

Abstract
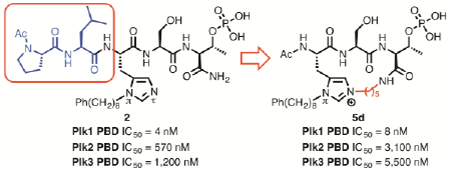
Transition toward peptide mimetics of reduced size is an important objective of peptide macrocyclization. We have previously shown that PLH*SpT (2a) (where H* indicates the presence of a –(CH2)8Ph group at the N(π) position and pT indicates phosphothreonine) is an extremely high affinity ligand of the polo-like kinase 1 (Plk1) polo-box domain (PBD). Herein we report that C-terminal macrocyclization of 2a employing N(π),N(τ)-bis-alkylated His residues as ring junctions can be achieved in a very direct fashion. The resulting macrocycles are highly potent in biochemical assays and maintain good target selectivity for the Plk1 PBD versus the PBDs of Plk2 and Plk3. Importantly, as exemplified by 5d, our current approach permits deletion of the N-terminal “Pro-Leu” motif to yield tripeptide ligands with decreased molecular weight, which retain high affinity and show improved target selectivity. These findings could fundamentally impact the future development of peptide macrocycles in general and Plk1 PBD-binding peptide mimetics in particular.
Keywords: Plk1 polo-box domain, protein-protein interaction, macrocyclic peptide mimetic, conformational constraint
Graphical Abstract
The field of protein-protein interaction (PPI) inhibitors has proven to be highly challenging, in part due to the extended protein interfaces that are often involved. However, significant progress can be made when key recognition features occur on the protein surface in localized “hot spot” regions, which may be amenable to disruption by reduced-size constructs.1, 2 Phospho-dependent PPIs can be viewed as being hot-spot in nature, wherein phosphorylamino acid components serve as critical binding determinants.3 There is wide interest in developing and applying new approaches to phospho-dependent PPI inhibitors, particularly given the importance of these interactions in cellular signal transduction.4 The serine/threonine-specific polo-like kinase 1 (Plk1) is a critical mediator in the initiation and progression of mitosis and it is recognized as a potential target for anticancer drug development.5–7 Plk1 localizes to mitotic structures through its C-terminal polo-box domain (PBD) by engaging in PPIs with phosphoserine and phosphothreonine (pThr)-containing epitopes.8 We have focused our attention on developing PBD-binding inhibitors based on peptidomimetic ligands starting from the pentapeptide PLHSpT (1).9 Through extensive studies, we have examined various structural aspects of PBD recognition and binding of the key pThr residue.10–14 We have also found by appending long-chain alkylphenyl moieties to different regions of the peptide, that we can achieve more than three orders-of-magnitude higher affinities by occupying a “cryptic” pocket on the PBD surface proximal to the pThr-binding site. This reflects a more general principle, that in addition to binding in the hot-spot region, a PPI inhibitor should interact with ancillary pockets on the protein surface and take advantage of protein adaptability.1 We have accessed the cryptic pocket by tethering fragments from N-terminal regions of the peptide15, 16 and from the N(π) position of the histidine residue located at the pThr-2 position.17–20 The peptide PLH*SpT (2a) (where H* indicates the presence of a –(CH2)8Ph group at the N(π) position) is a representative example of the latter class of construct. Peptide 2a exhibits > 1,000-fold enhanced PBD-binding affinity relative to the parent peptide 1.17
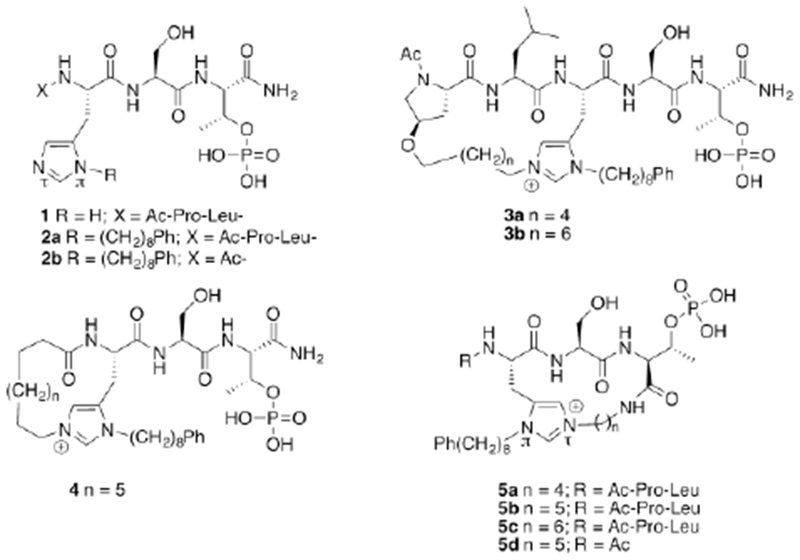
