

ABSTRACT
Clostridium difficile is a major nosocomial pathogen responsible for close to half a million infections and 27,000 deaths annually in the U.S. Preceding antibiotic treatment is a major risk factor for C. difficile infection (CDI) leading to recognition that commensal microbes play a key role in resistance to CDI. Current antibiotic treatment of CDI is only partially successful due to a high rate of relapse. As a result, there is interest in understanding the effects of microbes on CDI susceptibility to support treatment of patients with probiotic microbes or entire microbial communities (e.g., fecal microbiota transplantation). The results reported here demonstrate that colonization with the human commensal fungus Candida albicans protects against lethal CDI in a murine model. Colonization with C. albicans did not increase the colonization resistance of the host. Rather, our findings showed that one effect of C. albicans colonization was to enhance a protective immune response. Mice pre-colonized with C. albicans expressed higher levels of IL-17A in infected tissue following C. difficile challenge compared to mice that were not colonized with C. albicans. Administration of cytokine IL-17A was demonstrated to be protective against lethal murine CDI in mice not colonized with C. albicans. C. albicans colonization was associated with changes in the abundance of some bacterial components of the gut microbiota. Therefore, C. albicans colonization altered the gut ecosystem, enhancing survival after C. difficile challenge. These findings demonstrate a new, beneficial role for C. albicans gut colonization.
KEYWORDS: Candida albicans, Clostridium difficile, colonization, susceptibility, IL-17A
Introduction
In recent years, the bacterium Clostridium difficile has become an increasingly significant cause of human morbidity and mortality.1 The major risk factor for infection is the use of broad-spectrum antibiotics, with elderly and immunocompromised individuals at particularly high risk for disease.2-4 Antibiotic treatment disrupts the normally protective, resident bacterial community in the gut and leads to susceptibility to Clostridium difficile infection (CDI). This debilitating infection is treated with other antibiotics, all of which continue to disrupt the gut microbiota to some degree, and recurrence occurs in up to 25% of cases.3,4
An alternative approach to CDI, fecal microbiota transplantation (FMT), relies on restoring a normal gut microbial community in a CDI patient who has experienced more than one recurrence. A randomized-controlled clinical trial and other smaller studies show success rates of 80–90% for FMT in treatment of recurrent CDI.5-7 This approach is thus extremely promising but there is currently no consensus on the optimal source or makeup of donor microbiota. In addition, there have been instances of unintended consequences following FMT, including exacerbation of inflammatory bowel disease8,9 and unexplained weight gain.10 A deeper understanding of the effects of commensal organisms on CDI would contribute to identification of an optimal donor microbiota composition.
The study described in this communication focuses on the effects of the commensal fungus Candida albicans on the outcome of CDI. C. albicans colonizes the gut of most humans as a benign commensal. Huffnagle and co-workers demonstrated that, in mice, gut colonization by C. albicans influences the recovery of the bacterial gut community after antibiotic disruption.11 Therefore, C. albicans gut colonization might affect the susceptibility of a host to CDI following antibiotic treatment. In addition, C. albicans affects the immunological milieu. Gut colonization by C. albicans promotes accumulation of regulatory T cell populations.12 Cytokines such as IL-17 and IL-22 are up-regulated in the gut in response to C. albicans colonization.13-16 Monocytes exposed to C. albicans are reprogrammed to confer nonspecific protection from secondary infections, a process termed trained immunity.17 These properties of C. albicans led us to hypothesize that the presence of the fungus in the gut microbiota would protect an antibiotic-treated host from a C. difficile challenge.
The data described here show that mice pre-colonized with C. albicans exhibit an increased ability to survive a lethal challenge with C. difficile. These results highlight a new aspect of C. albicans biology and show that under some circumstances, the effects of C. albicans colonization are beneficial for the host.
Results
Prior colonization of mice with C. albicans reduces susceptibility to lethal challenge with C. difficile
We conducted a direct test of the central hypothesis that C. albicans pre-colonization would attenuate lethal CDI. Antibiotic treatment was used to increase the susceptibility of mice to lethal CDI using a modification of the model established by Young and co-workers (Fig. 1A).18 These investigators demonstrated that cefoperazone-treated mice remained susceptible to CDI for up to 6 weeks after cessation of cefoperazone treatment if given clindamycin on the day before spore challenge. Building on these key observations, we modified the procedure of Reeves, et al. to establish C. albicans colonization in mice prior to challenge with C. difficile spores. Briefly, C57BL/6 mice received cefoperazone in drinking water for 10 days. This antibiotic regimen was used for all mice except certain control mice, as described in Materials and Methods. On the tenth day, some mice were orally inoculated with C. albicans. All mice were then switched to standard water without antibiotics to permit the gut microbiota to recover from the antibiotic treatment for 3 weeks, in the presence or absence of C. albicans. Colonization over this time period was shown by plating homogenized fecal pellets collected at various times post-inoculation (Fig. S1). All mice were successfully colonized with C. albicans following a single inoculation.
Figure 1.
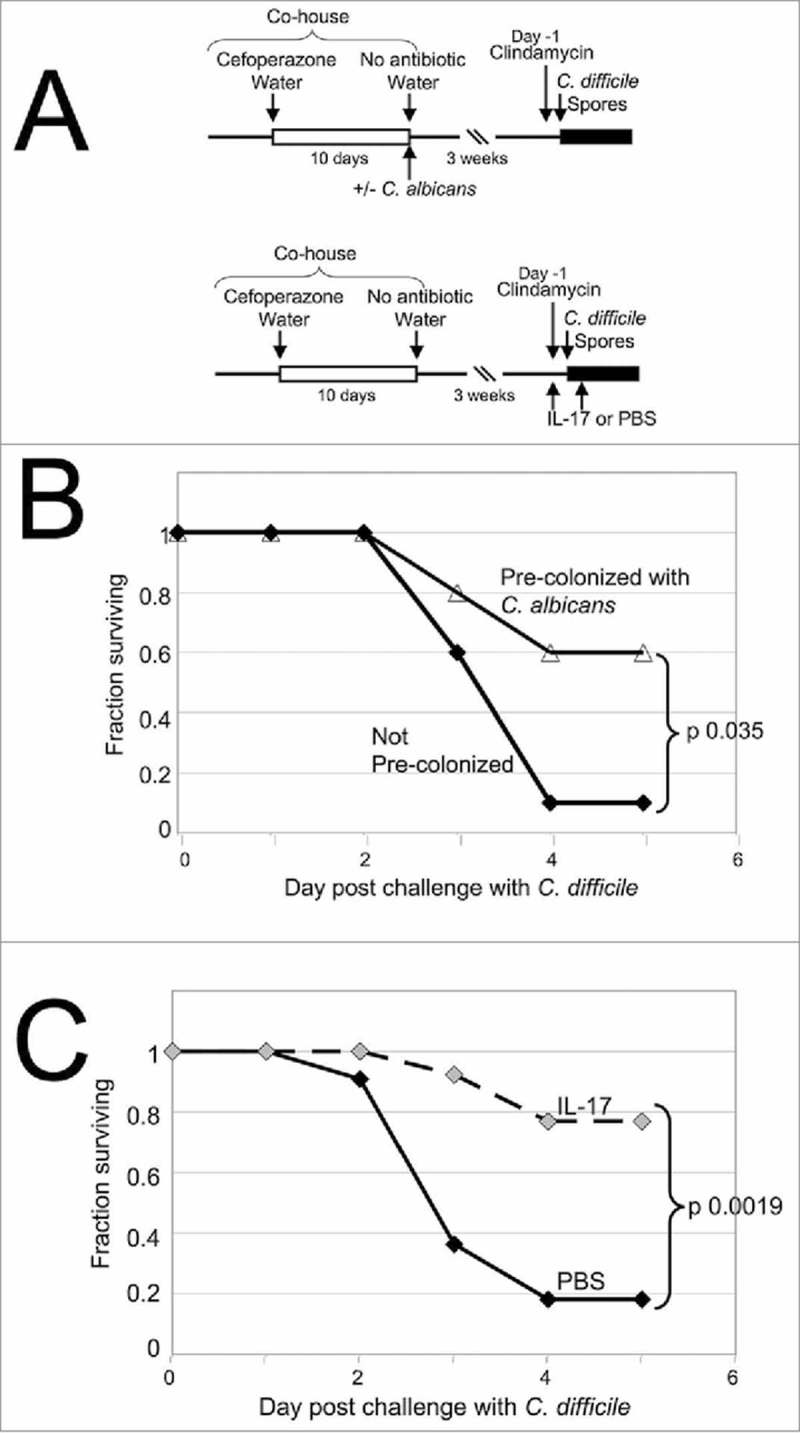
Enhanced survival of C. difficile-challenged mice that were pre-colonized with C. albicans. (A) Timeline of the experiments. All mice used in one experiment were co-housed in a large cage as shown. Mice were given cefoperazone in drinking water, shown as a white box. Mice were then split into smaller groups, given standard water and some mice were orally inoculated with C. albicans. Mice were housed for 3 weeks and then injected with clindamycin intraperitoneally (Day -1). C. difficile challenge was initiated by oral inoculation with C. difficile UK1 spores on the following day. Survival and weight loss were monitored for 5 days following inoculation, shown as a black box. In some experiments, mice treated as above were not colonized with C. albicans and instead received recombinant IL-17A or PBS by intraperitoneal injection on the day before and the day after C. difficile challenge. (B) The fraction of mice surviving on each day after challenge with C. difficile is plotted as a function of day post-challenge. Combined results are from 4 different experiments. Black diamonds, without C. albicans, n = 10; open triangles, pre-colonized with C. albicans, n = 10; p value, log rank test. (C) The fraction of mice surviving on each day after challenge with C. difficile is plotted as a function of day post-challenge. Combined results are from 3 different experiments. Black diamonds, treated with PBS, n = 11; grey diamonds, treated with IL-17A, n = 13. p value, log rank test.
