

SUMMARY
B-cell activation during normal immune responses and oncogenic transformation impose increased metabolic demands on B-cells and their ability to retain redox homeostasis. While the serine/threonine-protein phosphatase 2A (PP2A) was identified as tumor suppressor in multiple types of cancer, our genetic studies revealed an essential role of PP2A in B-cell tumors. Thereby, PP2A redirects glucose carbon utilization from glycolysis to the pentose phosphate pathway (PPP) to salvage oxidative stress. This unique vulnerability reflects constitutively low PPP activity in B-cells and transcriptional repression of G6PD and other key PPP enzymes by the B-cell transcription factors PAX5 and IKZF1. Reflecting B-cell-specific transcriptional PPP-repression, glucose carbon utilization in B-cells is heavily skewed in favor of glycolysis resulting in lack of PPP-dependent antioxidant protection. These findings reveal a gatekeeper function of the PPP in a broad range of B-cell malignancies that can be efficiently targeted by small molecule inhibition of PP2A and G6PD.
Graphical Abstract
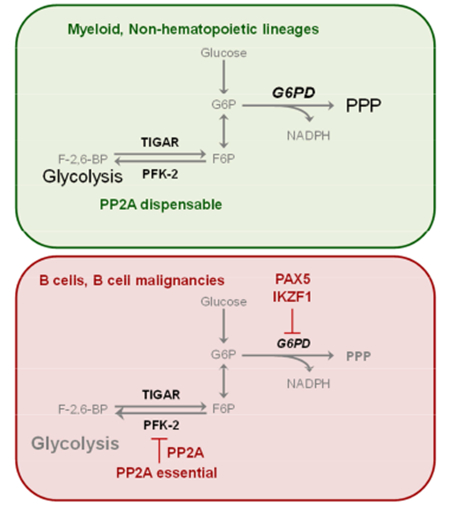
INTRODUCTION
The paradigm of targeted therapy of cancer is based on kinase inhibitors, e.g. the prototypic tyrosine kinase inhibitor (TKI) imatinib for the treatment of BCR-ABL1-driven (Ph+) leukemias (Hochhaus et al., 2017). Oncogenic signaling frequently involves aberrant activation of kinases, leading to the notion that phosphatases opposing oncogenic kinase signaling have tumor suppressive activity. For instance, the PTEN (Li et al., 1997) and INPP5D (Lakhanpal et al., 2010) phosphatases are classical tumor suppressors and frequently mutated across multiple types of cancer (http://cancer.sanger.ac.uk/cosmic). Likewise, multiple subunits of the serine-threonine phosphatase PP2A are recurrently deleted or mutated in a wide range of cancer subtypes (Wang et al., 1998), demonstrating that PP2A, like PTEN and INPP5D, represents a tumor suppressor. Recent studies from our group revealed that B cell-derived leukemias are uniquely dependent on PTEN and INPP5D function. Inducible deletion of Pten (Shojaee et al., 2016) and Inpp5d (Chen et al., 2015) phosphatases in B-lineage leukemia cells caused hyperactivation of B-cell receptor (BCR)-related tyrosine kinases, which mimics overwhelming signaling strength from an autoreactive BCR (Müschen 2018). Despite malignant transformation, B-lymphoid leukemia cells fully retain their sensitivity to negative selection and cell death at autoimmunity checkpoints. Interestingly, besides PTEN and INPP5D, also PP2A functions as a critical regulator of negative selection and autoimmunity (Apostolidis et al., 2016; Sharabi et al., 2017). Because of its role in cancer and autoimmunity, we studied here the function of PP2A in normal B cells and B-cell malignancies.
RESULTS
B-cell lineage-specific effects of PP2A ablation
PP2A functions as a dimeric holoenzyme composed of a scaffolding subunit A and a catalytic subunit C, which often associates with the regulatory subunit B. As a model for loss of PP2A holoenzyme activity, we studied here genetic ablation of the Ppp2r1a gene encoding the PP2A scaffold subunit Aα (PP2A Sub A), which is essential for PP2A activity (Ruediger et al., 2011). To test the function of PP2A in normal B-cell development and B-lymphoid leukemia and lymphoma, we crossed mice carrying floxed Ppp2r1a alleles (Ruediger et al., 2011) with the Mb1-Cre deleter strain (Hobeika et al., 2006). Genotyping of flow-sorted early B-cells from Ppp2r1afl/fl Mb1-Cre mice confirmed efficient Cre-mediated deletion from fraction B (pro-B-cell stage; Figure S1A). Heterozygous Ppp2r1a+/fl Mbl-Cre mice were phenotypically normal and studied as normal control. Phenotypic analyses of bone marrow and splenic B-cell populations revealed normal pro-B (fraction B) and early pre-B-cell (fraction C) numbers in Ppp2r1afl/fl Mb1-Cre mice. However, B-cell populations were diminished by >10-fold once they reached pre-B-cell receptor (pre-BCR)-dependent stages of development (Buchner et al., 2015; fraction C’; Figure 1A-B). Mature splenic B-cells were barely detectable upon deletion of Ppp2r1a and numbers were reduced by >20-fold, suggesting that PP2A is also required for survival of mature B-cells (Figure 1C). These results indicate a stage-specific function of PP2A throughout pre-BCR- and BCR-dependent stages of B-cell development. Detailed flow cytometry analyses revealed that ablation of Ppp2r1a resulted in selective loss of pre-BCR- (μ-heavy chain) and BCR- (IgM, IgD, Igk) dependent B-cell populations (Figure 1D). Cre-mediated deletion of Ppp2r1a not only abrogated expression of the PP2A scaffold Sub A but also destabilized protein expression of catalytic Sub C (Figure 1E; Chen et al., 2005). Conversely, retroviral expression of Sub A rescued normal expression levels of both Sub A and Sub C despite Cre-mediated deletion of Ppp2r1a. While Cre-mediated deletion of Ppp2r1a reduced enzymatic PP2A activity to background levels, retroviral expression of Ppp2r1a (Sub A) increased PP2A enzymatic activity by 7-fold (Figure 1F-G). Inducible ablation of Ppp2r1a under cell culture conditions resulted in acute cell death of both pre-BCR+ early and BCR+ mature B-cells (Figure 1H). Interestingly, inducible activation of Cre in Ppp2r1afl/fl myeloid progenitor cells from the same mice had no deleterious effect and slightly increased survival and proliferation, suggesting that dependency on PP2A function is B-cell-specific (Figure 1H). To directly validate that PP2A regulates cell survival in a strict B-cell lineage-dependent manner, Ppp2r1afl/fl B-cell acute lymphoblastic leukemia (ALL) cells were reprogrammed into the myeloid lineage by inducible overexpression of the myeloid transcription factor C/EBPα (Figure 1I; Xie et al., 2004). While deletion of Ppp2r1a in CD19+ B-ALL cells caused acute cell death, viability of CD11b+ C/EBPα-reprogrammed myeloid leukemia cells remained unchanged (Figure 1I).
Figure 1: PP2A function is essential for B cell survival.
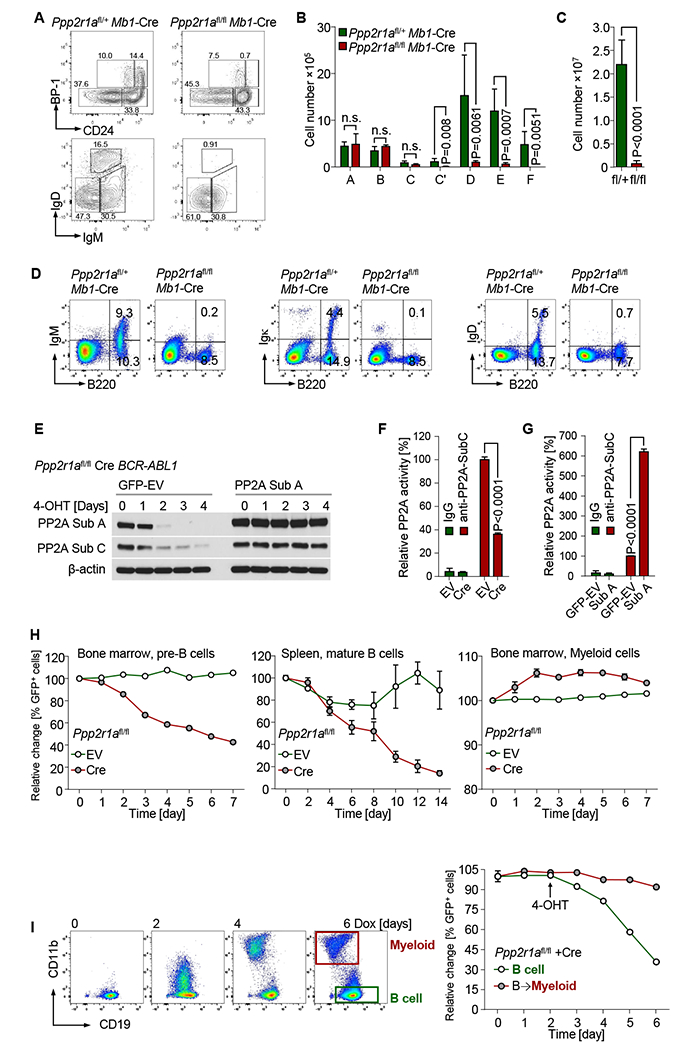
(A) B cell development in Ppp2r1afl/fl Mb 1-Cre mice was examined by flow cytometry. Hardy fractions of B cell subsets isolated from bone marrow were analyzed. Absolute cell numbers were calculated for B cell subsets from bone marrow (B) and spleen (C). Surface staining of BCR components was performed by flow cytometry (D). Ppp2r1afl/fl BCR-ABL1 ALL cells were transduced with 4-hydroxy-tamoxifen (4-OHT)-inducible Cre-ERT2 (Cre) or ERT2-vector (EV). Cells were subsequently transduced with PP2A-subA-IRES-GFP (PP2A Sub A) or GFP-Vector (GFP-EV). (E) PP2A expression in Cre-transduced cells after 4-OHT treatment was measured by Western blot. PP2A Thr/Ser phosphatase activity was measured in Cre- or EV-transduced cells after 3 days of 4-OHT treatment (F). Cre-GFP+ Cre cells transduced with Sub A or GFP-EV were sorted for PP2A-activity measurement after 3-days 4-OHT treatment (G). Bone marrow pre-B cells, splenic B cells and myeloid leukemia cells from Ppp2r1afl/fl mice were transduced with Cre or EV (H). Percentages of GFP+ cells were determined by flow cytometry at time points indicated following 4-OHT treatment. (I) Ppp2r1afl/fl BCR-ABL1 B-ALL cells were transduced with doxycycline (Dox)-inducible C/EBPα (TetOn-Cebpa) and subsequently transduced with Cre. Percentages of GFP+ cells in B-lymphoid (CD19+ CD11b−) and B→myeloid (CD19− CD11b+ ) populations were measured by flow cytometry at time points indicated following Dox (4-OHT treatment started at time point indicated by arrow). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
B-ALL cell survival requires PP2A activity.
Inducible ablation of Ppp2r1a induced rapid depletion of both BCR-ABLl and NRASG12D driven B-ALL cells (Figure S1B-C) and suppressed colony forming ability by >10-fold (Figure 2A-B). Conversely, overexpression of Ppp2r1a (Sub A) increased competitive fitness and induced outgrowth of Sub A-overexpressing cells (Figure 2C). Likewise, Sub A-overexpression quadrupled the number of colonies formed after plating in semisolid methylcellulose (Figure 2C). Loss of colony formation capacity upon Ppp2r1a deletion is consistent with DNA-damage (H2AX-phosphorylation) and activation of Arf and p53 (Figure 2D-E). Ablation of Ppp2r1a increased cell death but only slightly affected the cell cycle of B-ALL cells (Figure S1D-G). Mirroring the apoptotic phenotype of B-ALL cells upon Ppp2r1a deletion, upregulation of p53 was paralleled by downregulation of Bcl2 (Figure 2D). Taken together, these genetic in vitro studies show that PP2A activity is critical for the survival of B-ALL cells.
Figure 2: PP2A is required for the survival of BCR-ABL1 and NRASG12D ALL cells.
