

ABSTRACT
Bispecific antibody therapeutics can expand the functionality of a conventional monoclonal antibody drug because they can bind multiple antigens. However, their great potential is counterbalanced by the challenges faced in their production. The classic asymmetric bispecific containing an Fc requires the expression of four unique chains – two light chains and two heavy chains; each light chain must pair with its correct heavy chain, which then must heterodimerize to form the full bispecific. The light-chain pairing problem has several solutions, some of which require engineering and optimization for each bispecific pair. Here, we introduce a technology called EFab Domain Substitution, which replaces the Cϵ2 of IgE for one of the CL/CH1 domains into one arm of an asymmetric bispecific to encourage the correct pairing of the light chains. EFab Domain Substitution provides very robust correct pairing while maintaining antibody function and is effective for many variable domains. We report its effect on the biophysical properties of an antibody and the crystal structure of the EFab domain substituted into the adalimumab Fab (PDB ID 6CR1).
Keywords: Antibody engineering, asymmetric bispecific, Fab, domain substitution, IgE, stability, binding, heterodimerization
Introduction
There has been an increasing interest in the use of bispecific antibodies (bsAbs) as biologic drugs recently because they have the potential to harness novel mechanisms of action that cannot be achieved with a combination of two conventional monospecific antibodies.1,2 Therefore, efficient methods of generating bispecific antibodies are being pursued. Initial attempts to produce bsAbs as protein therapeutics involved chemical conjugation of monospecific antibodies and fusion of monoclonal antibody (mAb)-expressing cells,3,4 but low efficiency and the necessity of purification from abundant side products limited the widespread use of these strategies. Advancements in protein engineering and molecular biology have enabled the generation of a variety of new bsAb formats, and more than 60 such formats have been described in the literature.1,2 However, the altered biochemical/biophysical properties, serum half-life, and/or stability of these engineered bsAb formats can sometimes render them unsuitable as a therapeutic. Thus, new technologies for the generation of bsAbs are still being developed.
Two problems must be solved to efficiently express a bsAb in the form of an asymmetric IgG composed of 4 different chains: 1) the two heavy chains must form a heterodimer, and 2) each heavy chain must pair with its cognate light chain. The first solution to heavy chain heterodimerization was developed in the 1990s (“knobs-into-holes”), and several other solutions have been published since.5–7 Solutions to the light chain pairing problem, on the other hand, are not as well advanced and may require individual optimization.8,9 In order to drive correct assembly of LC:HC pairs in a bispecific antibody, the interface between the chains might be engineered so that steric clashes or repelling charges prevent incorrect assembly.10,11 The light chain of an antibody makes contact with the heavy chain in the variable and the constant domain and both contacts, VL:VH and CL:CH1, contribute to recognition and engagement. Consequently, point mutations in the constant domains are not sufficient to steer each light chain towards correct pairing, and further engineering of the variable domains is necessary.11 Therefore, we sought an alternative strategy to enable correct pairing.
The variable domains of an antibody can maintain their binding properties in the absence of constant regions, for example as a single-chain variable fragment (scFv),12 or when they are fused to heterologous peptides. For example, the variable domains of an IgG can be fused onto the constant domains of the T-cell receptor (TCR) and retain binding affinity in this chimeric construct.13,14 The substitution of the CH1 and CL domains with the constant domains of the TCR alpha and beta chain enables the expression of a functional bispecific antibody.8 This substitution is possible because Ig-fold domains share a similar geometry, which enables the variable domains to pair correctly and form a functional Fv when grafted onto the TCR constant domains. An additional example of domain substitution involves the exchange of the variable heavy and light domains within an antigen-binding fragment (Fab), which has been used in the Crossmab bispecific format to control chain pairing.15 Thus, we hypothesized that other Ig-fold domains may be used in place of CH1/CL as a strategy to prevent mispairing of light chains in a bispecific antibody.
The CH2 domain of IgE (Cɛ2) was identified as a candidate for domain substitution to solve the light chain pairing problem. This domain serves as the homodimerization interface between IgE heavy chains and is linked by two interchain disulfides. Thus, unlike the CH2 domain of IgG, the Cɛ2 domain is not involved in effector function and makes no contact with the FcɛRIα.16 Interestingly, the homodimers of IgM or IgE CH2 domains have been used as homodimerization scaffolds for fusion proteins.17 However, the use of Cɛ2 as a heterodimerization platform has not been reported. Here, we describe a novel solution called EFab domain substitution that is based on fusion of the variable domains of an antibody to the Cɛ2 domain of the IgE Fc. This format maintains antigen binding without the requirement of additional engineering of the variable domains of the bispecific antibody to robustly achieve correct pairing. To probe potential effects of fusing variable domains to Cɛ2 domains on the resulting bispecific antibodies, we investigated EFab variations with heterodimerization mutations, as well as different linker lengths and types. We also confirmed the proper assembly of an asymmetric bispecific with the EFab substitution by mass spectrometry and binding assays. Finally, we report the crystal structure of the antibody adalimumab Fab with the EFab domain substitution. In comparison with the previously reported adalimumab Fab and IgE Fc structures,18,19 the adalimumab EFab shows a high degree of similarity in the variable domains. However, clear differences in the relative domain orientation of variable domains with respect to constant domains are observed that do not appear to have a detrimental effect on target antigen binding.
Results
Generation of EFab Fab variants
We designed a hybrid Fab construct composed of the variable domains of an antibody fused to Cɛ2 domains via the native linkers from the variable domains. The VH domain was linked to the first β-strand of the Cɛ2 domain starting with Pro2 (IMGT unique numbering of C-domains) via the elbow sequence of the CH1 constant region (ASTKG) (Figure 1). The same strategy was used for fusing the VL to the Cɛ2 domain with the elbow sequence from the kappa constant region (RTVAA). The human Cɛ2 domain has one N-linked glycosylation site at Asn38 (IMGT numbering), which was mutated to Gln to prevent glycosylation of this site in the EFab. In this initial design, named EFab E0, the variable domains were grafted onto an unaltered Cɛ2 domain, except for the agly mutation, and therefore had identical heavy and light chains.
Figure 1.
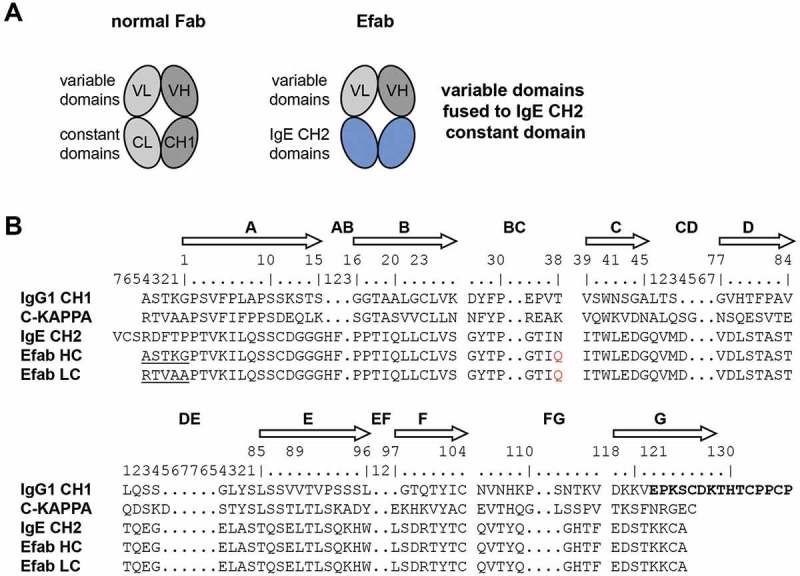
a) Cartoons of a normal Fab and an EFab, which has the CL and CH1 domains replaced by the two CH2 domains of IgE b) Sequence alignments of human IgG1 CH1 (including hinge in bold), kappa constant, IgE CH2 and EFab constant domains. IgG elbow sequences used in the EFab are underlined. IMGT constant domain numbering is shown above. The N-linked glycosylation site of IgE CH2 is mutated to Gln in EFabs (red).
