S1P levels and SphK1 expression and/or activity are increased in distinct cancer types, including solid tumors of the breast, colon, lung, ovary, stomach, uterus, kidney, liver, and melanoma and in leukemia, among others [7–9]. Moreover, upregulation of SphK1 has been associated with tumor angiogenesis and lymphangiogenesis [10–13] and correlates with poor prognosis in cancer patients [14,15]. Indeed, substantial evidence indicates that pharmacological or molecular inhibition of SphK1 has anti-cancer effects [11,16–25]. Thus, it is clear that SphK1 is a promising novel molecular target for therapeutic intervention in cancer and inflammatory diseases [26,27].
The first crystal structure of SphK1 was reported in 2013 [28]. More recently two SphK1-co-crystal structures (4l02 and 4v24) with potent inhibitory compounds have also been published [29,30]. These structures provide useful structural information on the interactions of ligands at the active site of SphK1.
Previously, various compounds have been reported with inhibitory activity on SphK1 [29,31–37]. The structural scaffolding of the best known compounds is shown in Fig. 1, with their respective references. Among these compounds, compound SLP7111228 has been recently reported [37]. Recently, the crystal structure of SphK1 with PF-543, the most potent and selective SphK1 inhibitor, was also revealed [30,36]. While numerous non-specific and several isozyme-specific inhibitors of SphK1 have been studied, considering the important role of this enzyme in inflammatory processes related to cancer [38–41], an important task is to develop new and more potent SphK1 inhibitors based on different structural scaffolds than those of the known inhibitors. Thus, the main objective of this work is to obtain new SphK1 inhibitors having a different structural basis than the well-known inhibitors. In the first step of our study, which was based on the crystal structures of SphK1 alone [28] and complexed with an inhibitor [29,30], we have carried out virtual screening that allowed us to identify potential new inhibitors. Next, we synthesized and tested the inhibitory activity of these novel compounds, and finally, we conducted a molecular modelling study that allowed us to understand interactions at the molecular level that stabilize the formation of different Ligand-Receptor (L-R) complexes. Fig. 2 shows in schematic form the various steps that have been carried out in this study.
Fig. 1.
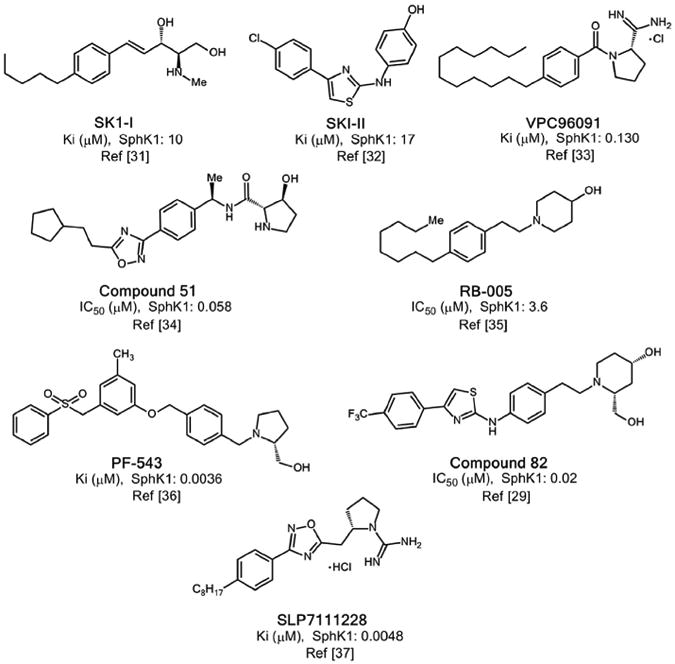
Main structural scaffolds previously reported for inhibitors of SphK1.
Fig. 2.
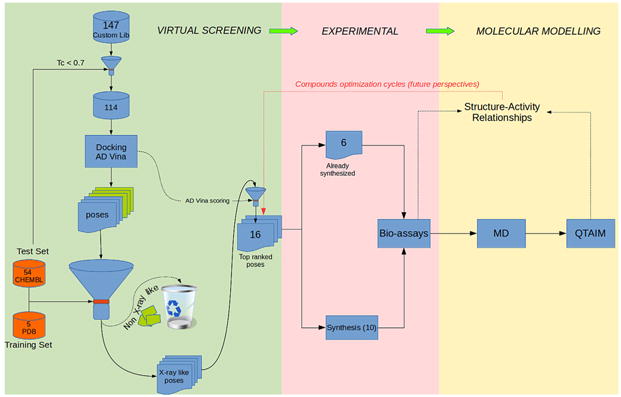
Flow chart showing the various steps and techniques carried out in our study. The numbers inside the boxes indicate the number of compounds evaluated.
2. Results and discussion
2.1. Structure-based virtual screening
Structure-Based Virtual Screening (SBVS) strategies rely on the three dimensional structure of a target and on the ability of docking algorithms to predict the binding mode and the binding affinities of different compounds obtained from libraries [42]. The docking process is usually divided into two major steps: first, the correct placement of the ligand at the protein binding-site; and then estimation of the ligand affinity by a scoring function [43].
Before undertaking the prospective Virtual Screening (VS) campaigns to search for novel inhibitors of SphK1, we considered it prudent to evaluate the performance of the docking algorithm in retrieving known inhibitors of SphK1 from a decoys library. There are two treatments for constructing these libraries. In the most common approach a few active compounds are seed in a larger database of randomly selected (and supposedly inactive) molecules with similar physico-chemical properties but dissimilar 2-D topologies. However, in real medicinal chemistry pipelines, molecules are often congeneric as they come from parallel synthesis and, consequently, are structurally more similar to one another. It would therefore be more reasonable to select decoys according to their similarity to the active molecules [44]. We constructed the decoy library to evaluate docking performance by following this second approach. Since in this approach decoys are more likely to be actives than the randomly selected molecules, one needs to be sure that they are truly inactive. Therefore, their biological activities against the target of interest must be known. Accordingly, we have compiled a dataset of 54 compounds from CHEMBL (www.ebi.ac.uk/chembl/) [45] for which their experimentally measured dissociation constants (Ki values) against SphK1 are known. The compounds were sorted from the most actives to the marginally active ones according to their Ki values. The cutoff was set to 10000 nM, compounds above (below) this threshold were flagged as active (inactive). The chosen cutoff value ensures a balanced population of active/inactive compounds.
After screening the library with AutoDock Vina (AD Vina) [46] we constructed a Receiver Operating Characteristic Curve (ROC) to asses the quality of the docking results [44]. Then the ROC curve was constructed by plotting the True Positive Rate (or Sensitivity) versus the False Positive Rate (or 1-Specificity) calculated at intervals over the ordered list of docking scores. The ROC is depicted in Fig. 3 for VS results on one of the experimentally solved SphK1 protein conformers, 3vzd_A_chainA (see cross-docking section below for details on protein conformers naming). In this graphic, the 45° diagonal (gray line) represents a random classification of the database with area under the curve (AUC) for the random case of 0.5. Any model with an AUC >0.5 performs better than random in discriminating the most active compounds from less active ones. As observed in the figure, the docking model performs very well in the classification of the dataset (AUC = 0.87). Similar results were obtained for the remaining available SphK1 structures.
Fig. 3.
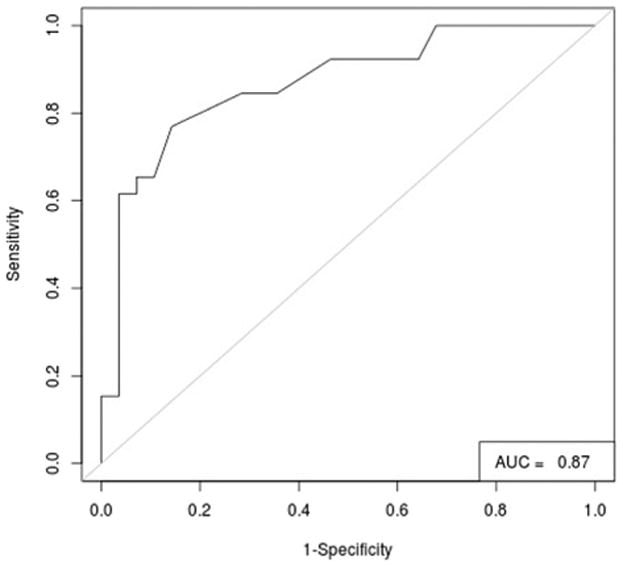
Performance for the docking of the 54 known inhibitors of SphK1 on the conformer 3vzd_A_chainB.
Fig. 4a shows the best score pose for the 54 known inhibitors of SphK1 as docked in the same protein conformer 3vzd_A_chainB. While there is no structural information on how these inhibitors actually bind to SphK1, they share the same structural scaffolds as the four ligands in the solved structures and therefore, it is assumed that they will bind also in the same binding mode. As depicted in Fig. 4, most of the inhibitors adopt the characteristic J-shaped binding mode observed for the crystallographic ligands. However, some outliers can be observed among the poses: some of them do not enter entirely into the J-shaped cavity overlapping with the ADP binding site and some other ligands are turned over, i.e. with their polar heads anchored at the bottom and the hydrophobic tail near the entrance of the J-shaped cavity.
Fig. 4.
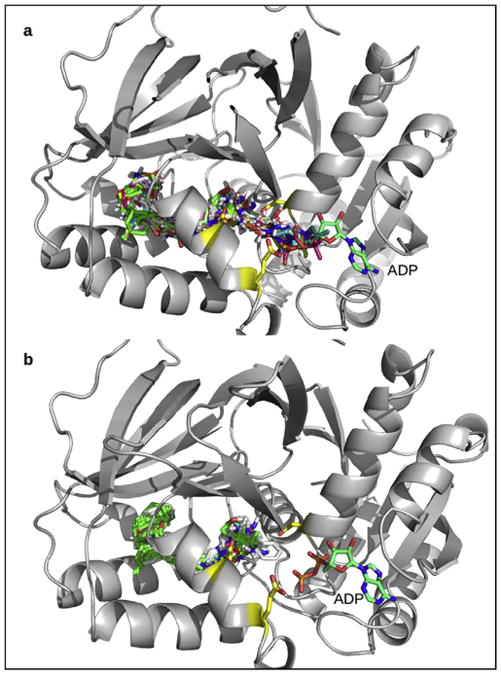
Binding modes of the 54 known inhibitors of SphK1 before (a) and after (b) the filtering out of the non-structure-like poses.
Therefore, although the AD Vina scoring function performs well in discriminating between more active and less active ligands, the ability of the docking algorithm in retrieving the correct ligand poses is somewhat deficient.
2.1.1. Cross-docking
Many studies have assessed the success rate of programs in self-docking tests, whereby a ligand is docked into the protein structure from which it was extracted (native docking). Cross-docking, or using a protein structure from a complex containing a different ligand, provides a more realistic assessment of a docking program's ability to reproduce X-ray results [47].
To evaluate the ability of the docking algorithm to retrieve the correct poses of known binders of SphK1, we performed a cross-docking analysis of the crystallographic complexes of SphK1 available to date. Currently, there are five SphK1 crystal structures deposited in the Protein Data Bank shown in Fig. 5a. In one, SphK1 was crystallized both in apo form (code 3vzb, chain C) and in complex with a substrate sphingosine-like lipid SQS (code 3vzb, chains A, B). In the remaining structures, SphK1 was solved in complex with a moderate, non-isozyme-specific inhibitor SKI-II in presence of ADP (3vzd) and without ADP (code 3vzc) and complexed with two potent inhibitors, PF543 (code 4v24) and 1V2 (code 4l02).
Fig. 5.
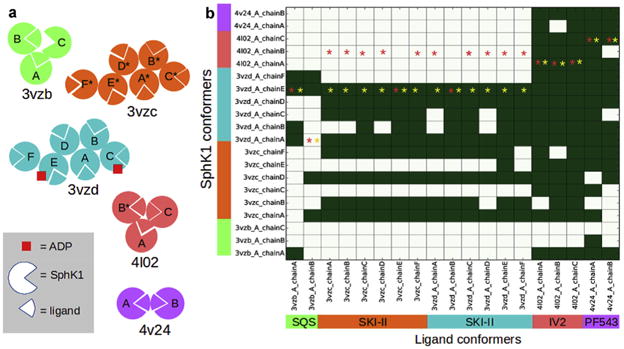
(a) Schematic representation of the 5 crystal structures available for SphK1. This scheme is intended to illustrate the number and arrangement of the chains within each structure as well as the composition stoichiometry of each chain (ligand-bound/unbound, ADP-bound/unbound states). In structures 3vzb, 4l02 and 4v24 the ligand binding sites from each chain are packed against each other, while in structures 3vzc and 3vzd, the binding sites point outward from the asymmetric unit. Chains labeled with a star lack some residues in the loop that connect helices α7 and α8 (see below). (b) Performance of the cross-docking experiment Top-ranked docking poses with a RMSD ≤2 with respect to the crystallographic binding mode in each chain are depicted in green. Red and yellow stars indicate the best scored protein conformer for each ligand before and after filtering out the “non X-ray like”, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Four of the structures, 3vzb, 3vzd, 4l02 and 4v24 displayed both “A” and “B” alternative conformations for some residues. Since none of the residues with alternate conformations were close to the substrate cavity, only conformation “A” of those structures was kept. Moreover, while SphK1 is monomeric (biological assembly), the asymmetric unit in the crystal structures contains from 2 to 5 molecules (chains) depending on the structure (see Fig. 5a). Since the asymmetric unit constitutes the smallest repetitive portion of the crystal and due to the different stoichiometry of the chains within the same structure (i.e. apo form, bound to ligand, bound to ligand + ADP, see Fig. 5a), structural differences can be observed among the chains in a single crystal structure. In 19 out of the 20 SphK1 chains or conformers, there was a ligand bound to the substrate cavity in the C-terminal domain. Therefore,we performed cross-docking of the 19 ligands against the 20 conformations of proteins from the five crystal structures.
For the naming of the different SphK1 conformers, we adopted the following convention: “PDB id_X_chainY”, where PDB id is the four digit code with which the 3D structure is deposited in the Protein Data Bank; X the alternate conformation (i.e: A, B, etc); and Y corresponds to the protein chain (chain A, chain B, etc).
Previous to the cross-docking calculations, the twenty SphK1 conformers were superimposed by their protein alpha carbons. Then, each ligand from each chain in the crystal structures was cross-docked against all of the protein conformers. After docking, the RMSD was calculated between the experimental binding mode of each ligand conformer and its docked pose in each one of the twenty protein conformers. The binary heatmap in Fig. 5b summarizes the results of the cross-docking experiment.
As can be seen, PF543 and 1V2 are docked in the proposed experimental binding pose in most of the protein conformers. On the other hand, SKI-II and SQS in general reproduces the experimental pose only in its native conformers and/or in a smaller subset of protein conformers.
The poor performance of the cross-docking of SQS and SKI-II into their non-native conformers suggests that conformational changes might take place in the enzyme that are driven by the ligand characteristics (induced fit effects). This is also evidenced by the fact that none of the ligands dock well in the apo form of the enzyme (molecule C from 3vzb).
Examination of the apo form (molecule C of 3vzb) revealed a similarly shaped cavity with a comparable entrance around the head group in comparison with the holo enzyme, i.e. the J-shaped cavity already exists in the apo form. However, its solvent-accessible volume decreased markedly on going from the apo to the holo form due to the inward movement of helices α7 and α8 (Fig. 6a). As suggested by Wang et al. [28], the α7-α8 segment in SphKs might act like a lipid gate that controls the in-and-out of lipid substrate and product.
Fig. 6.
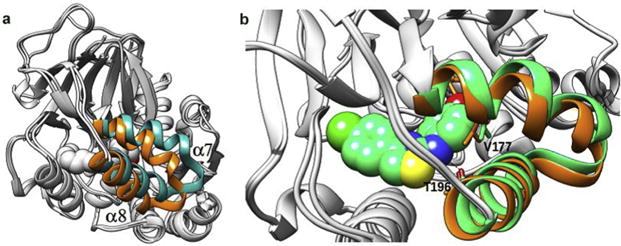
(a) Crystal structure of SphK1 bound to SQS (in orange) superimposed to the apo form of the enzyme (in cyan). (b) Crystal structure of the SQS bound SphK1 (in orange) superimposed to the X-ray structure of the SKI-II bound enzyme (in green). SKI-II atoms are represented with spheres to highlight the steric hindrance with V177 from the SQS-bound protein conformer. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The SQS-bound enzyme superimposed to the SKI-II-bound, which illustrates the importance of the conformational changes of SphK1, is shown in Fig. 6b. As can be seen, the “α7-α8 gate” shows a slightly different conformation in SKI-II-bound (green) and SQS-bound conformers (orange). As a consequence of this conformational difference, when SKI-II is docked into the SQS-bound protein conformer, it cannot approach close enough to the α8 helix to form the N-H⋯O H-bond with Thr196 because Val177 would be located too close to the aminophenol ring of SKI-II leading to steric hindrance. The inability of SKI-II to form that H-bond in the SQS-bound protein conformer, which is present in the SKI-II-bound native structure, might explain in part the poor performance of the cross-docking of SKI-II. Thus, the conformational changes in the α7-α8 segment, and the fact that the docking performance is very sensitive to those changes, highlight the necessity of employing an ensemble of protein conformers in the virtual screening campaigns against SphK1.
