

ABSTRACT
Monoclonal antibodies (mAbs) have become a major class of protein therapeutics that target a spectrum of diseases ranging from cancers to infectious diseases. Similar to any protein molecule, mAbs are susceptible to chemical modifications during the manufacturing process, long-term storage, and in vivo circulation that can impair their potency. One such modification is the oxidation of methionine residues. Chemical modifications that occur in the complementarity-determining regions (CDRs) of mAbs can lead to the abrogation of antigen binding and reduce the drug’s potency and efficacy. Thus, it is highly desirable to identify and eliminate any chemically unstable residues in the CDRs during the therapeutic antibody discovery process. To provide increased throughput over experimental methods, we extracted features from the mAbs’ sequences, structures, and dynamics, used random forests to identify important features and develop a quantitative and highly predictive in silico methionine oxidation model.
Keywords: Chemical stability, mass spectrometry, in silico modeling, protein structure, molecular modeling, structure property relationship, QSPR, algorithm, computer aided drug design, elastic network model
Introduction
Protein-based therapeutics are widely recognized for their potential1,2 in treating a range of diseases, with almost a quarter of the biopharmaceutical product approvals in the past 20 years being monoclonal antibodies (mAbs).3 In addition to possessing the desired antigen affinity and specificity, the successful therapeutic antibody development candidate needs to meet favorable developability criteria,4 such as an optimal in vivo clearance profile, low aggregation propensity, and high levels of physical (thermal/pH) and chemical stability.5 The absence of a favorable profile can cause attrition or delay in development of a therapeutic mAb candidate; thus, in silico prediction of chemical liabilities in mAbs early in the drug discovery process provides beneficial resource management, and has attracted considerable attention.
Substantial progress has been made in computational prediction of thermal/pH stability,6 aggregation propensity,7,8 viscosity9,10 and in-vivo clearance of mAbs.9,11 Other antibody liabilities due to chemical stress in the manufacturing process include the deamidation of asparagine (Asn), isomerization of aspartic acid (Asp) and oxidation of methionine (Met) and tryptophan (Trp) residues. To this end, studies on prediction models for Asn deamidation,12,13 Asp isomerization12 and Trp oxidation9 have been reported.
Oxidation of Met in proteins can result from the conversion of Met to methionine sulfoxide (MetO) by reactive oxygen species (ROS) over a broad pH range.14 Protection against Met oxidation can only be found in certain tissues and immune cells where this effect can be reversed by enzymes known as methionine sulfoxide reductases, which can reduce MetO back to Met via a thioredoxin-dependent reaction.15,16 It is believed that this reversible oxidation of Met plays a key role in the regulation of many enzymes and peptide hormones.17 Oxidized forms of proteins have been shown to exhibit decreased chemical and physical stability when compared to the unoxidized form,18,19 thereby possibly affecting their biological activity. In the case of mAbs, oxidation may interfere with the mAbs’ ability to bind to its target, especially if the oxidation occurs within the complementarity-determining region (CDR), thereby decreasing its efficacy.
Previous studies on predicting Met oxidation in proteins20-25 and antibodies26,27 have shown that measures of solvent exposure, degree of water coordination, and spatial distance between the Met sulfur atom and the closest aromatic residue28 are indicative of the oxidative susceptibility. However, these studies either considered a limited set of features, notably excluding dynamics features, or relied on expensive and time-consuming molecular dynamics (MD) simulations to obtain dynamics features, resulting in small sets of proteins.
Here, we extracted dynamic features from a relatively large number of mAbs using the more efficient coarse-grained elastic network models, and, along with features extracted from the primary sequence and predicted tertiary structure obtained using homology modeling, constructed a random forest (RF)-based machine learning model to quantitatively predict the risk of Met oxidation in the CDRs of mAbs. The model was generated using a benchmark dataset containing an experimentally determined susceptibility to Met oxidation of 172 Met residues in CDRs of 122 distinct mAbs, and was further validated on an independent hold-out set of 17 Met residues in 12 mAbs and a validation set of 121 clinical stage mAbs. We describe the experimental approach in identifying antibodies with methionine oxidation liability, how the various features used in the RF model were obtained, how the model was built, and the performance of the model.
The quantitative prediction model performs remarkably well according to the conventional performance metrics, suggesting that simple features extracted from the structure and dynamics of the molecule can quickly inform us about the stability of potential Met liabilities in the CDRs of mAbs. Our approach is different from previous work in that the analysis is performed over a larger dataset of antibodies, takes into consideration dynamics features using coarse-grained elastic network models that are much less expensive compared to MD, and adopts a rigorous machine-learning framework to develop a predictive regression model.
Results
Prediction of methionine oxidation risk using a random forest model
Susceptibility to oxidation upon 2,2ʹ-azobis(2-amidinopropane) dihydrochloride (AAPH) stress was measured as the relative change in percentage of oxidized Met species (with respect to control) for a set of 172 Met residues across 122 mAbs (Table S1) as described in Materials and Methods. Of the 172 Met in the training set, 18 were identified as ‘liable’ (positive class) to Met oxidation whereas 154 were identified as ‘non-liable’ (negative class).
RFs29 are one of the most popular machine learning (ML) methods for classification or regression because of their effectiveness, especially when the number of cases available for training is small. RFs belong to a class of ML methods known as ensemble methods; namely they use a combination of multiple regression trees and typically deliver better performance than individual regression trees alone by averaging the predictions of each tree.30 In addition, RFs offer two specific advantages: 1) they automatically provide an unbiased ‘out-of-bag’ (OOB) performance measure (all data points were predicted only using an ensemble of trees that were not generated using that data point) without the need to explicitly divide the dataset into training and test sets,31 and 2) they provide a reliable measure of the predictive power of each feature.
A RF regression model was trained to predict the relative change in percentage of oxidized species upon AAPH treatment on 172 Met residues using 4 (of a total of 18 considered) numeric descriptors (Table 1) extracted from the predicted structure and dynamics of the mAbs (see Materials and Methods for details and outlined protocol in Figure 1). Rather than using expensive MD simulations as in most previous works, we relied on coarse-grained representations of proteins, namely elastic network models to model the protein dynamics.
Table 1.