However, since the Cɛ2 domain normally functions as homodimerization module for the IgE heavy chains, additional mutations were introduced into the domains to prevent the formation of HC:HC or LC:LC homodimers; these designs were named EFab E1 and E2 (Table 1). These two variants of EFab have a bulky hydrophobic residue (tryptophan or isoleucine) introduced at a position in the heavy chain that would prevent homodimerization (E1: L7W, E2: S10I), and smaller residues were introduced on the opposing light chain positions to make room for the bulky side chain (E1: L22G, E2: T121G), similar to the knobs-into-holes strategy in the CH3 domain.5
Table 1.
Heterodimerization mutations in EFab variants.
| Variant | Light Chain* | Heavy Chain* |
|---|---|---|
| E0 | None | None |
| E1 | L22G | L7W |
| E2 | T121G | S10I |
*All three versions have N36Q mutation in each chain to remove glycosylation site
To evaluate the EFab designs in controlling light chain pairing, several expression vectors were built using the two Fabs M60-A02 (anti- epidermal growth factor receptor (EGFR)) and C06 (anti-IGF1R), which have been described previously.20 The test molecules were tagged with green fluorescent protein (GFP) (30 kDa, light chain of C06) or human serum albumin (HSA) (66 kDa, heavy chain of Fab 1 or EFab, M60-A02) to enable simple differentiation of correct versus incorrect Fab pairs by migration on non-reducing SDS-PAGE (Figure 2, for reducing SDS-PAGE, see Figure S1). The proteins were generated by transient expression in Chinese hamster ovary (CHO) cells. Analysis of the expressed proteins showed that the EFab was highly efficient in controlling the light chain pairing. When the two Fabs M60-A02 and C06 were co-expressed as regular Fabs with IgG1/kappa constant domains, mispairing of the M60-A02 light chain with the C06 heavy chain was readily detected (50 kDa band in Figure 2). The bands corresponding to correct pairing and mispairing were similar in intensity, suggesting that the chain pairing may in fact occur randomly. However, when the M60-A02 was converted into an EFab, no mispairing between the light chains was seen. Similarly, when the anti-IGF1R C06 was constructed as EFab, no mispairing was detected when it was co-expressed with another Fab (anti-IGF1R G11, Figure S2). Interestingly, while the Cɛ2 domain did not strongly induce the formation of homodimers between the chains of the EFab E0 in this experiment, the design EFab E1, which has a tryptophan introduced in the heavy chain at position 7, showed a band on SDS-PAGE that migrated at the molecular weight of a heavy chain homodimer (2x HC1 in Figure 2). However, the EFab design E2 showed no such heavy chain homodimers in this experiment and was slightly less prone to aggregation (data not shown). Therefore, this E2 Cɛ2 heterodimer design (Table 1) was subsequently used to further study EFab domain substitution.
Figure 2.
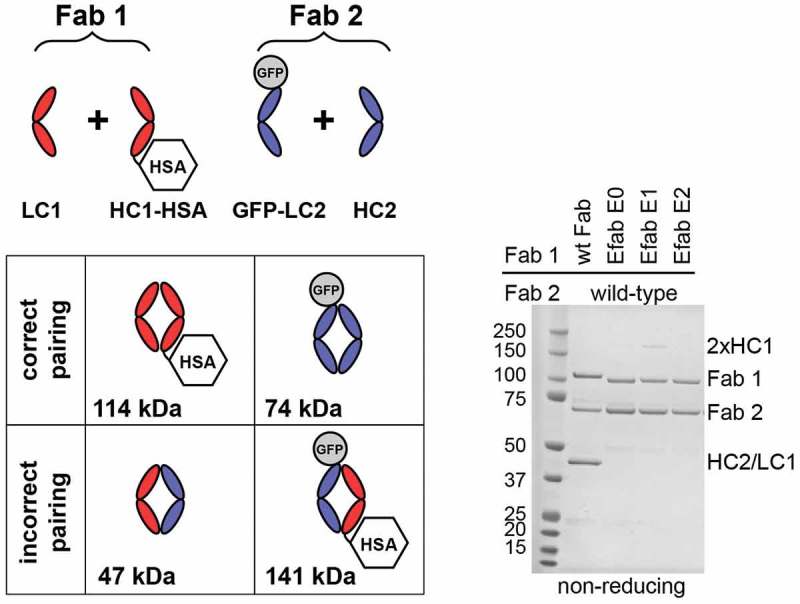
Fab fragment fusions of M60-A02 (anti-EGFR) and C06 (anti-IGF1R) were produced with HSA fused to the C-term of M60-A02 heavy chain (HC1-HSA) and GFP fused to the N-term of C06 light chain (GFP-LC2). The EFab is substituted for the CH1 domain of M60-A02 in the EFab-containing expressions. The schematic shows possible pairing options. Correct pairing of constructs produces bands at 114 and 74 kDa. Incorrect pairing results in bands at 47 and 141 kDa. The SDS-PAGE non-reducing gel shows that approximately equal amounts of correctly paired and incorrectly paired Fabs result when both Fabs have the WT CH1. EFab eliminates the incorrect pairing.
Incorporation of EFab into a bispecific antibody
Next, an EFab light chain solution was combined with an in-house heavy chain heterodimerization technology within the CH3 domain that causes steric clashing of homodimers and preferential formation of heterodimers (unpublished results) to generate full IgG-like bispecific antibodies. This bispecific antibody is composed of 4 different chains and contains one regular Fab and one EFab to induce correct light chain pairing (Figure 3A). Using the anti-EGFR antibody M60-A02 and the anti-IGF1R antibody C06 (two antibodies with the same framework), expression vectors for the bispecific antibody were generated with the EFab engineered into the M60-A02 arm, and the protein was transiently expressed in CHO cells. Analysis of the protein A purified material by mass spectrometry demonstrated that a bispecific antibody made with a single EFab assembled correctly from the four peptide chains and with no detectable heavy chain homodimers or mispaired light chains, the latter of which would result in an increase or decrease of 312 Da in mass, depending on the identity of the mispaired light chain (Figure 3B). In contrast, when the bispecific was generated with two normal Fab arms, mispairing of light chains occurred and was readily detected by mass spectrometry (Figure 3B). The same bispecific antibody was also constructed with the EFab on the C06 arm and M60-A02 as a regular Fab, but it was not analyzed by mass spectrometry.
Figure 3.
