

Abstract
Background
Metformin, as the first-line treatment anti-diabetic drug, represents increasing evidence of a potential efficacy in improving dyslipidemia. However, the exact molecular mechanism(s) by which metformin influences lipid metabolism remains incompletely understood.
Methods
The HepG2 cells were treated with metformin and the AMP-activated protein kinase (AMPK) inhibitor compound C or a dominant-negative form of AMPK plasmid. ELISA assay was employed to measure AMPK activity, and cellular cholesterol content was determined by enzymatic colorimetric method. RT-PCR and western blotting were used to detect SREBP-2 mRNA levels and its target protein levels.
Results
We found that metformin significantly stimulated AMPK activity and decreased intracellular total cholesterol contents in HepG2 cells. Metformin reduced the sterol regulatory element-binding protein-2 (SREBP-2) and its downstream target proteins and increased low-density lipoprotein receptor (LDLR) levels.
Conclusion
Our preliminary results demonstrate that metformin as a first-line and initial medication suppresses the synthesis of SREBP-2 and upregulates LDLR, and consequently decreases cholesterol production via activation of AMPK, at least partly. These findings suggest a therapeutic target and potential beneficial effects of metformin on the prevention of dyslipidemia or related diseases.
Keywords: type 2 diabetes mellitus, cholesterol, metformin, AMP-activated protein kinase, sterol regulatory element-binding protein-2
Introduction
Type 2 diabetes mellitus (T2DM) is a chronic, progressive disease accompanied by an increase in associated risk factors of cardiovascular diseases (CVD) and cerebrovascular diseases.1 Diabetes has become a leading public health problem affecting ~11.6% of the adult population in China.2 According to the United Kingdom Prospective Diabetes Study, dyslipidemia is an important risk factor for fatal and nonfatal myocardial infarction or angina in T2DM patients.3 Therefore, early detection and prevention of dyslipidemia in T2DM patients will play an important role in reducing cardiovascular and cerebrovascular events and mortality.
Dyslipidemia in T2DM patients is characterized by high plasma triglyceride (TG), increased serum total cholesterol, and low high-density lipoprotein-cholesterol (HDL-c).2,3 Due to its well-established efficacy, modest weight loss and good safety profile, metformin is the first-line treatment and the most prescribed anti-diabetic drug worldwide.1,4,5 Besides antihyperglycemic efficacy, metformin represents increasing evidence of a potential efficacy in improving dyslipidemia, and exerting weight loss, anticancer and cardioprotective effects.4,6–9 Studies have shown that metformin decreased plasma total cholesterol and TG levels and increased HDL-c level.6,10,11 However, the exact molecular mechanism(s) by which metformin influences lipid metabolism remains incompletely understood.
AMP-activated protein kinase (AMPK) is a cellular energy and nutrient sensor that plays a crucial role in regulating catabolic and anabolic pathways involving glycolysis, fatty acid oxidation, gluconeogenesis, and fatty acid biosynthesis.12 It has been shown that the mechanism by which metformin regulates glucose metabolism is mediated by AMPK signaling pathway.13 Xu et al have demonstrated that metformin-induced activation of AMPK suppressed sterol regulatory element-binding protein 1 c (SREBP1-c) and fatty acid desaturase leading to a decrease of low-density lipoprotein-cholesterol (LDL-c) levels.14 Peroxisome proliferator-activated receptor-α (PPAR-α) plays a critical role in the regulation of enzyme involvement in fatty acid oxidation, such as carnitine-palmitoyl transferase-1 and medium chain acyl-CoA dehydrogenase. Metformin could activate AMPK to inhibit PPAR-α activity, but the effect could be completely overcome with treatment of compound C (AMPK inhibitor) in H4IIEC3 cells.15
Sterol regulatory element-binding proteins (SREBPs) are key lipogenic transcription factors that comprise three subtypes, SREBP-1a, SREBP-1c, and SREBP-2. Whereas, SREBP-2 primarily controls cholesterol biosynthesis, and it is initially anchored (precursor protein) in the endoplasmic reticulum and then undergoes a cleavage process in the Golgi apparatus, changing into a nuclear active form. Finally, it transports to the nucleus to regulate its target genes such as 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), and low-density lipoprotein receptor (LDLR).16 Previous studies have shown that AMPK inhibited SREBP-2 by suppressing its cleavage process and nuclear destination.17 Our recent studies have shown that the 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR, an AMPK activator) could stimulate AMPK, which directly phosphorylated SREBP-2 and inhibited SREBP-2 expression and decreased HMGCR and HMGCS protein levels.18
Liver is a major organ regulating energy metabolism. This study aimed to investigate whether the molecular mechanism of cholesterol reduction by metformin in the liver is through activating AMPK to suppress SREBP-2. Therefore, elucidation of inhibitory effect of metformin on cholesterol and its mechanism might provide a basis and target for future therapeutic strategies to prevent dyslipidemia, cardiovascular events, and mortality.