2.1.2. Ensemble docking
In ensemble docking, the ligand is docked against a number of conformations of the protein. The highest scoring binding mode is then selected from the ensemble of dockings against all protein conformers. Accordingly, we selected the highest scoring pose obtained for each one of the 19 ligands, which are labeled with a red star on the binary heat map in Fig. 5b. As can be seen in that figure, only in 8 of the 19 ligands (42% of the cases) was a correct pose (i.e. a pose with RMSD ≤ 2 Å) selected by the standard scoring function of AD Vina [46]. Thus, it is evident that an ensemble docking campaign to find novel inhibitors of SphK1 would likely fail if the “non X-ray like” (i.e. the poses with RMSD ≥ 2 Å) are not first filtered out from the ensemble.
2.1.3. Model to filter out the “non X-ray like”
Taking advantage of the structural information available for SphK1, we trained a classification model that was able to distinguish the docking “X-ray like” from the “non X-ray like” according to a 2 Å RMSD cutoff, with ∼86% accuracy on the training set. Fig. 7a shows the form of the logistic regression hypothesis hθ(X) where X and θ are vectors containing the independent variables (or features) and the adjustable parameters of the model, respectively. The model, once the parameters were adjusted, estimated the probability (between 0 and 1) of the docking pose being a crystal structure-like pose or a “X-ray like” pose, based on the ligand binding modes from the experimentally resolved structures of SphK1.
Fig. 7.
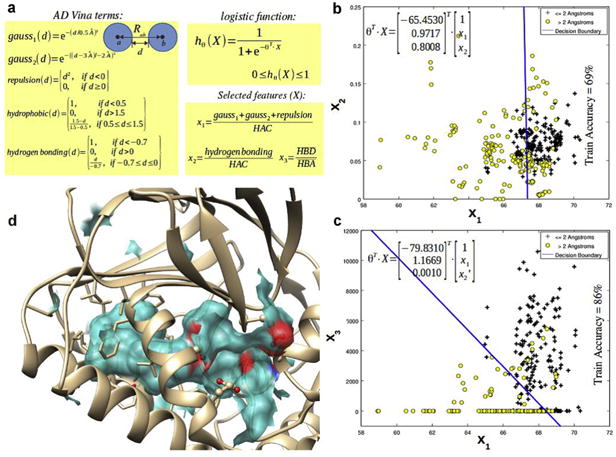
Construction of the logistic regression model to classify docking poses in crystal structure-like/non crystal structure-like poses.
The features that fit the model were selected from the conformation-dependent terms defined in the AD Vina scoring function. The conformation-dependent terms in the default AD Vina scoring function consist of three steric terms (gauss1, gauss2 and repulsion), a hydrogen bond term, and a hydrophobic term [46].
To make the features independent of the ligand size, each term was normalized by the number of heavy atoms of the ligand or heavy atom counts (HAC) so that the model might generalize to ligands other than the ones in the training set.
By plotting the size-independent AD Vina terms against each other, we found that the hydrophobic term was irrelevant for discriminating between “X-ray like” and “non X-ray like”, so it was removed from the set of potential features for the classification model. The hydrophobic term accounts for the desolvation penalty due to ligand binding. Since both “X-ray like” and “non X-ray like” are quite buried into the protein core (see Fig. 4a), the hydrophobic term does not differ appreciably between both kinds of poses. From the remaining four conformation-dependent terms, a new set of features was constructed by taking into account the physical meaning of these terms and the knowledge gained from the experimentally solved structures of SphK1.
Fig. 7a shows the new set of features constructed from the original AD Vina terms. Thus, the three steric terms (previously normalized by the HAC) were added together in a single term x1 based on the observation that these three terms together (i.e. the two gauss attractive terms for dispersion and the repulsive term) roughly behave like the van der Walls terms in force field based scoring functions (i.e. the 12-6 Lennard-Jones potential). The combined steric term (x1), similarly to the van der Waals terms, might be considered as a measure of the shape complementarity between the ligand and the receptor binding cavity. In addition to x1, x2 represents the AD Vina hydrogen bonding term normalized by the heavy atom count. Finally, alternative hydrogen bonding term x3 was constructed. Unlike x2, x3 discriminates between those interactions where the ligand acts as hydrogen bond donor and as hydrogen bond acceptor. Thus, x3 computes the AD Vina hydrogen bonding function for the interactions involving ligand donor atoms (HBD) versus ligand acceptors atoms (HBA), each one normalized by the total number of ligand H-bond donor and acceptor atoms of the ligand, respectively.
In Fig. 7b, x1 is plotted against x2 with the “X-ray like” (≤2 Å RMSD) and “non X-ray like” (>2 Å RMSD) depicted with black crosses and yellow circles, respectively, and the decision boundary between them is depicted in blue. This decision boundary discriminates between both kinds of poses with 69% accuracy. On the other hand, when x3 is used to fit the model instead of x2 (Fig. 7c), the resulting decision boundary performs much better in discriminating between crystal-structure like and non-crystal structure-like poses. This improvement in the model performance is related to the simple observation that the ligands in the experimentally solved structures act almost exclusively as hydrogen bond donors against the enzyme residues. This results in a very high value of x3 (and a very high HBD/HBA ratio) in the crystal structurelike poses and a very low value of x3, allowing a better discrimination between both kinds of poses.
Fig. 7d depicts the J-shaped tunnel occupied by the ligand in the enzyme binding site. As can be seen, the borders of the J-shaped tunnel are defined mostly by hydrophobic residues and also by a set of negatively charged residues near the entrance of the tunnel, including Asp81 and Asp178 among others. Thus, the molecular recognition event involves shape complementarity between the ligand and SphK1 which is evidenced by the overall higher values of x1 in the crystal structure-like poses and also charge complementarity which is better recovered by x3.
The yellow stars in Fig. 5b show the highest scoring poses for each one of the 19 ligands after filtering out the “non X-ray like” with the trained logistic model. In 18 out of the 19 ligands a crystal structure-like pose was selected that represented a clear improvement as compared with the ensemble docking performance without applying the filter.
2.1.4. Model validation
In order to validate the trained classification model we applied it to the docked poses of the 54 known inhibitors of SphK1 from CHEMBL database to filter out the “non X-ray like”. Fig. 4b shows the best scored poses for the protein conformer 3vzd_A_chainB, after applying the trained model. By visual inspection of Fig. 4b, one can actually see that most of the outlier poses were removed from the test set after filtering out the non-crystal structure-like poses.
2.1.5. Pre-selection of candidate compounds and results of the virtual screening
From the point of view of the structure, the compounds selected for further testing should bear some structural resemblance to the known ligands; i.e. have a polar head and a hydrophobic tail, as well as some conformational flexibility. Based on this, we selected a group of 147 compounds including chalcones, acetogenins, proto-berberines, nitrosopyrimidines and carbamates among others. The fingerprints of these 147 compounds were calculated using Open Babel [48], a chemical expert system mainly used for converting chemical file formats, and the results were compared with fingerprints obtained for the 54 compounds used previously to test the model. Those compounds having a Tanimoto index (Tc) [43] greater than 0.7 were discarded. Note that the fingerprints are a way to encode the structure of a molecule in order to compare it with other compounds; whereas the Tc is a measure of the similarity between two structures ranging from 0 to 1. Based on the Tc index, we selected 114 structurally diverse compounds which possess structural differences with respect to the known binders from the test set, justifying their further study.
2.2. Chemistry
Our model predicted sixteen different compounds as potential inhibitors of SphK1 (Table 1) that were tested in vitro for inhibition of recombinant SphK1. Synthesis of compounds 2–12, 14 and 15 have not been previously reported in the literature, and thus their characterization is now described (compounds 7–9 were reported only as moderate antimicrobial and/or antioxidant agents, but without their synthesis and analytical data [49–51]). Synthesis and characterization of compounds 1, 13 and 16 have been previously reported [52–54].
Table 1.
Structural features of compounds evaluated as SphK1 inhibitors.
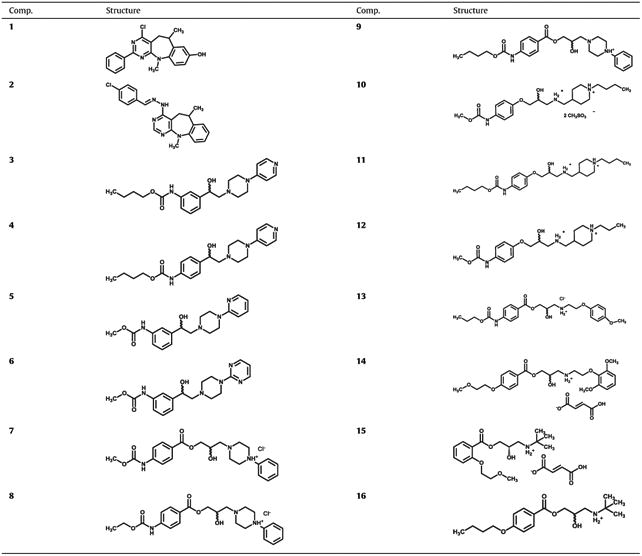
|
Compound 2 was synthesized in two steps from synthetically available 4-chloro-6,11-dimethyl-6,11-dihydro-5H-benzo[b]pyrimido [5,4-f]azepine [52] by treatment with excess of hydrazine monohydrate and subsequent condensation of the resulted hydrazinyl derivative with p-chlorobenzaldehyde. Both the above precursor of compound 1 and compound 2 were prepared following the general procedure shown in Scheme 1, which starts from the corresponding 5-allyl-4,6-dichloropyrimidine that suffers aminolysis by reaction with a substituted N-methylaniline to afford the corresponding 5-allyl-4-arylamino-4-chloropyrimidine which under a strong acid media provokes intramolecular Friedel-Crafts cyclization to afford the corresponding 6,11–dihydro–5H–benzo [b]pyrimido [5,4–f]azepine; in the case of starting from p-methoxy-N-methylaniline, the demethylation of the methoxy group is observed in the cyclization step and so rendering the compound 1.
Scheme 1.
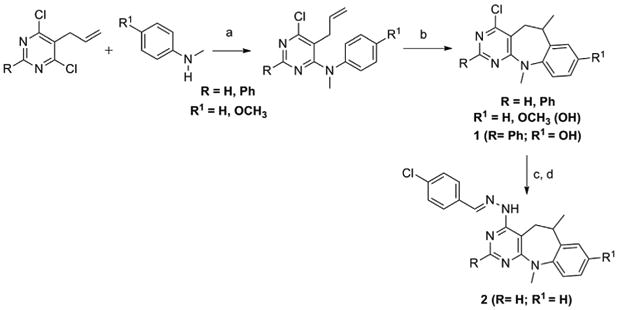
Synthesis of 4-chloro-6,11-dimethyl-6,11-dihydro-8-hydroxy-2-phenyl-5H-benzo[b]pyrimido[5,4-f]azepine, (1) (R = Ph; R1 = OH) [49] and (E)-4-(2-(4-chlorobenzylidene)hydrazinyl)-6,11-dimethyl-6,11-dihydro-5H-benzo[b]pyrimido[5,4-f]azepine (2) (R = R1 = H).
Reagents and conditions
a) DIPEA, EtOH, reflux, 48–72 h; b) CH3SO3H, 115–120 °C, 10–20 min; c) NH2NH2·H2O, EtOH, reflux, 24 h; d) p-chlorobenzaldehyde, EtOH, AcOH (cat.) reflux, 4 h.
Reaction of 3- or 4-aminoacetophenone and a suitable alkyl chloroformiate gave alkyl (3-/4-acetylphenyl)carbamates 3a–6a, which reacted with bromine in chloroform to yield alkyl [3-/4-(bromoacetyl)phenyl]carbamates 3b–6b; these compounds by treatment with different N-monosubstituted aryl(heteroaryl)pi-perazines provided the corresponding alkyl {3-/4-[(4-aryl(heteroaryl)piperazin-1-yl)acetyl]phenyl}carbamates 3c–6c. Subsequent reduction of keto group using NaBH4 provided target alkyl {3-/4-[1-hydroxy-2-(4-aryl(heteroaryl)piperazin-1-yl) ethyl] phenyl}carbamates 3–6, see Scheme 2.
Scheme 2.
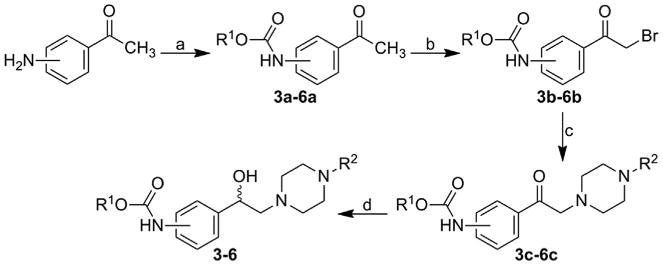
Synthesis of alkyl {3-/4-[1-hydroxy-2-(4-arylpiperazin-1-yl)ethyl] phenyl}carbamates 3–6. R1: 3-Me, 3-Bu, 4-Bu; R2: pyridine-2-yl, pyridine-4-yl, pyrimidine-2-yl.
Reagents and conditions
a) ClCOOR1, pyridine, acetone, reflux 3 h; b) Br2, CHCl3, ambient temperature 3 h; c) N-(hetero)arylpiperazine, TEA, THF, ambient temperature 3 h; d) NaBH4, MeOH, reflux 3 h.
Studied 1-(3-{4-[(alkoxycarbonyl)amino]benzoyloxy}-2-hydroxypropyl)-4-phenylpiperazin-1-ium chlorides 7–9 were prepared by multiple-step reaction described in Scheme 3. Epoxides 7a–9a and 13a (see below) were prepared from 4-aminobenzoic acid through reaction with methyl, ethyl, propyl and butyl chloroformiates giving appropriate 4-[(alkoxycarbonyl) amino]benzoic acids. Chlorides of these acids formed by thionyl chloride treatment gave desired epoxides 7a–9a and 13a after reaction with 2,3-epoxypropan-1-ol [53]. In the last step final compounds 7–9 were prepared by a reaction of the epoxides with 1-(4-phenyl)piperazine and then converted to the hydrochloride salts using ethereal HCl to enhance their solubility in water [55].
Scheme 3.
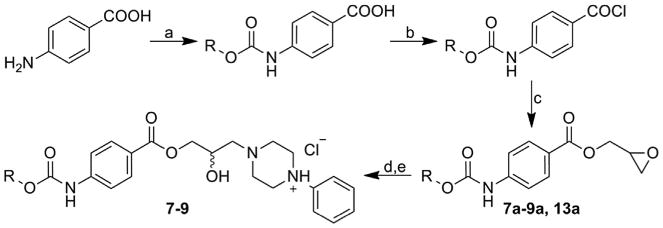
Synthesis of studied 1-(3-{4-[(alkoxycarbonyl)amino]benzoyloxy}-2-hydroxypropyl)-4-phenylpiperazin-1-ium chlorides 7–9. R: Me, Et, Bu and Pr (13a).
Reagents and conditions
a) ClCOOR, pyridine, acetone, reflux 3 h; b) SOCl2, toluene, reflux; c) 2,3-epoxypropan-1-ol, THF, TEA, 0 °C then ambient temperature; d) 1-phenylpiperazine, i-PrOH, reflux; e) HCl, Et2O, ambient temperature.
(1-Butylpiperidin-4-yl)methanamine (10b=11b) and (1-propylpiperidin-4-yl)methanamine (12b) were prepared according to the literature from piperidine-4-carboxamide by alkylation followed by reduction [56,57]. The preparation of target products 10–12 was conducted as follows: the commercially available 4-aminophenol was treated with alkyl chloroformates (R1 = Me, Bu) followed by epichlorohydrin addition to give requisite methyl [4-(oxiran-2-ylmethoxy)phenyl]carbamate (10a=12a) and butyl [4-(oxiran-2-ylmethoxy)phenyl]carbamate (11a) [58]. The carbamates were then coupled to previously prepared methanamines in MeOH at ambient temperature. The prepared final products as free bases were transformed to target 4-{[(2-hydroxy-3-{4-[(alkoxycarbonyl)amino]phenoxy}propyl)azaniumyl]methyl}-1-alkylpiperidin-1-ium dimethanesulfonates 10–12, see Scheme 4.
Scheme 4.