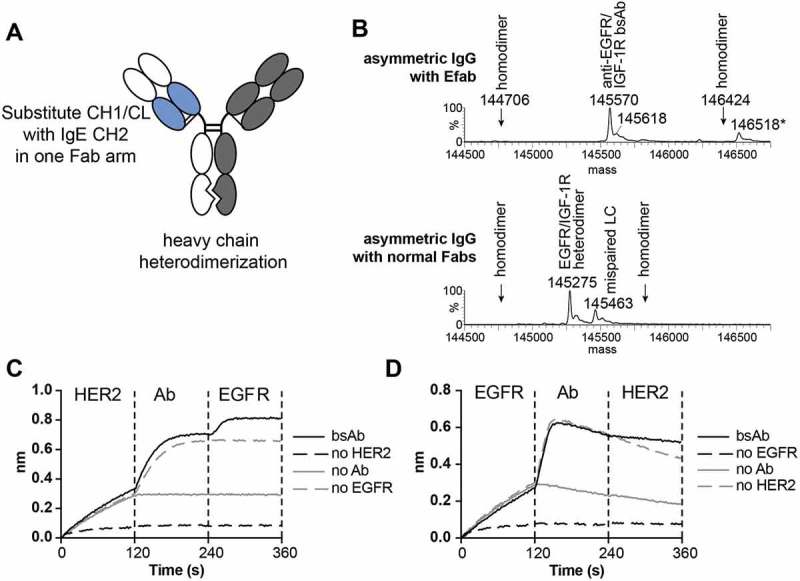
A) Cartoon of EFab-containing asymmetric IgG bispecific antibody. EFab domain substitutions are colored in blue. Heterodimerization mutations are present in CH3 domain. B) Deconvoluted mass spectra of asymmetric IgGs with and without EFab. Anti-EGFR M60-A02 and anti-IGF1R C06 make up the antibody pair, with the EFab on the anti-EGFR arm. The correctly formed EFab containing bispecific has an expected mass of 145,567 Da (145,570 Da observed; the peak labeled * is the correctly formed bispecific antibody plus an O-glycan). There is no observed mispairing of light chain for the EFab molecule, (+ 312 Da for two IGF1R light chains and – 312 Da for two EGFR light chains), but significant mispairing of the regular Fab-containing construct is observed, resulting in antibody containing two M60-A02 light chains. C) and D) Binding by BLI of bispecific antibody to HER2 and EGFR (trastuzumab/cetuximab) in a sandwich format in both directions. Anti-penta-His biosensors were loaded with His-tagged antigens at 5 µg/ml, followed by bispecific antibodies (20 nM) and second antigen (15 µg/ml).
An additional asymmetric EFab-containing bispecific antibody was generated using the therapeutic antibodies cetuximab and trastuzumab, in which case trastuzumab was formatted as the EFab and cetuximab as a regular Fab. Mass spectrometric analysis of this construct showed that the major component was the expected bispecific antibody composed of two different light chains and two different heavy chains (the detected mass was 147180 Da and the calculated mass is 147179.3 Da for the bispecific antibody containing an N-linked glycan, GlcNAc2Man3Hex2HexNAc2Fuc). This conclusion was reached despite the complicated mass spectrum obtained (Figure S3A) due to the presence of an N-linked complex glycan in the variable region of the cetuximab heavy chain that could not be removed by PNGase F treatment under non-reducing conditions. When the bispecific antibody was generated using cetuximab and trastuzumab without the EFab, the major component detected by mass spectrometry was the undesired product containing two light chains of trastuzumab (the detected mass was 147185 Da, with a calculated mass of 147187.9 Da for the antibody containing the N-linked glycan GlcNAc2Man3Hex2HexNAc2FucSia) (Figure S3B).
All bispecific antibodies produced were tested for binding to both targets using bio-layer interferometry (BLI). The trastuzumab EFab/cetuximab bispecific bound both antigens (EGFR and human epidermal growth factor receptor-2 (HER2)) simultaneously in a sandwich format (Figure 3C, D). First, his-tagged antigen was captured by the biosensor, followed by the bispecific antibody. Next, the complex was exposed to the second antigen. Each antibody of the M60-A02/C06 bispecifics did bind to their respective antigen (Figures S4 and S5), but did not exhibit simultaneous binding to both antigens in a sandwich format (Figure S4). Further, the His-tagged EGFR used for both sandwich BLI experiments utilizing anti-penta-His BLI biosensors exhibited a fast off-rate under the assay conditions, complicating interpretation.
The impact of EFab substitution on stability
To evaluate the effect of EFab substitution on antibody stability, we chose to engineer trastuzumab, which has a higher than average thermal stability compared to 137 clinical-stage and FDA-approved antibody therapeutics.21 Trastuzumab was engineered as a heterodimer with one Fab converted to an Efab and the other left as a normal Fab to make analysis of the impact of EFab substitution straightforward and not complicated by the presence of two different antibodies with differing biophysical properties. In addition to the trastuzumab EFab construct, trastuzumab and trastuzumab with the CH3 heterodimerization mutations (Het) were both produced. The antibodies were purified by Protein A chromatography followed by preparatory size-exclusion (SEC) chromatography to remove any aggregate and half-antibody (which is present in most transient expressions of heterodimers due to overproduction of one half of the antibody) (Figure S6). A small amount of aggregate was formed after storage at −80°C with one freeze/thaw cycle (Figure S7). These three constructs were also analyzed by differential scanning calorimetry (DSC) (Figure 4C). The trastuzumab IgG is the most thermally stable, with two transitions at 72.9 and 84.2°C, while the CH3 heterodimerization mutation causes a decrease in stability, resulting in a lower melting temperature (70.5 °C) for the entire Fc domain (Figure 4A). Substitution of the EFab further lowered the melting temperature of the entire antibody (67.5 and 82.8°C).
Figure 4.
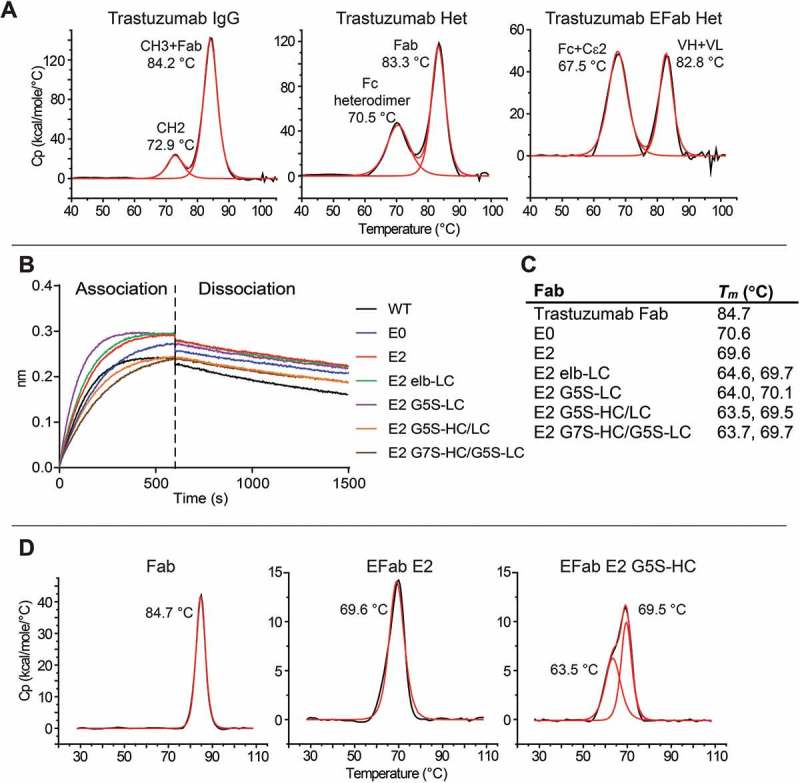
A) DSC thermograms of the trastuzumab constructs. B) Trastuzumab Fabs variants binding to HER2 by BLI. Binding experiments were performed on an OctetRED. HER2-hFc was bound at 10 µg/mL to anti-human IgG biosensors. Each Fab was tested at 10 nM. All constructs exhibit HER2 binding. C) Table of Tms of trastuzumab Fab variants by DSC. D) Select DSC thermograms of trastuzumab Fabs.
