

Abstract
Brown fat is a thermogenic tissue that generates heat to maintain body temperature in cold environments and dissipate excess energy in response to overfeeding. We have addressed the role of the IGFIR in the brown fat development and function. Mice lacking IGFIR exhibited normal brown adipose tissue/body weight in knockout (KO) vs control mice. However, lack of IGFIR decreased uncoupling protein 1 expression in interscapular brown fat and beige cells in inguinal fat. More importantly, the lack of IGFIR resulted in an impaired cold acclimation. No differences in the total fat volume were found in the KO vs control mice. Epididymal fat showed larger adipocytes but with a lower number of adipocytes in KO vs control mice at age 12 months. In addition, KO mice showed a sustained moderate hyperinsulinemia and hypertriglyceridemia upon time and hepatic insulin insensitivity associated with lipid accumulation, with the outcome of a global insulin resistance. In addition, we found that the expression of uncoupling protein 3 in the skeletal muscle was decreased and its expression was increased in the heart in parallel with the expression of beta-2 adrenergic receptors. Upon nonobesogenic high-fat diet, we found a severe insulin resistance in the liver and in the skeletal muscle, but unchanged insulin sensitivity in the heart. In conclusion, our data suggest that IGFIR it is not an essential growth factor in the brown fat development in the presence of the IR and very high plasma levels of IGF-I, but it is indispensable for full brown fat functionality.
There are two functionally and morphologically different types of adipose tissue in mammals: white adipose tissue (WAT), which is the primary site of triglyceride storage; and brown adipose tissue (BAT), which specializes in energy expenditure. Brown fat is primarily a thermogenic tissue that burns fat to generate heat and help maintain body temperature in cold environments. It also helps dissipate excess energy in response to overfeeding (1). In mammals, adaptive thermogenesis occurs primarily in brown fat and in skeletal muscle. Adaptive thermogenesis can be divided into three subtypes: 1) shivering thermogenesis, a function of skeletal muscle; 2) nonshivering thermogenesis, a function of brown fat (both in response to cold); 3) and diet-induced thermogenesis, which is also a function of brown fat and induced by overfeeding. Ablation of BAT in rodents by means of toxins or genome manipulation results in obesity (2). Although earlier studies of mice uncoupling protein 1 (UCP-1) ablation failed to demonstrate an obesogenic effect, recently it has been demonstrated that these mice do become obese and demonstrate a loss of diet-induced thermogenesis when maintained under thermoneutrality (3). Interscapular brown fat (iBAT) in humans largely disappears shortly after birth, and thus it has traditionally been assumed that there is no functional brown fat present in adult humans. However, this concept has been radically revised after active brown fat was identified in humans studies using positron emission tomography–computed tomography imaging (4–8). More importantly, activity of those brown fat depots correlates inversely with body mass index (BMI) (4), suggesting a critical role of brown fat in human adult energy metabolism and the potential of using brown fat–mediated energy expenditure as an antiobesity therapy. However, it is now recognized that there are at least two distinct types of brown fat cells. Classical BAT, epitomized by the interscapular depot in rodents, arises from a Myf5+, muscle-like cell lineage. Protein domain containing 16 (PRDM16), a transcriptional regulatory protein, seems to control this muscle–brown fat decision between days 9 and 12 of gestation in mice (9). In addition, UCP-1+ cells can also appear in white fat depots under certain stimuli. This has been termed beige or brite adipocytes and does not come from a Myf5+ lineage (9).
Several developmental signaling molecules implicated in the evolution of mesodermal tissues have been shown to influence the development of BAT. These include nodal, wingless, fibroblast growth factors, members of the TGF-β family (bone morphogenetic proteins or BMPs), and IGF-I/insulin signaling. The insulin receptor (IR)/(IGFIR) signaling that works on cell proliferation and adipogenesis, whereas TGF-beta like signaling pathway mostly stimulated by BMP-7 and 8B acts on the recruitment of mesenchymal cells into brown fat cell linage (10). We previously developed the first BAT-specific knockout targeting the insulin receptor (BATIRKO). Those mice showed a severe brown fat lipoatrophy and susceptibility to develop visceral adiposity upon aging (11, 12). More importantly, recently it has been demonstrated that the double Igf1r/Ir knockout (KO) in fat showed a severe brown fat atrophy, providing strong support to the concept that IGFIR/IR-signaling pathways plays an essential role in brown fat development (13). However, the role of IGFIR by itself in the brown fat development remains largely unknown. To address this important issue, we have developed the IGFIR BAT-specific knockout (BATIGFIRKO). Our data suggest that IGFIR it is not an essential growth factor in the brown fat development in the presence of the IR. However, the lack of IGFIR decreased the UCP-1 expression in iBAT and much affect the development of the beige cells bearing UCP-1 protein expression mostly in the inguinal white fat depots. Overall, IGFIR it is indispensable to maintain the full thermogenic capacity, the lack of IGFIR resulting in an impaired cold acclimation. In addition, brown fat driven by IGFIR plays an unrecognized role in the redistribution of the adipose organ and lipid mobilization that affect the insulin sensitivity in peripheral tissues as revealed by sustained moderate hyperinsulinemia and hypertriglyceridemia, hepatic insulin sensitivity being much impaired associated with lipid accumulation, with the outcome of a global insulin resistance.
Materials and Methods
Experimental models and mice genotyping
Igf1r L/L mice were created by homologous recombination using an Igf1r gene-targeting vector with loxP sites flanking exon 3 as previously described (14). To create a specific BAT inactivation of the Igf1r we bred Igf1r L/L with Ucp-1-Cre transgenic mice (11) to obtain BAT-specific Igf1r-knockout (BATIGFIRKO) mice. Experiments were performed on male Igf1r L/L and BATIGFIRKO mice maintained on the hybrid 129/SvJ C57BL/6 background. Mice were maintained in the Animal Care Facility under the standard conditions of temperature and 12-hour light/dark cycle and were fed on standard diet (3% calories from fat, A04) for 3 or 12 months and on high-fat diet (18.4% proteins, 21.3% carbohydrates 60.3% kcal from fat: high proportion of polyunsaturated: arachidonic, linoleic, and linolenic acids vs saturated and trans fatty acids) for 16 weeks.
All animal experimentation described in this article was conducted according with accepted standards of human animal care, as approved by the corresponding institutional committee.
Igf1r L/L and Ucp-1-Cre transgenic mice were genotyped by PCR. Tail DNA (100–200 ng) was amplified 30 cycles (40 s, 94°C; 40 s, 60°C; and 1 min, 75°C) by a thermal cycler. Two primers flanking the loxP site behind exon 3 of the Igf1r were used: the forward primer (5′-ATGAATGCTGGTGAGGGTTGTCTT-3) and the reverse primer (5′-ATCTTGGAGTGGTGGGTCTGTTTC-3′). A 310-base-pair (bp) band was obtained for the floxed allele or a 250-bp band for the wild-type (WT) allele. Ucp-1-Cre transgenic mice were genotyped as previously described (11).
Protein extraction and Western blot
Igf1r L/L and BATIGFIRKO mice (age 3 and 12 mo) were killed by cervical dislocation and the different tissues were lysed as described (15). Western blot analyses were performed in iBAT, inguinal white adipose tissue (iWAT), epididymal white adipose tissue (eWAT), gastrocnemius, heart, liver, and brain homogenates, as previously described (16). The antibodies used were anti-IGF1Rβ (C20, sc-713), IRβ (C19, sc-711), PI3K (Z8, sc-423), UCP-1 (M17, sc-6529), CPT1-M (H120, sc-20670), CYT C (7H8, sc-13560), UCP-2 (C20, sc-6525), UCP-3 (sc-7756), and β2 adrenergic receptor (M20, sc-570) were from Santa Cruz Biotechnology. Antibodies anti-HSL (#4107), ACVR1 (#4398), p-SMAD 1/5/8 1/5(S463/465)/8(S426/428) (#9511), FAS (#3180) were from Cell Signaling Technology. Antibodies anti-IRS1 (#06-248) and anti-IRS2 (#06-506) were from Merck-Millipore. Anti-BMP7 (#5626-1) was from Epitomics. Anti-Hsp60 (ADI-SPP-741) antibody was obtained from Enzo Life Sciences. Anti β-actin from Sigma-Aldrih was used as a loading control in all membranes and different membranes were subjected to each inmunodetection. All antibodies were prepared in 1 × TBS, 0.1% Tween (Table 1). Immunoreactive bands were visualized using the ClarityECL Western blotting protocol (Bio-Rad).