List of descriptors investigated in this study.
| No. | Descriptor Name | Explanation | Source | In Final Model? |
|---|---|---|---|---|
| 1 | NoverlResa | Number of overlaps between atoms of Met residue with spatial neighbors | Structure | Yes |
| 2 | TotSasaResa | Total solvent accessible surface area of Met residue | Structure | Yes |
| 3 | anmFluca | Mean square fluctuation of Met Cα atom based on Anisotropic Network Model | Dynamics | Yes |
| 4 | hnmFluca | Mean square fluctuation of Met Cα atom based on Hinsen’s Network Model | Dynamics | Yes |
| 5 | PhobSasaResa | Hydrophobic partition of the solvent accessible surface area of Met residue | Structure | No |
| 6 | PhilSasaResa | Hydrophilic partition of the solvent accessible surface area of Met residue | Structure | No |
| 7 | cdrLength | Length of CDR in which Met is located | Sequence | No |
| 8 | Centeredness | Location of Met with respect to center of CDR | Sequence | No |
| 9 | cdrLocation | CDR in which Met is located (CDR-H1/H2/H3/L1/L2/L3) | Sequence | No |
| 10 | IgGType | IgG type of the antibody | Sequence | No |
| 11 | lcFramework | Germline family of the light chain | Sequence | No |
| 12 | hcFramework | Germline family of the heavy chain | Sequence | No |
| 13 | QSasaResa | Ratio of exposed-to-total solvent accessible surface area of Met residue | Structure | No |
| 14 | dipoleMoment | Magnitude of the dipole Moment of the mAb | Structure | No |
| 15 | energyInt | Energy of interaction between VH and VL | Structure | No |
| 16 | protpI3D | 3D structure-based pI of the protein | Structure | No |
| 17 | chargeAtpH5 | Net charge of the mAb at pH 5.0 | Structure | No |
| 18 | chargeAtpH7 | Net charge of the mAb at pH 7.0 | Structure | No |
aThese descriptors were also calculated for the (N-1) th and (N + 1)th residues; but not identified to be useful; where N is the index of the Met residue.
Figure 1.
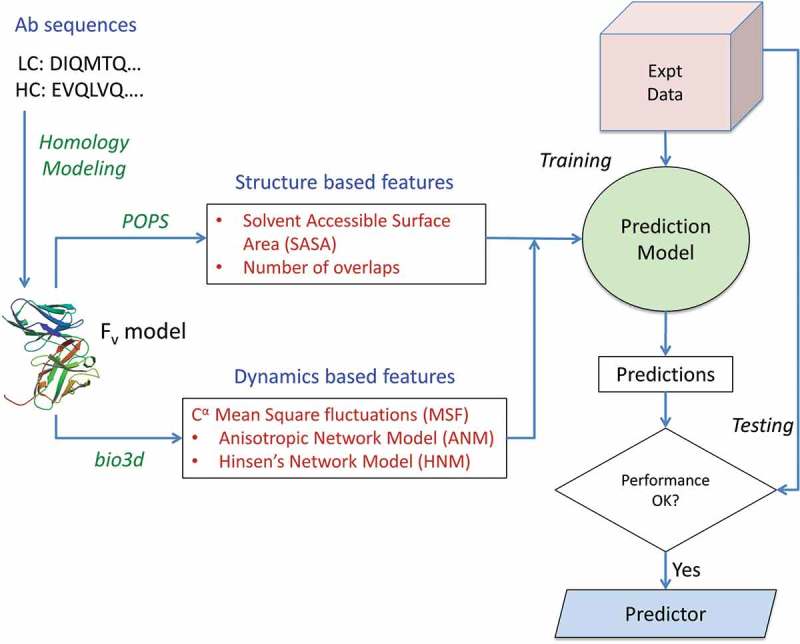
Schematic workflow of the methodology. Antibody sequences are obtained from an in-house database and the Fv regions for each structure modeled using MOE protocols. Features are extracted from the sequence, structure and dynamics of the mAb Fv regions and used to implement a random forest-based predictor in R. The performance of the model is assessed using the standard metrics of correlation and root mean square error for regressor model and accuracy, precision, sensitivity and specificity for the implicit classifier model.
The value of each feature for the 172 Met residues, as well as the experimental and predicted values, can be found in Table S2. Figure 2 shows a scatterplot of the predicted vs. experimental values of the relative changes in oxidized species. The correlation between the predicted and experimental values, = 0.77 and the root mean square error (RMSE) of the predictions was 11.39. The standard deviation of the errors, was 10.08.
Figure 2.
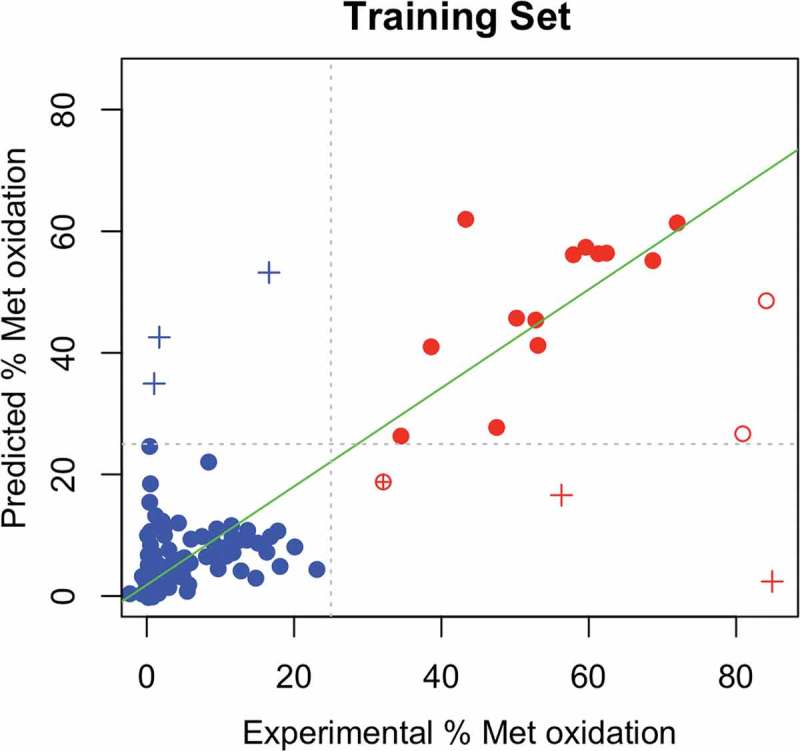
Scatterplot showing the predicted vs experimental % change in oxidized species for 172 Met residues. Abscissae represent the experimentally measured % change in oxidized species upon AAPH treatment whereas the ordinates represent the predicted values from the random forest regressor model. Residues with relative change < 25% (‘Non-liable’) as identified by experiment are colored in blue, while liable residues are colored in red. Outliers (having a prediction error > 3) which are mispredicted according to the classifier scheme are shown with a ‘+’ sign; and correct predictions as hollow circles. Non-outliers which are correctly predicted are shown as filled circles, and mispredicted ones with a ⊕ sign. The line of best fit (excluding outliers) is shown as a green line.
The predictions from the regression model were then used to construct an implicit classifier model to annotate whether the change in percentage of oxidized species is above or below a specific threshold (25%). When the relative change in oxidized species is above 25%, the residue is classified as ‘Liable’ and otherwise as ‘Non-liable’. The model was able to discriminate between liable and non-liable residues well (see confusion matrix in Table 2). There were six mispredictions, 3 false positives and 3 false negatives. Interestingly, all mis-predicted Met except one (M92, the only case in the CDR-L3 region) were located in the CDR-H3 region. Moreover, these mispredicted cases in the CDR-H3 region are also located on very long loops (length given in brackets): M97 (11), M100b (13), M100h (16), M100i (19) and M100* (20). This suggested that the failure of the model in these cases was most likely due to uncertainty in the CDR-H3 modeling,32,33 which would affect the features extracted from these structures.
Table 2.
