

ABSTRACT
Antitumor cytotoxic T lymphocytes (CTLs) are essential for immune surveillance, yet the blockade of eliciting such CTLs during oncolytic virotherapy remains incompletely understood. Here, we show that oncolysis of mesothelioma by modified vaccinia Tiantan (MVTT) induces damage-associated molecular patterns exposure. Although MVTT leads to regression of established mesothelioma dose-dependently, antitumor CTLs are rarely induced. Mechanistically, MVTT virotherapy generates C-X-C chemokines that recruit CXCR2-expressing polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) into tumor microenvironment, where they suppress dendritic cells (DCs) by producing IL-10 and halt CTL responses. During the virotherapy, however, depletion of PMN-MDSCs but not of monocytic (M)-MDSCs results in the induction of potent antitumor CTLs that not only eradicate established mesothelioma but also prevent the second tumor challenge. Our findings suggest that vaccinia virotherapy may combine strategies that prevent the chemotactic recruitment of PMN-MDSCs, block their suppression on DCs or deplete PMN-MDSCs in order to induce potent CTLs for tumor eradication.
KEYWORDS: Modified vaccinia Tiantan, virotherapy, mesothelioma, MDSCs, CTLs
Introduction
Mesothelioma is an asbestos-associated malignant form of cancer, which often has a poor prognosis in humans after disease onset.1 The current standard of care for this life-threatening malignancy only achieves suboptimal improvements in patient survival.2,3 Harnessing the host immune system to eradicate malignant cells has become a clinical strategy in cancer immunotherapy. However, although immune checkpoint inhibitors have improved the therapeutic efficacy in certain cancers, their effects are unsatisfactory in patients with mesothelioma.3 Novel strategies, therefore, are needed for treating mesothelioma. Recently, oncolytic virotherapy has emerged as a promising cancer immunotherapeutic strategy for the treatment of solid tumors including malignant mesothelioma, yet the mechanism underlying the limited virotherapeutic efficacy remain elusive.3,4
Direct virus-mediated oncolysis of cancer cells is one of the major mechanisms of oncolytic virotherapy. During oncolysis, danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) are released into the microenvironment, which can modulate the immunogenicity of released tumor antigens by creating an immune-activating environment and subsequently eliciting or reinforcing tumor-reactive T cell responses.5 The crucial role of adaptive T cell immunity in oncolytic virotherapy has been demonstrated in both preclinical and clinical studies.6,7 However, the tumor microenvironment (TME) is often an immunosuppressive environment that inhibits the activation of tumor-reactive T cells by inducing tolerogenic dendritic cells (DCs) and CD25+Foxp3+ regulatory T lymphocytes (Tregs).5,8,9 A number of studies indicated that bone marrow myeloid-derived suppressor cells (MDSCs) in the TME can dampen the responsiveness of cytotoxic T lymphocytes (CTLs),10 leading to limited efficacy in patients, especially when the TME is highly immunosuppressive.11-14 Because T cell immunity is indispensable for the efficacy of oncolytic virotherapy, the better understanding of restrictive mechanisms in the TME is particularly important for improving the clinical outcomes of the virotherapeutic strategy.
MDSCs represent one of the major immunosuppressive populations in the TME and a major obstacle to the effectiveness of cancer immunotherapy.10 In malignant mesothelioma models, we have previously reported that MDSCs expand quickly with the development of tumor lesions and contribute to the inhibition of tumor-reactive CTL responses.15,16 Consistently, decreased numbers of MDSCs in the TME are likely associated with the generation of antigen-specific CTL responses and therapeutic efficacy during oncolytic virotherapy in patients.11 MDSCs consist of monocytic (M) and polumophonuclear (PMN)-MDSCs. A recent study further indicated that targeting the COX-2-PGE2 pathway during vaccinia virotherapy is capable of decreasing PMN-MDSC levels while increasing antitumor CTL responses.17 Moreover, an earlier study using the COX-2 inhibitor celecoxib improved DC-based immunotherapy against mesothelioma by reducing the PMN-MDSC frequency.18 While these studies indicate the critical role of PMN-MDSCs in cancer immunotherapy, curing established tumors has rarely been observed. To date, the mechanism underlying MDSCs accumulation in the TME, the functional difference between MDSC subsets, and their impact on eliciting antitumor CTLs during oncolytic virotherapy remain incompletely understood.
In this study, we aimed to investigate the virotherapeutic effects of modified vaccinia Tiantan (MVTT) because MVTT readily induced DAMPs including calreticulin (CRT) exposure, HMGB1 and ATP release, as well as oncolysis of AB1 mesothelioma cells. We further determined the role of MVTT virotherapy in eliciting tumor-reactive CTLs, which are essential for curing malignant mesothelioma.15,19 We found that MVTT virotherapy alone had limited in vivo efficacy and was unable to induce tumor-reactive CTLs. Mechanistically, MVTT virotherapy induces chemotaxis that recruits IL-10-producing PMN-MDSCs into the TME, where they suppress DCs and therefore block the induction of antitumor CTLs. Lastly, depletion of PMN-MDSCs but not of M-MDSCs during MVTT virotherapy unleashes tumor-reactive CTLs, leading to the therapeutic cure of established mesothelioma.
Results
MVTT-mediated oncolysis of mesothelioma cells triggers exposure of CRT as well as the release of HMGB1 and ATP
To determine the oncolytic effects of MVTT, we generated a recombinant MVTT virus (rMVTT) to simultaneously express two detection markers, HIV-1 p24 and far-red fluorescent mutant HcRed (Supplementary Figure S1A), which facilitates the measurement of rMVTT replication and infected cells. We found that murine mesothelioma AB1 cells were susceptible to rMVTT infection, displaying red fluorescent syncytia (Supplementary Figure S1B) and expressing virus-encoded p24 protein (Supplementary Figure S1C). An increase in the HcRed signal and released free virus over time indicated that the rMVTT virus could infect and replicate in AB1 cells (Supplementary Figure S1D and E). We then determined the activity of rMVTT-mediated oncolysis. rMVTT infection significantly decreased AB1 cell viability (Figure 1A). Moreover, we measured the expression of calreticulin (CRT), a DAMP that is normally expressed in the lumen of the endoplasmic reticulum but is translocated to the surface of dying cells,5 in AB1 cells after rMVTT infection by flow cytometric analysis. When 0.2 MOI rMVTT was used for infection, less than 5% of AB1 cells showed exposure of CRT on their surface during the first 24 hours. Due to active viral replication, however, this percentage increased to 70% and 90% at 48 and 72 hours post infection, respectively (Figure 1B, left panel). Importantly, all CRT-positive cells showed expression of HcRed, suggesting that rMVTT infection resulted in CRT exposure on the cell surface (Figure 1B, right panel). Furthermore, western blot analysis demonstrated that rMVTT infection caused upregulated expression of CRT in AB1 cells (Figure 1C). In addition to CRT, the release of other DAMPs, such as high mobility group box 1 (HMGB1) protein and ATP, from dying cells may activate antigen-presenting cells (APCs) to mount antitumor immunity.5 We therefore measured the HMGB1 and ATP levels to address the possibility that rMVTT-mediated oncolysis would lead to immunogenic cell death. We found that HMGB1 was readily detected in the culture supernatants at 72 hours post rMVTT infection, but not in uninfected AB1 cell controls (Figure 1D). Moreover, the released ATP in the corresponding supernatants was also increased significantly (Figure 1E). Taken together, these data indicated that rMVTT infection not only resulted in effective oncolysis of AB1 cells but also induced upregulated expression and cell surface exposure of CRT as well as the release of ATP and HMGB1 from dying cells, which are commonly recognized as the three major hallmarks of immunogenic cell death for provoking adaptive antitumor immune responses.20
Figure 1.
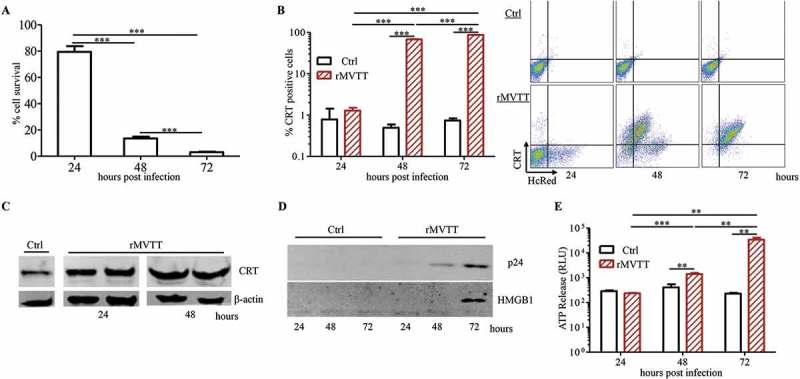
MVTT-mediated oncolysis of AB1 cells triggers exposure of CRT as well as release of HMGB1 and ATP. (A) AB1 cell viability upon infection with 0.2 MOI rMVTT. CRT expression on AB1 cells was detected with an anti-CRT antibody and analyzed either by flow cytometric analysis (B) or western blot analysis (C). β-actin served as the internal control showing that similar amounts of proteins were used for analysis. (D) Western blot analysis of released HMGB1 in culture supernatants after rMVTT infection. (E) Released ATP levels in the culture supernatant. Ctrl, culture medium alone. Data shown are representative of two independent experiments.
MVTT treatment eliminated established AB1 tumors dose-dependently yet failed to mount antitumor T cell immunity
To investigate the ability of rMVTT to treat established AB1 mesothelioma in BALB/c mice, we used intratumoral (i.t) viral injection as a means to determine its direct oncolytic efficacy. For this purpose, mice were subcutaneously (s.c) inoculated with wild-type AB1 cells, which express neither p24 nor HcRed, 7 days before they received different doses of rMVTT treatment, classified as the high- (total 5 × 108 PFU), medium- (5 × 107 PFU), and low-dose (2 × 107 PFU) groups (Figure 2A). We found that the growth of AB1 mesothelioma was significantly inhibited in all mice that received rMVTT treatment (Figure 2B). Furthermore, observations of tumor growth in individual mice showed that high-dose viral treatment completely eliminated tumor growth (Figure 2C), leading to 100% survival during our observation period (Figure 2D), while low- and medium-dose groups showed antitumor efficacies of only 37.5% and 50% tumor-free animals, respectively (Figure 2B-D). These findings suggested that rMVTT treatment was able to eliminate established AB1 mesothelioma in a dose-dependent way.