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised In; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| IGF1R β subunit | C terminus of IGF-IRβ of human origin | IGF-IRβ Antibody (C-20) | Santa Cruz Biotechnology, sc-713 | rabbit; polyclonal | 1:500 |
| PI3K p85 α subunit | Amino acids 333–430 of the 85 kDa subunit of PI3 kinase of human origin | PI3 kinase p85α | Santa Cruz Biotechnology, sc-423 | rabbit; polyclonal | 1:1000 |
| IR β subunit | C terminus of insulin Rβ of human origin | Insulin Rβ antibody (C-19) | Santa Cruz Biotechnology, sc-711 | rabbit; polyclonal | 1:1000 |
| UCP-1 | C terminus of UCP-1 of mouse origin | UCP-1 antibody (M-17) | Santa Cruz Biotechnology, sc-6529 | goat; polyclonal | 1:500 |
| CPTI-M | Amino acids 16–135 mapping near the N-terminus of CPTI-M of human origin | CPTI-M antibody (H-120) | Santa Cruz Biotechnology, sc-20670 | rabbit; polyclonal | 1:1000 |
| Cytochrome c | Against full-length denatured cytochrome c of human origin | Cytochrome c antibody (7H8) | Santa Cruz Biotechnology, sc-13560 | mouse; monoclonal | 1:1000 |
| UCP-2 | C terminus of UCP-2 of human origin | UCP-2 antibody (C-20) | Santa Cruz Biotechnology, sc-6525 | goat; polyclonal | 1:1000 |
| UCP-3 | Domain common to the C terminus of UCP-3 L and UCP-3S of human origin | UCP-3 antibody (C-20) | Santa Cruz Biotechnology, sc-7756 | goat; polyclonal | 1:1000 |
| HSL | Total HSL protein | HSL antibody | Cell Signaling Technology, #4107 | rabbit; polyclonal | 1:1000 |
| ACVR1 | Total ACVR1 protein | ACVR1 antibody | Cell Signaling Technology, #4398 | rabbit; polyclonal | 1:500 |
| Phospho SMAD 1/5/8 | Phospho-Smad1 (Ser463/465)/Smad5 (Ser463/465)/Smad8 (Ser426/428) | Phospho-Smad1 (Ser463/465)/Smad5 (Ser463/465)/Smad8 (Ser426/428) antibody | Cell Signaling Technology, #9511 | rabbit; polyclonal | 1:1000 |
| Phospho AKT (Ser473) | Endogenous levels of Akt1 only when phosphorylated at Ser473 | Phospho-Akt (Ser473) Antibody | Cell Signaling Technology, #9271 | rabbit; polyclonal | 1:1000 |
| Phospho AKT (Thr308) | Endogenous levels of Akt only when phosphorylated at Thr308. | Phospho-Akt (Thr308) (C31E5E) Rabbit mAb | Cell Signaling Technology, #2965 | rabbit; monoclonal | 1:1000 |
| AKT | Endogenous levels of total Akt1, Akt2 and Akt3 proteins | Akt Antibody | Cell Signaling Technology, #9272 | rabbit; polyclonal | 1:1000 |
| BMP7 | Synthetic peptide corresponding to residues in Human BMP7 | Anti-BMP7 antibody [EPR5897] | Epitomics, #5626-1 | rabbit; monoclonal | 1:500 |
| HSP60 | HSP60 (mouse), (recombinant) | Enzo Life Sciences, ADI-SPP-741 | mouse; | 1:1000 | |
| β2-adrenergic receptor | Against a peptide mapping at the C terminus of B2-AR of mouse origin | β2-AR (M-20) | Santa Cruz Biotechnology, sc-570 | rabbit; polyclonal | 1:1000 |
| UCP-1 | Synthetic peptide conjugated to KLH, corresponding to amino acids 145–159 of Human UCP-1, with N-terminal cysteine added | Anti-UCP-1 antibody | Abcam, ab 10983 | rabbit; polyclonal | IHC, 1:500 |
| IRS1 | Carboxy-terminal 14 amino acid peptide ([C]YASINFQKQPEDRQ) of rat liver IRS1 | Anti-IRS1 | Millipore #06-248 | rabbit; polyclonal | 1:500 |
| IRS2 | GST fusion protein containing amino acids 976–1094 of mouse IRS2 | Anti-IRS2 | Millipore #06-506 | rabbit; polyclonal | 1:500 |
| β-actin | N-terminal end of the β-isoform of actin | Anti-β-actin, clone AC-74 | Sigma-Aldrich, A2228 | mouse; monoclonal | 1:5000 |
| Tubulin | C-terminal end of the α-tubulin isoform (amino acids 426–430) | Anti-α-tubulin, clone DM1A | Sigma-Aldrich, T6199 | mouse; monoclonal | 1:5000 |
| GAPDH | Anti-GAPDH antibody, clone GAPDH 71.1 | Sigma-Aldrich, G8795 | mouse; monoclonal | 1:5000 |
Ex vivo lipolysis assay
BAT from Igf1r L/L and BATIGFIRKO mice (age 3 and 12 mo) was removed and weighted and placed in prewarmed Krebs buffer for 5 minutes. The samples were used to ex vivo lipolysis assays as previously described (17).
PCR and qRT-PCR
Total RNA was extracted from BAT, and iWAT and eWAT tissues by TRIzol method (Invitrogen). RNA (5 μg) was reverse transcribed with a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's instructions. The gene expression of Lep, Adipoq, Slc2a4 (Glut4), Dio2, Prdm16, Igf1, Tnf, Serpine (PAI-1), and Usp15 were analyzed by real- time qPCR using TaqMan probes and GAPDH as endogenous control using a StepOnePlus Real-Time PCR System (Applied Biosystem). The results were calculated using the 2−ΔΔCt method as previously described (18).
To corroborate BAT Igf1r deletion, 500 ng of BAT cDNA were amplified 1 cycle of 95°C for 5 minutes, 40 cycles (95°C for 30 s; 62°C for 30 s; 72°C for 1 min) followed by one cycle of 72°C for 10 minutes on a thermal cycler. Two primers flanking the loxP site behind exon 3 Igf1r were used: the forward primer (5′-CCGCTGCTGGACCACAAATC-3′) and reverse primer (5′-CTTCATCGCCGCAGACTTTG-3′). A 398-bp band was obtained for the floxed allele and an 85 bp band was obtained when the Igf1r exon 3 was deleted.
Immunohistochemistry
For UCP-1 immunohistochemistry, iWAT and eWAT sections from 3- and 12-month old mice were incubated with rabbit polyclonal UCP-1 antibody (Abcam, ab10983) at 2 mg/mL in PBS-T/1% BSA overnight at 4°C. Secondary antibody incubation and developing using diaminobenzidine substrate kit was performed as previously described (9).
Thermogenic response to cold exposure
For the acute cold exposure experiment, 3- and 12-month-old Igf1r L/L and BATIGFIRKO male littermates that had been acclimatized to thermoneutrality (28°C) for 3 days were transferred to 4°C for 12 hours with full access to water and food. Body temperature was measured periodically by using a digital thermometer with a colonic probe (BIO-9882; Bioseb). Three-month-old Igf1r L/L and BATIGFIRKO mice were euthanized under thermoneutrality conditions after 4 or 12 hours of cold exposure, and the BAT was dissected and processed for subsequent analysis.
Indirect calorimetry
LabMaster metabolic cages, from TSE Systems were used to quantify the oxygen consumption (VO2), carbon dioxide production (VCO2), heat production, physical activity, and food and water intake of individual mice. Three- and 12-month-old WT (n = 5) and IGFIRKO (n = 6) mice were individually housed in chambers maintained at 24 ± 2°C with free access to chow and water. Before the experiment, mice were readapted to the new environment for 24 hours. At 0900 hours, measurements were taken every 10 minutes during 72 hours. Respiratory exchange ratio was calculated as the quotient between VCO2 and VO2. Data were analyzed with Phenomaster software (TSE Systems).
Metabolic efficiency: energy storage by nuclear magnetic resonance
Igf1r L/L and BATIGFIRKO 3- and 12 month-old mice were anesthetized with ECG; respiration was continuously monitored, and body fat was measured using a BrukerBioSpin (BioSpec: 47/40) as previously shown (19). The results were represented as fat body volume vs total body volume using Image J Launcher 1.46 software.