Performance measures of the random forest classifier on the training dataset of 172 Met residues.
| Experimental ‘Liable’ | Experimental ‘Non-liable’ | ||
|---|---|---|---|
| Predicted ‘Liable’ | 15 (TP) | 3 (FP) | Precision = 0.83 TP/(TP+ FP) |
| Predicted ‘Non-liable’ | 3 (FN) | 151 (TN) | |
| Recall/Sensitivity = 0.83 TP/(TP+ FN) |
Specificity = 0.98 TN/(TN+ FP) |
Accuracy = 0.96 (TP+ TN)/Total |
|
| Matthew’s Correlation Coefficient (MCC) = = 0.81 | |||
TP = True Positive, TN = True Negative, FP = False Positive, FN = False Negative
All predictions are ‘out-of-bag’ (OOB); that is predictions on each data point were made only using the trees not generated using that point.
This implicit classifier model offers a high accuracy of 0.96 and a high specificity of 0.98. Sensitivity and precision are both at 0.83. Since the majority of the Met are not susceptible to oxidative stress, this is an unbalanced dataset. We thus also calculated the Matthews Correlation Coefficient (MCC), which was 0.81. In addition, we wanted to know how the predictions change if the cutoff for liability is changed. To understand this, we subjected the predictions to a receiver-operating characteristic (ROC) curve analysis (Figure S1) by plotting the true positive rate against the false positive rate for different cutoffs. The model gave a remarkable area under the curve (AUC) of 0.96, suggesting that it is very robust.
Identification of features important in determining liability of a met residue to oxidation
As described above, one of the advantages of using a RF model is the ability to estimate the importance of each of the extracted features in terms of its predictive power. Variable importance was estimated using the two measures as described in the Materials and Methods section, and the results based on a model of 8 descriptors are shown in Figure S2.
From a logical perspective, the set of important descriptors can be divided into three main categories: 1) descriptors that relate to the residue interactions; 2) descriptors that relate to the intrinsic dynamics of the residues; and 3) descriptors that relate to the solvent exposure of the residues. As can be seen from Figure S2, the set of the most important descriptors include the number of contacts the residue makes with its spatial neighbors (‘NoverlRes’) and the mean square fluctuation from the coarse-grained elastic network models (‘anmFluc’ and ‘hnmFluc’). The second set of important variables includes the ‘TotSasaRes’ and ‘PhobSasaRes’ of the residues, representing the total and the hydrophobic partition of the solvent accessible surface areas (SASA) of the residues, respectively.
When the distributions of ‘NoverlRes’ of liable versus non-liable methionines is visualized (Figure 3), it becomes increasingly apparent that this feature is indeed very powerful in discriminating between liable and non-liable residues. The scatterplot also highlights the mispredictions by the binary classifier model (M92, M97, M100b, M100h, M100i, M100*). It can be clearly seen that the false positives have a very high SASA compared to non-liable residues and the false-negatives have a very low SASA compared to liable residues. The separation of the two classes based on the solvent-accessible surface area of the residue (‘TotSasaRes’) (Figure 3) is less evident, although significant.
Figure 3.
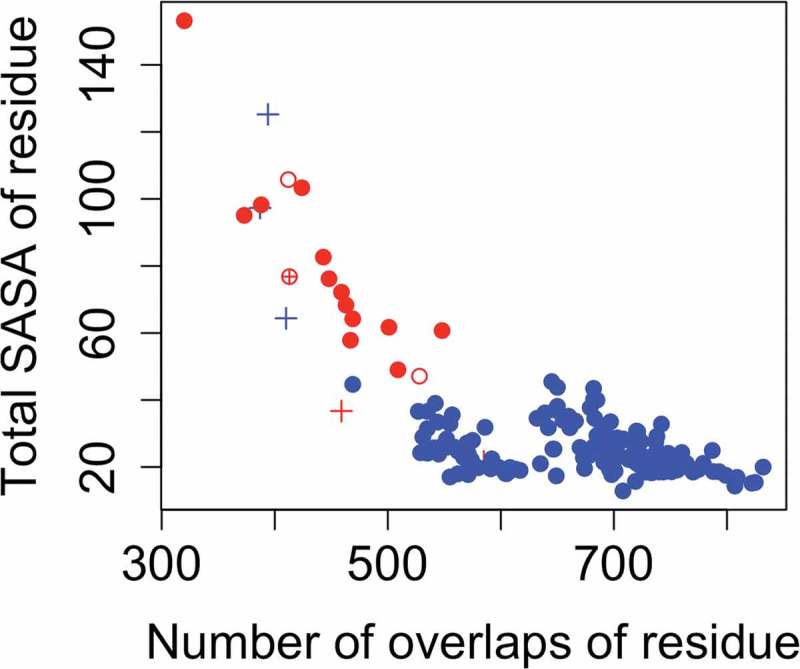
Scatterplot showing the distribution of important features for liable versus non-liable Met residues. The number of overlaps of the Met residue with atoms of spatial neighbors (the feature ‘NoverlRes’) is shown along the x-axis and the total solvent accessible surface area of the residue (the feature ‘TotSasaRes’) along the y-axis. Liable Met residues are shown in red and non-liable Met residues in blue. Outliers (having a prediction error > 3) which are mispredicted according to the classifier scheme are shown with a ‘+’ sign; and correct predictions as hollow circles in their respective colors. Non-outliers which are correctly predicted are shown as filled circles, and mispredicted ones with a ⊕ sign.
A complete list of descriptors investigated in this study is shown in Table 1. Many of these descriptors were eliminated based on the fact that their importance was ascertained to be much less compared to that of the 8 descriptors examined here, 4 of which were used to construct the final model.
Hold-out validation of the prediction model
To evaluate the model in a real-life scenario, we tested the performance on an independent validation set of 17 Met residues across 12 mAbs that went through the same oxidative stress and measurement as the training set. One of the 17 Met residues was ‘liable’ and 16 were non-liable. A summary of the dataset is provided in Table S3. Features for each mAb were also extracted as described in Materials and Methods (Table S4). Predictions from regression model gave a correlation of 0.94 and an RMSE of 8.18 (Figure S3). The standard deviation of the errors was 6.35. These values suggested that the model was robust, in accordance with the OOB performance measures obtained on the training set. Notably, there was one outlier prediction with an error of more than (M30).
It is also important to note that the level of performance offered by the RFs is better than that of simpler models like multiple linear regression (MLR). For example, a MLR model trained using the same set of descriptors on the same training set and tested on these 17 cases gave an R of 0.82 and an RMSE of 10.62, further reinforcing the fact that RFs are superior at descriptor selection, especially when applied to small datasets.
We then tested the RF classifier (by applying a cutoff of 25% on the regression predictions) on this test set. The confusion matrix of the results is shown in Table S5. The model performed well with no mis-predictions. Even the outlier prediction M30 was correctly predicted to be liable (i.e., a relative oxidation of > 25%), further demonstrating the robustness of the model developed in this work.