Materials and methods
Reagents and antibodies
Metformin hydrochloride, methyl thiazolyl tetrazolium (MTT) kit, and GAPDH antibody were from Sigma-Aldrich Co. (St Louis, MO, USA). RPMI-1640 medium, Trizol, and FBS were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Compound C was from Merck Calbiochem (Darmstadt, Germany). SREBP-2, LDLR, HMGCR, and HMGCS antibodies were obtained from Abcam (Cambridge, MA, USA). Cholesterol assay kits were obtained from Wako Pure Chemical Corporation (Osaka, Japan). AMPK Kinase Assay ELISA kit was from Cyclex (Nagano, Japan).
Cell culture
HepG2 cells (Cell Library of the Chinese Academy of Sciences, Shanghai, China) were cultured and maintained in RPMI-1640 supplemented with 10% FBS, 100 units/mL penicillin, and 100 µg/mL streptomycin with 5% CO2 incubator at 37°C. The cells were cultured to 80% confluence and exposed to metformin with or without compound C after overnight serum starvation.
MTT assay
The HepG2 cells were seeded in 96-well plates at a concentration of 1×105 cells/mL and incubated for 24 hours; this was followed by treatment with different concentrations (0, 5, 10, and 15 mmol/L) of metformin for indicated 1, 8, and 24 hours. After treatments, 20 µL MTT (5 mg/mL) was added for 4 hours following the manufacturer’s protocol. The media were removed and dissolved in 150 µL dimethyl sulfoxide by shaking for 10 minutes. The absorbance at 490 nm wave length was detected in an ELISA reader. MTT assay was carried out three times.
Cholesterol assay
Cells were lysed in a lysis buffer consisting of 1% Triton X-100 in PBS, and the intracellular cholesterol was measured using total cholesterol assay kits based on an enzymatic colorimetric method of cholesterol oxidase-peroxidase-4-aminoantipyrine (COD-PAP). The optical density was measured at 600 nm in an enzyme-labeled instrument (Thermo Fisher Scientific).
Real-time quantitative PCR (RT-qPCR)
Total RNA from cells was isolated by means of Trizol method, and real-time quantitative PCR was used to measure relative SREBP-2 mRNA expression. About 1 µg total RNA was reverse transcribed into cDNA with a PrimeScript RT reagent (TaKaRa, Kusatsu, Japan), and the cDNA amplification was performed with SYBR Premix Ex Taq II (TaKaRa, Kusatsu, Japan) in a Roche 480 detection system. The gene expression was calculated by 2−ΔΔCt method and β-actin gene was used as a control. The primer sequences were as follows: SREBP-2 forward primer 5′-CACCCCTATCCAGACGGC-3′, and reverse primer 5′-TCCGCCTTTCTCCTTCTTTG-3′; β-actin forward primer 5′-TGACGTGGACATCCGCAAAG-3′, and reverse primer 5′-CTGGAAGGTGGACAGCGAGG-3′.
Western blotting (WB) analysis
Nuclear and cytoplasmic proteins from HepG2 cells were isolated according to the instruction of Thermo NE-PER nuclear and cytoplasmic extraction reagent (Pierce, Rockford, IL, USA). Total protein isolation and WB analysis have been described previously.19 Quantification analysis of bands was performed by ImageJ 1.45 software (NIH, Bethesda, MD, USA).
ELISA for AMPK activity
Briefly, after metformin or compound C treatment, the cells were lysed and centrifuged at 14,000 ×g for 20 minutes. The cell supernatants were collected for determining intracellular AMPK activity via AMPK Kinase Assay ELISA kit.20
Plasmid and transfection
Transient transfection assays were performed with Lipofectamine 2000 reagent according to the manufacturer’s instructional guides.15 HepG2 cells in 6 cm dishes were cultured in complete RPMI-1640 medium, synchronized overnight in serum-free RPMI-1640, and then transfected with a dominant-negative form of AMPK (DN-AMPK, a generous gift from Prof Dave Carling, Imperial College London) plasmid. After 24 hours of incubation, the serum-free medium was replaced and administered with or without metformin.