Hematoxylin-eosin staining, Oil Red O staining, and adipocytes size and number quantification
Freshly isolated liver and brown and white fat depots collected from Igf1r L/L or BATIGFIRKO mice were fixed in 10% formalin for 24 hours and embedded in paraffin for histological analysis. Different sections (5–7 μm thick) were deparaffinated and rehydrated to be stained with hematoxylin and eosin. Liver, muscle, and heart tissues were optimal cutting temperature-embedded and sections of 7-μm interval were Oil-Red-O/hematoxylin stained to measure lipid depot. Positive staining was quantified as previously described (20). All images were taken at magnification, ×20 and individual adipocyte area was determined using image analysis software (ImageJ Launcher 1.46, US edition). Relative adipocyte size from five animals (WAT) or three (iBAT) per group was calculated in arbitrary fields, by quantitation of 200 adipocytes per mouse. Adipocyte number was calculated by counting all the adipocytes per image in at least eight images per mouse.
Glucose tolerance test and insulin tolerance test
Glucose and insulin tolerance tests were performed on Igf1r L/L (n = 17 at 3 mo and n = 10 at 12 mo) and BATIGFIRKO mice (n = 15 at 3 mo and n = 8 at 12 mo) as described (21). Mice were fasted the day previous to the glucose tolerance test and all tests were performed in the morning at the same hour.
Plasma analysis
Plasma insulin, leptin, adiponectin, TNF-α, T3, IGF1, and BMP7 levels were measured from blood collected from 3 and 12-month-old fed mice by ELISAs according to the manufacturer's instructions. Rat/mouse insulin ELISA kit (#EZRMI-13K), mouse leptin ELISA kit (#EZML-82K), mouse adiponectin ELISA kit (#EZMADP-60K), mouse TNF-α ELISA kit (EZMTNFA) were from Merck-Millipore. Mouse/rat T3 ELISA (SE120091) and mouse IGF1 ELISA kit (RAB0229) were from Sigma-Aldrich and mouse BMP7 ELISA kit (EMBMP7) were from ThermoScientific. Triglycerides were tested in serum samples from 3- and 12-month-old fed mice using an enzymatic colorimetric assay from Spinreact.
In vivo insulin-signaling analysis
For in vivo insulin signaling studies, fed Igf1r L/L and BATIGFIRKO 12-month-old mice were injected with 1 U/kg body weight of human insulin (Novo Nordisk) into the peritoneal cavity. After 15 minutes, liver, iBAT, gastrocnemius, heart, and different WAT depots were removed and immediately frozen in liquid nitrogen. We used p-AKT/Akt ratio as marker of insulin sensitivity. Duplicate blots of samples from five individuals (Igf1r L/L) or four (BATIGFIRKO) under standard diet and four individuals (Igf1r L/L) or three (BATIGFIRKO) under HFD were used. The antibodies used were anti-phospho-Akt (Ser 473, #9271), anti-phospho-Akt (Thr 308, #2965), total Akt (#9272) from Cell Signaling and IR-β (sc-711) antibody from Santa Cruz Biotechnology.
Data analysis
All values were expressed as means ± SEM. Statistical significance was tested with the unpaired Student t test or with the one-way or two-way ANOVA followed by the Bonferroni test if differences were noted (GraphPad Prism 5.0). In some cases, nonparametric analysis such as Kruskall Wallis and Mann-Whitney U test were needed after testing normality condition by one sample Kolmogorov-Smirnov test. The null hypothesis was rejected when P < .05.
Results
Role of IGFIR in the BAT and beige cells gene expression and phenotype
Mice carrying a floxed Igf1r allele were bred with mice carrying a Cre transgene driven by the Ucp-1 promoter to produce BATIGFIRKO mice. No expression of IGF1R was observed in the BAT taken from KO mice compared with their littermates controls (Figure 1A). By contrast, IGF1R levels were unchanged in white fat, gastrocnemius, brain, and liver (Figure 1B). Mice exhibited normal development up to 12 months of age (Figure 1C), and the amount of interscapular BAT/body weight in KO mice remained unchanged vs controls (Figure 1D). Hematoxylin-eosin staining of sections of interscapular BAT revealed no major histological change at age 3 months between KO and control mice. However, the number of brown adipocytes with large lipid droplets was decreased in KO vs control mice at age 12 months (Figure 1E). Cell size increased and cell number decreased in 12-month-old vs 3-month-old control or KO mice. No significant changes were observed in their cell size or cell number in KO vs control mice at age 12 months (Figure 1F). At this stage, we wonder about the rate of lipolysis in those mice. Thus, basal rate of lipolysis significantly increased by more than 1-fold in KO vs control mice at age 3 or 12 months, respectively (Figure 1G). Total UCP-1 protein expression per processed iBAT tissue decreased only at age 12 months (Figure 1H). However, relative UCP-1 protein expression decreased at age 3 or 12 months in the KO vs control mice (Figure 1I). Assessment of tissue gene expression was performed at age 3 and 12 months of age by qPCR. This revealed that in the younger mice the expression of Usp15 (ubiquitin-specific peptidase 15; the specific deubiquitinase of Smad 1/5/8 transducer protein) and iodothyronine deiodinase (Dio2) was significantly increased, whereas the expression of Scla4 (Glut4), Lep, adipoq, Tnf, and Igf1 was significantly decreased (Figure 1I). By 12 months, however, there was a significant increase in Prdm16 in the KO vs control mice (Figure 1J). Western-blot analysis of BAT at 3 months of age revealed that the expression of IR and p85alpha PI3 kinase were unchanged vs controls (Figure 2A). However, the expression of insulin receptor substrate-1 (IRS-1) and insulin receptor substrate-2 (IRS-2) (Figure 2A) and hormone sensitive lipase (HSL) (Figure 2B) significantly increased at that age. In addition, IRS-1, IRS-2, p85alpha PI3 kinase, and HSL were significantly increased in KO vs control mice at age 12 months (Figure 2, A and B). There was an increase in the expression of heat shock protein 60 (HSP-60) at age 3 months and cytochrome c at age 12 months (mitochondrial content markers) and an increase of carnitine palmitoyl coenzyme A transferase CPT1-M (mitochondrial fatty oxidation marker) at age 3 and 12 months (Figure 2B). Finally, we studied BMP-7 signaling pathway machinery, an early signal involved in the recruitment of the brown adipocyte cell lineage. Thus, we found no change in the precursor form of BMP-7 expression in KO vs control mice at any time of development studied. The expression of mature form of BMP-7 and Smad 1/5/8 was markedly decreased in KO mice at age 3 months, and the expression of mature form of BMP-7 returned toward normal by 12 months. The expression of ACVR1 (BMP7 type I receptor) and Smad 1/5/8 (BMP-7 specific signaling transducer protein) was significantly increased in KO mice vs control at age 12 months (Figure 2C). Regarding beige cells, we have studied the expression of UCP-1, a thermogenic marker, in iWAT or eWAT vs iBAT in a comparative manner upon development. Immunocytochemistry studies revealed immunopositivity for UCP-1 in the iWAT tissue slides from 3- or 12-month-old control mice compared with iBAT positive control. However, the presence of UCP-1 immunopositive beige cells in iWAT fat depots was negligible in KO mice (Figure 3A). The presence of UCP-1 immunoreactive beige cells from eWAT at age 3 or 12 months was undetectable in control and KO mice. (Figure 3B). To assure the expression of UCP-1, we performed Western blots from the corresponding fat depots and genotype. At age 3 and 12 months, densitometric analysis revealed that UCP-1 expression found in iWAT was impaired in KO vs control mice. At age 12 months, the expression of UCP-1 found in iWAT was almost impaired under the same experimental conditions (Figure 3D). Interestingly, Prdm16, a brown adipocyte differentiation marker, increased very significantly in the inguinal fat at all ages studied (Figure 3C). No expression of UCP-1 was found in the eWAT at age 3 or 12 months from control or KO mice, respectively (Figure 3D).
Figure 1.