Comparison of the prediction model with other methods on an independent test dataset
In order to obtain an unbiased estimate of the performance of the model, it is important to understand how well the model performs on an independent set. For this, we used a benchmark dataset of 121 clinical stage antibodies with experimentally characterized Met liability data from Adimab.26 We compared the performance of our model on this dataset with that of their machine-learning based SASA prediction model26 and another independent method called Methionine Oxidation Predictive Model (MOPM) developed by Aledo et al.28 using non-antibody proteins’ Met oxidation data.
The Adimab dataset is organized differently from our dataset. Instead of reporting oxidation status of each methionine residue as we did, the Adimab dataset reports the total number of methionine residues oxidized in the whole antibody, including both CDR and non-CDR frameworks, without specific oxidation status for each methionine residue. To facilitate comparisons between our method and other methods on this dataset, we converted our predictions into oxidation states as follows. First, we predict the relative percentages of oxidation for each Met residue in the heavy and light chains of the mAbs using our regression model. Then, we determined for each mAb, the number of Met residues that were classified as liable using different percentage cutoffs as thresholds (from 0% to 100%). If there was at least one liable Met in the mAb, then it was considered ‘Liable’ and otherwise ‘Non-liable’. These predictions were matched with the experimental data (similarly converted into binary class information). Thus, for each cutoff, the true positive rate (TPR) was plotted against the false positive rate (FPR) to construct the ROC curve (Figure 4), and the AUC (area under the curve) determined. Comparisons based on AUC also served the additional purpose of eliminating any bias introduced through the use of arbitrary cutoffs by the different methods for liability classification.
Figure 4.
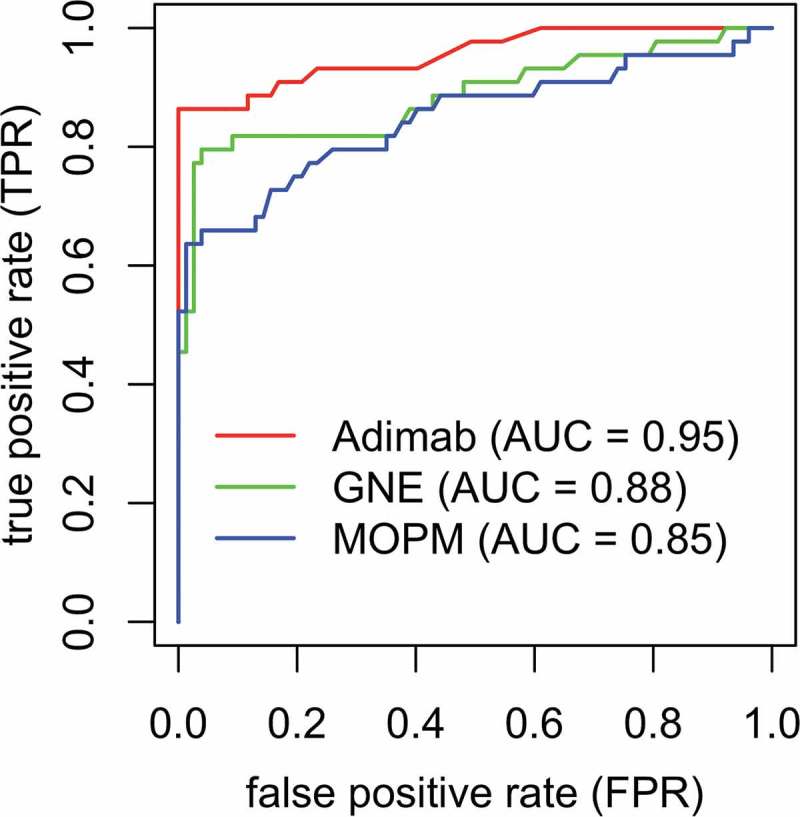
Comparison of Receiver operating characteristic (ROC) curves for different methods on the benchmark clinical mAb dataset. Plot of the true positive rate (TPR) against the false positive rate (FPR) for our random forest-based prediction model (green) in comparison with that of Adimab (red) and MOPM (blue).
Based on the AUC analysis (Figure 4), our prediction model (AUC = 0.88) performed better than the MOPM model (AUC = 0.85) in being able to correctly classify whether the mAbs had liability or not, but the Adimab method (AUC = 0.95) outperformed both of these methods on the dataset. However, we would like to emphasize four ways in which the data is different from our in-house data. First, the stress conditions are different. The Adimab dataset was characterized under hydrogen peroxide induced stress, whereas ours was under AAPH stress. Interestingly, although our model is trained using AAPH stress data, and the MOPM model is trained using H2O2 stress data (the same as the Adimab dataset), our model still outperforms the MOPM model, illustrating the importance of developing antibody-specific models. Second, the criteria to determine Met oxidation state is different. Third, the Adimab dataset includes Met in both CDRs and framework regions, while our model was trained using experimental data generated for Met in CDRs only. Finally, the Adimab dataset only contains the Met oxidation state for each chain, i.e., the number of oxidized Met in each chain and not residue-wise liability information.
Given these major differences, an AUC of 0.88 was quite encouraging. A different choice of stress condition or criteria to define whether a residue is liable or not could have resulted in an even better performance of our model. The Adimab method in fact uses SASA only for prediction, and we have shown in our dataset that our model outperforms prediction using SASA only. In an ideal scenario, a fairer comparison could be achieved by using another large and independent dataset.
Distribution of liable and non-liable residues on the mAb primary sequence
The available dataset also provided an opportunity to investigate whether there are any particular locations on the primary sequence of the mAbs where liable residues are more likely to be found. Figure 5 shows a mapping of the liable and non-liable residues in the training dataset onto the Kabat sequence of the mAbs. Our dataset contains Met residues present in CDR-L1 (M29, M30, M32, M33), CDR-L3 (M89, M92, M96), CDR-H1 (M34), CDR-H2 (M50, M51, M53, M57, M58, M62, M63, M64, M77, M78) and CDR-H3 (M96, M97, M100, M100a, M100b, M100c, M100d, M100e, M100f, M100h, M100i and M100*), all of which (except CDR-H1 where all Met residues are at M34 and non-liable) contain at least one liable Met residue and one non-liable residue. In addition, there are positions in the dataset at which Mets were identified experimentally to be both liable and non-liable (M100b), emphasizing the unbiased nature of the dataset, and the need for a prediction tool to interpret the dataset.
Figure 5.