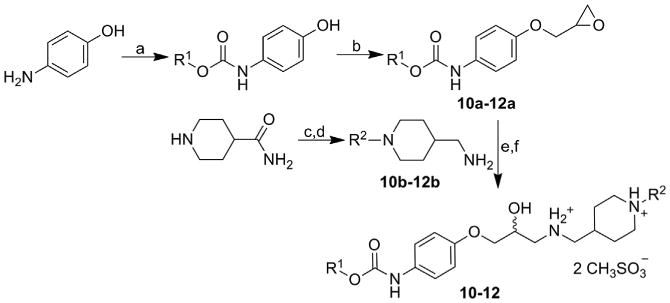
Synthesis of studied alkyl [4-(3-{[(1-alkylpiperidin-4-yl)methyl]amino}-2-hydroxypropoxy)phenyl]carbamate dimethanesulfonates 10–12. R1: Me, Bu; R2: Pr, Bu.
Reagents and conditions
a) ClCOOR1, Et2O, pyridine, ambient temperature; b) 2-(chloromethyl)oxirane, Et2O, KOH, ambient temperature; c) alkyl halides; d) LiAlH4; e) MeOH, ambient temperature; f) CH3SO3H, acetone.
Synthesis of 2-hydroxy-3-[2-(4-methoxyphenoxy)ethylamino] propyl 4-(propoxycarbonylamino)benzoate hydrochloride (13) was described in Tengler et al. [53]. The first main intermediate, oxiran-2-ylmethyl 4-[(propoxycarbonyl)amino]benzoate (13a) was synthesized according to Scheme 3. The epoxide ring was opened by addition of the second main intermediate, 2-(4-methoxyphenoxy) ethanamine (13b) prepared by Gabriel synthesis from 4-methoxyphenol via 1-(2-bromoethoxy)-4-methoxybenzene and 2-[2-(4-methoxyphenoxy)ethyl]-1H-isoindole-1,3(2H)-dione, see Scheme 5. The higher aqueous-soluble hydrochloride salt was prepared from the acquired base using ethereal HCl, as described in Tengler et al. [53].
Scheme 5.
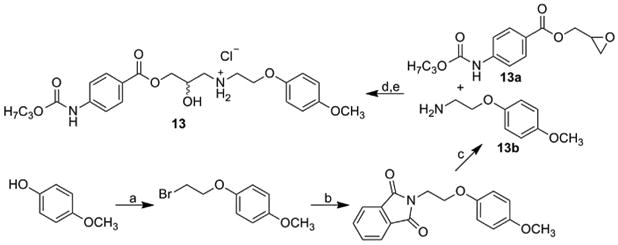
Synthesis of 2-hydroxy-3-[2-(4-methoxyphenoxy)ethylamino]propyl 4-(propoxycarbonylamino)benzoate hydrochloride (13).
Reagents and conditions
13a see Scheme 3; a) 1,2-dibromoethane, NaOH; b) potassium phtalimide, KI, DMF; c) NH2NH2·H2O, EtOH; d) i-PrOH; e) HCl, Et2O.
Final product 14 was prepared via multi-step synthesis as described in Scheme 6. Tosylate intermediates 2-methoxyethyl 4-methylbenzenesulfonate and oxiran-2-ylmethyl 4-methylbenzenesulfonate were prepared according to the published procedure by reaction of 4-methylbenzenesulfonyl chloride with 2-methoxyethanol and oxiran-2-ylmethanol, respectively, in dichloromethane [59,60]. 2-(2,6-Dimethoxyphenoxy)ethanamine was prepared via Gabriel synthesis (see Scheme 5) from 2-[2-(2,6-dimethoxyphenoxy)ethyl]-1H-isoindole-1,3(2H)-dione in two-step synthesis starting from 2,6-dimethoxyphenol that gave 2-(2-bromoethoxy)-1,3-dimethoxybenzene with 1,2-dibromoethane.Then the 2-(2-bromoethoxy)-1,3-dimethoxybenzene was reacted with potassium phthalimide [61]. The main synthesis was carried out as described by Marvanova et al. [62]. 4-(2-Methoxy)ethoxybenzoic acid (14a) was prepared from ethyl 4-hydroxybenzoate via reaction with 2-methoxyethyl 4-methylbenzenesulfonate and following hydrolysis of the ester. The final product, fumarate salt (14), was prepared from the potassium salt of acid 14a that gave (oxiran-2-yl)methyl 4-(2-methoxyethoxy)benzoate (14b) after reaction with oxiran-2-ylmethyl 4-methylbenzenesulfonate. The oxirane ring was then opened by 2-(2,6-dimethoxyphenoxy) ethanamine. The obtained base was finally converted to its fumarate salt using ethereal solution of fumaric acid to enhance the solubility of the compounds in water.
Scheme 6.
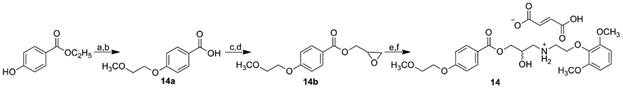
Synthesis of target [2-(2,6-dimethoxyfenoxy)ethyl]{2-hydroxy-3-[4-(2-methoxyethoxy)benzoyloxy]propyl} ammonium fumarate (14).
Reagents and conditions
a) 2-methoxyethyl 4-methylbenzenesulfonate, K2CO3, acetone, ambient temperature; b) i) NaOH, ii) HCl, CHCl3; c) MeOH, i-PrOH, KOH; d) oxiran-2-ylmethyl 4-methylbenzenesulfonate, DMF, 7 h, 70 °C; e) 2-(2,6-dimethoxyphenoxy)ethanamine, i-PrOH, 1 h at 85 °C and for 72 h at ambient temperature; f) fumaric acid, Et2O.
1-Chloro-2-methoxyethane by reaction with methyl 2-hydroxybenzoate gave methyl 2-(2-methoxyethoxy)benzoate (15a) that yielded acid 15b after hydrolysis. A reaction of the acid with SOCl2 provided 2-(2-methoxyethoxy)benzoyl chloride that with (2S)-oxiran-2-ylmethanol in the presence of 4-dimethylaminopyridine formed (2R)-oxiran-2-ylmethyl 2-(2-methoxyethoxy)benzoate (15c), a reaction of which with tert-butylamine resulted in the formation of (2R)-3-(tert-butylamino)-2-hydroxypropyl 2-(2-methoxyethoxy)benzoate that was converted to final fumarate salt 15, see Scheme 7.
Scheme 7.
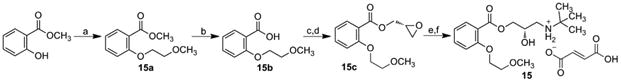
Synthesis of target [2-(2,6-dimethoxyfenoxy)ethyl]{2-hydroxy-3-[4-(2-methoxyethoxy)benzoyloxy]propyl} ammonium fumarate (15).
Reagents and conditions
a) 1-chloro-2-methoxyethane, K2CO3, KI, DMF, reflux, 6 h; b) i) NaOH, ii) HCl, CH3Cl; c) SOCl2, toluene; d) (2S)-oxiran-2-ylmethanol, DMAP, CH2Cl2; e) tert-butylamine, i-PrOH; f) fumaric acid, Et2O.
Target compound 16 was prepared by a multiple-step synthesis according to Scheme 8, as described in Tengler et al. [54]. Starting 4-butoxybenzoic acid was transformed to acid chloride that formed a suitable epoxide with 2,3-epoxypropan-1-ol. The epoxide was opened by reaction with tert-butylamine and gave 3-tert-butylamino-2-hydroxypropyl 4-butoxybenzoate. The obtained base was transformed to hydrochloride salt 16 with increased water solubility using ethereal HCl [54].
Scheme 8.
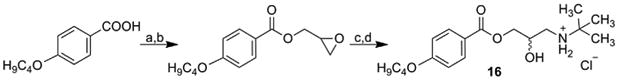
Synthesis of 3-tert-butylamino-2-hydroxypropyl 4-butoxybenzoate hydrochloride (16).
Reagents and conditions
a) toluene, PCl5; b) 2,3-epoxypropan-1-ol, CH2Cl2; c) tert-butylamine, i-PrOH; d) HCl, Et2O.
2.3. Binding affinities for sphingosine kinase 1 (bioassays)
Putative SphK1 inhibitors were evaluated in 384-well high-throughput format as described [63].
Only 3 of the 17 compounds predicted to be potential inhibitors of SphK1 using virtual screening showed activity as inhibitors (Fig. 8). One dihydrobenzo[b]pyrimido[5,4-f]azepine (2) and two alkyl {3-/4-[1-hydroxy-2-(4-arylpiperazin-1-yl)ethyl] phenyl}carbamates (3 and 4) were the compounds possessing significant inhibitory activities against SphK1; compound 2 had relatively strong inhibitory activity with an IC50 of 12 μM. While this inhibition was only moderate, it was considered significant for exploratory pre-screening.
Fig. 8.
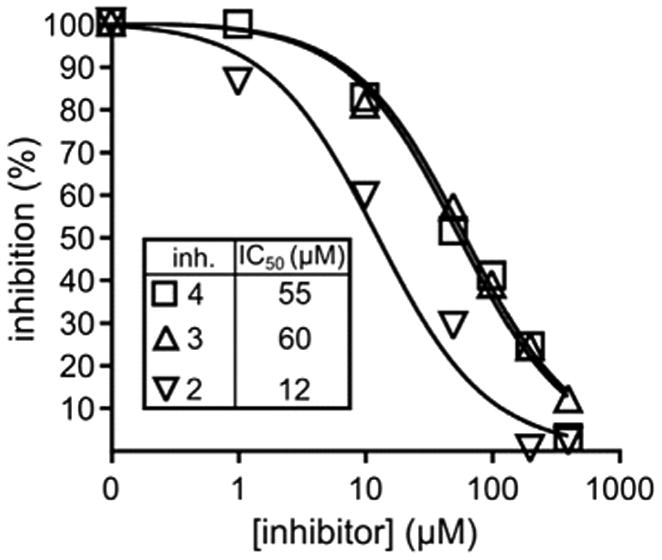
SphK1 percent inhibition versus concentration plot for the compounds 2, 3 and 4.
2.4. Molecular modelling
In order to examine why these compounds were much weaker inhibitors than PF543, one of the most potent SphK1 inhibitors reported so far [30,36], we next conducted a molecular modelling study in which we simulated the molecular interactions of the active compounds (2, 3 and 4) with SphK1 to analyze the different molecular interactions involved in the complexes of the new compounds with SphK1.
The main objective was to assess the molecular interactions that can stabilize and destabilize the different ligand-receptor complexes. We also included compound PF543 in our molecular modelling study, which therefore allowed us to perform a comparative analysis of the different activities displayed for these molecules in relation to their structural differences.
The molecular modelling study was conducted in three different stages. In the first step we performed a docking analysis using the Autodock program [64]. In the second stage of this study, we carried out molecular dynamics (MD) simulations using the AMBER software package [65]. From the trajectories obtained with the MD simulations, we performed an analysis per residue for the different compounds. Finally, to better understand the molecular interactions involved in the different L-R complexes, a Quantum Theory of Atoms In Molecules (QTAIM) study was carried out for the most representative structures of each complex. In previous work, we have demonstrated the importance of these QTAIM studies for understanding of the details of the different molecular interactions that stabilize or destabilize the various complexes [66–70].
From docking studies (although they might be considered exploratory and preliminary), it was already possible to see significant differences between PF543 and some of the new compounds reported here. For PF543, the docking studies suggest that it binds in the well-known J-shaped pocket (polar head in the polar zone of the receptor and the hydrophobic tail at the hydrophobic areas) (Fig. 9a). The same result was obtained for compound 2 (Fig. 9b). In contrast, the docking analysis of compounds 4, and 3 suggests that these compounds can bind in two ways: the known J form and in an inverted form in which the polar head interacts with the hydrophobic region of the acceptor and the hydrophobic tail is located at the polar region of the binding pocket (Fig. 9b and c). It is interesting to note that compound 2, which possessed the strongest inhibitory effect among the compounds obtained here, binds to the active site only in the so-called correct form, the same as PF543.
Fig. 9.
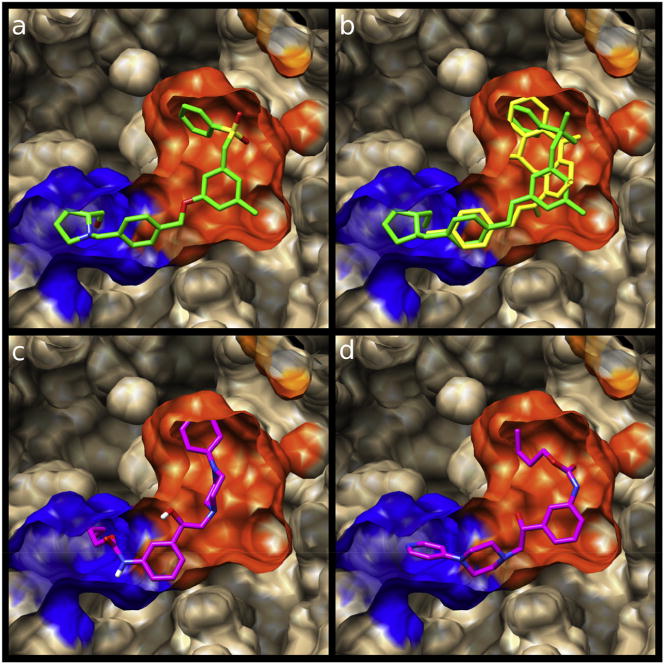
Spatial view of the different ligands bonded in the binding pocket of SphK1. The blue and orange zones represent the cationic and hydrophobic portions of the active site, respectively. a) PF543 (in green), b) PF543 (in green) superimposed on compound 2 (in yellow). c) and d) compound 3 (in magenta) bonded in the two different ways. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Interesting results were also obtained from the molecular dynamics simulations. Analysis per residue obtained from such simulations allowed us to define the main interactions that stabilize the different complexes among the simulations (Fig. 10). In general, the active compounds studied here displayed their pharmacophoric portions in a closely related spatial form to that displayed by PF543 [30] and other well-known inhibitors of SphK1 [28,29]. Consistent with previous experimental results [41,71,72], our simulations indicate the importance of the negatively charged D178, F192, L268 and F303 residues for binding of these ligands to SphK1 (Fig. 10). Superposition of interactions for compounds 2, 4 and 3 with those displayed by PF543 are shown in Fig. 10a–c. From this, it is evident that these compounds bind in a similar manner to PF543 because they interact with essentially the same amino acids. However, these interactions are generally weaker than those shown for PF543 along the simulations. These results are in agreement with the experimental data and might explain, at least in part, the lower inhibitory effects of the compounds reported here than PF543.
Fig. 10.
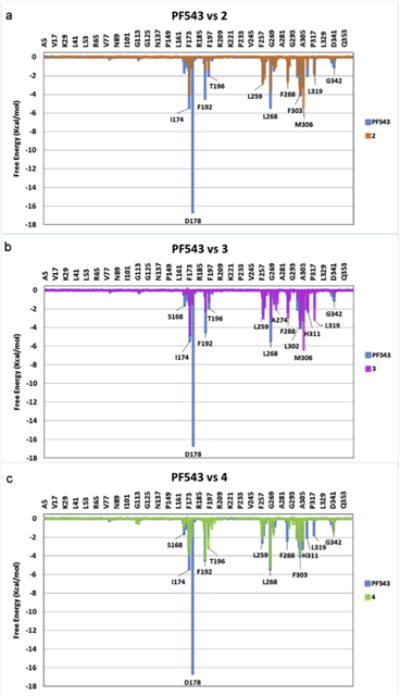
Overimposed Histograms showing the interaction energies of compounds 2 (orange), 3 (violet) and 4 (green) with the main amino acids involved in the complex formation. The histogram obtained for PF543 is shown in light blue for comparison. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
It is important to note that we are particularly interested in detecting and quantifying the interactions that stabilize and destabilize the formation of the inhibitor complexes with SphK1. While these interactions are mostly relatively weak, it is clear that molecular dynamics simulations are not accurate enough to compare with affinities observed experimentally.
Therefore, we decided to conduct a QTAIM study that can better quantify the molecular interactions obtained for the different complexes. We recently reported that the charge density value at the bond critical point (ρ(rb)), which is a descriptor of the strength of the different bonds, can be used to quantify the affinity of a ligand to form a complex ligand-receptor [67,70]. We have analyzed the complexes obtained for the three compounds reported here 2, 3 and 4 using this type of analysis and also included compound PF543 for comparison. For the sake of brevity, we only discuss the results obtained for compounds PF543 and 2, but similar results data were obtained for compounds 3 and 4.
Fig. 11 shows the sum of the ρ(rb) values corresponding to the interactions of the polar head (blue bars) and the hydrophobic tail (orange bars) obtained for compounds 2 and PF543. The sum of the ρ(rb) values for all the interactions of one part of the inhibitor (i.e., the polar head or hydrophobic tail) provides a measure of the anchoring strength of each moiety of the inhibitor to the binding pocket. This clearly shows that the hydrophobic tail of compound 2 binds to SphK1 with similar strength to that observed for PF543. However, the anchoring through the polar head is much weaker for the new inhibitor than for PF543, which is more strongly anchored in the binding pocket (Fig. 11).