Generation and development of BATIGFIRKO mice. The targeting strategy in BAT is shown. Thus, PCR analysis of iBAT cDNA showed that the Lox/Lox E3 allele present in the Igf1r L/L mice was absent in the BATIGFIRKO mice (A). As a result, IGF1R protein expression was absent in BAT (A), but present in both Igf1r L/L (n = 4) and BATIGFIRKO (n = 4) eWAT, iWAT, liver, gastrocnemius, and brain (B). C, Plot expressing no differences in male body weight comparing Igf1r L/L (n = 38) and BATIGFIRKO (n = 40) animals from age 4 to 52 wk. D, Graph indicating the iBAT weight/body weight ratio in Igf1r L/L (n = 5) and BATIGFIRKO (n = 6) animals age 3 and 12 mo. E, Representative image of hematoxylin-eosin staining of iBAT at age 3 and 12 mo comparing Igf1r L/L (n = 3) and BATIGFIRKO (n = 3) mice. Magnification, ×20. F, Brown adipocyte size from iBAT compartment is shown comparing Igf1r L/L (n = 3) and BATIGFIRKO (n = 3) mice from 3 and 12 mo (n = 200 adipocytes per group) at magnification, ×20. Adipocyte number quantification from iBAT compartment comparing Igf1r L/L (n = 3) and BATIGFIRKO (n = 3) mice at 3 and 12 mo is shown, respectively. G, Ex vivo iBAT lipolysis experiment comparing Igf1r L/L and BATIGFIRKO mice (n = 4 at 3 mo and n = 4 at 12 mo, respectively). H, Graph indicating the fold increase of UCP-1 protein levels per mg of processed iBAT comparing Igf1r L/L (n = 6 at 3 mo and n = 5 at 12 mo) and BATIGFIRKO (n = 5 at 3 mo and n = 5 at 12 mo). I, Representative UCP-1 Western blotting from in iBAT comparing Igf1r L/L (n = 6 at 3 mo and n = 5 at 12 mo) and BATIGFIRKO (n = 5 at 3 mo and n = 5 at 12 mo) mice with its corresponding quantitation. Statistical significance assessed by two-tailed Student t test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 mo, respectively. J, Plot indicating fold-increased mRNA levels of different genes in BAT at age 3 and 12 mo comparing Igf1r L/L (n = 5 at 3 mo and n = 6 at 12 mo) and BATIGFIRKO (n = 5 at 3 mo and n = 5 at 12 mo). Statistical significance assessed by 1-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 mo; #, P < .05; ##, P < .01; ###, P < .001 between WT groups; and §, P < .05; §§, P < .01; §§§, P < .001 between KO groups.
Figure 2.

Brown fat differential protein expression in BATIGFIRKO mice. A, Representative western-blotting from iBAT showing different regulators of insulin signaling comparing Igf1r L/L (n = 6 at 3 mo and n = 5 at 12 mo) and BATIGFIRKO (n = 5 at 3 mo and n = 5 at 12 mo) mice with its corresponding quantitation. B, Representative Western blotting from different components involved in mitochondriogenesis and its corresponding quantitation. C, Representative Western blotting from different components involved in BMP7 signaling with its corresponding quantitation. Statistical significance assessed by two-tailed Student t test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 months, respectively.
Figure 3.
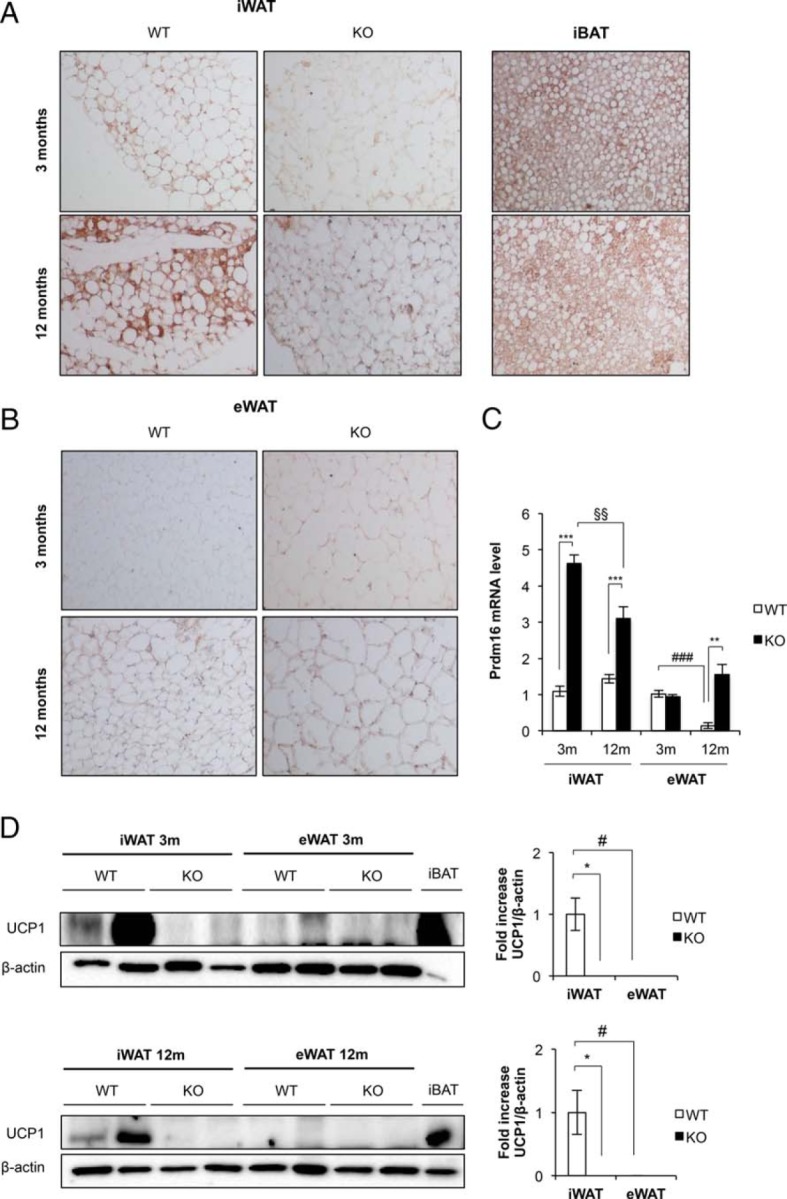
Beiging process in inguinal and epididymal white adipose tissue in BATIGFIRKO mice. A, Immunohistochemistry for Ucp-1 protein (brown stain) in sections of iWAT, iBAT was used as a positive control. B, Immunohistochemistry for Ucp-1 protein in sections of eWAT. All images were taken at magnification, ×20. C, mRNA levels of Prdm16 in iWAT and eWAT at 3 and 12 mo from Igf1r L/L (n = 6 at 3 mo and n = 4 at 12 mo) and BATIGFIRKO (n = 4 at 3 mo and n = 4 at 12 mo) mice respectively. Statistical significance assessed by one-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 months; #, P < .05; ##, P < .01; ###, P < .001 between WT groups; and §, P < .05; §§, P < .01; §§§, P < .001 between KO groups. D, Representative blots from UCP-1 protein expression in iWAT and eWAT (45 μg) at 3 and 12 mo and its corresponding quantitation (n = 3–4 per group). In both blots 8 μg of iBAT at 3 or 12 mo were used as a positive control of UCP-1 protein expression. Statistical significance assessed by Kruskall Wallis and Mann-Whitney U test after testing normality condition by one sample Kolmogorov-Smirnov test, *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 mo; #, P < .05; ##, P < .01; ###, P < .001 between WT groups.
Role of brown fat IGF-IR in cold acclimation and energy expenditure
Mice maintained for 3 days at thermoneutrality (28°C) were exposed to cold acclimation at 4°C. At age 3 months, KO mice failed to maintain rectal body temperature compared with their controls. Thus, KO mice decrease their body temperature to approximately 34.5°C upon 12 hours of cold exposure vs 36°C observed in control mice (Figure 4A, left panel). At age 12 months, we observed a similar thermogenic failure upon 12 hours of cold exposure in KO vs control mice (Figure 4A, right panel). In addition to the statistical analysis depicted in Figure 4A, we also confirmed by two-way ANOVA test the multivariate significance upon 6 or 12 hours of cold exposure. However, 4 hours cold-induced acute response of UCP-1 mRNA or protein expression remained unchanged in KO vs control mice at age 3 months, suggesting an intact sympathetic innervation and intracellular signaling mechanisms in the absence of IGFIR (Figure 4, B and C). Regarding energy expenditure, we submitted KO mice to indirect calorimetry studies in metabolic cages. Daily food intake was indistinguishable between KO mice and control mice as measured in metabolic cages (Figure 4D). BATIGFIRKO vs control mice, at age 3 months, showed that VO2 (Figure 4E) or heat production (Figure 4F) was significantly diminished in the dark period. However, respiratory exchange ratio remained unchanged vs controls (results not shown). At age 12 months, no significant changes were observed in VO2 or heat production in KO vs control mice (Figure 4E and F).
Figure 4.