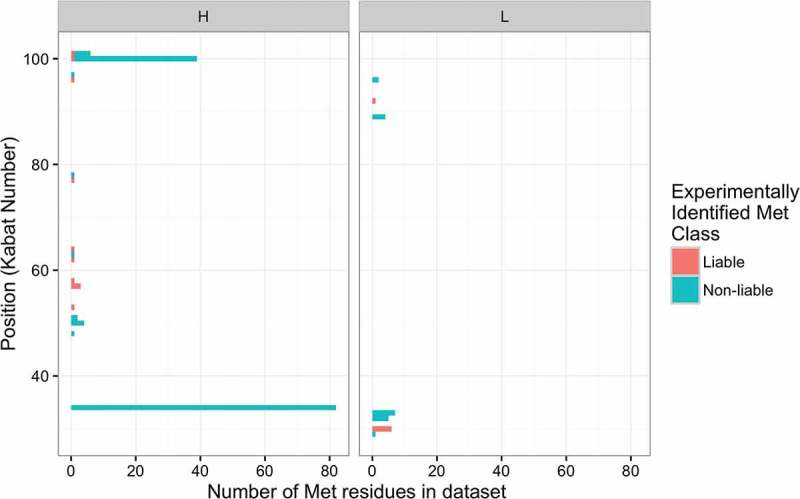
Map of liable and non-liable Met residues on the variable region of the antibodies. Histogram showing the frequencies of Met in the experimental dataset of 122 mAbs at various positions identified to be liable (red bars) and non-liable (green bars) based on Kabat numbering in different complementarity-determining regions of the heavy (left panel) and light chains (right panel). M100b is observed to be liable in one mAb and non-liable in another and there shows a green + red bar.
In order to investigate whether there are certain positions that are naturally more susceptible than others, we analyzed the natural frequency of occurrence of Met in human34 and mouse35 antibody repertoires. The naturally observed probability of Met at the different Kabat positions in the dataset is shown in Table S6. Our analysis showed that the natural propensity of occurrence of Met at liable positions is not significantly different from that at non-liable positions at a 95% confidence level (Wilcoxon rank-sum test; see p-values in Table S7). This lends further support to our observations that susceptibility to Met oxidation is structure-mediated rather than evolution-governed; thereby requiring a prediction model like the one presented here.
Discussion
In this work, we developed a quantitative predictor for Met oxidation liability in mAbs by analyzing a large dataset, based on a RF machine learning model. The quantitative regressor can potentially be used across the industry, where different cutoff values can be applied. We find that simple features extracted from the structure of the molecule, as well as flexibility information from coarse-grained representation of the mAb, are sufficient to identify oxidative susceptibility of Met residues in mAbs. The number of contacts with neighboring residues, average fluctuations of the Met residue and solvent-accessible surface area were identified as the primary features to predict percent oxidation of a Met residue, suggesting that a higher solvent exposure and higher fluctuation at a Met residue accentuate its susceptibility to oxidation. Notably, coarse-grained elastic network-based calculations for estimating residue fluctuations used in this study are significantly less expensive compared to MD or Monte Carlo simulations.
The remarkable accuracy of the prediction model sheds light on the mechanism of Met oxidation and susceptibility of the residues to various ROS. The important descriptors in the model are in a way related to each other. For example, residues with fewer spatial contacts with neighbors are likely more exposed to the solvent, and this property has been measured in some studies as the water coordination number (WCN).27 In turn, these residues will exhibit higher average fluctuations. Increased fluctuation of the Met residue (partly arising from exposure to solvent) will also contribute to increased susceptibility to oxidation as manifested by the importance of the features ‘anmFluc’ and ‘hnmFluc’. Fewer residue contacts, larger fluctuations and a larger solvent exposure results in a higher chance of the residue coming in contact with ROS. The observation that the hydrophobic partition of SASA (‘PhobSasaRes’) is more important than the hydrophilic partition (‘PhilSasaRes’) can be explained by the fact that the oxidation process results in the energetically favorable conversion of an exposed hydrophobic patch on Met to a hydrophilic patch in contact with water. The RF model is a perfect framework to dissociate the contributions of these descriptors and offers robust predictions.
Although SASA has been found to be predictive of Met oxidation in previous studies,26 we examined the performance of using SASA only and found that the RF model is superior. For example, in the training set, a linear model of oxidation percentage vs. SASA only gave an R of 0.68 and RSME of 13.18 (see predictions in Table S2), compared to an OOB R of 0.77 and RSME of 11.48 for the RF model. As for the hold-out test set, the linear model of oxidation percentage vs. SASA gave an R of 0.91 and RMSE of 8.31 (see predictions in Table S4), compared to R of 0.96 and RSME of 7.74 for RF model. The corresponding SASA-only implicit classification model using 25% relative oxidation as a cutoff gave reduced sensitivity of 0.72, precision of 0.81 and MCC of 0.74 (Table S8) compared to sensitivity and precision both of 0.83 and MCC of 0.81 for the RF model using the 4 features. The reduced sensitivity is especially of concern, as increased false negatives would mean higher probability of encountering a previously predicted non-liable molecule tested as liable at a later stage, a costly event that should be avoided even if rare.
Another important point is that our model has been trained specifically on antibodies. In addition to the independent dataset, the MOPM model also gave a lower correlation with the experimental data (R = 0.69) compared to our model (OOB R = 0.77) on our training dataset of 172 Met. In terms of the classifier model, MOPM also gave a much higher number of mispredictions (8 false negatives and 2 false positives) compared to our model (3 false negatives and 3 false positives). Again, the higher number of false-negatives is of concern. The improved performance of our model over the MOPM model suggests that training the models specifically on antibodies can provide gains in the ability to identify these liabilities.
In addition to RFs, support vector machines (SVM) and neural networks (NN) are the other popular ML methods. We also compared the performance of our RF-based model with SVMs and NNs (using the same four features). While the methods were able to perform slightly better on the training dataset based on cross-validated performance (Table S9); the RF model outperformed the other two on the independent benchmark test dataset (Table S10). Hence, we have presented results with the RF-based model only.
Another question is the applicability of the various prediction models to different datasets. The behaviors of Met residues under different oxidative stress conditions (e.g., thermal, light, numerous chemical reagents) can be different,36 but the protocol presented in the paper can be easily adapted to develop stress-specific prediction models, and it would be interesting to compare prediction models trained under different stress conditions to determine if they are similar or how they differ from each other. For example, the performance of our model on the Adimab benchmark dataset may have been better if a different threshold was chosen for identifying liable residues, or if the experiments were performed under a different oxidative stress condition. These issues will be addressed in future studies.
Our highly predictive Met oxidation model now offers early in silico molecular assessment that enables prioritization of candidate mAbs for lead selection, or allows engineering efforts to substitute the liable Met residues prior to experimental confirmation of chemical liability. Both strategies expedite and streamline resource utilization in the development process. The regression nature of the model also allows potential wide use across the industry, where each company or institute can determine their own cutoff value as liability threshold.
One caveat is that our model depends on the quality of the input 3D structure used for extracting the features. This becomes especially important for long CDR-H3 loops, where even state-of-the art models still lack reliability.32,33 It is also worth noting that predictive power is directly influenced by dataset size; therefore, the current model can be further improved (especially to minimize the number of false positives), with increased dataset size (specifically, liable Met) in the future. Some studies have shown that residues that fall outside of the traditionally defined CDRs can also be important to antigen binding,37 which suggests that molecular assessment studies may need to be further extended to these residues.
Another important consideration is the chemical environment around the Met residues. Previous works have shown that the differential rates of oxidation of various Met on the same protein can be attributed to differences in interactions with the residues in the structural environments or with the solvent.38–40 For example, a previous study showed that oxidized Met residues were located in closer proximity to phosphorylation sites than non-oxidized ones.41 In addition, Met residues are often located in spatial proximity to aromatic rings, contributing to protein stability through the hydrophobic effect.42 The oxidation of such Met residues can lead to conformational changes that result in the exposure of previously unexposed hydrophobic residues.43,44 In other words, the oxidation of different Met residues on a protein may not be independent of each other. However, a detailed understanding of the mechanisms behind such correlations is still lacking. Improvement of existing methods could focus on capturing such aspects of the Met residues in the context of the overall structure.
