

Abstract
Juvenile myelomonocytic leukemia (JMML) is an uncommon myeloproliferative neoplasm driven by Ras pathway mutations and hyperactive Ras/MAPK signaling. Outcomes for many children with JMML remain dismal with current standard-of-care cytoreductive chemotherapy and hematopoietic stem cell transplantation. We used patient-derived induced pluripotent stem cells (iPSCs) to characterize the signaling profiles and potential therapeutic vulnerabilities of PTPN11-mutant and CBL-mutant JMML. We assessed whether MEK, JAK, and PI3K/mTOR kinase inhibitors (i) could inhibit myeloproliferation and aberrant signaling in iPSC-derived hematopoietic progenitors with PTPN11 E76K or CBL Y371H mutations. We detected constitutive Ras/MAPK and PI3K/mTOR signaling in PTPN11 and CBL iPSC-derived myeloid cells. Activated signaling and growth of PTPN11 iPSCs were preferentially inhibited in vitro by the MEKi PD0325901 and trametinib. Conversely, JAK/STAT signaling was selectively activated in CBL iPSCs and abrogated by the JAKi momelotinib and ruxolitinib. The PI3Kδi idelalisib and mTORi rapamycin inhibited signaling and myeloproliferation in both PTPN11 and CBL iPSCs. These findings demonstrate differential sensitivity of PTPN11 iPSCs to MEKi and of CBL iPSCs to JAKi, but similar sensitivity to PI3Ki and mTORi. Clinical investigation of mutation-specific kinase inhibitor therapies in children with JMML may be warranted.
INTRODUCTION
Juvenile myelomonocytic leukemia (JMML) is an uncommon, frequently fatal myeloproliferative neoplasm (MPN) of early childhood characterized by splenomegaly, thrombocytopenia, peripheral monocytosis, elevated hemoglobin F, and hypersensitivity to granulocyte macrophage colony-stimulating factor (GM-CSF).1–4 Chemotherapy decreases splenomegaly and reduces transfusion-dependent cytopenias, but is not curative. Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only effective therapy for achieving long-term disease control.1 However, five-year event-free and overall survival in children with JMML remain suboptimal at 52% and 64%, respectively, largely due to post-HSCT relapse and end-organ infiltration with JMML cells leading to respiratory failure and death.5–7 Clinical predictors of inferior outcomes include age >2 years at diagnosis, severe thrombocytopenia (<33,000 platelets/dL), and hemoglobin F≥10%.8 Interestingly, spontaneous disease resolution has been reported in rare patients and may be more common in specific genetic subtypes.1, 9, 10 JMML remains a biologically heterogeneous and unsolved clinical problem for which new therapeutic approaches are needed.
More than 90% of JMML is now known to be driven by germline or somatic loss-of-function or gain-of-function mutations in Ras pathway and associated genes, including NF1 (neurofibromatosis type 1), KRAS (Kirsten rat sarcoma virus), NRAS (neuroblastoma RAS viral oncogene homolog), PTPN11 (protein-tyrosine phosphatase, non-receptor-type, 11; encoding SHP2), and CBL (Casitas b-lineage lymphoma).1, 2 All of these mutations induce constitutive activation of intracellular kinase signaling, including Ras/MAPK (mitogen-activated protein kinase), STAT5 (signal transducer and activator of transcription 5), and/or PI3K/Akt/mTOR (phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin) networks. Accordingly, small molecule kinase inhibitors targeting aberrant signaling proteins may have therapeutic potential in JMML and other Ras-driven diseases (‘Rasopathies’). Single cell phosphoflow cytometric analysis of primary JMML cells with PTPN11, NRAS, or KRAS mutations demonstrated marked STAT5 phosphorylation upon low dose GM-CSF stimulation,11 and this hyperactive signaling could be abrogated by JAK inhibition. Other preclinical studies have also demonstrated in vivo activity of MEK or PI3K inhibitors in genetically-modified murine models of Ras-mutant MPNs.12–16 While murine models have provided valuable insights about JMML biology, these preclinical models may not fully recapitulate human disease or predict clinical response to therapies. The therapeutic potential of kinase inhibitors in patients with JMML is unknown, although the Children’s Oncology Group trial ADVL1521 is currently testing the safety and efficacy of the MEK inhibitor trametinib in children with relapsed JMML (clinicaltrials.gov NCT03190915).
Hyperactive Ras/MAPK signaling is a well-known biochemical feature of JMML,12, 14 and aberrant JAK/STAT and/or PI3K/Akt/mTOR signaling have also been implicated in its pathogenesis.11, 15–17 While the protein tyrosine phosphatase SHP-2 (encoded by PTPN11) and the guanosine trisphophate/guanosine diphosphate-regulated Ras family proteins encoded by KRAS, NRAS, and NF1 are signaling molecules involved in cellular growth, differentiation, and survival, the precise role of the E3 ubiquitin ligase CBL in JMML pathogenesis and intracellular signaling networks is poorly understood. JMML remains a difficult disease to study given its rare incidence and paucity of primary specimens obtainable from very young children. Our group and others have generated patient-derived induced pluripotent stem cell (iPSC) lines using hematopoietic cells from children with PTPN11-mutant JMML to provide a renewable cell source for mechanistic and therapeutic investigation.18, 19 In earlier studies, we and other demonstrated that PTPN11-mutant iPSC-derived myeloid cells show characteristic GM-CSF hypersensitivity and constitutive ERK phosphorylation that is abrogated with in vitro MEK inhibition.18, 19
We extend these observations in the current studies to investigation of CBL-mutant JMML iPSCs. We examined whether CBL-mutant versus PTPN11-mutant iPSC-derived myeloid progenitors have distinct signaling profiles that may be differentially targetable with specific kinase inhibitors. We observed hyperactive JAK/STAT signaling in CBL-mutant myeloid cells, while aberrant Ras/MAPK activation was most pronounced in PTPN11-mutant cells. Accordingly, CBL-mutant cells showed preferential sensitivity to the JAK inhibitors momelotinib and ruxolitinib, while PTPN11-mutant myeloproliferation was suppressed by the MEK inhibitors PD0325901 and trametinib. Constitutive phosphorylation of PI3K/Akt/mTOR signaling was observed in both PTPN11-mutant and CBL-mutant JMML lines and abrogated with the PI3Kδ inhibitor idelalisib and the mTOR inhibitor rapamycin. We thus report for the first time biochemical characterization of a CBL-mutant JMML iPSC line and demonstrate that patient-derived iPSC models can be used to study mutation-specific signaling profiles of genetic subtypes of JMML and to test the therapeutic potential of Ras/MAPK, JAK/STAT, and PI3K/Akt/mTOR pathway kinase inhibitors.