Figure 2.
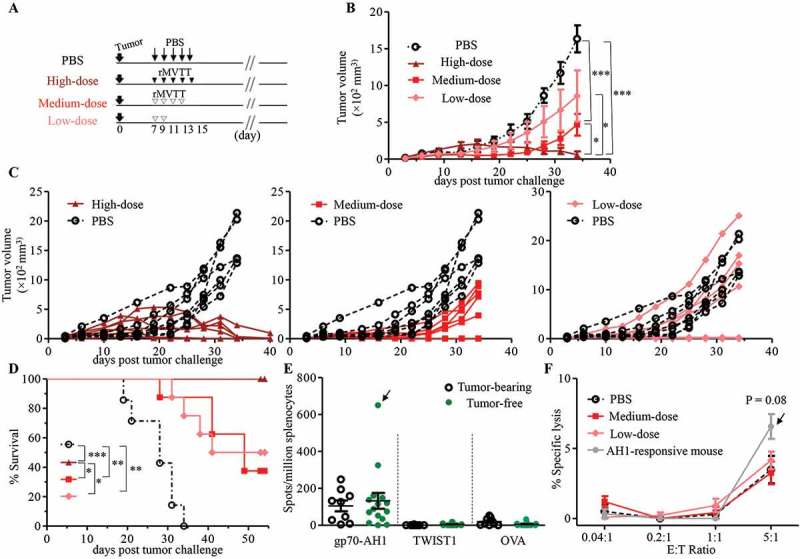
MVTT treatment eliminated established AB1 tumors in a dose-dependent manner, yet failed to induce antitumor T cell immunity in BALB/c mice. (A) Schematic representation of our therapeutic study of AB1 tumor-bearing mice using different doses of rMVTT. Solid AB1 mesothelioma tumors were established with s.c inoculation of 5 × 105 AB1 cells 7 days prior to treatment. In the high-dose group (n = 8), 1 × 108 PFU rMVTT per dose was delivered i.t every 2 days for 5 injections, whereas in the medium-dose group (n = 8) 1 × 107 PFU per injection was given i.t 4 times, and 2 injections were given to the low-dose group (n = 8). (B) Tumor volumes of each group were measured over time with a caliper. (C) Individual tumor growth curve. Every line represents one mouse. (D) Survival curve, taken as time to tumor length > 15 mm, was determined by caliper measurement. (E) T cell responses in splenocytes. Secreted IFN-γ was quantified by ELISpot assay after ex vivo stimulation of splenocytes with gp70-AH1 and TWIST1 peptides or the control peptide ovalbumin (OVA257-264). Only one tumor-free mouse from the medium-dose group showed a strong response against the gp70-AH1 epitope, as indicated by the arrow. (F) CTL assay of CD3+ T cells isolated from tumor-free mice towards AB1 cells at different effector:target (E:T) ratios. The grey line represents CTL activity of CD3+ T cells from the mouse with strong AH1 responses. P = 0.08, compared to the PBS group. The experiment was repeated 2 times.
Since the in vitro experiments described above showed that rMVTT infection triggered immunogenic cell death in AB1 cells, we speculated that the oncolytic effect of rMVTT would create an immune-stimulatory environment to induce anti-mesothelioma immune responses in vivo. To investigate anti-mesothelioma T cell responses, two tumor antigens found in AB1 mesothelioma, immunodominant AH1 (gp70423–431) and Twist-related protein 1 (TWIST1) peptides, were used in an ELISpot assay. The peptide gp70-AH1 is a well-characterized immunodominant CTL epitope derived from glycoprotein 70 (gp70) of endogenous murine leukemia virus.21,22 The TWIST1 is a transcription factor that is crucial to the tumor metastatic process and tumor resistance to drug treatment.23 Since both gp70-AH1 and TWIST1 are present in wild-type AB1 cells, we sought to determine the relationship between antitumor T cell responses and the tumor elimination efficacy. Surprisingly, we found that splenocytes from only one treated and tumor-free mouse displayed an AH1-specific ELISpot response (Figure 2E) and cytotoxic effect against AB1 cells (Figure 2F). There was no statistical significance between tumor-bearing and tumor-free mice (Figure 2E and F). Furthermore, high-dose viral treatment reduced AB1 tumor burden and eventually led to the rejection of established tumors in SCID mice, suggesting that high-dose rMVTT induced tumor regression independent of the adoptive antitumor T cell immunity (Supplementary Figure S2A-C). These data suggested that although rMVTT treatment eliminated established AB1 mesothelioma in a dose-dependent manner, oncolysis of tumors did not readily induce antitumor T cell immunity.
MVTT treatment recruited PMN-MDSCs into tumor microenvironment
Since the initiation of adaptive antitumor immunity after oncolysis primarily occurs inside the tumor, we sought to examine the TME after rMVTT treatment. Analysis of rMVTT-injected AB1 mesothelioma revealed that expression of virus-encoded HcRed was readily detected 2 days after i.t injection and rapidly decreased thereafter (Supplementary Figure S3A). Consistently, immunohistochemical staining of vaccinia viral proteins was only found in tumor tissues at 2 days but not at 4 days after rMVTT treatment, with visible necrotic areas within and adjacent to the zones of infection (Supplementary Figure S3B). These results demonstrated rapid but limited rMVTT replication in the TME. We then measured different tumor resident immune cells, including the proportions of CD3+ T cells, natural killer (NK) cells, CD4+ Tregs (CD4+CD25+Foxp3+) and MDSC subsets (PMN-MDSCs, CD11b+Ly6G+Ly6Clow/int; M-MDSCs, CD11b+Ly6G−Ly6Chi) as well as the expression of the exhaustion surface markers PD-1 and Tim-3 on CD3+ T cells by flow cytometry (Supplementary Figure S3C). We observed that the overall levels of MDSCs in the spleens appeared to decrease over the course of rMVTT treatment, while the frequencies of tumor-infiltrating MDSCs were maintained at similar levels (Figure 3A). We further examined the two major subsets of MDSCs, PMN-MDSCs and M-MDSCs, because they have remarkable differences in their morphology and suppressive activities.24 Consistent with findings by others,25 PMN-MDSCs were largely expanded in peripheral lymphoid organs, whereas M-MDSCs preferentially accumulated inside tumors of untreated control mice (Figure 3B). Furthermore, rMVTT treatment did not influence the frequencies of M-MDSCs either in spleens or in tumors; however, PMN-MDSCs decreased significantly in spleens and increased significantly in the TME (Figure 3B and C). The absolute cell number of PMN-MDSCs in tumors also increased significantly after rMVTT treatment (Figure 3D). For comparison, although rMVTT treatment decreased the frequencies of CD4+ Tregs in the spleen, no significant difference was found in their frequency or cell number in tumors (Figure 3E). Interestingly, in contrast to the remarkable accumulation of PMN-MDSCs in tumors as early as day 2 post rMVTT treatment, the frequency and cell number of NK cells were significantly decreased (Figure 3F), implying a possible counteraction between these two cell types.26,27 Infection-induced inflammatory responses have been shown to increase lymphocyte infiltration into the TME.6,28 Indeed, we observed strikingly increased CD3+ T cells inside tumors at day 4 after rMVTT treatment (Supplementary Figure S3D). The increased T cell infiltration, however, was coupled with significantly elevated expression of the exhaustion markers PD-1 and Tim-3 (Supplementary Figure S3E). Collectively, our results demonstrated that rMVTT treatment changed the local and systemic distributions of immune cells, particularly the accumulation of PMN-MDSCs in the TME.
Figure 3.
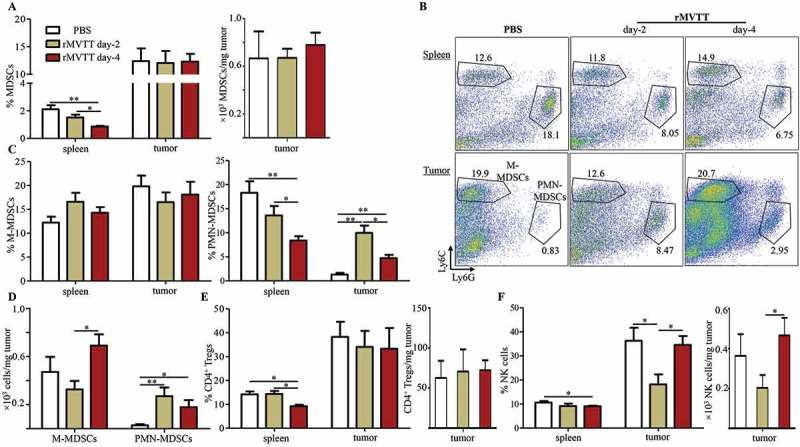
MVTT treatment recruited PMN-MDSCs into the TME. (A) Percentage of total MDSCs in the spleen and tumor (left panel) and absolute cell number of MDSCs in the tumor (right panel). Numbers of MDSCs per milligram of tumor at the indicated time points were calculated. (B) Representative dot plots showing populations of PMN-MDSCs and M-MDSCs within CD11b+ cells in the spleen and tumor. Numbers indicate cell proportions. (C) Percentages of MDSC subsets were calculated for M-MDSCs (left panel) and PMN-MDSCs (right panel). (D) Absolute cell numbers of M-MDSCs and PMN-MDSCs in the tumor. Numbers of PMN-MDSCs or M-MDSCs per milligram of tumor at the indicated time points were calculated. (E) Percentage of NK cells in the spleen and tumor (left panel) and absolute cell number of NK cells in the tumor (right panel). (F) Percentage of CD4+ Tregs in the spleen and tumor (left panel) and absolute cell number of CD4+ Tregs in the tumor (right panel). Graphs show cumulative data from two separate experiments (n = 7).