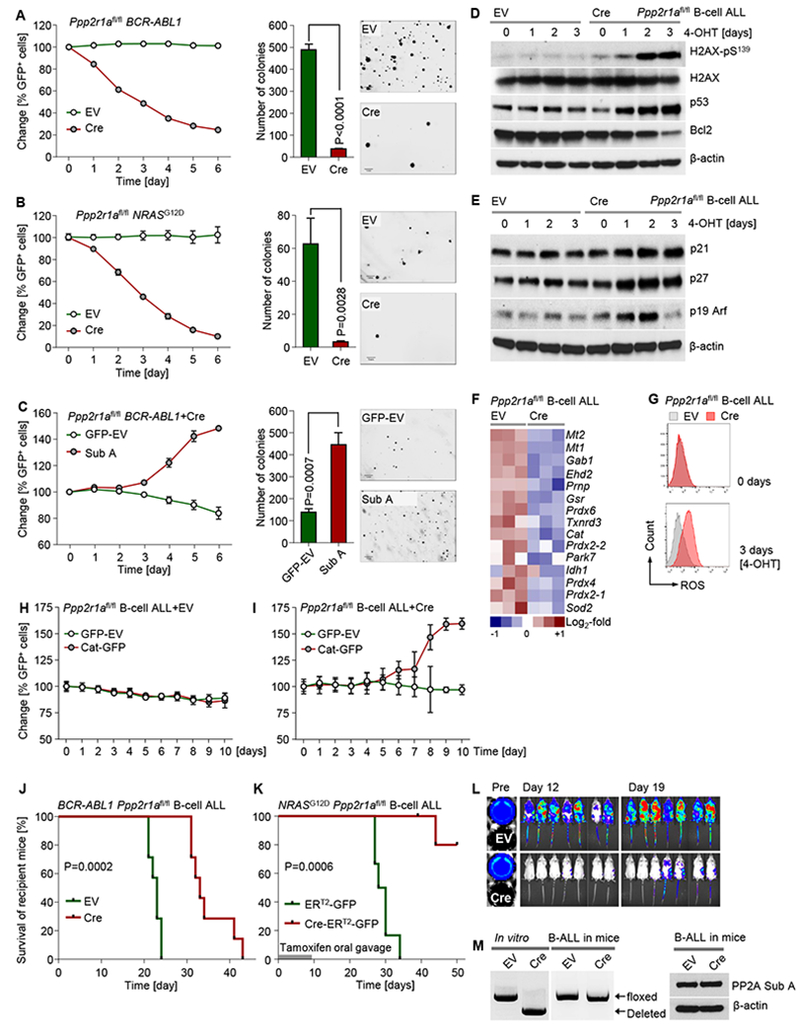
Ppp2r1afl/fl BCR-ABL1 (A) and NRASG12D B-ALL (B) cells were transduced with Cre-ERT2-IRES-GFP (Cre) or ERT2-IRES-GFP-vector control (EV). Percentages of GFP+ cells were measured following 4-OHT treatment. Cre or EV-transduced ALL cells were treated for 3 days with 4-OHT and plated in methylcellulose to perform colony formation assays. Photomicrographs of colonies were taken for colony counting (scale bar indicating 1 mm). (C) The fractions of GFP+ BCR-ABL1 B-ALL cells transduced with Sub A or GFP-EV were measured following 4-OHT treatment. GFP+-sorted cells transduced with Sub A or GFP-EV were studied by colony formation assay. Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with Cre or EV were treated with 4-OHT. (D) Western blot to measure phosphorylation of H2AX and expression of p53, Bcl2 (D) and p21, p27 and p19 Arf (E) was performed after 4-OHT treatment. (F) A heatmap to depict gene expression changes of antioxidant genes (microarray data; GSE83742) of Ppp2r1afl/fl B-ALL cells after 3-days 4-OHT treatment (Cre-mediated deletion) is shown. (G) ROS levels were measured by flow cytometry as DCF-fluorescence. Ppp2r1afl/fl BCR-ABL1 ALL cells transduced with EV (H) or Cre (I) were transduced with Catalase-IRES-GFP (Cat-GFP) or GFP-vector (EV). Percentages of GFP+ cells were measured following 4-OHT treatment. (J) Ppp2r1afl/fl BCR-ABL1 ALL cells were labeled with firefly luciferase and transduced with Cre or EV. 2 days after transduction, GFP+ cells were injected intravenously into recipient NSG mice (1×106 cells/mouse; 7 mice in each group). B-ALL cell burden was monitored by luciferase bioimaging (L). Ppp2r1afl/fl NRASG12D B-ALL cells were transduced with inducible Cre or EV, sorted for GFP+ cells and injected intravenously into recipient NSG mice (3×106 cells/mouse; 6 mice in each group) followed by 7 days of tamoxifen treatment via oral gavage to induce deletion in vivo (K). In (J-K), Kaplan-Meier estimates were used to plot the survival probabilities for mice that were transplanted with Cre- and EV-transduced cells. Log-rank test was used to assess statistical significance. In experiment (J), mice that showed signs of illness from leukemia were sacrificed to collect splenic B-ALL cells. GFP+ cells from recipient mice were used for PCR and Western blot to determine Ppp2r1a genotype and PP2A expression, respectively (M). Except survival analyses, other experimental data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Functional consequences of PP2A deletion in B-ALL cells
To study the mechanistic basis of the unexpected dependency of B-ALL cells on PP2A enzymatic activity, we determined the biochemical consequences of Ppp2r1a-deletion. Measurement of changes of the phosphorylation status of signaling molecules in a phospho-protein array revealed moderately increased phosphorylation of Erk and Akt in response to Ppp2r1a-deletion (Figure S2A). Increased activation of PI3K and AKT-mTOR may indeed contribute to toxicity upon deletion of Ppp2r1a, because inhibition of PI3K (Idelalisib) and mTOR (Rapamycin) signaling partially rescued cell death in response to Ppp2r1a-deletion (Figure S2B-D). Downstream of AKT, deletion of Ppp2r1a not only increased activation of mTOR-S6 kinase signaling but also massive phosphorylation and inactivation of FoxO1 and FoxO3a (Figure S3A), which are important mediators of redox equilibrium (Nemoto, 2002). To elucidate the significance of FoxO1- and FoxO3a-phosphorylation and inactivation, we determined the effects of inducible Foxo1-deletion in B-cell ALL cells. FoxO factors negatively regulate B-cell proliferation and are inactivated by AKT-mediated phosphorylation (Yusuf et al., 2004). However, tamoxifen-inducible deletion of Foxo1 in BALL cells abrogated colony formation and caused acute cell death (Figure S4B-C). These findings demonstrate an important role of Foxo1 in B cell leukemogenesis.
Antioxidant protection in B cells depends on PP2A-function
Since FoxO proteins promote an antioxidant transcriptional program (Nogueira et al., 2008), we examined whether Ppp2r1a-deletion affects antioxidant functions of transformed B cells. Multiple antioxidant genes including several known FoxO targets (catalase, Cat; superoxide dismutase 2, Sod2) were down-regulated upon inducible Ppp2r1a deletion (Figure 2F). Gene set enrichment analyses showed a global reduction of expression levels for antioxidant genes (P<0.01; Table S1 and Figure S3F), which was paralleled by intracellular accumulation of ROS (8-fold increased; Figure 2G). For functional validation of a potential mechanistic contribution of FoxO factors and the antioxidant catalase, we performed rescue experiments based on overexpression of constitutively active Foxo1 and Foxo3a as well as catalase. Cat-overexpression conferred a significant survival advantage upon inducible Ppp2r1a deletion (Figure 2H-I). In contrast, overexpression of constitutively active Foxo1 and Foxo3a mutants was toxic in B-ALL cells regardless of PP2A-ablation (Figure S3D-E). Since PP2A-mediated activation of Foxo1 induces apoptosis through activation of Bim (Yan et al., 2008), overexpression of catalase may predominantly mediate antioxidant protection while Foxo1 and Foxo3 exacerbate the pro-apoptotic phenotype of B-ALL cells upon deletion of Ppp2r1a.
Validation of PP2A as therapeutic target in B cell lineage leukemia
To test whether ablation of Ppp2r1a is sufficient to prevent development of B-ALL in an in vivo setting, Ppp2r1afl/fl BCR-ABL1- and NRASG12D-driven B-ALL cells were intravenously injected into sublethally irradiated NOD-scid Il2rg-/- (NSG) mice in the presence or absence of Cre (Figure 2J-K). In vivo leukemogenesis was monitored through bioimaging of luciferase labeled ALL cells (Figure 2L). In line with in vitro results, recipient mice injected with Ppp2r1afl/fl B-ALL cells showed reduced leukemia burden upon deletion of Ppp2r1a and significantly prolonged overall survival in both BCR-ABL1- and NRASG12D-driven B-ALL models (Figure 2J-K). To determine whether mice in the Ppp2r1afl/fl +Cre group succumbed to leukemia because B-ALL can develop in the absence of PP2A (B-ALL with deleted Ppp2r1a alleles) or because some B-ALL clones escaped Cre-mediated deletion (B-ALL retaining Ppp2r1a alleles), we studied the genotype of B-ALL cells that were isolated from enlarged spleens when the mice were euthanized (Figure 2M). Genomic and protein expression studies revealed that in all 7 cases, B-ALL developed from Ppp2r1afl/fl clones that evaded Cre-mediated deletion and expressed normal levels of PP2A. Given that <0.1% of Cre-GFP+ cells escape deletion, these findings demonstrate the strong selective pressure in favor of B-ALL clones that retained Ppp2r1afl/fl alleles.
PP2A restricts glycolytic activity in B-lymphoid but not myeloid leukemia cells
AMPK is negatively regulated by PP2A (Wu et al., 2007) and the active phospho-AMPK-T172 was identified among the upregulated phosphoproteins upon Ppp2r1a-deletion (Figure S2A). We recently found that AMPK-activation in B-ALL cells promotes glycolysis (Chan et al., 2017), suggesting that PP2A negatively regulates glycolysis in B cells. Here we showed that inducible deletion of Ppp2r1a amplified mTORC1 signaling (Figure S3A), which was previously shown to stimulate glycolysis as a result of PI3K signaling (Düvel et al., 2010). For this reason, we studied the impact of Ppp2r1a-deletion on glucose and energy metabolism in B-cell and myeloid lineage leukemias. Inducible ablation of Ppp2r1a in B cells substantially increased glucose consumption, ATP levels and lactate production (by 3-fold; Figure S4A). Seahorse metabolic analysis revealed that Ppp2r1a-deletion increased glycolytic activity in B-lymphoid but not myeloid leukemia cells (Figure 3A-B). Mitochondrial respiration remained unchanged after Ppp2r1a-deletion in both B-lymphoid and myeloid leukemia cells (Figure S4B-C). While glycolysis primarily generates metabolic intermediates and ATP, the pentose phosphatase pathway (PPP) has a critical antioxidant function to generate reducing equivalents, in the form of NADPH (Cairns et al., 2011). Reduced nicotinamide adenine dinucleotide phosphate (NADPH) represents a critical product of the PPP and has important reducing potential to maintain redox balance (Patra and Hay, 2014). For instance, NADPH can reduce glutathione disulfide (GSSG) to the sulfhydryl form glutathione (GSH), which can salvage oxidative stress (Cairns et al., 2011). In addition, the PPP is critical for DNA replication in proliferating leukemia cells and generates ribose 5-phosphate (R5P), used in the de novo synthesis of ribonucleotides and later nucleic acids.
Figure 3: PP2A is essential in B-lymphoid but not myeloid cells to balance glucose carbon utilization via glycolysis and PPP.
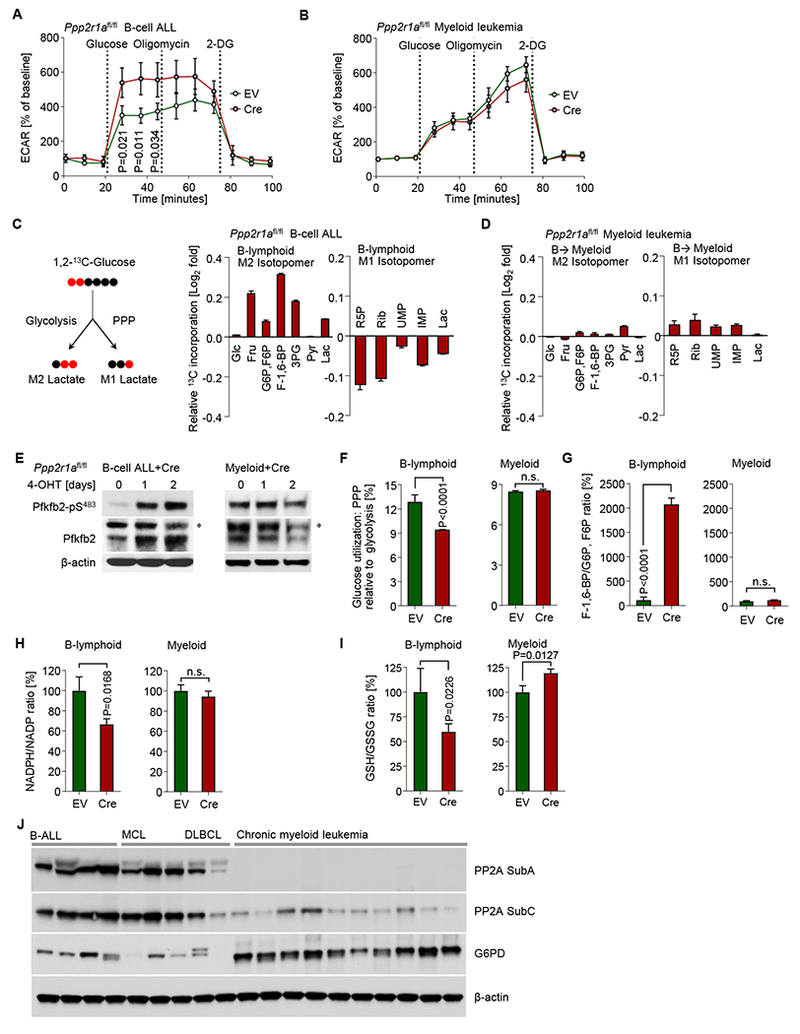
Extracellular acidification rates (ECAR) were measured in (A) B-lymphoid and (B) myeloid Ppp2r1afl/fl BCR-ABL1 leukemia cells that were transduced with Cre-ERT2 (Cre) or ERT2-vector (EV). Cells were treated with 4-OHT for 2 days prior to ECAR measurements. (C-D) Ppp2r1afl/fl BCR-ABL1 ALL cells (B-lymphoid) were cultured with 25 mmol/l 1,2-213 C D-glucose for 24 hours, then harvested to extract metabolites for LC-MS based profiling. Ppp2r1afl/fl BCR-ABL1 ALL cells carrying TetOn-Cebpa were transduced with Cre or EV and were treated for 5-days with Dox for B→myeloid reprogramming. Cre-induced changes of cellular amounts of metabolites that were utilized in glycolysis (M2 13C isotopomer) or the pentose phosphate pathway (PPP; M1 13C isotopomer) are shown on a Log2-fold scale (Cre relative to EV) in B-lymphoid (C) and B→myeloid (D) cells. Phosphorylation levels of Pfkfb2 at S-483 were measured by Western blot in Ppp2r1afl/fl B-lymphoid and myeloid leukemia cells after 4-OHT treatment for Cre-mediated deletion of Ppp2r1a. Asterisks denote non-specific bands. (F) Cellular lactate M1/M2 ratio is measured from metabolites profiling data and indicates the relative amount of glucose carbon utilization via PPP and glycolysis pathways, respectively. Relative cellular F1,6BP/G6P, F6P (G) and GSH/GSSG ratios (I) were calculated in B-lymphoid and B→myeloid cells. (H) Relative NADPH/NADP ratios were measured in Ppp2r1afl/fl B-lymphoid and myeloid cells after 2-days 4-OHT treatment for Cre-mediated deletion of Ppp2r1a. (J) Western blot to compare PP2A and G6PD expression in patient-derived B-ALL (n=4; MXP2,LAX2, PDX2 and BLQ5), mantle cell lymphoma (MCL; n=3; BOS4, BOS5 and BOS6), diffuse large B cell lymphoma (DLBCL; n=2; BOS10 and BOS12), and chronic myeloid leukemia (n=10; Table S4) samples. Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
PP2A is required to redirect glucose carbon utilization towards the pentose phosphate pathway in B cells
We examined how loss of PP2A activity affected glucose utilization via glycolysis relative to PPP in B-ALL cells. To trace the activity of glycolysis relative to the PPP pathway upon Ppp2r1a-deletion, we performed mass spectrometry-based metabolite profiling and carbon fate analyses based on 13C isotopic labels. Carbon fate tracing showing the M1 and M2 isotopomer distribution of lactate derived from [1,2-13C] glucose was used to distinguish 13C atoms directly passing through the glycolytic pathway (M2) from 13C atoms passing through the oxidative arm of the PPP and cycled back to glycolysis (M1). Interestingly, Ppp2r1a-deletion significantly reduced overall carbon utilization through the PPP in B-lymphoid cells (Figure 3C) while the distribution of M1 (PPP) and M2 (glycolysis) 13C atoms remained unchanged when Ppp2r1a was deleted in B→Myeloid reprogrammed cells (Figure 3D; inducible expression of Cebpa). A detailed analysis of individual glycolysis (M2) and PPP (M1) metabolites revealed that Ppp2r1a-deletion in B-lymphoid cells strongly increased labeling of 13C atoms in multiple glycolytic intermediates (in particular F-1,6-BP) while carbon labeling was consistently reduced for all PPP metabolites studied (Figure 3C). Conversely, for B→Myeloid reprogrammed cells, inducible deletion of Ppp2r1a had only minor effects and caused modest stimulation of 13C atoms through both glycolytic (M2) and PPP (M1) intermediates (Figure 3D).
