

Abstract
Currently incurable, Charcot-Marie-Tooth (CMT) disease is the most commonly inherited neurological disorder, which affects a small percentage of the population. The most common cause of CMT is the duplication of a region on the short arm of chromosome 17, which includes the gene PMP22. We report a thirty-seven-year-old man with CMT disease having sleep, memory and attention disorders characterized by brief retrograde amnesia at early age. The patient has no genetic disease in the family, but was diagnosed with diabetes mellitus, which emphasizes the sensory loss and prolonged infections. Diabetes mellitus emphasizes the sensory symptomatology and predisposes to the development of infections with delayed healing.
Keywords: Charcot-Marie-Tooth, inherited neuromuscular disorder, sleep disorders
Introduction
Charcot-Marie-Tooth (CMT) disease is an inherited affection, also known as a hereditary sensory and motor neuropathy (HMSN), characterized by muscle weakness affecting the distal limbs, feet and hands, sensory abnormalities, which are caused by the degeneration of the peripheral nerves [1]. Abnormalities of the amplitude and of the velocity of the nerve conduction also characterized this disease [2,3]. Firstly described in 1886 by JM Charcot, P Marie and HH Tooth, CMT affects 1 in 2500 patients, being the most common inherited neuromuscular disorder, whose onset is most frequently in childhood but it can also occur in the late adulthood, the severity of the symptoms varying between the patients [1].
The classificationof CMT was made taking into account the genetic transmission (autosomal dominant transmission, rarely autosomal recessive and sometimes there are cases with transmission linked by chromosome X) and the axonal or demyelinating form [4]. Dych et al classified in 1975 CMT in types 1-7, but the most frequently encountered forms are type 1 (demyelinating) characterized by lower median nerve conduction velocityand type 2 (axonal) which is characterized by close to normal nerve conduction velocity, having fibrillation on the detection test; there are also forms, also known as intermediate forms, that have features of both types [4,5].
CMT type 1 is the most frequent one representing about 80% of all the cases of CMT but there are regions such as Japan where the prevalence of CMT type 2 is higher than the prevalence of CMT type 1 [6,7,8]. The intermediate forms represent about 4% of all the cases of CMT [4].
The classic CMT’s clinical symptoms include foot drop and deformity, weakness of the lower extremities, sensory deficits, muscular atrophy which are usually accompanied by a family history [9].
Case report
A thirty-seven-year-old man, from the urban area, smoker, alcohol and coffee consumer, suffering from CMT disease was admitted to the first Neurology Clinic of the Neuropsychiatric Hospital of Craiova, in January 2016 with memory, attention and sleeping disorders. The symptoms appeared approximately two months before along with emphasized sensitivity disorders in the lower third part of the legs accompanied by muscular fatigability and paresthesia in the proximal half of the lower limbs.
Personal physiological and heredocolateral antecedents: He was the first child of a non consanguineous marriage with normal birth and development and none of his family members were found to have any similar history. However his father suffers from insulin requiring diabetes mellitus and essential hypertension, grade 2.
Personal pathological antecedents: CMT at the age of seven years old, bacterial meningitis one year later, static foot disorders (splay foot) surgically corrected in 2005, 2006, and 2007, right olecranon fracture with ablation of material in 2011, diabetes mellitus type II in treatment with Metformin 2000mg per day, and Glimepiride 5mg per day, pilonidal chyst surgery in March 2013, multiple respiratory infections, clavus which became infected on left sole in 2015 with wound dehiscence until now.
Anamnesis: The onset was atypical at the age of seven years old with muscle weakness in the lower limbs accompanied by transitory episodes of diplopia and one episode of loss of consciousness. The patient has been investigated by electroencephalogram (EEG), which did not reveal arguments to support a neurological disease and also nerve and muscle biopsy were performed (right sural nerve and right gastrocnemius muscle) which confirmed the diagnostic. Over the course of the disease multiple bone and joint deformities appeared, touching the metacarpophalangeal joints, both proximal and distal interphalangeal joints of the lower and upper limb; fractures and metabolic disorders also appeared.
