

ABSTRACT
Background: Although programmed death-1 (PD-1) blockade is effective in treating several types of cancer, the efficacy of this agent in glioblastoma (GBM) is largely unknown.
Methods: We evaluated therapeutic effects of anti-PD-1, temozolomide (TMZ), and their combination in an orthotopic murine GBM model. The phenotype, number, and composition of lymphocytes were evaluated using flow cytometry. Transcriptional profiles of tumor tissues were analyzed using microarrays. Generation of antitumor immunological memory was investigated upon rechallenge.
Results: Combined treatment with anti-PD-1 and TMZ yielded synergistic antitumor efficacy in the presence of donor-originated PD-1+CD8+ T cells in vitro, necessitating in vivo validation. Whereas TMZ did not rescue GBM-implanted mice, anti-PD-1 completely eradicated GBM in 44.4% of mice, and combination of anti-PD-1 and TMZ in all mice. Anti-PD-1 significantly increased the number of tumor-infiltrating lymhpocytes (TILs), and reduced frequencies of exhausted T cells and regulatory T cells. However, combining TMZ with anti-PD-1 significantly decreased the number of TILs, which was also observed with TMZ treatment alone. A transcriptome analysis of tumor tissues revealed that anti-PD-1 monotherapy perturbed immune-related genes, distinctly with combined therapy. Upon rechallenge, tumor growth was not observed in mice cured by anti-PD-1 monotherapy, whereas tumors regrew in the combination group. Furthermore, an analysis of peripheral blood revealed that antitumor memory T cells were generated in mice cured by anti-PD-1 monotherapy, not in the combination group.
Conclusion: PD-1 blockade induces long-term therapeutic response, and combination with TMZ further enhances antitumor efficacy. However, immunological memory is provoked by anti-PD-1 monotherapy, not by combined therapy.
Keywords: checkpoint inhibitor, combination therapy, glioblastoma, PD-1, temozolomide
Introduction
Glioblastoma (GBM) is the most common and aggressive malignancy of the central nervous system (CNS).1 Despite multimodal interventions, GBM is associated with a dismal prognosis and a high recurrence rate, which necessitate the development of novel therapeutic approaches.2,3 However, recent studies targeting angiogenic pathways4 and cell-matrix interactions5 have failed to improve patient outcomes.
The CNS has traditionally been considered as an immune-privileged organ. However, growing evidence suggests the existence of interactions between the host immune system and the CNS.6 In addition, several studies have demonstrated immunosuppressive microenvironment in human GBM,7,8 leading to the expectation that the patient outcomes could be improved by immunotherapy. Immune checkpoint blockers have garnered significant recent attention with encouraging clinical data observed in various malignancies including malignant melanoma,9 non-small-cell lung cancer,10 and Hodgkin’s lymphoma.11 Likewise, preclinical investigations on the anti-glioma effects of immune checkpoint blockade have shown promising results,12 providing a rationale for incorporation of immunotherapy in the treatment of GBM.
Blocking the interactions between programmed death-1 (PD-1) and its ligand PD-L1, a crucial regulator of T cell exhaustion, reinvigorates exhausted T cells and reduces tumor burden.13 To further improve the efficacy of anti-PD-1 or anti-PD-L1 blocking antibodies, combinational strategies with other immunomodulatory agents,14 chemotherapy,15 and radiotherapy16 have been investigated in GBM. However, they are clinically used without precise mechanisms due to urgent clinical needs, and few studies have provided insight into the combinatorial effects of immunotherapy and temozolomide (TMZ), the most widely used chemotherapeutic alkylating agent in GBM.
In this study, we investigated the antitumor efficacy, immune response, and immunological memory induced by anti-PD-1 alone and in combination with TMZ in GBM. Our findings elucidate the combinational effects and working mechanisms of immunotherapy and chemotherapy, helping to guide the development of effective therapeutic strategies against GBM. As this study suggests a plausible answer for optimized use of anti-PD-1, which is very expensive for long-term use, we believe that our data suggest the possibility of adding immunotherapy to standard GBM treatment modalities minimizing overdose of antibodies.
Results
Anti-PD-1 alone and in combination with TMZ exerts antitumor effects in human GBM co-culture system and orthotopic murine GBM model
To test antitumor effects of anti-PD-1 and/or TMZ, in vitro co-culture system of PD-1+CD8+ T cells and human GBM cells were established. In this assay, HLA-A*0201 (+) U87MG cells with MGMT promoter methylation and HLA-A*0201 (+) T98G cells without MGMT promoter methylation were used as target cells. In the presence of HLA-A*0201(+) donor-originated PD-1+CD8+ T cells (Supplementary Fig. S1A and B), anti-PD-1 blocking antibodies induced cell death more efficiently in both GBM cell lines (Supplementary Fig. S1C and D). Compared to anti-PD-1 or TMZ treatment alone, combined treatment with anti-PD-1 and TMZ induced higher levels of cell death in both GBM cell lines. These findings prompted us to conduct further in vivo experiments.