MATERIALS AND METHODS
Generation of control and JMML iPSCs
Peripheral blood or bone marrow specimens were obtained from study participants (Table 1) under institutional review board-approved research protocols at the Children’s Hospital of Philadelphia and the Benioff Children’s Hospital at the University of California, San Francisco in accordance with the Declaration of Helsinki. Patients with JMML met World Health Organization 2016 diagnostic criteria20 and had received standard-of-care treatment with chemotherapy and allogeneic stem cell transplantation. One patient with JMML had a heterozygous PTPN11 E76K mutation (somatic), and one had a homozygous CBL Y371H mutation (germline). A normal male donor bone marrow specimen was used to generate control iPSCs and was confirmed to have wild-type PTPN11 and CBL by Sanger sequencing.21, 22 No additional mutations were identified in diagnostic bone marrow specimens from the JMML patients by whole exome sequencing analysis.
Table 1.
Genetic characteristics of primary patient specimens used for iPSC generation.
| iPSC line abbreviation | USI | Patient age at diagnosis | Cell source | Karyotype | JMML-associated mutation |
|---|---|---|---|---|---|
| Control | CHOP-WT618 | adult | BM | 46,XY | none |
| PTPN11 | CHOP-JMML219218 CHOP-JMML2193 |
3.67 years | PB BM |
46,XY | PTPN11 E76K/wt |
| CBL | CHOP-JMML185422 | 7 months | BM | 46,XX | CBL Y371H/Y371H |
BM = bone marrow, PB = peripheral blood, USI = unique specimen identifier, wt = wild-type
Ficoll-purified mononuclear cells from these patients were reprogrammed via transduction with a STEMCCA lentivirus expressing human OCT4, KLF4, MYC, and SOX2 to create JMML and control iPSC lines for downstream experimental studies as previously described.18 All iPSCs studied fulfilled standard pluripotency criteria, including expression of endogenous pluripotency markers, silencing of lentivirally encoded reprogramming genes, and formation of all three germ-cell layers.23, 24 Lines were passaged at least 20 times to minimize lineage bias caused by potential memory effects.18, 25 Two independently-derived clones of each control and JMML iPSC line were used for the downstream experimental studies described below.
Hematopoietic Differentiation of iPSCs
Control and JMML iPSCs were differentiated with cytokines to support multipotent hematopoietic progenitor formation via embryoid body (EB) formation as previously described.18, 25 In brief, mouse feeder cell-depleted iPSCs were cultured in serum-free StemPro-34 medium (Invitrogen) containing glutamine 2 mM, ascorbic acid 50 μg/mL, transferrin 150 μg/mL, and monothioglycerol 0.4 mM (Sigma). Sequential addition of cytokines included bone morphogenic protein 4 (BMP4) 25 ng/mL, vascular endothelial growth factor (VEGF) 50 ng/mL (days 0-4); stem cell factor (SCF) 50 ng/mL, thrombopoietin (TPO) 50 ng/mL, FLT3-ligand (FLT3-L) 50 ng/mL, basic fibroblast growth factor (bFGF) 20 ng/mL (days 2-4); VEGF 50 ng/mL, SCF 50 ng/mL, TPO 50 ng/mL, FLT3-L 50 ng/mL, bFGF 20 ng/mL (days 4-8); SCF 50 ng/mL, interleukin-3 (IL-3) 10 ng/mL, and GM-CSF 10 ng/mL (day 8+). All cytokines were from R&D Systems except bFGF (Invitrogen). Cell cultures were maintained in at 37ºC with 5% CO2, 5% O2, and 90% N2.
GM-CSF hypersensitivity assays were performed with day 8 hematopoietic progenitors cultured in methylcellulose with increasing concentrations of human GM-CSF (R&D Systems). Colonies were enumerated after 14 days as described.18, 26 Representative colonies were viewed with a Leica DMI 4000B microscope, and digital images were captured with Application Suite software (Leica Microsystems). May-Grünwald-Giemsa-stained cytospins were prepared as described.18
To assess the ability of kinase inhibitors to impair colony formation, day 8 hematopoietic progenitor control, CBL-mutant, and PTPN11-mutant cells were cultured in methylcellulose with saturating doses of GM-CSF (10 ng/mL) and increasing concentrations of the following drugs: MEK inhibitor (i) PD0325901 (PD901), MEKi trametinib, JAKi momelotinib, JAKi ruxolitinib, PI3Kδi idelalisib, and mTORi rapamycin. Kinase inhibitors were purchased from LC Labs (PD901, trametinib, ruxolitinib), Selleck Chemicals (momelotinib, rapamycin), or Active Biochem (idelalisib). The half-maximal effective concentration (EC50) for each kinase inhibitor was determined, then used in subsequent methylcellulose colony assays with dose titration of GM-CSF. Colonies were counted after 14 days. All assays were performed in triplicate using two clones of each iPSC line. Data were analyzed and displayed graphically with Prism (GraphPad).