Cold acclimation and energy expenditure in BATIGFIRKO and (WT) mice. A, Representative plots of cold-exposure analysis showing the temperature from 0 to 12 h comparing Igf1r L/L (n = 8 at 3 mo and n = 5 at 12 mo) and BATIGFIRKO (n = 6 at 3 mo and n = 11 at 12 mo) mice at 3 mo (left plot) and 12 mo (right plot). B, Ucp-1 mRNA levels detected by q-PCR after 4 h of cold-exposure comparing 3-month-old Igf1r L/L (n = 4) and BATIGFIRKO (n = 4) mice. C, Comparison of UCP-1 protein levels at 4 and 12 h of cold exposure and its corresponding quantitation comparing 3-month-old Igf1r L/L (n = 4) and BATIGFIRKO (n = 4) mice. Statistical significance assessed by two-tailed Student t test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 mo. D, Daily food intake under standard diet; E,VO2; and F, heat production were measured comparing Igf1r L/L (n = 4 at 3 mo and n = 4 at 12 mo) and BATIGFIRKO (n = 4 at 3 mo and n = 4 at 12 mo) mice. Mice were housed individually and all measurements were taken every 10 minutes during 72 h. Statistical significance assessed by one-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 mo; #, P < .05; ##, P < .01; ###, P < .001 between WT groups; and §, P < .05; §§, P < .01; §§§, P < .001 between KO groups, respectively.
Role of brown fat lacking IGFIR in the adipose organ development
We explored the body fat mass by nuclear magnetic resonance (NMR). Our data clearly suggest that the total fat volume progressively increased from 3 to 12 months of age. No differences were found in the KO vs control mice (Figure 5, A and B). Then we analyzed the fat mass of several tissues of the adipose organ. Thus, we found that epididymal, inguinal, and mesenteric fat mass contribute the most to fat mass enhancement above described in the control and KO mice at age 3 and 12 months, retroperitoneal fat mass playing a minor contribution (Figure 5C). Although fat depots histology revealed differential adipocyte size vs cell number at 12 months of age. Thus, epididymal fat cells showed larger adipocytes but with a lower number of adipocytes in KO vs control mice at age 12 months. No significant changes were observed in the inguinal fat cell size/cell number at age 12 months. Retroperitoneal fat histology revealed larger adipocytes but lower cell number in KO vs control mice at age 3 and 12 months, respectively. Finally, mesenteric fat histology showed no differences in the adipocyte size/cell number at age 12 months. (Figure 5, D and E). At this stage, we wonder whether changes in the adipocyte size/cell number in the overall adipose organ might affect the global expression of putative adipokines. Our data showed no significant changes in the circulating levels of leptin or adiponectin (as fat mass markers) upon development, in any group of mice studied. However, plasma levels of TNF-alpha (an adipose organ local inflammation marker) significantly decreased at 12- vs 3-month-old mice down to very low plasma levels in KO and control mice, respectively (Table 2).
Figure 5.

The adipose organ in BATIGFIRKO mice. A, Representative images from NMR comparing Igf1r L/L and BATIGFIRKO mice at 3 and 12 mo. B, Graph representing the percentage of fat volume per total volume at 3 and 12 mo in Igf1r L/L (n = 4 at 3 mo and n = 4 at 12 mo) and BATIGFIRKO (n = 4 at 3 mo and n = 4 at 12 mo) mice. C, Graph representing the eWAT, iWAT, rWAT and mWAT weights per total body weight comparing Igf1r L/L (n = 12) and BATIGFIRKO (n = 13) mice at 3 and 12 mo, respectively. D, Adipocyte size from eWAT, iWAT, rWAT, and mWAT compartments are shown comparing Igf1r L/L (n = 5) and BATIGFIRKO (n = 5) mice from 3 and 12 mo (n = 250 adipocytes per group) at magnification, ×20. The corresponding quantitation is presented below. E, Adipocyte number quantification from eWAT, iWAT, rWAT, and mWAT compartments comparing Igf1r L/L (n = 5) and BATIGFIRKO (n = 5) mice at 3 and 12 mo, respectively. Statistical significance assessed by one-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups at 3 and 12 mo; #, P < .05; ##, P < .01; ###, P < .001 between WT groups; and §, P < .05; §§, P < .01; §§§, P < .001 between KO groups, respectively.
Table 2.
Metabolic and Endocrine Status of Control and BATIGFIRKO Mice
| WT 3 mo | KO 3 mo | WT 12 mo | KO 12 mo | |
|---|---|---|---|---|
| T3, ng/mL | 0.49 ± 0.04 | 0.56 ± 0.03 | 0.53 ± 0.05 | 0.76 ± 0.03a,g |
| IGF-I, ng/mL | 141.88 ± 24.05 | 111.05 ± 18.69 | 113.51 ± 13.29 | 257.73 ± 36.83b,g |
| BMP7, pg/mL | 2.44 ± 0.29 | 1.99 ± 0.17 | 4.22 ± 0.55b | 2.45 ± 0.30a |
| Insulin, ng/mL | 0.75 ± 0.05 | 1.44 ± 0.27 | 1.17 ± 0.17 | 1.31 ± 0.12 |
| Glucose, mg/dL | 143.06 ± 3.17 | 150.48 ± 3.01 | 137.62 ± 4.76 | 143 ± 3.99 |
| TG, mg/dL | 72.96 ± 6.64 | 104.52 ± 12.58a | 65.21 ± 10.85 | 133.41 ± 26.56a |
| Leptin, ng/mL | 2.74 ± 0.51 | 2.58 ± 0.45 | 2.05 ± 0.45 | 3.00 ± 0.38 |
| Adiponectin, ng/mL | 23.93 ± 0.58 | 22.94 ± 0.54 | 21.36 ± 1.12 | 21.74 ± 0.76 |
| TNF-α, pg/mL | 2.63 ± 0.37 | 1.01 ± 0.1 | 0.83 ± 0.09b | 0.67 ± 0.06 |
Changes in fed plasma levels of endocrine signals such as IGFI, T3, BMP-7, insulin, leptin, adiponectin, and TNF-α and metabolites such as glucose or triglycerides are shown comparing L/L (n = 5–9) and BATIGFIRKO (n = 4–9) mice, at 3 and 12 mo, respectively. Statistical significance assessed by one-way ANOVA followed by the Bonferroni test.
P < .05;
P < .01;
P < .001 between KO and WT groups at 3 and 12 mo, respectively.
P < .05;
P < .01;
P < .001 between WT groups.
P < .05;
P < .01;
P < .001 between KO groups, respectively.
Metabolic and endocrine status of control and BATIGFIRKO mice
To assess the glycemic homeostasis and whole-body insulin sensitivity of KO vs control mice, we studied the glucose and insulin tolerance tests. Thus, glucose tolerance remained unchanged upon development in 3- and 12-month-old KO vs control mice (Figure 6A). However, insulin tolerance showed differential results. Thus, at age 3 months KO mice showed normal insulin sensitivity vs controls. At age 12 months, however, KO mice showed a manifest insulin resistance compared with their controls (Figure 6B). The significance of the statistical analysis depicted in Figure 6B upon several time points was confirmed by two-way ANOVA test. Consistently, we found no significant changes in the postprandial glycemia throughout development. However, at fed status insulin revealed a sustained moderate hyperinsulinemia in the KO vs control mice upon 12 months' development. (Table 2). In parallel, a progressive hypertriglyceridemia was observed in the KO vs control mice. These results are consistent with the progressive insulin resistance status observed in KO vs control mice described above. Regarding brown fat and adipose organ development and the adaptive/nonadaptive thermogenesis, three putative candidates such as T3, IGF-I, and BMP-7 were studied in fed status. T3 plasma levels significantly increased by 50% in KO vs control mice at age 12 months. In parallel, IGF-I plasma levels increased by 2.5-fold under the same experimental conditions. BMP-7 plasma levels slightly increased at age 12 vs 3 months in control or KO mice, KO mice showing a lower plasma level (Table 2).
Figure 6.

Glucose homeostasis and insulin sensitivity in BATIGFIRKO mice. A, Glucose-tolerance test and fasted glucose levels. B, insulin-tolerance test and fed glucose levels were measured in Igf1r L/L (n = 17 at 3 mo and n = 10 at 12 mo) or BATIGFIRKO (n = 15 at 3 mo and n = 8 at 12 mo) mice. Statistical significance was assessed by two-tailed Student t test *, P < .05; and **, P < .01; and ***, P < .001 between WT and KO groups at 3 and 12 mo. For insulin-tolerance test at 12 mo statistical significance was assessed by two-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; *** P < .001 between WT and KO groups at 3 and 12 mo.