When admitted: no fever, BMI=20.7, cold skin with normal color, postoperative scar on the external part of the right ankle with trophic disorders, sinistro-concave scoliosis(Fig. 1), joint deformities, ”hammer” like fingers, claw-looking hand (Fig. 2,3), deformities of the proximal and distal interphalangeal joints of the feet (Fig. 4), muscle atrophy touching both lower legs, minor atrophy in the lower third of the thighs. He also presented weakness of right shoulder abductors, elbow extensors, wrist flexors and thenar muscles; normal breathing sounds, rhythmic heart sounds, blood pressure 130/80mmHg. Neurological examination: language and visual-spatial functions were intact, eye movements kept on both axes, nystagmus in both left and right horizontal gaze, steppage gait, without comparative motor deficits. Muscular tonus was normal in all extremities, the tendon reflexes were absent in both upper and lower limbs, bilateral flatfoot, paresthesia in the lower half of the lower limbs, superficial sensitivity abolished in the legs and diminished in both lower legs, deep sensitivity diminished; memory and attention disorders characterized by brief retrograde amnesia and sleeping disorders.
Figure 1.

Sinistro-concave scoliosis
Figure 2.

Joint deformities, ”hammer” like fingers, claw-looking hand (dorsal view)
Figure 3.
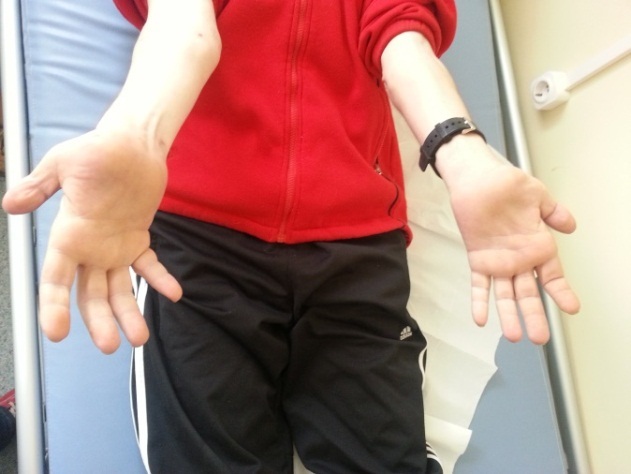
Joint deformities, ”hammer” like fingers, claw-looking hand (palmar view)
Figure 4.

Deformities of the proximal and distal interphalangeal joints of the feet
Biological tests revealed normal count blood cells, high levels of glucose and cholesterol. On the electrocardiography: q wave without medical significance in DIII.
Right sural nerve biopsy showed very severe neuropathy with myelinated fibers density of 1748/mm2, mostly affecting large myelinated fibers. Hypertrophic segmental demyelination was the primitive lesion. Amyelinated fibers were less affected. The regenerative activity was very intense.
Right gastrocnemius muscle biopsy showed neurogenic muscle injury.
Electromyography (EMG): Motor velocity (motor nerve conduction velocity): Compound Muscle Action Potential (CMAP) and Velocity Conduction Motor (VCM) with amplitudes severely diminished for the following nerves: external popliteal sciatic, internal popliteal sciatic, median, ulnar and sural nerves. Sensitive velocity (sensory nerve conduction velocity): Sensory Nerve Action Potential (SNAP) with amplitudes severely diminished for the following nerves: median, ulnar, peroneal and sciatic nerves.
Both the EMG and the biopsy plead for Charcot-Marie-Tooth type I characterized by: myelopathy polyneuropathy, segmental demyelination, concentric aberrant remyelination.
At the Memory Mental State Examination Test (MMSE) the patient obtained 26 points, confirming a mild cognitive disorder.
Discussion
In our case report the patient does not have a family history and he was diagnosed with CMT1. Normally according to Mendelian inheritance patterns, in what CMT is concerned, one can talk about dominant, recessive and X-linked types but sometimes mutations de novo can occur in some patients as the case we presented [1]. In CMT1 mutations in PMP22, MPZ, EGR2, LITAF, PRX, MTMR2 and SBF2, genes found in the myelin membrane negatively influence both the architecture and the electrical properties of the axons leading to the slowdown of the nervous velocity conduction [10,11].
The product of PMP22 gene, which is located on chromosome 17p11.2-12, is a membrane glycoprotein, that is found in a percentage of 2-5% in the peripheral myelin, whose function still remains incompletely elucidated [12]. Her duplication is responsible for the occurrence of CMT1 A, which affects about 79% of the patients diagnosed with CMT [12]. Another protein belonging to the immunoglobulins, involved in CMT1’s pathogenesis is represented by the product of MPZ gene, situated on chromosome 1q22-23, protein that is also part of the peripheral myelin with an important role in adhesion [13]. More than 70 mutations of this gene were described, the phenotype being an axonal or a demyelinating one depending on the phenotype’s mutation [14]. Other genes that have minor involvements in CMT1’s pathogenesis were also described.