Signaling analyses
Phosphoflow cytometry analysis of control and JMML iPSCs was performed as described18, 27 with minor modifications. Day 14-16 iPSC-derived myeloid cells (0.25-0.5 × 106 cells/condition) were incubated in vitro with human GM-CSF (R&D Systems) 10 ng/mL, PD901 100 nM, trametinib 100 nM, momelotinib 1 uM, ruxolitinib 1 uM, idelalisib 1 uM, or rapamycin 10 nM for 60 minutes at 37 °C. Dimethyl sulfoxide (DMSO; ThermoFisher) 0.01% was used as a negative control. Pervanadate (reagents from ThermoFisher) 125 uM was used as a positive signaling control as described.27 Cells were fixed in 1.5% paraformaldehyde (Electron Microscopy Services), permeabilized in 90% ice-cold methanol (VWR), and stained with surface and intracellular antibodies prior to flow cytometry analysis as described and delineated in Supplemental Methods.27, 28 Digital data were acquired on LSRII or FACSVerse flow cytometers (BD Biosciences) and analyzed in Cytobank.29 Phosphoflow cytometry experiments were performed in triplicate for each iPSC line. Graphic data display and statistical analyses were performed with Prism with normalization of data to mean phosphoprotein levels in control iPSCs.
RESULTS
JMML iPSCs recapitulate human biology
We reprogrammed bone marrow mononuclear cells obtained from a child with JMML harboring a homozygous CBL Y371H mutation associated with chromosome 11q isodisomy.22 Control and PTPN11 E76K-mutant JMML iPSCs were derived as described from bone marrow and peripheral blood specimens from an unrelated healthy donor and a child with JMML,18 respectively (Table 1). DNA sequencing of iPSC lines confirmed presence of specific PTPN11 and CBL mutations originally detected in the corresponding patient specimens (not shown). Hematopoietic differentiation and GM-CSF hypersensitivity assays were performed in 2 independent clones of PTPN11 and CBL-mutant iPSCs.
Control (CHOPWT6), PTPN11 E76K-mutant JMML (PTPN11), and CBL Y371H-mutant JMML (CBL) iPSCs were differentiated via EB formation to generate multipotent hematopoietic progenitors.18, 25 By day 8 of EB formation, a defined CD43+41+235+ population was present in similar frequencies among all three iPSC lines (not shown). Day 8 CD43+41+235+ progenitor cells plated in vitro in methylcellulose demonstrated 1.6-1.9 fold increased myeloid colony formation of PTPN11 and CBL cells compared to growth of control CHOPWT6 cells (Figure 1A). The PTPN11- and CBL-mutant iPSC-derived progenitors demonstrated classic growth hypersensitivity with low doses of human GM-CSF (Figure 1B-C and Supplemental Figure 2), consistent with prior observations of constitutive activation of GM-CSF signaling in primary JMML cells.4, 11 Qualitatively, PTPN11 and CBL iPSC myeloid colonies were larger and more dispersed than CHOPWT6 colonies (not shown), demonstrating their increased proliferative capacity. No significant differences in colony formation or GM-CSF hypersensitivity were observed between PTPN11 and CBL iPSCs (Figure 1B–1C), suggesting overlap of JMML biologic phenotypes despite distinct mutations. By microscopic cell morphology analyses, macrophages were the predominant cell type comprising the myeloid colonies in both JMML and control cell lines (Figure 1D).
Figure 1. GM-CSF hypersensitivity of patient-derived PTPN11- and CBL-mutant JMML iPSCs.
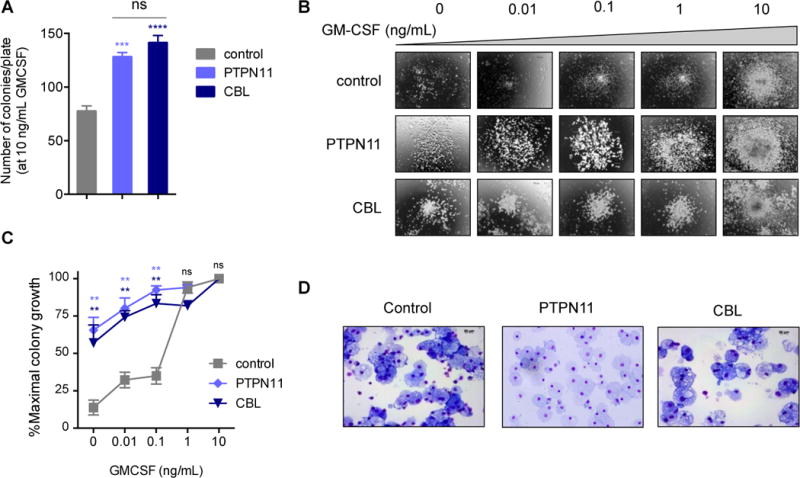
(A) Total number of myeloid colonies per 1500 day 8 progenitor cells derived from control, PTPN11-, and CBL-mutant iPSCs were cultured with 10 ng/mL GM-CSF after 14 days. Assays were performed in triplicate. (B) Representative photomicrographs of day 8 control, PTPN11, and CBL iPSC-derived myeloid colonies cultured with increasing doses of GM-CSF. (C) Percent of maximal colony number is displayed for each iPSC-derived progenitor cell line incubated with increasing GM-CSF concentrations. (D) Representative cytospin photomicrographs of cells from control, PTPN11, and CBL myeloid colonies with May-Grünwald-Giemsa staining (shown at 40×). PTPN11 and CBL colony data were compared to control data by one-way ANOVA with the Dunnett post-test for multiple comparisons. **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant.
Constitutive signaling activation in PTPN11-mutant and CBL-mutant JMML iPSCs
To investigate signaling activation and kinase inhibitor sensitivities in CBL-mutant and PTPN11-mutant JMML, we first characterized basal phosphorylation of ERK, JAK2, STAT5, S6, and Akt proteins within CD45+18+14+ myeloid cells from control and JMML iPSCs by in vitro phosphoflow cytometry analysis (Supplemental Figure 1). We observed constitutive activation of Ras/MAPK signaling in PTPN11 myeloid cells with high basal levels of phosphorylated ERK (pERK) compared to control cells (Figure 2). While increased pERK was also observed in CBL myeloid cells versus control cells, levels were appreciably higher in PTPN11 cells (Figure 2). In contrast, CBL myeloid cells had markedly elevated levels of pJAK2 and pSTAT5, while more modest activation of JAK/STAT signaling was detected in PTPN11 myeloid cells. Similar hyperactivation of PI3K/Akt/mTOR signaling with high pS6 and pAkt levels was observed in both PTPN11 and CBL myeloid cells. Taken together, these results demonstrate activation of multiple canonical oncogenic signaling networks in JMML, but also suggest important signaling differences between the PTPN11 and CBL genetic subtypes.