We attempted to rescue the destabilizing effect of EFab by engineering the linker region between the variable domain and the EFab. A panel of C-terminally His-tagged EFab Fabs were generated from trastuzumab. This panel included the E0 and E2 variants, which were fused to the variable region of trastuzumab via the native linker from the antibody, as well as a series with variations in the linker regions in either the heavy chain or light chain. The E2-elb light chain variant contained the N-terminal linker region native to the Cɛ2 domain (SRDFTP) with the heavy chain containing the linker from trastuzumab. Flexible G5S or G7S linkers were also engineered into the EFab on the heavy and light chain (E2 G5S-LC, E2 G5S-HC/LC, E2 G7S-HC/G5S-LC). Binding to HER2 was first confirmed by BLI (Figure 4B). Binding was similar for all the constructs, with some of them appearing to associate faster than wild type (WT), although this result may be due to slightly different Fab concentrations. All constructs have equivalent dissociation rates. To determine the effect of different linkers on the thermal stability of the Fabs, the melting temperature of each variant was measured by DSC (Figure 4C and D). Like the IgG, trastuzumab Fab has the highest melting temperature of 84.7 °C, while the EFab containing constructs have a significantly lower thermal stability. The melting temperatures of E0 and E2 are about 15°C lower than the trastuzumab Fab and the linker variants impart a slight decrease in thermal stability, gaining an additional thermal transition at 64°C that is not present in the original two EFab constructs. The effect of changing the linkers to either a flexible linker or that of the Cϵ2 domain appears to cause one of the domains to unfold at a lower temperature than with the native VH/L to CH1/CL linker. Therefore, the tested linkers did not improve the thermal stability of the EFab.
Crystallography
To gain more insight into the molecular features of the EFab platform, particularly the orientation of the Cϵ2 domain to the variable domain, we produced and performed crystallization screening on a panel of EFab Fabs of antibodies with existing apo structures deposited in the Protein Data Bank (PDB). We successfully crystallized the EFab of adalimumab and determined its structure at 1.52 Å resolution (Table 2). The VH and VL domains exhibit typical geometry and the overall variable domain structure within the framework regions and complementarity-determining regions is very similar between the adalimumab EFab and adalimumab Fab (PDB id: 4NYL, 3WD5) (Figure 5A).19 A superposition of the VH and VL domains of EFab and Fab show a root mean square deviation (rmsd) of 0.37 Å (Cα atoms only) (Figure 5B), demonstrating the high structural similarity. In contrast, when the Cϵ2 domains of the EFab are superimposed to the Cϵ2 domains of the IgE Fc (PDB id: 2WQR)16 the rmsd of the Cα atoms is 1.32 Å, indicating less structural similarity (Figure 5B).
Table 2.
Crystallographic data collection and refinement statistics.
| adalimumab EFab | |
|---|---|
| Data Collection | |
| Space group | C2221 |
| Cell dimensions | |
| a, b, c (Å) | 125.4, 195.8, 48.8 |
| Resolution (Å) | 30.0–1.52 (1.54–1.52)a |
| Rsym | 11.5 (75.7) |
| I/σ(I) | 9.3 (2.0) |
| Completeness (%) | 99.6 (99.2) |
| Redundancy | 7.4 |
| Refinement b | |
| Resolution (Å) | 25 – 1.52 |
| No. of reflections | 92,219 |
| Rwork/Rfree | 0.153/0.179 |
| No. of residues | 436 |
| No. of waters/ions | 496/2 |
| RMSD bond lengths (Å) | 0.015 |
| RMSD bond angles (°) | 1.427 |
| B-factors (Å2) | 34.2 |
| Ramachandran regions (%) | |
| Favored/Allowed/Disallowed | 98/2/0 |
a Values in parenthesis are for highest resolution shell. b TLS groups were used in the refinement.
Figure 5.
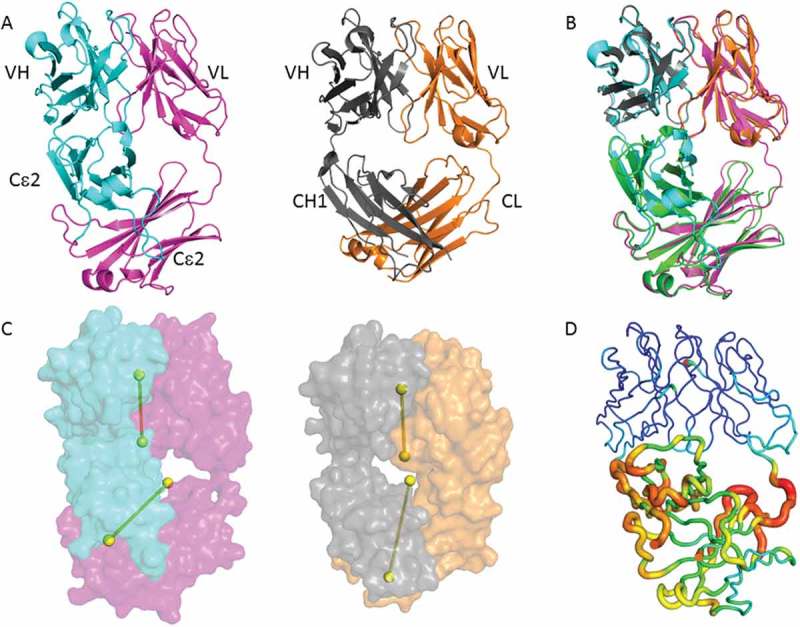
Adalimumab EFab crystal structure and relative domain organization. (A) Side-by-side structural comparison of the adalimumab EFab with its heavy chain in cyan and light chain in magenta alongside with the previously reported adalimumab Fab (PDB ID: 4NYL) with its heavy chain in grey and light chain in orange. (B) Top, superposition of variable domains in EFab and Fab with a C-alpha rmsd of 0.37 Å. Bottom, superposition of the Cϵ2 domains in EFab and IgE Fc (green, PDB ID: 2WQR)16 with an rmsd of 1.32 Å. (C) Relative domain orientation of variable and constant domains in EFab and Fab. The pseudo-twofold axes of variable and constant domains are shown as dumbbells. Molecules are oriented so that the axes of the Fab are parallel to the paper plane. (D) B-factor putty representation of the adalimumab EFab structure. Orange to red colors and a wider tube indicate regions with higher B-factors, whereas shades of blue and a narrower tube indicate regions with lower B-factors. The lowest B-value is observed in the VH and VL domains (dark blue). The largest B-factor is observed in the region of Cϵ2 dimer (red) that rests just below the variable domains, where the electron density clearly shows more disorder than elsewhere in the structure.
The most noteworthy feature in the EFab structure is the asymmetric bending of the Cϵ2 dimer with respect to the VH/VL pseudo-axis, giving it an overall lopsided appearance (Figure 5C). The focal point of asymmetry is at the VH/Cϵ2 elbow region, which is noticeably closer and more compact than the equivalent VL/Cϵ2 elbow. In typical Fab structures, the pseudo two-fold axes between the VH/VL domains and CH1/CL domains are roughly aligned with each other, as is found in the adalimumab Fab (Figure 5C). To further investigate the EFab domain organization, we measured the elbow angles, which in a Fab is the angle between VH/VL and CH1/CL domains, or in the case of the EFab is the angle of VH/VL with Cϵ2 dimer. The WT adalimumab Fab structure has elbow angle of 145 degrees, whereas the adalimumab EFab molecule is 113 degrees. A study of the distribution of elbow angles in experimental X-ray structures shows that possible elbow angles cover a wide range (127 to 220 degrees in the reported set of examples).22 Therefore the elbow angle of adalimumab EFab falls just outside the typical range exhibited by most Fabs with natural domain organization.