After 3 weeks, mice received a single dose of clindamycin by intraperitoneal injection and, on the following day, were orally inoculated with approximately 4 × 105 C. difficile spores. C. difficile strain UK1, a NAP1/027/BI human epidemic strain19 was used for all studies. The response of inoculated mice to the C. difficile challenge was monitored over 5 days. Weight loss was often evident at day 2 post-inoculation and some mice died or became moribund by day 3. There were slight differences in the timing and synchrony of deaths between experiments and more deaths were observed when higher numbers of spores were used.
In the absence of C. albicans pre-colonization, only 1 of 10 mice with CDI survived to day 5 (Fig. 1B), consistent with previous results.18 Of the 9 who succumbed to severe CDI within 5 days, 5 died, 2 became moribund and were sacrificed and 2 were sacrificed due to weight loss. The average relative weight of surviving mice on day 2 post-inoculation was 0.92 ± 0.06 and on day 3 post-inoculation was 0.84 ± 0.03 of each mouse's pre-inoculation weight (p < 0.00004; paired t test, day 3 versus day of inoculation), demonstrating weight loss due to CDI. Mice injected intraperitoneally with PBS on the day before and the day after C. difficile challenge (as a control for cytokine treatment, see below) were also highly susceptible to lethal CDI (Fig. 1C). Only 2 of the 11 mice in this group survived to day 5 and of the other 9, 7 died, 1 was sacrificed when moribund and 1 was sacrificed due to weight loss. The relative weight of these surviving mice on day 3 post-inoculation was 0.84 ± 0.05 (p < 0.00098; paired t test), demonstrating weight loss due to CDI.
In contrast, the majority of mice pre-colonized with C. albicans survived challenge with C. difficile as compared to those not pre-colonized (Fig. 1B; p = 0.035; log rank test). Six of the 10 mice pre-colonized with C. albicans survived to day 5, while only 1 died, 1 was sacrificed when moribund and 2 were sacrificed due to weight loss. Mice pre-colonized with C. albicans thus showed increased survival of CDI compared to mice without C. albicans pre-colonization.
There was no statistically significant difference in weight loss due to CDI with or without C. albicans pre-colonization. For C. albicans pre-colonized mice infected with C. difficile, average relative weight was 0.91 ± 0.05 of their starting weight on day 2 post-C. difficile inoculation and 0.84 ± 0.06 on day 3 post-C. difficile-inoculation, a statistically significant difference (p < 0.00003; paired t test, day 3 vs day of inoculation) indicating weight loss. Relative weights for surviving mice generally increased at the end of the experiment. The average relative weight at day 5 post-inoculation was 0.96 ± 0.07 (p < 0.003, t test, relative weight on day 3 vs. day 5) consistent with recovery from infection.
Histological analysis of cecum and colon tissues demonstrated that C. difficile-challenged (Cd-challenged) mice exhibited histological features of murine CDI, regardless of the presence of C. albicans. Regions exhibiting edema with leukocyte influx into the lamina propria (Fig. S2, panel E,F) had a patchy distribution. Eighteen sections of cecal tissue from 7 Cd-challenged mice without C. albicans at day 2 post-infection (with or without PBS) were examined. Each cecum section exhibited multiple regions showing edema and leukocyte influx (Fig. S2, panel E; 125 10x microscope fields examined). Ten cecal sections from 5 Cd-challenged mice with C. albicans pre-colonization (with or without PBS) at day 2 post-infection revealed multiple regions with edema and leukocyte influx (Fig. S2, panel F; 74 10x microscope fields examined). Eleven cecal sections from 6 control mice that were antibiotic treated but not challenged with C. difficile, either with or without C. albicans did not exhibit these features (Fig. S2, panel A,B).
Additional parameters of CDI were similar in mice with or without C. albicans even though C. albicans pre-colonized mice had increased survival of CDI. For example, the levels of C. difficile spores detected in the GI tracts of mice at day 2 post-C. difficile inoculation were similar in mice with or without pre-colonization with C. albicans (Fig. 2A; p = 0.76, t test with log transformed data). For this and subsequent experiments, mice were injected intraperitoneally with PBS on the day before and the day after C. difficile challenge. Regardless of pre-colonization with C. albicans, spore levels were lower on day 1 (Fig. S3) than on day 2 (Fig. 2A; median 5,000-fold lower on day 1 than on day 2), showing that spores detected on day 2 reflected growth and new spore production within the GI tract of the host and were not solely the spores fed to the mice.
Figure 2.