Figure 2. Constitutive signaling activation in PTPN11-mutant and CBL-mutant JMML iPSC-derived myeloid cells.
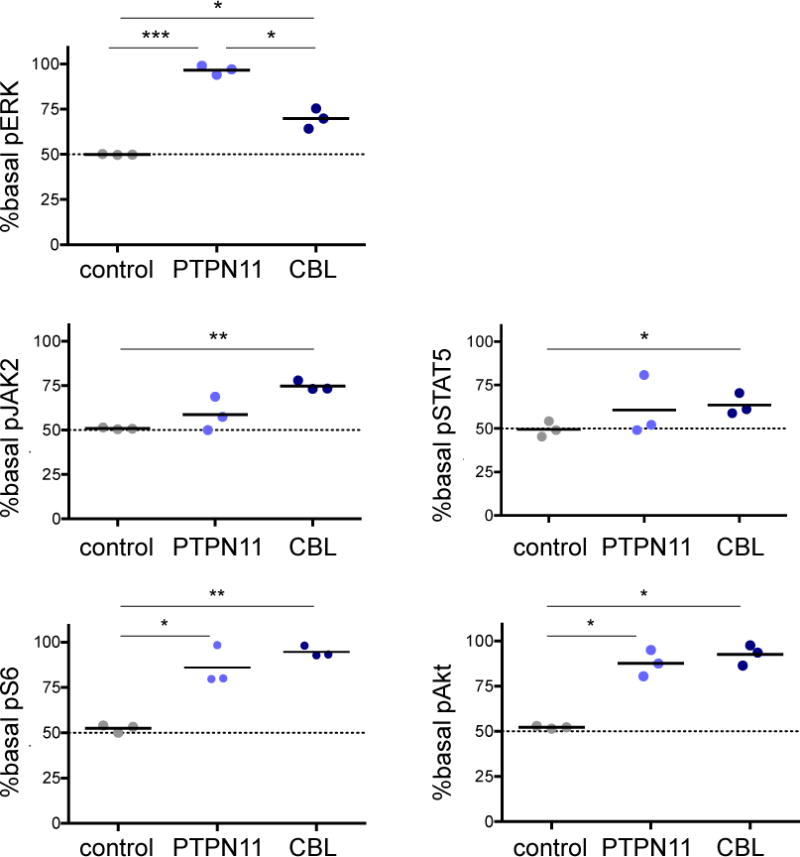
Phosphoflow cytometric analysis of day 14 CD45+14+18+ myeloid cells from control, PTPN11 and CBL iPSCs was performed. Basal levels of phosphorylated (p) ERK, JAK2, STAT5, S6, and AKT in CD45+14+18+ control or JMML iPSCs were measured and normalized to the median level of each phosphoprotein in control cells for comparison of signaling activation. Data points denote three independent experiments with means (thick black lines). Gating strategy of surface markers and median control phosphoprotein levels is depicted in Supplemental Figure 1. Data were analysed by one-way ANOVA with the Tukey post-test for multiple comparisons. *p<0.05, **p<0.01, ***p<0.001.
Kinase inhibition abrogates signaling hyperactivation
Based upon our observations of constitutive Ras/MAPK, JAK/STAT, and PI3K/Akt/mTOR signaling in JMML iPSC-derived myeloid cells, we then sought to determine mutation-specific sensitivities of JMML iPSCs to clinically-relevant kinase inhibitors (Figure 3A). We first assessed GM-CSF-inducible phosphorylation of ERK, JAK2, STAT5, S6, and Akt (S473) in control and JMML cells and defined maximal signaling levels for each iPSC line and phosphoprotein with pervanadate as a positive control (not shown). GM-CSF stimulation resulted in increased pERK, pJAK2, pSTAT5, pS6, and pAkt proteins above basal levels in control myeloid cells (Figure 3B, 3C, 3D). However, increased phosphoprotein levels were not observed with GM-CSF stimulation in PTPN11 and CBL myeloid cells, confirming near-maximal basal activation of Ras/MAPK, JAK/STAT, and PI3K/Akt/mTOR signaling and suggesting therapeutic potential of targeted kinase inhibitors.
Figure 3. Inhibition of signaling phosphoproteins in JMML iPSCs.