Classical clinical description of CMT includes weakness which affects the anterior distal leg muscles responsible of the foot’s dorsiflexion causing “foot drop” with important consequences on the locomotor function [15,16], weakness of the peroneal muscles leading to ankle inversion and instability, that makes the patient prone to falls [17]. The majority of the patients suffering from CMT present hammertoes, deformities of the hand leading to the particular aspect of “claw-hand” [18]. Although in the early stages of the disease the patient say they do not have loss of sensation, at the clinical examination different degrees of sensation loss including loss of proprioception, light touch or even pain can be observed [4]. However, there are cases that present ataxia, spasticity or even atrophy of the optic nerve [4]. The involvement of cranial nerves, clinically represented by facial muscle weakness and trigeminal neuralgia, has also been mentioned in previous studies [19].
Although data from the literature show that patients suffering from CMT2 also have sleep disorders due to sleep fragmentation [20], the patient from our presentation, diagnosed as CMT1, also presents this problem. In patients suffering from CMT sleep disorders are directly correlated with breathing problems such as collapsibility of the upper airways caused by pharyngeal neuropathy, it has also been demonstrated that these patients suffer from obstructive sleep apneea [21,22]. Patients suffering from CMT also present chest abnormalities, phrenic, laringeal and diaphragmatic disfunctions [23,24].
Conclusion
The particularities of the case were: the absence of any genetic disease in the family and presence of chronic diseases such as: diabetes mellitus and dyslipidemia which influence the quality of life. Diabetes mellitus emphasizes the sensory symptomatology and predisposes to the development of infections with delayed healing. The patient also presented minor memory, attention and sleeping disorders.
Acknowledgments
All authors equally contributed in the research and drafting of this paper.
The authors declare that they have no conflict of interests.
References
- 1.Timmerman V, Strickland AV, Züchner S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes (Basel) 2014;5(1):13–32. doi: 10.3390/genes5010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jerath NU, Gutmann L, Reddy CG, Shy ME. Charcot-marie-tooth disease type 1X in women: Electrodiagnostic findings. Muscle Nerve. 2016;54(4):728–732. doi: 10.1002/mus.25077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie C, Zhou X, Zhu D, Liu W, Wang X, Yang H, Li Z, Hao Y, Zhang GX, Guan Y. CNS involvement in CMTX1 caused by a novel connexin 32 mutation: a 6-year follow-up in neuroimaging and nerve conduction. NeurolSci. 2016;37(7):1063–1070. doi: 10.1007/s10072-016-2537-6. [DOI] [PubMed] [Google Scholar]
- 4.McCorquodale D, Pucillo EM, Johnson NE. Management of Charcot-Marie-Tooth disease: improving long-term care with a multidisciplinary approach. J Multidiscip Healthc. 2016;9:7–19. doi: 10.2147/JMDH.S69979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholson G, Myers S. Intermediate forms of Charcot-Marie-Tooth neuropathy: a review. Neuromolecular Med. 2006;8(1-2):123–130. doi: 10.1385/nmm:8:1-2:123. [DOI] [PubMed] [Google Scholar]
- 6.Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB. Genetic epidemiology of Charcot-Marie-Tooth in the general population. Eur J Neurol. 2011;18(1):39–48. doi: 10.1111/j.1468-1331.2010.03037.x. [DOI] [PubMed] [Google Scholar]
- 7.Braathen GJ. Genetic epidemiology of Charcot-Marie-Tooth disease. Acta Neurol Scand Suppl. 2012;193(iv):22–22. doi: 10.1111/ane.12013. [DOI] [PubMed] [Google Scholar]
- 8.DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, Braastad CD, Bishop CM, Jaremko M, Wang Z, Liaquat K, Hoffman CA, York MD, Batish SD, Lupski JR, Higgins JJ. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. 2014;2(6):522–529. doi: 10.1002/mgg3.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudnik-Schöneborn S, Tölle D, Senderek J, Eggermann K, Elbracht M, Kornak U, von der Hagen M, Kirschner J, Leube B, Müller-Felber W, Schara U, von Au K, Wieczorek D, BuBmann C, Zerres K. Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: experiences from a German genetic laboratory on the basis of 1206 index patients. Clin Genet. 2016;89(1):34–43. doi: 10.1111/cge.12594. [DOI] [PubMed] [Google Scholar]