Fusing the VH to the Cϵ2 creates an interface in the EFab that is not typically observed in Fabs. To investigate the extent of the interdomain contacts, we measured buried surface area (BSA) and shape complementarity between the VH domain and the Cϵ2 domain. When the VH rests upon the Cϵ2 surface, it results in an overall BSA of 688Å2 with a shape complementarity statistic of Sc = 0.49, indicating only minor surface complementarity. Conversely, no inter-domain contacts are observed between the VL domain and the Cϵ2 domain. In contrast, the adalimumab Fab has a less extensive and more complemental interface between its VH and CH1, as evident by a BSA of 328Å2 and Sc = 0.72. Furthermore, the high average temperature factor for Cϵ2 (47 Å2) is considerably higher than the most ordered part of the molecule at the variable domains (average B-factor 18 Å2), suggesting that the interdomain contacts destabilize this region of Cϵ2 (Figure 5D). In particular, the area in close proximity to the variable domain is the most flexible part of the Cϵ2 where the electron density clearly shows more disorder than elsewhere in the structure. It is worthy of note that the Cϵ2 dimer is well defined in the IgE Fc structure.16 It is unlikely that this disorder is caused by the knob-and-hole heterodimerization mutations in the EFab design because the Cϵ2 heterodimer interface shows clear electron density in an unbiased composite omit maps (Figure 6), indicating a defined geometry that matches well with the WT Fab. The disordered region is more likely to be explained by the structural constraints imposed by the Cϵ2 domains being fused to the variable region.
Figure 6.
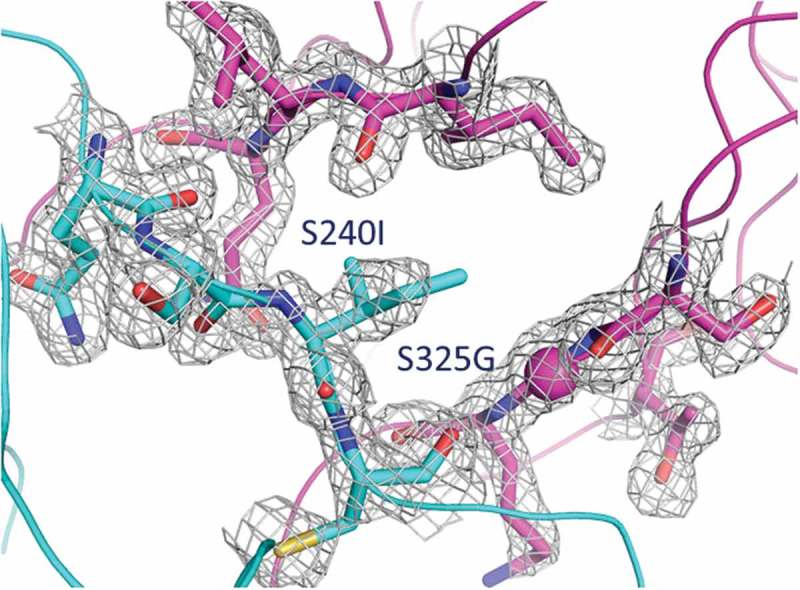
Composite omit map around the knob-and-hole mutations at the Cϵ2 – Cϵ2 interface of the EFab, contoured at 1.2 sigma using a carve distance of 1.6 Å. The color code used is as in Figure 5.
Binding kinetics of adalimumab EFab to tumor necrosis factor
Because of the significant impact of EFab domain substitution on the structure of adalimumab, we measured the impact of EFab substitution on antigen binding. The monovalent binding kinetics of adalimumab EFab to human tumor necrosis factor (TNF) were determined using surface plasmon resonance (SPR) and compared to the WT Fab (Figure 7), which has been previously reported.19 The substitution of the EFab domain slowed the association rate slightly from 3.9 x 106 M−1s−1 to 2.5 x 106 M−1s−1 while it increased the off rate from 5.8 x 106 M−1s−1 to 8.0 x 106 M−1s−1 (Table S1). The impact on affinity was two-fold lower (from 0.15 nM to 0.32 nM).
Figure 7.
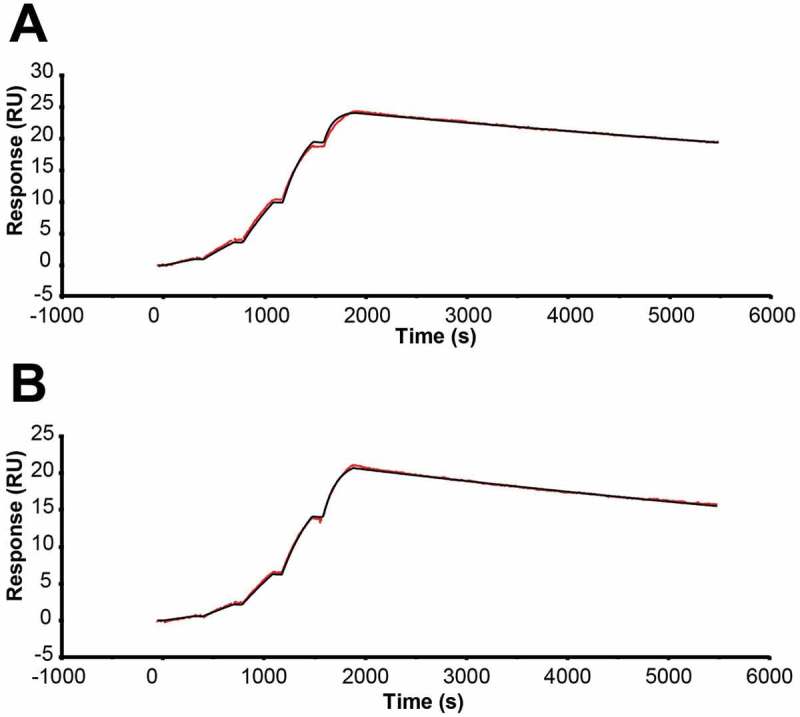
SPR sensograms performed on a Biacore T200 at 25°C in HBS-EBP buffer with a CM5 chip, with hTNFalpha-Fc capture by immobilized anti-hFc antibody. Single-cycle kinetics were used to measure monovalent binding of a) adalimumab Fab and b) adalimumab EFab. The EFab exhibited a two-fold reduction in affinity from 0.15 nM to 0.32 nM.
Limited proteolysis comparison
The large amount of disorder caused by the EFab domain substitution, as observed in the crystal structure of adalimumab EFab, could potentially make the Fab more labile to proteolytic cleavage. Additionally, limited proteolysis can provide some insight into the effect of this domain substitution on the solution conformation of the Fab. Therefore, adalimumab Fab, EFab, and the Cϵ2 of IgE (agly) were subjected first to a protease screen and then to limited proteolysis. The initial screen identified proteinase K and subtilisin as effective proteases (data not shown) and proteinase K was used in the limited proteolysis experiment (Figure 8). The highest concentration of proteinase K (50 µg/mL) caused complete cleavage of all three proteins. The EFab was the most proteolytically labile, with no full-length material remaining at the second-highest concentration (16.7 µg/mL). The adalimumab Fab was completely cleaved at only the highest concentration. IgE CH2, being a single domain, was the most stable.