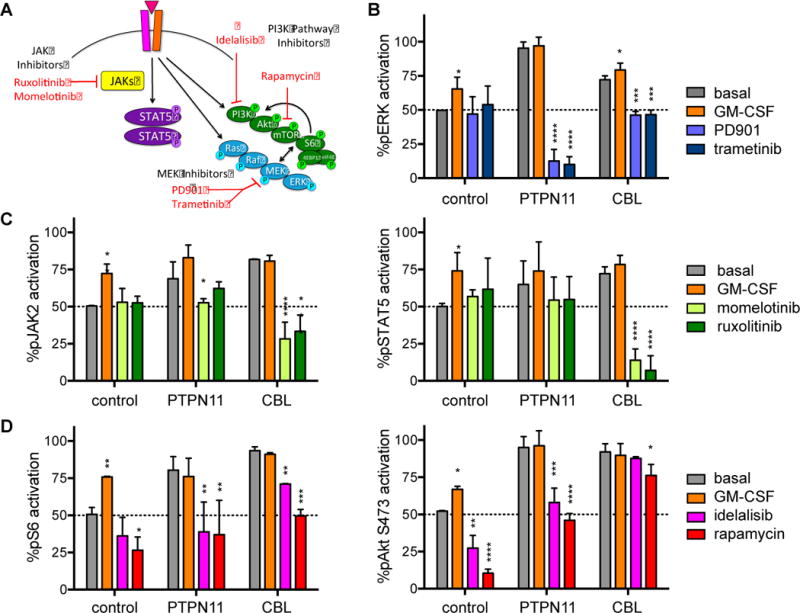
Day 14 control, PTPN11, and CBL iPSC-derived CD45+14+18+ myeloid cells were washed and incubated in vitro in serum-free medium with GM-CSF 10 ng/mL (positive signaling control) or (A) MEK inhibitors (PD901 100 nM, trametinib 100nM), JAK inhibitors (momelotinib 1 uM, ruxolitinib 1 uM), or PI3K pathway inhibitors (idelalisib 1 uM, rapamycin 10 nM) for 60 minutes at 37°C prior to antibody staining and phosphoflow cytometry analysis. Flow cytometry data for each phosphoprotein and iPSC line were gated as in Figure 2 and Supplemental Figure 1. (B) Percent pERK, (C) pJAK2 and pSTAT5, and (D) pS6 and pAkt activation for each cell line and inhibitor condition are displayed relative to mean basal phosphoprotein levels of control CD45+14+18+ myeloid cells. Experiments were performed in triplicate. Statistical analyses were performed with ANOVA and the Dunnett post-test for multiple comparisons. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
We next tested whether MEK, JAK, or PI3K pathway kinase inhibitors could abrogate constitutive signaling activation in JMML iPSCs. In vitro incubation of PTPN11 and CBL myeloid cells with MEK inhibitors PD901 and trametinib markedly decreased pERK levels, but inhibition was most effective in PTPN11 cells. No significant pERK inhibition was observed in control iPSC myeloid cells treated with PD901 or trametinib (Figure 3B). Incubation of CBL myeloid cells with JAK inhibitors momelotinib or ruxolitinib significantly decreased pJAK2 and pSTAT5, while minimal or no effects of JAK inhibition were observed in PTPN11 or control cells (Figure 3C). Lastly, incubation of iPSC-derived myeloid cells with the PI3Kδ inhibitor idelalisib or the mTOR inhibitor rapamycin suppressed pS6 activation in all cell types. Inhibition was more pronounced in PTPN11 compared to CBL cells. Despite similar basal pAkt elevation in both JMML cell lines, little pAkt inhibition was observed in CBL iPSCs incubated with idelalisib or rapamycin (Figure 3D). Taken together, data from these phosphosignaling analyses demonstrate discrete sensitivities of JMML iPSCs to specific kinase inhibitors with preferential sensitivity of PTPN11 cells to MEK inhibition and CBL cells to JAK inhibition.
Kinase inhibitors inhibit cellular growth
To validate the kinase inhibitor sensitivity patterns observed in phosphoflow cytometric analyses of PTPN11- and CBL-mutant iPSC-derived myeloid cells, we tested whether these drugs could inhibit myeloproliferation in methylcellulose colony assays. We first identified the half-maximal effective concentration (EC50) of each drug by plating control, PTPN11, and CBL iPSC-derived hematopoietic progenitors in methylcellulose with saturating doses of GM-CSF (10 ng/mL) (Supplemental Figure 3). Control and JMML iPSC-derived progenitors were then plated with DMSO, PD901, trametinib, momelotinib, ruxolitinib, idelalisib, or rapamycin at EC50 concentrations with increasing concentrations of GM-CSF for each cell type (Figure 4). Concordant with our in vitro phosphosignaling data, the MEK inhibitors PD901 and trametinib preferentially inhibited colony formation from PTPN11 iPSC-derived hematopoietic progenitors at sub-saturating concentrations (<10 ng/mL) of GM-CSF (Figure 4B), but had no appreciable effect upon CBL progenitor colony formation (Figure 4C). The JAK inhibitors momelotinib and ruxolitinib modestly diminished colony growth in PTPN11 progenitors at most doses of GM-CSF, but more effectively inhibited CBL progenitor colony formation (Figure 4B and 4C). The PI3Kδ inhibitor idelalisib decreased colony formation preferentially in PTPN11 compared to CBL cells and was only rescued by the highest tested dose of GM-CSF. The mTOR inhibitor rapamycin potently inhibited colony formation in control, PTPN11, and CBL progenitors at all concentrations of GM-CSF (Figure 4A–4C). In summary, PTPN11 progenitors were most sensitive to MEK and PI3K/mTOR inhibition, while CBL progenitor colony growth was best inhibited by JAK and mTOR inhibitors (Figure 4A, 4B, 4C).
Figure 4. Inhibition of PTPN11-mutant and CBL-mutant hematopoietic progenitor myeloproliferation with kinase inhibitors.