Trafficking of PMN-MDSCs into the TME by MVTT-induced chemotaxis
To understand the mechanism underlying the increase in PMN-MDSCs in the TME, we hypothesized that PMN-MDSCs are preferentially recruited to the TME after rMVTT treatment. To study this, the expression of chemokine receptors on both MDSC subsets and the levels of chemokines in rMVTT-treated tumors were examined.29,30 Flow cytometric analysis of chemokine receptor expression revealed that CXCR2 was expressed only on PMN-MDSCs but not on M-MDSCs. Conversely, high levels of CCR2 expression were found on M-MDSCs but not on PMN-MDSCs (Figure 4A). We then measured the levels of various chemokines in tumor homogenates after rMVTT treatment. We found that a panel of C-X-C chemokines, including CXCL5, CXCL9 and CXCL13, were significantly upregulated in AB1 mesothelioma as early as 2 days after treatment (Figure 4B), whereas upregulated C-C chemokine production was only observed 4 days after treatment (Figure 4C). These results suggested that CXCR2-expressing PMN-MDSCs might migrate into and adhere to the tumor bed primarily in response to the rapidly increased C-X-C chemokines in the TME. To test this hypothesis, CFSE-labelled MDSCs derived from mesothelioma-bearing mice were adoptively transferred into recipient mice that also bore mesothelioma tumors but were treated with either rMVTT or PBS following the MDSC transfer. CFSE-labelled MDSCs in both the spleen and mesothelioma were then quantified by flow cytometry 24 hours after rMVTT treatment (Supplementary Figure S4). Compared to PBS-treated recipients, we observed a significant increase in both the percentage and absolute number of CFSE+ MDSCs in tumors of rMVTT-treated recipients (Figure 4D). Migrated PMN-MDSCs in tumors were distinguished from M-MDSCs by the expression of Ly6G (Figure 4E). Among the rMVTT-treated recipients, we found that their spleens showed slightly decreased PMN-/M-MDSCs ratios, while their tumors displayed strikingly elevated PMN-/M-MDSCs ratios and absolute numbers of PMN-MDSCs (Figure 4E and F). Overall, these results demonstrated that PMN-MDSCs preferentially migrated from the peripheral lymph system into the TME in response to chemotaxis induced by rMVTT treatment.
Figure 4.
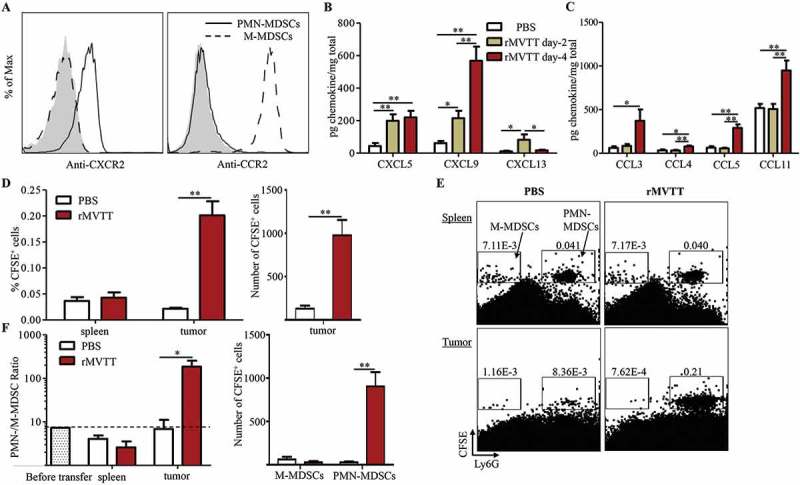
Trafficking of PMN-MDSCs into the TME by MVTT-induced chemotaxis. (A) Flow cytometric analysis of chemokine receptor expression on splenic PMN-MDSCs and M-MDSCs from AB1 tumor-bearing mice. Representative histogram plots are shown from 3 independent experiments; the shaded region represents an isotype control. Expression of C-X-C chemokines (B) and C-C chemokines (C) in tumor homogenates after rMVTT treatment. (D) Frequencies (left panel) and absolute numbers (right panel) of CFSE-labelled total MDSCs in both spleen and tumor 24 hours after treatment. Each mouse received 50 µl PBS or rMVTT (1 × 107 PFU). (E) Migrated M-MDSCs and PMN-MDSCs in the tumor 24 hours after treatment. Representative dot plots are shown with numbers indicating gated cell proportions relative to total singlets. (F) Changes in the ratio of the PMN-MDSC proportion to the M-MDSC proportion were analyzed (left panel). PMN-/M-MDSC ratio measured before adoptive transfer was shown as baseline. Changes in the absolute numbers of M-MDSCs and PMN-MDSCs in the tumor are shown (right panel). Graphs show the cumulative data from two separate experiments (n = 6).
Preferential depletion of MDSC subsets by antibody and peptibody
To investigate the role of MDSCs in the rMVTT treatment, two MDSC-depleting agents, anti-Ly6G monoclonal antibody 1A8 and the specific depleting peptibody H6-pep, were explored in our mesothelioma model. 1A8 is routinely used to deplete Ly6G+ cells, primarily PMN-MDSCs, whereas H6-pep and G3-pep are two peptibodies with binding specificity to both PMN-MDSCs and M-MDSCs.31 Accordingly, we manufactured these two peptibodies by a transient expression system in 293F cells using expression plasmids (Supplementary Figure S5A). We found that H6-pep showed a relatively higher binding affinity than G3-pep to total MDSCs derived from AB1-mesothelioma-bearing mice (Supplementary Figure S5B and C); we therefore used H6-pep in our depletion experiments. When AB1 tumor-bearing mice were treated with 1A8 or H6-pep by i.t injection, only 1A8-treated mice had a significantly decreased frequency of splenic MDSCs, yet both 1A8 and H6-pep did not seem to reduce total MDSC accumulation in tumors (Supplementary Figure S5D). As expected, however, 1A8 diminished Ly6G+ PMN-MDSCs selectively in both spleens and tumors at day 2 after injection (Figure 5A and B). While this effect was maintained in the tumor at day 4, splenic but not TME PMN-MDSCs started to reappear. Unlike PMN-MDSCs, tumor M-MDSCs were not affected by 1A8, whereas a marked increase in splenic M-MDSCs was observed compared with an isotype control, probably due to the continuous generation of MDSCs from bone marrow.10,24 Conversely, with its higher binding affinity to M-MDSCs, H6-pep treatment significantly depleted M-MDSCs but not PMN-MDSCs, especially in the TME; this effect was maintained through day 4 (Supplementary Figure S5E and F). Following depletion of M-MDSCs, a significant compensatory increase in the frequency of splenic PMN-MDSCs was observed.
Figure 5.
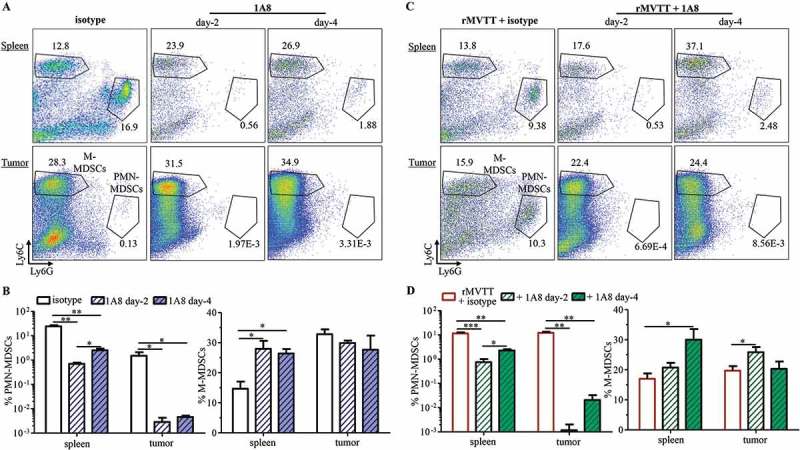
Preferential depletion of MDSC subsets by antibody and peptibody treatment. (A) Representative dot plots gated on CD11b+ cells showing populations of PMN-MDSCs and M-MDSCs in the spleen and tumor 2 days and 4 days after receiving i.t injection of 100 µg of either 1A8 or anti-rat IgG2a (clone: 2A3) isotype control. Numbers within dot plots represent cell proportions. (B) Changes in frequencies were calculated with PMN-MDSCs (left panel) and M-MDSCs (right panel). (C) Representative dot plots showing population of PMN-MDSCs and M-MDSCs in the spleen and tumor 2 days and 4 days after combination treatment. 100 µg of either 1A8 or isotype 2A3 were combined with 1 × 107 PFU rMVTT and i.t injected into AB1 mesothelioma. (D) Analysis of frequencies of PMN-MDSCs (left panel) and M-MDSCs (right panel) after combination treatment. Graphs show cumulative data from two separate experiments (n = 5).
We then investigated the efficacy of 1A8 and H6-pep during rMVTT treatment. Consistent with the findings described above, rMVTT treatment resulted in the increased recruitment of PMN-MDSCs in tumors (Figure 5A and C). This increased population, however, was nearly cleared by 1A8 antibody treatment at day 2 (Figure 5C and D). 1A8 also prevented tumor recruitment of PMN-MDSCs at day 4, despite a significantly elevated frequency of splenic PMN-MDSCs. By contrast, H6-pep treatment decreased M-MDSCs while increasing PMN-MDSCs in both the spleens and tumors (Supplementary Figure S5G and H). These results demonstrated that administration of 1A8 and H6-pep preferentially depleted PMN-MDSCs and M-MDSCs, respectively, and their depletion effects were maintained even after rMVTT administration, which allowed us to study the impact of PMN-MDSCs and M-MDSCs on the induction of antitumor immunity during MVTT-based oncolytic virotherapy.