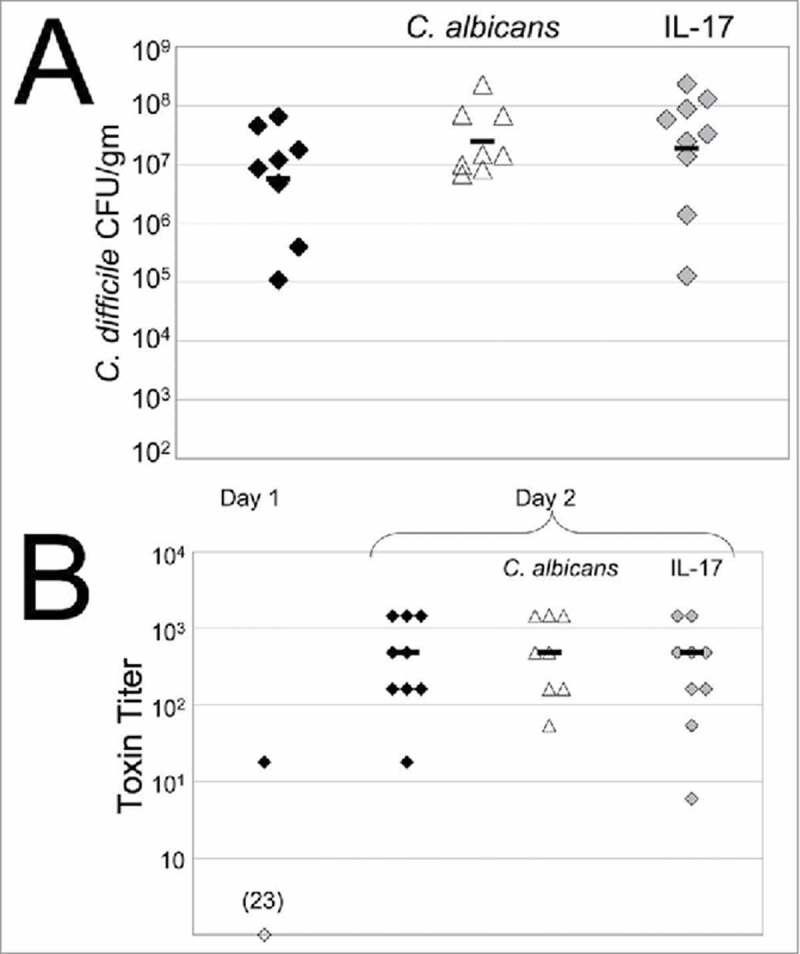
C. difficile spore levels and toxin production are unaffected in pre-colonized or IL-17A-treated mice. (A) Mice were treated as described in Fig. 1A. Mice were euthanized 2 days post-C. difficile inoculation and cecal contents were collected. Samples were heated at 60°C for 10 min and C. difficile spores were enumerated by plating on TCCFA. Black diamonds, received PBS and C. difficile; open triangles, received C. albicans, PBS and C. difficile; grey diamonds, received IL-17A and C. difficile. Each symbol shows CFU/gm from an individual mouse; the bar shows the geometric mean. (B) C. difficile toxin titer in fecal or cecal extracts was measured by a cell-rounding assay. Fecal pellets were collected 1 day post-inoculation. Cecal contents were collected after mice were sacrificed on day 1 or 2 post-inoculation. Toxin titer is defined as the inverse of the greatest dilution that produced 100% cell rounding. Black diamonds, received PBS and C. difficile; open diamond, samples that yielded no detectable toxin activity (23 indicates that 23 of the 24 samples yielded undetectable toxin levels); open triangles, received C. albicans, PBS and C. difficile; grey diamonds, received IL-17A and C. difficile. Each sample shows results from an individual mouse and the bar shows the median.
C. difficile toxin levels were also similar in infected mice with or without C. albicans pre-colonization. C. difficile produces glucosylating toxins that act on small G-proteins of the host, such as Rho and Rac, during growth in the GI tract.20 Toxin activity was measured in extracts of the cecum or fecal pellets from mice using a mammalian cell-rounding assay.21 Toxin titer was defined as the inverse of the greatest dilution that produced 100% cell rounding. The results showed that toxin levels in cecum contents of infected mice sacrificed 1 day post-C. difficile inoculation or in fecal pellets collected on the same day were mostly undetectable (Fig. 2B). Higher levels of toxin activity were present in cecum contents of infected mice sacrificed 2 days post-C. difficile inoculation (Fig. 2B). Levels of toxin were comparable in mice with or without C. albicans pre-colonization (p = 0.91; Mann Whitney test). Toxin activity was not detected in samples from mice that were not inoculated with C. difficile. Therefore, pre-colonization with C. albicans did not reduce the ability of C. difficile to grow and produce toxins in the mouse GI tract. Thus, the presence of C. albicans did not increase the colonization resistance of the host.
Combined, our observations indicate that mice pre-colonized with C. albicans were better able to resist lethal disease due to C. difficile despite the growth of C. difficile in the GI tract, the production of C. difficile toxin and the presence of inflammation and tissue damage.
Altered host response to C. difficile challenge in pre-colonized mice.
A significant fraction of hospitalized patients colonized with C. difficile do not develop symptomatic CDI. Host factors such as immunological status are therefore thought to affect the risk of CDI.3 Based on this and on the increased survival of the C. albicans pre-colonized mice, we hypothesized that C. albicans altered the immune response to C. difficile challenge, which contributed to resistance to lethal CDI.
We therefore measured the expression of genes encoding IL-17A, IL-22, TNF-α and IFN-γ in colonic tissue of Cd-challenged mice with or without C. albicans pre-colonization. Results showed that two days post challenge with C. difficile, mice pre-colonized with C. albicans expressed higher levels of Il17a mRNA than mice without C. albicans (Fig. 3B). In contrast, expression of the other cytokines was not significantly altered by pre-colonization with C. albicans.
Figure 3.
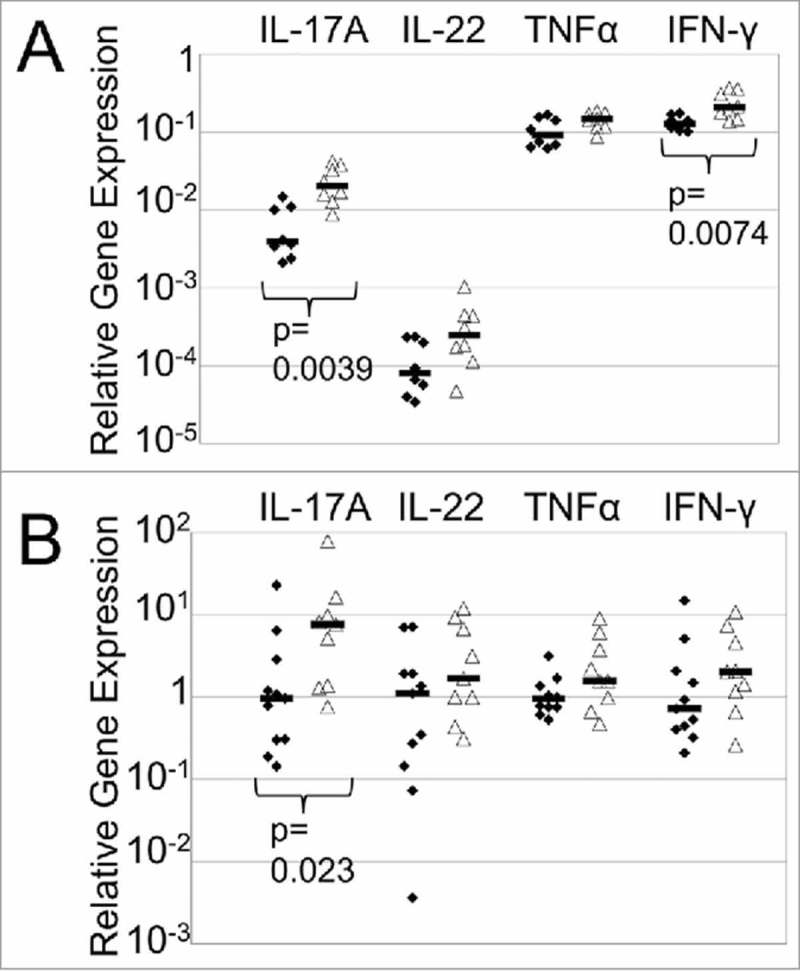
Increased expression of Il17a in the colon of C. albicans-precolonized, Cd-challenged mice. All mice were treated with cefoperazone for 10 days and received clindamycin. Mice were euthanized on the day of challenge before receiving spores (A) or 2 days post challenge with C. difficile (B). RNA was extracted from colon tissue, converted to cDNA and expression of transcripts was measured in cDNA by qRT-PCR and was expressed in arbitrary units using GAPDH for normalization. The average for Cd-challenged mice was set to 1 for each experiment and samples were expressed relative to this value. Combined results of two experiments are shown. Each symbol represents a sample from an individual mouse and the bar indicates the geometric mean. Black diamond, received PBS but no C. albicans; open triangle, received PBS and C. albicans. p values, Mann Whitney test.
The genes encoding IL-17A and IFN-γ were also expressed at higher levels in C. albicans pre-colonized mice prior to inoculation with C. difficile (Fig. 3A). These results demonstrated that the presence of C. albicans altered the host response both before and after the initiation of C. difficile infection.