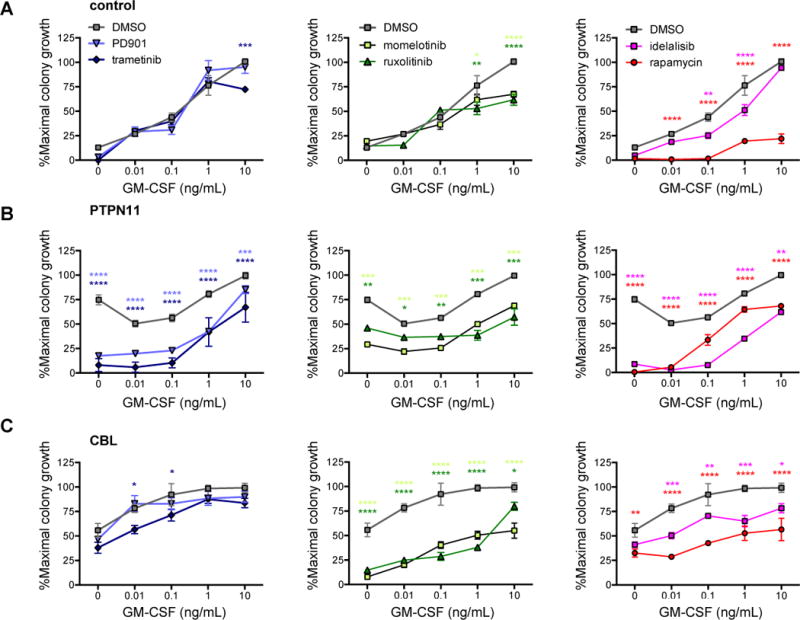
Day 8 CD43+41+235+ hematopoietic progenitors from (A) control, (B) PTPN11, and (C) CBL iPSC lines were cultured in methylcellulose with DMSO, MEK inhibitors (PD901, trametinib), JAK inhibitors (momelotinib, ruxolitinib), or PI3K pathway inhibitors (idelalisib, rapamycin) at EC50 doses and with the specified concentrations of GM-CSF. Colonies were enumerated after 14 days in culture, and data were normalized to maximal colony growth for the DMSO control for each iPSC line (y-axes). Experiments were performed in triplicate with data depicted as means (symbols) with standard error (bars). Statistical analyses were performed with two-way ANOVA and the Dunnett post-test for multiple comparisons. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
DISCUSSION
Children with JMML have suboptimal outcomes with current best available therapy comprised of cytoreductive chemotherapy and allogeneic HSCT. The major cause of JMML-associated mortality is relapse post-HSCT. Given the mutational landscape of JMML and activation of oncogenic signaling networks, it is hypothesized that treatment with targeted kinase inhibitors may improve pre-HSCT disease control and augment clinical outcomes in these young patients. However, definitive biochemical characterization of potential therapeutic vulnerabilities in JMML has been extremely difficult due to lack of JMML cell lines and disease rarity that limits primary cells for study. While >90% of JMML cases are caused by a small number of Ras pathway mutations, disease phenotypes and severity vary based on specific somatic versus germline mutations.1, 2, 30 In particular, children with CBL-mutant JMML usually experience spontaneous disease resolution and may be closely observed without need for intensive chemotherapy or HSCT, although it is unknown whether HSCT can prevent the associated vasculopathy prevalent in patients with germline CBL mutations.1, 31, 32
Robust preclinical models are still necessary to advance understanding of the biology of and new treatment strategies for JMML. While outcomes may correlate with specific underlying JMML-associated mutations,1, 30, 33, 34 patient-specific biologic and clinical variabilities are imperfect predictors of long-term relapse-free survival. Chemotherapy agents commonly utilized in children with JMML (e.g., cytarabine, fludarabine, 5-azacytidine, 6-mercatopurine) are not curative and are frequently ineffective. It is plausible that use of kinase inhibitors for disease stabilization in children with JMML could minimize need for cytotoxic chemotherapy and that kinase inhibitor maintenance therapy post-HSCT minimize relapse. Our current studies suggest potential mutation-specific activity of kinase inhibitors that may be new adjunctive treatment strategies for children with JMML.
To our knowledge, these studies are the first to compare directly the constitutive signaling activation and potential therapeutic vulnerabilities of PTPN11- versus CBL-mutant JMML. Preclinical activity of MEK inhibitors such as PD901 and trametinib has been well-documented in genetic mouse models of Ras-driven MPNs and solid tumors.12, 14, 35 However, while MEK inhibition has reduced splenomegaly and dysplastic myelopoiesis and prolonged survival in MPN models, complete elimination of Ras pathway-mutant cells has not been observed.12 Subsequent studies have also implicated constitutive PI3K/Akt/mTOR signaling with potential Ras/MAPK crosstalk in KRAS-, NF1-, PTPN11-, or CBL-mutant JMML models and shown preclinical activity of PI3K pathway signaling inhibitors.15–17, 33, 36 Concordant with these data, our results demonstrate that mTOR inhibition may be a useful adjuvant therapy for patients with JMML regardless of underlying genetic lesion. Rapamycin is commonly utilized in children for a variety of indications, including treatment of post-HSCT graft-versus-host disease, immunosuppression after solid organ transplantation, and management of autoimmune lymphoproliferative syndrome.37–39
JAK inhibition is another attractive potential therapeutic strategy based on characteristic basal and GM-CSF-inducible STAT5 signaling in JMML, particularly for children with CBL mutations. The JAK1/2 inhibitor ruxolitinib is a Food and Drug Administration-approved therapy for adults with MPNs and is currently under study in children and adults with JAK pathway-mutant acute lymphoblastic leukemia (clinicaltrials.gov NCT02723994, NCT03117751, NCT02420717).40, 41 Treatment of patients with chronic myelomonocytic leukemia with ruxolitinib in a phase 1 trial also reduced splenomegaly, improved hematologic dysfunction, and abrogated pSTAT5 activation in pharmacodynamic assays.42 A preclinical study demonstrated prolonged animal survival in NF1-deficient MPN mice treated with ruxolitinib, as well as reduced colony formation of primary KRAS-mutant JMML cells plated in vitro with ruxolitinib.43 In our prior work, however, we observed minimal inhibition of JAK/STAT signaling in PTPN11-mutant JMML iPSC-derived cells treated with ruxolitinib.18
Our current study conversely shows significant JAK/STAT signaling inhibition in CBL iPSC-derived hematopoietic cells. This observation is concordant with a recent report of in vivo activity of ruxolitinib in a mouse model of CBL-deleted myeloid leukemia.44 In that study, genetic deletion of SH2B3 (encoding LNK, a negative regulator of JAK2) or CBL directly stabilized JAK2 proteins, leading to upregulated JAK/STAT signaling that was sensitive to JAK inhibition. Taken together, these data provide unique mechanistic insight into the discrete leukemogenic dependencies of CBL- versus PTPN11-mutant JMML and the potential differential sensitivities of JMML genetic subtypes to kinase inhibitors. It is plausible that pharmacologic JAK inhibition may have particular cytoreductive potential in children with CBL-mutant JMML. We postulate that ruxolitinib (with established pediatric dosing45) could be used as a minimally toxic ‘temporizing’ therapy to reduce splenomegaly while awaiting usual spontaneous disease resolution. Furthermore, simultaneous pharmacologic inhibition of PI3K/Akt/mTOR and Ras/MAPK pathways or PI3K/Akt/mTOR and JAK/STAT pathways may have enhanced efficacy in patients with PTPN11-mutant and CBL-mutant JMML, respectively.