Figure 8.
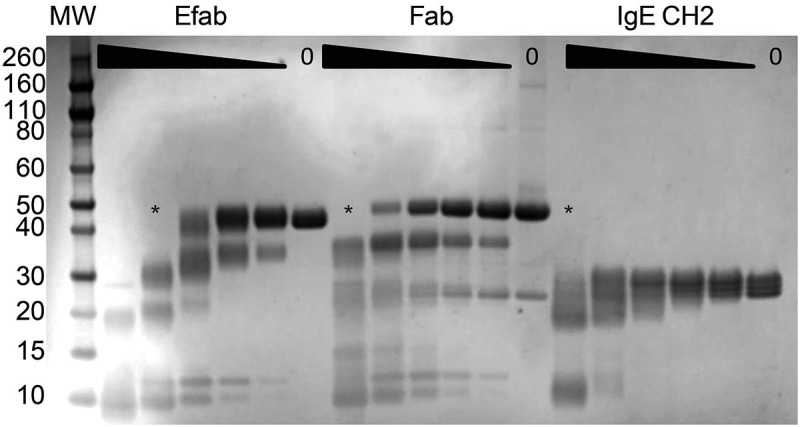
SDS-PAGE of the limited proteolysis experiment of adalimumab EFab and Fab and the IgE CH2 domain with proteinase K. Proteins were incubated with the protease for 30 minutes at 25°C starting at 50 µg/mL, with half-log dilutions to 0.62 µg/mL. Asterisks (*) indicates the lane where no more fully intact material is detectable.
Discussion
EFab domain substitution offers a novel solution to the light-chain pairing problem without the need to engineer the variable regions into an scFv or to identify a common light chain. A variety of antibodies were produced as EFabs and tested first for expression and next for antigen binding. While most antibodies could be expressed as EFabs with varying amounts of aggregate (Figure S2), we did observe a small number of antibodies with very poor expression when we converted them into EFabs, as was the case for C06 and pembrolizumab (the latter of which yielded no purified product). Importantly, EFabs that could be expressed consistently exhibited the correct pairing of light chains with the appropriate heavy chain. Trastuzumab, adalimumab, M60-A02, and C06 all maintained binding to their respective antigens by BLI assay (see Figure S4 for C06-EFab binding), although we did observe one chimeric antibody (cetuximab; data not shown) that could be expressed as an EFab but did not bind its antigen. In the constructs tested, EFab substitution for the CH1:CL of one of the Fabs in an asymmetric bispecific antibody format maintained antigen binding for both Fabs for each of the pairs. Here, we show that a trastuzumab/cetuximab bispecific with EFab substituted on the trastuzumab arm binds both antigens simultaneously in a BLI sandwich format assay. Another pair tested, M60-A02/C06, did not exhibit simultaneous binding, although a M60-A02/C06 bispecific was previously reported to bind both antigens simultaneously in a tetravalent bispecific format, with two C06 scFvs on the C-term of the Fc.20 The lack of binding observed here is likely a result of steric hindrance caused by the asymmetric bispecific format we used to study EFab domain substitution.
Substitution of the EFab led to a decrease in thermal stability of the Fabs as measured by DSC. A series of trastuzumab constructs were produced containing various linkers, including the linker from the Cɛ2, which was engineered onto the light chain, as well as GlySer linkers on both the light and heavy chains. These constructs exhibited slightly decreased thermal stability, indicating that linker length and flexibility are not critical parameters for improving stability and that the native linker from the starting antibody may be best. Nevertheless, based on what was observed from the structure of adalimumab EFab, it may be necessary to engineer an even longer linker on the heavy chain to increase the angle of the elbow region to relieve the clashing at the interface of VH and Cɛ2. The high amount of disorder as indicated by the high B-factors of this region may be a result of this steric clashing or may be inherent to the interaction of the two domains. In the elbow region of a Fab, the variable and constant domains meet to form a tightly associated molecular ball-and-socket joint.23 Mutations in this region distal from the antigen-binding site have been shown to negatively affect thermo-stability, binding affinity, and bispecific activity by modulating the interdomain conformational dynamics.24,25 In spite of the structural effects of EFab domain substitution, the monovalent binding kinetics of adalimumab EFab was only two-fold worse than adalimumab to its antigen, human TNF.
It has long been known that the constant domains exert a positive effect on the stability of a Fab.26 This stabilizing effect of the constant domains has been attributed to the fixed distance and orientation of the domains rather than the formation of a tightly packed interface between variable and constant domains, which is loosely packed and rather small.26 Consistent with this notion, the tethering of antibody variable domains by coiled-coil domains has been shown to result in increased thermal stability of Fv.27 However, a recent study on the stability of chimeric Fab molecules has demonstrated that the positive effect on thermal stability was limited to the kappa constant domain and was not observed in Fabs comprising lambda or hybrid light chains.28 Here, we found that the Cɛ2 domain-substituted EFabs did not maintain the thermal stability of the parental Fabs, despite the fact that the spacing and orientation of the variable domains were maintained. Our results provide evidence that the constant domain provides a stabilizing effect on a Fab that goes beyond the spacing and orientation of the domains.
EFab domain substitution represents a solution to the light-chain pairing problem and is a useful tool that can be used to recombinantly produce two antibodies on the same molecule with correct light-chain pairing. EFab substitution was successful for the antibody pairs reported here, although it was tolerated in some antibodies better than others and should be tested on both Fabs of a bispecific to determine which provides the optimal properties. Though EFab domain substitution had a negative impact on thermal stability, our structural and biochemical data indicate that the EFab approach, along with a heavy chain heterodimerization solution, represents a viable option for the generation of bispecific heterodimeric IgG antibodies.
Materials and methods
Molecular biology
Expression vectors were generated by an assembly method using PCR amplification and the NEBuilder HiFi DNA assembly kit (New England Biolabs), or by standard DNA cloning methods using T4 ligase (New England Biolabs). Variable domains of antibodies were generated from synthetic double-stranded DNA (Integrated DNA Technologies). All plasmids constructs were verified by DNA sequencing. Protein expression in small to medium-scale (30–300 ml) suspension CHO transient transfection was performed with Fectopro (Polyplus) followed by batch production for 9 to 12 days. Supernatants were cleared by centrifugation and filtered through 0.45 µm filters.
Protein purification
Small-scale purification (1 ml) was done using Capturem Protein A (Takara) or Nab Protein A Plus Spin Kit (Pierce) according to manufacturer’s instructions. Bound proteins were eluted using IgG Elution Buffer (Thermo Scientific), neutralized with 1/10th volume 1 M Tris/HCl pH 9, and buffer exchanged three times into phosphate-buffered saline (PBS) using 10,000 MWCO Amicon concentrators (Millipore).
Larger-scale purification of full antibodies was done using HiTrap rProtein A FF columns (GE Healthcare). CHO conditioned media was loaded onto columns, followed by washing with 10 column volumes of PBS. Protein was eluted with 25 mM H3P04, 0.1 M NaCl, pH 2.8 and fractions were neutralized with 1:40 dilution neutralization buffer (0.5 M NaPi, pH 8.6). Overexpressed half-antibody and aggregate were removed by preparatory scale size-exclusion chromatography. The protein was concentrated to < 5 mL and purified on an AKTA Pure over a HiLoad 16/600 Superdex 200 pg column in PBS at 1 mL/min.