Depletion of PMN-MDSCs enhances MVTT treatment efficacy by inducing antitumor T cell immunity
Considering that MDSCs are one of the major immunosuppressive cells that inhibit antitumor T cell responses, we sought to explore whether the depletion of PMN-MDSCs enhanced the therapeutic efficacy of MVTT-based oncolytic virotherapy. In a similar setting as described above, BALB/c mice bearing 7-day-old wild-type AB1 mesothelioma were simultaneously injected with low-dose rMVTT (1 × 107 PFU) in combination with either 100 µg of 1A8 or H6-pep for the specific depletion of PMN-MDSCs and M-MDSCs, respectively (Supplementary Figure S6A). Consistent with our findings described above, a single delivery of low-dose rMVTT did not control tumor growth. The incorporation of MDSC depletion in this setting, however, did not slow tumor progression or prolong survival (Supplementary Figure S6B and C). Given the known dose-dependent effect of the rMVTT treatment, we attempted to improve the antitumor effect via an additional low-dose 2 days later (Figure 6A). We found that two rMVTT treatments alone slowed tumor growth and resulted in tumor regression in 1/7 mice, whereas 1A8 alone did not impact tumor growth at all (Figure 6B and C). Strikingly, however, the second combined low-dose rMVTT and 1A8 treatment effectively controlled tumor growth and eventually led to complete elimination of established AB1 mesothelioma (Figure 6B and C). By contrast, the combined rMVTT and H6-pep treatment did not show significant antitumor activity or synergistic effects in mesothelioma elimination (Supplementary Figure S6D and E). To determine whether prolonged antitumor T cell immunity was generated in these controller mice, we re-challenged them with a much higher dose (2 × 106 cells) of AB1-Luc cells on their opposite flank 40 days after complete tumor rejection (Figure 6A). Complete rejection of AB1-Luc mesothelioma was observed 11 days later in these controller mice, leading to tumor-free survival > 30 weeks, while all mice from the control group developed tumors (Figure 6D and E). These results demonstrated that depletion of PMN-MDSCs but not of M-MDSCs could improve rMVTT treatment efficacy significantly, probably by inducing prolonged antitumor immunity.
Figure 6.
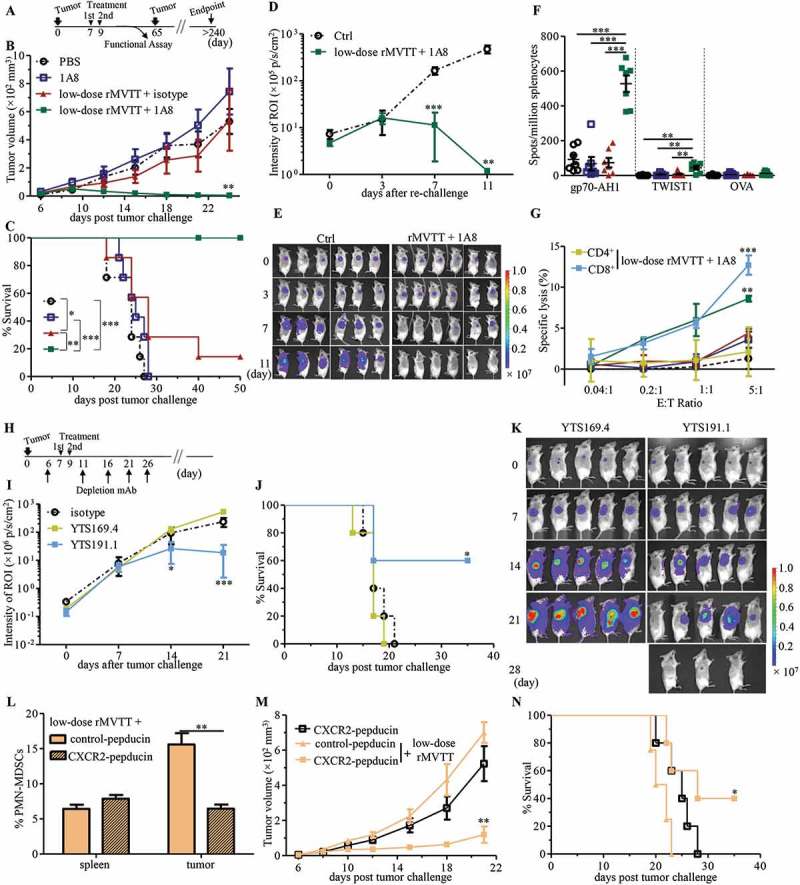
Depletion of PMN-MDSCs enhances MVTT treatment efficacy by inducing antitumor T cell immunity. (A) Schematic representation of treatment schedule. 5 × 105 AB1 cells were s.c inoculated into BALB/c mice and grown for 7 days, following i.t administration of rMVTT (n = 7), 1A8 antibody (n = 7), combined rMVTT and 1A8 (n = 6) or PBS control (n = 7). An additional treatment was scheduled at day 9 in each group. Tumor growth (**P = 0.0069 compared to PBS-treated group) (B) and survival curves (C) of mice were calculated. 40 days after tumor ablation, protected mice (n = 6) in the combined rMVTT and 1A8 group were re-challenged and measured for tumor growth (D) with representative bioluminescence images of AB1-Luc tumors (E). Ctrl, naïve BALB/c mice. (F) T cell responses in splenocytes measured by ELISpot assay. (G) In vitro cytotoxic activity of CD3+ T cells in each group, or CD4+ and CD8+ T cells from the combined rMVTT and 1A8 group, towards AB1 cells at different effector:target (E:T) ratios. (H) Schematic representation for T cell depletion experiments with 2 administrations of combined rMVTT and 1A8 therapy (n = 5). 5 × 105 AB1-Luc cells were s.c inoculated. AB1-Luc tumor growth (I) and survival curve (J) of combined rMVTT and 1A8-treated mice with CD8+ T cell depletion (YTS169.4) or CD4+ T cell depletion (YTS191.1), or AB1-Luc tumor-bearing mice receiving only LTF-2 control antibody (isotype). (K) Representative bioluminescence images of AB1-Luc tumors in T cell depletion groups. BALB/c mice bearing 6-day AB1 tumors were treated with 2.5 mg/kg control-pepducin or CXCR2-pepducin, followed by 1 mg/kg pepducin daily for the duration of the study. rMVTT were given i.t at day 7 and percentages of PMN-MDSCs in the spleen and tumor were analyzed at day 9 (L). An additional rMVTT were administrated at day 9. Tumor growth (**P = 0.0063 compared to CXCR2-pepducin-treated group) (M) and survival curves (N) of mice (n = 5 for each group) were calculated.
To further test this hypothesis, we measured tumor-reactive T cell responses in our experimental animals. Murine splenocytes were harvested and tested against gp70-AH1 or TWIST1 peptides (Figure 6A). We found that the T cell responses against both gp70-AH1 and TWIST1 were significantly increased among mice treated twice with the low-dose rMVTT and 1A8 combination (Figure 6F). This enhancement was not found with the double rMVTT and H6-pep combination that depleted M-MDSCs (Supplementary Figure S6F). In addition, in vitro cytotoxic assays demonstrated enhanced CD8+ CTLs in controller mice in comparison to other groups (Figure 6G). Furthermore, CD4+ or CD8+ T cells were depleted using the monoclonal antibodies YTS191.1 and YTS169.4, respectively, before AB1 tumor-bearing mice received the rMVTT and 1A8 combination therapy (Figure 6H). Remarkably, the depletion of CD8+ T cells by YTS169.4 completely diminished the antitumor activity of the combination therapy, resulting in uncontrolled tumor outgrowth, and all mice died within 21 days. By contrast, depletion of CD4+ T cells by YTS191.1 preserved partial therapeutic effects and caused tumor regression in 3/5 mice (Figure 6I-K). To determine whether our discovery could be applied to other malignant tumors, the efficacy of the combined rMVTT and 1A8 therapy was tested in a distinct syngeneic C57BL/6 melanoma model. Similarly, we found that this combination therapy resulted in enhanced B16F10 tumor regression, prolonged survival and augmented antitumor T cell responses (Supplementary Figure S6G-I). Furthermore, it was reported that CXCR2 blockade impaired the migration of myeloid lineage cells.32,33 Given the high level of CXCR2 expression on PMN-MDSCs in our model, we assessed antitumor T cell responses after CXCR2 blockade during MVTT virotherapy. In established AB1 tumors, blocking of CXCR2 by pepducin reduced PMN-MDSCs in the tumor microenvironment (Supplementary Figure S6J and K) and also counteracted MVTT-mediated PMN-MDSCs recruitment (Figure 6L), while it had no effect on recruitment of M-MDSCs, CD4+ Treg and NK cells (Supplementary Figure 6L and M). Importantly, combined CXCR2-pepducin and low-dose rMVTT therapy promoted tumor regression and enhanced tumor-free survival (Figure 6M and N). These results provide additional evidence that PMN-MDSCs play a key role in suppressing antitumor T cell immunity during MVTT virotherapy, and suggested targeting CXCR2 signaling may serve as an alternative approach to counteract PMN-MDSCs-mediated immunosuppression. Collectively, our results demonstrated that depletion of PMN-MDSCs during localized MVTT-based oncolytic virotherapy elicited potent systemic and long lasting antitumor T cell immunity.