Fig. 11.
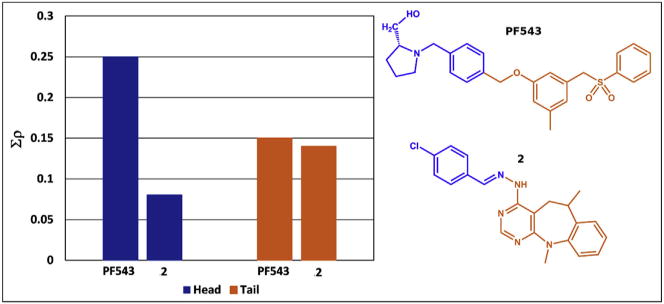
Charge density values for the total interactions of the polar head (blue stacked bars) and the hydrophobic portion (orange stacked bars) for PF543 and compound 2 at the binding pocket. The repulsive short C–H⋯H–C contacts were not included. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The strength and weakness of the different molecular interactions might be better appreciated in Fig. 12 which shows a comparative analysis of the different interactions per residue. It should be noted that the strongest interaction for PF543 is with D178 (denoted by a red arrow). It seems that a highly conserved aspartic acid is important for the binding of the ligands, indicating that the terminal carboxyl group may function as an anchoring point for molecules possessing strong inhibitory activity against SphK1 [28,71,72]. After 1.5 ns of MD simulations, the ligand has moved somewhat compared with its initial position; however, the strong interaction with D178 was maintained, supporting the suggestion that this aspartate residue might function as an anchoring point for this type of ligands. This interaction is not present in compound 2. From these results it appears that introduction of structural changes to enhance this interaction might lead to more active inhibitors.
Fig. 12.
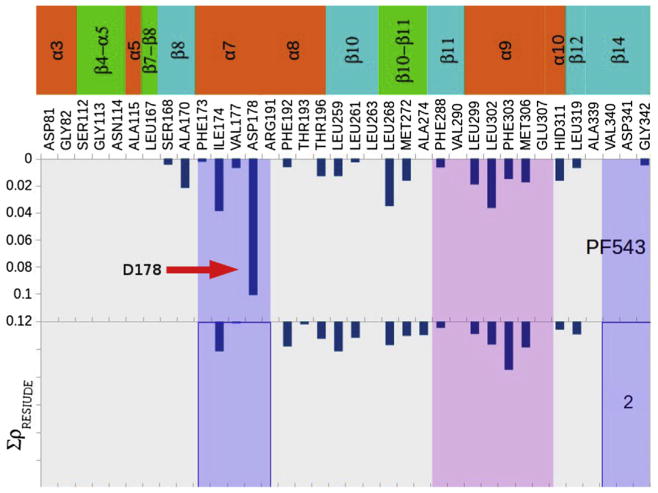
Charge density values obtained for the total interactions of PF543 and compound 2, showing the different interacting amino acids. The strong interaction of PF543 with Asp178 is denoted with a red arrow. This interaction is missing in compound 2. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 9b shows a spatial overlap of compounds 2 and PF543. This figure clearly shows that the hydrophobic portions of both compounds superimposed very well and fit perfectly into the hydrophobic portion of the active site. However, it should be noted that compound 2 is shorter than PF543, and therefore cannot interact with Asp178 (the interatomic distance between the polar head 2 and Asp178 is about 8 Å).
We evaluated the different molecular interactions obtained for the complexes in detail using molecular graphs. We only discuss here the results obtained for compounds PF543 and 2 (Fig.13), but similar data were obtained for compounds 4 and 3 (Figs. S1 and S2 in supporting information). The molecular size of these complexes is large and therefore it is not possible to visualize in detail the different interactions that stabilize and destabilize such complexes and we focused on the different molecular interactions that take place in the cationic head of the ligands. It must be remarked that the molecular interactions observed at the hydrophobic tails of all the ligands are very similar in all the complexes studied here (not examined).
Fig. 13.
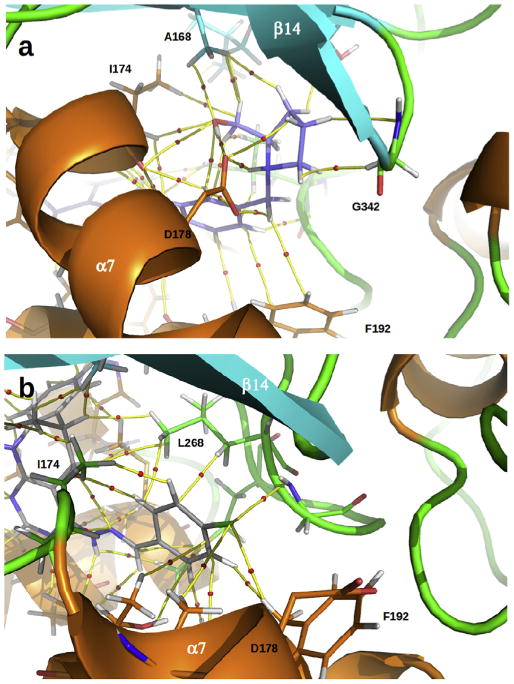
a) Molecular graph of charge density obtained for the complex of SphK1 with PF543 (in violet). Yellow lines connecting the nuclei are the bond paths, and the small red spheres on them are the bond critical points (BCPs). b) Molecular graph obtained for the complex of SphK1 with compound 2 (in gray). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 13a shows the main interactions, A170, I174, D178, F192, L268 and G342, which stabilize the polar head of PF543 complexed with SphK1. A bond critical point (BCP) and the corresponding bond paths that connect the protonated amine group of PF543 with the carboxylate group in D178 can be observed in the molecular graph of Fig. 13a. The local charge density value (ρb) at this BCP is 0.0415 a.u. which is on the border between a moderate to strong H-bond [73]. PF543 is also engaged to D178 through another moderate hydrogen bond O–H⋯O=CO (ρb = 0.0365 a.u.) and two weak C–H⋯O=CO contacts (Σρb = 0.0129 a.u.). Moreover, PF543 also forms several intermolecular interactions with I174, most of them involving the primary alcohol of the inhibitor. All together, the interactions with I174 contribute in 0.03880 a.u. of charge density to the anchoring of PF543 at the enzyme cavity. Residue A170 at the back wall of the J-shaped cavity also contribute appreciably (Σρb = 0.0217 a.u.) to the anchoring of PF543 into the binding pocket (see Fig. 12). The benzene ring of F192 forms stacking interactions with the aromatic ring from the polar head of PF543. While these interactions together only contribute to the anchoring of PF543 with the strength of a weak hydrogen bond (Σρb = 0.0064 a.u.) they seem to be critical for the proper positioning of the inhibitor into the enzyme cavity.
Fig. 13b displays the most relevant interactions observed for compound 2. It should be noted that this compound has less and weaker interactions than those of PF543. Unlike PF543, compound 2 does not form any interaction with D178. Moreover, Compound 2 virtually does not form interactions with residues from helix α7 and from sheet β14 that conform the front and back walls of the entrance to the SphK1 cavity, respectively. This is because polar part of compound 2 is not big enough and so the anchoring is driven by its hydrophobic tail that tends to occupy the back of the J-shaped cavity. This is highlighted in the complex structure at the right of the decomposition profile for this compound shown in Fig. 12. The polar head of compound 2 is anchored at the enzyme cavity mainly due to interactions with residues from helix α8 (F192, T193, T196) and α9 (L268, M302, F306), which are halfway between the entrance and the bottom of the J-shaped cavity.
In summary, our molecular modelling study clearly indicates that PF543 provides stronger molecular interactions with SphK1 than the new compounds reported here. These results are in a complete agreement with the experimental data and could at least partly explain the significantly lower inhibitory effects observed for these compounds.
3. Conclusion
This theoretical and experimental study has allowed us to find two new structural scaffolds (three new compounds), which could be used as starting structures for the design and then the development of new inhibitors of SphK1. It was carried out in several steps: virtual screening, synthesis, bioassays and molecular modelling and has allowed us to propose compound 2 as an excellent starting structure for the development of new SphK1 inhibitors. The dihydrobenzo[b]pyrimido[5,4-f]azepine motif represents a novel core for SphK1 inhibitors and should find application to the design and development of new inhibitors of this enzyme. In future publications from our group, we will detail the evolution of this pharmacophore to new core structures with greater intrinsic potency.
On the other hand, although compounds 4 and 3 showed less activity as inhibitors, considering that they have been obtained from a primary screening, these compounds are promising and also deserve to be further analyzed as alternatives initial structures. Another interesting contribution of this work is the insight in to details of certain structural aspects which are essential for understanding the formation of the complex ligand-SphK1 interactions.
On the basis of our molecular modelling results, it seems that from the results already obtained in the first steps, it is possible to obtain useful information and guidance for the design of new inhibitors. It seems that those structures, in which it is possible to distinguish more clearly the portion corresponding to the cationic head and the hydrophobic tail, are more likely to be good ligands for the active site of the SphK1. It is important to remark that such information cannot be obtained using very simple methods like the docking techniques for example. To obtain more detailed information on these molecular complexes, it is necessary to use more specific techniques. Thus, using QTAIM calculations enabled us to describe the molecular interactions that stabilize the different L-R complexes and to draw conclusions regarding two aspects; on the one hand, to explain why these novel compounds are significantly less potent SphK1 inhibitors than PF543, and on the other hand to determine what portion of the compounds should be changed in order to increase their affinity with the SphK1. From our results, it is clear that the cationic head group must be enhanced in order to obtain an increased in the binding of these ligands.
4. Experimental section
4.1. General
Commercially available compounds were used as received, unless stated otherwise. Melting points were measured by a Barstead electrothermal 9100 apparatus or a Kofler hot plate apparatus HMK (Franz Kustner Nacht GK, Dresden, Germany) are uncorrected. TLC was performed on silica gel 60 F254 on aluminium plates (Merck, Darmstadt, Germany) and visualized with UV light (254 nm). Residues were purified by silica gel 60 (40–63 mm, Merck 9385) column chromatography. 1H NMR and 13C NMR spectra were standardly recorded at 25 °C with CDCl3, methanol-d4 or DMSO-d6 as solvents on Bruker AC-300, AC-400, AC-500 or Avance III 400 MHz FT-NMR spectrometers (Bruker, Karlsruhe, Germany). The carbon typology (C, CH, CH2 or CH3) was deduced from 13C NMR DEPT experiments, which along with the 2D experiments, COSY, HSQC and HMBC correlations, permitted the fully assignation of all carbons and hydrogens. Chemical shifts are relative to the solvent peaks used as reference and reported in δ parts per million (ppm), and J values in Hz. High-resolution mass spectra (HRMS) were measured using a high-performance liquid chromatograph Dionex UltiMate® 3000 (Thermo Scientific, West Palm Beach, FL, USA) coupled with a LTQ Orbitrap XL™ Hybrid Ion Trap-Orbitrap Fourier Transform Mass Spectrometer (Thermo Scientific) with injection into HESI II in the positive or negative mode, or on a Waters Micromass AutoSpect NT (equipped with a direct inlet probe) by electronic impact operating at 70 eV.
4.2. Chemistry
4.2.1. Synthesis of compound 2
(E)-4-(2-(4-chlorobenzylidene)hydrazinyl)-6,11-dimethyl-6,11-dihydro-5H-benzo[b]pyrimido[5,4-f]azepine (2)
Hydrazine monohydrate (0.15 mL, 3.0 mmol) was added to a stirred solution of 4-chloro-6,11-dihydro-5H-benzo[b]pyrimido[5,4-f]azepine (100 mg, 0.39 mmol) [52] in ethanol (10 mL). The mixture was heated at reflux for 24 h, and then ethanol and excess of hydrazine were removed under reduced pressure. The solid residue was washed with water (2 × 50 mL), and then dried and used without further purification in the next step. To a solution of above intermediate 6,11-dimethyl-4-hydrazinyl-6,11-dihydro-5H-benzo[b] pyrimido[5,4-f]azepine (99.6 mg, 0.39 mmol) in ethanol (3 mL), p-chlorobenzaldehyde (68.3 mg, 0.49 mmol) and two drops of glacial acetic acid were added. The mixture was heated at reflux for 4 h. The reaction mixture was cooled to ambient temperature and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (hexane/EtOAc 50:50) to afford the hydrazine 1 as a white solid in yield 66%; m.p. > 150 °C. 1H NMR (CDCl3) δ 8.38 (s, 1H, CH-2), 8.00 (br s, 1H, NH-4), 7.79 (br s, 1H, -N=CH), 7.58 (d, J = 8.4 Hz, 2H, CH-2′ and CH-6′), 7.31 (d, J = 8.4 Hz, 2H, CH-3′ and CH-5′), 7.22–7.27 (m, 2H, CH-7 and CH-9), 7.13–7.19 (m, 2H, CH-8 and CH-10), 3.67–3.75 (m, 1H, CH-6), 3.52 (s, 3H, CH3-11), 2.90 (dd, J = 15.6, 2.2 Hz, 1H, CHA-5), 2.68 (dd, J = 15.6, 11.2 Hz, 1H, CHB-5), 1.47 (d, J = 7.0 Hz, 3H, CH3-6). 13C NMR (CDCl3) δ 160.2, 157.9, 154.5, 146.4, 141.4, 140.4, 135.2, 132.6, 128.9, 128.2, 126.9, 124.9, 123.9, 122.2, 98.6, 39.9, 38.7, 32.4, 18.8. HRMS (EI, 70 eV): C21H18ClN5 [M – 2H]+ calculated 375.1251 m/z, found 375.1242 m/z.
4.2.2. Synthesis of compounds 3–6
4.2.2.1. General procedure for the preparation of alkyl (3-/4-acetylphenyl)carbamates (3a–6a)
A solution of an appropriate alkyl chloroformiate (37 mmol) in acetone (5 mL) was added dropwise to a stirred solution of 3-aminoacetophenone (5.00 g; 37 mmol) or 4-aminoacetophenone (5.00 g; 37 mmol) and pyridine (3.0 mL; 37 mmol) in acetone (20 mL), and then the mixture was heated to reflux for 3 h. The solvent was removed at reduced pressure, and the resulting solid was washed with water, and recrystallized from EtOH.
4.2.2.1.1. Butyl (3-acetylphenyl)carbamate (3α)
White solid, Yield 91%, m.p. 58–59 °C [53–55 °C [74]. 1H NMR (DMSO-d6) δ 0.92 (t, 3H, -CH3 J = 6.2 Hz), 1.33–1.46 (m, 2H, -CH2-), 1.54–1.64 (m, 2H, -CH2-), 2.54 (s, 3H, -CH3), 4.09 (t, 2H, -CH2-, J = 6.6 Hz), 7.42 (t, 1H, ArH, J = 7.9 Hz), 7.60 (d, 1H, ArH, J = 7.7 Hz), 7.68 (d, 1H, ArH, J = 7.7 Hz), 8.07 (s, 1H, ArH), 9.83 (s, 1H, NH). 13C NMR (DMSO-d6) δ 197.5, 153.6, 139.6, 137.4, 129.0, 122.6, 122.3, 117.3, 63.9, 30.5, 26.6, 18.5, 13.5.
4.2.2.1.2. Butyl (4-acetylphenyl)carbamate (4a)
White solid, Yield 95%, m.p. 89–91 °C [87–88.5 °C [74]. 1H NMR (DMSO-d6) δ 0.92 (t, 3H, -CH3, J = 7.3 Hz), 1.30–1.48 (m, 2H, -CH2-), 1.55–1.69 (m, 2H, -CH2-), 2.51 (s, 3H, -CH3), 4.11 (t, 2H, -CH2-J = 6.6 Hz), 7.60 (d, 2H, ArH, J = 8.8 Hz), 7.91 (d, 2H, ArH, J = 8.8 Hz), 10.05 (s, 1H, NH). 13C NMR (DMSO-d6) δ 196.3, 153.4, 143.7, 130.9, 129.4, 117.2, 64.1, 30.4, 26.2, 18.47, 13.5.
4.2.2.1.3. Methyl (3-acetylphenyl)carbamate (5a=6a)
White solid, Yield 95%, m.p. 103–104 °C. 1H NMR (DMSO-d6) δ 2.55 (s, 3H, -CH3), 3.69 (s, 3H, -OCH3), 7.44 (t, 1H, ArH, J = 8.2 Hz), 7.61 (d, 1H, ArH, J = 7.3 Hz), 7.71 (d, 1H, ArH, J = 7.7 Hz), 8.06 (s, 1H, ArH), 9.87 (s, 1H, NH). 13C NMR (DMSO-d6) δ 197.5, 153.9, 139.5, 137.4, 129.0, 122.6, 122.4, 117.3, 51.5, 26.5.