These results raised the possibility that higher levels of IL-17 are protective against lethal CDI. We therefore hypothesized that administration of IL-17A, in the absence of C. albicans pre-colonization, would protect mice from lethal CDI. To test this hypothesis, mice were given cefoperazone antibiotic treatment for 10 days and then standard water for 20 days, as shown in Fig. 1A. The day before C. difficile challenge, mice were injected intraperitoneally with recombinant IL-17A (or PBS) and clindamycin. Mice were orally inoculated with C. difficile UK1 spores and one day later injected intraperitoneally again with IL-17A (or PBS). Survival and weight loss were monitored for up to 5 days post-C. difficile infection (Fig. 1C). Only 2 of the 11 mice that were injected with PBS survived, while 7 died, 1 was sacrificed when moribund and 1 was sacrificed due to weight loss, as noted above. In contrast, 10 of the 13 mice that were injected with IL-17A survived, while 2 died and 1 was sacrificed due to weight loss. IL-17A treated mice thus showed a significant and striking improved survival of CDI in comparison to mice that were not treated (Fig. 1C, p = 0.0019; log rank test). Average relative weight of surviving IL-17A-treated mice on day 3 post-inoculation was 0.85 ± 0.04, significantly different from the starting weight (p < 0.000009; paired t test). Also, IL-17A treatment did not significantly alter the levels of spores or C. difficile toxin in the infected mice (Fig. 2). Histological analysis of 9 cecal sections from 4 IL-17A treated, Cd-challenged mice showed multiple regions exhibiting edema with leukocyte influx (Fig. S2, panel H; 80 10x microscope fields examined) but not in uninfected mice (Fig. S2, panel D). These results demonstrated that higher levels of IL-17A enhanced survival following challenge with C. difficile spores although IL17A-treated mice had similar levels of weight loss, spore counts and inflammation due to CDI when compared to mice not treated with IL-17A.
Effects of pre-treatments on microbiota composition.
In humans, susceptibility to CDI is strongly affected by the state of the gut bacterial microbiota and antibiotic treatment is a major risk factor for infection.2-4 Therefore, we analyzed the composition of the cecal bacterial microbiota in Cd-challenged mice with or without C. albicans pre-colonization and with or without additional IL-17A treatment. We did not detect a statistically significant difference in the overall microbiota composition between groups; however, there were some differences between groups for specific genera.
The cecal microbiota of mice treated with cefoperazone, colonized (or not) with C. albicans and then given clindamycin, PBS or IL-17A, and finally, C. difficile spores was analyzed. Bacterial community composition was characterized by sequencing the V4 region of the 16S rRNA gene and analyzed using QIIME.22 Principal Coordinate Analysis (PCoA) of weighted UniFrac distance for the bacterial communities in Cd-challenged mice with or without C. albicans and with or without additional IL-17A is shown in Fig. 4A. Permanova analysis of these results did not detect a statistically significant difference between the treatment groups (p = 0.148).
Figure 4.
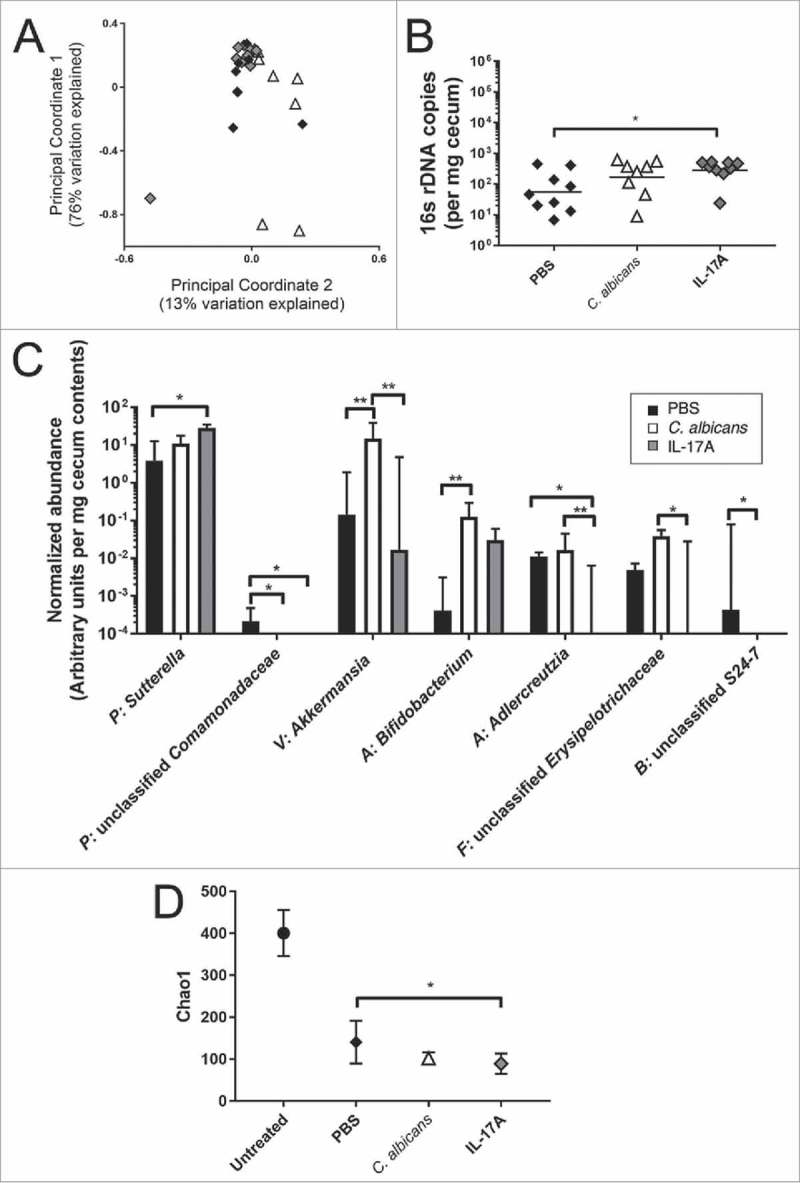
Bacterial microbiota of Cd-challenged mice with or without C. albicans pre-colonization or IL-17A treatment. Cecal bacterial microbiota composition from mice euthanized on day 2 post-C. difficile challenge was analyzed by Illumina sequencing of the V4 region of bacterial 16S rRNA gene. Relative abundance of bacterial taxa was determined using QIIME. (A) Results were analyzed by determining weighted UniFrac distances and performing Principal Coordinate Analysis using QIIME. Black Diamond, PBS-treated, Cd-challenged mice without C. albicans; white triangle, PBS-treated, Cd-challenged mice pre-colonized with C. albicans; grey diamond, IL-17A-treated, Cd-challenged mice without C. albicans. (B) Total levels of bacteria per cecal tip sample were measured by qPCR using eubacterial primers and normalized to milligrams of cecum sample used for DNA extraction. Symbols, as in (A); bar indicates the geometric mean. (C) Normalized abundance per mg of cecum sample (arbitrary units) for bacterial genera in the cecal microbiota of mice treated with PBS and C. difficile (black bars; n = 9); with PBS, C. albicans and C. difficile (white bars; n = 8); or with IL-17A and C. difficile (grey bars; n = 9). All genera with a statistically significant difference between at least two groups are shown. Phyla are indicated with a one letter abbreviation as follows: P = Proteobacteria, V = Verrucomicrobia, A = Actinobacteria, F = Firmicutes, B = Bacteroidetes. Column indicates the median value, bar indicates the upper quartile, brackets indicate statistically significant comparisons (*p<0.05 **p<0.01, Kruskal Wallis Test followed by Dunn's multiple comparisons test) D) Chao1 rarefaction analysis indicates that gut microbial community diversity for C. difficile-inoculated mice is low. Diversity of each sample was calculated by averaging 10 rarefactions taken at a depth of 11,500 sequences. For comparison, community diversity in untreated mice (no antibiotics and not inoculated with C. albicans or C. difficile) is shown (Grey circle). Black diamond, PBS and C. difficile; open triangle, PBS, C. albicans and C. difficile; grey diamond, IL-17A and C. difficile. Symbol indicates mean for mice in each group, with standard error of the mean.