Our current studies extend the scope of available patient-derived preclinical models of JMML and provide further insight into potential genotype-phenotype correlations and drug sensitivities. While results from in vitro experiments may not fully predict treatment responses in patients, our renewable iPSC cell lines provide novel biologic tools that allowed efficient evaluation of six clinically relevant kinase inhibitors in two distinct genetic models of a rare and often fatal childhood MPN. Further biochemical characterization of signaling dependencies in the existing PTPN11 and CBL iPSC lines and study of additional iPSCs from patients with other JMML-associated mutations will likely facilitate testing of additional drugs, alone or in combination. Introduction of cooperating mutations by gene editing of iPSCs may also further delineate correlations between JMML-associated driver mutations and signaling defects.
Based on our current data and results from other studies, we predict that children with CBL-mutant JMML could be sensitive to JAK inhibitors, while MEK inhibition may be more appropriate for patients with PTPN11 mutations. Use of mTOR inhibitors may have broader therapeutic potential in multiple JMML subtypes given the similarly potent effects we observed in both CBL and PTPN11 cell lines. Ideally, results from these and future studies will lead to further refinement of molecularly-targeted treatment strategies to improve outcomes for children with JMML.
Supplementary Material
Acknowledgments
We thank the human pluripotent stem cell core at the Children’s Hospital of Philadelphia for technical assistance. This work was supported by the National Institutes of Health/National Cancer Institute K08CA184418 (SKT), National Institute of Diabetes and Digestive and Kidney Diseases 5P30DK090969 (MJW) and R01DK100854 (STC), National Heart Lung Blood Institute T32HL0007150 (JC) and 5RC2HL101606 (MJW), the Leukemia Lymphoma Society 6466-15 (MLL), and Hyundai Hope on Wheels (MLL). MLL is the Benioff Chair of Children’s Health at the University of California, San Francisco and the Deborah and Arthur Ablin Endowed Chair of Pediatric Molecular Oncology. MJW is the Arthur Nienhuis Endowed Chair in Hematology at St Jude Children’s Research Hospital.
SKT receives research funding from Gilead Sciences for unrelated studies. SKT and MLL receive research funding from the Incyte Corporation for unrelated studies.
Footnotes
Conflicts of interest: The authors declare no competing financial interests relevant to this work.
AUTHOR CONTRIBUTIONS
SKT designed and directed the study, performed experiments, analyzed data, and wrote the manuscript. JC designed the study, performed experiments, and analyzed data. SG-B, DP, ALG, and GL performed experiments and analyzed data. MLL provided critical clinical specimens. MLW contributed to study design and oversight. STC and DLF designed and directed the study, analyzed data, and edited the manuscript. All authors reviewed the final version of the manuscript.
References
- 1.Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015 Feb 12;125(7):1083–1090. doi: 10.1182/blood-2014-08-550483. [DOI] [PubMed] [Google Scholar]
- 2.Chang TY, Dvorak CC, Loh ML. Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood. 2014 Oct 16;124(16):2487–2497. doi: 10.1182/blood-2014-03-300319. [DOI] [PubMed] [Google Scholar]
- 3.Loh ML. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol. 2011 Mar;152(6):677–687. doi: 10.1111/j.1365-2141.2010.08525.x. [DOI] [PubMed] [Google Scholar]
- 4.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991 Mar 01;77(5):925–929. [PubMed] [Google Scholar]
- 5.Woods WG, Barnard DR, Alonzo TA, Buckley JD, Kobrinsky N, Arthur DC, et al. Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children’s Cancer Group. J Clin Oncol. 2002 Jan 15;20(2):434–440. doi: 10.1200/JCO.2002.20.2.434. [DOI] [PubMed] [Google Scholar]
- 6.Locatelli F, Nollke P, Zecca M, Korthof E, Lanino E, Peters C, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005 Jan 01;105(1):410–419. doi: 10.1182/blood-2004-05-1944. [DOI] [PubMed] [Google Scholar]
- 7.Yabe M, Ohtsuka Y, Watanabe K, Inagaki J, Yoshida N, Sakashita K, et al. Transplantation for juvenile myelomonocytic leukemia: a retrospective study of 30 children treated with a regimen of busulfan, fludarabine, and melphalan. Int J Hematol. 2015 Feb;101(2):184–190. doi: 10.1007/s12185-014-1715-7. [DOI] [PubMed] [Google Scholar]
- 8.Niemeyer CM, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, Creutzig U, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood. 1997 May 15;89(10):3534–3543. [PubMed] [Google Scholar]
- 9.Flotho C, Kratz CP, Bergstrasser E, Hasle H, Stary J, Trebo M, et al. Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood. 2008 Jan 15;111(2):966–967. doi: 10.1182/blood-2007-09-111831. author reply 967-968. [DOI] [PubMed] [Google Scholar]
- 10.Matsuda K, Shimada A, Yoshida N, Ogawa A, Watanabe A, Yajima S, et al. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood. 2007 Jun 15;109(12):5477–5480. doi: 10.1182/blood-2006-09-046649. [DOI] [PubMed] [Google Scholar]
- 11.Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008 Oct 07;14(4):335–343. doi: 10.1016/j.ccr.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang T, Krisman K, Theobald EH, Xu J, Akutagawa J, Lauchle JO, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013 Jan;123(1):335–339. doi: 10.1172/JCI63193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyubynska N, Gorman MF, Lauchle JO, Hong WX, Akutagawa JK, Shannon K, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011 Mar 30;3(76):76ra27. doi: 10.1126/scitranslmed.3001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009 Sep 17;461(7262):411–414. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akutagawa J, Huang TQ, Epstein I, Chang T, Quirindongo-Crespo M, Cottonham CL, et al. Targeting the PI3K/Akt pathway in murine MDS/MPN driven by hyperactive Ras. Leukemia. 2016 Jun;30(6):1335–1343. doi: 10.1038/leu.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodwin CB, Li XJ, Mali RS, Chan G, Kang M, Liu Z, et al. PI3K p110delta uniquely promotes gain-of-function Shp2-induced GM-CSF hypersensitivity in a model of JMML. Blood. 2014 May 01;123(18):2838–2842. doi: 10.1182/blood-2013-10-535104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunda S, Qin K, Kommaraju K, Heir P, Ohh M. Juvenile myelomonocytic leukaemia-associated mutation in Cbl promotes resistance to apoptosis via the Lyn-PI3K/AKT pathway. Oncogene. 2015 Feb 05;34(6):789–797. doi: 10.1038/onc.2013.596. [DOI] [PubMed] [Google Scholar]