Differential organ-specific insulin sensitivity and UCP expressions
At this stage, we wonder about the insulin sensitivity in the adipose organ and other major insulin target tissues. Our results in vivo suggest that insulin sensitivity. Our results in vivo suggest that p-Akt/Akt ratio remained unchanged in epididymal fat, but increased in inguinal fat, in KO vs control mice (Figure 7A). Concurrently, no changes were observed in the insulin sensitivity in iBAT in KO vs control mice (Figure 7A). However, insulin response was significantly impaired in the liver of KO mice (Figure 7B, upper panel). Liver histology revealed a moderate liver steatosis in the KO vs control mice at age 12 months (Figure 7B, bottom panel). In addition, at that time, insulin sensitivity in the skeletal muscle or in the heart remained unchanged (Figure 7C). However, either in the muscle or in the heart there was no lipid deposition in KO vs control mice at age 12 months (results not shown). At this point, we wonder whether the failure in the thermogenic capacity in canonical brown fat and beige cells, related to a significant loss of UCP-1 expression, might induce changes in the expression of the uncoupling proteins in peripheral tissues. In this regard, hepatic UCP-2 protein expression remained unchanged at very low levels in KO vs control mice at age 12 months (Figure 8A). In addition, we found a robust expression of UCP-3 in the skeletal muscle upon development at age 12 months, its expression being significantly much lower in the KO mice. In the heart, however, the expression of UCP-3 was much higher in the KO vs control mice at age 12 months (Figure 8A). To address the potentially overimposed sympathetic innervation effect on the nonoxidative and oxidative muscles, we assayed the protein expression of β-2 adrenergic receptors. Thus, the expression of the β-2 adrenergic receptors paralleled the expression of UCP-3 in the muscles. Thus, its expression declined in skeletal muscle, but robustly increased in the heart, in KO vs control mice at age 12 months (Figure 8B). At this point, it is well known that UCP-3 might regulate β-oxidation and lipid accumulation in skeletal muscle and in the vasculature. To address the important issue of the protective role played by this differential expression of UCP-3 against the lipid insult, in oxidative and nonoxidative muscles, we studied the insulin signaling in those tissues under HFD. For this purpose we used a nonobesogenic high-fat diet that contains a high proportion of polyunsaturated vs saturated fatty acids. Thus, control and KO mice fed a high-fat diet did not increase their body weight, or the visceral adiposity, or the plasma leptin levels, compared with their corresponding mice fed on chow diet. Insulin sensitivity in the epididymal or inguinal fat was only slightly significantly decreased in the KO vs control mice. However, we found severe insulin resistance in the liver or in the skeletal muscle in the KO mice. Noteworthy, insulin sensitivity remained unchanged in the heart in the KO vs control mice (Figure 8C).
Figure 7.

Insulin sensitivity in major insulin target tissues. A, After 1 U/kg body weight of human insulin injection, insulin signaling was measured in iWAT and eWAT and iBAT compartments at 12 mo of age comparing Igf1r L/L (iWAT and eWAT n = 5; iBAT n = 4) and BATIGFIRKO (iWAT and eWAT n = 4; iBAT n = 3) mice. The corresponding quantitation is presented below indicating the fold-increase of phospho-Akt/Akt. B, Representative blots from insulin signaling experiments in liver of 12 mo of age in Igf1r L/L (n = 5) and BATIGFIRKO (n = 4) and its corresponding quantitation indicating the fold-increase observed in the ratio phospho-Akt/Akt (upper panel). Representative hematoxylin-eosin and Oil-red O staining from liver of 12 months of age in WT (n = 5) and BATIGFIRKO (n = 4) mice and its corresponding quantitation is presented below. Magnification, ×20 (botton panel). Statistical significance assessed by two-tailed Student t test. *, P < .05; and **, P < .01; and ***, P < .001 between WT and IGF1R KO. C, Representative blots from insulin signaling experiments in gastrocnemius muscle and heart of 12 mo of age in Igf1r L/L (n = 5) and BATIGFIRKO (n = 4). It is represented in the corresponding plots the fold-increase observed in the ratio phospho-Akt/Akt. Statistical significance assessed by one-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; and ***, P < .001 between WT and KO in control vs insulin; #, P < .05; ##, P < .01; and ###, P < .001 comparing insulin from WT vs KO.
Figure 8.
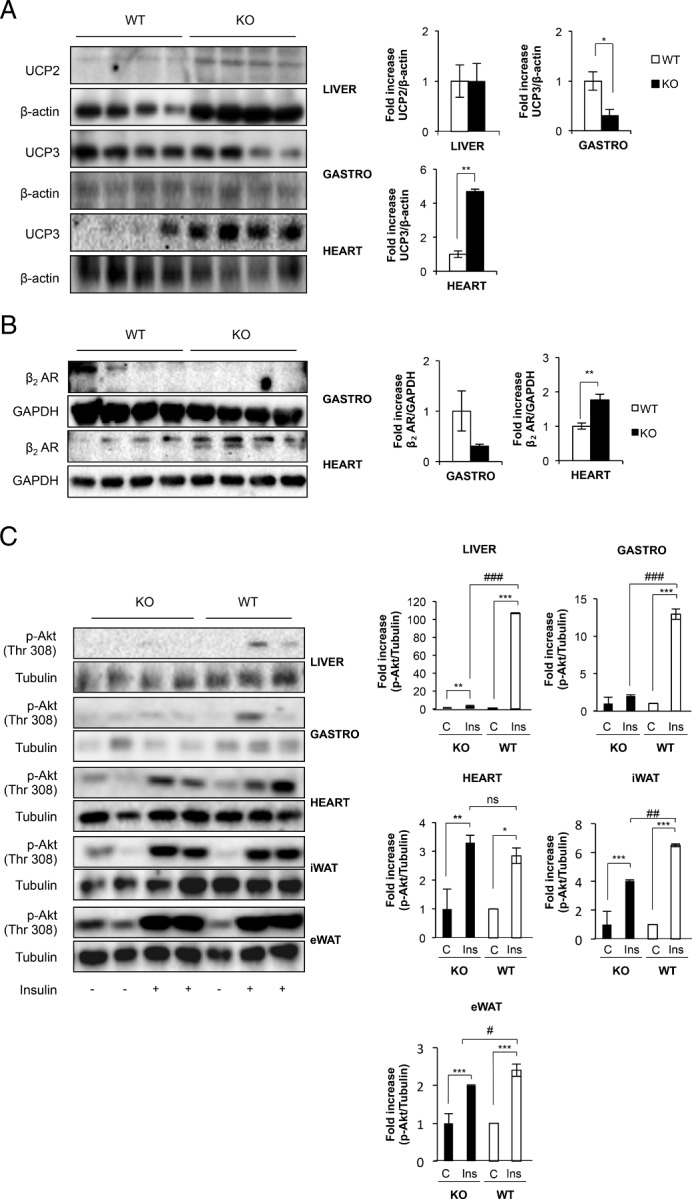
Protective effect of UCPs on insulin-target tissues against exogenous lipid insult. A, Representative blots from UCP-2 (in liver) and UCP-3 (in gastrocnemius muscle and heart) protein levels observed comparing Igf1r L/L (n = 4) and BATIGFIRKO (n = 4) mice from age 12 mo. The corresponding quantitation is presented below indicating the fold-increase of UCP-2 or UCP-3/β-actin. B, Representative blots from β2 adrenergic receptors in gastrocnemius muscle and heart of 12 months of age comparing Igf1r L/L (n = 4) and BATIGFIRKO (n = 4) mice. Statistical significance assessed by two-tailed Student t test. *, P < .05; **, P < .01; ***, P < .001 between WT and KO groups, respectively. C, Insulin signaling in liver, gastrocnemius muscle, heart, iWAT and eWAT in Igf1r L/L (n = 3) and BATIGFIRKO (n = 4) upon 16 wk of HFD. It is represented in the corresponding plots the fold-increase observed in the ratio phospho-Akt/α-tubulin. Statistical significance assessed by one-way ANOVA followed by the Bonferroni test. *, P < .05; **, P < .01; and ***, P < .001 between WT and KO in control vs insulin; #, P < .05; ##, P < .01; and ###, P < .001 comparing insulin from WT vs KO, respectively.