In vivo therapeutic effects of anti-PD-1 and/or TMZ were evaluated in a murine GBM model using luciferase-transfected GL261 cells (GL261-luc). Experimental schedule (15-wk course) was designed as follows (Figure 1(a)): One week after tumor implantation in the right hemisphere, engraftment was confirmed using in vivo imaging system (IVIS). TMZ was administered once daily for five days. One day after completion of TMZ administration (day 13), anti-PD-1 antibodies were administered twice a week. Treatment was maintained until week 15, and IVIS images were obtained every two weeks (Supplementary Fig. S2). Bioluminescence imaging revealed that all treatment regimens exhibited antitumor efficacy compared with untreated controls (Figure 1(b,c)). Consistently, prolonged survival was demonstrated in all treatment groups (Figure 1(d)). Notably, bioluminescent imaging analysis showed complete responses in 44.4% of subjects (4/9) treated with anti-PD-1 monotherapy and in all subjects (9/9) treated with the combination of anti-PD-1 and TMZ (Figure 1(b)), indicating a synergistic effect of the combination.
Figure 1.
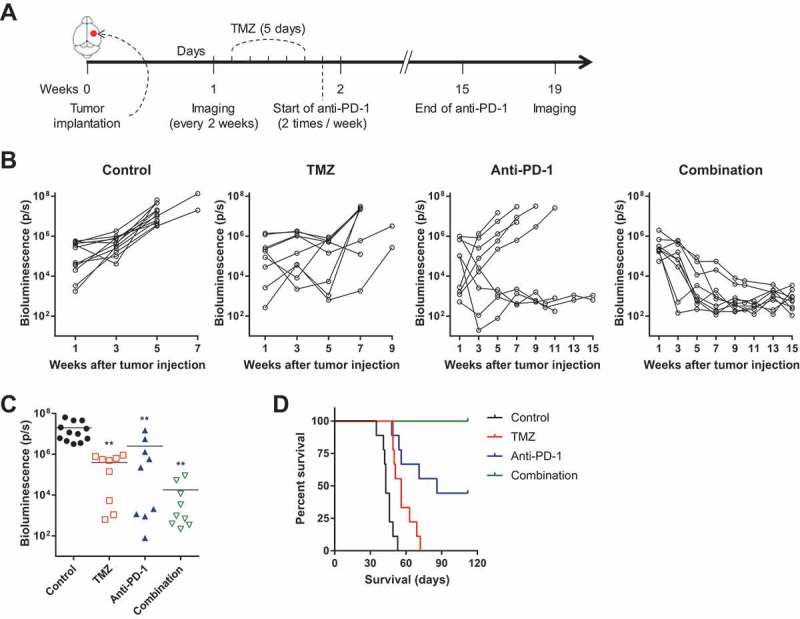
In vivo antitumor efficacy of anti-PD-1 and/or TMZ.
In vivo therapeutic effects of anti-PD-1 and/or TMZ on tumor growth were tested in a mouse orthotopic model using GL261-luc cells (15-wk course). (A) Experimental schedule; applies to Figure 1 and Supplementary Fig. S2. (B) Tumor volume (signal intensity of bioluminescence images) for each mouse is depicted as spider plots. Signal intensity was quantified as total luminescence flux (photon/s). (C) Differences in tumor volume among treatment groups at 5 wk were compared by one-way ANOVA with Tukey’s post hoc test for multiple comparisons (**P < 0.01, compared to control). (D) Survival probability for each group was estimated based on Kaplan-Meier curves. Log-rank test was performed to calculate statistical significance (P < 0.001).
Anti-PD-1 reduces the relative frequency of tumor-infiltrating t cells with exhausted phenotypes
To examine immunological responses, we sacrificed tumor-implanted mice early after treatment (Figure 2(a)) and confirmed no differences in tumor volume among groups (Supplementary Fig. S3A and B). Prior to the main flow cytometry analysis, we found that therapeutically administered anti-PD-1 (clone RMP1-14) was bound to tumor-infiltrating lymphocytes (TILs) across blood-brain barrier (Supplementary Fig. S4A). No interference between therapeutic antibodies and detection antibodies (clone 29F.1A12) was observed, enabling further analyses (Supplementary Fig. S4B).
Figure 2.

Effects of anti-PD-1 and/or TMZ on the frequency of tumor-infiltrating CD8+ and CD4+ T cells with exhausted phenotypes early after the treatment.
Effects of anti-PD-1 and/or TMZ on immune microenvironement were assessed in a mouse orthotopic model using GL261-luc cells (16-day course). (A) Experimental schedule; applies to Figures 2–5 and Supplementary Fig. S3-6 (16-day course). (B) Representative flow cytometry data for tumor-infiltrating CD8+ and CD4+ T cells. X-axis indicates PD-1 expression; Y-axis indicates LAG-3 expression. (C and D) The relative frequency of PD-1+ (left) or LAG-3+ (right) cells among tumor-infiltrating CD8+ (C) and CD4+ (D) T cells. Differences among groups were compared by one-way ANOVA with Tukey’s post hoc test for multiple comparisons; means ± SD; *P < 0.05, **P < 0.01, ***P < 0.001, where asterisks over treatment groups denote significant differences compared with controls.