- 10.Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, Kirk CA, Herrmann DN, Laurá M, Li J, Lloyd T, Sumner CJ, Muntoni F, Piscosquito G, Ramchandren S, Shy R, Siskind CE, Yum SW, Moroni I, Pagliano E, Zuchner S, Scherer SS, Shy ME, Inherited Neuropathies Consortium. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. 2015;86(8):873–878. doi: 10.1136/jnnp-2014-308826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bird TD. Charcot-Marie-Tooth Neuropathy X Type 1 . In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews. 2. University of Washington, Seattle: Seattle (WA); 2016. pp. 1993–2017. [Google Scholar]
- 12.Watila MM, Balarabe SA. Molecular and clinical features of inherited neuropathies due to PMP22 duplication. J Neurol Sci. 2015;355(1-2):18–24. doi: 10.1016/j.jns.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 13.Hayasaka K, Himoro M, Wang Y, Takata M, Minoshima S, Shimizu N, Miura M, Uyemura K, Takada G. Structure and chromosomal localization of the gene encoding the human myelin protein zero (MPZ) Genomics. 1993;17(3):755–758. doi: 10.1006/geno.1993.1400. [DOI] [PubMed] [Google Scholar]
- 14.Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, Ohnishi A, Hayasaka K, Onodera O, Baba M, Yasuda H, Saito T, Nakashima K, Kira J, Kaji R, Oka N, Sobue G; Study Group for Hereditary Neuropathy in Japan. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain. 2003;126(Pt 1):134–151. doi: 10.1093/brain/awg012. [DOI] [PubMed] [Google Scholar]
- 15.McDonald CM. Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):495–563. doi: 10.1016/j.pmr.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abresch RT, Carter GT, Han JJ, McDonald CM. Exercise in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):653–673. doi: 10.1016/j.pmr.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Skalsky AJ, McDonald CM. Prevention and management of limb contractures in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):675–687. doi: 10.1016/j.pmr.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yagerman SE, Cross MB, Green DW, Scher DM. Pediatric orthopedic conditions in Charcot-Marie-Tooth disease: a literature review. Curr Opin Pediatr. 2012;24(1):50–56. doi: 10.1097/MOP.0b013e32834e9051. [DOI] [PubMed] [Google Scholar]
- 19.Das N, Kandalaft S, Wu X, Malhotra A. Cranial nerve involvement in Charcot-Marie-Tooth Disease. J Clin Neurosci. 2016;5868(16):30740–30740. doi: 10.1016/j.jocn.2016.10.049. [DOI] [PubMed] [Google Scholar]
- 20.Souza CC, Hirotsu C, Neves EL, Santos LC, Costa IM, Garcez CA, Nunes PS, Araujo AA. Sleep pattern in charcot-marie-tooth disease type 2: report of family case series. J Clin Sleep Med. 2015;11(3):205–211–205–211. doi: 10.5664/jcsm.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dziewas R, Waldmann N, Böntert M, Hor H, Müller T, Okegwo A, Ringelstein EB, Young P. Increased prevalence of obstructive sleep apnoea in patients with Charcot-Marie-Tooth disease: a case control study. J Neurol Neurosurg Psychiatry. 2008;79(7):829–831. doi: 10.1136/jnnp.2007.137679. [DOI] [PubMed] [Google Scholar]
- 22.Dematteis M, Pépin JL, Jeanmart M, Deschaux C, Labarre-Vila A, Lévy P. Charcot-Marie-Tooth disease and sleep apnoea syndrome: a family study. Lancet. 2001;357(9252):267–272. doi: 10.1016/S0140-6736(00)03614-X. [DOI] [PubMed] [Google Scholar]
- 23.Thomas RJ, Mietus JE, Peng CK, Gilmartin G, Daly RW, Goldberger AL, Gottlieb DJ. Differentiating obstructive from central and complex sleep apnea using an automated electrocardiogram-based method. Sleep. 2007;30(12):1756–1769. doi: 10.1093/sleep/30.12.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevilla T, Jaijo T, Nauffal D, Jaijo T, Nauffal D, Collado D, Chumillas MJ, Vilchez JJ, Muelas N, Bataller L, Domenech R, Espinós C, Palau F. Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain. 2008;131(Pt11):3051–3061. doi: 10.1093/brain/awn228. [DOI] [PubMed] [Google Scholar]