Materials and methods
Experimental identification of met liabilities
To determine oxidative liability, mAbs were oxidatively stressed by AAPH45 and evaluated using peptide mapping LC/MS techniques as previously described.36,46 Briefly, control and oxidatively stressed samples were tryptically digested; and the digested peptides were subsequently separated using LC or UHPLC, detected using an Orbitrap mass spectrometer and identified by accurate mass (MS1), and sequence and modification locations were verified from fragmentation patterns (MS2). For peptides containing methionine, extracted ion chromatograms (XICs) of MS1 mass–to-charge ratios () for the most abundant charge states for the non-oxidized, the mono-oxidized (plus 15.9944 Da) and the double-oxidized species (plus 31.9893 Da) were created for each peptide for both the control and stressed samples. Peaks from the XICs were integrated and areas were used to determine the percent relative oxidation for each methionine of interest. The relative percent oxidation for a site of interest was calculated by taking the sum of the areas of the oxidized species, dividing by the sum of the areas of the non-oxidized and oxidized species, and multiplying by 100. The relative percent oxidation for each methionine site was then compared for the control and oxidatively stressed samples. A methionine site having a relative percentage above a historically determined threshold (25%) was considered to be an oxidation liability.
Sequence based feature extraction
Several features were extracted from the primary sequence of the mAbs. The CDR definition was broadened to ensure sufficient representation of the antigen-binding site on the various mAbs. Therefore, Chothia, Kabat CDR definitions, and Vernier zone47 were augmented to provide the broader CDRs definition used in this study. In order to facilitate comparisons between mAbs consistently, Kabat numbering48 was used despite the use of modified CDR definition described above (For purposes of this study, residues 93 and 94 in the heavy chain are also considered part of CDR-H3 in accordance with in-house definitions). The location of the residue within the variable fragment (Fv) is specified as: CDR-H1/H2/H3 or CDR-L1/L2/L3. The germline family of light chain or heavy chain was obtained by aligning the corresponding sequences to an in-house database of sequences obtained from the IMGT database (http://www.imgt.org) where the gene family with the highest similarity to the query sequences was assigned.49 The length of the CDR in which the residue is located, referred to as ‘cdrLength’, was included as a feature. The ‘centeredness’ of the residue within the CDR was measured as a value ranging between 0 and 1 with 0 corresponding to either end of the CDR and 1 to the center of the CDR loop. No mAb in the dataset is identical to any other mAb in the dataset. When CDRs are identical between any two mAbs, their overall sequence identity is less than 94%. A detailed summary of the frequencies of Met according to CDR and IgG subtype is available in Table S1.
Structure-based feature extraction
The tertiary structures of the Fv regions in the mAbs panel were modeled using the automated AutoFv/CCG3 protocol50 in Molecular Operating Environment (MOE) obtained from the Chemical Computing Group as described previously.50 The 3D homology models of the Fv regions were used to extract several features for machine learning, including the SASA at the residue level. SASA values were calculated empirically using the POPS software.51 In addition to using the total SASA of the whole residue (‘TotSasaRes’), the hydrophobic partition (‘PhobSasaRes’) and the hydrophilic partition (‘PhilSasaRes’) of the SASA of each residue in the context of the mAb structure were also measured. These values were included under the assumption that high SASA values indicate exposure to water and hence higher susceptibility to ROS and oxidation. Another parameter ‘NoverlapRes’ (also obtained from the POPS software) measures the total number of overlaps between atoms in the query Met residue and its proximal residues’ atoms in the Fv structure. Two atoms are considered to overlap if the distance between them is more than the sum of their van der Waals radii and two times the solvent radius. This parameter was included under the assumption that higher number of overlaps would mean a higher number of interactions and lesser chance of interaction with ROS.
Dynamics-based feature extraction
In order to obtain a reliable measure of residue fluctuations in the mAbs, we adopted the coarse-grained models of protein dynamics referred to as elastic network models (ENMs).52–54 In ENMs, the molecules are represented in a simplified manner using a bead-spring model. Figuratively, in the case of proteins, the beads are the C-alpha (Cα) atoms; with one bead per residue. Bead interactions are assumed to be restricted to only nearby beads (within a specified distance cutoff, here 13 Å). Interactions between beads in the model are simulated by springs. Despite their coarse-grained nature, ENMs capture the overall geometry of proteins efficiently and modes from ENMs have been shown to be significantly accurate at reproducing experimental temperature factors for a number of crystal structures.55–58 All the elastic network models and fluctuations were implemented using the ‘bio3d’ package59 in R. We specifically used two variations of elastic network models implemented in the bio3d package: the anisotropic network model (ANM)60 and the Hinsen’s network model61 (referred to as HNM in this paper). In the ANM, all springs between residue i and j are assumed to have the same stiffness (spring constant ) whereas in the HNM, springs between sequentially adjacent Cα atoms are represented as and those between non-adjacent Cα atoms as where is the distance between residues and ; and and are constants as previously discussed.61 In both models, the potential energy of the system is measured to be proportional to the sum of squares of displacements of the beads from their equilibrium positions. The hessian matrix of the double derivatives of the potential function is then constructed and eigen-decomposed to derive the modes and their frequencies (square root of the eigenvalues). For both network models, the mean square fluctuation of residues (Cα atoms) can be obtained from the corresponding elements of the pseudo inverse of the hessian matrix.
Random forest prediction model
RF model was implemented using the ‘randomForest’ package62 in R (available from the Comprehensive R Archive Network (CRAN) repository), which is an implementation of Leo Breiman’s algorithm.29 All adjustable parameters were set to default values and all predictions shown on the training set are OOB,31 equivalent to a cross-validated performance. The model thus generated is validated by using it to predict the cases in the independent test set and measuring the performance therein.
Assessing the importance of features
One of the main advantages of RFs is that they can easily provide a measure of feature importance. The importance of a variable (feature) can be measured directly from how the performance of the model is affected when values of this variable are perturbed, or from how tightly the variable fits the data in the process of constructing the decision tree, as explained below. Accordingly, the ‘randomForest’ package provides two such measures: 1) ‘%IncMSE’: the percentage increase in mean square error (MSE) when the values on that feature are randomly permuted (averaged across all trees); and 2) ‘IncNodePurity’: the percentage increase in node purity of the descendant nodes with respect to the parent nodes when split using that variable. Impurity at a node is measured as the residual sum of squares (deviations from the actual experimental values) and the total decrease in node impurities from splitting on that variable is averaged across all trees to obtain the second measure.
Funding Statement
This work was supported by Genentech, Inc., a member of the Roche Group.