Statistical analyses
Results were expressed as mean ± SD and analyzed by Prism v5.0 (GraphPad Software Inc, San Diego, CA, USA). Differences between multiple groups were determined by one-way ANOVA (Tukey’s tests). P<0.05 was indicated to be statistically significant.
Results
Metformin exhibited an inhibitory effect on viability of HepG2 cells
To examine the effects of metformin on the cell viability of HepG2 cells, HepG2 cells were administered with increasing concentrations (0, 5, 10, and 15 mmol/L) of metformin for indicated 1, 8, and 24 hours. MTT assay was used to evaluate the cell viability. As shown in Figure 1, metformin treatment exhibited an inhibitory effect on HepG2 cell viability with a significant dose- and time-dependent manner. The HepG2 cell survival was unaffected in low dose (5 mmol/L) after schedule times (1 and 8 hours) incubation except 24 hours. In contrast, HepG2 cell survival was dramatically inhibited with the increasing high-dose (10 and 15 mmol/L) metformin treatments.
Figure 1.
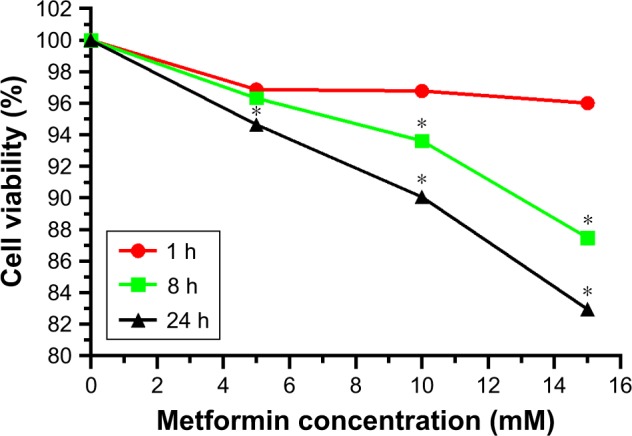
Effects of different metformin concentrations and treatment times on the cell viability of HepG2 cells.
Notes: HepG2 cells were administered with increasing concentrations (0, 5, 10, and 15 mmol/L) of metformin for 1, 8, and 24 hours. Cell viability was determined by the MTT assay. Data are listed as the mean ± SD (n=3). *P<0.05 vs control groups.
Abbreviation: MTT, methyl thiazolyl tetrazolium.
Metformin induced AMPK activation in HepG2 cells
To explore the involvement of metformin in AMPK activity, HepG2 cells were incubated with 15 mmol/L metformin with or without 20 µmol/L AMPK inhibitor compound C (an AMPK inhibitor) for 24 hours.21 As shown in Figure 2, a striking increase of AMPK activity in HepG2 cells was observed after treatment with metformin. The metformin-induced AMPK activation was decreased when the AMPK inhibitor compound C was added. These data demonstrated that metformin could stimulate AMPK activity.
Figure 2.
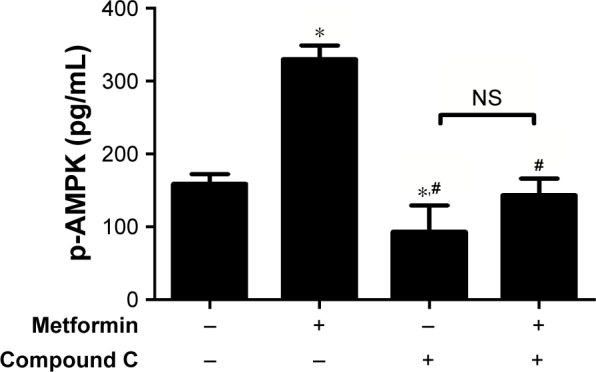
Metformin stimulated the AMPK activity in HepG2 cells.
Notes: HepG2 cells were treated with 15 mmol/L metformin in the absence or presence of 20 µmol/L compound C for 24 hours. AMPK activity was measured by an AMPK Kinase Assay kit. The data are presented as the mean ± SD (n=3). *P<0.05 vs control group, #P<0.05 vs metformin-treated group.