Total levels of bacteria were measured using qPCR and universal 16s rRNA primers. The total number of bacteria detected per milligram of cecum sample was not significantly different between mice that received PBS versus mice that were pre-colonized with C. albicans (Fig. 4B). Cecum samples from mice that received IL-17A contained higher numbers of bacteria per milligram of cecum sample in comparison to mice receiving only PBS (Fig. 4B; geometric mean for IL-17A treated mice 5-fold higher than PBS-treated mice; p = 0.0385, ANOVA with Tukey's multiple comparisons test, using log transformed data).
To test for possible differences in specific genera within the microbiota between groups, we compared the relative abundance of bacterial genera, normalized to the total level of bacteria per milligram of cecum sample. Normalization was used to account for differences in the total levels of bacteria per mg of sample. All bacterial genera with a median fraction greater than 0 in at least one of the three groups were included in order to identify the consistently observed genera. These mice had received multiple antibiotic treatments and were infected with C. difficile, and they were colonized with relatively few genera. We detected a total of 67 genera and 28 exhibited a median greater than 0 in at least one group.
The normalized abundance of the 28 genera was compared between Cd-challenged mice with or without C. albicans or IL-17A using the Kruskal-Wallis nonparametric test followed by Dunn's multiple comparisons test (Table S1). Of the 28 genera that met the criterion for analysis, 7 exhibited a statistically significant difference between at least two of the three experimental groups (Fig. 4C and bold text within Table S1). These included the abundant genera Akkermansia sp. (phylum Verrucomicrobia) and Sutterella sp. (phylum Proteobacteria) and the relatively rare genera Bifidobacterium sp. (phylum Actinobacteria), Adlercreutzia sp. (phylum Actinobacteria), and unidentified genera within the family Comamonadaceae (phylum Proteobacteria), the family Erysipelotrichaceae (phylum Firmicutes) and the family S24-7 (phylum Bacteroidetes).
To detect a possible effect of C. albicans or IL-17A on the relative abundance of rare taxa, we measured community diversity using the Chao1 estimator (Fig. 4D). The microbiota of Cd-challenged mice exhibited relatively low diversity in comparison to mice not treated with antibiotics or microbes, consistent with previous results,23-25 and this decrease was statistically significant. However, the microbiota of mice that were more likely to survive C. difficile challenge (C. albicans precolonized or IL-17A treated) did not exhibit increased diversity in comparison to the highly susceptible, antibiotic-treated infected mice using the Chao1 or other measures (Fig. 4D; Simpson index and Shannon index shown in Fig. S4). The above results showed that the communities present in C. difficile-challenged mice with or without C. albicans or IL-17A exhibited some differences in composition and overall low diversity.
To summarize, the results of this study demonstrate that increased levels of IL-17A were protective against CDI and that C. albicans pre-colonization enhanced expression of IL-17A following C. difficile challenge. Differences in microbiota composition or amounts were detected in association with C. albicans colonization or IL-17A administration and might also play a role in protection. Thus, C. albicans pre-colonization altered the GI tract ecosystem, leading to reduced susceptibility to C. difficile challenge.
Discussion
The results of this study demonstrated significant attenuation of lethal murine CDI conferred by pre-colonization with the commensal fungus C. albicans. Previous studies have shown that bacterial microbiota influence susceptibility to CDI by affecting the metabolism of bile acids that serve as spore germinants2 and by increasing colonization resistance.26,27 By contrast, the protective effect of C. albicans did not alter growth of C. difficile in the GI tract or the production of C. difficile toxin. Thus, C. albicans protects against lethal murine CDI through a mechanism other than colonization resistance.
The results reported here used an experimental mouse model to demonstrate protection against lethal CDI due to C. albicans colonization. Some evidence suggestive of a protective effect of Candida colonization against CDI has been observed in humans. Manian and Bryant found that only 10 of 60 patients with CDI (16.7%) were colonized with high levels of Candida spp., statistically significantly different from the rate in patients who tested negative for CDI (30.5%).28 Similarly low frequencies of Candida colonization in CDI patients were observed by Nerandzic et al. (16%)29 and Blanco et al. (18%).30 Interestingly, Blanco et al. observed that CDI patients who carried the hypervirulent C. difficile 027 ribotype were more likely than patients carrying other ribotypes to exhibit high level C. albicans colonization.30 This observation may reflect the association between high level Candida colonization and situations that produce higher levels of inflammation.31 In another study, recent antifungal use (within 6 weeks) was found to be a risk factor for CDI.32 Perhaps related, administration of the probiotic yeast, Saccharomyces boulardii, to human patients has also shown some efficacy in ameliorating symptoms of CDI.33-35 Combined, the observations from these studies are consistent with fungal colonization being protective against CDI. However, one investigation of CDI and Candida colonization contradicts this showing a significant correlation between CDI and Candida colonization.36 Since the histories of the patients in the various studies are not identical, other variables may have contributed to the finding of a correlation between CDI and Candida colonization in the latter study. Our results are consistent with those human studies in which fungal colonization appears to protect against symptomatic CDI and support a role for C. albicans in altering the intestinal environment and preventing the most serious consequences of C. difficile infection.
Pre-colonization with C. albicans had multiple effects on the GI tract ecosystem. Pre-colonized mice exhibited higher IL-17A expression in the colon both before and after challenge with C. difficile, demonstrating that the activities of the host were altered. Further, we demonstrated that direct administration of IL-17A resulted in reduced lethality following challenge with C. difficile spores. Despite the strong effect of IL-17A administration seen in these studies, loss of IL-17 does not exacerbate CDI; an IL-17A IL-17F double null mutant mouse exhibits normal (or even slightly reduced) susceptibility to CDI.37 These results suggest that high levels of IL-17A as a result of administration can improve host resistance but that basal levels of IL-17A are dispensable.
In contrast, cytokine IL-22 is necessary for normal susceptibility to CDI; IL-22-deficient mice are highly susceptible to lethal infection by C. difficile.38 IL-27 receptor deficient mice also exhibit high susceptibility to CDI.39 Further, administration of either IL-27 or IL-25 increases resistance of mice to lethal CDI.39,40 These cytokines likely have related but not identical effects. In the absence of IL-17, the functions of other cytokines may compensate for the lack of IL-17.
IL-17 has a well-known role in promoting inflammation. IL-17 stimulates the recruitment of neutrophils to the site of inflammation41 and is involved in neutrophil recruitment during CDI.37 In addition, IL-17 plays a role in promoting the repair of epithelia. IL-17A deficient mice exhibit enhanced intestinal permeability and mislocalization of the tight junction protein occludin after treatment with dextran sodium sulfate (DSS).42 Either or both of these effects, enhancing neutrophil recruitment or repair processes, might contribute to attenuation of C. difficile infection.
We explored potential differences in the bacterial microbiota in the different groups of mice because alterations in bacterial microbiota increase susceptibility to CDI. Our results do not exclude a role for the bacterial microbiota in the protection from lethal CDI seen with C. albicans pre-colonization. However, we did not observe a major shift in gut bacterial microbiota in the different groups of mice. Pre-colonized mice did exhibit statistically significantly increased abundance of two bacterial genera, Bifidobacterium and Akkermansia, both of which are associated with beneficial effects on mammalian host health. Probiotic strains of Bifidobacterium sp. have been shown to decrease inflammation, protect against infection by intestinal pathogens and improve gut mucosa integrity in various mouse models.43-47 Akkermansia sp. are mucin-degrading bacteria and a member of the normal human gut microbiota.48 Gastrointestinal pre-colonization with Akkermansia sp. can decrease colonic inflammation and tissue damage,49 improve gut barrier function50 and promote immune tolerance.51 It is possible that the increased abundance of these two bacterial genera in the C. albicans-pre-colonized mice might contribute to the protective effects of C. albicans pre-colonization against C. difficile infection. These genera did not exhibit increased abundance in IL-17A-treated mice; Bifidobacterium and Akkermansia may be less important for protection when IL-17A is administered.