- 18.Gandre-Babbe S, Paluru P, Aribeana C, Chou ST, Bresolin S, Lu L, et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013 Jun 13;121(24):4925–4929. doi: 10.1182/blood-2013-01-478412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulero-Navarro S, Sevilla A, Roman AC, Lee DF, D’Souza SL, Pardo S, et al. Myeloid Dysregulation in a Human Induced Pluripotent Stem Cell Model of PTPN11-Associated Juvenile Myelomonocytic Leukemia. Cell Rep. 2015 Oct 20;13(3):504–515. doi: 10.1016/j.celrep.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May 19;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 21.Archambeault S, Flores NJ, Yoshimi A, Kratz CP, Reising M, Fischer A, et al. Development of an allele-specific minimal residual disease assay for patients with juvenile myelomonocytic leukemia. Blood. 2008 Feb 01;111(3):1124–1127. doi: 10.1182/blood-2007-06-093302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009 Aug 27;114(9):1859–1863. doi: 10.1182/blood-2009-01-198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010 Oct 15;24(20):2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills JA, Paluru P, Weiss MJ, Gadue P, French DL. Hematopoietic differentiation of pluripotent stem cells in culture. Methods Mol Biol. 2014;1185:181–194. doi: 10.1007/978-1-4939-1133-2_12. [DOI] [PubMed] [Google Scholar]
- 25.Byrska-Bishop M, VanDorn D, Campbell AE, Betensky M, Arca PR, Yao Y, et al. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J Clin Invest. 2015 Mar 02;125(3):993–1005. doi: 10.1172/JCI75714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, et al. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012 Oct 23;109(43):17573–17578. doi: 10.1073/pnas.1211175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012 Jul 26;120(4):833–842. doi: 10.1182/blood-2011-12-389932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017 Jan 12;129(2):177–187. doi: 10.1182/blood-2016-05-707653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom. 2010 Jul; doi: 10.1002/0471142956.cy1017s53. Chapter 10: Unit10 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshida N, Yagasaki H, Xu Y, Matsuda K, Yoshimi A, Takahashi Y, et al. Correlation of clinical features with the mutational status of GM-CSF signaling pathway-related genes in juvenile myelomonocytic leukemia. Pediatr Res. 2009 Mar;65(3):334–340. doi: 10.1203/PDR.0b013e3181961d2a. [DOI] [PubMed] [Google Scholar]
- 31.Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010 Sep;42(9):794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyakuna N, Muramatsu H, Higa T, Chinen Y, Wang X, Kojima S. Germline mutation of CBL is associated with moyamoya disease in a child with juvenile myelomonocytic leukemia and Noonan syndrome-like disorder. Pediatr Blood Cancer. 2015 Mar;62(3):542–544. doi: 10.1002/pbc.25271. [DOI] [PubMed] [Google Scholar]
- 33.Caye A, Strullu M, Guidez F, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet. 2015 Nov;47(11):1334–1340. doi: 10.1038/ng.3420. [DOI] [PubMed] [Google Scholar]
- 34.Stieglitz E, Troup CB, Gelston LC, Haliburton J, Chow ED, Yu KB, et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood. 2015 Jan 15;125(3):516–524. doi: 10.1182/blood-2014-09-601690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013 Jan;123(1):340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu W, Yu WM, Zhang J, Chan RJ, Loh ML, Zhang Z, et al. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia. 2017 Jan 03; doi: 10.1038/leu.2016.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tasian SK, Teachey DT, Rheingold SR. Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Front Oncol. 2014;4:108. doi: 10.3389/fonc.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teachey DT, Greiner R, Seif A, Attiyeh E, Bleesing J, Choi J, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009 Apr;145(1):101–106. doi: 10.1111/j.1365-2141.2009.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016 Jan 7;127(1):17–28. doi: 10.1182/blood-2015-07-657981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012 Mar 01;366(9):799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015 Jan 29;372(5):426–435. doi: 10.1056/NEJMoa1409002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Padron E, Dezern A, Andrade-Campos M, Vaddi K, Scherle P, Zhang Q, et al. A Multi-Institution Phase I Trial of Ruxolitinib in Patients with Chronic Myelomonocytic Leukemia (CMML) Clin Cancer Res. 2016 Aug 01;22(15):3746–3754. doi: 10.1158/1078-0432.CCR-15-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sachs Z, Been RA, DeCoursin KJ, Nguyen HT, Mohd Hassan NA, Noble-Orcutt KE, et al. Stat5 is critical for the development and maintenance of myeloproliferative neoplasm initiated by Nf1 deficiency. Haematologica. 2016 Oct;101(10):1190–1199. doi: 10.3324/haematol.2015.136002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lv K, Jiang J, Donaghy R, Riling CR, Cheng Y, Chandra V, et al. CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies. Genes Dev. 2017 May 15;31(10):1007–1023. doi: 10.1101/gad.297135.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, et al. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: A Children’s Oncology Group phase 1 consortium study (ADVL1011) Pediatr Blood Cancer. 2015 Oct;62(10):1717–1724. doi: 10.1002/pbc.25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.