4.2.2.2. General procedure for the preparation of alkyl [3-/4-(bromoacetyl)phenyl]carbamates (3b–6b)
Into a stirred solution of an appropriate alkyl (3-/4-acetylphenyl)carbamate (36 mmol) in chloroform (80 mL), a solution of bromine (1.9 mL; 36 mmol) in chloroform (10 mL) was added dropwise and stirred for 3 h at ambient temperature. The solvent was removed under reduced pressure. Solid crude products were recrystallized from i-PrOH.
4.2.2.2.1. Butyl [3-(bromoacetyl)phenyl]carbamate (3b)
White solid, Yield 85%, m.p. 80–86 °C. 1H NMR (DMSO-d6) δ 0.92 (t, 3H, -CH3, J = 7.3 Hz), 1.34–1.47 (m, 2H, -CH2-), 1.54–1.64 (m, 2H, -CH2-), 4.10 (t, 2H, -CH2-, J = 6.6 Hz), 4.88 (s, 2H, -CH2-), 7.46 (t, 1H, ArH, J = 7.9 Hz), 7.65–7.77 (m, 2H, ArH), 8.10 (s, 1H, ArH), 9.89 (s, 1H, NH). 13C NMR (DMSO-d6) δ 191.4, 153.6, 139.8, 134.5, 129.2, 123.3, 122.9, 117.6, 63.9, 33.6, 30.4, 18.5, 13.4.
4.2.2.2.2. Butyl [4-(bromoacetyl)phenyl]carbamate (4b)
White solid, Yield 85%, m.p. 152–154 °C. 1H NMR (DMSO-d6) δ 0.92 (t, 3H, -CH3, J = 7.3 Hz), 1.30–1.48 (m, 2H, -CH2-), 1.56–1.70 (m, 2H, -CH2-), 4.12 (t, 2H, -CH2-, J = 6.6 Hz), 4.84 (s, 2H, -CH2-), 7.62 (d, 2H, ArH, J = 8.8 Hz), 7.96 (d, 2H, ArH, J = 8.8 Hz), 10.13 (s, 1H, NH). 13C NMR (DMSO-d6) δ 190.1, 153.3, 144.4, 130.1, 127.8, 117.3, 64.2, 33.4, 30.4, 18.5, 13.5.
4.2.2.2.3. Methyl [3-(bromoacetyl)phenyl]carbamate (5b=6b)
White solid, Yield 75%, m.p. 99–103 °C. 1H NMR (DMSO-d6) δ 3.68 (s, 3H, -CH3), 4.89 (s, 2H, -CH2-), 7.47 (t, 1H, ArH, J = 8.1 Hz), 7.66–7.78 (m, 2H, ArH), 8.08 (s, 1H, ArH), 9.92 (s, 1H, NH). 13C NMR (DMSO-d6) δ 191.4, 153.9, 139.7, 134.5, 129.2, 123.3, 122.9, 117.6, 51.6, 33.6.
4.2.2.3. General procedure for preparation of alkyl {3-/4-[(4-arylpiperazin-1-yl)acetyl]phenyl} carbamates (3c–6c)
A solution of arylpiperazine (5.5 mmol) and triethylamine (0.8 mL; 5.5 mmol)in anhydrous THF (20 mL) was added dropwise to a stirred solution of an appropriate alkyl [3-/4-(bromoacetyl)phenyl]carbamate (5.5 mmol) in anhydrous THF (30 mL), and the mixture stirred for 3 h at ambient temperature. The solvents were removed under reduced pressure, and added chloroform (100 mL) and water. The organic phase was washed with additional water, dried over anhydrous sodium sulfate and the solvent removed under reduced pressure, to give a solid crude product, which was recrystallized from acetone.
4.2.2.3.1. Butyl {3-[(4-(pyridine-4-yl)piperazin-1-yl)acetyl] phenyl}carbamate (3c)
White solid, Yield 37%, m.p. 154–158 °C. 1H NMR (DMSO-d6) δ 0.92 (t, 3H, -CH3, J = 7.0 Hz), 1.29–1.47 (m, 2H, -CH2-), 1.54–1.67 (m, 2H, -CH2-), 2.50–2.75 (m, 4H, -CH2-), 3.20–3.40 (m, 4H, -CH2-), 3.88 (s, 2H, -CH2-), 4.09 (t, 2H, -CH2-, J = 6.6 Hz), 6.79–6.87 (m, 2H, ArH), 7.42 (t, 1H, ArH, J = 7.9 Hz), 7.63–7.82 (m, 2H, ArH), 8.13–8.16 (m, 2H, ArH), 8.32 (s, 1H, ArH), 9.85 (s, 1H, NH). 13C NMR (DMSO-d6) δ 196.4, 154.5, 153.6, 149.6, 139.6, 136.4, 128.9, 122.7, 122.1, 117.3, 108.3, 63.9, 63.4, 52.0, 45.4, 30.5, 18.5, 13.5.
4.2.2.3.2. Butyl {4-[(4-(pyridine-4-yl)piperazin-1-yl)acetyl] phenyl}carbamate (4c)
White solid, Yield 19%, m.p. 113–114 °C. 1H NMR (DMSO-d6) δ 0.91 (t, 3H, -CH3, J = 7.3 Hz), 1.30–1.50 (m, 2H, -CH2-), 1.52–1.70 (m, 2H, -CH2-), 2.55–2.70 (m, 4H, -CH2-), 3.22–3.35 (m, 4H, -CH2-), 3.83 (s, 2H, -CH2-), 4.10 (t, 2H, -CH2-, J = 6.6 Hz), 6.80 (d, 2H, ArH, J = 6.6 Hz), 7.58 (d, 2H, ArH, J = 8.8 Hz), 7.95 (d, 2H, ArH, J = 8.8 Hz), 8.14 (d, 2H, ArH, J = 6.2 Hz), 10.05 (s, 1H, NH). 13C NMR (DMSO-d6) δ 195.2, 154.5, 153.3, 149.6, 143.8, 129.9, 129.4, 117.1, 108.3, 63.4, 63.2, 52.0, 45.4, 30.5, 18.5, 13.5.
4.2.2.3.3. Methyl {3-[(4-(pyridine-2-yl)piperazin-1-yl)acetyl] phenyl}carbamate (5c)
White solid, Yield 89%, m.p. 130–133 °C. 1H NMR (DMSO-d6) δ 2.58–2.62 (m, 4H, -CH2-), 3.47–3.51 (m, 4H, -CH2-), 3.68 (s, 3H, -CH3), 3.86 (s, 2H, -CH2-), 6.62 (dd, 1H, ArH, J = 6.9,J = 5.0 Hz), 6.81 (d, 1H, ArH, J = 8.1 Hz), 7.43 (t, 1H, ArH, J = 7.8 Hz), 7.51 (ddd, 1H, ArH, J = 8.7, J = 7.1, J = 2.1 Hz), 7.66 (d, 1H, ArH, J = 7.8 Hz), 7.71 (dd, 1H, ArH, J = 6.9, J = 5.0 Hz), 8.09–8.11 (m, 2H, ArH), 9.87 (s, 1H, NH). 13C NMR (DMSO-d6) δ 159.1, 154.0, 147.6, 145.4, 138.9, 137.5, 128.3, 120.2, 116.8, 115.9, 112.9, 107.1, 69.9, 66.4, 52.9, 51.6, 44.7.
4.2.2.3.4. Methyl {3-[(4-(pyrimidine-2-yl)piperazin-1-yl)acetyl] phenyl}carbamate (6c)
White solid, Yield 97%, m.p. 94–96 °C. 1H NMR (DMSO-d6) δ 2.50–2.68 (m, 4H, -CH2-), 3.68 (s, 3H, -CH3), 3.72–3.77 (m, 4H, -CH2-), 3.87 (s, 2H, -CH2-), 6.61 (t, 1H, ArH, J = 4.8 Hz), 7.43 (t, 1H, ArH, J = 8.1 Hz), 7.60–7.80 (m, 2H, ArH), 8.10 (s, 1H, ArH), 8.34 (d, 2H, ArH, J = 4.8 Hz), 9.84 (s, 1H, NH). 13C NMR (DMSO-d6) δ 196.4, 161.1, 157.8, 153.9, 139.5, 136.4, 128.9, 122.7, 122.2, 117.3, 109.9, 63.6, 52.3, 51.6, 43.1.
4.2.2.4. General procedure for preparation of alkyl {3-/4-[1-hydroxy-2-(4-arylpiperazin-1-yl)ethyl] phenyl}carbamates (3–6)
Solid sodium borohydride (0.30 g; 8.0 mmol) was added in small portions to a solution of the appropriate alkyl {3-/4-[(4-arylpiperazin-1-yl) acetyl]phenyl} carbamate (4.0 mmol) in hot methanol (50 mL), and then the mixture was refluxed for 1 h. The solvent was removed under reduced pressure, and the residue was treated with distilled water (100 mL) and chloroform (100 mL). The organic phase was washed with additional water, dried over anhydrous sodium sulfate and solvent removed under reduced pressure to give a crude product, which was recrystallized from acetone.
4.2.2.4.1. Butyl {3-[1-hydroxy-2-(4-(pyridine-4-yl)piperazin-1-yl)ethyl]phenyl}carbamate (3)
White solid, Yield 96%, m.p.147–149 °C. 1H NMR (DMSO-d6) δ 0.90 (t, 3H, -CH3, J = 7.1 Hz),1.26–1.45 (m, 2H, -CH2-), 1.52–1.66 (m, 2H, -CH2-), 2.36–2.48 (m,2H, -CH2N), 2.48–2.59 (m, 4H, -CH2-), 3.20–3.35 (m, 4H, -CH2-),4.05 (t, 2H, -CH2-, J = 6.6 Hz), 4.64–4.72 (m, 1H, -CH-), 5.09 (d, 1H,OH, J = 3.7 Hz), 6.77–6.85 (m, 2H, ArH), 6.96 (d, 1H, ArH, J = 7.7 Hz),7.19 (t, 1H, ArH, J = 7.7 Hz), 7.31 (d, 1H, ArH, J = 7.7 Hz), 7.50 (s, 1H,ArH), 8.12–8.15 (m, 2H, ArH), 9.56 (s, 1H, NH). 13C NMR (DMSO-d6) δ 154.5, 153.6, 149.7, 145.2, 138.1, 128.1, 120.0, 116.8, 116.0, 108.2, 69.9, 66.1, 63.7, 52.6, 45.2, 30.6, 18.5, 13.5. HR-MS (Orbitrap):C22H31N4O3 [M+H]+ calculated 399.2391 m/z, found 399.2385 m/z.
4.2.2.4.2. Butyl {4-[1-hydroxy-2-(4-(pyridine-4-yl)piperazin-1-yl)ethyl]phenyl}carbamate (4)
White solid, Yield 53%, m.p. 175–179 °C. 1H NMR (DMSO-d6) δ 0.90 (t, 3H, -CH3, J = 7.3 Hz), 1.28–1.46 (m, 2H, -CH2-), 1.53–1.66 (m, 2H, -CH2-), 2.35–2.55 (m, 2H, -CH2N), 2.55–2.65 (m, 4H, -CH2-), 3.15–3.35 (m, 4H, -CH2-), 4.05 (t, 2H, -CH2-, J = 6.6 Hz), 4.64–4.73 (m, 1H, -CH-), 5.00 (d, 1H, OH, J = 3.7 Hz), 6.79 (d, 2H, ArH, J = 6.3 Hz), 7.24 (d, 2H, ArH, J = 8.8 Hz), 7.39 (d, 2H, ArH J = 8.8 Hz), 8.13 (d, 2H, ArH, J = 6.3 Hz), 9.55 (s, 1H, NH). 13C NMR (DMSO-d6) δ 154.5, 153.6, 149.7, 138.4, 137.8, 126.3, 117.9, 108.2, 69.9, 66.1, 63.7, 52.6, 45.4, 30.5, 18.5, 13.5. HR-MS (Orbitrap): C22H31N4O3 [M+H]+ calculated 399.2391 m/z, found 399.2399 m/z.
4.2.2.4.3. Methyl {3-[1-hydroxy-2-(4-(pyridine-2-yl)piperazin-1-yl)ethyl]phenyl}carbamate (5)
White solid, Yield 69%, m.p. 174–177 °C. 1H NMR (DMSO-d6) δ 2.38–2.55 (m, 2H, -CH2N), 2.55–2.58 (m, 4H, -CH2-), 3.44–3.48 (m, 4H, -CH2-), 3.65 (s, 3H, -CH3), 4.68–4.72 (m, 1H, -CH-), 5.07 (d, 1H, OH, J = 3.2 Hz), 6.62 (dd, 1H, ArH, J = 7.1, J = 5.3 Hz), 6.80 (d, 1H, ArH, J = 8.7 Hz), 6.98 (d, 1H, ArH, J = 7.8 Hz), 7.21 (t, 1H, ArH, J = 7.8 Hz), 7.34 (d, 1H, ArH, J = 8.2 Hz), 7.48 (s, 1H, ArH), 7.51 (ddd, 1H, ArH, J = 8.7, J = 7.1, J = 2.1 Hz), 8.09–8.11 (m, 1H, ArH), 9.60 (s, 1H, NH). 13C NMR (DMSO-d6) δ 159.1, 154.0, 147.6, 145.4, 138.9, 137.5, 128.3, 120.2, 116.8, 115.9, 112.9, 107.1, 69.9, 66.4, 52.9, 51.6, 44.7. HR-MS (Orbitrap): C19H25N4O3 [M+H]+ calculated 357.1921 m/z, found 357.1939 m/z.
4.2.2.4.4. Methyl {3-[1-hydroxy-2-(4-(pyrimidine-2-yl)piperazin-1-yl)ethyl]phenyl}carbamate (6)
White solid, Yield 55%, m.p. 157–159 °C. 1H NMR (DMSO-d6) δ 2.36–2.48 (m, 2H, -CH2N), 2.50–2.60 (m, 4H, -CH2-), 3.64 (s, 3H, -CH3), 3.70–3.80 (m, 4H, -CH2-), 4.64–4.72 (m, 1H, -CH-), 5.07 (d, 1H, OH, J = 3.7 Hz), 6.60 (t, 1H, ArH, J = 4.8 Hz), 6.97 (d, 1H, ArH, J = 7.3 Hz), 7.20 (t, 1H, ArH, J = 7.7 Hz), 7.33 (d, 1H, ArH, J= 7.9 Hz), 7.50 (s, 1H, ArH), 8.30–8.40 (m, 2H, ArH), 9.59 (s, 1H, NH). 13C NMR (DMSO-d6) δ 161.2, 157.8, 153.9, 145.3, 138.8, 128.2, 120.1, 116.8, 116.0, 109.9, 69.8, 66.3, 52.9, 51.4, 43.3. HR-MS (Orbitrap): C18H24N5O3 [M+H]+ calculated 358.1874 m/z, found 358.1894 m/z.
4.2.3. Synthesis of compounds 7–9
4.2.3.1. General procedure for preparation of 1-(3-{4-[(alkoxycarbonyl)amino]benzoyloxy}-2-hydroxypropyl)-4-phenylpiperazin-1-ium chlorides (7–9)
A mixture of epoxides 7a–9a [53] (0.2 mol) and 1-(4-phenyl)piperazine (0.2 mol) in i-PrOH (150 mL) was heated at 80 °C for 4 h. The solvent was evaporated under reduced pressure, and the residing oil was dissolved in Et2O. The solution of the base was converted to its chloride salt by addition of ethereal HCl. The amine salt was collected by filtration and recrystallized from i-PrOH to give white crystals.
4.2.3.1.1. 1-(2-Hydroxy-3-{4-[(methoxycarbonyl)amino]benzoyloxy}propyl)-4-phenylpiperazin-1-ium chloride (7)
Yield 62%, Rf: 0.83 (acetone/toluene 3:1), m.p. 197–200 °C. 1H NMR (DMSO-d6)δ 10.74 (s,1H, -NH+-), 10.17 (s,1H, -NH), 7.98 (d, 3J= 8.7, 2H, ArH2,6),7.63 (d, 3J = 8.7, 2H, ArH3,5), 7.30–7.22 (m, 2H, ArH-Npip), 7.02–6.98(m, 2H, ArH-Npip), 6.89–6.82 (m, 1H, ArH-Npip), 6.07 (s, 1H, -OH),4.49–4.46 (m, 1H, -CH(OH)), 4.24–4.21 (m, 2H, -COOCH2-),3.83–3.56 (m, 4H, Hpip), 3,69 (s, 3H, -CH3), 3.38–3.11 (m, 6H,Hpip + -CH2Npip). 13C NMR (DMSO-d6) δ 165.1, 153.7, 149.5, 143.9, 130.6, 129.0, 122.9, 119.8, 117.3, 115.8, 66.0, 63.3, 58.2, 52.1, 51.8, 50.7, 45.2. HR-MS (Orbitrap): C23H26N3O5 [M – H]- calculated 412.1877 m/z, found 412.1885 m/z.