Finally, C. albicans promotes the ability of C. difficile to grow in aerobic conditions in the laboratory.52 Thus, there may be direct effects of C. albicans on the growth of C. difficile in the GI tract. Attenuation of disease likely resulted from these multiple effects of C. albicans colonization on the GI tract ecosystem. Taken together, the results of this study establish a new facet of C. albicans-host interaction, demonstrating that this fungal organism, a common colonizer of humans, can have a beneficial effect on the host through increased survival of CDI.
Materials and methods
Strains and growth conditions
C. albicans strain CKY101,53 a virulent strain derived from the sequenced strain SC5314, was used for all studies. For mouse inoculations, cells were grown at 37°C in YPD (1% yeast extract (BD 212750), 2% peptone (Difco 0118-17-0), 2% glucose (Sigma G8270))54 for 21–24 hrs.
C. difficile strain UK1, a NAP1/027/BI human epidemic strain,19 was used for all studies. Spores were isolated as previously described55 except that gradient purification was omitted. For enumeration of C. difficile spores in extracts from mice, samples were plated on pre-reduced TCCFA plates (Taurocholate (Calbiochem 580217), cycloserine (Sigma C6880), cefoxitin (Sigma C4786), fructose (Macron Fine Chemicals 7756-12))56 and incubated at 35°C for 2 days in an anaerobic chamber.
GI colonization in mice
Five-week-old female C57BL/6 mice (Jackson Laboratory) were co-housed in a large cage (24” x 17”). Mice were treated with antibiotic (cefoperazone (Sigma C4292), 0.5 gm/L) in drinking water for 10 days. All mice were treated with cefoperazone for 10 days, except for the no antibiotics, no C. difficile mice used in Figs. 4D and S4.
Prior to inoculation with C. albicans, mice were tested and shown to be negative for cultivable fungi on YPD-SA agar medium [YPD agar plus 100 μg/ml streptomycin (Sigma S6501) and 50 μg/ml ampicillin (Sigma A9518)] incubated for 2 days at 37°C. On the 10th day of antibiotic exposure, some mice were inoculated orally with 5 × 107 C. albicans cells in 25 μl (n = 10 mice). All of the inoculated mice described in this study became colonized with C. albicans following a single inoculation.
All mice were transferred from the large cage to standard sized cages, housed 2 mice per cage and switched to water without cefoperazone. C. albicans colonization was measured over time by collecting fresh fecal pellets and plating homogenates on YPD-SA agar. Mice that were not inoculated with C. albicans (n = 10 mice), but had been treated with cefoperazone in the same large cage as the C. albicans-inoculated mice, were housed in the same room as the C. albicans-inoculated mice.
After 3 weeks of C. albicans colonization, mice were orally inoculated with C. difficile spores (∼3–8 × 105 spores per mouse). On the day before inoculation with C. difficile, mice were inoculated intraperitoneally with clindamycin (Sigma C5269) (10 mg/kg). Some mice were inoculated intraperitoneally with IL-17A (R&D Systems 421-ML-025, 1 μg in 100 μl PBS) or with PBS on the day before and the day after C. difficile inoculation. All mice used in these experiments were shown to be negative for C. difficile colonization prior to inoculation with C. difficile spores by collecting fecal pellets and plating on pre-reduced TCCFA.
Mice were weighed daily and sacrificed 5 days post-inoculation or when moribund. Mice exhibiting severe signs of illness (extreme inactivity, hunched posture, ruffled fur) were considered moribund and were sacrificed. Mice were also sacrificed if their weight loss exceeded 20%. Within one experiment (i.e., one batch of mice), some of the mice were sacrificed on day 1 or 2 post-inoculation for the various analyses while other mice were monitored for survival over 5 days. The survival data show combined results of mice from 3–4 experiments.
Relative weights were compared using the t test. Survival was compared using the log rank test. The cecum was dissected for further studies. Samples of cecum contents were plated on both YPD-SA to detect C. albicans and on pre-reduced TCCFA after heating at 60°C for 10 min. to detect C. difficile.
All experiments were done in compliance with Tufts University IACUC guidelines. C. difficile susceptibility experiments were performed during the winter months to avoid the occasional presence of cultivable fungi in the GI tracts prior to inoculation with C. albicans.
Microbiota analysis
The distal portion of the cecum, including contents, was dissected from mice two days post-inoculation with C. difficile. For comparison, ceca from control, untreated mice that did not receive antibiotic treatment and were not inoculated with microbes were collected. Mice from 4–5 different experiments were analyzed. The mucosa was scraped using a glass slide and the scraped material plus the luminal contents were combined in PBS. The sample was centrifuged at 16,100 rcf in a refrigerated Eppendorf microcentrifuge for 5 min. and the pellet was weighed and frozen at -80°C. Microbial DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen 51504) with an additional beadbeating step. Briefly, cecum samples were lysed by beadbeating in Qiagen lysis buffer ASL, and the lysate was treated with InhibitEX tablets followed by enzymatic digestion with proteinase K (20mg/ml) and RNaseA (1mg/ml) and column DNA purification.
Libraries were prepared from each sample and sequenced as described.57 Briefly, PCR amplification of the V4 region of the 16S rRNA gene was performed with primers that included adapters for Illumina sequencing and twelve base barcodes. Two hundred fifty bp paired-end sequencing was performed using an Illumina MiSeq. Base calling was performed using CASAVA 1.8 and the resulting fastq files were used as input for downstream analysis using QIIME (1.8.0).22 Briefly, the paired-end reads from the fastq files were joined, barcodes extracted and then demultiplexed. The operational taxonomic units (OTUs) were determined using a closed reference approach by aligning reads to the Greengenes Database (version 13_8) at 99% identity. The Greengenes phylogenetic tree was used to define the phylogenetic relationship between OTUs. The resultant OTU tables contained the relative abundance of bacterial taxa in each sample. These tables were used to calculate overall diversity within each sample. To compare the composition and diversity of samples to each other taking into account phylogenetic relatedness, the OTU tables were used to calculate the weighted UniFrac distance matrix, which was summarized with Principal Coordinate Analysis. Permanova analysis was performed using Qiime.
Total levels of bacteria per cecal tip sample were measured by qPCR using eubacterial primers (Table S2). qPCR reactions were conducted using SYBR Green PCR Master Mix (Applied Biosystems 4364346) and a LightCycler 480 II (Roche) instrument. Normalized abundance of bacterial genera was calculated by multiplying the fraction of total reads for a genus by the total level of bacteria per mg of cecum sample (in arbitrary units).
Histology
Cecum tissue was dissected from mice sacrificed prior to or two days post-inoculation with C. difficile. Tissue was fixed in buffered formalin and processed for staining with Hematoxylin and Eosin (H & E) by the Tufts Animal Histology Core facility (http://sites.tufts.edu/histopath/animal-histology-core/).
C. difficile toxin assay
Cecum contents from mice sacrificed one or two days post-inoculation with C. difficile or fecal pellets collected one day post-inoculation were weighed and diluted with 10 times the volume of PBS. Serial three-fold dilutions were made in DMEM (Corning Cell Gro MT10-013CV) and samples were applied to monolayers of mouse embryonic fibroblast cells (MEF) cells grown in DMEM with 10% heat-inactivated Fetal Bovine Serum (Atlanta Biologicals S11150). Cells were incubated for 24 hours at 37°C and 5% CO2 and scored visually at 10X magnification for cell rounding. Toxin titer is defined as the inverse of the greatest dilution that resulted in 100% cell rounding.