Abbreviations
- AAPH
2,2ʹ-Azobis(2-amidinopropane) dihydrochloride
- ANM
anisotropic network model
- AUC
area under the curve
- CDR
complementarity-determining region
- ENM
elastic network models
- HNM
Hinsen’s network model
- LC
liquid chromatography
- mAb
monoclonal antibody
- MCC
Matthews Correlation Coefficient
- MDA
mean decrease in accuracy
- MDG
mean decrease in Gini
- MetO
methionine sulfoxide
- MLR
multiple linear regression
- MOE
Molecular Operating Environment
- MS
mass spectrometer
- MSE
mean square error
- OOB
‘out-of-bag’
- RF
random forest
- RMSE
root mean square error
- ROC
receiver-operating characteristic
- ROS
reactive oxygen species
- SASA
solvent accessible surface area
- UHPLC
ultra-high performance liquid chromatography
- XIC
extracted ion chromatograms
Acknowledgments
We thank Samarkand Estee, Yilma Adem, Norman Shih and Benny Freistadt for stressing the molecules; Michael Madonna for help in collecting and compiling the LC/MS peptide map molecule assessment data; and Tom Patapoff, Lydia Beasley, Jeff Blaney, Bill Galush, Isidro Hotzel, Bill Forrest, Binqing Wei, Greg Lazar and Paul Carter for fruitful discussions & support.
Supplemental material
Supplemental data for this article can be accessed here.
Disclosure statement
All authors were paid employees of Genentech Inc., when this work was conducted. No potential conflict of interest was reported by the authors.
References
- 1.Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1–26. doi: 10.1007/978-1-61779-921-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2017;9:86–120. doi: 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh G. Biopharmaceutical Benchmarks 2014. Nat Biotechnol. 2014;32:992–1000. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 4.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci. 2017;114:944–949. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swann PG, Tolnay M, Muthukkumar S, Shapiro MA, Rellahan BL, Clouse KA. Considerations for the development of therapeutic monoclonal antibodies. Curr Opin Immunol. 2008;20:493–499. doi: 10.1016/j.coi.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 6.King AC, Woods M, Liu W, Lu Z, Gill D, Krebs MRH. High-throughput measurement, correlation analysis, and machine-learning predictions for pH and thermal stabilities of Pfizer-generated antibodies. Protein Sci. 2011;20:1546–1557. doi: 10.1002/pro.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voynov V, Chennamsetty N, Kayser V, Helk B, Trout BL. Predictive tools for stabilization of therapeutic proteins. MAbs. 2009;1:580–582. doi: 10.4161/mabs.1.6.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Prediction of aggregation prone regions of therapeutic proteins. J Phys Chem B. 2010;114:6614–6624. doi: 10.1021/jp911706q. [DOI] [PubMed] [Google Scholar]
- 9.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111:18601–18606. doi: 10.1073/pnas.1421779112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomar DS, Li L, Broulidakis MP, Luksha NG, Burns CT, Singh SK, Kumar S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. MAbs. 2017;9:476–489. doi: 10.1080/19420862.2017.1285479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs. 2016;8:43–48. doi: 10.1080/19420862.2016.1196521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Mølhøj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One. 2014;9:e100736. doi: 10.1371/journal.pone.0100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Q, Huang M, Lewis MJ, Hu P. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs. 2018;1–12. doi: 10.1080/19420862.2018.1478646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50:301–307. doi: 10.1080/713803739. [DOI] [PubMed] [Google Scholar]
- 15.Moskovitz J, Weissbach H, Brot N. Cloning the expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proc Natl Acad Sci U S A. 1996;93:2095–2099. doi: 10.1073/pnas.93.5.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- 18.Kim YH, Berry AH, Spencer DS, Stites WE. Comparing the effect on protein stability of methionine oxidation versus mutagenesis: steps toward engineering oxidative resistance in proteins. Protein Eng. 2001;14:343–347. doi: 10.1093/protein/14.5.343. [DOI] [PubMed] [Google Scholar]
- 19.Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, Li L, Brems DN, Remmele RL Jr.. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47:5088–5100. doi: 10.1021/bi702238b. [DOI] [PubMed] [Google Scholar]
- 20.Niu S, Hu -L-L, Zheng -L-L, Huang T, Feng K-Y, Cai Y-D, Li H-P, Li Y-X, Chou K-C. Predicting protein oxidation sites with feature selection and analysis approach. J Biomol Struct Dyn. 2012;29:650–658. doi: 10.1080/07391102.2011.672629. [DOI] [PubMed] [Google Scholar]
- 21.Chu JW, Brooks BR, Trout BL. Oxidation of methionine residues in aqueous solutions: free methionine and methionine in granulocyte colony-stimulating factor. J Am Chem Soc. 2004;126:16601–16607. doi: 10.1021/ja047044i. [DOI] [PubMed] [Google Scholar]
- 22.Chu JW, Yin J, Brooks BR, Wang DIC, Ricci MS, Brems DN, Trout BL. A comprehensive picture of non-site specific oxidation of methionine residues by peroxides in protein pharmaceuticals. J Pharm Sci. 2004;93:3096–3102. doi: 10.1002/jps.20207. [DOI] [PubMed] [Google Scholar]
- 23.Chu JW, Yin J, Wang DIC, Trout BL. A structural and mechanistic study of the oxidation of methionine residues in hPTH(1-34) via experiments and simulations. Biochemistry. 2004;43:14139–14148. doi: 10.1021/bi049151v. [DOI] [PubMed] [Google Scholar]
- 24.Chu J-W, Yin J, Wang DIC, Trout BL. Molecular dynamics simulations and oxidation rates of methionine residues of granulocyte colony-stimulating factor at different pH values. Biochemistry. 2004;43:1019–1029. doi: 10.1021/bi0356000. [DOI] [PubMed] [Google Scholar]
- 25.Chennamsetty N, Quan Y, Nashine V, Sadineni V, Lyngberg O, Krystek S. Modeling the Oxidation of Methionine Residues by Peroxides in Proteins. J Pharm Sci. 2015;104:1246–1255. doi: 10.1002/jps.24340. [DOI] [PubMed] [Google Scholar]
- 26.Yang R, Jain T, Lynaugh H, Nobrega RP, Lu X, Boland T, Burnina I, Sun T, Caffry I, Brown M, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs. 2017;9:646–653. doi: 10.1080/19420862.2017.1290753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agrawal NJ, Dykstra A, Yang J, Yue H, Nguyen X, Kolvenbach C, Angell N. Prediction of the hydrogen peroxide induced methionine oxidation propensity in monoclonal antibodies. J Pharm Sci. 2018;107:1282–1289. doi: 10.1016/j.xphs.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Aledo JC, Cantón FR, Veredas FJ. A machine learning approach for predicting methionine oxidation sites. BMC Bioinformatics. 2017;18:430. doi: 10.1186/s12859-017-1848-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breiman L. Random forests. Mach Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 30.Breiman L. Bagging predictors. Mach Learn [Internet]. 1996;24:123–140. http://link.springer.com/10.1007/BF00058655. [Google Scholar]
- 31.Breiman L. Out-of-bag estimation [Internet]. 1996. https://www.stat.berkeley.edu/~breiman/OOBestimation.pdf.