Abbreviations: AMPK, AMP-activated protein kinase; NS, no significance.
Metformin decreased intracellular total cholesterol contents in HepG2 cells
To assess the action of metformin on intracellular cholesterol contents, HepG2 cells were administered with 15 mmol/L metformin, and then 20 µmol/L compound C was added or not added for 24 hours. As shown in Figure 3, the cholesterol contents were significantly lower in metformin-treated HepG2 cells, and compound C treatment inversed these effects. These data demonstrated that metformin activated AMPK and decreased the cholesterol contents.
Figure 3.
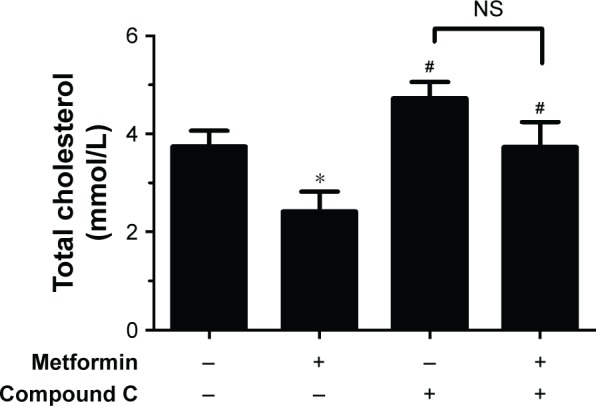
Metformin decreased the intracellular total cholesterol contents of HepG2 cells.
Notes: HepG2 cells were treated with 15 mmol/L metformin and then 20 µmol/L compound C was added or not added for 24 hours. The intracellular cholesterol contents were measured using cholesterol assay kits based on enzymatic methods. The data are presented as the mean ± SD (n=3) of three independent experiments. *P<0.05 vs control group, #P<0.05 vs metformin-treated group.
Abbreviation: NS, no significance.
Metformin downregulated SREBP-2 expression in HepG2 cells
As a key nuclear transcription factor, SREBP-2 plays an important role in cholesterol biosynthesis in liver. RT-PCR assays showed that the SREBP-2 mRNA expression significantly decreased after metformin treatment in HepG2 cells compared with those in the control cells and the compound C-treated cells (Figure 4). These data indicated that metformin treatment enhanced AMPK activity and downregulated SREBP-2 expression.
Figure 4.
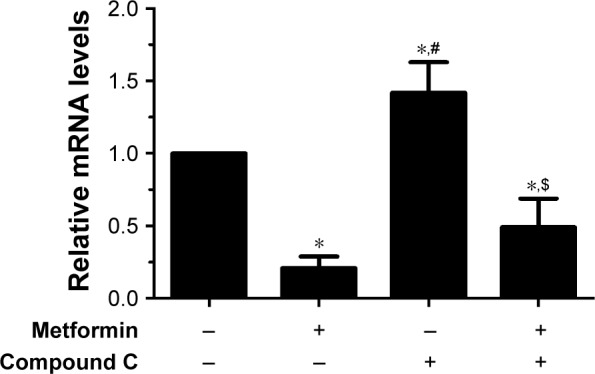
Metformin downregulated SREBP-2 expression in HepG2 cells.
Notes: HepG2 cells were exposed to 15 mmol/L metformin with or without 20 µmol/L compound C for 24 hours. The relative SREBP-2 mRNA levels were normalized to those of β-actin in the RT-PCR data. Experiments were conducted in triplicates. *P<0.05 vs control group, #P<0.05 vs metformin-treated group, $P<0.05 vs compound C-treated group.
Abbreviations: RT-PCR, real-time PCR; SREBP, sterol regulatory element-binding protein.
Metformin reduced SREBP-2 and downstream target protein levels in HepG2 cells
To further examine the inhibitory effects of metformin on SREBP-2 and its downstream target proteins, HepG2 cells were administered with metformin for 24 hours with or without compound C. Both SREBP-2 precursor and its nuclear active forms were obviously reduced in the metformin-treated HepG2 cells compared with those in the control and compound C-treated cells, and the metformin-induced decrease in SREBP-2 was not completely inhibited by compound C (Figure 5A and B). Compound C is a reversible, selective AMPK inhibitor. To further examine whether AMPK was involved in the pathway of metformin action, as shown in Figure 5C, overexpression of the dominant-negative AMPK (DN-AMPK) completely abrogated the effect of metformin to decrease SREBP-2 in HepG2 cells. Paralleled with the decrease in SREBP-2 protein levels, there was a sharp decline in the levels of the SREBP-2 downstream target proteins HMGCR and HMGCS by metformin treatment. Surprisingly, the LDLR protein level conversely increased; the correlation between metformin with this change remains unclear (Figure 5D and E). These results revealed that metformin-induced AMPK activation directly inhibited SREBP-2 activity, at least partly.