Among tumor-infiltrating CD8+ T cells, the relative frequency of PD-1+ cells was significantly decreased by anti-PD-1 monotherapy and combined treatment, but not by TMZ monotherapy. We also examined the expression of lymphocyte-activation gene 3 (LAG-3), another marker of T cell exhaustion.17 The relative frequency of LAG-3+ cells among tumor-infiltrating CD8+ T cells showed similar patterns with those of PD-1+ cells (Figure 2(b,c)). The frequency of PD-1+ or LAG-3+ cells among tumor-infiltrating CD4+ T cells also showed similar results (Figure 2(b,d)). Moreover, anti-PD-1 increased the frequency of interferon-γ- (IFN-γ) or tumor necrosis factor- (TNF) producing TILs in intracellular cytokine staining assays, whereas addition of TMZ decreased their frequency (Supplementary Fig. S5A-C). There were no differences in the frequency of PD-1+ cells among splenic CD8+ or CD4+ T cells (Supplementary Fig. S6A). LAG-3+ cells were not detected among splenic CD8+ or CD4+ T cells (data not shown). These data indicate that anti-PD-1 treatment reduces the frequency of exhausted T cells in the tumor microenvironment.
Anti-PD-1 monotherapy, but not combined treatment, increases the number of tils
Next, we examined the number of TILs early after treatment. Anti-PD-1 monotherapy increased the numbers of total CD3+, CD8+, CD4+, γδ T cells, and natural killer (NK) cells in the tumor compared with untreated controls, whereas TMZ treatment decreased them (Figure 3(a)). Intriguingly, addition of TMZ to anti-PD-1 reduced the number of TILs compared with anti-PD-1 alone without any effects on the composition of T cells (Figure 3(b)). The relative frequency and composition of T cells and NK cells in the spleen were not changed by anti-PD-1 and/or TMZ treatment (Supplementary Fig. S6B and C), although systemic lymphodepletion was observed in TMZ-treated mice (Figure 3(c,d)). These results indicate that anti-PD-1 results in immune-favorable microenvironment by increasing the number of TILs; however, addition of TMZ to anti-PD-1 therapy abolishes these favorable effects of anti-PD-1.
Figure 3.

Effects of anti-PD-1 and/or TMZ on the number and composition of lymphocytes from tumor microenvironment and peripheral blood.
(A) The numbers of immune cells among TIL subsets (CD3+, CD8+, CD4+, γδ T cells, and NK cells). (B) The ratios of immune cell subsets (CD8+, CD4+, and γδ T cells) to tumor-infiltrating CD3+ T cells. (C) The numbers of immune cells in 1 μL of peripheral blood (CD3+, CD8+, CD4+, γδ T cells, and NK cells). (D) The ratios of immune cell subsets (CD8+, CD4+, and γδ T cells) to peripheral CD3+ T cells. Differences among groups were compared by one-way ANOVA with Tukey’s post hoc test for multiple comparisons; means ± SD; **P < 0.01, ***P < 0.001, where asterisks over treatment groups denote significant differences compared with controls.
Anti-PD-1 reduces relative frequency and function of tumor-infiltrating regulatory t cells
We also investigated the relative frequency and phenotypes of tumor-infiltrating CD4+CD25+Foxp3+ regulatory T (Treg) cells. Anti-PD-1 monotherapy significantly decreased the frequencies of Treg cells among tumor-infiltrating CD4+ T cells, whereas TMZ increased them (Figure 4(a)). Addition of TMZ to anti-PD-1 partially abrogated the effect of anti-PD-1 on Treg cell frequency. The expression levels of CD39 and cytotoxic T-lymphocyte associated protein 4 (CTLA-4), other markers of Treg cell function,18,19 were also influenced by the treatments. The relative frequencies of CD39+ or CTLA-4+ Treg cells were significantly reduced by anti-PD-1 monotherapy and combination with TMZ, but not by TMZ monotherapy (Figure 4(b,c)). In contrast to its effect on the tumor microenvironment, TMZ did not affect the relative frequency or function of splenic Treg cells (Supplementary Fig. S6D and E). Taken together, TMZ monotherapy results in immune-suppressive tumor microenvironment by increasing the relative frequency of Treg cells, whereas anti-PD-1 treatment induces immune-favorable microenvironment by decreasing relative frequency and function of Treg cells.
Figure 4.

Effects of anti-PD-1 and/or TMZ on the frequency and function of tumor-infiltrating CD4+CD25+FoxP3+ Treg cells.