PMN-MDSCs prevent the induction of antitumor T cell immunity by restricting dendritic cell activation
Although rMVTT-induced oncolysis created an immune-activating environment with the production of CRT, HMGB1 and ATP, anti-mesothelioma specific T cell responses were not readily induced (Figure 2E and F). This situation, however, was completely changed when PMN-MDSCs were depleted during the rMVTT treatment (Figure 6F and G). We therefore speculated that PMN-MDSCs might have suppressive effects on DCs through direct cross-talk in the TME of our model.34 To test this hypothesis, we sought to determine the direct impact of PMN-MDSCs on DCs. We first tested the ability of bone marrow-derived DCs (BMDCs)35 to process and present antigens for activating CD3+ T cells derived from controller mice that received combined rMVTT and 1A8 treatment. In this experiment, rMVTT-treated AB1 cell supernatants were used as a supply of tumor antigens to pulse BMDCs. We observed remarkably increased production of the proinflammatory cytokine IL-6 in co-cultures when BMDCs were pulsed with antigens (Supplementary Figure S7A). Meanwhile, antigen-loaded BMDCs greatly enhanced the production of TNF-α and IFN-γ (Figure 7A), as well as the Th17 cytokines IL-17A and IL-22 (Supplementary Figure S7A), in co-cultures with CD3+ T cells of controller mice but not of naïve mice, suggesting T cell activation in response to tumor antigens. Previously, surface-exposed CRT protein has been shown to chaperone tumor antigens to facilitate their uptake by DCs.36 Indeed, an anti-CRT antibody significantly reduced the production of both TNF-α and IFN-γ (Figure 7A). To confirm these findings, we further measured T cell proliferation. Antigen-pulsed BMDCs effectively induced controller CD4+ and CD8+ T cell proliferation (Figure 7B), demonstrating activation of tumor antigen-specific T cells. Once again, the presence of an anti-CRT antibody inhibited T cell proliferation (Figure 7B), suggesting a role for CRT in the activation of the DC-T cell axis. Therefore, in the absence of PMN-MDSCs, rMVTT-induced CRT exposure enhances the activation of BMDCs to elicit potent antitumor T cell immunity.
Figure 7.
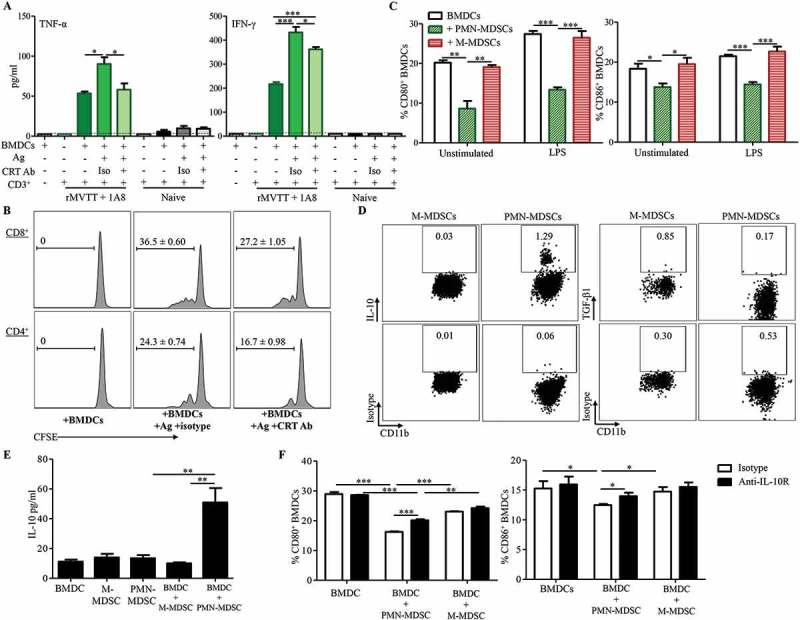
PMN-MDSCs prevent the induction of antitumor T cell immunity by restricting DC activation. (A) Cytokine production following incubation of CD3+ T cells with antigen-pulsed BMDCs. BMDCs were pulsed with rMVTT-treated AB1 cell supernatants overnight, following washing with culture medium. Then, purified CD3+ T cells were added and culture supernatants were collected for analysis of cytokine production. Anti-CRT antibody or isotype control were present in several of the cultures during antigen-pulsing. Naïve, purified CD3+ T cells from naïve BALB/c mice. (B) Proliferation of CFSE-labelled CD3+ T cells after co-culture with antigen-pulsed BMDCs. Representative histogram plots are shown with numbers in each plot indicating proliferating populations. (C) Expression of CD80 and CD86 on BMDCs pulsed with culture medium (Unstimulated) or LPS. Purified PMN-MDSCs or M-MDSCs were labelled with CFSE and were present in the culture at a ratio of 2:1 with BMDCs. Graphs from (A) to (C) show cumulative data from two separate experiments. (D) Frequencies of IL-10+ and TGF-β1+ PMN-MDSCs and M-MDSCs. Representative dot plots from 3 independent experiments are shown with numbers indicating positive cell populations in each gate. (E) Production of IL-10 was enhanced by crosstalk between PMN-MDSCs and BMDCs. 5 × 104 purified PMN-MDSCs or M-MDSCs were present in the culture with or without 1 × 105 BMDCs (BMDC:MDSC = 1:2). Supernatant were collected at 4 days post incubation and measured for cytokine production. (F) Expression of CD80 and CD86 on LPS-activated BMDCs in the presence of IL-10 receptor blocking antibody or isotype control. Purified PMN-MDSCs or M-MDSCs were labelled with CFSE and were present in the culture at a ratio of 2:1 with BMDCs. IL-10 receptor was blocked by anti-IL-10R antibody (5 µg/ml) before BMDCs were stimulated with 100 ng/ml LPS. Graphs from (E) to (F) show representative data from two separate experiments.
Subsequently, we also measured the direct interaction between AB1-induced MDSCs and BMDCs. BMDCs were co-cultured with AB1-induced MDSCs in the presence or absence of LPS. We found that CD80 and CD86 expression on BMDCs was significantly upregulated by LPS stimulation (P < 0.001 for CD80, P < 0.05 for CD86, Unstimulated versus LPS), suggesting BMDC maturation (Figure 7C). Notably, when MDSCs were present in the co-culture, PMN-MDSCs but not M-MDSCs significantly suppressed expression of CD80 and CD86 on both unstimulated and LPS-stimulated BMDCs (Figure 7C). LPS-induced changes in cytokine production were also analyzed. Supernatants collected from BMDCs without LPS showed very low levels of cytokines consistently. In contrast, culture supernatants with LPS resulted in marked increases of the proinflammatory cytokines IL-6 and TNF-α, as well as type 1 cytokine IL-12p70 (Supplementary Figure S7B). In consistency with PMN-MDSC’s ability of down-regulating BMDC activation, the presence of PMN-MDSCs in the co-culture significantly inhibited the induction of IL-6, TNF-α and IL-12p70, further supporting the role of PMN-MDSCs in suppressing BMDCs activation (Supplementary Figure S7B). We then asked whether PMN-MDSCs have similar suppressive effects when BMDCs were pulsed with rMVTT-treated AB1 cell supernatants rather than LPS. By measuring cytokines related to BMDC activation, we found that PMN-MDSCs but not M-MDSCs significantly inhibited IL-6 and TNF-α production in co-cultures, and the inhibitory effect of PMN-MDSCs on TNF-α production was dose-dependent (Supplementary Figure S7C).
In order to understand the underlying mechanism of PMN-MDSC-mediated immunosuppression, productions of IL-10 and TGF-β were examined in our model.34,37 We found that the production of IL-10 was enhanced in tumor following intratumoral rMVTT treatment in vivo (Supplementary Figure S7D). Further intracellular staining analyses showed that PMN-MDSCs but not M-MDSCs exhibited an IL-10-producing subset (Figure 7D), and the secreted IL-10 in culture supernatant of PMN-MDSCs was further increased when being co-cultured with BMDCs in vitro (Figure 7E), without contribution from BMDCs (Supplementary Figure S7E). The immunosuppressive cytokine IL-10 is well-known to inhibit DC maturation and prevent DCs from initiating Th1 responses.38 Crosstalk between MDSC and macrophage has been reported to reduce macrophage production of IL-12 and increase MDSC production of IL-10 to promote tumor progression.39,40 Therefore, we speculated that the suppressive capacity of PMN-MDSCs might be dependent on their IL-10 production in our experiments. To determine this, purified PMN-MDSCs or M-MDSCs derived from AB1-bearing mice were co-cultured with LPS-activated BMDCs in the presence of IL-10 receptor blocking antibody or isotype control. When compared the expression of activation markers on BMDCs, we found that the presence of PMN-MDSCs consistently down-regulated CD80 and CD86 expression on BMDCs (Figure 7F). However, PMN-MDSC-mediated suppression can be partially alleviated by the blockade of IL-10 receptor (Figure 7F). In addition, we examined secreted cytokines in the supernatant and found that blocking IL-10 receptor also significantly elevated production of TNF-α and IL-12p70 (Supplementary Figure S7F), suggesting IL-10 production by PMN-MDSCs appeared to be a direct means of suppression in our in vitro suppression assay. Collectively, our results demonstrated that while rMVTT treatments facilitate CRT-dependent antigen uptake, as well as activation and antigen-presentation of BMDCs, PMN-MDSCs likely directly inhibit DC activation and lead to the reduced efficacy or failure of oncolytic viral treatment.
Discussion
Cancer virotherapy using oncolytic viruses is a promising therapeutic strategy with demonstrated clinical benefits.14,41 Following the approval of T-vec (also known as Imlygic), a recombinant herpes simplex virus expressing the immune-activating cytokine GM-CSF for treating skin and lymph node melanoma in the USA and Europe, a variety of oncolytic viruses have progressed to clinical development.42-44 Among these, the use of ONCOS-102 adenovirus for treating malignant mesothelioma was able to induce tumor-infiltration by CD8+ T cells, systemic antitumor CD8+ T cells and Th1-type polarization in a clinical setting.12 Although the therapeutic effects of T-vec and ONCOS-102 are promising, only a small fraction of treated patients experienced clinical responses in these studies. Therefore, investigating how to induce potent antitumor immune responses is essential for enhancing the therapeutic efficacy of virotherapy in patients.3,45 Most of the viruses that are currently being tested in clinical trials were designed to acquire the capability to trigger immune responses.4,5,41,42 To this end, understanding the mechanism underlying the blockade and regulation of systemic antitumor immunity is critical for the further improvement of oncolytic virotherapy. Here, we demonstrated that the combined use of oncolytic vaccinia MVTT with PMN-MDSC depletion resulted in complete remission of mesothelioma in mice. Specifically, our findings demonstrated that intratumoral PMN-MDSCs are key DC suppressors in the mesothelioma TME that restrict the induction of antitumor CTLs, compromising the efficacy of MVTT-based virotherapy.