4.2.3.1.2. 1-(3-{4-[(ethoxycarbonyl)amino]benzoyloxy}-2-hydroxypropyl)-4-phenylpiperazin-1-ium chloride (8)
Yield 68%, Rf: 0.86 (acetone/toluene 3:1), m.p. 192–194 °C. 1H NMR (DMSO-d6)δ 10.75 (s, 1H, -NH+-), 10.12 (s, 1H, -NH), 7.98 (d, 3J = 8.6, 2H, ArH2,6), 7.63 (d, 3J = 8.6, 2H, ArH3,5), 7.30–7.22 (m, 2H, ArH-Npip), 7.02–6.98(m, 2H, ArH-Npip), 6.89–6.82 (m, 1H, ArH-Npip), 6.07 (s, 1H, -OH), 4.49–4.46 (m, 1H, -CH(OH)-), 4.23–4.10 (m, 4H, -COOCH2-+ -CH2CH3), 3.83–3.61 (m, 4H, Hpip), 3.38–3.11 (m, 6H, Hpip +-CH2Npip), 1,25 (t, 3J = 7.1, 3H, -CH3). 13C NMR (DMSO-d6) δ 165.1, 153.3, 149.5, 144.0, 130.6, 129.0, 122.8, 119.8, 117.3, 115.8, 66.0, 63.3, 60.5, 58.2, 52.1, 50.8, 45.2, 14.4. HR-MS (Orbitrap): C24H28N3O5[M – H]- calculated 426.2034 m/z, found 426.2042 m/z.
4.2.3.1.3. 1-(3-{4-[(butoxycarbonyl)amino]benzoyloxy}-2-hydroxypropyl)-4-phenylpiperazin-1-ium chloride (9)
Yield 57%, Rf: 0.92 (acetone/toluene 3:1), m.p. 178–182 °C. 1H NMR (DMSO-d6)δ 10.71 (s, 1H, -NH+-), 10.11 (s, 1H, -NH), 7.98 (d, 3J = 8.6,2H, ArH2,6),7.63 (d, 3J = 8.6, 2H, ArH3,5), 7.30–7.22 (m, 2H, ArH-Npip), 7.02–6.98(m, 2H, ArH-Npip), 6.89–6.82 (m, 1H, ArH-Npip), 6.07 (s, 1H, -OH),4.51—4.43 (m, 1H, -CH(OH)-), 4.23–4.21 (m, 2H, -COOCH2-), 4,11 (t,J = 6.5, 2H, -CH2(CH2)2CH3), 3.83–3.61 (m, 4H, Hpip), 3.41–3.10 (m,6H, Hpip + -CH2Npip), 1.68–1.54 (m, 2H, -CH2CH2CH2CH3), 1.47–1.29 (m, 2H, -(CH2)2CH2CH3), 0.91 (t, J = 7.2, 3H, -(CH2)3CH3). 13C NMR(DMSO-d6) δ 165.1, 153.4, 149.5, 144.0, 130.6, 129.0, 122.8, 119.8, 117.3, 115.8, 66.0, 63.3, 61.9, 58.2, 52.1, 50.8, 45.2, 30.4, 18.5, 13.5.HR-MS (Orbitrap): C26H32N3O5 [M – H]- calculated 454.2347 m/z, found 454.2355 m/z.
4.2.4. Synthesis of compounds 10–12
4.2.4.1. General procedure for the syntheses of 10–12
A solution of the corresponding (1-alkylpiperidin-4-yl)methanamine 10b–12b [56,57] (11.60 mmol) in methanol (30 mL) was added dropwise to a solution of the appropriate alkyl [4-(oxiran-2-ylmethoxy)phenyl] carbamate 10a–12a [58] (9.00 mmol) in methanol (40 mL). The resulting mixture was left stirring for two days at ambient temperature and then concentrated under reduced pressure. The respective free bases obtained as yellowish oils were dissolved in acetone (10 mL) and treated with a solution of methanesulfonic acid (2 equiv.) in acetone (5 mL) by dropwise addition at 0 °C. The resulting mixtures were stirred at ambient temperature overnight and then concentrated in vacuo to furnish dimesylates 10–12 as solids that were recrystallized as described below.
4.2.4.1.1. 4-{[(2-hydroxy-3-{4-[(methoxycarbonyl)amino]phenoxy}propyl)azaniumyl]methyl}-1-butylpiperidin-1-ium dimethanesulfonate (10)
The compound was prepared from methyl [4-(oxiran-2-ylmethoxy)phenyl]carbamate (10a=12a) and (1-butylpiperidin-4-yl)methanamine (10b=11b). Crystallization from i-PrOH afforded 10 as white crystalline powder (1.10 g, overall yield (2 steps): 33%), m.p. 94–97 °C. 1H NMR (DMSO-d6) δ 0.91 (t, 1JHH = 7.3 Hz, 3H, CH3 (CH3CH2CH2CH2)), 1.20–1.75 (m, 6H, CH2 (CH3CH2CH2CH2 + piperidine)), 1.88–2.09 (m, 3H, CH + CH2 (piperidine)), 2.37 (s, 6H, CH3 (2 × CH3SO2)), 2.74–3.25 (m, 8H, CH2 (piperidine + CH2NH2CH2 + CH3CH2CH2CH2NH)), 3.42–3.59 (m, 2H, CH2 (NH2CH2CH(OH))), 3.63 (s, 3H, CH3 (CH3OCONH)), 3.79–4.04 (m, 2H, CH2 (CH2CH(OH)CH2)), 4.10–4.26 (m, 1H, CH (CH2CH(OH) CH2)), 5.81 (d, V1 = 3.5 Hz, 1H, OH), 6.89 (d, 1JHH = 9.0 Hz, 2H, CHarom), 7.36 (d, HHhh = 9.0 Hz, 2H, CHarom), 8.51 (br. s., 2H, NH2 (CH2NH2CH2)), 9.09 (br. s., 1H, NH (piperidine)), 9.43 (s, 1H, NH (CH3OCONH)). 13C NMR (DMSO-d6) δ 13.4, 19.4, 25.2, 26.6, 30.4, 39.7, 49.9, 51.1, 51.40, 51.44, 55.7, 64.7, 70.0, 114.7, 119.8, 132.6, 153.7, 154.1. HR-MS (Orbitrap): C21H35N3O4 [M+H]+ calculated 394.2706 m/z, found 394.2701 m/z.
4.2.4.1.2. 4-{[(2-hydroxy-3-{4-[(butoxycarbonyl)amino]phenoxy} propyl)azaniumyl]methyl}-1-butylpiperidin-1-ium dimethanesulfonate (11)
The compound was prepared from butyl [4-(oxiran-2-ylmethoxy)phenyl]carbamate (11a) and (1-butylpiperidin-4-yl) methanamine (10b=11b). Crystallization from i-PrOH/i-Pr2O afforded 11 as white crystalline powder (0.67 g, overall yield (2 steps): 26%), m.p. 108–110 °C. 1H NMR (DMSO-d6) δ 0.91 (t, 1JHH = 7.3 Hz, 6H, CH3 (2 × CH3CH2CH2CH2)), 1.14–1.73 (m, 10H, CH2 (2 × CH3CH2CH2CH2 + piperidine)), 1.85–2.08 (m, 3H, CH + CH2 (piperidine)), 2.37 (s, 6H, CH3 (2 × CH3SO2)), 2.76–3.26 (m, 8H, CH2 (piperidine + CH2NH2CH2 + CH3CH2CH2CH2NH)), 3.43–3.60 (m, 2H, CH2 (NH2CH2CH(OH))), 3.84–3.99 (m, 2H, CH2 (CH2CH(OH)CH2)), 4.05 (t, JHH = 6.6 Hz, 2H, CH2 (CH3CH2CH2CH2O)), 4.09–4.26 (m, 1H, CH (CH2CH(OH)CH2)), 5.81 (d, 1JHH = 4.1 Hz, 2H, OH), 6.88 (d, 1JHH = 9.0 Hz, 2H, CHarom.), 7.37 (d, 1JHH = 9.0 Hz, 2H, CHarom.), 8.51 (br. s., 2H, NH2 (CH2NH2CH2)), 9.09 (br. s., 1H, NH (piperidine)), 9.39 (s, 1H, NH (CH3OCONH)). 13C NMR (DMSO-d6) δ 13.4, 13.5, 18.6, 19.4, 25.2, 26.6, 30.4, 30.6, 39.7, 49.9, 51.1, 51.4, 55.7, 63.7, 64.7, 70.0, 114.7, 119.7, 132.7, 153.6, 153.7. HR-MS (Orbitrap): C24H41N3O4 [M+H]+ calculated 436.3175 m/z, found 436.3170 m/z.
4.2.4.1.3. 4-{[(2-hydroxy-3-{4-[(methoxycarbonyl)amino]phenoxy}propyl)azaniumyl]methyl}-1-propylpiperidin-1-ium dimethanesulfonate (12)
The compound was prepared from methyl [4-(oxiran-2-ylmethoxy)phenyl]carbamate (10a=12a) and (1-propylpiperidin-4-yl)methanamine (12b). Crystallization from MeOH/i-PrOH afforded 12 as white crystalline powder (0.83 g, overall yield (2 steps): 42%), m.p. 158–160 °C. 1H NMR (DMSO-d6) δ 0.90 (t, 1JHH = 7.3 Hz, 3H, CH3 (CH3CH2CH2)), 1.34–1.77 (m, 4H, CH2 (CH3CH2CH2 + piperidine)), 1.85–2.12 (m, 3H, CH + CH2 (piperidine)), 2.39 (s, 6H, CH3 (2 × CH3SO2)), 2.65–3.23 (m, 8H, CH2 (piperidine + CH2NH2CH2 + CH3CH2CH2NH)), 3.35–3.55 (m, 2H, CH2 (NH2CH2CH(OH))), 3.63 (s, 3H, CH3 (CH3OCONH)), 3.88–4.00 (m, 2H, CH2 (CH2CH(OH)CH2)), 4.17–4.22 (m, 1H, CH (CH2CH(OH)CH2)), 5.81 (br. s., 2H, OH), 6.90 (d, 1JHH = 6.0 Hz, 2H, CHarom.), 7.37 (d, 1JHH = 8.8 Hz, 2H, CHarom.), 8.53 (br. s., 1H, NH2 (CH2NH2CH2)), 9.12 (br. s., 1H, NH (piperidine)), 9.44 (s, 1H, NH (CH3OCONH)). 13C NMR (DMSO-d6) δ 10.9, 16.8, 26.6, 30.4, 39.7, 49.9, 51.1, 51.4, 51.5, 57.5, 64.8, 70.0, 114.7, 119.8, 132.6, 153.7, 154.1. HR-MS (Orbitrap): C20H33N3O4 [M+H]+ calculated 380.2549 m/z, found 380.2544 m/z.
4.2.5. Synthesis of compound 14
4.2.5.1. 4-(2-Methoxy)ethoxybenzoic acid (14a)
A mixture of ethyl-4-hydroxybenzoate (3.32 g, 0.02 mol), 2-methoxyethyl 4-methylbenzenesulfonate [59] (0.02 mol) and potassium carbonate (0.06 mol) in acetone (25 mL) was heated at 70 °C for 8 h and then stirred for 12 h at ambient temperature. The precipitation was then filtered and acetone was evaporated. The residue was dissolved in EtOAc and washed with 2 M NaOH and water, dried over anhydrous MgSO4, and then the solvent was evaporated. The crude product was heated for 2 h with 5 M NaOH (50 mL) at 100 °C. The reaction mixture was washed with CH3Cl and neutralized with concentrated HCl. The resulting white precipitate was collected by filtration. Yield: 71%, Rf: 0.67 (EtOAc), m.p. 148–151 °C. 1H NMR (DMSO-d6) δ 12.63 (bs, 1H, COOH), 7.88 (d, J = 8.9 Hz, 2H, Ar-H2,6), 7.01 (d, J = 8.9 Hz, 2H, Ar-H3,5), 4.18–4.13 (m, 2H, ArOCH2), 3.69–3.64 (m, 2H, -CH2OCH3), 3.30 (s, 3H, -OCH3). 13C NMR (DMSO-d6) δ 166.8, 161.9, 131.2, 122.9, 114.2, 70.1, 67.1, 58.1.
4.2.5.2. (Oxiran-2-yl)methyl 4-methoxyethoxybenzoate (14b)
A mixture of acid 14a (0.028 mol) in methanol (75 mL) and KOH (2.2 g, 0.042 mol) in i-PrOH (50 mL) was stirred for 1 h at ambient temperature, and after that, i-PrOH (175 mL) was added for the final ratio MeOH/i-PrOH 1:3. The resulting white precipitate was collected by filtration and dried under low pressure. This potassium salt with (oxiran-2-yl)methyl 4-methylbenzensulfonate [60] (3.5 g, 0.015 mol) in DMF (50 mL) was heated for 7 h at 70 °C. The solvent was removed under reduced pressure, and the residue was dissolved in EtOAc and washed with water. The organic layer was dried over anhydrous MgSO4, and solvent removed under reduced pressure and a crude compound was purified by column flash chromatography. Yield: 69%, Rf: 0.60 (EtOAc/petroleum ether 1:1). 1H NMR (DMSO-d6) δ 7.92 (d, J = 8.9 Hz, 2H, Ar-H2,6), 7.07 (d, J = 8.9 Hz, 2H, Ar-H3,5), 4.60 (dd, J = 12.4, 2.7 Hz, 1H, COOCH2), 4.20–4.16 (m, 2H, ArOCH2), 4.04 (dd, J = 12.04, 6.4 Hz, 1H, COOCH2), 3.69–3.65 (m, 2H, CH2OCH3), 3.34–3.30 (m, 4H, -OCH3 + CH-oxirane), 2.86–2.81 (m, 1H, CH2-oxirane), 2.72 (dd, J = 5.0, 2.6 Hz, 1H, CH2-oxirane). 13C NMR (DMSO-d6) δ 165.0, 162.5, 131.2, 121.5, 114.4, 70.1, 67.2, 64.9, 58.1, 48.9, 43.8.
4.2.5.3. [2-(2,6-Dimethoxyfenoxy)ethyl]{2-hydroxy-3-[4-(2-methoxyethoxy)benzoyloxy]propyl} ammonium fumarate (14)
Oxirane 14b (0.004 mol) was added to the solution of the corresponding phenoxyethylamine (0.004 mol) in i-PrOH (15 mL). The reaction mixture was heated at 80 °C for 1 h and stirred then for 72 h at ambient temperature. Afterwards, the reaction mixture was cooled for at least 48 h at −18 °C. The precipitate was filtered off and dissolved in Et2O and transformed to its fumaric salt by addition of an excess of a saturated solution of fumaric acid in Et2O. This new precipitate was filtered off and recrystallized from i-PrOH, if necessary. Yield: 49%, Rf: 0.49 (MeOH), m.p. 118–121 °C. 1H NMR(DMSO-d6) δ 7.95 (d, 3J = 8.6 Hz, 2H, Ar-H2,6), 7.04–7.01 (m, 3H, OAr-H3,5 + NAr-H4), 6.67 (d, 3J = 8.4 Hz, 2H, NAr-H3,5), 6.50 (s, 2H,fumarate), 4.22–4.04 (m, 7H, −CH2OAr + −COOCH2CH− + −NH2+CH2CH2O−), 3.77 (s, 6H, ArOCH3), 3.68–3.67 (m, 2H,-CH2CH2OAr), 3.31 (s, 3H, -OCH3), 3.07–2.92 (m, 4H, CH2NH2+CH2−). 13C NMR (DMSO-d6) δ 167.6, 165.3, 162.4, 153.1, 135.7, 134.9, 131.4, 124.0, 121.9, 114.3, 105.4, 70.1, 69.9, 67.2, 66.5, 65.9, 58.2, 55.8, 50.6, 47.7. HR-MS (Orbitrap): C25H33NO10 [M – H]- calculated 506.2043 m/z, found 506.2044 m/z.
4.2.6. Synthesis of compound 15
4.2.6.1. Methyl 2-(2-methoxyethoxy)benzoate (15a)
1-chloro-2-methoxyethane (0.48 mol) was added to the mixture of methyl 2-hydroxybenzoate (0.40 mol), K2CO3 (0.80 mol), KI (0.04 mol) and DMF (150 mL). The mixture was heated for 6 h at 150 °C. The solution was cooled and filtered, and the solvent was evaporated under reduced pressure. The residue was dissolved in CHCl3 and extracted with water and 10% NaOH. The organic phase was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. Yield: 96%, RF: 0.78 (EtOAc/ petroleum ether 1:1).