(A) The ratio of tumor-infiltrating Treg cells to total tumor-infiltrating CD4+ T cells. (B) Representative flow cytometry analysis of tumor-infiltrating Treg cells. X-axis indicates CTLA-4 expression; Y-axis indicates CD39 expression. (C) The relative frequency of CD39+ (left) or CTLA-4+ (right) cells among tumor-infiltrating Treg cells. Differences among groups were compared by one-way ANOVA with Tukey’s post hoc test for multiple comparisons; means ± SD; **P < 0.01, ***P < 0.001, where asterisks over treatment groups denote significant differences compared with controls.
Transcriptional profiles following anti-PD-1 and/or TMZ treatment
We next used microarrays to examine the ability of anti-PD-1 and TMZ to reprogram transcriptional profiles of tumor tissues. Hierarchical clustering of whole differentially expressed genes (DEGs) showed strong intragroup clustering and distinct expression patterns compared with controls (Supplementary Data S1 and Figure 5(a)). Among 1,952 DEGs, 289 and 547 genes were uniquely perturbed by TMZ and anti-PD-1 monotherapy, respectively, and a total of 928 genes were altered by combined treatment (Figure 5(c)). Functional annotation of these DEGs using the Gene Ontology (GO) database revealed distinct enriched gene sets across the treatment groups. Most immune response-related gene sets were exclusively enriched by anti-PD-1 monotherapy, whereas combined therapy affected other groups of gene sets, including cell cycle, proliferation, death (Supplementary Data S2 and Figure 5(c)). Similar results were reproduced in enrichment maps using GO hierarchy, in that DEGs by anti-PD-1 monotherapy were enriched in immune-associated modules, whereas DEGs by combined therapy were enriched in cell cycle-related GO terms (Supplementary Fig. S7). Differential transcriptional reprogramming effects of each therapeutic regimen were recapitulated by functional annotation using ImmuneSigDB gene sets. Each DEG group showed mutually exclusive patterns in enrichment score (Supplementary Data S3 and Figure 5(d)), suggesting that anti-PD-1 monotherapy results in immune responses distinct from those caused by combination treatment.
Figure 5.

Changes in gene expression profiles induced by treatment with anti-PD-1 and/or TMZ.
(A) Microarray data were obtained from tumor masses of each treatment group; expression levels of DEGs are displayed as a heat map. Differences among groups were compared by one-way ANOVA with Tukey’s post hoc test for multiple comparisons; DEGs were defined as those with P < 0.05. Hierarchical clustering results are depicted as a dendrogram. (B) The numbers of DEGs in each group are presented as a Venn diagram. (C and D) Functional annotation of DEGs was performed by ORA using gene sets obtained from GO (C) and ImmuneSigDB (D). Statistical significance was determined using Fisher’s exact test; enrichment scores are presented as a heat map.
Anti-PD-1 monotherapy, but not combined treatment, generates antitumor immunological memory
An in vivo evaluation of antitumor efficacy showed that anti-PD-1 monotherapy and combined treatment resulted in complete remission of GBM in 44.4% and 100% of mice, respectively. We confirmed the absence of tumors on IVIS images at week 19 (Supplementary Fig. S2), and tested whether cured mice acquired immunological memory against the tumor. To this end, we rechallenged these mice by implanting GL261-luc cells in the contralateral hemisphere at week 22 (Figure 6(a)), and monitored tumor formation without any further treatment. Interestingly, no tumor growth was observed in mice cured by anti-PD-1 monotherapy, whereas all mice cured by combined treatment developed progressively growing tumors (Figure 6(b,c)), similar to naïve mice challenged with GL261-luc cells. These results were confirmed by survival analysis (Figure 6(d)). To determine the presence of memory CD8+ or CD4+ T cells with antitumor immunity, ex vivo co-culture experiments of peripheral blood mononuclear cells and GL261 cells were performed in cured mice before rechallenge. IFN-γ- or TNF-producing CD8+ or CD4+ T cells were detected in mice cured by anti-PD-1 monotherapy (Figure 6(e)) and these cells largely belonged to effector/memory phenotypes (Figure 6(f)). In contrast, memory T cells were totally absent in mice cured by combined treatment, similar to mice never implanted GL261 (Figure 6(e)). To dissect the underlying mechanisms, we compared DEGs between anti-PD-1 and combination groups (Supplementary Data S4 and Figure 6(g)). Consistent with Figure 5(d), 145 genes upregulated in the anti-PD-1 group and 84 genes upregulated in the combination group showed an almost exclusive pattern of enrichment score calculated by over-representation analysis (ORA) using ImmuneSigDB gene sets. Notably, genes upregulated in memory T cells were enriched in the anti-PD-1 group, whereas downregulated genes were enriched in the combination group (Supplementary Data S5 and Figure 6(h)). These data indicate that anti-PD-1 monotherapy results in antitumor immunological memory following successful eradication of the tumor, preventing tumor relapse. However, combined treatment did not induce antitumor immunological memory, even after successful initial eradication of the tumor.
Figure 6.