PP2A regulates glucose carbon utilization in B cells at the level of Pfkfb2
The relative contribution of glycolysis and PPP flux is controlled by the bifunctional 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase 2 (Pfkfb2) kinase or phosphatase activity (Ros and Schulze, 2013). Importantly, Ppp2r1a-deletion in B cells but not myeloid cells increased phosphorylation of Pfkfb2 at S-483 (Figure 3E), which promotes Pfkfb2 kinase (6-phosphofructo-2-kinase; PFK-2) at the expense of its phosphatase activity (Pozuelo Rubio et al., 2003). The consequence of enhanced PFK-2 activity is allosteric activation of the rate-limiting enzyme PFK-1 which catalyzes the conversion of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F-1,6-BP) and thereby promotes glycolysis at the expense of the PPP. While F6P can still be utilized in the PPP via G6P as substrate for G6PD, F-1,6-BP is committed to the glycolysis pathway and gives rise to lactate. These findings suggest that PP2A regulates the phosphorylation status of Pfkfb2-S483 and, thereby, a critical metabolic switch between glycolysis and PPP. Indeed, massively increased S-483 phosphorylation of Pfkfb2 following Ppp2r1a deletion (Figure 3E), redirects glucose carbon flux from the PPP to glycolysis pathway (Ros et al. 2012). Functional consequences of this switch in B cells include increased utilization of glycolysis at the expense of the PPP (Figure 3F), >20-fold increased conversion of F6P to F-1,6-BP (Figure 3G), reduced NADPH/NADP ratios (Figure 3H). Consistent with diminished NADPH/NADP ratios, inducible deletion of Ppp2r1a substantially decreased GSH/GSSG ratios, indicating exhaustion of reductive reserves in B cells upon loss of PP2A function (Figure 3I). Interestingly, none of these changes occurred in myeloid cells. While patient-derived B cell leukemia and lymphoma cells express PP2A protein at high levels, protein levels for PP2A subunit C are much lower in myeloid leukemia samples (Table S2-S4) and virtually undetectable for PP2A subunit A (Figure 3J). PP2A expression levels in B-cell and myeloid leukemia match the phosphorylation status of Pfkfb2-S483 (Figure 3E), which provides an explanation for the strikingly different outcome of PP2A-deletion in B cell- and myeloid leukemia cells.
The fructose-2,6-bisphosphate 2-phosphatase TIGAR efficiently rescues cell death upon deletion of PP2A
The TP53 Induced Glycolysis Regulatory Phosphatase (TIGAR) can reverse the effects of PFKFB2 S-483 phosphorylation and stimulate carbon flux through the pentose phosphate pathway (PPP) at the expense of glycolysis (Bensaad et al., 2006; Cheung et al., 2013). For this reason, we tested the ability of Tigar overexpression (Figure 4A-B) to rescue cell death upon inducible ablation of Ppp2r1a in B-cell ALL. In BALL cells with intact PP2A expression, overexpression of Tigar resulted in a minimal survival advantage. However, when Ppp2r1a was deleted by inducible expression of Cre, overexpression of Tigar rescued BALL cells from cell death and induced a strong survival advantage (Figure 4C-D). Inducible deletion of Ppp2r1a in B-cell ALL substantially decreased NADPH/NADP ratios (Figure 4E). As expected, normal NADPH/NADP ratios were restored by reconstitution of Ppp2r1a expression using a retroviral overexpression vector (compared to empty vector, EV; Figure 4E). Overexpression of Tigar in B-ALL cells with intact PP2A-function induced a slight increase of NADPH/NADP ratios. Inducible deletion of Ppp2r1a substantially decreased NADPH/NADP ratios, which was fully restored by Tigar overexpression, comparable to restoration of Ppp2r1a itself (Figure 4F). The important role of TIGAR in the diverting glucose carbon flux from glycolysis to the PPP (Bensaad et al., 2006; Cheung et al., 2013) and the ability of Tigar overexpression to fully substitute for PP2A (Figure 4) support the concept that PP2A serves an essential role in the survival of B cells by balancing carbon utilization of glycolysis versus PPP. Given the complexity of redox regulation, it is likely that factors other than PPP also contribute to loss of NADPH upon Ppp2r1a deletion of in B cells.
Figure 4: The fructose-2,6-bisphosphate 2-phosphatase TIGAR rescues cell death upon PP2A-deletion.
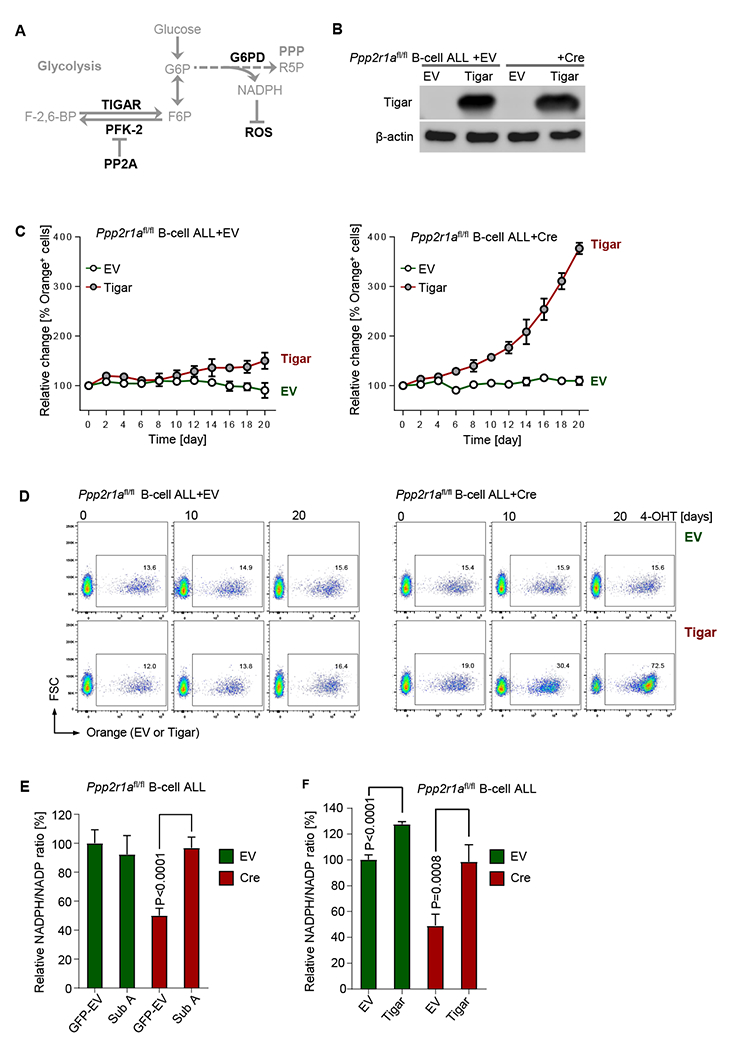
(A) Schematic diagram of TIGAR and PP2A in balancing glucose carbon flux via glycolysis and PPP. (B) Ppp2r1afl/fl BCR-ABL1 B-ALL cells carrying inducible Cre or EV were subsequently transduced with Tigar-IRES-Orange (Tigar) or Orange-empty vector (EV). Tigar expression was measured by Western blot. (C) Percentages of Orange+ B-ALL cells were monitored in a competitive growth assay following 4-OHT treatment. Representative FACS plots are shown at the times indicated (D). (E) Ppp2r1afl/fl B-ALL cells carrying inducible Cre or EV were transduced with vectors for reconstitution or overexpression of PP2A Sub A (Sub A) or GFP-EV and assayed for cellular NADPH/NADP ratios after 2-days of 4-OHT treatment for inducible activation of Cre or EV for deletion of Ppp2r1a. Likewise, Orange+ Ppp2r1afl/fl B-ALL cells carrying inducible Cre or EV were transduced with Tigar or EV and assayed for cellular NADPH/NADP ratios after 2-days 4-OHT treatment (F). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
PP2A and pentose phosphate pathway enzymes represent B-cell specific vulnerabilities
Recent work by our group showed that genetic lesions of PTEN occur throughout virtually all types of human cancer with the exception of B-ALL (Shojaee et al., 2016). Likewise, we found that mutations and deletions of PPP2R1A occur frequently throughout multiple cancer types of cancer but were not detected in B-ALL and B cell lymphoma. In solid tumors across 16 types of cancer (23,009 samples studied) we found genetic lesions of PPP2R1A on average in 3% of cases studied (range 0.5 to 14%). On the other hand, genetic lesions of PPP2R1A were also found in myeloid leukemia (187 samples studied) but not in B cell leukemia and lymphoma (323 samples studied; Figure S5A). These genetic findings show that malignant clones in B cell tumors are -unlike other types of cancer- not positively selected for loss of PP2A function. On the premise that PP2A regulates a central metabolic switch between glycolysis and PPP, we studied expression levels of glycolysis and PPP-related enzymes in B-cell compared to myeloid malignancies (Figure S4D; Figure S5B). Studying 807 B-lymphoid leukemia patient samples and 355 myeloid samples, we found substantially higher mRNA levels of glycolytic PFK-1 enzymes in B-lymphoid compared to myeloid samples (PFKL, P<0.0001; PFKM, P=0.0091; PFKP, P<0.0001). Interestingly, while we did not detect mutations in PP2A subunits, we found 19 somatic mutations in PFK-1 enzymes in a panel of 56 BALL and B cell lymphoma cell lines (Table S5). Conversely, mRNA levels of PPP enzymes were substantially downregulated in B-lymphoid compared to myeloid samples (G6PD, PGD, RPIA, RPE, TKT and TALDO1, P<0.0001 for each gene; Figure S4D). Glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (PGD) catalyze the rate-limiting steps of PPP and promote PPP relative to glycolysis (Hitosugi et al., 2012; Lin et al., 2015). Compared to normal myeloid progenitor cells (n=43) and myeloid leukemia (n=484), mRNA levels of G6PD are low in normal (n=47) and malignant (n=405) B cell populations (Figure S5B).
B-lymphoid transcription factors repress G6PD and PPP activity
Cebpa-mediated reprogramming of B cells into the myeloid lineage gradually increased mRNA and protein levels of G6PD and PGD (Figure 5A-B). ChIP-seq data from human B-cells showed enriched binding of these B-lymphoid transcription factors on G6PD promoters, which was confirmed by single-locus quantitative ChIP (Figure 5C-D). The B-lymphoid transcription factors PAX5 and IKZF1 negatively regulate glucose uptake and are frequently deleted in B-lineage ALL (Chan et al., 2017). Interestingly, restoration of PAX5 expression in a Pax5-deficient mouse model of B-lineage ALL (Liu et al., 2014) significantly repressed G6pdx and G6pd2 expression levels (Figure 5E). Likewise, restoration of PAX5 and IKZF1 in PAX5- and IKZF1-deficient patient-derived B-lineage ALL xenografts reduced G6PD by >50-fold to levels at or below the detection limit (Figure 5F). In some cases, genetic lesions of PAX5 and IKZF1 result in expression of dominant-negative (DN) transcription factors (PAX5-ETV6; intragenic deletion of IKZF1 zinc fingers). Overexpression of DN-PAX5 and DN-IKZF1 in patient-derived B-lineage ALL xenografts expressing functional PAX5 and IKZF1 induced strong upregulation of G6PD (Figure 5F) demonstrating that functional G6PD deficiency in B cells is the result of B cell-specific transcriptional repression. Interestingly, higher than median expression levels of G6PD are associated with poor clinical outcome and shorter overall survival in all four clinical trials that we studied for patients with B cell lymphomas but not in patients with acute myeloid leukemia (Figure 5G-H).
Figure 5: B cell-specific transcriptional repression of rate-limiting PPP enzymes.
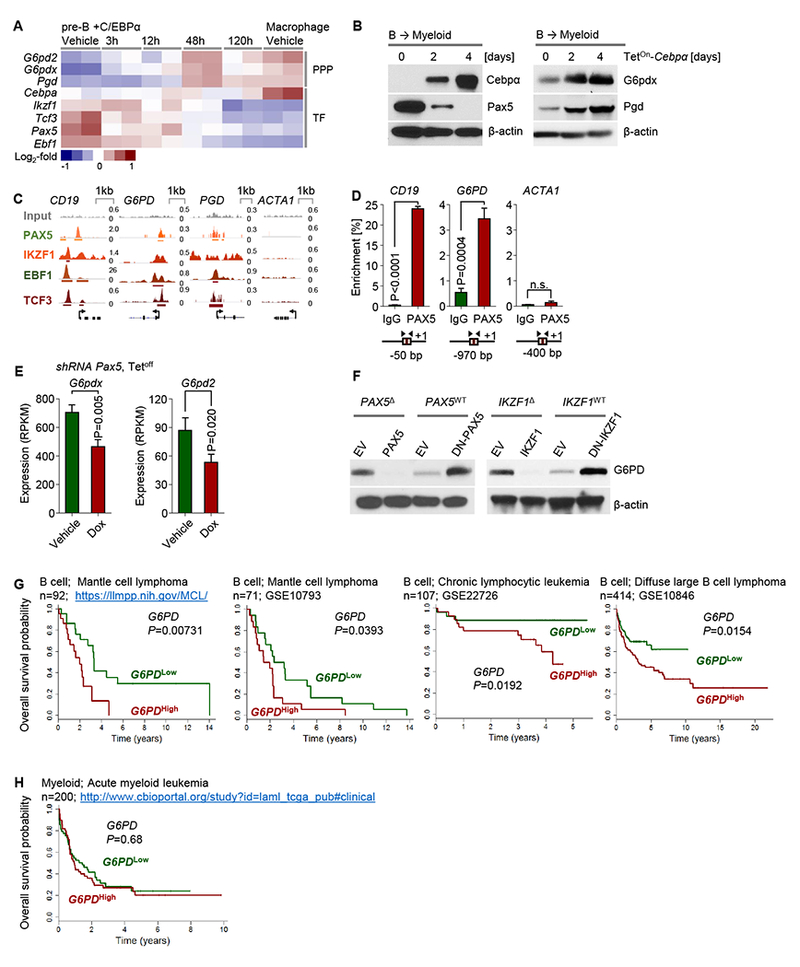
(A) Gene expression changes in response to Dox-inducible Cebpa-mediated reprogramming of mouse pre-B cells into myeloid cells (GSE32330) as a heatmap. (B) Murine B-cell ALL cells were transduced with TetOn-Cebpa and induced with Dox. Western blots measuring Cebpα, Pax5, G6pdx and Pgd expression after Dox induction are shown. (C) ChIP-seq data for promoter-binding of transcription factors in human B cells (ENCODE GM12878) of G6PD and PGD are shown. CD19 and ACTA1 are shown as positive and negative controls for PAX5, respectively. (D) Single-locus quantitative ChIP in patient-derived Ph+ ALL cells (ICN1) was performed to confirm the recruitment of PAX5 to the G6PD promoter. (E) G6pdx and G6pd2 expression from RNA-seq data (GSE52870) of Dox-induced Pax5 restoration in a mouse model for B-ALL is shown as bar graph. (F) Patient-derived Ph+ ALL cells (MXP2 and ICN1) carrying PAX5 deletions (PAX5Δ) or wildtype (PAX5WT) alleles were transduced with wildtype PAX5 or dominant-negative ETV6-PAX5 (DN-PAX5) respectively. IKZF1Δ (BV173) and IKZF1WT (ICN1) Ph+ ALL cells were transduced with wildtype IKZF1 or DN-IKZF1 respectively. Protein lysates were used for Western blots to measure G6PD expression. Patients with MCL, DLBCL and CLL (G) or AML (H) were divided into two groups based on 25% highest- or lowest- mRNA levels of G6PD. Overall survival of patients was assessed in the two groups by Kaplan–Meier analysis. Log-rank test was used to assess statistical significance. Besides clinical outcome studies, experimental data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Studying PP2A and G6PD protein levels in patient-derived B-ALL (n=4), diffuse large B cell lymphoma (DLBCL; n=2) and mantle cell lymphoma (MCL; n=3) xenografts, we confirmed that B cell tumors generally express high levels of PP2A subunit A and C and low levels of the PPP rate-limiting enzyme G6PD compared to patient-derived myeloid leukemia samples (n=10; Figure 3J). These findings suggest that B-cell intrinsic repression of the rate-limiting PPP enzyme G6PD may serve as a gatekeeper function to safeguard against malignant transformation, while higher expression levels of G6PD are associated with a more aggressive course of disease in B-cell malignancies. To test whether G6PD was sufficient to rescue inducible ablation of Ppp2r1a in B-cell ALL cells, we overexpressed murine G6pdx and human G6PD and verified increased expression levels by Western blot (Figure S6A). Unlike TIGAR, which redirects carbon glucose flux from glycolysis to the PPP, the PPP key enzyme G6PD (murine G6pdx and human G6PD) was not sufficient to rescue B-ALL cell death upon inducible deletion of Ppp2r1a (Figure S6B-C). This is a central difference from TIGAR (Figure 4), which unlike G6PD has no direct function in the PPP but controls supply of the PPP with glucose carbon flux (Bensaad et al., 2006).