Cytokine gene expression
For measurement of cytokine gene expression, mice were sacrificed prior to or two days post-inoculation with C. difficile and colon tissue was frozen in RNALater (Invitrogen AM7021) at -80°C. RNA was purified with Trizol (Invitrogen 15596026) extraction and column purification, using the Ambion Purelink RNA mini kit (Invitrogen 12183018A). DNA was eliminated with on-column DNase treatment. cDNA preparation with Superscript III (Invitrogen 18080051) was performed using manufacturer's protocol. qRT-PCR reactions were performed as described above for qPCR. Triplicate samples were measured; controls lacking template did not yield products. Standard curves were generated and all results normalized to the level of GAPDH expression in each sample. Primers are listed in Table S2.
Supplementary Material
Funding Statement
HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) HHS | NIH | National Institute of Allergy and Infectious DisPlease note that the Funding section(s) has/have been created from information provided through CATS. Please correct if this is inaccurate.eases (NIAID) Information LM and LS were supported by NIH training grant T32AI07422. CAK was supported in part by NIH NIAID R01 AI081794 and R01 AI118898 (to Carol A. Kumamoto). ERG and JM were supported by NIH NIAID R01113166 (to Joan Mecsas) and KPL by NIH NIAID R01 AI101018 (to Katherine P. Lemon).
Disclosure statement
The authors have no conflicts of interest to declare. The funders of this research had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors gratefully acknowledge the generous contributions of Drs. Linc Sonenshein, Laurent Bouillaut and Shonna McBride, whose knowledge of C. difficile biology was critical to the success of this project. We also thank Anne Kane, Albert Tai and Max Isberg for assistance with measurements of microbiota composition and analysis of those results. We are grateful to Mihai Netea and Aimee Shen for discussion, Robin Ruthazer for statistical analysis and Verna Manni for critical insight at an early stage in this project.
References
- 1.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, et al.. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825–34. doi: 10.1056/NEJMoa1408913. PMID:25714160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schenck L P, Beck P L, MacDonald J A. Gastrointestinal dysbiosis and the use of fecal microbial transplantation in Clostridium difficile infection. World J Gastrointest Pathophysiol. 2015;6:169–80. doi: 10.4291/wjgp.v6.i4.169. PMID:26600975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon K. The host immune response to Clostridium difficile infection. Ther Adv Infect Dis. 2013;1:19–35. doi: 10.1177/2049936112472173. PMID:25165542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shields K, Araujo-Castillo RV, Theethira TG, Alonso CD, Kelly CP. Recurrent Clostridium difficile infection: From colonization to cure. Anaerobe. 2015;34:59–73. doi: 10.1016/j.anaerobe.2015.04.012. PMID:25930686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al.. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–15. doi: 10.1056/NEJMoa1205037. PMID:23323867. [DOI] [PubMed] [Google Scholar]
- 6.Youngster I, Russell GH, Pindar C, Ziv-Baran T, Sauk J, Hohmann EL. Oral, capsulized, frozen fecal microbiota transplantation for relapsing Clostridium difficile infection. JAMA. 2014;312:1772–8. doi: 10.1001/jama.2014.13875. PMID:25322359. [DOI] [PubMed] [Google Scholar]
- 7.Kassam Z, Lee CH, Yuan Y, Hunt RH. Fecal microbiota transplantation for Clostridium difficile infection: systematic review and meta-analysis. Am J Gastroenterol. 2013;108:500–8. doi: 10.1038/ajg.2013.59. PMID:23511459. [DOI] [PubMed] [Google Scholar]
- 8.Li YT, Cai HF, Wang ZH, Xu J, Fang JY. Systematic review with meta-analysis: long-term outcomes of faecal microbiota transplantation for Clostridium difficile infection. Aliment Pharmacol Ther. 2016;43:445–57. doi: 10.1111/apt.13492. [DOI] [PubMed] [Google Scholar]
- 9.De Leon LM, Watson JB, Kelly CR. Transient flare of ulcerative colitis after fecal microbiota transplantation for recurrent Clostridium difficile infection. Clin Gastroenterol Hepatol. 2013;11:1036–8. doi: 10.1016/j.cgh.2013.04.045. PMID:23669309. [DOI] [PubMed] [Google Scholar]
- 10.Alang N, Kelly CR. Weight gain after fecal microbiota transplantation. Open Forum Infect Dis. 2015;2:ofv004. doi: 10.1093/ofid/ofv004. PMID:26034755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erb Downward JR, Falkowski NR, Mason KL, Muraglia R, Huffnagle GB. Modulation of post-antibiotic bacterial community reassembly and host response by Candida albicans. Sci Rep. 2013;3:2191. doi: 10.1038/srep02191. PMID:23846617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Luca A, Montagnoli C, Zelante T, Bonifazi P, Bozza S, Moretti S, D'Angelo C, Vacca C, Boon L, Bistoni F, et al.. Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J Immunol. 2007;179:5999–6008. doi: 10.4049/jimmunol.179.9.5999. PMID:17947673. [DOI] [PubMed] [Google Scholar]
- 13.Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, Belladonna ML, Vacca C, Conte C, Mosci P, et al.. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol. 2007;37:2695–706. doi: 10.1002/eji.200737409. PMID:17899546. [DOI] [PubMed] [Google Scholar]
- 14.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D'Angelo C, Massi-Benedetti C, Fallarino F, et al.. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–85. doi: 10.1016/j.immuni.2013.08.003. PMID:23973224. [DOI] [PubMed] [Google Scholar]
- 15.Carvalho A, Giovannini G, De Luca A, D'Angelo C, Casagrande A, Iannitti R G, Ricci G, Cunha C, Romani L. Dectin-1 isoforms contribute to distinct Th1/Th17 cell activation in mucosal candidiasis. Cell Mol Immunol. 2012;9:276–86. doi: 10.1038/cmi.2012.1. PMID:22543832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Luca A, Zelante T, D'Angelo C, Zagarella S, Fallarino F, Spreca A, Iannitti RG, Bonifazi P, Renauld J C, Bistoni F, et al.. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 2010;3:361–73. doi: 10.1038/mi.2010.22. PMID:20445503. [DOI] [PubMed] [Google Scholar]
- 17.Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, et al.. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–32. doi: 10.1016/j.chom.2012.06.006. PMID:22901542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reeves A E, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2:145–58. doi: 10.4161/gmic.2.3.16333. PMID:21804357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorg J A, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol. 2010;192:4983–90. doi: 10.1128/JB.00610-10. PMID:20675492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen A. Clostridium difficile toxins: mediators of inflammation. J Innate Immun. 2012;4:149–58. doi: 10.1159/000332946. PMID:22237401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol. 2008;8:192. doi: 10.1186/1471-2180-8-192. PMID:18990232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. PMID:20383131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schubert A M, Sinani H, Schloss PD. Antibiotic-Induced Alterations of the Murine Gut Microbiota and Subsequent Effects on Colonization Resistance against Clostridium difficile. MBio. 2015;6:e00974. doi: 10.1128/mBio.00974-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schubert AM, Rogers MA, Ring C, Mogle J, Petrosino JP, Young VB, Aronoff DM, Schloss PD. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio. 2014;5:e01021–01014. doi: 10.1128/mBio.01021-14. PMID:24803517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahinas D, Silverman M, Sittler T, Chiu C, Kim P, Allen-Vercoe E, Weese S, Wong A, Low DE, Pillai DR. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. MBio. 2012;3:e00338–12. doi: 10.1128/mBio.00338-12. PMID:23093385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al.. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–8. doi: 10.1038/nature13828. PMID:25337874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun. 2012;80:3786–94. doi: 10.1128/IAI.00647-12. PMID:22890996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manian FA, Bryant A. Does Candida species overgrowth protect against Clostridium difficile infection?. Clin Infect Dis. 2013;56:464–5. doi: 10.1093/cid/cis854. PMID:23042967. [DOI] [PubMed] [Google Scholar]