His6-tagged Fabs were purified using Ni Excel Sepharose resin (GE Healthcare) in batch mode. Resin was washed with 3 column volumes of 20 mM HEPES, pH 7.5, 0.5 M NaCl. The resin was added to CHO conditioned media and incubated for 1–2 hr with gentle rocking at room temperature. The resin was poured into a column and washed with 10 column volumes of 20 mM HEPES, pH 7.5, 0.5 M NaCl, 10 mM imidazole. His-tagged Fab was eluted with 20 mM HEPES, pH 7.5, 0.5 M NaCl, 500 mM imidazole and fractions containing protein (as measured by A280 or Bradford assay) were combined. The protein was concentrated, buffer exchanged three times into PBS, and sterile filtered through 0.22 um PVDF membrane. Fabs were evaluated for purity by SDS-PAGE and analytical SEC.
For purification of adalimumab EFab Fab for crystallization, following the Ni Excel Sepharose step, the protein was concentrated to < 5 mL and injected onto a 16/600 Superdex 200 column on an AKTA Pure with 20 mM HEPES pH 7, 0.1 M NaCl, 0.02% azide buffer and run at 1 mL/min. Fractions containing pure EFab (with no aggregate) were identified by analytical SEC (Acquity UPLC, Zenix SEC-300 Sepax column, 3 um, 300 Å, 7.8 x 300 mM with 100 mM sodium phosphate, 200 mM NaCl, pH 6.5 with 0.04% sodium azide), combined, concentrated to 10 mg/mL, and sterile filtered.
Intact mass measurement
Approximately 30 µg of protein in each protein A purified sample was deglycosylated with PNGase F (Prozyme) at 37°C overnight. Approximately 10 µg of the deglycosylated protein was then analyzed on an LC‒MS system composed of a UPLC (ACQUITY, Waters Corp.), a TUV dual wavelength UV detector (Waters Corp.), and an LCT mass spectrometer (Waters Corp.). Separation of components was achieved on a TSKgel Phenyl-5PW column (2.0 x 75 mm, 10 μm, TOSOH Bioscience) with a 25-min water/acetonitrile gradient (2–80% acetonitrile) containing 0.03% trifluoroacetic acid at a flow rate of 0.07 mL/min at 70 ºC. Capillary voltage was set to 2500 V and sample cone voltage was 55 V. Molecular masses were obtained by deconvolution of raw mass spectra using the MaxEnt 1 program embedded in MaxLynx 4.1 software (Waters Corp.).
Bio-layer interferometry determination of binding
Binding of trastuzumab EFab variants to HER2 was measured by BLI on an OctetRed96 (Pall ForteBio) at 25°C. Anti-human Fc biosensors were presoaked for 10 minutes in Octet buffer (PBS, 1 mg/mL bovine serum albumin, 0.02% TWEEN 20, 0.01% azide). The biosensors were dipped into a well of buffer for 60 s, then loaded with 10 ug/mL HER2-hFc-His6 (cat# 1129-ER R&D Systems) for 300 s. The biosensors were blocked with hIgG1 at 50 ug/mL for 300s, followed by acquisition of another baseline for 60 s. Next, the biosensors were dipped into 10 nM EFab for 600 s of association, followed by 900 s of dissociation in Octet buffer. The data were aligned to the second baseline measurement. Binding of the cetuximab/trastuzumab EFab bispecific was done in a similar manner. Using anti-penta his biosensors, the first antigen was loaded at 5 ug/mL (HER2-Fc-His6, cat# 1129-ER R&D Systems or sEGFR-ectodomain-His10, made in-house), followed by 200 nM antibody, and then the second antigen at 50 ug/mL (EGFR-Fc, cat#344-ER R&D Systems or HER2-Fc-His, cat# 1129-ER R&D Systems), for 120 s each.
Differential scanning calorimetry (DSC) for thermal stability analysis
Measurements were made on a VP-DSC Capillary Cell Microcalorimeter (MicroCal Inc.). Proteins were prepared at 1 mg/mL in PBS, pH 7.2. Each sample plus a buffer reference was scanned from 20 to 110°C at 200°C/h with a 5 min prescan hold. The data was analyzed using Origin 7.0 Software. The buffer reference was subtracted from the data, which was next normalized based on concentration. The data was fit using the non-2-state model.
Crystallization and data collection
The adalimumab EFab was dialyzed into a buffer containing 0.02 M HEPES pH 7.2, 0.1 M sodium chloride and 0.02% sodium azide. Crystallization was performed by the nanodroplet vapor diffusion method at a temperature of 277 K (4°C), by mixing 200 nL of EFab (10 mg/ml) with 200 nL of a reservoir solution containing 15% PEG 3350 and 0.25M potassium nitrate. For data collection, crystals were cryoprotected by consecutively soaking with the reservoir solutions containing increased concentrations (5%, 10%, 15% and 20%) of PEG200 prior to flash frozen in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source (APS) at Lilly Research Laboratories Collaborative Access Team (LRL-CAT) at 100 K using MAR225 CCD detector, and were processed using the program HKL2000.29 Data statistics are summarized in Table 1.
Structure determination and refinement
The structure of adalimumab EFab was solved by molecular replacement (MR) methods using the program MOLREP.30 Separate search models were the Fab variable domains of adalimumab Fab (PDB id 4NYL) and IgE Cϵ2 domains from an IgE Fc (PDB id 2WQR).16 The adalimumab EFab model was rebuilt using with Coot and refinement using REFMAC resulted in placement of nearly all the adalimumab EFab residues except for the His tag. The final stages of refinement employed TLS refinement with anisotropic motion tensors refined for each of the domains, using REFMAC. 31–33 Refinement statistics are summarized in Table 1. Coordinates and experimental structure factors of the adalimumab EFab have been deposited with the Protein Data Bank (PDB id 6CR1).
Using the program Phenix, cross-validated, sigma-A weighted 2Fo–Fc composite omit maps were calculated to verify the accuracy of the structural model.34 According to Stanfield et al. (see ref. 16), the elbow angle is defined as the angle between the pseudo-twofold axes between the light and heavy chain variable and constant domains, respectively. Elbow angles were calculated using Phenix. Analysis of the stereochemical quality of the models was accomplished using the AutoDepInputTool (http://deposit.pdb.org/adit/). Figures were prepared with PyMOL (Schrodinger LLC).
Surface plasmon resonance binding analysis of adalimumab Fab and EFab to TNF
SPR analysis was performed on a Biacore T200 (GE Healthcare) at 25°C in HBS-EBP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% bovine serum albumin, and 0.005% TWEEN 20) with a CM5 chip. The chip was prepared by NHS/EDC activation to immobilize anti-hFc antibody (in HBS-EP buffer). hTNFalpha-hFc (cat# 10,602-H01H Sino Biological) was flowed over for 30 s @ 10 uL/min at 1 ug/mL, resulting in an Ru of ~ 55. Single-cycle kinetics were used to make affinity measurements, with five Fab concentrations (0.37, 1.1, 3.3, 10, 30 nM), 300 s per analyte concentration with a 3600 s dissociation after the highest concentration with a flow rate of 30 uL/min. The chip was regenerated with 3 mM MgCl2 for 60 s. Binding of both Fabs was measured in triplicate and data was reference and buffer subtracted. Kinetic constants were determined by fitting to 1:1 binding model with RI parameter setting set to 0.