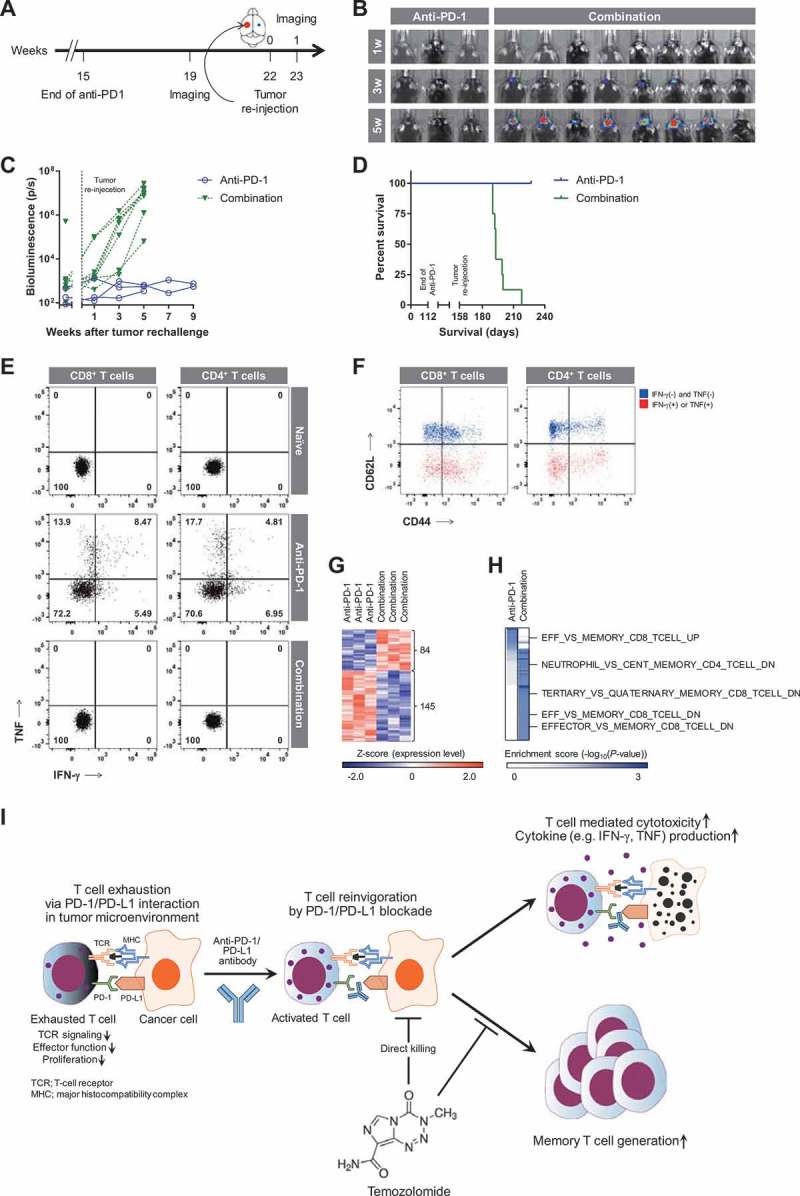
Evaluation of tumorigenesis after tumor rechallenge.
Mice in which tumors were completely eradicated were rechallenged. (A) Experimental schedule after initial tumor implantation (continues from Figure 1(a)). (B and C) Tumor volume was measured by bioluminescence imaging (B), and depicted as a spider plot (C). (D) Survival probability of rechallenged mice was estimated based on Kaplan-Meier curves. Log-rank test was performed to calculate statistical significance (P < 0.05). (E) Representative flow cytometry data for the presence of IFN-γ- or TNF-producing memory CD8+ (left) and CD4+ (right) T cells with antitumor immunity. Effector cells (peripheral blood mononuclear cells) were co-cultured with target cells (GL261) at effector:target ratio of 1:10. X-axis indicates IFN-γ production; Y-axis indicates TNF production. (F) Representative flow cytometry data for the phenotypes of CD8+ (left) and CD4+ (right) T cells in mice cured by anti-PD-1 monotherapy. Red dots indicate IFN-γ- or TNF-producing T cells; Blue dots indicates IFN-γ- and TNF-negative T cells; X-axis indicates CD44 expression; Y-axis indicates CD62L expression. (G and H) Expression levels of DEGs (84 upregulated and 145 downregulated genes in the combination group) between anti-PD-1 and combination groups are depicted as a heat map (G). Functional annotation of these genes was performed by ORA using ImmuneSigDB gene sets (H). Statistical significance was determined using Fisher’s exact test; enrichment scores are presented as a heat map. (I) Summary of action mechanisms of anti-PD-1 and/or TMZ in treatment of GBM.