Transcription factors that determine B cell identity regulate PPP-activity and dependency on PP2A
We tested here the hypothesis that B cell-specific transcription factors are responsible for low PPP activity in B cells, hence PP2A as a B cell-specific vulnerability. To this end, we replaced the four stem cell factors Oct4, Sox2, Nanog and Myc in a vector that was previously used to generate iPS cells (Takahashi & Yamanaka 2006) with the four B cell transcription factors Pax5, Ikaros, Ebf1 and Tcf3. Inducible activation of this polycistronic vector in human embryonic kidney cells resulted in expression of all four B-lymphoid transcription factors and substantial reduction of NADPH/NADP ratios (Figure 6A-B). In addition, doxycycline-inducible expression was sufficient to reprogram murine myeloid leukemia cells into B cells (Figure 6C-D). Myeloid→B cell reprogramming downregulated G6PD and upregulated PP2A (Figure 6 E) and hence recapitulated metabolic characteristics of B cells (Figure 3J). In a third approach, we studied small cell lung cancer cells (SCLC). Unlike other types of lung cancer, SLCL cells often aberrantly express PAX5 (Kanteti et al. 2009). Compared to PAX5− SCLC (e.g. DMS273), PAX5+ SCLC (e.g. H69) cells expressed G6PD at low and PP2A at high protein levels, respectively (Figure 6F), and had much lower NADPH/NADP ratios (Figure 6G). Interestingly, lentiviral expression of PAX5 in PAX5− SCLC (DMS273) reduced NADPH/NADP ratios down to levels in PAX5+ SCLC cells (H69). Overexpression of PAX5 in SCLC (H69) reduced NADPH/NADP ratios even further (Figure 6G). These effects depend on PAX5 and likely other B-lymphoid transcription factors and can occur in non-B cells (e.g. kidney, myeloid and SLCL cells) when PAX5 is aberrantly expressed. While not the focus of this study, these findings suggest a previously unexplored rationale for specific targeting of PP2A and G6PD in SCLC cells.
Figure 6: Transcription factors that determine B cell identity regulate activity of the pentose phosphate pathway and dependency on PP2A.
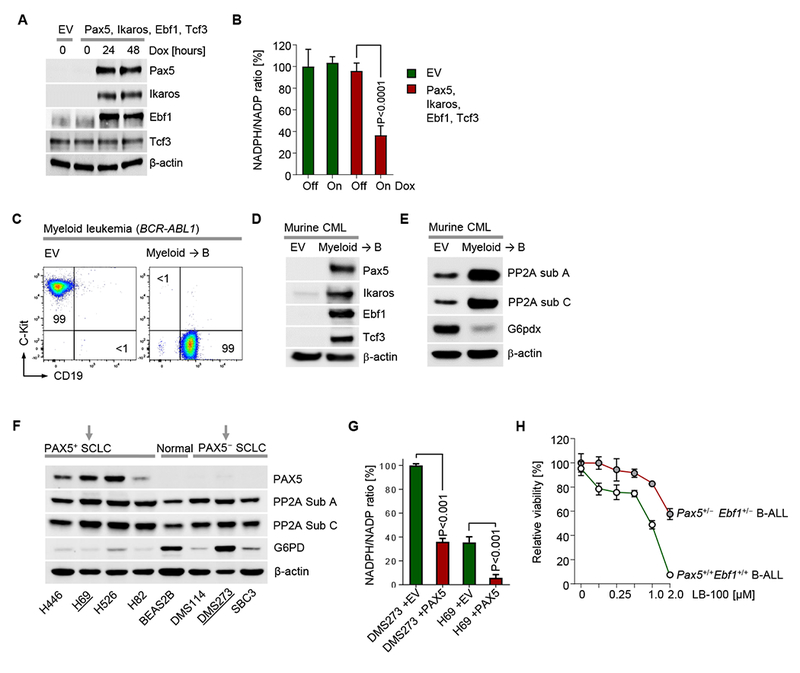
(A) Human embryonic kidney cells (HEK-293) were transduced with a polycistronic doxycycline (Dox)-inducible vector to express the B-lineage transcription factors Pax5, Ikaros, Ebf1 and Tcf3, as confirmed by Western blot after Dox treatment for the times indicated. (B) Relative NADPH/NADP ratios were measured before and after 24 hours of Dox-induction of the polycistronic vector (Pax5, Ikaros, Ebf1 and Tcf3) or EV. (C) Murine myeloid leukemia cells were transduced with the inducible polycistronic vector for myeloid→B cell reprogramming. Phenotypic changes were monitored by flow cytometry 48 hours after induction of Dox and expression of Pax5, Ikaros, Ebf1 and Tcf3 was confirmed by Western blot. (E) In the same cells, protein levels for PP2A Sub A, PP2A Sub C and G6pdx were measured by Western blot. (F) Human small cell lung cancer (SCLC) and normal lung epithelial (BEAS2B) cell lines were used to compare PAX5, PP2A and G6PD expression by Western blot. (G) NADPH/NADP ratios were measured in PAX5+ and PAX5− SLCL cell lines and upon expression of PAX5 or EV. (H) Murine Pax5+/−Ebf1+/− BCR-ABL1 B-ALL cells were treated with the PP2A-inhibitor LB-100 at the concentrations indicated. Dose response curves depicting relative cell viability after 72 hours are depicted. Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Target validation of PP2A in human B cell malignancies.
In proof-of-concept experiments, we have tested here the PP2A small molecule inhibitor LB-100 (4-Methylpiperazin-1-yl-7-oxabicyclo-heptane-2-carboxylic acid) and the G6PD-inhibitor by 6-aminonicotinamide (6-AN; Figure S7A) in B cell leukemia and lymphoma samples. LB-100, the first-in-class PP2A inhibitor studied here is currently in two clinical trials (NCI designated NCT03027388 and NCT01837667). A phase 1 safety study with 7 dose-escalations did not note relevant toxicity (Chung et al., 2017). LB-100 is a water-soluble small molecule inhibitor of PP2A (IC50 0.9 μmol/l) and was initially designed to sensitize solid tumors to chemotherapy (Lu et al., 2009). To document feasibility, we injected C57B/6J mice with LB-100 (1.5 mg/kg) five times over the course of 9 days and monitored PP2A inhibition in normal T cell subsets (Figure S7C-E) and myeloid subsets, which showed minimal impact. Interestingly, a recent study showed that proarthritogenic T cells in rheumatoid arthritis are dependent on PPP-mediated antioxidant protection (Yang et al., 2016), suggesting that targeting of PP2A alone or in combination with G6PD may be useful for these patients. Here we used LB-100 to study pharmacological PP2A ablation in the context of Pax5 and Ebf1 haploinsufficiency (hemizygous Pax5 and Ebf1 deletions). To this end, Pax5+/−Ebf1+/− B-ALL cells and wildtype control B-ALL cells were treated with LB-100 at the concentrations indicated and cell viability was measured. Interestingly, partial or complete loss of B cell-specific transcription factors (e.g. Pax5 and Ebf1) relieved dependency on PP2A and resulted in reduced sensitivity to LB-100 in B cells (Figure 6H).
CRISPR/Cas9-mediated genetic ablation of PPP2R1A in patient-derived B-cell ALL
We transduced patient-derived Ph+ ALL cells with inducible Cas9 and three different guide-RNAs directed against PPP2R1A and a non-targeting guide (Figure 7A; Table S6). Western blot analysis revealed that deletion of PPP2R1A was achieved at varying levels. The extent of PPP2R1A deletion correlated with loss of the catalytic PP2A subunit C and phosphorylation of H2AX (S139; Figure 7A). Compared to patient-derived Ph+ ALL cells carrying Cas9 and a non-targeting guide RNA, three different guide-RNAs directed against PPP2R1A induced depletion of B-ALL cells from cell culture (Figure 7B and S7B), confirming dependency of patient-derived B-ALL cells on PP2A function.
Figure 7: Target validation of PP2A and the PPP in human B cell malignancies.
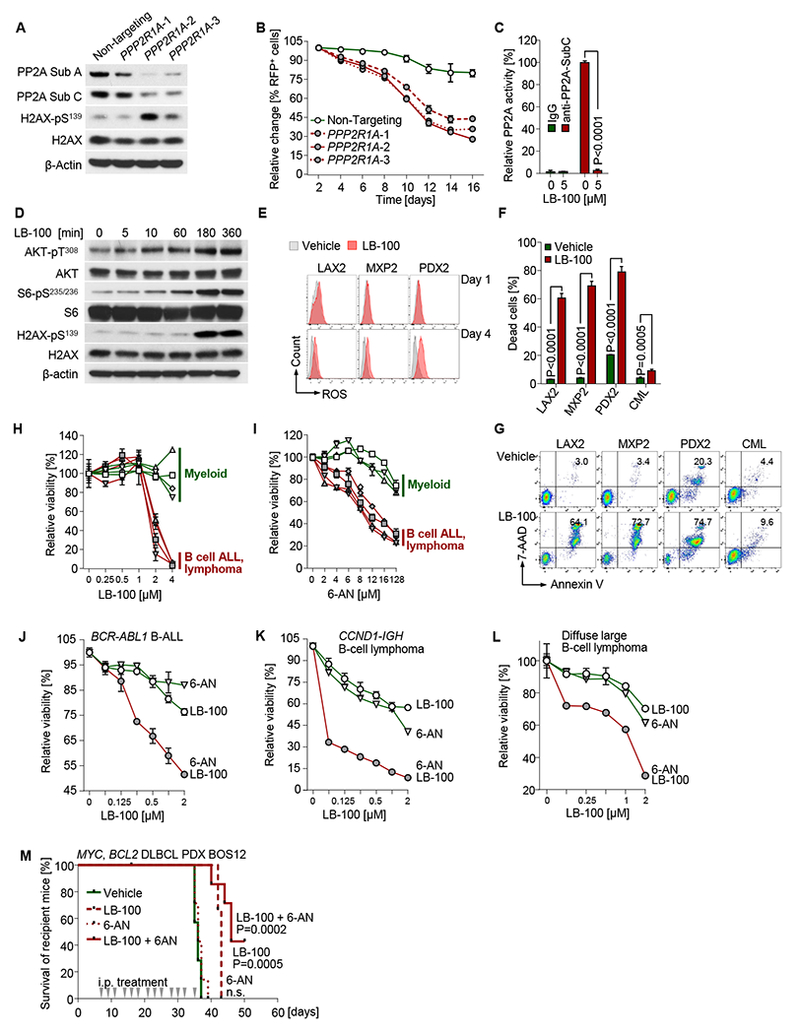
(A) Patient-derived Ph+ ALL xenograft (PDX2) cells were used for doxycycline (Dox)-inducible CRISPR-Cas9 mediated gene disruption of PPP2R1A (3 different gRNAs were used). Western blot was performed to measure expression of PP2A and phosphorylation of H2AX after 3 days of Dox-treatment. (B) Percentages of targeted cells were measured following Dox-treatment. (C) Patient-derived Ph+ ALL xenograft (LAX2) cells were treated with the PP2A inhibitor LB-100 (5 μmol/l) for 12 hours. Cell lysates were assayed for PP2A Thr/Ser phosphatase activity assay. (D) The same cells were cultured with 2 μmol/L LB-100 and harvested at time points indicated. Western blot was performed to measure phosphorylation of AKT, S6 and H2AX. (E) Intracellular ROS levels were measured in patient-derived Ph+ ALL xenografts cells on days 1 and 4 of treatment with LB-100 (2 μmol/L). (F) Cell viability measurements of human B-lymphoid Ph+ ALL (LAX2, MXP2, PDX2) and myeloid leukemia cells (JURL-MK1) cells after 4-days of treatment with 2 μmol/L LB-100 are plotted. Representative flow cytometry plots are shown (G). (H-I) Relative cell viabilities were measured after treatment of LB-100 (H) or 6-AN (I) for 72 hours with gradients of concentrations as indicated in B-lineage Ph+ ALL (LAX2, MXP2, PDX2 and BLQ5; n=4) and myeloid leukemia (KYO-1, EM-2, JURL-MK1 and MV4-11; n=4) cells. Y-axis shows percentages of viable cells relative to vehicle-treated cells (set to 100%). (J) Patient-derived Ph+ ALL (PDX2) cells, (K) mantle cell lymphoma cells (SP-53) and (L) diffuse large B-cell lymphoma cells (OCI-Ly10) were treated with LB-100, 6-AN or a combination of both compounds for 72 hours. Relative cell viability was assessed to measure synergistic effect of combination treatment. (M) Patient-derived diffuse large B cell lymphoma cells (BOS12) were intravenously injected to recipient NSG mice (3×106 cells/mouse; 7 mice in each group). Intraperitoneal injection of LB-100 (1.5 mg/kg), 6-AN (1 mg/kg) or combination (1.5 mg/kg LB-100 + 1 mg/kg 6-AN) started 7 days after cell injection at indicated time points. Kaplan-Meier survival curves are shown. Log-rank test was used to assess statistical significance of each treated group compared to vehicle group. Except for Kaplan-Meier survival analyses, other experimental data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Feasibility and efficacy of pharmacological PP2A inhibition in B cell malignancies.