MVTT virotherapy alone is insufficient for efficient tumor clearance. Replication of the oncolytic virus in the tumor releases the danger signals CRT, HMGB1 and ATP, as well as tumor antigens for DCs, to trigger antitumor immune responses.5 However, we found that complete mesothelioma eradication was only achieved by i.t administration of extremely high doses of rMVTT at multiple sites of the solid tumors, yet even in protected mice, antitumor T cell responses were rarely elicited. It is possible that persistent viral infection and recruitment of innate immune cells can lead to the observed tumor regression,46,47 but this was not tested here. In attempts to clarify the mechanism underlying the failed CTL induction, we found that MVTT virotherapy significantly expanded MDSCs in the mesothelioma TME. Expansion of MDSCs has been regarded as a key immune evasion mechanism in various human cancers, such as renal cell carcinoma, squamous cell carcinoma, breast cancer, and non-small cell lung carcinoma.48-50 In mice with mesothelioma, we previously reported that the tumor induced a rapid increase of MDSCs as early as 7 days after AB1 cell inoculation, and the elimination of MDSCs during immunotherapy was closely related to tumor rejection.15,16 Here, we clarified that the expanded PMN-MDSCs in the mesothelioma TME during MVTT virotherapy were due to the production of C-X-C chemokines associated with the viral infection of tumor cells. C-X-C chemokines then preferentially recruit CXCR2+ PMN-MDSCs from peripheral lymphoid organs to tumor sites by chemotaxis. These results are in agreement with previous reports that emphasized the critical role of the C-C and C-X-C axes in the trafficking of M-MDSCs and PMN-MDSCs, respectively.30,51 Previously, viral infection-recruited PMN-MDSCs were found to be responsible for either suppression of NK cells by reactive oxygen species (ROS) production or augmentation of local immune suppression by PD-L1 expression.26,52 In the present study, our data suggested that PMN-MDSCs exhibited potent immunosuppressive function against DC activation. Because similar immunosuppressive effects on DCs were not found with M-MDSCs, our results likely indicate a functional difference between these two MDSC subsets in the mesothelioma TME.
Depletion of PMN-MDSCs alone is also insufficient for efficient tumor clearance. Previous studies have demonstrated that targeted depletion of PMN-MDSCs allowed modest CTL responses in pancreatic ductal adenocarcinoma and lung cancer models.53,54 AB1 mesothelioma in mice, however, has been recognized as a poorly immunogenic model.55 This notion was in agreement with our previous results. For example, AB1 mesothelioma displayed similar growth kinetics in immunodeficient SCID mice compared to immunocompetent BALB/c mice.15 Moreover, purified T cells from mesothelioma-bearing mice did not contain antigen-specific T cells with potent cytotoxic activity. In this study, to better define the function of PMN-MDSCs and M-MDSCs in modulating antitumor immunity, we conducted depletion experiments using anti-Ly6G or H6-pep monotherapy, respectively. Interestingly, depletion of either PMN-MDSCs or M-MDSCs did not induce any inhibitory effects on mesothelioma growth. Additionally, no measurable antitumor CTLs were detected. We therefore demonstrated that depletion of MDSC subsets alone did not promote the exposure of mesothelioma antigens to trigger DC activation during these experiments. These results indirectly implied that MVTT virotherapy is necessary to promote tumor antigen exposure and subsequent induction of systemic antitumor T cell responses.
Curing established mesothelioma requires a combination of MVTT virotherapy and PMN-MDSC depletion to potentiate DC function for the induction of potent antitumor CTLs, which can overcome immunosuppression despite of compensatorily increasing intratumoral M-MDSCs. Previous studies have indicated that PMN-MDSCs play a critical role in modulating antitumor CTL responses.17,18,56 Using the PMN-MDSC-depleting antibody 1A8 and M-MDSC-depleting peptibody H6-pep, we showed that PMN-MDSCs but not M-MDSCs are essential for the TME to restrict the induction of tumor-reactive CTL responses during MVTT virotherapy. Moreover, only the combination of MVTT virotherapy and depletion of PMN-MDSCs was able to activate endogenous T cells to elicit antitumor CTLs with broad-reactive spectrum, cytolytic activity and protective long-term memory responses. During this process, the compensatorily increased intratumoral M-MDSCs were unable to block T cell activation and antitumor CTLs. Mechanistically, intratumoral PMN-MDSCs but not M-MDSCs suppressed DC activation by preventing CD80 and CD86 upregulation and IL-6, TNF-α and IL-12p70 secretion. Therefore, in addition to the suppressive effects of MDSCs on T cells that we have previously described,15,16 our current study highlighted the mechanism by which mesothelioma-derived PMN-MDSCs exhibit immune suppressive activity on DCs. Cross-talk between PMN-MDSCs and DCs demolished antitumor immunity by increasing IL-10 production and decreasing DC activation. Previous reports suggested that tumor-derived MDSCs upregulated IL-10 production and neutralization of IL-10 abrogated the suppressive effect of MDSCs in mouse models.57-59 Given the plasticity of the immune suppressive myeloid compartment under various tumors and infectious agents, one pioneer study demonstrated that acute phase response protein induced the expansion and polarization of IL-10-secreting tumor associated neutrophils to suppress antigen specific T cell responses in melanoma patients.60 Thus, our study together with others strongly suggested that IL-10-sereting PMN-MDSCs act as a barricade to protect tumors from immune surveillance. We further provided new evidence that chemotactically recruited IL-10-sereting PMN-MDSCs are critical DC suppressors to halt T cell activation during the MVTT virotherapy.
Combination therapy has become a useful strategy to improve the efficacy of oncolytic virotherapy in fighting various types of tumors, including malignant mesothelioma.61 This type of therapy includes augmenting host antitumor responses through the incorporation of immune activating molecules (e.g., GM-CSF), immune-regulatory drugs (e.g., cyclophosphamide) or immune checkpoint inhibitors.62 In addition to the rapidly increased use of immune checkpoint inhibitors, a GM-CSF-incorporated herpes simplex virus (T-vec) has also received regulatory approval for treating patients with late-stage melanoma.41 Decreasing immune suppression of MDSCs and Tregs by sunitinib has been shown in clinical trials to augment anti-renal cell carcinoma immune responses during oncolytic reovirus treatment.63 In terms of malignant mesothelioma, the use of first-line chemotherapeutic agents (cisplatin or pemetrexed) during oncolytic adenovirus treatment has been shown to enhance virus-mediated cytotoxicity in mice.64 Here, we provide evidence that depletion of tumor-induced PMN-MDSCs during oncolytic virotherapy is potentially a new approach to treat malignant mesothelioma and melanoma. Since recruitment of PMN-MDSCs is a common phenomenon caused by local tumor inflammation following tumor cell lysis in both oncolytic virus and poly(I:C) treatment (data not shown), targeting PMN-MDSCs could be a novel approach to provoke antitumor immunity in these settings.65 To this end, however, the biomarker(s) of human PMN-MDSCs remain to be identified. Here, we showed that CXCR2 inhibition reduced PMN-MDSCs recruitment during MVTT virotherapy and promoted tumor regression. Since human CXCR2+ PMN-MDSCs were reported to be associated with multiple immunosuppressive mechanisms,66 our study suggested that CXCR2 inhibition may serve as an alternative way for PMN-MDSCs blockade in clinical setting. In addition, we recently discovered that inhibiting cell cycle-related kinase (CCRK) signaling diminished PMN-MDSC-mediated immunosuppression and inhibited tumorigenicity of hepatocellular carcinoma.67 Therefore, an epigenetic modulatory approach targeting this druggable CCRK to specifically disrupt PMN-MDSC accumulation would be also important in the development of combination therapy with MVTT for treating a variety of human tumors, including mesothelioma.
Methods
Mice
All mice were maintained according to standard operational procedures at HKU Laboratory Animal Unit (LAU) and all procedures were approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR) of HKU (license #3654–15). 6–8 week-old female BALB/c, C57BL/6N and SCID mice were used.
Cell culture
Vero cells, purchased from ATCC, and B16F10 cells, a kind gift from Dr. Kevin Ng (Department of Surgery, HKU), were maintained in complete Dulbecco’s modified Eagle’s medium (DMEM, Gibco; supplemented with 10% FBS and antibiotics). AB1 cell line, purchased from European Collection of Cell Cultures, was maintained in complete Roswell Park Memorial Institute-1640 medium (RPMI, Gibco; supplemented with 10% FBS, 2 mM L-glutamine and antibiotics). Luciferase-expressing cells were maintained in complete RPMI supplemented with 1 µg/ml puromycin (Invitrogen).15 T cells and splenocytes were cultured in complete RPMI supplemented with 50 µM 2-mercaptoethanol (Sigma).
Virus and in vitro infection
rMVTT viral stocks were prepared as previously described and virus titers were determined by plaque forming assay in Vero cells.68 In vitro infection was performed in 24-well plate with 2 × 105 AB1 mesothelioma cells each well. 0.2 MOI recombinant rMVTT was added into the culture to allow 1 hour attachment before cells were washed and incubated with 1ml fresh medium. Culture supernatant were harvested 24, 48 and 72 hours after infection, and virus titers were measured by plaque forming assay in Vero cells. Released HMGB1 were examined by western blotting using anti-HMGB1 antibody (Abcam, ab79823). Released ATP and cell viability were determined by CellTiter-Glo luminescent cell viability assay (Promega) per the manufacturer’s instructions. Anti-CRT antibody (Abcam, ab92516) was used to detect CRT expression by flow cytometric analysis and western blotting. rMVTT-treated AB1 cell supernatants used for antigen-presentation assay was collected at 48 hours after infection. Cell debris was removed by centrifuge, passed through a 0.2 µm low-protein binding membrane (Millipore) and heat-inactivated at 60°C for 1 hour. Successful elimination of live virus was confirmed by plaque forming assay.