When peptides serve as starting points for the development of PPI inhibitors, macrocyclization is often used to advance linear peptides toward peptide mimetics by restraining and preorganizing important components to orientations favorable for binding, increasing proteolytic stability and improving general druggability.21–24 Application of head-to-tail cyclization strategies on a 2a-derived peptide has resulted in a three orders-of-magiitude or greater loss of PBD-binding affinity relative to 2a.25 Sidechain-to-tail ring closure offers a potential alternative method of cyclization. In this regard, we found that N(π),N(τ)-bis-alkylated His residues can serve as bifurcated macrocycle ring junctions, in which the cryptic pocket-accessing –(CH2)8Ph group projects from the imidazole N(π) position, while the N(τ) site serves as the site of attachment for ring closure with the N-terminus (see peptides of type 3 and 4, Figure 1).26 We have shown that cyclic pentapeptides of type 3 can be equipotent to the linear parent peptide 2a. However, these macrocycles are synthetically difficult to obtain. In addition, an important goal of peptide macrocyclization is transition toward more compact peptide mimetics. Yet, by utilizing the full pentapeptide sequence, macrocycles of type 3 do not advance this objective. In contrast, while cyclic tripeptides of type 4 do afford the potential for size reduction, they have significantly attenuated potencies in biochemical assays.26 Accordingly, the goal of our current work was to utilize bis-alkyl His residues as ring junctions in a way that affords ready access to a new genre of macrocycles, which may constitute potential starting points for the further development of high affinity peptidomimetics of decreased molecular size.
Figure 1.
Structures of peptides discussed in the text.
Macrocycle Design and Synthesis.
The X-ray co-crystal structure of 2a bound to the PBD (PDB: 3RQ717) indicates that the His N(τ)-nitrogen is approximately 5.7 Å from the C-terminal carboxamide nitrogen (Figure 2). We envisioned that the most direct approach to a new family of cyclic ligands would be to use amide-forming macrocyclization reactions that employ methylene linkers of various sizes between the imidazole N(τ)-nitrogen and the C-terminus. We designed a series of macrocycles having ring-closing chains of four, five and six methylenes (5a, 5b and 5c, respectively, Figure 1). We synthesized the required initial linear resin-bound pentapeptides (6a and 6b) on hyper-acid labile (4-hydroxymethyl-3-methoxy-phenoxyjbutanoic acid (HMPB) resin using Fmoc-based solid-phase peptide synthesis (SPPS) and our previously reported reagent, N-Fmoc-His[N(π)-(CH2)8Ph]-OH27 (Scheme 1). We found that alkylation of the His N(τ)-nitrogen could be achieved using a microwave-assisted on-resin SN2 reaction with N-Boc-protected aminoalkyl iodides in similar yields as our previously reported Mitsunobu conditions.26, 28, 29 Treatment of the resulting N(π),N(τ)-bis-alkylated resins (7a – 7c) with dilute TFA (25% in DCM) provided the linear N(π),N(τ)-bis-alkylated His-containing pentapeptides (8a – 8c), while maintaining benzyl pThr phosphoryl protection. Macrocyclization was performed using PyBOP/HOBt and the phosphoryl benzyl protection was then removed to provide the desired type 5 ligands containing 4, 5, or 6 methylene linkers (5a – 5c, respectively). This protocol is significantly more direct than what we previously employed to prepare macrocycles of type 3.26 More importantly, we were able to synthesize the cyclic tripeptide 5d, which lacks the N-terminal “Pro-Leu” residues. This allowed us to evaluate the PBD-binding affinity of a reduced-size construct (Scheme 1).
Figure 2.