Studying biochemical activity of LB-100 in patient-derived Ph+ ALL cells, we found that near-complete loss of PP2A activity at 5 μmol/l (Figure 7C) was paralleled by strong activation of AKT-mTOR signaling and H2AX-phosphorylation (Figure 7D), which is consistent with increased ROS-levels in patient-derived Ph+ ALL cells (Figure 7E). Consistent with lineage-specific effects of Ppp2r1a deletion (Figure 1I), LB-100 selectively induced cell death in B-cell leukemia and lymphoma PDX but not myeloid leukemia cells (CML, AML; Figure 7F-H). Owing to B-lymphoid transcriptional repression of G6PD and other central PPP enzymes (Figure 5A-F), B cells are uniquely vulnerable to PPP inhibition. In agreement with these results, we found that pharmacological inhibition of G6PD by 6-AN further exacerbated B cell-intrinsic G6PD deficiency and induced cell death in B-cell leukemia and lymphoma but not myeloid leukemia cells (Figure 7I). Interestingly, combination studies of LB-100 and 6-AN revealed that direct (6-AN via G6PD) and indirect (LB-100 via PP2A) inhibition of PPP strongly synergized in patient-derived Ph+ ALL, mantle cell lymphoma and DLBCL cells (Figure 7J-L; Table S7). To test this therapeutic concept in a preclinical transplant experiment, we injected patient-derived DLBCL cells (Table S3) carrying concurrent MYC and BCL2 gene rearrangements into NSG mice and treated the mice with LB-100 (1.5 mg/kg), 6-AN (1 mg/kg) or a combination of both via i.p. injections. The Kaplan-Meier analysis on day 50 showed a significant difference in survival for the LB-100 (P=0.0005) or LB-100 +6AN (P=0.0002) arms compared to vehicle controls (Figure 7M; Log-rank test). In summary, we propose that targeting of PP2A in B-cell malignancies is immediately translatable given the evidence supporting safety of LB-100. Our studies provide a strong rationale for agents that will target PP2A, TIGAR or G6PD as lineage-specific and previously unrecognized vulnerabilities in B cell malignancies (B-ALL and B cell lymphoma).
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Markus Müschen (mmuschen@coh.org).
Experimental Model and Subject Details
Cell lines
Leukemia cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (GIBCO) with GlutaMAX supplemented with 10-20% fetal bovine serum (FBS), 100 IU/ml penicillin and 100 μg/mL streptomycin (GIBCO). Small cell lung cancer cell lines provided by Dr. Ravi Salgia, were cultured in the same condition as for leukemia cell lines. All cell lines were cultured at 37°C in a humidified incubator with 5% CO2. All cell lines were tested to be mycoplasma free by MycoAlert PLUS kit (LONZA).
Patient-derived B-ALL and B-cell lymphoma samples
Patient samples were obtained in compliance with the internal review board of the Beckman Research Institu te of City of Hope. Patient samples were collected from biopsy of ALL (bone marrow) or lymphoma (periph eral blood) patients at the time of diagnosis or relapse. All samples were transplanted into sublethally irradiat ed NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG; The Jackson Laboratory) mice through tail vein injection. Aft er collection of cells, lymphoma xenografts were frozen in aliquots and ALL xenografts were cultured on OP 9 stroma cells in Alpha Minimum Essential Medium (MEMα; GIBCO) with GlutaMAX supplemented with 20% FBS, 1mM sodium pyruvate, 100 IU/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidifie d incubator with 5% CO2. Before experimental usage, all the human xenografts were tested to be mycoplasm a free (MycoAlert PLUS, LONZA).
Murine primary and leukemia cells
Bone marrow and splenic cells were harvested from mice between 6-8 weeks of age without signs of inflammation. After filtration through a 70 μm cell strainer, cells were supplied to lysis buffer based erythrocytes depletion (RBC Lysis Buffer, BioLegend). Resuspended cells were then used for further experiments. Bone marrow cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM; GIBCO) with GlutaMAX supplemented with 20% FBS, 100 IU/ml penicillin, 100 μg/mL streptomycin and 50 μmol/l β-mercaptoethanol (GIBCO). Bone marrow cells, for IL-7-dependent pre-B cell culture were cultured in full IMDM supplemented with 10 ng/ml recombinant mouse IL-7 (PeproTech). To generated murine B-lineage Ph+ ALL cell, retroviral transduction of BCR-ABLI was performed to cultured pre-B cells. After transformation with BCR-ABL1, ALL cells were cultured in IL-7-withdrawed IMDM. NRASG12D B-ALL cells were selected with puromycin (GIBCO) and were maintained with IL-7 supplemented IMDM. Murine B-ALL cells were cultured no longer than 2 months to avoid accumulation of additional genomic mutations. Murine BCR-ABL1 B-ALL cells used in 1,2-213C D-Glucose tracing and metabolites profiling experiments, were cultured in glucose-free DMEM with 20% dialyzed FBS, 100 IU/ml penicillin, 100 μg/mL streptomycin and 50 μmol/l β-mercaptoethanol (GIBCO). The same medium with 25 mmol/l 1,2-213C D-Glucose (Cambridge Isotope Laboratories, Inc.) was used labeling during a 24 hours interval. For myeloid leukemia (CML-like) cells, the myeloid-specific protocol described previously (Chan et al., 2017) was used. In brief, murine bone marrow cells were cultured in IMDM containing 10 ng/ml recombinant mouse IL-3, 25 ng/ml recombinant mouse IL-6 and 50 ng/ml recombinant mouse stem cell factor (SCF; PeproTech). After BCR-ABL1 mediated transformation, murine CML-like cells were cultured in IMDM without additive cytokines. Lineage-specific characterization of BCR-ABL1-transformed Ph+ ALL and CML-like cells was performed by flow cytometry (LSRFortessa, BD Biosciences). Mature splenic B cells were cultured in IMDM with 20% FBS, 100 IU/ml penicillin, 100 μg/mL streptomycin and 50 μmol/l β-mercaptoethanol (GIBCO), supplemented with 10 ng/ml mouse IL-4 (Pepro Tech), 100 ng/ml human BAFF (Cell Guidance Systems) and 1 μg/ml mouse CD40L (Cell Guidance Systems). After 2 days in cell culture, more than 90% of the cells were CD19+ B cells and were used for further experiments.
Animals
C57BL/6, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Mb1-Cre and Ppp2r1afl/fl mice were purchased from The Jackson Laboratory. Ppp2r1afl/fl mice were backcrossed to C57BL/6 background for more than 8 generations. Vav-tTAxTetoff-shPax5 mice were obtained from Dr. Ross A. Dickins. For animals bred in house, littermates of the same sex were randomized to experimental groups. All mouse breeding and experiments were subject to institutional approval by the Beckman Research Institute Animal Care and Use Committee.
Method details
Flow cytometry
Cells were washed in PBS and subjected to Fc receptor blocking (TruStain, Biolegend) for 10 min at room temperature. 106 cells per ml were used to perform staining with antibodies listed for 15-30 min at 4°C. After washing with PBS, cells were suspended in PBS with propidium iodide (PI, 0.2 μg/mL) or DAPI (0.75 μg/mL) to exclude dead cells. For cell-cycle analysis, the Click-iT EdU Flow Cytometry Assay Kit (Life Technologies) was used according to the manufacturer’s instructions. To measure intracellular ROS levels, cells were incubated for 7 min with 1 μmol/L 5-(and-6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Life Technologies) at 37°C. After washing with PBS, the cells were incubated for additional 15 min at 37°C in PBS. Fluorescence signals from oxidated CM-H2DCFDA in viable cells were measured by flow cytometry. All flow cytometry assays were performed on a LSRFortessa (BD Biosciences). All the fluorescence-activated cell sorting procedures were performed on a FACSAria II (BD Biosciences). Flow cytometry data were exported and analyzed with FlowJo (FlowJo, LLC).
Western blotting
PBS washed cells were lysed in CelLytic buffer (Sigma-Aldrich) supplemented with 1% protease inhibitor cocktail (Fisher Scientific). 20 μg of protein lysates per sample were separated on Criterion TGX (Bio-Rad) 4-20% Tris-Glycine extended gradient gels and transferred on nitrocellulose membranes (Bio-Rad). The Western Breeze Immunodetection System (Life Technologies) was used for signal development, and chemiluminescent signal was detected by X-ray film exposure. Primary antibodies used for western blotting are listed below.
PP2A phosphatase activity measurement
2×107 leukemia cells were used for measuring PP2A activity following manufacturer’s protocol. PP2A Immunoprecipitation Phosphatase Assay Kit (17-313, Millipore) was used to perform this assay.
Reprogramming of leukemia cells from pre-B into myeloid lineage by C/EBPα
Ppp2r1afl/fl BCR-ABL1 ALL cells were transduced with rtTA C/EBPα. After antibiotic selection, those cells were transduced with Cre-ERT2-GFP or EV. For growth competition assays, cells were induced with doxycycline for 2 days, 4-OHT (Sigma-Aldrich) was used to induce Cre-ERT2 activity. The percentages of GFP+ cells of pre-B (CD19+ CD11b−) and reprogrammed myeloid (CD19− CD11b+) populations were measured by flow cytometry at the same time. For metabolites extraction, transduced cells were sorted for GFP+ cells before induction. 5 days after induction with doxycycline, reprogrammed cells were used for 1,2-213C D-glucose tracing and metabolites profiling.
Retroviral and lentiviral transduction
Constructs used for retroviral or lentiviral transduction are listed in the key resources Table. Virus production was performed with HEK-293FT cells (Life Technologies) using Lipofectamine 2000 (Life Technologies). Cells were co-transfected with the plasmid of interest, pHIT60 (gag-pol) and pHIT123 (ecotropic env; kindly provided by Donald B Kohn, UCLA). HEK-293FT cells were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) with GlutaMAX supplemented with 10% FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 25 mM HEPES, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids (GIBCO). 16h after liposomal transfection, cell culture medium was replaced by full grow media, supplemented with 10 mM sodium butyrate. After 8 hours, cell culture medium was replaced with full grow media. 24 hours later, the virus-containing supernatant was collected and filtered through 0.45 μm filters. Cell transduction was performed with RetroNectin (TAKARA) according to manufactures protocol. Lentiviral particles were produced via co-transfection of plasmid of interest with pCD/NL-BH*DDD and EM140 like the retroviral particle production. Antibiotic selection was performed on transduced cells according to the specific antibiotic resistance carried by the respective construct.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal (C4) anti-β-actin | Santa Cruz Biotechnology | Cat# sc-47778 |
| Monoclonal (81G5) anti-PP2A subunit A | Cell Signaling Technology | Cat# 2041 |
| Monoclonal (52F8) anti-PP2A subunit C | Cell Signaling Technology | Cat# 2259 |
| Polyclonal anti-STAT5 | Cell Signaling Technology | Cat# 9363 |
| Monoclonal (D47E7) anti-p-STAT5 (Y694) | Cell Signaling Technology | Cat# 4322 |
| Polyclonal anti-Erk | Cell Signaling Technology | Cat# 9102 |
| Monoclonal (D13.14.4E) anti-p-Erk (T202/Y204) | Cell Signaling Technology | Cat# 4370 |
| Polyclonal anti-Akt | Cell Signaling Technology | Cat# 9272 |
| Monoclonal (D25E6) anti-p-Akt (T308) | Cell Signaling Technology | Cat# 13038 |
| Monoclonal (C29H4) anti-FoxO1 | Cell Signaling Technology | Cat# 2880 |
| Polyclonal anti-p-FoxO1 (T24) | Cell Signaling Technology | Cat# 9464 |
| Polyclonal anti-p-FoxO1 (S256) | Cell Signaling Technology | Cat# 9461 |
| Monoclonal (75D8) anti-FoxO3a | Cell Signaling Technology | Cat# 2497 |
| Monoclonal (D18H8) anti-p-FoxO3a (S253) | Cell Signaling Technology | Cat# 13129 |
| Monoclonal (2217) anti-S6 | Cell Signaling Technology | Cat# 2217 |
| Monoclonal (4856) anti-p-S6 (S235/236) | Cell Signaling Technology | Cat# 4856 |
| Polyclonal anti-p70 S6 kinase | Cell Signaling Technology | Cat# 9202 |
| Polyclonal anti-p-p70 S6 kinase (T389) | Cell Signaling Technology | Cat# 9205 |
| Polyclonal anti-PFKFB2 | Milipore | Cat# 07-1530 |
| Monoclonal (D4R1W) anti-p-PFKFB2 (S483) | Cell Signaling Technology | Cat# 13064 |
| Polyclonal anti-H2AX | Abcam | Cat# Ab10475 |
| Polyclonal anti-p-H2AX (S139) | Abcam | Cat# Ab2893 |
| Monoclonal (2524) anti-p53 | Cell Signaling Technology | Cat# 2524 |
| Monoclonal (C-2) anti-Bcl2 | Santa Cruz Biotechnology | Cat# sc-7382 |
| Polyclonal anti-p21 | Santa Cruz Biotechnology | Cat# sc-397 |
| Monoclonal (F-8) anti-p27 | Santa Cruz Biotechnology | Cat# sc-1641 |