Tumor models and treatment
AB1 or B16F10 cells were harvested and single cell suspensions of 5 × 105 cells in 100 µl PBS were injected s.c into right hind flank of BALB/c or C57BL/6N mice, respectively. Tumor volumes were measured by caliper (Tumor volume = 1/2(length × width2)). Luciferase-expressing tumors were measured with IVIS spectrum (PerkinElmer) and presented as photons/s/cm2/sr within regions of interest (ROI) using Living Image software (version 4.0, PerkinElmer), as previously described.15,16 Established tumors were treated i.t with rMVTT, antibodies or pepducin as indicated in figure legends. CXCR2-pepducin (RTLFKAHMGQKHR, palmitoyl N-terminal, amidation C-terminal) and control-pepducin (TRFLAKMHQGHKR, palmitoyl N-terminal, amidation C-terminal) were synthesized by GL Biochem (Shanghai) with > 95% purity. For pepducin treatment, mice were injected s.c by the side of solid tumors with 2.5 mg/kg pepducin in PBS, followed by 1 mg/kg pepducin daily for the duration of the study.69
Ex vivo cell preparation and MDSC adoptive transfer
Splenocytes were isolated as previously described.15,16 Tumors were cut into pieces and digested with 1mg/ml collagenase IV (Sigma) and 0.5 U/ml DNase I (Roche) for 1.5 hours at 37°C. Cells were passed through a 70 µm strainer and then subjected to 40%/80% Percoll gradient (Sigma). Leukocytes at the interphase were recovered after centrifuge at × 800g 20min. Bone-marrow leukocytes were flushed out from tibia and femur, passed through a 70 µm strainer and following removing red blood cells using Lysing Buffer (BD Biosciences). Single-cell suspensions of splenocytes were used for cell isolation. CD3+ T cells were isolated using Dynabeads Untouched T Cell Kits (Thermo Scientific). CD4+ and CD8+ T cells were isolated using T Cell Isolation Kit (Miltenyi). MDSCs were isolated using MDSCs Isolation Kit (Miltenyi), according to manufacturer’s instructions. Purified MDSCs were labelled with CFSE (Thermo Scientific). 4 × 106 MDSCs were intravenously transferred through tail vein and were detected 24 hours later.
In vivo cell depletion
CD4+ and CD8+ T cells were depleted by intraperitoneal injection of 250 µg anti-CD4 (YTS191.1, BioXcell) or anti-CD8 (YTS169.4, BioXcell), respectively, every 5 days, starting 1 day before therapy. Successful depletion was confirmed by flow cytometric analysis of peripheral blood mononuclear cell (PBMC). i.t treatment of established tumors was started at 7 days after tumor inoculation, with 100 µl of rMVTT, anti-Ly6G antibody (clone 1A8, BioXCell) or combination of the two. 1A8 was administered at 100 μg per dose and rat IgG2a (clone 2A3, BioXcell) was injected alone or in combination with rMVTT as an isotype control. Re-challenge was done with 2 × 106 AB1-Luc cells s.c on their opposite flank. Animals were euthanized when tumor length > 15mm, according to LAU guidelines.
Measurement of cytokine and chemokine production
IL-21p70 was measured by Mouse IL-12p70 DuoSet ELISA kit (R&D). All the other cytokine concentrations were measured by LEGENDplex T Helper Cytokine Panel (BioLegend). Tumors were cut into pieces and homogenized in T-PER Tissue Protein Extraction Reagent (Thermo Scientific) supplemented with Protease Inhibitor Cocktail (Roche). Chemokine concentrations were determined by LEGENDplex Proinflammatory Chemokine Panel (BioLegend) and normalized against total proteins determined by BCA protein assay (Thermo Scientific).
BMDCs culture, in vitro antigen-presentation and suppression assays
Following a standard protocol,35 isolated bone-marrow cells were plated in 6-well plate at 3 × 106 cell per well in the presence of 40 ng/ml GM-CSF and IL-4. Half of the differentiation medium was replaced every 2 days. On day 9, loosely adherent cells were resuspended by repeated pipetting and collected together with non-adherent cells for flow cytometric analysis with anti-CD3, anti-CD11c and anti-MHC II, resulting in > 90% CD11c+MHC II+ BMDCs. For BMDCs-T cells co-culture, BMDCs were seeded into 96-well V-bottom plate at 2 × 104 cells per well in the presence of 100 µl rMVTT-treated AB1 cell supernatants or culture medium. In some cultures, anti-CRT antibody (Abcam, ab92516) or rabbit IgG were added at 100 ng/ml. After incubation overnight, BMDCs were thoroughly washed with culture medium and CFSE labelled CD3+ T cells were added at a ratio of 1:1, for an additional culture of 10 days, with replacement of half of the culture medium every 4 days. Culture supernatant collected on day 7 and cells collected on day 10 were subjected to analysis of cytokine secretion and T cell proliferation, respectively. For BMDCs-MDSCs co-culture, BMDCs were seeded in 96-well U-bottom plate at 5 × 104 cells per well, stimulated by 100 ng/ml LPS (Sigma) or 100 µl rMVTT-treated AB1 cell supernatants, in the presence of 1 × 105 purified MDSCs (BMDC:MDSC = 1:2). To distinguish BMDCs from MDSCs by flow cytometry, purified MDSCs subsets were labelled with CFSE prior to incubation. 48 hours after LPS-stimulation, BMDCs maturation was assessed via flow cytometry. Culture supernatant were collected for measurement of cytokine production.
IL-10 receptor blocking assay
BMDCs were seeded in 96-well U-bottom plate at 5 × 104 cells per well and were subjected to incubate with 5 µg/ml anti-mouse CD210 (IL-10R, clone 1B1.3a, BioLegend) antibody for 30 min at 37°C. Then 1 × 105 CFSE labelled PMN-MDSCs or M-MDSCs were added into the culture at a ratio of 2:1 with BMDCs, following stimulation with 100 ng/ml LPS for 48 hours in the incubator. Culture volume was maintained at 100 µl each well and rat IgG1 (eBioscience) was used as isotype control.
Flow cytometry
Cell surface and intracellular immunostaining were performed as previously described.15 The following antibody were purchased from eBioscience: anti-CD11b (clone M1/70), anti-Ly6C (clone HK1.4), anti-Ly6G (clone 1A8-Ly6g), anti-CD3 (clone 17A2), anti-CD4 (clone GK1.5), anti-CD8 (clone 53–6.7), anti-PD1 (clone J43), anti-Tim3 (clone RMT3-23), anti-CD11c (clone N418), anti-MHC II (clone M5/114.15.2), anti-CD80 (clone 16-10A1), and anti-CD49b (clone DX5). The following antibody were purchased from BioLegend: anti-CD25 (clone 3C7), anti-Foxp3 (clone 150D), anti-CXCR2 (clone SA045E1), anti-CXCR3 (clone CXCR3-173), anti-LAP (TGF-β1, clone TW7-16B4) and anti-IL-10 (clone JES5-16E3). Anti-CCR2 (clone REA538) was purchased from Miltenyi. Samples were run on a BD FACSAria II cell sorter (BD Biosciences) and analyzed using FlowJo (Tree Star, v10).
ELISpot and T cell cytotoxicity assay
IFN-γ-producing T cells in isolated splenocytes was assessed by ELISpot assay.15,16 gp70-AH1 (SPSYVYHQF), OVA257-264(SIINFEKL), GP100 (EGPRNQDWL), TRP2 (SVYDFFVWL), and TWIST1 peptides (15-mers spanning the entire amino acid sequence with 11 amino acids overlapping) were synthesized by GL Biochem (Shanghai). Cytotoxic effect of purified T cells against AB1 cells was determined using LIVE/DEAD Viability/Cytotoxicity Kit (Thermo Scientific), as previously described.15
Statistical analyses
All data are presented as mean ± s.e.m. Significance was determined by the two-tailed Student t-test and P-value < 0.05 was considered statistically significant. Survival of all animals was plotted on Kaplan-Meier survival curve and the log-rank test was performed to analyze differences in GraphPad Prism 5 software.
Supplementary Material
Funding Statement
This work was supported by the Hong Kong Health and Medical Research Fund [03142666, 04151266]; Hong Kong Pneumoconiosis Compensation Fund Board; Hong Kong Research Grant Council [TRS T11-706/18-N, HKU5/CRF/13G, RGC17122915].
Acknowledgments
We sincerely thank Dr. Allen KL Cheung for critical reading and editing of the manuscript and Michael YC Wong for providing critical discussion.
Conflict of interest
The authors declare that they have no competing financial interests.