- 32.Almagro JC, Beavers MP, Hernandez-Guzman F, Maier J, Shaulsky J, Butenhof K, Labute P, Thorsteinson N, Kelly K, Teplyakov A, et al. Antibody modeling assessment. Proteins Struct Funct Bioinforma. 2011;79:3050–3066. doi: 10.1002/prot.23130. [DOI] [PubMed] [Google Scholar]
- 33.Almagro JC, Teplyakov A, Luo J, Sweet RW, Kodangattil S, Hernandez-Guzman F, Gilliland GL. Second Antibody Modeling Assessment (AMA-II). Proteins Struct Funct Bioinforma. 2014;82:1553–1562. doi: 10.1002/prot.24567. [DOI] [PubMed] [Google Scholar]
- 34.Ippolito GC, Hoi KH, Reddy ST, Carroll SM, Ge X, Rogosch T, Zemlin M, Shultz LD, Ellington AD, VanDenBerg CL, et al. Antibody repertoires in humanized NOD-scid-IL2Rγnull mice and human B cells reveals human-like diversification and tolerance checkpoints in the mouse. PLoS One. 2012;7:e35497. doi: 10.1371/journal.pone.0035497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins AM, Wang Y, Roskin KM, Marquis CP, Jackson KJL, Schroeder H, Cavacini L, Tonegawa S, Alt F, Baltimore D, et al. The mouse antibody heavy chain repertoire is germline-focused and highly variable between inbred strains. Philos Trans R Soc Lond B Biol Sci. 2015;370:S41–52. doi: 10.1098/rstb.2014.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dion MZ, Wang YJ, Bregante D, Chan W, Andersen N, Hilderbrand A, Leiske D, Salisbury CM. The use of a 2,2ʹ-Azobis (2-Amidinopropane) dihydrochloride stress model as an indicator of oxidation susceptibility for monoclonal antibodies. J Pharm Sci. 2017;107:550–558. doi: 10.1016/j.xphs.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Kunik V, Peters B, Ofran Y. Structural consensus among antibodies defines the antigen binding site. PLoS Comput Biol. 2012;8:e1002388. doi: 10.1371/journal.pcbi.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu HS, Fausset PR, Narhi LO, Horan T, Shinagawa K, Shimamoto G, Boone TC. Chemical modification and site-directed mutagenesis of methionine residues in recombinant human granulocyte colony-stimulating factor: effect on stability and biological activity. Arch Biochem Biophys. 1999;362:1–11. doi: 10.1006/abbi.1998.1032. [DOI] [PubMed] [Google Scholar]
- 39.Griffiths SW, Cooney CL. Relationship between protein structure and methionine oxidation in recombinant human α1-antitrypsin. Biochemistry. 2002;41:6245–6252. doi: 10.1021/bi025599p. [DOI] [PubMed] [Google Scholar]
- 40.Meyer JD, Ho B, Manning MC. Effects of conformation on the chemical stability of pharmaceutically relevant polypeptides In: Carpenter JF, Manning MC, editors. Rational design of stable protein formulations. Pharmaceutical biotechnology. Vol. 13 Boston (MA): Springer; 2002. p. 85–107. [DOI] [PubMed] [Google Scholar]
- 41.Veredas FJ, Cantón FR, Aledo JC. Methionine residues around phosphorylation sites are preferentially oxidized in vivo under stress conditions. Sci Rep. 2017;7:40403. doi: 10.1038/srep40403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem. 2012;287:34979–34991. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chao CC, Ma YS, Stadtman ER. Modification of protein surface hydrophobicity and methionine oxidation by oxidative systems. Proc Natl Acad Sci U S A. 1997;94:2969–2974. doi: 10.1073/pnas.94.7.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji JA, Zhang B, Cheng W, Wang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: mechanisms and stabilization. J Pharm Sci. 2009;98:4485–4500. doi: 10.1002/jps.21746. [DOI] [PubMed] [Google Scholar]
- 46.Andersen N, Vampola L, Jain R, Alvarez M, Chamberlain S, Hilderbrand A. Rapid UHPLC HRMS Peptide Mapping for Monoclonal Antibodies. Am Pharm Rev. 2014;17(6) https://www.americanpharmaceuticalreview.com/Featured-Articles/169253-Rapid-UHPLC-HRMS-Peptide-Mapping-for-Monoclonal-Antibodies/ [Google Scholar]
- 47.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol. 1992;224:487–499. doi: 10.1016/0022-2836(92)91010-M. [DOI] [PubMed] [Google Scholar]
- 48.Te WT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 1970;132:211–250. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. IMGT®, the international ImMunoGeneTics information system®. Nucleic Acids Res. 2009;37:D1006–12. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maier JKX, Labute P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins Struct Funct Bioinforma. 2014;82:1599–1610. doi: 10.1002/prot.24576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fraternali F, Cavallo L. Parameter optimized surfaces (POPS): analysis of key interactions and conformational changes in the ribosome. Nucleic Acids Res. 2002;30:2950–2960. doi: 10.1093/nar/gkf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanejouand Y-H. Elastic Network Models: theoretical and Empirical Foundations In: Monticelli L, Salonen E, editors. Biomolecular simulations. Methods in molecular biology (Methods and Protocols). New York, NY: Humana Press; 2013. p. 601–616. [DOI] [PubMed] [Google Scholar]
- 53.Jernigan RL, Yang L, Song G, Kurkckuoglu O, Doruker P. Elastic network models of coarse-grained proteins are effective for studying the structural control exerted over their dynamics In: Voth GA, editor. Coarse-graining of condensed phase and biomolecular systems. Boca Raton (FL): CRC Press; 2009. p. 237–254. [Google Scholar]
- 54.Bahar I, Lezon TR, Yang L-W, Eyal E. Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kundu S, Melton JS, Sorensen DC, Phillips GN. Dynamics of proteins in crystals: comparison of experiment with simple models. Biophys J. 2002;83:723–732. doi: 10.1016/S0006-3495(02)75203-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, Song G, Jernigan RL. Comparisons of experimental and computed protein anisotropic temperature factors. Proteins. 2009;76:164–175. doi: 10.1002/prot.v76:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sankar K, Mishra SK, Jernigan RL. Comparisons of protein dynamics from experimental structure ensembles, molecular dynamics ensembles, and coarse-grained elastic network models. J Phys Chem B. 2018;122:5409–5417. doi: 10.1021/acs.jpcb.7b11668. [DOI] [PubMed] [Google Scholar]
- 58.Mishra SK, Sankar K, Jernigan RL. Altered dynamics upon oligomerization corresponds to key functional sites. Proteins Struct Funct Bioinforma. 2017;85:1422–1434. doi: 10.1002/prot.25302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 60.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hinsen K, Petrescu AJ, Dellerue S, Bellissent-Funel MC, Kneller GR. Harmonicity in slow protein dynamics. Chem Phys. 2000;261:25–37. doi: 10.1016/S0301-0104(00)00222-6. [DOI] [Google Scholar]
- 62.Liaw A, Wiener M. Classification and Regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.