| Polyclonal anti-p19 ARF (mouse) | Abcam | Cat# ab80 |
| Polyclonal anti-G6PD | Cell Signaling Technology | Cat# 8866 |
| Monoclonal (G-2) anti-PGD | Santa Cruz Biotechnology | Cat# sc-398977 |
| Polyclonal anti-TIGAR | Invitrogen | Cat# PA5-23053 |
| Polyclonal anti-Pax5 (for ChIP) | Santa Cruz Biotechnology | Cat# sc-1974 |
| Monoclonal anti-Pax5 (for western blot) | Cell Signaling Technology | Cat# 8970S |
| Monoclonal anti-Ikaros | Cell Signaling Technology | Cat# 14859S |
| Monoclonal anti-EBF1 | Abcam | Cat# ab108369 |
| Poly clonal anti-E2A (TCF3) | Santa Cruz Biotechnology | Cat# sc-349 |
| Monoclonal (1D3) anti-Mouse CD19 | BD Biosciences | Cat# 557655 |
| Monoclonal (RA3-6B2) anti-Mouse CD45R | Biolegend | Cat# 103224 |
| Monoclonal (M1/70) anti-Mouse CD11b | Biolegend | Cat# 101208 |
| Monoclonal (6C3) anti-Mouse Ly-51 (BP-1) | Biolegend | Cat# 108305 |
| Monoclonal (M1/69) anti-Mouse CD24 | BD Biosciences | Cat# 562477 |
| Monoclonal (11-26c.2a) anti-Mouse IgD | Biolegend | Cat# 405716 |
| Monoclonal (R6-60.2) anti-Mouse IgM | BD Biosciences | Cat# 550881 |
| Monoclonal (JC5-1) anti-Mouse Ig Light Chain, λ | Southern Biotech | Cat# 1175-09 |
| Monoclonal (187.1) anti-Mouse Ig Light Chain, κ | BD Biosciences | Cat# 562476 |
| Monoclonal (17A2) anti-Mouse CD3 | Biolegend | Cat# 555275 |
| Monoclonal (RM4-5) anti-Mouse CD4 | BD Biosciences | Cat# 553049 |
| Monoclonal (53-6.7) anti-Mouse CD8a | BD Biosciences | Cat# 553033 |
| Monoclonal (PK136) anti-Mouse NK1.1 | BD Biosciences | Cat# 553165 |
| Monoclonal (RB6-8C5) anti-Mouse Gr-1 (Ly-6G/Ly-6C) | BD Biosciences | Cat# 553128 |
| Monoclonal (TER-119) anti-Mouse TER-119 | BD Biosciences | Cat# 553673 |
| Monoclonal (2B8) anti-Mouse CD117 (c-Kit) | BD Biosciences | Cat# 553356 |
| Monoclonal (HIB19) anti-Human CD19 | BD Biosciences | Cat# 555413 |
| Biological Samples | ||
| Patient-derived pre-B ALL samples, see Table S2 | Müschen Laboratory | http://lymphoblasts.org/ |
| Patient-derived B-cell lymphoma samples, see Table S3 | Müschen Laboratory | http://lymphoblasts.org/ |
| Patient-derived B-cell lymphoma samples, see Table S3 | Weinstock Laboratory | https://www.proxe.org/ |
| Patient-derived chronic myeloid leukemia (CML) samples, see Table S4 | Thomas Ernst and Andreas Hochhaus | Jena University Hospital, Germany |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imatinib | LC Laboratories | Cat# I-5577 |
| Idelalisib | Selleck Chemicals LLC | Cat# S2226 |
| Rapamycin | Sigma-Aldrich | Cat# 37094 |
| 4-hydroxy-tamoxifen | Sigma-Aldrich | Cat# 94873 |
| Doxycycline | Clontech Laboratories | Cat# 631311 |
| Tamoxifen | Sigma-Aldrich | Cat# 85256 |
| LB-100 | Selleck Chemicals LLC | Cat# S7537 |
| 6-AN | Sigma-Aldrich | Cat# A68203 |
| Critical Commercial Assays | ||
| Click-iT EdU Flow Cytometry Assay Kit | Life Technologies | Cat# C10635 |
| PP2A Immunoprecipitation Phosphatase Assay Kit | Millipore | Cat# 17-313 |
| APC Annexin V | BD Biosciences | Cat# 550474 |
| Guava TUNEL Kit for Flow Cytometry | Millipore | Cat# 4500-0121 |
| Phospho-Kinase Antibody Array | R&D Systems | Cat# ARY003B |
| NADP/NADPH Assay Kit | Abcam | Cat# Ab65394 |
| Amplex™ Red Glucose/Glucose Oxidase Assay Kit | Thermo Fisher Scientific | Cat# A22189 |
| L-Lactate Assay Kit | Cayman Chemical | Cat# 700510 |
| ATP Bioluminescence Assay Kit CLS II | Roche | Cat# 11699709001 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent Technologies | Cat# 103020-100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | Cat# 103015-100 |
| Deposited Data | ||
| Affymetrix Gene Chip expression data, Ppp2r1a-deletion | This paper | GSE83742 |
| Affymetrix Gene Chip expression data, CEBPA-mediated reprogramming | Dr. Thomas Graf | GSE32330 |
| Chromatin IP sequencing data | ENCODE | GM12878 |
| Expression profiling by high throughput sequencing, Pax5 restoration | Dr. Ross A. Dickins | GSE52870 |
| Affymetrix Gene Chip expression data, mantle cell lymphoma | Dr. Andreas Rosenwald | GSE10793 |
| Affymetrix Gene Chip expression data, mantle cell lymphoma | LLMPP | http://llmpp.nih.gov/MCL |
| Affymetrix Gene Chip expression data, diffuse large B cell lymphoma (DLBCL) | Dr. Louis M. Staudt | GSE10846 |
| Affymetrix Gene Chip expression data, chronic lymphocytic leukemia (B-CLL) | Dr. Tobias Herold | GSE22762 |
| Affymetrix Gene Chip expression data, acute myeloid leukemia | TCGA | N/A |
| Experimental Models: Cell Lines | ||
| KASUMI-2 | DSMZ | Cat# ACC 526 |
| KYO-1 | DSMZ | Cat# ACC 601 |
| EM-2 | DSMZ | Cat# ACC 135 |
| JURL-MK1 | DSMZ | Cat# ACC 532 |
| KCL-22 | DSMZ | Cat# ACC 519 |
| K-562 | ATCC | Cat# CCL-243 |
| MV4-11 | ATCC | Cat# CRL-9591 |
| MOLM-13 | DSMZ | Cat# ACC 554 |
| U-937 | ATCC | Cat# CRL-1593.2 |
| MEG-01 | ATCC | Cat# CRL-2021 |
| BV-173 | DSMZ | Cat# ACC 20 |
| OCI-Ly10 | From Dr. Daniel Hodson | CVCL_8795 |
| SP-53 | From Dr. Ngo Vu | CVCL_C122 |
| H446 | ATCC | Cat# HTB-171 |
| H69 | ATCC | Cat# HTB-119 |
| H526 | ATCC | Cat# CRL-5811 |
| H82 | ATCC | Cat# HTB-175 |
| DMS114 | ATCC | Cat# CRL-2066 |
| DMS273 | Sigma | Cat# 95062830 |
| SBC3 | JCRB | Cat# JCRB0818 |
| BEAS2B | ATCC | Cat# CRL-9609 |
| HEK 293T | Clontech Laboratories | Cat# 632180 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ppp2r1afl/fl: Ppp2r1atm1.1Wltr | The Jackson Laboratory | Cat# 017441 |
| Mouse: Mb1-Cre: Cd79atm1(cre)Reth | The Jackson Laboratory | Cat# 020505 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Cat# 005557 |
| Mouse: Pax5+/− Ebf1+/− | Dr. Michael Farrar | N/A |
| Oligonucleotides | ||
| Primers for genotyping and qChIP, see Table S6 | Eurofins Scientific | N/A |
| sgRNA for CRISPR-Cas9 mediated gene disruption, see Table S6 | transOMIC Technologies | TEVH-1259980 TEVH-1192838 TEVH-1125696 |
| Recombinant DNA | ||
| MSCV-BCR-ABL1 | Rick Van Etten Laboratory | BCR-ABL1 (p210) |
| MSCV-BCR-ABL1-IRES-Neo | Rick Van Etten Laboratory | BCR-ABL1 (p210) |
| MSCV-NRASG12D-Puro | Müschen Laboratory | NRASG12D |
| MSCV-Luc-Puro | Müschen Laboratory | Firefly luciferase |
| pCL6-Luc-Blast | Müschen Laboratory | Firefly luciferase |
| MSCV-IRES-GFP | Müschen Laboratory | GFP |
| MSCV-Cre-IRES-GFP | Müschen Laboratory | GFP |
| MSCV-PPP2R1A-IRES-GFP | Müschen Laboratory | PP2A subunit A |
| MSCV-CAT-IRES-GFP | Müschen Laboratory | Catalase |
| MSCV-ERT2-IRES-Puro | Müschen Laboratory | Puromycin |
| MSCV-Cre-ERT2-IRES-Puro | Müschen Laboratory | GFP |
| MSCV-ERT2-IRES-GFP | Müschen Laboratory | GFP |
| MSCV-Cre-ERT2-IRES-Puro | Müschen Laboratory | GFP |
| MSCV-mG6pdx-IRES-GFP | Müschen Laboratory | G6PD |
| MSCV-G6PD-IRES-GFP | Müschen Laboratory | G6PD |
| MSCV-FoxO1-ADA-IRES-GFP | Müschen Laboratory | FOXO1 |
| pMIThy1.1(CD90) | From Dr. David A. Fruman | CD90 |
| pMIThy1.1(CD90)-FoxO3a-CA | From Dr. David A. Fruman | CD90 |
| pCGDNsam-IRES-Kusabira Orange | From Dr. H. Phillip Koeffler | K-Orange |
| pCGDNsam-Tigar-IRES-Kusabira Orange | Müschen Laboratory | K-Orange |
| pRetroX-Tet3G-Neo | Clontech Laboratories | Cat# 631188 |
| pRetroX-TRE3G-c/EBPα-Puro | Thomas Graf, Barcelona | CEBPA |
| pRetroX-TRE3G-Pax5-Ikaros-Ebf1-Tcf3 | Müschen Laboratory | Ali’s vector |
| pCL6-ERT2-IRES-GFP | Hassan Jumaa Laboratory | GFP |
| pCL6-PAX5-ERT2-IRES-GFP | Müschen Laboratory | PAX5 |
| pCL6-PAX5-ETV6-ERT2-IRES-GFP | Müschen Laboratory | PAX5-ETV6 |
| pLVX-TRE3G-GFP | Clontech Laboratories | Cat# 631361 |
| pLVX-TRE3G-IKZF1-GFP | Hilde Schjerven laboratory, UCSF | Ikaros |
| pLVX-TRE3G-IK6-GFP | Hilde Schjerven laboratory, UCSF | Dominant-negative Ikaros |
| pTOL-hCMV-TET3G-Hygro | transOMIC Technologies | Cat# CCIHS1001 |
| pCLIP-TRE3G-Cas9-ZsGreen | transOMIC Technologies | Cat# CCIMS1001 |
| pCLIP-hCMV-gRNA-tRFP | transOMIC Technologies | Cat# CCIRS1001 |
| Software and Algorithms | ||
| BRB Array Tool | http://linus.nci.nih.gov/BRB-ArrayTools.html | |
| Gene Cluster | http://www.eisenlab.org/software.html | |
| Java Treeview | http://jtreeview.sourceforge.net/ | |
| Gene Set Enrichment Analysis (GSEA) | http://software.broadinstitute.org/gsea/index.jsp | |
| CompuSyn | http://www.combosyn.com/ | |
| R package “survival” Version 2.35-8 | N/A | |
| Other | ||
Colony forming assay for murine ALL cells
10,000 B-ALL cells were resuspended in 1 mL murine MethoCult medium (M3231 or M3630, STEMCELL Technologies) and cultured in 35 mm dishes at 37°C in a humidified incubator with 5% CO2. Colony numbers were counted after 7-14 days using an automated colony counter.
NADPH/NADP ratio measurement
2×107 cells were harvested and subjected to measure NADPH/NADP ratio by NADP/NADPH Assay Kit (ab65349, Abcam). NADPH/NADP ratios relative to control cells were calculated in each experiment.
Affymetrix GeneChip expression analysis
Total RNA was isolated using the NucleoSpin RNA kit (MACHEREY-NAGEL). RNA quality was determined with the Agilent Bioanalyzer (Agilent Technologies). Biotinylated cRNA was generated and fragmented based on the Affymetrix protocol and hybridized to mouse Gene 1.0 ST (Affymetrix). After scanning (GeneChip Scanner 3000 7G; Affymetrix) of the arrays, the CEL files generated were analyzed using BRB Array Tool (http://linus.nci.nih.gov/BRB-ArrayTools.html) and processed using the RMA algorithm (robust multi-array average) for normalization and summarization. Gene expression ratios were processed and visualized as a heat map utilizing Gene Cluster and Java Treeview. Gene Set Enrichment Analysis (GSEA) was performed using list in Table S1 with GSEA software. Raw data are available via GEO accession number GSE83742.
In vivo leukemia and lymphoma cell transplantation
Ppp2r1afl/fl BCR-ABL1 B-ALL cells were transduced with retrovirus to overexpress firefly luciferase, selected with puromycin, and then transduced with Cre-IRES-GFP or GFP empty vector. 2 days after transduction, GFP+ cells were sorted by FACSAria II (BD Biosciences) and genotyping was performed to verify Cre mediated deletion. 1×106 viable cells were intravenously injected into sublethally irradiated (2 Gy) NSG mice (6-8 weeks old females). Comparable viability and luciferase activity of Cre-IRES-GFP and GFP transduced cells were confirmed by flow-cytometry and D-Luciferin (Gold Bio) induced bioluminescence signal. Leukemia progression in mice was monitored at the times indicated using an in vivo IVIS Spectrum bioluminescence/optical imaging system (PerkinElmer Inc). 2.5 mg D-Luciferin (dissolved in PBS) per mouse was injected intraperitoneally 15 min before measuring. General anesthesia was induced with 5% isoflurane and continued during the procedure with 2% isoflurane introduced through a nose cone. Ppp2r1afl/fl NrasG12D ALL cells were transduced with Cre-ERT2-IRES-GFP or ERT2-IRES-GFP empty vector. 2 days after transduction, GFP+ cells were sorted by FACSAria II (BD Biosciences). 3×106 viable cells after 24 hours’ 4-OHT treatment were intravenously injected into sublethally irradiated (2 Gy) NSG mouse (6-8 weeks female). After transplantation, 5 mg/mouse/day Tamoxifen (Sigma-Aldrich) was administrated to recipient mice with NrasG12D ALL cells for 7 days through oral gavage. 3×106 viable patient-derived diffuse large B cell lymphoma cells (BOS12; Table S3) were intravenously injected into sublethally irradiated (2 Gy) NSG mouse (6-8 weeks female). Treatment of vehicle, LB-100 (1.5 mg/kg), 6- AN (1 mg/kg) or combination (1.5 mg/kg LB-100 + 1 mg/kg 6-AN) started 7 days after transplantation by intraperitoneal injection. Mice were euthanized when they reached the criteria of terminal sickness caused by leukemia. Bone marrow and spleen cells were collected for flow cytometry analysis. All mouse experiments were subject to institutional approval by the Beckman Research Institute Animal Care and Use Committee. 6-8 weeks old female NSG mice were randomly allocated into each group. The minimal number of mice in each group was calculated by using the ‘cpower’ function in R/Hmisc package. No blinding was used.
Extracellular glucose, lactate and intracellular ATP measurements
Glucose and lactate levels in cell culture medium were measured with the Amplex Red Glucose/Glucose Oxidase Assay Kit (Life Technologies) and the L-Lactate Assay Kit (Cayman Chemical). Medium at indicated time points was collected and stored at −80°C and were later processed according to the manufacturers’ instructions. Intracellular ATP levels were measured using the ATP Bioluminescence Assay Kit CLS II (Roche). Cell pellets were harvested at different time points and processed as instructed by manufacturer’s protocol.