4.2.6.2. 2-(2-Methoxyethoxy)benzoic acid (15b)
Ester 15a (0.27 mol) was heated for one hour with an excess of 10% NaOH (0.58 mol). After cooling down to room temperature, the reaction mixture was neutralized with HCl and extracted into CHCl3. The organic phase was dried over anhydrous MgSO4, and solvent removed under reduced pressure. The crude product was purified by fractional vacuum distillation. Yield: 93%, Rf: 0.52 (acetone/petroleum ether 1:1), b.p. (3–6 torr) 170–171 °C. 1H NMR(DMSO-d6) δ 10.94 (bs, 1H, -COOH), 8.18–8.13 (m, 1H, -H6), 7.59–7.51 (m, 1H, -H4), 7.18–7.04 (m, 2H, -H3 + -H5), 4.39–4.35 (m, 2H, ArOCH2-), 3.83–3.79 (m, 2H, -CH2OMe), 3.46 (s,3H, -OMe). 13C NMR (DMSO-d6) δ 165.4, 157.4, 134.7, 133.6, 122.5, 118.7, 113.5, 69.9, 69.3, 59.0.
4.2.6.3. (2R)-oxiran-2-ylmethyl 2-(2-methoxyethoxy)benzoate (15c)
2-(2-methoxyethoxy)benzoyl chloride (0.028 mol) was dissolved in dry CH2Cl2 and was added to the mixture of 4-dimethylaminopyridine (DMAP, 0.028 mol), (S)-(–)-glycidol (0.028 mol) in anhydrous CH2Cl2 (20 mL) under an inert atmosphere. The solution was stirred at ambient temperature for 3 h. The reaction mixture was then filtered and the filtrate washed with 10% HCl. The organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure. The crude compound was purified by column flash chromatography. Yield: 88%, Rf: 0.63 (acetone/CH2Cl2 1:20), [α]D – 14.82. 1H NMR(DMSO-d6) δ 7.87–7.82 (m, 1H, -H6), 7.51–7.42 (m, 1H, -H4), 7.04–6.95 (m, 2H, -H3 + -H5), 4,64–4.57 (m, 1H, -H7), 4.22–4.13 (m, 3H, ArOCH2 + -H8), 3.84–3.79 (m, 2H, -CH2OMe), 3.46 (s, 3H, -OMe), 3.37–3.29 (m, 1H, -H9), 2.90–2.86 (m, 1H, -H10), 2.78–2.74 (m, 1H,-H11). 13C NMR (DMSO-d6) δ 165.9, 158.7, 133.62, 131.8, 120.65, 120.45, 114.0, 71.0, 68.9, 65.11, 59.2, 49.5, 44.7.
4.2.6.4. (2R)-N-tert-butyl-2-hydroxy-3-{[2-(2-methoxyethoxy)benzoyl]oxy}propan-1-aminium hemifumarate (15)
Epoxide 15c (2.8 mmol) was dissolved in i-PrOH, and an excess of tert-butylamine was added to the solution. The mixture was heated at 70 °C for 1.5 h, after that the reaction mixture was stirred at ambient temperature for 72 h. The solvent was removed under reduced pressure, and the resulting amines were converted to the fumarate salts by dissolving the amine bases in diethylether and mixing with a solution of fumaric acid in diethylether. Crystalline products were filtered and recrystallized from i-PrOH. Yield: 57%, Rf: 0.43 (EtOAc/Et2NH 10:1), [α]D 14.64, m.p. 123–125 °C. 1H NMR (DMSO-d6) δ 7.74–7.69 (m, 1H, -H6), 7.57–7.48 (m, 1H, -H4), 7.18–7.14 (m, 1H, -H3), 7.07–6.99 (m, 1H, -H5), 6.42 (s, 2H, CH=CHfumarate), 4.21–4.05 (m, 5H, Ar-OCH2 + COOCH2CH(OH)-), 3.69–3.64 (m, 2H, -CH2OMe), 3.31 (s, 3H, -OMe), 3.06–2.75 (m, 2H, -CH2NH-), 1,24 (s, 9H, -CH(CH3)3). 13C NMR (DMSO-d6) δ 168.9, 165.4, 157.55, 135.4, 133.4, 130.8, 120.4, 120.3, 114.0, 70.2, 68.2, 66.4, 65.45, 58.3, 54.3, 44.2, 25.65. HR-MS (Orbitrap): C21H32NO9 [M – H]- calculated 440.1926 m/z, found 440.1934 m/z.
4.3. SphK1 inhibition assays
Putative inhibitors were evaluated with fluorescence SphK assays in 384-well plate format as described [63]. Briefly, compounds were dissolved in DMSO and initially screened at 650 μM; those showing inhibition were further characterized to obtain IC50s. Assays contained 100 nM recombinant SphK1, 30 mM Tris-HCl [pH 7.4], 0.05% Triton X-100,150 mM NaCl, 10% glycerol, 0.05% triton X-100, and 1% DMSO. All reactions were prepared as master mixes, dispensed into 384-well polypropylene plates (Greiner Bio-One, Frickenhausen, Germany), and allowed to pre-equilibrate at 37 °C for 10 min. Reactions were initiated with 20 × ATP-Mg (20 mM ATP, 200 mM MgCl2, 900 mM Tris-HCl, pH 7.4), and were followed in a TECAN Infinite M1000 fluorescence plate reader (Mannedorf, Switzerland) at 37 °C. Excitation wavelength was 550 nm and emission wavelength was 584 nm with a 5 nm band-pass. All data were analyzed using Prism (GraphPad, La Jolla, USA).
4.4. Molecular modelling
4.4.1. Virtual screening
Receptor preparation and docking setup were carried out with AutoDock Tools [64]. All operations involving format conversion, filtering, and manipulation of ligand molecules (including ligand preparation for docking) were performed with OpenBabel [48].
Docking calculations were all performed with Autodock Vina [46]. ROC curves were constructed by using the ROCR package [75] from R software [76]. The logistic regression model to filter out the “non X-ray like” poses from the ensemble of docking poses was constructed within Octave scientific programming environment [77].
4.4.2. Molecular docking
AutoDock4 [64] was used to dock each compound to the SphK1 active site using a Lamarckian genetic algorithm with pseudo-Solis and Wets local search [78]. The following parameters were used: the initial population of trial ligands was constituted by 150 individuals; the maximum number of generations was set to 2.7 × 104. The maximum number of energy evaluations was 25.0 × 106. For each docking job, 100 conformations were generated. All other run parameters were maintained at their default setting. The resulting docked conformations were clustered into families by considering the backbone rmsd. The lowest docking-energy conformation was considered the most favorable orientation [79].
4.4.3. MD simulations
The complex geometries from docking were soaked in boxes of explicit water using the TIP3P model [80] and subjected to MD simulation. All MD simulations were performed with the Amber software package [65] using periodic boundary conditions and cubic simulation cells. The particle mesh Ewald method (PME) [81] was applied using a grid spacing of 1.2 Å, a spline interpolation order of 4 and a real space direct sum cutoff of 10 Å. The SHAKE algorithm was applied allowing for an integration time step of 2 fs. MD simulations were carried out at 310 K temperature. Three MD simulations of 50 ns were conducted for each system under different starting velocity distribution functions; thus, in total 150 ns were simulated for each complex. The NPT ensemble was employed using Berendsen coupling to a baro/thermostat (target pressure 1 atm, relaxation time 0.1 ps). Post MD analysis was carried out with program PTRAJ.
4.4.4. Quantum calculations setup
Reduced 3D model systems including the tested compound and the interacting residues from Sphk1 binding pocket were constructed from the MD simulation. In this work, we identified the binding site residues of the receptors by using the free energy decomposition approach (MM/GBSA). The side chains of the binding site residues that contributed with a |ΔG| higher than 1.0 kcal/ mol in the per residue energy decomposition together with each inhibitor were included in the reduced model.
4.4.5. Atoms in molecules theory
The reduced models were used as input for quantum theory of atoms in molecules (QTAIM) analysis [82], which was performed with the help of Multiwfn software [83]. The wave function used as input for these calculations were computed with the Gaussian 09 package [84] by employing the B3LYP functional with dispersion correction (B3LYP-D) and 6-31G(d) as basis set. The empirical dispersion correction for the B3LYP functional was applied by invoking the IOp 3/124 = 3 keyword in Gaussian 09. This type of calculations have been used in recent works because it ensures a reasonable compromise between the wave function quality required to obtain reliable values of the derivatives of ρ(r) and the computer power available, due to the extension of the system in study [85,86].
4.5. Additional materials
FITC-annexin and propidium iodide staining solution were from BD Bioscience (San Jose, CA). Unless stated, all other reagents were from Sigma-Aldrich.
Supplementary Material
Acknowledgments
Grants from Universidad Nacional de San Luis: Número (UNSL): PROICO 2-1214 partially supported this work. Marcela Vettorazzi thanks a doctoral fellowship of CONICET-Argentina. This work was supported in part by NIH grants (K22 CA187314-01 to S.L and R01GM043880 to S.S), by IGAVFU Brno 315/2015/FaF, 323/2016/FaF, 323/2017/FaF, by the institutional support RVO: 68081715 of the Institute of Analytical Chemistry of the CAS, v.v.i., by the Slovak Research and Development Agency (Grant No. APVV-0516-12), by Colombian Institute for Science and Research (COLCIENCIAS, Grant No. 1102-658-42651) and by Universidad de Jaén. This publication utilizes also research results of the CEBV project (ITMS 26240120034). Technical and human support provided by CICT of Universidad de Jaén (UJA, MINECO, Junta de Andalucía, FEDER) is gratefully acknowledged. This study was also supported by SANOFI-AVENTIS Pharma Slovakia, s.r.o.
Footnotes
Appendix A. Supplementary data: Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2017.08.017.
References
- 1.Blaho VA, Hla T. Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem Rev. 2011;111:6299–6320. doi: 10.1021/cr200273u. http://dx.doi.org/10.1021/cr200273u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. http://dx.doi.org/10.1038/nrm2329 [DOI] [PubMed] [Google Scholar]
- 3.Ponnusamy S, Meyers-Needham M, Senkal CE, Saddoughi SA, Sentelle D, Selvam SP, Salas A, Ogretmen B. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010;6:1603–1624. doi: 10.2217/fon.10.116. http://dx.doi.org/10.2217/fon.10.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–415. doi: 10.1038/nri2974. http://dx.doi.org/10.1038/nri2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. doi: 10.1038/nature13475. http://dx.doi.org/10.1038/nature13475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyne S, Pyne NJ. Translational aspects of sphingosine 1-phosphate biology. Trends Mol Med. 2011;17:463–472. doi: 10.1016/j.molmed.2011.03.002. http://dx.doi.org/10.1016/j.molmed.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 7.Pchejetski D, Bohler T, Stebbing J, Waxman J. Therapeutic potential of targeting sphingosine kinase 1 in prostate cancer. Nat Rev Urol. 2011;8:569–678. doi: 10.1038/nrurol.2011.117. http://dx.doi.org/10.1038/nrurol.2011.117 [DOI] [PubMed] [Google Scholar]
- 8.Heffernan-Stroud LA, Obeid LM. Sphingosine kinase 1 in cancer. Adv Cancer Res. 2013;117:201–235. doi: 10.1016/B978-0-12-394274-6.00007-8. http://dx.doi.org/10.1016/B978-0-12-394274-6.00007-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez YI, Campos LE, Castro MG, Aladhami A, Oskeritzian CA, Alvarez SE. Sphingosine-1 phosphate: a new modulator of immune plasticity in the tumor microenvironment. Front Oncol. 2016;6:218. doi: 10.3389/fonc.2016.00218. http://dx.doi.org/10.3389/fonc.2016.00218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gault CR, Obeid LM. Still benched on its way to the bedside: sphingosine kinase 1 as an emerging target in cancer chemotherapy. Crit Rev Biochem Mol Biol. 2011;46:342–351. doi: 10.3109/10409238.2011.597737. http://dx.doi.org/10.3109/10409238.2011.597737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pitman MR, Pitson SM. Inhibitors of the sphingosine kinase pathway as potential therapeutics. Curr Cancer Drug Targets. 2010;10:354–367. doi: 10.2174/156800910791208599. http://dx.doi.org/10.2174/1568210203706850096 [DOI] [PubMed] [Google Scholar]
- 12.Pyne NJ, Tonelli F, Lim KG, Long J, Edwards J, Pyne S. Targeting sphingo-sine kinase 1 in cancer. Adv Biol Regul. 2012;52:31–38. doi: 10.1016/j.advenzreg.2011.07.001. http://dx.doi.org/10.1016/j.advenzreg.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 13.Pyne NJ, Tonelli F, Lim KG, Long JS, Edwards J, Pyne S. Sphingosine 1-phosphate signalling in cancer. Biochem Soc Trans. 2012;40:94–100. doi: 10.1042/BST20110602. http://dx.doi.org/10.1042/BST20110602 [DOI] [PubMed] [Google Scholar]
- 14.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. http://dx.doi.org/10.1038/nrc2875 [DOI] [PubMed] [Google Scholar]
- 15.Long JS, Edwards J, Watson C, Tovey S, Mair KM, Schiff R, Natarajan V, Pyne NJ, Pyne S. Sphingosine kinase 1 induces tolerance to human epidermal growth factor receptor 2 and prevents formation of a migratory phenotype in response to sphingosine 1-phosphate in estrogen receptor-positive breast cancer cells. Mol Cell Biol. 2010;30:3827–3841. doi: 10.1128/MCB.01133-09. http://dx.doi.org/10.1128/MCB.01133-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antoon JW, Beckman BS. Sphingosine kinase: a promising cancer therapeutic target. Cancer Biol Ther. 2011;11:647–650. doi: 10.4161/cbt.11.7.14921. [DOI] [PubMed] [Google Scholar]
- 17.Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, Hannun YA, Obeid LM. Role for sphingosine kinase 1 in colon carcinogen-esis. FASEB J Off Publ Fed Am Soc Exp Biol. 2009;23:405–414. doi: 10.1096/fj.08-117572. http://dx.doi.org/10.1096/fj.08-117572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weigert A, Schiffmann S, Sekar D, Ley S, Menrad H, Werno C, Grosch S, Geisslinger G, Brune B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int J Cancer. 2009;125:2114–2121. doi: 10.1002/ijc.24594. http://dx.doi.org/10.1002/ijc.24594 [DOI] [PubMed] [Google Scholar]
- 19.Shirai K, Kaneshiro T, Wada M, Furuya H, Bielawski J, Hannun YA, Obeid LM, Ogretmen B, Kawamori T. A role of sphingosine kinase 1 in head and neck carcinogenesis. Cancer Prev Res (Phila) 2011;4:454–462. doi: 10.1158/1940-6207.CAPR-10-0299. http://dx.doi.org/10.1158/1940-6207.CAPR-10-0299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao M, Chen Z, Xu Y, Zhao Y, Zha R, Huang S, Liu L, Chen T, Li J, Tu H, He X. Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver Int. 2012;32:331–338. doi: 10.1111/j.1478-3231.2011.02666.x. http://dx.doi.org/10.1111/j.1478-3231.2011.02666.x [DOI] [PubMed] [Google Scholar]
- 21.Heffernan-Stroud LA, Helke KL, Jenkins RW, De Costa AM, Hannun YA, Obeid LM. Defining a role for sphingosine kinase 1 in p53-dependent tumors. Oncogene. 2012;31:1166–1175. doi: 10.1038/onc.2011.302. http://dx.doi.org/10.1038/onc.2011.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albinet V, Bats ML, Huwiler A, Rochaix P, Chevreau C, Segui B, Levade T, Andrieu-Abadie N. Dual role of sphingosine kinase-1 in promoting the differentiation of dermal fibroblasts and the dissemination of melanoma cells. Oncogene. 2014;33:3364–3373. doi: 10.1038/onc.2013.303. http://dx.doi.org/10.1038/onc.2013.303 [DOI] [PubMed] [Google Scholar]
- 23.Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM, Shao Y, Sabbadini R, Ogretmen B. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol Med. 2012;4:761–775. doi: 10.1002/emmm.201200244. http://dx.doi.org/10.1002/emmm.201200244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, Ai Y, Ristimaki AP, Fyrst H, Sano H, Rosenberg D, Saba JD, Proia RL, Hla T. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26:7211–7223. doi: 10.1128/MCB.02341-05. http://dx.doi.org/10.1128/MCB.02341-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truman JP, Garcia-Barros M, Obeid LM, Hannun YA. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim Biophys Acta. 2014;1841:1174–1188. doi: 10.1016/j.bbalip.2013.12.013. http://dx.doi.org/10.1016/j.bbalip.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orr Gandy KA, Obeid LM. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway in disease: review of sphingosine kinase inhibitors. Bio-chim Biophys Acta. 2013;1831:157–166. doi: 10.1016/j.bbalip.2012.07.002. http://dx.doi.org/10.1016/j.bbalip.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. http://dx.doi.org/10.1124/pr.107.07113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Min X, Xiao SH, Johnstone S, Romanow W, Meininger D, Xu H, Liu J, Dai J, An S, Thibault S, Walker N. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure. 2013;21:798–809. doi: 10.1016/j.str.2013.02.025. http://dx.doi.org/10.1016/j.str.2013.02.025 [DOI] [PubMed] [Google Scholar]