Discussion
Our data show that mice with a complete knockout of IGFIR in brown fat still have normal tissue development. In contrast, muscle-specific IGFIR KO mice showed a compensatory hyperplasia (22). In pancreatic β-cells lacking IGFIR, however, the beta cell mass was unchanged (14). Also, the lack of IGFIR in the brown fat does not result in macrosomic mice as previously reported in mice lacking IGFIR in the fat (23). In addition, it is solidly established that IGFIR/IR signaling mostly controls BAT development (13). In this regard, at age 3 months the insulin signaling machinery is robustly expressed in the KO mice vs control mice. However, BMP-7 signaling machinery as reflected by BMP-7 mature form, and more importantly, by Smad 1/5/8, was impaired in the KO mice. At age 12 months, BAT from KO mice showed a very significant increase of both insulin (IRS-2, IRS-1, and PI3 Kinase) and BMP-7 (ACVR1 and Smad 1/5/8, but not endogenous BMP-7) protein machinery compared with controls. Thus, the lack of IGFIR may be mostly compensated by putative candidates such as IGF1, insulin, BMP-7, or T3 among others. Our data showed a very significant increase in the circulating IGF-I or T3 and a trend to moderate hyperinsulinemia in KO vs control mice upon 1-year development. However, no significant changes were found in the circulating levels of BMP-7 under the same experimental conditions. This robust increase in circulating plasma IGF-I is consistent with the elevated plasma levels described in mice lacking IGFIR in the adipose organ (23), and might account through the enhanced insulin/IGFIR signal transduction machinery for the overall normal growth of brown fat lacking IGFIR. In fact, fetal brown adipocytes lacking IGFIR enhanced their insulin signaling (24). More importantly, elevated circulating levels of IGF-I in response to severe hepatic insulin resistance contribute to the pancreatic beta cell expansion, this effect being mediated by the A isoform of the IR (25). In addition, iLIRKO mice showed a manifest iBAT hypertrophy (unpublished observation). However, a putative compensatory mechanism to the loss of the full thermogenic capacity by brown fat and beige cells by BMP-7 can be ruled out. Thus, our results suggest that IGFIR it is not an essential growth regulator of BAT in the presence of IR and very high circulating levels of IGF-I.
Despite the normal BAT development, brown fat thermogenesis depends on lipid utilization as a fuel provided by endogenous lipolysis, long-chain fatty acyl CoA accessible to the mitochondria, and the mitochondrial machinery involved in fatty oxidation and respiratory chain and the uncoupling of ATP synthesis. In this regard, the expression of brown fat HSL protein increased in 3 months KO mice. In addition, basal lipolysis was also significantly increased. Concurrently, the expression of UCP-1 diminished, but the amount of UCP-1 per processed iBAT tissue remained unchanged, in the KO vs control mice. In addition, its expression in beige cells from iWAT was totally impaired. Thus, the lack of IGFIR in the KO mice at age 3 months may be partially compensated by an enhanced sympathetic innervation effect rather than by an enhanced IGF1/insulin effect. The outcome of these changes was an impaired thermogenesis in response to cold exposure and significantly lower energy expenditure (heat production) in KO vs control mice at age 3 months. At age 12 months, there was a robust increase in cytosolic HSL content, increase in the rate of basal lipolysis, diminution of large lipid droplets, increase in mitochondrial CPT-1 M protein expression, and a decrease in UCP-1 protein expression despite the increased gene expression of Prdm16. More importantly, the total amount on UCP-1 per processed iBAT tissue and the UCP-1 expression in beige cells from iWAT were diminished. The outcome of these changes was an unchanged basal energy expenditure. However, the thermogenic response to cold exposure was impaired in KO mice. Thus, at age 12 months, an increased compensatory IGF1/insulin effect through the increased IGF1/insulin signaling machinery together with an increased sympathetic innervation effect might occur in the absence of the IGFIR. Alternatively, the elevated level of T3 interacting synergistically with the sympathetic nervous system may directly increase basal metabolic rate (obligatory thermogenesis) and partially compensate the failure in the adaptive thermogenesis (26).
Regarding the adipose organ, the overall total body mass throughout development remained unchanged in KO vs control mice. By contrast, mice with a reduction in both IR and IGFIR produced using the aP2 Cre transgene showed a significant decrease in white and brown fat tissue mass, with no changes in the adipocyte size in the epididymal and inguinal compartments (13). However, there was a redistribution of the adipose organ as revealed by the changes in the ratio of adipocyte size/cell number occurred in several fat compartments in the KO vs control mice. More importantly, our data showed no significant changes in the circulating levels of leptin or adiponectin (as fat mass markers) or TNF-α (as a marker of local inflammation) upon development, in any group of mice studied. In the balance, the endocrine capacity of the adipose organ as related with the total fat mass was unchanged in the response to the loss of the thermogenic capacity of brown fat and beige cells lacking the IGFIR.
The redistribution of fat content within the adipose organ might affect lipid mobilization and content in peripheral tissues and subsequently their insulin sensitivity. In fact, KO vs control mice maintained a glucose tolerance, but a progressive insulin resistance, as revealed by the sustained hyperinsulinemia upon development. More importantly, lipid mobilization was increased as revealed by plasma triglycerides content in the KO vs control mice upon development. Thus, lipid content was enhanced in the liver and the hepatic insulin signaling was significantly impaired in KO mice, comparatively. In this regard, liver at age 12 months poorly expressed HSL (results not shown) and UCP-2 in both groups of mice. Noteworthy, overexpression of HSL by means of adenovirus infection in liver-specific PGC-1α transgenic mice ameliorated by 40–60% the severe fatty liver observed in ob/ob HFD-induced obese mice (27). Thus, liver it is particularly unprotected against lipid accumulation in mice at 12 months of age. In fact, it has been recently described that UCP-2 protects the vasculature against lipid accumulation, insulin resistance, and lesion in the mice (28). Thus, our data suggest an ectopic accumulation of lipids within the liver and in the circulation, which in fact might explain to some extent the hepatic insulin resistance found in the KO mice. However, the expression of UCP-3 as a controller of fatty acid oxidation in the heart and skeletal muscle were differentially regulated in parallel with the expression of the β-2 adrenergic receptors, suggesting a differential over imposed sympathetic effect. In contrast, down-regulation of the UCP-3 was associated with the absence of lipid droplets in the skeletal muscle in KO vs control mice (data not shown). These results are consistent with the putative role of UCP-3 negatively regulating fatty acid β-oxidation in the skeletal muscle. More importantly, UCP-3 lowering in the skeletal muscle has been associated with the insulin resistance showed in prediabetic states in the transition to type 2 diabetes (29). However, it is controversial whether the failure of brown fat- and beige-cell-dependent nonshivering thermogenesis could not be compensated by the putative nonshivering thermogenesis by the skeletal muscle (30). In the heart, however, there was a robust up-regulation of the UCP-3 expression in the KO mice. These results are consistent with elevated concentration of circulating lipids associated with increased concentration of UCP-3 in the heart, protecting against mitochondrial damage, in prediabetic and diabetic states (31).
Upon nonobesogenic high-fat diet–fed mice, we found a moderate loss of insulin sensitivity in the epididymal or inguinal fats, but a severe insulin resistance in the liver, in the KO vs control mice. Also, we found a severe insulin resistance in the skeletal muscle, but unchanged insulin sensitivity in the heart, under the same experimental conditions. These later results are consistent with the expression of UCP-3 in the KO mice, decreased in the skeletal muscle and increased in the heart, comparatively. In fact, UCP-3 increased fatty oxidation and protect against lipid accumulation in muscle cells (32). UCP-3, in contrast, may protect cardiac cells inducing autophagy to prevent apoptosis (33), protect against stress-induced myocardial injury (34), and protect heart against lipid input by increasing fatty oxidation to prevent insulin resistance. Our results, then, suggest a differential protection mechanism by UCP-3 to confront lipid accumulation, lowered in the skeletal muscle and enhanced in the heart, as a compensatory response to the significant loss of full thermogenic capacity in brown fat and beige cells within the iWAT in KO vs control mice. Overall, our data unravel a novel role of brown fat lacking IGFIR in the differential protection mechanisms by UCP-3 against lipids input in nonoxidative and oxidative muscles.
In conclusion, as summarized in Figure 9, our data suggest that IGFIR it is not an essential growth factor in the brown fat development in the presence of the IR and very high plasma levels of IGF-I. However, the lack of IGFIR decreased the UCP-1 expression in iBAT and much affect the development of beige cells bearing UCP-1 protein expression mostly in the inguinal white fat depots. Overall, IGFIR is indispensable to maintain the full thermogenic capacity, the lack of IGFIR resulting in an impaired cold acclimation. In addition, brown fat driven by IGFIR plays an unrecognized role in the redistribution of the adipose organ and lipid mobilization that affects the insulin sensitivity in peripheral tissues as revealed by sustained moderate hyperinsulinemia and hypertriglyceridemia, hepatic insulin sensitivity being much impaired associated with lipid accumulation, with the outcome of a global insulin resistance. The failure of the thermogenic capacity owing to the lack of IGFIR in the BAT and in the beige cells cannot be compensated by putative T3-enhanced mediated basal metabolic thermogenesis or by the putative T3-enhanced effect on UCP-3 expression and mitochondrial oxidation in skeletal muscle (35). However, the large enhancement of UCP-3 in heart emerges as potential cardio protector. This singularity protects heart against insulin resistance showed in the major insulin target tissues upon exogenous lipid insult.