Glycolysis and mitochondrial respiration measurements
Seahorse XFe24 extracellular flux analyzer (Agilent Technologies) was used to measure oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of mouse B-ALL and myeloid leukemia cells. Cells treated with 4-OHT or vehicle at indicated time points were subsequently processed to measure glycolysis and mitochondrial function with Seahorse XF Glycolysis Stress Test and Cell Mito Stress Test Kit (Agilent Technologies). In brief, 1.5×105 cells/well were plated in Cell-Tak (Corning) coated XF 24 micro culture plate (Agilent Technologies). For glycolysis stress assay, cells were incubated in XF-Base Medium supplemented with GlutaMAX for 1 h at 37°C in non-CO2 incubator, basal ECAR was then measured at the glucose-deprived stage. After injection of saturated glucose (10 mmol/l), glycolysis induced ECAR was measured. Oligomycin (1 μmol/l) and 2-deoxyglucose (2-DG, 0.1 mol/l) were sequentially injected and realtime ECAR was measured to reflect glycolytic capacity and specific inhibition of glycolysis. Non-glycolytic acidification (glucose deprived), glycolysis and glycolytic capacity were determined. ECAR values were normalized to cell numbers and are shown as fold change relative to non-glycolytic ECAR. Similarly, cells kept in XF-Base Medium supplemented with glucose (10 mmol/l) and GlutaMAX were used to measure OCR at basal respiration stage. After injection of ATP synthase inhibitor oligomycin (1 μmol/l), mitochondrial uncoupler Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP, 5 μmol/l) and respiratory chain inhibitor antimycin and rotenone (1 μmol/l), real-time OCR was measured to indicate mitochondrial function. All ECAR and OCR values were normalized to cell number and are shown as fold change relative to basal levels.
Metabolite Extraction and Mass Spectrometry-based Analysis
Metabolites were extracted from 2×106 cells per sample using the methanol based method. After incubation at 37°C for the indicated time, cells were rinsed with 150 mmol/l ammonium acetate, pH 7.3, and 100 μl cold 80% methanol (Optima* LC/MS, Fisher Scientific) was added to cells. 10 nmol norvaline (Sigma-Aldrich) was added as internal control. Samples were vortexed 3 times over 15 minutes and spun down at top speed for 5 minutes at 4°C. The supernatant was transferred to a new Eppendorf tube, and pellet was resuspended in 50 μl cold 80% methanol for second extraction. Combined 150 μl supernatant was dried on Vacufuge Plus (Eppendorf) at 30°C. Extracted metabolites were stored at −80°C. For mass spectrometry-based analysis, the metabolites were resuspended in 70% ACN and 5 μl used for analysis with a mass spectrometer. The mass spectrometer (Q Exactive, Thermo Scientific) was coupled to an UltiMate3000 RSLCnano HPLC. The chromatography was performed with 5 mmol/l NH4AcO (pH 9.9) (A) and ACN (B) at a flow rate of 300 μl/min starting at 85% B, going to 5% B at 18 min, followed by an isocratic step to 27 min and reequilibration to 34 min. The separation was achieved on a Luna 3u NH2 100A (150 × 2 mm) (Phenomenex). The Q Exactive was run in polarity switching mode (+3 kV / −2.25 kV). Metabolites were detected based on retention time (tR) and on accurate mass (± 3 ppm). Metabolite quantification was performed as area-under-the-curve (AUC) with TraceFinder 3.1 (Thermo Scientific). Data were normalized to the number of cells. Log2-fold changes relative to controls were plotted.
ChIP-seq data analysis
ChIPseq tracks for PAX5, IKZF1, EBF1 and TCF3 antibodies in a normal B cell sample are downloaded from (http://genome.ucsc.edu/ENCODE/dataMatrix/encodeChipMatrixHuman.html). Y-axes represent the normalized number of reads per million for peak summit for each track. The ChIP-seq peaks were called by the MACS peak caller (Zhang et al., 2008) by comparing read density in the ChIP experiment relative to the input chromatin control reads, and shown as bars under each wiggle track. Gene models were shown in UCSC genome browser hg19.
Single-locus quantitative chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously (Chan et al., 2017). Chromatin from fixed patient-derived Ph+ B-ALL cells (ICN1) was isolated and sonicated to 100-500 base pair DNA fragments. Chromatin fragments were immunoprecipitated with either IgG (as a control) or anti-Pax5 antibody. Following reversal of crosslinking by formaldehyde, specific DNA sequences were analyzed by quantitative real-time PCR. Primers were designed according to ChIP-seq tracks for PAX5 antibodies in B lymphocytes (ENCODE, Encyclopedia of DNA Elements, GM12878).
CRISPR-Cas9 mediated gene disruption
Patient-derived xenografts (PDX) were transduced with a set of plasmids for inducible Cas9 expression by lentiviral transduction. All Cas9-ZsGreen transduced cells were induced with doxycycline and then sorted for ZsGreen+ using a FACSAria II. After sorting, cells were cultured in tetracycline-free medium and later transduced with gRNAs by lenti-viral transduction. All gRNA-RFP transduced cells were FACS sorted for RFP+ reporter expression. Cas9-RFP expression was induced by doxycycline. ZsGreen+ RFP+ cells were purified for further analysis including target-gene expression measurement and functional assay. For growth competition assay, percentage of targeted cells was calculated as ratios between ZsGreen+RFP+ doublepositive and ZsGreen+ populations. CRISPR-Cas9 plasmid set including all gRNAs for target genes were obtained from transOMIC technologies. The gRNA sequences that were used are listed in Table S6.
Cell viability assays
4×104 human or mouse leukemia cells were seeded in a volume of 50 μl in complete growth medium on 96-well plates (Eppendorf). Compounds were added at the indicated concentrations in a total volume of 100 μl per well. After culturing for 3 days, cells were subjected to CCK-8 (Dojindo) or CellTiter-Glo Luminescent Cell Viability Assay (Promega). Medium without cells was used as blank. Relative viability was calculated using baseline values of untreated cells as a reference. For LB-100 (S7537, Selleck Chemicals) and 6-AN (A68203, Sigma-Aldrich) single treatment shown in Figure 7H-I, Ph+ ALL xenografts including LAX2, MXP2, PDX2 and BLQ5 (n=4) and myeloid leukemia including KYO-1, EM-2, JURL-MK1 and MV4-11 (n=4) were used.
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software Inc.) and were shown as mean ± SD as indicated. Unpaired 2-tailed Student’s t test was used to evaluate difference between two groups and P-values were plotted (P< 0.05 was considered statistically significant). For in vivo transplantation experiments, the minimal number of mice in each group was calculated through use of the ‘cpower’ function in the R/Hmisc package. Kaplan-Meier survival analysis was used to estimate OS with GraphPad Prism 7 and log-rank test was used to compare the difference between two groups. For patient overall survival analysis, patients in each dataset were segregated into 2 groups based on 25% highest or 25% lowest mRNA levels of the gene of interest. The datasets used include mantle cell lymphomas (GSE10793, n=71 and http://llmpp.nih.gov/MCL, n = 92), diffuse large B cell lymphoma (DLBCL; GSE10846, n=414), B cell lineage CLL (GSE22726, n=107) and acute myeloid leukemia (https://tcga-data.nci.nih.gov/; n=200).
Log-rank test was used to compare survival differences between patient groups. R package “survival” Version 2.35-8 was used for the survival analysis and Cox proportional hazards regression model in R package for the multivariate analysis (R Development Core Team, 2009). To determine synergism effect of LB-100 and 6-AN in B-ALL and B-cell lymphoma cells, combination index (CI) values were calculated using the CompuSyn software to determine interaction (synergistic, CI<1; additive, CI=1; or antagonistic, CI>1).
Data and Software Availability
The GEO accession number for the gene expression arrays in this study is GSE83742.
Supplementary Material
Figure S1: Effects of PP2A ablation on survival in B-cell lineage leukemia, Related to Figure 1
Early pro-B cells (Hardy fractions A and B) from bone marrow of Ppp2r1afl/fl Mb1-Cre mice were sorted and genotyped by PCR based on primers (see Table S6) depicted in the schematic (A). IL7-dependent B cell precursors propagated from Ppp2r1afl/fl mice were transformed with BCR-ABL1 or NRASG12D. BCR-ABL1 (B) or NRASG12D (C) driven B-ALL cells were transduced with 4-OHT-inducible Cre-ERT2-IRES-GFP (Cre) or ERT2-IRES-GFP-Vector control (EV). Percentages of GFP+ cells were measured by flow cytometry at different time points following 4-OHT treatment. Ppp2r1afl/fl BCR-ABL1 (D) or NRASG12D (E) B-ALL cells transduced with 4-OHT-inducible Cre or EV were treated with 4-OHT for 3 days and viable cells were identified by flow cytometry. Other cells treated under these conditions were subjected to EdU-based cell cycle analysis and TUNEL assay and analyzed by flow cytometry after 3 days of 4-OHT treatment (F-G). The high baseline levels of TUNEL+ cells likely reflect DNA double-strand breaks owing to ongoing V(D)J recombination in B-cell precursor ALL cells (Swaminathan et al., 2015). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S2: Inducible deletion of PP2A causes imbalances of oncogenic signaling in B cell leukemia cells, Related to Figure 2
Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with 4-OHT-inducible Cre-ERT2 (Cre) or ERT2-vector (EV) were treated with 4-OHT for 3 days and cell lysates were studied by phospho-protein array analysis (A). Quantitative differences between EV- and Cre-transduced cells are depicted in the bar graph (left). Ppp2r1afl/fl BCR-ABL1 ALL cells were transduced with 4-OHT-inducible Cre or EV control and incubated in the presence of the small molecule signaling inhibitors imatinib (1 μmol/l; B), idelalisib (32 μmol/l; C) or rapamycin (50 nmol/l; D). Percentages of GFP+ cells were measured by flow cytometry at the times indicated following 4-OHT-treatment. Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S3: PP2A is essential for FoxO1 and FoxO3a activity but neither FoxO1 nor FoxO3 can rescue cell death upon PP2A-ablation in B-ALL, Related to Figure 2
(A) Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with inducible Cre-ERT2 (Cre) or ERT2-vector (EV) were treated with 4-OHT for 3 days and cell lysates were used to measure phosphorylation of FoxO1, FoxO3a, p70 S6K and S6. (B) FoxO1fl/fl BCR-ABL1 B-ALL cells transduced with inducible Cre or EV were treated with 4-OHT and dead cells were defined by Annexin V and 7-AAD double positive population through flow cytometry. (C) FoxO1fl/fl BCR-ABL1 B-ALL cells were treated for 3-days 4-OHT and then were plated in methylcellulose to perform colony formation assays. Photomicrographs of colonies at 1× (scale bar indicating 1 mm) and 6.3× (scale bar indicating 0.1 mm) magnification are shown. Colony numbers were counted after 7 days of cell culture. (D) Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with EV or Cre were subsequently transduced with FoxO1-ADA-IRES-GFP (FoxO1-ADA) or GFP-Vector (GFP-EV). Percentages of GFP+ cells were measured by flow cytometry and at different time points following 4-OHT treatment and time course data are depicted. (E) Ppp2r1afl/fl BCR-ABL1 B-ALL cells were transduced with FoxO3a-CA-IRES-CD90 (FoxO3a-CA) or CD90-Vector (CD90-EV). Percentages of CD90+ cells were measured by flow cytometry and at time points indicated following 4-OHT treatment. (F) Gene set enrichment analysis (GSEA) was performed using a set of 69 antioxidant genes based on the gene expression data from microarray in Figure 2F (GSE83742). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S4: Metabolic consequences of PP2A-deletion and expression levels of glycolytic and PPP enzymes in B-lymphoid and myeloid cells, Related to Figure 3
(A) Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with Cre-ERT2 (Cre) or ERT2-Vector (EV) were treated with 4-OHT for 2 days then seeded in fresh medium for measurements of glucose consumption, lactate production or directly lysed to measure ATP levels. Relative levels (to EV) were shown. 2-days 4-OHT treated Ppp2r1afl/fl B-ALL (B) or myeloid CML-like cells (C) transduced with 4-OHT-inducible Cre or EV were seeded for real-time oxygen consumption rate (OCR) measurement to assess mitochondrial function. OCR measurements were performed before and after treatment of oligomycin, FCCP and antimycin + rotenone. OCR values were normalized to cell numbers and are shown as fold change relative to basal level OCR. (D) Gene expression profile of enzymes of glycolysis and pentose phosphate pathway (PPP) in healthy bone marrow, myeloid and B cell malignancies (collected from BloodSpot data base using dataset of Leukemia MILE study http://servers.binf.ku.dk/bloodspot/). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S5: Spectrum of genetic PPP2R1A lesions in human cancer and PPP2R1A, PPP2CA and G6PD mRNA levels in normal and malignant hematopoietic populations, Related to Figure 5
(A) Frequencies of PPP2R1A genetic lesion identified in cohorts of 16 different cancer types (collected from COSMIC http://cancer.sanger.ac.uk/cosmic). Mutations and deletions of PPP2R1A occur at an average frequency of ~3% throughout most types of solid tumors and myeloid leukemia (range 0.5% to 14%; 23,009 samples studied in COSMIC) but were not found in 323 B-cell tumor samples (B-ALL and mature B cell lymphoma). (B) mRNA levels for PPP2R1A, PPP2CA and G6PD across human normal and malignant myeloid and B-lymphoid samples (B-ALL and mature B cell lymphoma); gene expression data collected from http://amazonia.transcriptome.eu/.
Figure S6: Overexpression of G6PD is not sufficient to rescue cell death upon PP2A deletion Related to Figure 6
(A) Ppp2r1afl/fl BCR-ABL1 B-ALL cells carrying inducible Cre-ERT2 (Cre) or ERT2-vector (EV) were subsequently transduced with G6PD-IRES-GFP (murine G6pdx or human G6PD) or GFP empty vector (EV). (A) G6PD expression was measured in these cells by Western blot. Percentages of GFP+ cells were measured by flow cytometry and at the time points indicated following 4-OHT treatment (B-C). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S7: Target validation of PP2A in human B cell malignancies Related to Figure 7
(A) Chemical structures of LB-100 and 6-AN are shown. (B) Representative flow cytometry plots of growth competition assay of patient-derived Ph+ B-ALL cells (PDX2) after CRISPR-Cas9 mediated targeting of PPP2R1A (gRNA-2) or non-targeting gRNA as indicated in Figure 7B. (C-E) To test potential side-effects of LB-100 on normal T cells, C57/B6J mice were intraperitoneally injected with 1.5 mg/kg LB-100 or vehicle every 2 days over a period of 9 days. After 4 injections, mice were sacrificed and T cell compartments in the thymus (C, D), spleen and lymph nodes (E) were analyzed by flow cytometry. While thymic DN subsets were slightly reduced, LB-100 did not affect more mature thymic, splenic or lymph node T cell subsets. Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
HIGHLIGHTS.
PP2A redirects glucose flux from glycolysis to the PPP to salvage oxidative stress
B-lymphoid transcription factors repress rate-limiting PPP enzymes in B-cell tumors
The PPP-agonist TIGAR efficiently rescues B-cells upon genetic ablation of PP2A
PP2A small molecule inhibition can overcome drug-resistance in B-cell tumors
ACKNOWLEDGMENTS.