Discussion
Despite recent progress in understanding the pathogenesis of GBM, currently available treatment options are largely limited. Here, we report that the combination of anti-PD-1 and TMZ showed better antitumor efficacy than anti-PD-1 or TMZ alone. However, favorable immunological effects of anti-PD-1 therapy, such as increases in the number of TILs and decreases in the frequency of Treg cells, were abrogated by addition of TMZ. Importantly, the combination of anti-PD-1 and TMZ did not result in antitumor immunological memory, which was only observed with anti-PD-1 monotherapy.
One major limitation of anti-PD-1 monotherapy in GBM is relatively low response rates in preliminary results of prospective trials (NCT0205480, NCT02017717). To enhance therapeutic effects of anti-PD-1, combinations of other therapeutic agents with anti-PD-1 have been actively investigated in preclinical or clinical setting, including mitochondrial uncouplers,20 antiangiogenic agents,21 and oncogenic PI3K/Akt and MAPK signaling pathway inhibitors.22,23 In this study, we employed TMZ as a combination partner of anti-PD-1 based on its widespread use against GBM and manageable toxicities. Like other chemotherapeutic agents, TMZ induces lymphopenia and myelosuppression.24 Consistent with this, our study demonstrated that TMZ resulted in intratumoral and systemic lymphodepletion. In addition, TMZ increased the frequency of Treg cells among TILs, causing more immunosuppressive tumor microenvironment.
Given that frequent relapse is one of the major challenges in improving outcomes in GBM patients, we also tested how different treatment modalities impacted recurrence upon rechallenge. Intriguingly, mice in which tumors were completely eradicated by anti-PD-1 monotherapy showed no tumor formation following rechallenge, whereas mice previously cured by the combinatorial regimen succumbed to tumor rechallenge. This result suggests that antitumor immunological memory is established by PD-1 blockade, but diminished by TMZ combination. Anti-PD-1 monotherapy may promote long-term therapeutic responses through generation of immunological memory and prevention of recurrence, an issue of particular relevance to GBM.
In principle, reinvigoration of PD-1+ T cells by anti-PD-1 antibodies results in increased proliferation as well as enhancement of immediate effector functions, such as cytotoxicity and cytokine production. TMZ is a DNA-alkylating agent that suppresses the proliferation of rapidly dividing cells, e.g., tumor cells and reinvigorated T cells. Therefore, addition of TMZ to anti-PD-1 therapy might suppress the proliferation of reinvigorated T cells while preserving the enhanced immediate effector functions such as cytotoxicity. This might explain the poor generation of antitumor immunological memory after addition of TMZ to anti-PD-1 treatment, in contrast to the excellent primary efficacy of the combinatory strategy (Figure 6(i)). The distinct transcription profiles elicited by the combination of anti-PD-1 and TMZ compared with anti-PD-1 monotherapy provide evidence in support of this. Specifically, genes related to immunological memory were upregulated in the anti-PD-1 monotherapy group and downregulated in the combination group.
In conclusion, concurrent targeting of antitumor immune responses (by PD-1 blockade) and the tumor itself (by TMZ) exerted the greatest antitumor efficacy and survival improvement in a murine GBM model. However, inclusion of TMZ abrogated the favorable immune responses and antitumor immunological memory induced by anti-PD-1 monotherapy. When considering the use of combination regimens of chemo-immunotherapy for the treatment of cancer, the immediate antitumor efficacy is not the only assessment criterion; the generation of antitumor immunological memory, which prevents tumor recurrence long after initial successful treatment, is also important. It may be possible to achieve these two different goals by selecting appropriate chemotherapeutic agents and optimizing delivery methods and order of administration, thereby maximizing clinical outcomes in the treatment of GBM.
Materials and methods
Cell lines
GL261-luc cells were constructed using lentivirus-mediated transduction and puromycin-based selection, and cultured in tumorsphere complete media as previously described.25,26 U87MG and T98G cells were purchased from Korean Cell Line Bank and grown in media according to American Type Culture Collection recommendations. U87MG cells and T98G cells revealed methylated and unmethylated MGMT promoter region, respectively. Peripheral blood mononuclear cells obtained from HLA-A*0201(+) donors were used to generate cytomegalovirus (CMV) pp65-specific CD8+ T cell lines. Briefly, CD8+ T cells were isolated by CD8+ T Cell Isolation kit (Miltenyi Biotec), and CMV pp65495-503 (NLVPMVATV)-specific CD8+ T cells were enriched with HLA-A2-pp65495-503 (NLVPMVATV) pentamer (Proimmune Ltd.). Selected CMV pp65-specific CD8+ T cells were maintained in IL-2 (200 IU/mL; PeproTech), IL-7 (10 ng/mL; PeproTech), and IL-15 (100 ng/mL; PeproTech)-containing media. Anti-CD3 (Miltenyi Biotec) was used to expand or stimulate CD8+ T cell lines. Cell yield and viability were measured by dual-fluorescence method with acridine orange and propidium iodide (Nexcelom Bioscience).