Figure 5.

Metformin reduced the protein levels of SREBP-2 and its downstream targets HMGCR and HMGCS in HepG2 cells.
Notes: (A) HepG2 cells were administered with 15 mmol/L metformin with or without 20 µmol/L compound C for 24 hours. SREBP-2 proteins in nucleus and cytoplasm were determined by WB; (P) and (N) indicate the precursor and nuclear active forms of SREBP-2, respectively. (B) Densitometric quantification of SREBP-2 (P) and SREBP-2 (N) bands is shown using ImageJ (version 1.45) with normalization to GAPDH. Data are mean ± SD. All the experiments were performed in duplicate. *P<0.05, #P<0.05 vs the other groups; $P<0.05 vs the control and compound C-treated groups. (C) HepG2 cells were transfected with a dominant-negative form of AMPK (DN-AMPK) plasmid for 24 hours and then stimulated with or without 15 mmol/L metformin for another 24 hours of incubation. Nuclear and cytoplasmic extracts were analyzed by WB, and β-actin was used as internal marker. (D) HepG2 cells were administered with 15 mmol/L metformin with or without 20 µmol/L compound C for 24 hours. HMGCR, HMGCS, LDLR, and GAPDH protein levels were measured by WB. (E) Densitometric quantification of the HMGCR, HMGCS, and LDLR bands. The data are presented as the mean ± SD. All pictures are representative of three independent experiments. *P<0.05 vs control group, #P<0.05 vs metformin-treated group, $P<0.05 vs compound C-treated group.
Abbreviations: AMPK, AMP-activated protein kinase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HMGCS, 3-hydroxy-3-methylglutaryl-CoA synthase; LDLR, low-density lipoprotein receptor; NS, no significance; SREBP, sterol regulatory element-binding protein; WB, Western blotting.
Discussion
T2DM is now recognized as a risk equivalent for CVD, leading to DM being a major cause of CVD mortality.22,23 Dyslipidemia is a major risk factor of CVD. Clinical trials have shown that metformin as the first-line medication improved dyslipidemia and decreased the likelihood of cardiovascular events and death in obese patients with T2DM.3,11,14,24 Several clinical studies have indicated that metformin effectively improved nonalcoholic fatty liver disease (NAFLD) and its related dyslipidemia.25 However, mechanisms underlying the effect of metformin on dyslipidemia are still not clear at present.
Several studies demonstrated that the effect of metformin is mediated by the activation of AMPK signal pathway.6,17,19,26–28 AMPK controls cellular lipid metabolism in several different ways. On the one hand, AMPK as a serine/threonine kinase could inhibit lipogenesis by directly phosphorylating and inactivating acetyl-CoA carboxylase and HMGCR, which are rate-limiting enzymes involved in fatty acid and cholesterol synthesis.29 On the other hand, AMPK could inhibit transcription factors such as SREBP-1c activity, leading to reduced lipogenesis in liver.17,30–32 Lee et al have demonstrated that Ginsenoside Rg3, a ginseng extract, reduced cholesterol and TG accumulation by inhibiting SREBP-2 expression and stimulating AMPK in HepG2 cells.20 The mechanisms of AMPK regulating nuclear transcription factors maybe involved in post-translation modification that modulate protein stability to regulate target gene expression.17,33–35