We would like to thank Dr. John S. Kovach (Lixte Biotechnology), Dr. Arthur Riggs (Beckman Research Institute), Dr. Daniel Hodson (University of Cambridge), Dr. David A. Fruman (University of California Irvine). Dr. Michael Farrar (University of Minnesota) and Dr. H. Phillip Koeffler (University of California Los Angeles) and all members of the Muschen laboratory for support and helpful discussions. Research is funded by the NIH/NCI through Outstanding Investigator Award R35CA197628, R01CA137060, R01CA157644, R01CA172558 and R01CA213138 (to M.M.), a Wellcome Trust Senior Investigator Award and a Leukemia and Lymphoma Scholar award (to M.M.), the Howard Hughes Medical Institute HHMI-55108547 (to M.M.), the Norman and Sadie Lee Foundation (for Pediatric Cancer, to M.M.), and the Dr. Ralph and Marian Falk Medical Research Trust (to M.M.), Cancer Research Institute through a Clinic and Laboratory Integration Program grant (to M.M.) and the California Institute for Regenerative Medicine (CIRM) through DISC2-10061. H.J. is supported by Deutsche Forschungsgemeinschaft (SFB1074, P10) and ERC Advanced Grant #694992. M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
All authors declare no competing interests.
REFERENCES
- Apostolidis SA, Rodríguez-Rodríguez N, Suárez-Fueyo A, Dioufa N, Ozcan E, Crispín JC, Tsokos MG, and Tsokos GC (2016). Phosphatase PP2A is requisite for the function of regulatory T cells. Nat. Immunol. 17, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, and Vousden KH (2006). TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 126, 107–120. [DOI] [PubMed] [Google Scholar]
- Buchner M, Swaminathan S, Chen Z, and Müschen M (2015). Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol. Rev 263, 192–209. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, and Mak TW (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95. [DOI] [PubMed] [Google Scholar]
- Chan LN, Chen Z, Braas D, Lee J-W, Xiao G, Geng H, Cosgun KN, Hurtz C, Shojaee S, Cazzaniga V, et al. (2017). Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 542, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Arroyo JD, Timmons JC, Possemato R, and Hahn WC (2005). Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 65, 8183–8192. [DOI] [PubMed] [Google Scholar]
- Chen Z, Shojaee S, Buchner M, Geng H, Lee JW, Klemm L, Titz B, Graeber TG, Park E, Tan YX, et al. (2015). Signalling thresholds and negative B-cell selection in acute lymphoblastic leukaemia. Nature 521, 357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, and Vousden KH (2013). TIGAR Is Required for Efficient Intestinal Regeneration and Tumorigenesis. Dev. Cell 25, 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung V, Mansfield AS, Braiteh F, Richards D, Durivage H, Ungerleider RS, Johnson F, and Kovach JS (2017). Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res 23, 3277–3284. [DOI] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Pan L, Groen RWJ, Baleydier F, Kentsis A, Marineau J, Grebliunaite R, Kozakewich E, Reed C, Pflumio F, et al. (2014). Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Invest 124, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Elf S, Fan J, Kang H-B, Seo JH, Shan C, Dai Q, Zhang L, Xie J, et al. (2012). Phosphoglycerate Mutase 1 Coordinates Glycolysis and Biosynthesis to Promote Tumor Growth. Cancer Cell 22, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. U. S. A. 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus A, Saussele S, Rosti G, Mahon F-X, Janssen JJWM, Hjorth-Hansen H, Richter J, and Buske C (2017). Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol 28, iv41–iv51. [DOI] [PubMed] [Google Scholar]
- Jeon S-M, Chandel NS, and Hay N (2012). AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanteti R, Nallasura V, Loganathan S, Tretiakova M, Kroll T, Krishnaswamy S, Faoro L, Cagle P, Husain AN, Vokes EE, et al. (2009). PAX5 is expressed in small-cell lung cancer and positively regulates c-Met transcription. Lab. Investig. 89, 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhanpal GK, Vecchiarelli-Federico LM, Li Y-J, Cui J-W, Bailey ML, Spaner DE, Dumont DJ, Barber DL, and Ben-David Y (2010). The inositol phosphatase SHIP-1 is negatively regulated by Fli-1 and its loss accelerates leukemogenesis. Blood 116, 428–436. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- Lin R, Elf S, Shan C, Kang H-B, Ji Q, Zhou L, Hitosugi T, Zhang L, Zhang S, Seo JH, et al. (2015). 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat. Cell Biol 17, 1484–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GJ, Cimmino L, Jude JG, Hu Y, Witkowski MT, McKenzie MD, Kartal-Kaess M, Best SA, Tuohey L, Liao Y, et al. (2014). Pax5 loss imposes a reversible differentiation block in B-progenitor acute lymphoblastic leukemia. Genes Dev. 28, 1337–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Kovach JS, Johnson F, Chiang J, Hodes R, Lonser R, and Zhuang Z (2009). Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl. Acad. Sci. U. S. A 106, 11697–11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müschen M (2018). Autoimmunity checkpoints as therapeutic targets in B cell malignancies. Nat. Rev. Cancer 18, 103–116. [DOI] [PubMed] [Google Scholar]
- Nemoto S, and Finkel T (2002). Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 295, 2450–2452. [DOI] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen C-C, Xu P-Z, Chen M-L, Tonic I, Unterman T, and Hay N (2008). Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 14, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, and Hay N (2014). The pentose phosphate pathway and cancer. Trends Biochem. Sci. 39, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozuelo Rubio M, Peggie M, Wong BHC, Morrice N, and MacKintosh C (2003). 14-3-3s regulate fructose-2,6-bisphosphate levels by binding to PKB-phosphorylated cardiac fructose-2,6-bisphosphate kinase/phosphatase. EMBO J. 22, 3514–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros S, and Schulze A (2013). Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger R, Ruiz J, and Walter G (2011). Human cancer-associated mutations in the Aα subunit of protein phosphatase 2A increase lung cancer incidence in Aα knock-in and knockout mice. Mol. Cell. Biol. 31, 3832–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, Izadmehr S, Mazhar S, Wiredja DD, O’Connor CM, et al. (2017). Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Invest 127, 2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharabi A, Kasper IR, and Tsokos GC (2017). The serine/threonine protein phosphatase 2A controls autoimmunity. Clin. Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaee S, Chan LN, Buchner M, Cazzaniga V, Cosgun KN, Geng H, Qiu YH, von Minden MD, Ernst T, Hochhaus A, et al. (2016). PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 22, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon S-M, Trageser D, Hasselfeld B, Henke N, Mooster J, et al. (2015). Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat. Immunol 16, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, Evans GA (1998). Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 282: 284–287. [DOI] [PubMed] [Google Scholar]
- Wu Y, Song P, Xu J, Zhang M, and Zou M-H (2007). Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 282, 9777–9788. [DOI] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, and Graf T (2004). Stepwise Reprogramming of B Cells into Macrophages. Cell 117, 663–676. [DOI] [PubMed] [Google Scholar]
- Yan L, Lavin VA, Moser LR, Cui Q, Kanies C, and Yang E (2008). PP2A regulates the proapoptotic activity of FOXO1. J. Biol. Chem 283, 7411–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Huang Q, Lu Y, Li X, and Huang S (2012). Reactivating PP2A by FTY720 as a Novel therapy for AML with C-KIT tyrosine kinase domain mutation. J. Cell. Biochem 113, 1314–1322. [DOI] [PubMed] [Google Scholar]
- Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, and Weyand CM (2016). Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med 8, 331ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, and Staudt LM (2013). Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov 12, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf I, Zhu X, Kharas MG, Chen J, and Fruman DA (2004). Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood 104, 784–787. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Effects of PP2A ablation on survival in B-cell lineage leukemia, Related to Figure 1
Early pro-B cells (Hardy fractions A and B) from bone marrow of Ppp2r1afl/fl Mb1-Cre mice were sorted and genotyped by PCR based on primers (see Table S6) depicted in the schematic (A). IL7-dependent B cell precursors propagated from Ppp2r1afl/fl mice were transformed with BCR-ABL1 or NRASG12D. BCR-ABL1 (B) or NRASG12D (C) driven B-ALL cells were transduced with 4-OHT-inducible Cre-ERT2-IRES-GFP (Cre) or ERT2-IRES-GFP-Vector control (EV). Percentages of GFP+ cells were measured by flow cytometry at different time points following 4-OHT treatment. Ppp2r1afl/fl BCR-ABL1 (D) or NRASG12D (E) B-ALL cells transduced with 4-OHT-inducible Cre or EV were treated with 4-OHT for 3 days and viable cells were identified by flow cytometry. Other cells treated under these conditions were subjected to EdU-based cell cycle analysis and TUNEL assay and analyzed by flow cytometry after 3 days of 4-OHT treatment (F-G). The high baseline levels of TUNEL+ cells likely reflect DNA double-strand breaks owing to ongoing V(D)J recombination in B-cell precursor ALL cells (Swaminathan et al., 2015). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S2: Inducible deletion of PP2A causes imbalances of oncogenic signaling in B cell leukemia cells, Related to Figure 2
Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with 4-OHT-inducible Cre-ERT2 (Cre) or ERT2-vector (EV) were treated with 4-OHT for 3 days and cell lysates were studied by phospho-protein array analysis (A). Quantitative differences between EV- and Cre-transduced cells are depicted in the bar graph (left). Ppp2r1afl/fl BCR-ABL1 ALL cells were transduced with 4-OHT-inducible Cre or EV control and incubated in the presence of the small molecule signaling inhibitors imatinib (1 μmol/l; B), idelalisib (32 μmol/l; C) or rapamycin (50 nmol/l; D). Percentages of GFP+ cells were measured by flow cytometry at the times indicated following 4-OHT-treatment. Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S3: PP2A is essential for FoxO1 and FoxO3a activity but neither FoxO1 nor FoxO3 can rescue cell death upon PP2A-ablation in B-ALL, Related to Figure 2
(A) Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with inducible Cre-ERT2 (Cre) or ERT2-vector (EV) were treated with 4-OHT for 3 days and cell lysates were used to measure phosphorylation of FoxO1, FoxO3a, p70 S6K and S6. (B) FoxO1fl/fl BCR-ABL1 B-ALL cells transduced with inducible Cre or EV were treated with 4-OHT and dead cells were defined by Annexin V and 7-AAD double positive population through flow cytometry. (C) FoxO1fl/fl BCR-ABL1 B-ALL cells were treated for 3-days 4-OHT and then were plated in methylcellulose to perform colony formation assays. Photomicrographs of colonies at 1× (scale bar indicating 1 mm) and 6.3× (scale bar indicating 0.1 mm) magnification are shown. Colony numbers were counted after 7 days of cell culture. (D) Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with EV or Cre were subsequently transduced with FoxO1-ADA-IRES-GFP (FoxO1-ADA) or GFP-Vector (GFP-EV). Percentages of GFP+ cells were measured by flow cytometry and at different time points following 4-OHT treatment and time course data are depicted. (E) Ppp2r1afl/fl BCR-ABL1 B-ALL cells were transduced with FoxO3a-CA-IRES-CD90 (FoxO3a-CA) or CD90-Vector (CD90-EV). Percentages of CD90+ cells were measured by flow cytometry and at time points indicated following 4-OHT treatment. (F) Gene set enrichment analysis (GSEA) was performed using a set of 69 antioxidant genes based on the gene expression data from microarray in Figure 2F (GSE83742). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S4: Metabolic consequences of PP2A-deletion and expression levels of glycolytic and PPP enzymes in B-lymphoid and myeloid cells, Related to Figure 3
(A) Ppp2r1afl/fl BCR-ABL1 B-ALL cells transduced with Cre-ERT2 (Cre) or ERT2-Vector (EV) were treated with 4-OHT for 2 days then seeded in fresh medium for measurements of glucose consumption, lactate production or directly lysed to measure ATP levels. Relative levels (to EV) were shown. 2-days 4-OHT treated Ppp2r1afl/fl B-ALL (B) or myeloid CML-like cells (C) transduced with 4-OHT-inducible Cre or EV were seeded for real-time oxygen consumption rate (OCR) measurement to assess mitochondrial function. OCR measurements were performed before and after treatment of oligomycin, FCCP and antimycin + rotenone. OCR values were normalized to cell numbers and are shown as fold change relative to basal level OCR. (D) Gene expression profile of enzymes of glycolysis and pentose phosphate pathway (PPP) in healthy bone marrow, myeloid and B cell malignancies (collected from BloodSpot data base using dataset of Leukemia MILE study http://servers.binf.ku.dk/bloodspot/). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S5: Spectrum of genetic PPP2R1A lesions in human cancer and PPP2R1A, PPP2CA and G6PD mRNA levels in normal and malignant hematopoietic populations, Related to Figure 5
(A) Frequencies of PPP2R1A genetic lesion identified in cohorts of 16 different cancer types (collected from COSMIC http://cancer.sanger.ac.uk/cosmic). Mutations and deletions of PPP2R1A occur at an average frequency of ~3% throughout most types of solid tumors and myeloid leukemia (range 0.5% to 14%; 23,009 samples studied in COSMIC) but were not found in 323 B-cell tumor samples (B-ALL and mature B cell lymphoma). (B) mRNA levels for PPP2R1A, PPP2CA and G6PD across human normal and malignant myeloid and B-lymphoid samples (B-ALL and mature B cell lymphoma); gene expression data collected from http://amazonia.transcriptome.eu/.
Figure S6: Overexpression of G6PD is not sufficient to rescue cell death upon PP2A deletion Related to Figure 6
(A) Ppp2r1afl/fl BCR-ABL1 B-ALL cells carrying inducible Cre-ERT2 (Cre) or ERT2-vector (EV) were subsequently transduced with G6PD-IRES-GFP (murine G6pdx or human G6PD) or GFP empty vector (EV). (A) G6PD expression was measured in these cells by Western blot. Percentages of GFP+ cells were measured by flow cytometry and at the time points indicated following 4-OHT treatment (B-C). Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.
Figure S7: Target validation of PP2A in human B cell malignancies Related to Figure 7
(A) Chemical structures of LB-100 and 6-AN are shown. (B) Representative flow cytometry plots of growth competition assay of patient-derived Ph+ B-ALL cells (PDX2) after CRISPR-Cas9 mediated targeting of PPP2R1A (gRNA-2) or non-targeting gRNA as indicated in Figure 7B. (C-E) To test potential side-effects of LB-100 on normal T cells, C57/B6J mice were intraperitoneally injected with 1.5 mg/kg LB-100 or vehicle every 2 days over a period of 9 days. After 4 injections, mice were sacrificed and T cell compartments in the thymus (C, D), spleen and lymph nodes (E) were analyzed by flow cytometry. While thymic DN subsets were slightly reduced, LB-100 did not affect more mature thymic, splenic or lymph node T cell subsets. Data are shown as mean ± standard deviation (SD) and representative of at least three independent experiments.