Orthotopic, immunocompetent models of murine GBM
Male, 6–8 wk old C57BL/6 mice (Central Lab. Animal Inc.) were used in this study. Dissociated GL261-luc cells (2 × 105) were orthotopically implanted (day 0) as previously described.27,28 TMZ (Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO), and the anti-PD-1 therapeutic antibody (Bio X Cell) was diluted with phosphate-buffered saline (PBS). TMZ (30 mg/kg) was intraperitoneally administered to mice; anti-PD-1 (10 mg/kg) was intravenously injected. Bioluminescence image acquisition and analyses were performed using an IVIS imaging system and Living Image v4.2 software (Caliper Life Sciences).29,30 Mice were injected intraperitoneally with 100 μL D-luciferin (30 mg/mL, dissolved in PBS; Promega) 15 min prior to signal acquisition, conducted under 2.5% isoflurane anesthesia. Mice showing more than a 15% decrease in body weights compared with their maximum were euthanized according to the approved protocol. Cured mice were rechallenged; the same number of dissociated tumor cells was injected into the contralateral side of the brain. All experimental procedures were approved by the Yonsei University College of Medicine Institutional Animal Care and Use Committee under GLP principles.
Cytotoxicity assay
U87MG and T98G were treated with DMSO or TMZ (10 μM) for 3 days, labeled with PKH26 dye (Sigma-Aldrich), and pulsed with 10 μg/mL CMV pp65495-503 (NLVPMVATV; PeproTech). T cell-mediated cytotoxicity was evaluated by co-culture of U87MG or T98G and PD-1+ CMV pp65-specific CD8+ T cells (effector:target ratio 1:1) with isotype control (10 μg/mL; BioLegend) or anti-PD-1 antibody (10 μg/mL; BioLegend). After then, cells were labeled with TO-PRO-3 dye (Thermo Fisher Scientific) and TO-PRO-3+ cells in PKH26+ cells were considered as dead target cells.
Flow cytometry
Fresh peripheral blood, spleens, and tumors were obtained to examine immune cell population. Peripheral blood was obtained via retro-orbital bleeding. Spleen and brain were mechanically dissociated using gentleMACSTM Dissociator (Miltenyi Biotec). RBC lysis buffer (BioLegend) was applied to isolate lymphocytes from peripheral blood or spleen. Gradient separation was performed using Percoll solution (GE Healthcare Life Sciences) to isolate lymphocytes from brain. For cell-surface staining, single cell suspensions were incubated with LIVE/DEAD Fixable Red Dead Cell Stain kit or ethidium monoazide (Thermo Fisher Scientific) to exclude dead cells. Cells were then stained with fluorochrome-conjugated antibodies for 15 min at room temperature. For intracellular staining, cells were incubated with a fixation/permeabilization solution (Thermo Fisher Scientific) and stained with fluorochrome-conjugated antibodies. For intracellular cytokine staining, cells were stimulated in the presence of brefeldin A (GolgiPlug, BD Biosciences) and monensin (GolgiSTOP, BD Biosciences) for 6 hr according to the manufacturer’s instructions. All flow cytometry analyses were performed using an LSR II system (BD Biosciences) and FlowJo software (Tree Star Inc.). The antibodies used for flow cytometry are described in Supplementary Table S1. Detailed gating strategies are provided in Supplementary Fig. S8.
Gene expression microarray datasets and analysis
Total RNA was extracted from the tumor mass using RNeasy Plus Mini kit (Qiagen), and loaded onto GeneChip Mouse Gene 2.0 ST Array (Affymetrix). The raw microarray data was deposited in the Gene Expression Omnibus under accession number GSE110890. Average linkage hierarchical clustering was performed with Euclidian distance, and expression levels were depicted as heat maps using GENE-E. Functional annotation was performed by ORA using gene sets obtained from ImmuneSigDB 31 and GO databases. GO gene sets were manually categorized according to the functional similarity. Additionally, ORA results with GO terms were visualized as enrichment maps using Cytoscape and ClueGO 32 plug-in. Enriched GO terms were functionally clustered based on their kappa scores (> 0.36) and GO hierarchy, and clusters with at least three nodes were displayed. Statistical significance was determined using two-sided hypergeometric test.
Statistical analysis
Levels of significance for comparisons among treatment groups were determined using one-way ANOVA with Tukey’s post hoc test for multiple comparisons. ORA results were evaluated using Fisher’s exact test with Bonferroni adjustment. Survival analysis was performed using Kaplan-Meier curves with log-rank test. Results were considered statistically significant at a P-value < 0.05. GraphPad Prism 6 (GraphPad Software Inc.) was used for quantitative analysis.
Funding Statement
This work was supported by grants from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI17C2586 to Seok-Gu Kang), the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology, Republic of Korea (NRF-2014R1A2A1A10053662 to Eui-Cheol Shin), and the NRF grant funded by the MSIP: Ministry of Science, ICT and Future Planning, Republic of Korea (NRF-2017M2A2A7A01071036 to Seok-Gu Kang).