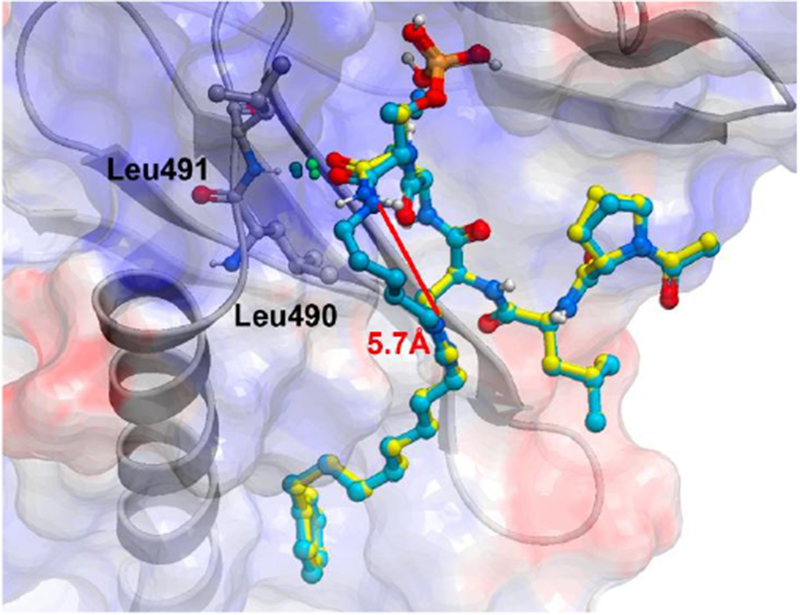
X-ray co-crystal strictures of Plk1 PBD-bound macrocyclic ligand 5b (cyan) superimposed onto the structure of PBD-bound linear peptide 2a (yellow, PDB: 3RQ717). The protein ribbon and semi-transparent electrostatic surface are associated with the 5b structure.
Scheme 1.
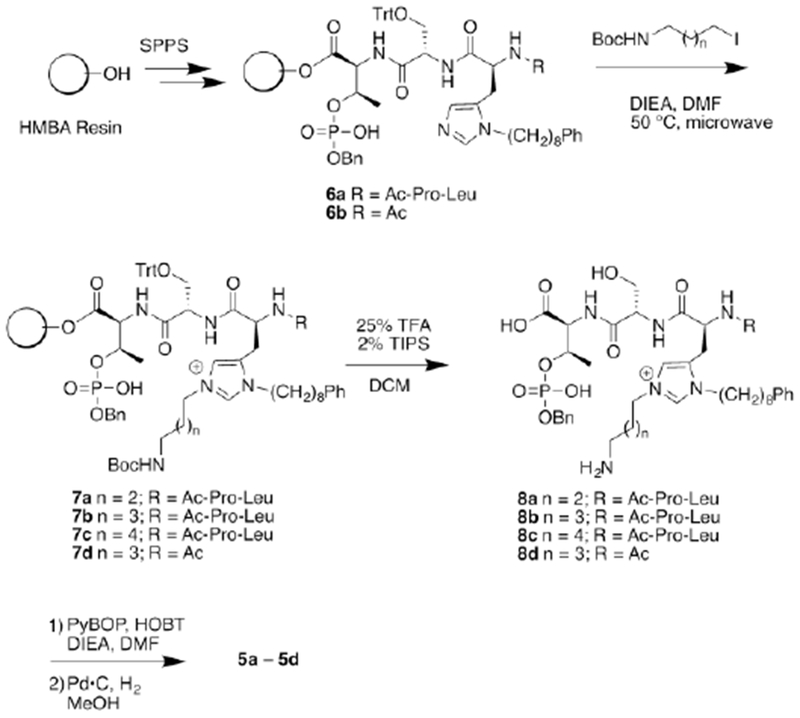
Synthesis of final products 5a – 5d.
Biological Evaluation.