Similar to prior observations, our current study supported that metformin stimulated AMPK activity. Cellular cholesterol contents were obviously reduced after metformin treatment. After activation by metformin, AMPK suppressed SREBP-2 expression and decreased the level of its target proteins HMGCR and HMGCS; DN-AMPK attenuated the cholesterol-lowering effect of metformin, which is obvious evidence suggesting that metformin effects are primarily mediated by AMPK. The mammalian target of rapamycin (mTOR) has been linked to many cancers, and findings reveal that mTOR/SREBP-2 pathway plays an essential role in lipogenesis and proliferation of cancer cells.36–38 AMPK stimulated by AICAR could induce apoptosis and suppress proliferation of human cancer cells via phosphorylating and activating the tuberous sclerosis complex 2, resulting in an inhibition of phospholipase D (PLD) activity, which is required for the stimulation of mTOR signaling.39,40 These findings suggest that metformin has an important therapeutic implication for cancer treatment.41
Previous studies have reported that there was an upregulation of SREBP-2 and SREBP-1c in NAFLD, which has been known as a risk factor of CVD,42–44 and metformin could activate AMPK and attenuate SREBP-1-induced intracellular lipid accumulation of fatty liver.45 Statins as HMGCR inhibitors were widely used to prevent CVD and reduce LDL-c;46 nevertheless, SREBP-2 serving as an upstream regulator of HMGCR maybe a potential drug target to combat CVD and NAFLD in future.47
Unexpectedly, the LDLR protein level did not decrease but instead increased. We guess that the net effect of metformin on lipid metabolism is to reduce the cholesterol level, which is consistent with the clinical outcome.11,14,48 Such effect of metformin on LDLR was consistent with the effect of berberine which as a new cholesterol-lowering drug upregulated LDLR and inhibited lipid synthesis in the liver through the activation of AMPK.49,50 A probable explanation for upregulated LDLR might be that LDLR was regulated not only by SREBP-2 but also by other mediators that upregulate LDLR expression.16,51–54 Further studies will be needed to restate the relevance and the critical mediators. Nevertheless, the clinically relevant findings of this study have important implications. Metformin decreases cholesterol contents via AMPK activity, at least partly, which has a potential beneficial value for prevention of CVD, NAFLD, and cancer.
Conclusion
Our preliminary results demonstrate that metformin as a first-line and initial medication suppresses the synthesis of SREBP-2 and upregulates LDLR, and consequently decreases cholesterol production via activation of AMPK, at least partly (Figure 6).
Figure 6.
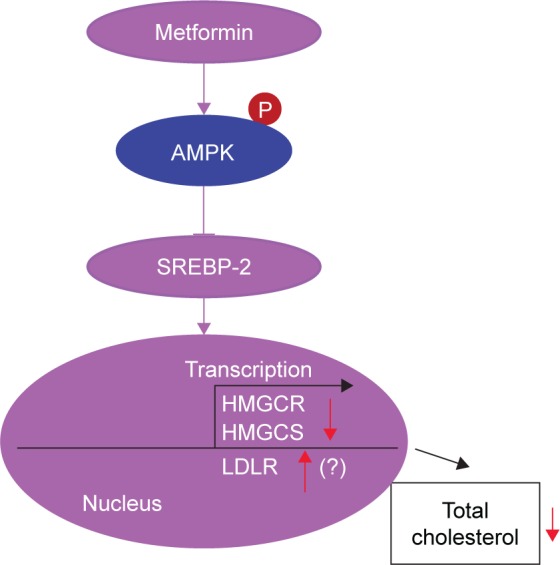
Proposed mechanism of metformin-induced reduction of cholesterol via AMPK.
Abbreviations: AMPK, AMP-activated protein kinase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HMGCS, 3-hydroxy-3-methylglutaryl-CoA synthase; LDLR, low-density lipoprotein receptor; SREBP-2, sterol regulatory element-binding protein-2.