Limited proteolysis
Adalimumab Fab, EFab and the CH2 of IgE were subjected to limited proteolysis. First, 1 mg/mL of each were subjected to a screen of six different proteases from the Proti-Ace Kit (Hampton Research), including α-chymotrypsin, trypsin, subtilisin, proteinase K, pepsin, and thermolysin. The proteins were incubated with the proteases at 0.05 mg/mL in PBS, pH 7.2 for 1 hr at 25°C. The proteolysis was stopped by the addition of non-reducing sample buffer and boiling for 1 min, and was analyzed by SDS-PAGE. Proteinase K and subtilisin both resulted in significant proteolysis, therefore proteinase K was used for in the limited proteolysis experiment.
Each of the three proteins was incubated with five different concentrations of proteinase K (0.05 to 0.0062 mg/mL, ½ log dilutions) at 25°C for 30 min. The extent of proteolysis was determined by SDS-PAGE and quantified using densitometry in the Bio-Rad Gel Doc EZ Software.
Acknowledgments
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LRL Collaborative Access Team (LRL-CAT) beam line facilities at Sector 31 of the Advanced Photon Source was provided by Eli Lilly & Company, which operates the facility. Thank you to Mia Rushe for discussions during the preparation of this manuscript and to Melissa Geddie for critical reading of the manuscript.
Competing financial interests
All authors are current or former employees of Biogen, Inc.
Supplemental material
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the authors.
Abbreviations
- BLI
Bio-layer interferometry
- BSA
buried surface area
- bsAbs
bispecific antibodies
- Cϵ2
CH2 (constant region 2, heavy chain) of IgE
- CHO
Chinese hamster ovary
- CH1
constant region 1, heavy chain
- CL
constant region, light chain
- DSC
Differential Scanning Calorimetry
- EGFR
epidermal growth factor receptor
- Fab
antigen-binding fragment
- GFP
green fluorescent protein
- HER2
human epidermal growth factor receptor-2
- HAS
human serum albumin
- PDB
Protein Data Bank
- RMSD
root mean square deviation
- scFv
single-chain variable fragment
- SPR
surface plasma resonance
- TCR
T-cell receptor
- TNF
tumor necrosis factor
- VH
variable region, heavy chain
- VL
variable region, light chain
- WT
wild-type
References
- 1.Brinkmann U, Kontermann RE.. The making of bispecific antibodies. mAbs. 2017;9:182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–106. doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Brennan M, Davison PF, Paulus H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science. 1985;229:81–83. [DOI] [PubMed] [Google Scholar]
- 4.Milstein C, Cuello AC. Hybrid hybridomas and their use in immunohistochemistry. Nature. 1983;305:537–540. [DOI] [PubMed] [Google Scholar]
- 5.Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270:26–35. doi: 10.1006/jmbi.1997.1116. [DOI] [PubMed] [Google Scholar]
- 6.Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285:19637–19646. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Von Kreudenstein TS, Escobar-Carbrera E, Lario PI, D’Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods RJ, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. mAbs. 2013;5:646–654. doi: 10.4161/mabs.25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu X, Sereno AJ, Huang F, Lewis SM, Lieu RL, Weldon C, Torres C, Fine C, Batt MA, Fitchett JR, et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. mAbs. 2015;7:470–482. doi: 10.1080/19420862.2015.1022694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dillon M, Yin Y, Zhou J, McCarty L, Ellerman D, Slaga D, Junttila TT, Han G, Sandoval W, Ovacik MA, et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. mAbs. 2017;9:213–230. doi: 10.1080/19420862.2016.1267089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. 2014;32:191–198. doi: 10.1038/nbt.2797. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z, Leng EC, Gunasekaran K, Pentony M, Shen M, Howard M, Stoops J, Manchulenko K, Razinkov V, Liu H, et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. J Biol Chem. 2015;290:7535–7562. doi: 10.1074/jbc.M114.620260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science. 1988;242:423–426. [DOI] [PubMed] [Google Scholar]
- 13.Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149:960–968. [DOI] [PubMed] [Google Scholar]
- 14.Seimiya H, Naito M, Mashima T, Yasui H, Kuwana Y, Kurosawa Y, Tsuruo T. T cell receptor-extracellular constant regions as hetero-cross-linkers for immunoglobulin variable regions. J Biochem. 1993;113:687–691. [DOI] [PubMed] [Google Scholar]
- 15.Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108:11187–11192. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holdom MD, Davies AM, Nettleship JE, Bagby SC, Dhaliwal B, Girardi E, Hunt J, Gould HJ, Beavil AJ, McDonnell JM, et al. Conformational changes in IgE contribute to its uniquely slow dissociation rate from receptor FcvarepsilonRI. Nat Struct Mol Biol. 2011;18:571–576. doi: 10.1038/nsmb.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seifert O, Plappert A, Fellermeier S, Siegemund M, Pfizenmaier K, Kontermann RE. Tetravalent antibody-scTRAIL fusion proteins with improved properties. Mol Cancer Ther. 2014;13:101–111. doi: 10.1158/1535-7163.MCT-13-0396. [DOI] [PubMed] [Google Scholar]
- 18.Wan T, Beavil RL, Fabiane SM, Beavil AJ, Sohi MK, Keown M, Young RJ, Henry AJ, Owens RJ, Gould HJ, et al. The crystal structure of IgE Fc reveals an asymmetrically bent conformation. Nat Immunol. 2002;3:681–686. doi: 10.1038/ni811. [DOI] [PubMed] [Google Scholar]
- 19.Hu S, Liang S, Guo H, Zhang D, Li H, Wang X, Yang W, Qian W, Hou S, Wang H, et al. Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor alpha-associated diseases from a molecular view. J Biol Chem. 2013;288:27059–27067. doi: 10.1074/jbc.M113.491530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong J, Sereno A, Aivazian D, Langley E, Miller BR, Snyder WB, Chan E, Cantele M, Morena R, Joseph IBJK, et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. mAbs. 2011;3:273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114:944–949. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J Mol Biol. 2006;357:1566–1574. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 23.Landolfi NF, Thakur AB, Fu H, Vasquez M, Queen C, Tsurushita N. The integrity of the ball-and-socket joint between V and C domains is essential for complete activity of a humanized antibody. J Immunol. 2001;166:1748–1754. [DOI] [PubMed] [Google Scholar]
- 24.Koenig P, Lee CV, Walters BT, Janakiraman V, Stinson J, Patapoff TW, Fuh G. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc Natl Acad Sci U S A. 2017;114:E486–E95. doi: 10.1073/pnas.1613231114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sampei Z, Igawa T, Soeda T, Funaki M, Yoshihashi K, Kitazawa T, Muto A, Kojima T, Nakamura S, Hattori K. Non-antigen-contacting region of an asymmetric bispecific antibody to factors IXa/X significantly affects factor VIII-mimetic activity. mAbs. 2015;7:120–128. doi: 10.4161/19420862.2015.989028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347:773–789. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Zhong P, Luo PP, Wang KC. Antibody engineering using phage display with a coiled-coil heterodimeric Fv antibody fragment. PLoS One. 2011;6:e19023. doi: 10.1371/journal.pone.0019023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toughiri R, Wu X, Ruiz D, Huang F, Crissman JW, Dickey M, Froning K, Conner EM, Cujec TP, Demarest SJ. Comparing domain interactions within antibody Fabs with kappa and lambda light chains. mAbs. 2016;8:1276–1285. doi: 10.1080/19420862.2016.1214785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- 30.Vagin A, Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. J Appl Crystallogr. 1997;30:1022–1025. doi: 10.1107/S0021889897006766. [DOI] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 33.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. [DOI] [PubMed] [Google Scholar]
- 34.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.