Acknowledgments
Authors would like to thank members of Brain Tumor Center, Severance Hospital (Ju Hyung Moon, Eui Hyun Kim, and Sun Ho Kim) and Graduate School of Medical Science and Engineering, KAIST (Kyung Hwan Kim, Hyung-Don Kim, and Ji Won Han) for scientific advice and useful discussion.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
- CMV
cytomegalovirus
- CTLA-4
cytotoxic T-lymphocyte associated protein 4
- DEG
differentially expressed gene
- GBM
glioblastoma
- GO
Gene Ontology
- IVIS
in vivo imaging system
- LAG-3
lymphocyte-activation gene 3
- NK
natural killer
- ORA
over-representation analysis
- PD-1
programmed death-1
- TIL
tumor-infiltrating lymphocyte
- TMZ
temozolomide
- TNF
tumor necrosis factor
- Treg
regulatory T
Supplementry material
Supplemental data for this article can be accessed here.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Roh TH, Park HH, Kang SG, Moon JH, Kim EH, Hong CK, Ahn SS, Choi HJ, Cho J, Kim SH, et al. Long-term outcomes of concomitant chemoradiotherapy with temozolomide for newly diagnosed glioblastoma patients: A single-center analysis. Medicine (Baltimore). 2017;96(27):e7422. doi: 10.1097/MD.0000000000007422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong Y-K, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1100–1108. doi: 10.1016/S1470-2045(14)70379-1. [DOI] [PubMed] [Google Scholar]
- 6.Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–131. doi: 10.1038/ni.3666. [DOI] [PubMed] [Google Scholar]
- 7.Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17(8):1064–1075. doi: 10.1093/neuonc/nou307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 9.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 11.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. doi: 10.1158/2326-6066.CIR-15-0151. [DOI] [PubMed] [Google Scholar]
- 13.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20(20):5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, Garzon-Muvdi T, Kim E, Theodros D, Polanczyk M, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8(370):370ra180. doi: 10.1126/scitranslmed.aag2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen J-F, Enjyoji K, Linden J, Oukka M, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, Granger L, Kruhlak M, Lindsten T, Thompson CB, et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood. 2012;119(22):5155–5163. doi: 10.1182/blood-2011-11-388918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A. 2017;114(5):E761–E70. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garber K. Promising early results for immunotherapy-antiangiogenesis combination. J Natl Cancer Inst. 2014;106:11. doi: 10.1093/jnci/dju392. [DOI] [PubMed] [Google Scholar]
- 22.Gray MJ, Gong J, Hatch MM, Nguyen V, Hughes CC, Hutchins JT, Freimark BD. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res. 2016;18(1):50. doi: 10.1186/s13058-016-0708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, Pineda C, Geukes Foppen MH, Scolyer R, Song JY, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology. 2016;5(12):e1238557. doi: 10.1080/2162402X.2016.1238557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085–4091. doi: 10.1200/JCO.2013.49.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwak J, Shim JK, Kim DS, Lee JH, Choi J, Park J, Shin KJ, Kim SH, Kim P, Huh YM, et al. Isolation and characterization of tumorspheres from a recurrent pineoblastoma patient: feasibility of a patient-derived xenograft. Int J Oncol. 2016;49(2):569–578. doi: 10.3892/ijo.2016.3554. [DOI] [PubMed] [Google Scholar]
- 26.Kong BH, Park NR, Shim JK, Kim BK, Shin HJ, Lee JH, Huh YM, Lee SJ, Kim SH, Kim EH, et al. Isolation of glioma cancer stem cells in relation to histological grades in glioma specimens. Childs Nerv Syst. 2013;29(2):217–229. doi: 10.1007/s00381-012-1964-9. [DOI] [PubMed] [Google Scholar]
- 27.Lal S, Lacroix M, Tofilon P, Fuller GN, Sawaya R, Lang FF. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000;92(2):326–333. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 28.Kong BH, Moon JH, Huh YM, Shim JK, Lee JH, Kim EH, Chang JH, Kim DS, Hong YK, Kim SH, et al. Prognostic value of glioma cancer stem cell isolation in survival of primary glioblastoma patients. Stem Cells Int. 2014;2014(838950. doi: 10.1155/2014/838950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Shim JK, Kang JH, Choi J, Chang JH, Kim SY, Kang SG. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro Oncol. 2017. doi: 10.1093/neuonc/nox243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eom H, Kaushik N, Yoo KC, Shim JK, Kwon M, Choi MY, Yoon T, Kang SG, Lee SJ. MerTK mediates STAT3-KRAS/SRC-signaling axis for glioma stem cell maintenance. Artif Cells Nanomed Biotechnol. 2018;1–9. doi: 10.1080/21691401.2018.1452022. [DOI] [PubMed] [Google Scholar]
- 31.Godec J, Tan Y, Liberzon A, Tamayo P, Bhattacharya S, Butte AJ, Mesirov JP, Haining WN. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity. 2016;44(1):194–206. doi: 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.