We evaluated the peptides in ELISA assays that measured their ability to inhibit the interaction between full-length Plk1 and an immobilized pThr-containing peptide.14 Relative to the linear pentapeptide 2a (IC50 = 150 nM), all cyclic pentapeptides retained good potencies [5a (n = 4), IC50 = 160 nM; 5b (n = 5), IC50 = 45 nM and 5c (n = 6), IC50 = 44 nM) (Table 1). An important objective of our current work was to reduce overall size. Interaction of the N-terminal region of 1 within a “pyrrolidine-binding pocket” on the surface of the PBD had been postulated to be important for overall binding affinity and deletion of these residues from the open-chain 2a to yield 2b has been reported to result in more than 200-fold loss of inhibitory potency.30, 31 In order to examine the effects of macrocyclization on 2b, we synthesized the cyclic tripeptide 5d, which lacks the “Pro-Leu” residues of 5b. We were gratified to find that the tripeptide 5d retains high PBD-binding affinity (IC50 = 370 nM). This represents an approximate 35-fold enhancement relative to open-chain 2b (IC50 = 13000 nM) (Table 1).
Table 1.
IC50 values for biochemical inhibition of full-length Plk1 by ELISA or isolated PBDs of Plks 1 – 3 by fluorescence polarization (FP).
| ELISA IC50, (nM)a,b | FP IC50, (nM)a,b | |||
|---|---|---|---|---|
| Cnd | Plk1 | Plk1 PBD | Plk2 PBD | Plk3 PBD |
| 2a | 150 ± 21 | 4 ± 0.4 | 570 ± 20 (142x)c | 1,200 ± 120 (300x) |
| 2b | 13000 ± 1700 | n.d.d | n.d. | n.d. |
| 5a | 160 ± 37 | 10 ± 1.0 | 610 ±20 (61x) | 970 ± 40 (97x) |
| 5b | 45 ± 12 | 6 ± 0.5 | 150 ± 10 (25x) | 130 ± 1 (22x) |
| 5c | 44 ± 6 | 5 ± 0.5 | 110 ± 10 (21x) | 80 ± 10 (16x) |
| 5d | 370 ± 25 | 8 ± 0.8 | 3,100 ± 17 (387x) | 5,500 ± 250 (687x) |
Values determined as reported in the Supporting Information14;
IC50 values from multiple independent experiments averaged to provide values ± standard error of the mean (SEM);
Fold-change relative to the Plk1 PBD;
Not determined.
An important aspect of developing peptidomimetic PBD-binding inhibitors is to achieve selectivity for the Plk1 PBD versus the highly homologous PBDs of Plk2 and Plk3. Accordingly, we measured the potency with which the most active cyclic ligands inhibit the binding of optimized pThr-containing fluorescence probes to the isolated PBDs of Plks 1 – 3.14 Consistent with previously reported results,17 the linear peptide 2a demonstrated more than 100-fold selectivity for Plk1 versus Plks 2 and 3. Compound 5a displayed similar selectivity as 2a with approximately 50- to 100-fold selectivity (Table 1). However, compounds 5b and 5c showed reduced selectivity of 16- to 25-fold for Plk1. Interestingly, the cyclic tripeptide 5d demonstrated an improved profile versus 2a, with approximately 400- and 700-fold selectivity for Plk1 versus Plk2 and Plk3, respectively. Given the very low affinity of 2b, we did not think it meaningful to determine the selectivity of this peptide. It is important to note that ELISA assays are conducted using full-length Plk1 protein containing cell lysate, whereas selectivity is evaluated using a fluorescence polarization (EP) assay with the isolated PBDs of Plks 1 – 3 (see Biological Methods in the Supporting Information). Due to inhibitory inter-domain interactions between the KD and PBD, IC50 values for full-length Plk1 can be increased in comparison to isolated PBD, as evidenced by the data in Table 1. This is consistent with previously reported results.8
X-ray Co-crystal Studies.