- 29.Gustin DJ, Li Y, Brown ML, Min X, Schmitt MJ, Wanska M, Wang X, Connors R, Johnstone S, Cardozo M, Cheng AC, Jeffries S, Franks B, Li S, Shen S, Wong M, Wesche H, Xu G, Carlson TJ, Plant M, Morgenstern K, Rex K, Schmitt J, Coxon A, Walker N, Kayser F, Wang Z. Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg Med Chem Lett. 2013;23:4608–4616. doi: 10.1016/j.bmcl.2013.06.030. http://dx.doi.org/10.1016/j.bmcl.2013.06.030 [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Knapp S, Pyne NJ, Pyne S, Elkins JM. Crystal structure of sphingosine kinase 1 with PF-543. ACS Med Chem Lett. 2014;5:1329–1333. doi: 10.1021/ml5004074. http://dx.doi.org/10.1021/ml5004074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, Spiegel S. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. http://dx.doi.org/10.1182/blood-2008-02-138958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 33.Mathews TP, Kennedy AJ, Kharel Y, Kennedy PC, Nicoara O, Sunkara M, Morris AJ, Wamhoff BR, Lynch KR, MacDonald TL. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem. 2010;53:2766–2778. doi: 10.1021/jm901860h. http://dx.doi.org/10.1021/jm901860h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiang Y, Hirth B, Kane JLJ, Liao J, Noson KD, Yee C, Asmussen G, Fitzgerald M, Klaus C, Booker M. Discovery of novel sphingosine kinase-1 inhibitors Part 2. Bioorg Med Chem Lett. 2010;20:4550–4554. doi: 10.1016/j.bmcl.2010.06.019. http://dx.doi.org/10.1016/j.bmcl.2010.06.019 [DOI] [PubMed] [Google Scholar]
- 35.Baek DJ, MacRitchie N, Pyne NJ, Pyne S, Bittman R. Synthesis of selective inhibitors of sphingosine kinase 1. Chem Commun. 2013;49:2136–2138. doi: 10.1039/c3cc00181d. http://dx.doi.org/10.1039/C3CC00181D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schnute ME, McReynolds MD, Kasten T, Yates M, Jerome G, Rains JW, Hall T, Chrencik J, Kraus M, Cronin CN, Saabye M, Highkin MK, Broadus R, Ogawa S, Cukyne K, Zawadzke LE, Peterkin V, Iyanar K, Scholten JA, Wendling J, Fujiwara H, Nemirovskiy O, Wittwer AJ, Nagiec MM. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J. 2012;444:79–88. doi: 10.1042/BJ20111929. http://dx.doi.org/10.1042/BJ20111929 [DOI] [PubMed] [Google Scholar]
- 37.Patwardhan NN, Morris EA, Kharel Y, Raje MR, Gao M, Tomsig JL, Lynch KR, Santos WL. Structure–Activity relationship studies and in vivo activity of guanidine-based sphingosine kinase inhibitors: discovery of SphK1- and SphK2-selective inhibitors. J Med Chem. 2015;58:1879–1899. doi: 10.1021/jm501760d. http://dx.doi.org/10.1021/jm501760d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitman MR, Costabile M, Pitson SM. Recent advances in the development of sphingosine kinase inhibitors. Cell Signal. 2016;28:1349–1363. doi: 10.1016/j.cellsig.2016.06.007. http://dx.doi.org/10.1016/j.cellsig.2016.06.007 [DOI] [PubMed] [Google Scholar]
- 39.Gao Y, Gao F, Chen K, Tian M, Zhao D. Sphingosine kinase 1 as an anticancer therapeutic target. Drug Des devel Ther. 2015;9:3239–3245. doi: 10.2147/DDDT.S83288. http://dx.doi.org/10.2147/DDDT.S83288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newton J, Lima S, Maceyka M, Spiegel S. Revisiting the sphingolipid rheostat: evolving concepts in cancer therapy. Exp Cell Res. 2015;333:195–200. doi: 10.1016/j.yexcr.2015.02.025. http://dx.doi.org/10.1016/j.yexcr.2015.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos WL, Lynch KR. Drugging sphingosine kinases. ACS Chem Biol. 2015;10:225–233. doi: 10.1021/cb5008426. http://dx.doi.org/10.1021/cb5008426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellecchia M. Fragment-based drug discovery takes a virtual turn. Nat Chem Biol. 2009;5:274–275. doi: 10.1038/nchembio0509-274. http://dx.doi.org/10.1038/nchembio0509-274 [DOI] [PubMed] [Google Scholar]
- 43.Zheng W, Johnson SR, Baskin I, Bajorath J, Horvath D, Laggner C, Langer T, Schneider G, Filimonov D, Poroikov V, Tetko I, Van De Waterbeemd H, Oprea T, Radchenko E, Palyulin V, Zefirov N, Peltason L, Wolber G, Schuster D, Kirchmair J, Proschak E, Tanrikulu Y, Varnek A, Tropsha A. Chemoinformatics approaches to virtual screening. R Soc Chem. 2008 http://dx.doi.org/10.1039/9781847558879
- 44.Triballeau N, Bertrand HO, Acher F. Pharmacophores and Pharmacophore Searches. Wiley-VCH Verlag GmbH & Co KGaA; 2006. Are you sure you have a good model? pp. 325–364.http://dx.doi.org/10.1002/3527609164.ch15 [Google Scholar]
- 45.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. http://dx.doi.org/10.1093/nar/gkr777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. http://dx.doi.org/10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sutherland JJ, Nandigam RK, Erickson JA, Vieth M. Lessons in molecular recognition. 2. Assessing and improving cross-docking accuracy. J Chem Inf Model. 2007;47:2293–2302. doi: 10.1021/ci700253h. http://dx.doi.org/10.1021/ci700253h [DOI] [PubMed] [Google Scholar]
- 48.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. http://dx.doi.org/10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanzel L, Malik I, Mokry P. Preliminary in vitro investigation of antioxidant potential of ultra short acting arylcarbamoyloxy-aminopropanols containing N-phenylpiperazine moiety. Dhaka Univ J Pharm Sci. 2016;15:235–239. http://dx.doi.org/10.3329/dujps.v15i2.30943 [Google Scholar]
- 50.Malík I, Janoscova M, Mokrý P, Csollei J, Andriamainty F. Basic physicochemical characterization of new potential ultrashort acting b 1-adre-noceptor blockers. Acta Fac Pharm Univ Comen. 2009;56:119–127. [Google Scholar]
- 51.Malik I, Bukovsky M, Mokrý P, Csollei J. Antimicrobial profile investigation of potential ultrashort acting beta-adrenoceptor blocking compounds containing N-phenylpiperazine moiety. Glob J Med Res. 2013;13:1–4. [Google Scholar]
- 52.Acosta-Quintero LM, Jurado J, Nogueras M, Palma A, Cobo J. Synthesis of pyrimidine-fused benzazepines from 5-Allyl-4,6-dichloropyrimidines. Eur J Org Chem. 2015;2015:5360–5369. http://dx.doi.org/10.1002/ejoc.201500632 [Google Scholar]
- 53.Tengler J, Kapustíkova I, Pesko M, Govender R, Keltosova S, Mokrý P, Kollar P, OMahony J, Coffey A, Kralova K. Jampílek, Synthesis and Biological evaluation of 2-Hydroxy-3-[(2-aryloxyethyl)amino]propyl 4-[(alkox-ycarbonyl)amino]benzoates. Sci World J. 2013;2013:274570. doi: 10.1155/2013/274570. http://dx.doi.org/10.1155/2013/274570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tengler J, Kapustíkova I, Stropnický O, Mokrý P, Oravec M, Csollei J, Jampílek J. Synthesis of new (arylcarbonyloxy)aminopropanol derivatives and the determination of their physico-chemical properties. Cent Eur J Chem. 2013;11:1757–1767. http://dx.doi.org/10.2478/s11532-013-0302-8 [Google Scholar]
- 55.Marvanova P, Padrtova T, Pekarek T, Brus J, Czernek J, Mokry P, Humpa O, Oravec M, Jampilek J. Synthesis and characterization of new 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-propoxybenzoates and their hydrochloride salts. Molecules. 2016;21 doi: 10.3390/molecules21060707. http://dx.doi.org/10.3390/molecules21060707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaster LM, Jennings AJ, Joiner GF, King FD, Mulholland KR, Rahman SK, Starr S, Wyman PA, Wardle KA. (1-Butyl-4-piperidinyl)methyl 8-amino-7-chloro-1,4-benzodioxane-5-carboxylate hydrochloride: a highly potent and selective 5-HT4 receptor antagonist derived from metoclopramide. J Med Chem. 1993;36:4121–4123. doi: 10.1021/jm00077a018. http://dx.doi.org/10.1021/jm00077a018 [DOI] [PubMed] [Google Scholar]
- 57.Lalut J, Tournier BB, Cailly T, Lecoutey C, Corvaisier S, Davis A, Ballandonne C, Since M, Millet P, Fabis F, Dallemagne P, Rochais C. Synthesis and evaluation of novel serotonin 4 receptor radiotracers for singlephoton emission computed tomography. Eur J Med Chem. 2016;116:90–101. doi: 10.1016/j.ejmech.2016.03.059. http://dx.doi.org/10.1016/j.ejmech.2016.03.059 [DOI] [PubMed] [Google Scholar]
- 58.Kettmann V, Csollei J, Račanská E, Švec P. Synthesis and structure-activity relationships of new β-adrenoreceptor antagonists. Evidence for the electrostatic requirements for β-adrenoreceptor antagonists. Eur J Med Chem. 1991;26:843–851. http://dx.doi.org/10.1016/0223-5234(91)90127-9 [Google Scholar]
- 59.Koelsch ST, Rolfson CF. Synthesis of certain 3,4-disubstituted piperidines. J Amer Chem Soc. 1950;72:1871–1873. [Google Scholar]
- 60.Ammazzalorso A, Amoroso R, Bettoni G, Fantacuzzi M, De Filippis B, Giampietro L, Maccallini C, Paludi D, Tricca ML. Synthesis and antibacterial evaluation of oxazolidin-2-ones structurally related to linezolid. Farmaco. 2004;59:685–690. doi: 10.1016/j.farmac.2004.05.002. http://dx.doi.org/10.1016/j.farmac.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 61.Dewar DR, Kapur GH, Mottram H. Some potential α-adrenoreceptor blocking 1,4-benzodioxanes and 2,6-dimethoxyphenoxyethylamines. Eur J Med Chem. 1983;18:286–290. [Google Scholar]
- 62.Marvanova P, Padrtova T, Odehnalova K, Hosik O, Oravec M, Mokry P. Synthesis and determination of physicochemical properties of new 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-alkoxyethoxybenzoates. Molecules. 2016;21 doi: 10.3390/molecules21121682. http://dx.doi.org/10.3390/molecules21121682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lima S, Milstien S, Spiegel S. A real-time high-throughput fluorescence assay for sphingosine kinases. J Lipid Res. 2014;55:1525–1530. doi: 10.1194/jlr.D048132. http://dx.doi.org/10.1194/jlr.D048132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. http://dx.doi.org/10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12 OR. University of California; San Francisco: 2012. citeulike-article-id:10779586. [Google Scholar]
- 66.Andújar SA, Tosso RD, Suvire FD, Angelina E, Peruchena N, Cabedo N, Cortés D, Enriz RD. Searching the “biologically relevant” conformation of dopamine: a computational approach. J Chem Inf Model. 2012;52:99–112. doi: 10.1021/ci2004225. http://dx.doi.org/10.1021/ci2004225 [DOI] [PubMed] [Google Scholar]
- 67.Tosso RD, Andújar SA, Gutierrez L, Angelina E, Rodríguez R, Nogueras M, Baldoni H, Suvire FD, Cobo J, Enriz RD. Molecular modeling study of dihydrofolate reductase inhibitors. Molecular dynamics simulations, quantum mechanical calculations, and experimental corroboration. J Chem Inf Model. 2013;53:2018–2032. doi: 10.1021/ci400178h. http://dx.doi.org/10.1021/ci400178h [DOI] [PubMed] [Google Scholar]
- 68.Parraga J, Cabedo N, Andújar S, Piqueras L, Moreno L, Galan A, Angelina E, Enriz RD, Ivorra MD, Sanz MJ, Cortes D. 2,3,9- and 2,3,11-trisubstituted tetrahydroprotoberberines as D2 dopaminergic ligands. Eur J Med Chem. 2013;68:150–166. doi: 10.1016/j.ejmech.2013.07.036. http://dx.doi.org/10.1016/j.ejmech.2013.07.036 [DOI] [PubMed] [Google Scholar]
- 69.Angelina EL, Andújar SA, Tosso RD, Enriz RD, Peruchena NM. Non-covalent interactions in receptor–ligand complexes. A study based on the electron charge density. J Phys Org Chem. 2014;27:128–134. http://dx.doi.org/10.1002/poc.3250 [Google Scholar]
- 70.Parraga J, Andújar SA, Rojas S, Gutié LJ, El Aouad N, Sanz MJ, Enriz RD, Cabedo N, Cortes D. Dopaminergic isoquinolines with hexahydrocyclopenta[ij]-isoquinolines as D2-like selective ligands. Eur J Med Chem. 2016;122:27–42. doi: 10.1016/j.ejmech.2016.06.009. http://dx.doi.org/10.1016/j.ejmech.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 71.Adams DR, Pyne S, Pyne NJ. Sphingosine kinases: emerging structure-function insights. Trends biochem Sci. 2016;41:395–409. doi: 10.1016/j.tibs.2016.02.007. http://dx.doi.org/10.1016/j.tibs.2016.02.007 [DOI] [PubMed] [Google Scholar]
- 72.Pulkoski-Gross MJ, Donaldson JC, Obeid LM. Sphingosine-1-phosphate metabolism: a structural perspective. Crit Rev Biochem Mol Biol. 2015;50:298–313. doi: 10.3109/10409238.2015.1039115. http://dx.doi.org/10.3109/10409238.2015.1039115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parthasarathi R, Subramanian V, Sathyamurthy N. Hydrogen bonding without Borders: an atoms-in-molecules perspective. J Phys Chem A. 2006;110:3349–3351. doi: 10.1021/jp060571z. http://dx.doi.org/10.1021/jp060571z [DOI] [PubMed] [Google Scholar]
- 74.Witek S, Bielawski J, Bielawska A. Synthesis of N-(Formylphenyl)- and N-(Acetophenyl) derivatives of urea and carbamic acid. J Für Prakt Chem. 1979;321:804–812. http://dx.doi.org/10.1002/prac.19793210512 [Google Scholar]
- 75.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. http://dx.doi.org/10.1093/bioinformatics/bti623 [DOI] [PubMed] [Google Scholar]
- 76.R Development Core Team. R Found Stat Comput. 2011. R: a language and environment for statistical computing. http://www.r-project.org/
- 77.John RW, Eaton W. David Bateman, Søren Hauberg, GNU Octave version 3.8.1 manual: a high-level interactive language for numerical computations. Creat Indep Publ Platf. 2014 Item 26.4 http://www.gnu.org/software/octave/doc/interpreter/
- 78.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. http://dx.doi.org/10.1002/(SICI)1096-987X(19981115)19 , 14<1639∷AID-JCC10>3.0.CO;2-B. [Google Scholar]
- 79.Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem. 1997;18:1175–1189. http://dx.doi.org/10.1002/(SICI)1096-987X(19970715)18 , 9<1175∷AID-JCC6>3.0.CO;2-O. [Google Scholar]
- 80.Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/e water models at 298 K. J Phys Chem A. 2001;105:9954–9960. http://dx.doi.org/10.1021/jp003020w [Google Scholar]
- 81.Darden T, York D, Pedersen L. Particle mesh Ewald: an N,log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98 [Google Scholar]
- 82.Bader RFW. Atoms in molecules. Acc Chem Res. 1985;18:9–15. http://dx.doi.org/10.1021/ar00109a003 [Google Scholar]
- 83.Lu T, Chen F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. http://dx.doi.org/10.1002/jcc.22885 [DOI] [PubMed] [Google Scholar]
- 84.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Barone GW, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Li MW, Li X, Hratchian HP, Izmaylov AF, JA, JEP, FO, MB, JJH, EB, KNK, VNS, RK, JN, KR, AR, JCB, SSI, JT, MC . Gaussian 09. Vol. 2009 Gaussian Inc; Wallingford CT: 2009. [Google Scholar]
- 85.Barrera Guisasola EE, Gutierrez LJ, Andujar SA, Angelina E, Rodriguez AM, Enriz RD. Pentameric models as alternative molecular targets for the design of new antiaggregant agents. Curr Protein Pept Sci. 2016;17:156–168. doi: 10.2174/1389203716666151102104926. [DOI] [PubMed] [Google Scholar]
- 86.Luchi AM, Angelina EL, Andujar SA, Enriz RD, Peruchena NM. Halogen bonding in biological context: a computational study of D2 dopamine receptor. J Phys Org Chem. 2016;29:645–655. http://dx.doi.org/10.1002/poc.3586 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.