- 29.Nerandzic MM, Mullane K, Miller MA, Babakhani F, Donskey CJ. Reduced acquisition and overgrowth of vancomycin-resistant enterococci and Candida species in patients treated with fidaxomicin versus vancomycin for Clostridium difficile infection. Clin Infect Dis. 2012;55(2):S121–126. doi: 10.1093/cid/cis440. PMID:22752860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blanco N, Walk S, Malani AN, Rickard A, Benn M, Eisenberg M, Zhang M, Foxman B. Clostridium difficile shows no trade-off between toxin and spore production within the human host. J Med Microbiol. 2018;67:631–640. doi: 10.1099/jmm.0.000719. PMID:29533173. [DOI] [PubMed] [Google Scholar]
- 31.Kumamoto CA. Inflammation and gastrointestinal Candida colonization. Curr Opin Microbiol. 2011;14:386–91. doi: 10.1016/j.mib.2011.07.015. PMID:21802979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu MS, Wang JT, Huang WK, Liu YC, Chang SC. Prevalence and clinical features of Clostridium difficile-associated diarrhea in a tertiary hospital in northern Taiwan. J Microbiol Immunol Infect. 2006;39:242–8. PMID:24889895 [PubMed] [Google Scholar]
- 33.McFarland LV. Deciphering meta-analytic results: a mini-review of probiotics for the prevention of paediatric antibiotic-associated diarrhoea and Clostridium difficile infections. Benef Microbes. 2015;6:189–94. doi: 10.3920/BM2014.0034. PMID:24889895. [DOI] [PubMed] [Google Scholar]
- 34.Johnson S, Maziade PJ, McFarland LV, Trick W, Donskey C, Currie B, Low DE, Goldstein EJ. Is primary prevention of Clostridium difficile infection possible with specific probiotics?. Int J Infect Dis. 2012;16:e786–792. doi: 10.1016/j.ijid.2012.06.005. PMID:22863358. [DOI] [PubMed] [Google Scholar]
- 35.O'Horo JC, Jindai K, Kunzer B, Safdar N. Treatment of recurrent Clostridium difficile infection: a systematic review. Infection. 2014;42:43–59. doi: 10.1007/s15010-013-0496-x. PMID:23839210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raponi G, Visconti V, Brunetti G, Ghezzi M C. Clostridium difficile infection and Candida colonization of the gut: is there a correlation?. Clin Infect Dis. 2014;59:1648–9. doi: 10.1093/cid/ciu637. PMID:25091308. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa T, Mori N, Kajiwara C, Kimura S, Akasaka Y, Ishii Y, Saji T, Tateda K. Endogenous IL-17 as a factor determining the severity of Clostridium difficile infection in mice. J Med Microbiol. 2016;65:821–7. doi: 10.1099/jmm.0.000273. PMID:27166143. [DOI] [PubMed] [Google Scholar]
- 38.Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz-Planillo R, Do N, Nunez G, Inohara N. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity. 2014;41:620–32. doi: 10.1016/j.immuni.2014.09.010. PMID:25367575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Cao J, Li C, Zhang L. IL-27/IL-27receptor signaling provides protection in Clostridium difficile-induced colitis. J Infect Dis. 2018;217:198–207. doi: 10.1093/infdis/jix581. [DOI] [PubMed] [Google Scholar]
- 40.Buonomo EL, Cowardin CA, Wilson MG, Saleh MM, Pramoonjago P, Petri WA Jr.. Microbiota-Regulated IL-25 Increases Eosinophil Number to Provide Protection during Clostridium difficile Infection. Cell Rep. 2016;16:432–43. doi: 10.1016/j.celrep.2016.06.007. PMID:27346351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubino SJ, Geddes K, Girardin SE. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends Immunol. 2012;33:112–8. doi: 10.1016/j.it.2012.01.003. PMID:22342740. [DOI] [PubMed] [Google Scholar]
- 42.Lee JS, Tato CM, Joyce-Shaikh B, Gulan F, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, et al.. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity. 2015;43:727–38. doi: 10.1016/j.immuni.2015.09.003. PMID:26431948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caplan MS, Miller-Catchpole R, Kaup S, Russell T, Lickerman M, Amer M, Xiao Y, Thomson R Jr. Bifidobacterial supplementation reduces the incidence of necrotizing enterocolitis in a neonatal rat model. Gastroenterology. 1999;117:577–83. doi: 10.1016/S0016-5085(99)70450-6. PMID:10464133. [DOI] [PubMed] [Google Scholar]
- 44.Fanning S, Hall LJ, Cronin M, Zomer A, MacSharry J, Goulding D, Motherway MO, Shanahan F, Nally K, Dougan G, et al.. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc Natl Acad Sci U S A. 2012;109:2108–13. doi: 10.1073/pnas.1115621109. PMID:22308390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Hara A M, O'Regan P, Fanning A, O'Mahony C, Macsharry J, Lyons A, Bienenstock J, O'Mahony L, Shanahan F. Functional modulation of human intestinal epithelial cell responses by Bifidobacterium infantis and Lactobacillus salivarius. Immunology. 2006;118:202–15. doi: 10.1111/j.1365-2567.2006.02358.x. PMID:16771855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Underwood MA, Arriola J, Gerber CW, Kaveti A, Kalanetra KM, Kananurak A, Bevins CL, Mills DA, Dvorak B. Bifidobacterium longum subsp. infantis in experimental necrotizing enterocolitis: alterations in inflammation, innate immune response, and the microbiota. Pediatr Res. 2014;76:326–33. doi: 10.1038/pr.2014.102. PMID:25000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Zhang W, Zuo L, Zhu W, Wang B, Li Q, Li J. Bifidobacteria may be beneficial to intestinal microbiota and reduction of bacterial translocation in mice following ischaemia and reperfusion injury. Br J Nutr. 2013;109:1990–8. doi: 10.1017/S0007114512004308. PMID:23122253. [DOI] [PubMed] [Google Scholar]
- 48.Derrien M, Vaughan E E, Plugge C M, de Vos W M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–76. doi: 10.1099/ijs.0.02873-0. PMID:15388697. [DOI] [PubMed] [Google Scholar]
- 49.Kang C S, Ban M, Choi EJ, Moon HG, Jeon JS, Kim DK, Park SK, Jeon SG, Roh TY, Myung SJ, et al.. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS One. 2013;8:e76520. doi: 10.1371/journal.pone.0076520. PMID:24204633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reunanen J, Kainulainen V, Huuskonen L, Ottman N, Belzer C, Huhtinen H, de Vos W M, Satokari R. Akkermansia muciniphila Adheres to Enterocytes and Strengthens the Integrity of the Epithelial Cell Layer. Appl Environ Microbiol. 2015;81:3655–62. doi: 10.1128/AEM.04050-14. PMID:25795669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derrien M, Van Baarlen P, Hooiveld G, Norin E, Muller M, de Vos W M. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front Microbiol. 2011;2:166. doi: 10.3389/fmicb.2011.00166. PMID:21904534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Leeuwen PT, van der Peet JM, Bikker FJ, Hoogenkamp M, Oliveira Paiva AM, Kostidis S, Mayboroda OA, Smits WK, Krom BP. Interspecies Interactions between Clostridium difficile and Candida albicans. mSphere. 2016;1:e00187–00116. doi: 10.1128/mSphere.00187-16. PMID:27840850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown DH Jr., Giusani AD, Chen X, Kumamoto CA. Filamentous growth of Candida albicans in response to physical environmental cues and its regulation by the unique CZF1 gene. Mol Microbiol. 1999;34:651–62. doi: 10.1046/j.1365-2958.1999.01619.x. PMID:10564506. [DOI] [PubMed] [Google Scholar]
- 54.Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-V. PMID:2005794. [DOI] [PubMed] [Google Scholar]
- 55.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–12. doi: 10.1128/JB.01765-07. PMID:18245298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson KH, Kennedy MJ, Fekety FR. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J Clin Microbiol. 1982;15:443–6. PMID:22402401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al.. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. PMID:22402401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.