To examine the effects of macrocyclization on binding geometries, we solved the X-ray co-crystal structure of 5b bound to the isolated Plk1 PBD at 1.45 Å resolution. We observed that macrocyclic 5b overlays very well with PBD-bound linear peptide 2a (Figure 2). In 5b, the ring-closing methylene chain is sufficiently long to allow the C-terminal pThr carboxamide to adopt a favorable trans amide conformation, as well as conserving hydrogen bonding interaction with the backbone amide of Leu491 (Figure 2). Importantly, the polymethylene ring-closing chain of 5b makes hydrophobic contacts with the sidechain of Leu490, which are not possible in the parent peptide 2a. This interaction may also contribute to improved PBD-binding since contacts involving leucine residues are among the most frequent hydrophobic interactions observed in protein-ligand complexes.32
Our current work with C-terminal cyclization extends our earlier studies examining N-terminal macrocyclized constructs of type 3 and 4.20 The resulting peptidomimetics of type 5 can be counted among a very limited number of macrocycles that employ the His imidazole group for ring closure and more specifically, that use N(π),N(τ)-bis-alkylated His residues as ring junctions. This is particularly relevant to Plk1 PBD-binding constructs, since employing His as a bifurcated ring junction permits simultaneous ring closure with extremely efficient access of a critical cryptic binding pocket. We have now found that ring closure in the manner exemplified by type 5 macrocycles can retain affinities in biochemical assays against full-length Plk1, while also maintaining adequate target selectivity for the Plk1 PBD versus the PBDs of Plk2 and Plk3. The Plk1 PBD co-crystal structures of macrocyclic ligands of types 3, 4, or 5 all show remarkably similar overlays with the parent linear peptide, particularly in the phosphoryl-binding site. Given this observation, it is possible that the improved binding affinity for type 5 macrocycles is at least partially derived from better preorganization of favorable binding interactions and decreased conformational rotation of the unbound ligand with a corresponding decrease in entropic loss upon binding. Based on the SAR, the 5- and 6-methylene linkers of 5b and 5c provide optimal length for pre-organization of the ligand, resulting in similarly increased affinities to all Plk isoforms at the cost of decreased selectivity. The 4-methylene linker of 5a is not beneficial to affinity, likely due to less efficient pre-organization of the pentapeptide.
One important objective of peptide macrocyclization is advancement toward reduced-size peptidomimetics. In the linear pentapeptide 1, previous work has shown that accessing a “pyrrolidine-binding” region31 by the N-terminal residues, such as Pro-Leu, is important for high affinity binding and removing these residues from 2a has been reported to result in approximately two orders-of-magnitude losses of affinity. In our current work, we show that C-terminal macrocyclization employing N(π),N(τ)-bis-alkylated His residues as ring junctions with optimal linker length, permits deletion of the “Pro-Leu” to yield the tripeptide ligand 5d, which retains high affinity with good target selectivity and significantly decreased molecular size. This is an important finding, since “SpT” serves as a critical PBD-recognition element, while the “H*” residue affords the most efficient access of the key cryptic binding pocket yet reported. Accordingly, “H*SpT” represents an essential core motif within 2a and peptide 5d may be viewed as a representing a minimal high affinity macrocyclic construct. We believe that our current work should fundamentally impact the future development of peptide-based Plk1 PBD inhibitors.
Supplementary Material
Highlights.
Peptide macrocyclization is an important step in advancing peptides to peptidomimetics
Size reduction of Plk1 PBD-binding peptides with retention of affinity is desired
Histidine N(τ)-cyclized macrocyclization has resulted in a tripeptide with high affinity
Acknowledgments
Our studies are supported by the NIH Intramural Program, Center for Cancer Research (ZIA BC 006198), National Cancer Institute (DH, KT and TB) and NIH grants ES015339 and GM104047 (RG and MY). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the members of the Drennan lab (MIT) for collecting the data. This work was supported in part by a JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH (KT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
The Supporting Information is available free of charge on the journal website. Included are synthetic procedures and analytical data for peptides, biological procedures and binding curves, X-ray crystallography procedures with a table of data collection and refinement statistics.
Accession Code
Atomic coordinates and structure factors for Plk1 PBD-bound 5b have been deposited in the RCSB Protein Data Bank (PDB code 6AX4). Authors will release the atomic coordinates and experimental data upon article publication.