Acknowledgments
This work was supported by grants from the Projects of Medical and Health Technology Development Program in Shandong Province (2016WS0427) and the National Natural Science Foundation of China (81573945).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Weng J, Ji L, Jia W, et al. Standards of care for type 2 diabetes in China. Diabetes Metab Res Rev. 2016;32(5):442–458. doi: 10.1002/dmrr.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu Y, Wang L, He J, et al. China Noncommunicable Disease Surveillance Group Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310(9):948–959. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 3.Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23) BMJ. 1998;316(7134):823–828. doi: 10.1136/bmj.316.7134.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm – 2017 Executive Summary. Endocr Pract. 2017;23(2):207–238. doi: 10.4158/EP161682.CS. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60(9):1586–1593. doi: 10.1007/s00125-017-4336-x. [DOI] [PubMed] [Google Scholar]
- 6.Geerling JJ, Boon MR, van der Zon GC, et al. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes. 2014;63(3):880–891. doi: 10.2337/db13-0194. [DOI] [PubMed] [Google Scholar]
- 7.Wurm R, Resl M, Neuhold S, et al. Cardiovascular safety of metformin and sulfonylureas in patients with different cardiac risk profiles. Heart. 2016;102(19):1544–1551. doi: 10.1136/heartjnl-2015-308711. [DOI] [PubMed] [Google Scholar]
- 8.Liu F, Yan L, Wang Z, et al. Metformin therapy and risk of colorectal adenomas and colorectal cancer in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Oncotarget. 2017;8(9):16017–16026. doi: 10.18632/oncotarget.13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Messaoudi S, Rongen GA, Riksen NP. Metformin therapy in diabetes: the role of cardioprotection. Curr Atheroscler Rep. 2013;15(4):314. doi: 10.1007/s11883-013-0314-z. [DOI] [PubMed] [Google Scholar]
- 10.Wei W, Zhao H, Wang A, et al. A clinical study on the short-term effect of berberine in comparison to metformin on the metabolic characteristics of women with polycystic ovary syndrome. Eur J Endocrinol. 2012;166(1):99–105. doi: 10.1530/EJE-11-0616. [DOI] [PubMed] [Google Scholar]
- 11.Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med. 2008;121(2):149–157.e2. doi: 10.1016/j.amjmed.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 12.Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. AMPK activation: a therapeutic target for type 2 diabetes? Diabetes Metab Syndr Obes. 2014;7:241–253. doi: 10.2147/DMSO.S43731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu T, Brandmaier S, Messias AC, et al. Effects of metformin on metabolite profiles and LDL cholesterol in patients with type 2 diabetes. Diabetes Care. 2015;38(10):1858–1867. doi: 10.2337/dc15-0658. [DOI] [PubMed] [Google Scholar]
- 15.Sozio MS, Lu C, Zeng Y, Liangpunsakul S, Crabb DW. Activated AMPK inhibits PPAR-{alpha} and PPAR-{gamma} transcriptional activity in hepatoma cells. Am J Physiol Gastrointest Liver Physiol. 2011;301(4):G739–G747. doi: 10.1152/ajpgi.00432.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Jing F, Yu C, Gao L, Qin Y, Zhao J. AICAR-induced activation of AMPK inhibits TSH/SREBP-2/HMGCR pathway in liver. PLoS One. 2015;10(5):e0124951. doi: 10.1371/journal.pone.0124951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephenne X, Foretz M, Taleux N, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. 2011;54(12):3101–3110. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S, Lee MS, Kim CT, Kim IH, Kim Y. Ginsenoside Rg3 reduces lipid accumulation with AMP-Activated Protein Kinase (AMPK) activation in HepG2 cells. Int J Mol Sci. 2012;13(5):5729–5739. doi: 10.3390/ijms13055729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen QW, Gerrard DE, Du M. Compound C, an inhibitor of AMP-activated protein kinase, inhibits glycolysis in mouse longissimus dorsi postmortem. Meat Sci. 2008;78(3):323–330. doi: 10.1016/j.meatsci.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 22.World Health Organisation Global report on diabetes. Working Papers. 2016. [Accessed December 4, 2018]. Available from: http://www.who.int/diabetes/global-report/en/
- 23.Gadi R, Samaha FF, Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Curr Diab Rep. 2007;7(3):228–234. doi: 10.1007/s11892-007-0036-0. [DOI] [PubMed] [Google Scholar]
- 24.Group UPDS. Effect of intensive blood-glucose control with met-formin on complications in overweight patients with type 2 diabetes (UKPDS 34) The Lancet. 1998;352(9131):854–865. [PubMed] [Google Scholar]