References
- 1.Yap TA, Aerts JG, Popat S, Fennell DA.. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer. 2017;17:475–488. doi: 10.1038/nrc.2017.42 [DOI] [PubMed] [Google Scholar]
- 2.Rusch VW. Pemetrexed and cisplatin for malignant pleural mesothelioma: a new standard of care? J Clin Oncol. 2003;21:2629–2630. doi: 10.1200/JCO.2003.02.043 [DOI] [PubMed] [Google Scholar]
- 3.Dozier J, Zheng H, Adusumilli PS. Immunotherapy for malignant pleural mesothelioma: current status and future directions. Transl Lung Cancer Res. 2017;6:315–324. doi: 10.21037/tlcr.2017.05.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–1379. doi: 10.1111/cas.13027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranki T, Pesonen S, Hemminki A, Partanen K, Kairemo K, Alanko T, Lundin J, Linder N, Turkki R, Ristimäki A, et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer. 2016;4:17. doi: 10.1186/s40425-016-0121-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 9.Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 2012;209:495–506. doi: 10.1084/jem.20111413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17:718–730. doi: 10.1245/s10434-009-0809-6 [DOI] [PubMed] [Google Scholar]
- 12.Ranki T, Joensuu T, Jäger E, Karbach J, Wahle C, Kairemo K, Alanko T, Partanen K, Turkki R, Linder N, et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8+ T-cell response, prominent infiltration of CD8+ lymphocytes and Th1 type polarization. Oncoimmunology. 2014;3:e958937. doi: 10.4161/21624011.2014.958937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M, Nokisalmi P, Raki M, Laasonen L, Särkioja M, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010;70:4297–4309. doi: 10.1158/0008-5472.CAN-09-3567 [DOI] [PubMed] [Google Scholar]
- 14.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan Z, Zhou J, Cheung AK, Yu Z, Cheung KW, Liang J, Wang H, Lee BK, Man K, Liu L, et al. Vaccine-elicited CD8+ T cells cure mesothelioma by overcoming tumor-induced immunosuppressive environment. Cancer Res. 2014;74:6010–6021. doi: 10.1158/0008-5472.CAN-14-0473 [DOI] [PubMed] [Google Scholar]
- 16.Yu Z, Tan Z, Lee BK, Tang J, Wu X, Cheung K-W, Lo NTL, Man K, Liu L, Chen Z. Antigen spreading-induced CD8+T cells confer protection against the lethal challenge of wild-type malignant mesothelioma by eliminating myeloid-derived suppressor cells. Oncotarget. 2015;6:32426–32438. doi: 10.18632/oncotarget.5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic virus-mediated targeting of PGE2 in the tumor alters the immune status and sensitizes Established and resistant tumors to immunotherapy. Cancer Cell. 2016;30:108–119. doi: 10.1016/j.ccell.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veltman JD, Lambers MEH, Van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. Bmc Cancer. 2010;10:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada N, Oizumi S, Kikuchi E, Shinagawa N, Konishi-Sakakibara J, Ishimine A, Aoe K, Gemba K, Kishimoto T, Torigoe T, et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol Immunother. 2010;59:1543–1549. doi: 10.1007/s00262-010-0881-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380 [DOI] [PubMed] [Google Scholar]
- 21.Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G, Sehgal S, Raj A, Matthaiou E, Coukos G, et al. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J Clin Invest. 2014;124:1497–1511. doi: 10.1172/JCI67382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreiter S, Vormehr M, Van De Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90–106. doi: 10.1038/cr.2011.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Youn J-I, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–2975. doi: 10.1002/eji.201040895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortin C, Huang XP, Yang YP. NK Cell Response to Vaccinia Virus Is Regulated by Myeloid-Derived Suppressor Cells. J Immunol. 2012;189:1843–1849. doi: 10.4049/jimmunol.1200584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, Lehner F, Manns MP, Greten TF, Korangy F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807. doi: 10.1002/hep.23054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, Merghoub T, Wolchok JD, Allison JP. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340. doi: 10.1038/ncomms14340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh R, Lillard JW Jr., Singh S. Chemokines: key players in cancer progression and metastasis. Front Biosci (Schol Ed). 2011;3:1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6:237ra67. doi: 10.1126/scitranslmed.3007974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin H, Lerman B, Sakamaki I, Wei GW, Cha SCC, Rao SS, Qian J, Hailemichael Y, Nurieva R, Dwyer KC, et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med. 2014;20:676–681. doi: 10.1038/nm.3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29:832–845. doi: 10.1016/j.ccell.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steele CW, Karim SA, Foth M, Rishi L, Leach JDG, Porter RJ, Nixon C, Jeffry Evans TR, Carter CR, Nibbs RJB, et al. CXCR2 inhibition suppresses acute and chronic pancreatic inflammation. J Pathol. 2015;237:85–97. doi: 10.1002/path.4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22:275–281. doi: 10.1016/j.semcancer.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis E Sousa C. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. 2015;42:1197–1211. doi: 10.1016/j.immuni.2015.05.018 [DOI] [PubMed] [Google Scholar]
- 36.Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med. 1999;189:797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt SV, Nino-Castro AC, Schultze JL. Regulatory dendritic cells: there is more than just immune activation. Front Immunol. 2012;3:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977 [DOI] [PubMed] [Google Scholar]
- 40.Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukocyte Biol. 2014;96:1109–1118. doi: 10.1189/jlb.3A0414-210R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 42.Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22:1048–1054. doi: 10.1158/1078-0432.CCR-15-2667 [DOI] [PubMed] [Google Scholar]
- 43.Pol J, Buqué A, Aranda F, Bloy N, Cremer I, Eggermont A, Erbs P, Fucikova J, Galon J, Limacher J-M, et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology. 2016;5:e1117740. doi: 10.1080/2162402X.2015.1117740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krug LM, Zauderer MG, Adusumili PS, Mcgee E, Sepkowitz K, Klang M, Yu YA, Scigalla P, Rusch VW. Phase I study of intra-pleural administration of GL-ONC1, an oncolytic vaccinia virus, in patients with malignant pleural effusion. J Clin Oncol. 2015;33:7559. [Google Scholar]
- 45.Boisgerault N, Achard C, Delaunay T, Cellerin L, Tangy F, Grégoire M, Fonteneau J-F. Oncolytic virotherapy for human malignant mesothelioma: recent advances. Oncolytic Virother. 2015;4:133–140. doi: 10.2147/OV.S66091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Research. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974 [DOI] [PubMed] [Google Scholar]
- 47.Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K, et al. Coxsackievirus B3 Is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Research. 2012;72:2609–2621. doi: 10.1158/0008-5472.CAN-11-3185 [DOI] [PubMed] [Google Scholar]
- 48.Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–6s. doi: 10.1158/1078-0432.CCR-06-2197 [DOI] [PubMed] [Google Scholar]
- 49.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Almand B, Clark JI, Nikitina E, Van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. [DOI] [PubMed] [Google Scholar]
- 51.Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72:876–886. doi: 10.1158/0008-5472.CAN-11-1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. 2017;8:14754. doi: 10.1038/ncomms14754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Srivastava MK, Zhu L, Harris-White M, Kar UK, Huang M, Johnson MF, Lee JM, Elashoff D, Strieter R, Dubinett S, et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS One. 2012;7:e40677. doi: 10.1371/journal.pone.0040677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63:1769–1781. doi: 10.1136/gutjnl-2013-306271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, Harken JA, Robinson BW, Lake RA, De Mello RA. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One. 2013;8:e61895. doi: 10.1371/journal.pone.0061895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glodde N, Bald T, Van Den Boorn-Konijnenberg D, Nakamura K, O’Donnell JS, Szczepanski S, Brandes M, Eickhoff S, Das I, Shridhar N, et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-met limit cancer immunotherapy. Immunity. 2017;47:789–802.e9. doi: 10.1016/j.immuni.2017.09.012 [DOI] [PubMed] [Google Scholar]
- 57.Van Valckenborgh E, Schouppe E, Movahedi K, De Bruyne E, Menu E, De Baetselier P, Vanderkerken K, Van Ginderachter JA. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia. 2012;26:2424–2428. doi: 10.1038/leu.2012.113 [DOI] [PubMed] [Google Scholar]
- 58.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi: 10.1084/jem.20131916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen S-H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299 [DOI] [PubMed] [Google Scholar]
- 60.De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, Salio M, Middleton M, Cerundolo V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol. 2010;11:1039–1046. doi: 10.1038/ni.1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer. 2018;18:419–432. doi: 10.1038/s41568-018-0009-4 [DOI] [PubMed] [Google Scholar]
- 62.Meyers DE, Wang AA, Thirukkumaran CM, Morris DG. Current Immunotherapeutic strategies to enhance oncolytic virotherapy. Front Oncol. 2017;7:114. doi: 10.3389/fonc.2017.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lawson KA, Mostafa AA, Shi ZQ, Spurrell J, Chen W, Kawakami J, Gratton K, Thakur S, Morris DG. Repurposing Sunitinib with Oncolytic Reovirus as a Novel Immunotherapeutic Strategy for Renal Cell Carcinoma. Clin Cancer Res. 2016;22:5839–5850. doi: 10.1158/1078-0432.CCR-16-0143 [DOI] [PubMed] [Google Scholar]
- 64.Yamanaka M, Tada Y, Kawamura K, Li Q, Okamoto S, Chai K, Yokoi S, Liang M, Fukamachi T, Kobayashi H, et al. E1B-55 kDa-defective adenoviruses activate p53 in mesothelioma and enhance cytotoxicity of anticancer agents. J Thorac Oncol. 2012;7:1850–1857. doi: 10.1097/JTO.0b013e3182725fa4 [DOI] [PubMed] [Google Scholar]
- 65.Bianchi F, Pretto S, Tagliabue E, Balsari A, Sfondrini L. Exploiting poly(I:C) to induce cancer cell apoptosis. Cancer Biol Ther. 2017;18:747–756. doi: 10.1080/15384047.2017.1373220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Najjar YG, Rayman P, Jia X, Pavicic PG Jr., Rini BI, Tannenbaum C, Ko J, Haywood S, Cohen P, Hamilton T, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and Mip-1α. Clin Cancer Res. 2017;23:2346–2355. doi: 10.1158/1078-0432.CCR-15-1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, Tong JH, Wong J, Chong CCN, Lai PBS, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018; 67:931–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu W, Fang Q, Zhu W, Wang H, Tien P, Zhang L, Chen Z. One time intranasal vaccination with a modified vaccinia Tiantan strain MVTT(ZCI) protects animals against pathogenic viral challenge. Vaccine. 2010;28:2088–2096. doi: 10.1016/j.vaccine.2009.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJB, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122:3127–3144. doi: 10.1172/JCI61067 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.