References and notes
- 1.Guo W; Wisniewski JA; Ji H Bioorg. Med. Chem. Lett 2014, 24, 2546. [DOI] [PubMed] [Google Scholar]
- 2.Scott DE; Bayly AR; Abell C; Skidmore J Nat. Rev. Drug Discov 2016, 15, 533. [DOI] [PubMed] [Google Scholar]
- 3.Grossmann A; Benlasfer N; Birth P; Hegele A; Wachsmuth F; Apelt L; Stelzl U Mol. Syst. Biol 2015, 11, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe N; Osada H Bioorg. Med. Chem 2016, 24, 3246. [DOI] [PubMed] [Google Scholar]
- 5.Archambault V; Lepine G; Kachaner D Oncogene 2015, 34, 4799. [DOI] [PubMed] [Google Scholar]
- 6.Lee KS; Burke TR Jr.; Park J-E; Bang JK; Lee E Trends Pharmacol. Sci 2015, 36, 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JE; Hymel D; Burke TR Jr.; Lee KS F1000 Research 2017, 6, 1024 (http://dx.doi.org/10.12688/f1000research.11398.1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elia AE; Rellos P; Haire LF; Chao JW; Ivins FJ; Hoepker K; Mohammad D; Cantley LC; Smerdon SJ; Yaffe MB Cell 2003, 115, 83. [DOI] [PubMed] [Google Scholar]
- 9.Yun S-M; Moulaei T; Lim D; Bang JK; Park J-E; Shenoy SR; Liu F; Kang YH; Liao C; Soung N-K; Lee S; Yoon D-Y; Lim Y; Lee D-H; Otaka A; Appella E; McMahon JB; Nicklaus MC; Burke TR Jr.; Yaffe MB; Wlodawer A; Lee KS Nat. Struct. Mol. Biol. 2009, 16, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F; Park J-E; Lee KS; Burke TR Jr. Tetrahedron 2009, 65, 9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian W-J; Park J-E; Liu F; Lee KS; Burke TR Jr. Bioorg. Med. Chem. 2013, 21, 3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian W-J; Burke TR Jr. Amino Acids 2013, 45, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian W-J; Lai CC; Kelley JA; Burke TR Jr. Chem. Biodiversity 2014, 11, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hymel D; Burke TR Jr. ChemMedChem 2017, 12, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F; Park J-E; Qian W-J; Lim D; Scharow A; Berg T; Yaffe MB; Lee KS; Burke TR Jr. ACS Chem. Biol 2012, 7, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu F; Park J-E; Qian W-J; Lim D; Scharow A; Berg T; Yaffe MB; Lee KS; Burke TR Jr. ChemBioChem 2012, 13, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu F; Park J-E; Qian W-J; Lim D; Graber M; Berg T; Yaffe MB; Lee KS; Burke TR Jr. Nat. Chem. Biol 2011, 7, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian W-J; Park J-E; Lee KS; Burke TR Jr. Bioor. Med. Chem. Lett 2012, 22, 7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao XZ; Hymel D; Burke TR Jr. Bioorg. Med. Chem. Lett 2016, 26, 5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao XZ; Hymel D; Burke TR Jr. Bioorg. Med. Chem 2017, 25, 5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelay-Gimeno M; Glas A; Koch O; Grossmann TN Angew. Chem. Int. Ed. Eng. 2015, 54, 8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White AM; Craik DJ Expt. Opin. Drug Disc 2016, 11, 1151. [DOI] [PubMed] [Google Scholar]
- 23.Gao M; Cheng K; Yin H Biopolymers 2015, 104, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardote TAF; Ciulli A ChemMedChem 2016, 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murugan RN; Park J-E; Lim D; Ahn M; Cheong C; Kwon T; Nam K-Y; Choi SH; Kim BY; Yoon D-Y; Yaffe MB; Yu D-Y; Lee KS; Bang JK Bioorg. Med. Chem 2013, 21, 2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian W-J; Park J-E; Grant R; Lai CC; Kelley JA; Yaffe MB; Lee KS; Burke TR Jr. Biopolymers Pept. Sci. 2015, 104, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian W; Liu F; Burke TR Jr. J. Org. Chem 2011, 76, 8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian W-J; Park J-E; Lim D; Lai CC; Kelley JA; Park S-Y; Lee KW; Yaffe MB; Lee KS; Burke TR Jr. Biopolymers Pept. Sci 2014, 102, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian W-J; Burke TR Jr. Org. Biomol. Chem 2015, 13, 4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srinivasrao G; Park J-E; Kim S; Ahn M; Cheong C; Nam K-Y; Gunasekaran P; Hwang E; Kim N-H; Shin SY; Lee KS; Ryu E; Bang JK PLoS One 2014, 9, e107432/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn M; Han Y-H; Park J-E; Kim S; Lee WC; Lee SJ; Gunasekaran P; Cheong C; Shin SY Sr.; Kim H-Y; Ryu EK; Murugan RN; Kim N-H; Bang JK J. Med. Chem 2015, 58, 294. [DOI] [PubMed] [Google Scholar]
- 32.Ferreira de Freitas R; Schapira M bioRxiv 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.