- 25.Mazza A, Fruci B, Garinis GA, Giuliano S, Malaguarnera R, Belfiore A. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012:1–13. doi: 10.1155/2012/716404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zang M, Zuccollo A, Hou X, et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279(46):47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- 27.Lv Q, Zhen Q, Liu L, et al. AMP-kinase pathway is involved in tumor necrosis factor alpha-induced lipid accumulation in human hepatoma cells. Life Sci. 2015;131:23–29. doi: 10.1016/j.lfs.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Howell JJ, Hellberg K, Turner M, et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25(2):463–471. doi: 10.1016/j.cmet.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 30.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yap F, Craddock L, Yang J. Mechanism of AMPK suppression of LXR-dependent Srebp-1c transcription. Int J Biol Sci. 2011;7(5):645–650. doi: 10.7150/ijbs.7.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madsen A, Bozickovic O, Bjune J-I, Mellgren G, Sagen JV. Metformin inhibits hepatocellular glucose, lipid and cholesterol biosynthetic pathways by transcriptionally suppressing steroid receptor coactivator 2 (SRC-2) Sci Rep. 2015;5(1):16430. doi: 10.1038/srep16430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker AK, Yang F, Jiang K, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010;24(13):1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sundqvist A, Bengoechea-Alonso MT, Ye X, et al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell Metab. 2005;1(6):379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Ricoult S, Yecies J, Manning B. Oncogenic signaling upstream of mTORC1 drives lipogenesis and proliferation through SREBP. Cancer Metab. 2014;2(Suppl 1):P62. [Google Scholar]
- 37.Griffiths B, Lewis CA, Bensaad K, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1(1):3. doi: 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis CA, Griffiths B, Santos CR, Pende M, Schulze A. Regulation of the SREBP transcription factors by mTORC1. Biochem Soc Trans. 2011;39(2):495–499. doi: 10.1042/BST0390495. [DOI] [PubMed] [Google Scholar]
- 39.Mukhopadhyay S, Saqcena M, Chatterjee A, Garcia A, Frias MA, Foster DA. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J Biol Chem. 2015;290(11):6986–6993. doi: 10.1074/jbc.M114.622571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukhopadhyay S, Chatterjee A, Kogan D, Patel D, Foster DA. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle. 2015;14(20):3331–3339. doi: 10.1080/15384101.2015.1087623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fay JR, Steele V, Crowell JA. Energy homeostasis and cancer prevention: the AMP-activated protein kinase. Cancer Prev Res. 2009;2(4):301–309. doi: 10.1158/1940-6207.CAPR-08-0166. [DOI] [PubMed] [Google Scholar]
- 42.Min HK, Kapoor A, Fuchs M, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(5):665–674. doi: 10.1016/j.cmet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohjima M, Higuchi N, Kato M, et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. 2008;21(4):507–511. [PubMed] [Google Scholar]
- 44.Liu X, Green RM. Beyond Farnesoid X receptor to target new therapies for NAFLD. Hepatology. 2017;66(6):1724–1726. doi: 10.1002/hep.29411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung EJ, Kwon SW, Jung BH, Oh SH, Lee BH. Role of the AMPK/SREBP-1 pathway in the development of orotic acid-induced fatty liver. J Lipid Res. 2011;52(9):1617–1625. doi: 10.1194/jlr.M015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cholesterol Treatment Trialists (CTT) Collaborators. Mihaylova B, Emberson J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380(9841):581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahmed MH, Byrne CD. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD) Drug Discov Today. 2007;12(17–18):740–747. doi: 10.1016/j.drudis.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 48.Sáenz Calvo A, Fernández Esteban I, Mataix Sanjuán A, Ausejo Segura M, Roqué M, Moher D. Metformin for type-2 diabetes mellitus. Systematic review and meta-analysis. Aten Primaria. 2005;36(4):183–191. doi: 10.1157/13078602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10(12):1344–1351. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]
- 50.Brusq JM, Ancellin N, Grondin P, et al. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J Lipid Res. 2006;47(6):1281–1288. doi: 10.1194/jlr.M600020-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Urban D, Pöss J, Böhm M, Laufs U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62(16):1401–1408. doi: 10.1016/j.jacc.2013.07.056. [DOI] [PubMed] [Google Scholar]
- 52.Li C, Briggs MR, Ahlborn TE, Kraemer FB, Liu J. Requirement of Sp1 and estrogen receptor alpha interaction in;17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology. 2001;142(4):1546–1553. doi: 10.1210/endo.142.4.8096. [DOI] [PubMed] [Google Scholar]
- 53.Yashiro T, Nanmoku M, Shimizu M, Inoue J, Sato R. 5-Aminoimidazole-4-carboxamide ribonucleoside stabilizes low density lipoprotein receptor mRNA in hepatocytes via ERK-dependent HuR binding to an AU-rich element. Atherosclerosis. 2013;226(1):95–101. doi: 10.1016/j.atherosclerosis.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y, Peng L, Yang LC, et al. Wedelolactone regulates lipid metabolism and improves hepatic steatosis partly by AMPK activation and up-regulation of expression of PPARα/LPL and LDLR. PLoS One. 2015;10(7):e0132720. doi: 10.1371/journal.pone.0132720. [DOI] [PMC free article] [PubMed] [Google Scholar]