Figure 9.

A scheme of the proposed mechanism involved in the major changes of the phenotype in BATIGFIRKO mice. The lack of IGFIR decreased the UCP-1 expression in iBAT and much affect the development of beige cells bearing UCP-1 protein expression mostly in the iWAT depots. As a result, BATIGFIRKO mice showed an impaired nonshivering thermogenesis in response to cold adaptation. In addition, brown fat driven by IGFIR plays an unrecognized role in the redistribution of the adipose organ and lipid mobilization that affect the insulin sensitivity in peripheral tissues as revealed by sustained moderate hyperinsulinemia, hepatic insulin sensitivity being much impaired associated with lipid accumulation, with the outcome of a global insulin resistance. The failure of the thermogenic capacity owing to the lack of IGFIR in the BAT and in the beige cells cannot be compensated by putative T3-enhanced mediated basal metabolic thermogenesis or by skeletal muscle nonshivering thermogenesis, as revealed by the robust loss of UCP-3. However, cardiac muscle emerges as thermogenic tissue owing to the large enhancement of UCP-3, which it confers a cardio protection.
Acknowledgments
This work was supported by Grants SAF2008/00031 and SAF2011/22555 from Ministerio de Ciencia e Innovación, and CIBER de Diabetes y Enfermedades Metabólicas Asociadas, Spanish Diabetes and Metabolic Research Network, Instituto de Salud Carlos III, Spain.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- BAT
brown adipose tissue
- BMP
bone morphogenetic protein
- eWAT
epididymal white adipose tissue
- HSL
hormone-sensitive lipase
- iBAT
interscapular brown fat
- BATIGFIRKO
BAT-specific knockout
- BATIRKO
brown adipose tissue-specific knockout targeting the insulin receptor Igf1r
- HSP-60
heat shock protein 60
- IR
insulin receptor
- IRS
insulin receptor substrate
- iWAT
inguinal white adipose tissue
- KO
knockout
- mWAT
mesenteric white adipose tissue
- NMR
nuclear magnetic resonance
- rWAT
retroperitoneal white adipose tissue
- UCP
uncoupling protein
- VCO2
carbon dioxide production
- VO2
oxygen consumption
- WAT
white adipose tissue
- WT
wild type.
References
- 1. Cannon B, Nedergaard J. Brown adipose tissue: Function and physiological significance. Physiol Rev. 2004;84:277–359. [DOI] [PubMed] [Google Scholar]
- 2. Lowell BB, S-Susulic V, Hamann A, et al. . Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. [DOI] [PubMed] [Google Scholar]
- 3. Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209. [DOI] [PubMed] [Google Scholar]
- 4. Cypess AM, Lehman S, Williams G, et al. . Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saito M, Okamatsu-Ogura Y, Matsushita M, et al. . High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. . Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. [DOI] [PubMed] [Google Scholar]
- 7. Virtanen KA, Lidell ME, Orava J, et al. . Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. [DOI] [PubMed] [Google Scholar]
- 8. Zingaretti MC, Crosta F, Vitali A, et al. . The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009;23:3113–3120. [DOI] [PubMed] [Google Scholar]
- 9. Cohen P, Levy JD, Zhang Y, et al. . Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156:304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tseng YH, Kokkotou E, Schulz TJ, et al. . New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guerra C, Navarro P, Valverde AM, et al. . Brown adipose tissue-specific insulin receptor knockout shows diabetic phenotype without insulin resistance. J. Clin Invest. 2001;108:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gómez-Hernández A, Otero YF, de las Heras N, et al. . Brown fat lipoatrophy and increased visceral adiposity through a concerted adipocytokines overexpression induces vascular insulin resistance and dysfunction. Endocrinology. 2012;153:1242–1255. [DOI] [PubMed] [Google Scholar]
- 13. Boucher J, Mori MA, Lee KY, et al. . Impaired thermogenesis and adipose tissue development in mice with fat-specific disruption of insulin and IGF-1 signalling. Nat Commun. 2012;3:902–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kulkarni RN, Holzenberger M, Shih DQ, et al. . β-cell–specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter β-cell mass. Nat Genet. 2002;31:111–115. [DOI] [PubMed] [Google Scholar]
- 15. Brüning JC, Michael MD, Winnay JN, et al. . A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Molec Cell. 1998;2:559–569. [DOI] [PubMed] [Google Scholar]
- 16. Valverde AM, Lorenzo M, Pons S, White MF, Benito M. Insulin receptor substrate (IRS) proteins IRS-1 and IRS-2 differential signaling in the insulin/insulin-like growth factor-I pathways in fetal brown adipocytes. Molec Endocrinol. 1998;12:688–697. [DOI] [PubMed] [Google Scholar]
- 17. Turpin SM, Nicholls HT, Willmes DM, et al. . Obesity-induced CerS6-dependent C16:0 Ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20:678–686. [DOI] [PubMed] [Google Scholar]
- 18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 19. Garcia-Guerra L, Nieto-Vazquez I, Vila-Bedmar R, et al. . G protein-coupled receptor kinase 2 plays a relevant role in insulin resistance and obesity. Diabetes. 2010;59:2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc. 2013;8:1149–1154. [DOI] [PubMed] [Google Scholar]
- 21. Guillen C, Bartolome A, Vila-Bedmar R, García-Aguilar A, Gomez-Hernandez A, Benito M. Concerted expression of the thermogenic and bioenergetic mitochondrial protein machinery in brown adipose tissue. J Cell Biochem. 2013;114:2306–2313. [DOI] [PubMed] [Google Scholar]
- 22. Fernández AM, Dupont J, Farrar RP, Lee S, Stannard B, Le Roith D J. Muscle-specific inactivation of the IGF-I receptor induces compensatory hyperplasia in skeletal muscle. J. Clin Invest. 2002;109:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klöting N, Koch L, Wunderlich T, et al. . Autocrine IGF-1 action in adipocytes controls systemic IGF-1 concentrations and growth. Diabetes. 2008;57:2074–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mur C, Valverde AM, Kahn CR, Benito M. Increased insulin sensitivity in IGF-I receptor–deficient brown adipocytes. Diabetes. 2002;51:743–754. [DOI] [PubMed] [Google Scholar]
- 25. Escribano O, Guillén C, Nevado C, Gómez-Hernández A, Kahn CR, Benito M. β-Cell hyperplasia induced by hepatic insulin resistance: Role of a liver-pancreas endocrine axis through insulin receptor A isoform. Diabetes. 2009;58:820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silva JE. Physiological importance and control of non-shivering facultative thermogenesis. Front Biosci (Schol Ed). 2011;25:352–371. [DOI] [PubMed] [Google Scholar]
- 27. Reid BN, Ables GP, Otlivanchik OA, et al. . Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem. 2008;283:13087–13099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gómez-Hernández A, Perdomo L, de las Heras N, et al. . Antagonistic effect of TNF-alpha and insulin on uncoupling protein 2 (UCP-2) expression and vascular damage. Cardiovasc Diabetol. 2014;13:108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schrauwen P, Mensink M, Schaart G, et al. . Reduced skeletal muscle uncoupling protein-3 content in prediabetic subjects and type 2 diabetic patients: Restoration by rosiglitazone treatment. J Clin Endocrinol Metab. 2006;91:1520–1525. [DOI] [PubMed] [Google Scholar]
- 30. Bal NC, Maurya SK, Sopariwala DH, et al. . Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat Med. 2012;18:1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cole MA, Murray AJ, Cochlin LE, et al. . A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol. 2011;106:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. MacLellan JD, Gerrits MF, Gowing A, Smith PJ, Wheeler MB, Harper ME. Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes. 2005;54:2343–2350. [DOI] [PubMed] [Google Scholar]
- 33. Riehle C, Abel ED. Insulin Regulation of Myocardial Autophagy. Circ J. 2014;78:2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X, Gong J, Liu X, et al. . Expression of uncoupling protein 3 in mitochondria protects against stress-induced myocardial injury: A proteomic study. Cell Stress Chaperones. 2010;15:771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silvestri E, Moreno M, Lombardib A, et al. . Thyroid-hormone effects on putative biochemical pathways involved in UCP3 activation in rat skeletal muscle mitochondria. FEBS Letters. 2005;579:1639–1645. [DOI] [PubMed] [Google Scholar]