

ABSTRACT
Ovarian cancer is frequently diagnosed as peritoneal carcinomatosis. Unlike other tumor locations, the peritoneal cavity is commonly exposed to gut-breaching and ascending genital microorganisms and has a unique immune environment. IL-33 is a local cytokine that can activate innate and adaptive immunity. We studied the effectiveness of local IL-33 delivery in the treatment of cancer that has metastasized to the peritoneal cavity. Direct peritoneal administration of IL-33 delayed the progression of metastatic peritoneal cancer. Prolongation in survival was not associated with a direct effect of IL-33 on tumor cells, but with major changes in the immune microenvironment of the tumor. IL-33 promoted a significant increase in the leukocyte compartment of the tumor immunoenvironment and an allergic cytokine profile. We observed a substantial increase in the number of activated CD4+ T-cells accompanied by peritoneal eosinophil infiltration, B-cell activation and activation of peritoneal macrophages which displayed tumoricidal capacity. Depletion of CD4+ cells, eosinophils or macrophages reduced the anti-tumor effects of IL-33 but none of these alone were sufficient to completely abrogate its positive benefit. In conclusion, local administration of IL-33 generates an allergic tumor environment resulting in a novel approach for treatment of metastatic peritoneal malignancies, such as advanced ovarian cancer.
KEYWORDS: Immunotherapy, ovarian cancer, IL-33, tumor immunology, Peritoneal cavity
Introduction
Ovarian cancer is the deadliest gynecological cancer which has an incidence of approximately 22,200 women/year, 60% of the patients being diagnosed after the disease has spread outside of the ovaries, which is associated with a dismal 5-year survival rate of 28.9%.1,2 Because of this frequently metastatic stage at diagnosis, surgery and chemotherapy have barely improved survival over the past 40 years. However, metastatic spreading of ovarian cancer is typically limited to the peritoneal cavity. Therefore, local administration of therapeutic agents is a preferred approach for this cancer. Local injection can achieve higher in-tumor drug concentrations with less systemic adverse effects. Intraperitoneal (local) chemotherapy has shown improved overall survival and progression-free survival for ovarian cancer when compared with intravenous administration.3,4 Due to its benefits, local cytokine therapy has also been studied in peritoneal tumors using IL-2, IFNγ and IFNα.5-7
Interleukin-33 (IL-33) is a cytokine of the IL-1 superfamily that exerts its functions through the ST2 receptor (IL-1R-like1).8 Depending on its context, IL-33 has been reported to promote either Th1 9,10 or Th2 like immunity.8 We have previously reported that IL-33 can be used as an immunoadjuvant to enhance DNA vaccine induced anti-tumor immunity by enhancing a Th1 response as an adjuvant in the periphery, particularly when expressed in muscle.9 However, as ovarian cancer is often restricted to the peritoneal cavity, which is subject to heightened immunosurveilance, we hypothesized that IL-33 alone may promote a local immune response that could impact the prognosis of advanced ovarian cancer.
In this paper we study the effects of direct intraperitoneal therapy with IL-33. We report that intraperitoneal treatment with IL-33 delays tumor progression in peritoneal carcinomatosis, including a murine model of metastatic ovarian cancer. Furthermore, we show that this protection depends on the promotion of an allergic like environment, through the action of peritoneal CD4+ T-cells, eosinophils and macrophages.
Results
IL-33 delays ovarian cancer progression
Advanced ovarian cancer is considered a peritoneal disease, and local treatment is recommended due to advantages in drug delivery into the local tumor environment.11 IL-33 is a short-lived, locally active cytokine. We hypothesized that peritoneal immunotherapy using IL-33 could impact peritoneally-metastasized ovarian cancer by activating tumor-associated inflammation. Therefore, we challenged mice with ID8-Defb29/Vegf-a tumor cells, an aggressive ovarian cancer model engineered to accelerate peritoneal carcinomatosis and ascites accumulation in vivo. Following tumor challenge, we treated the mice with weekly administration of either IL-33 or PBS starting on day 7 (Figure 1a). Only 5 administrations were needed to reproducibly improve median survival by approximately 140% from 50 to 68 days (Figure 1b). To better understand the mechanism by which IL-33 delays tumor progression, we analyzed and ruled out that ID8-Defb29/Vegf-a tumor cells expressed the ST2 receptor (Figure 1c) thus eliminating that IL-33 plays a direct effect on the tumor. In addition, we found no difference in in vitro proliferation of ID8-Defb29/Vegf-a in the presence or absence of IL-33 (Figure 1d), supporting that this antitumor effect was highly unlikely to be a direct effect of IL-33 on the tumor cells. Furthermore, the effectiveness of local administration of IL-33 was not limited to ovarian tumors, as in a second study, intraperitoneal LLC tumor progression was similarly delayed by IL-33 treatment (Figure 1e).
Figure 1.
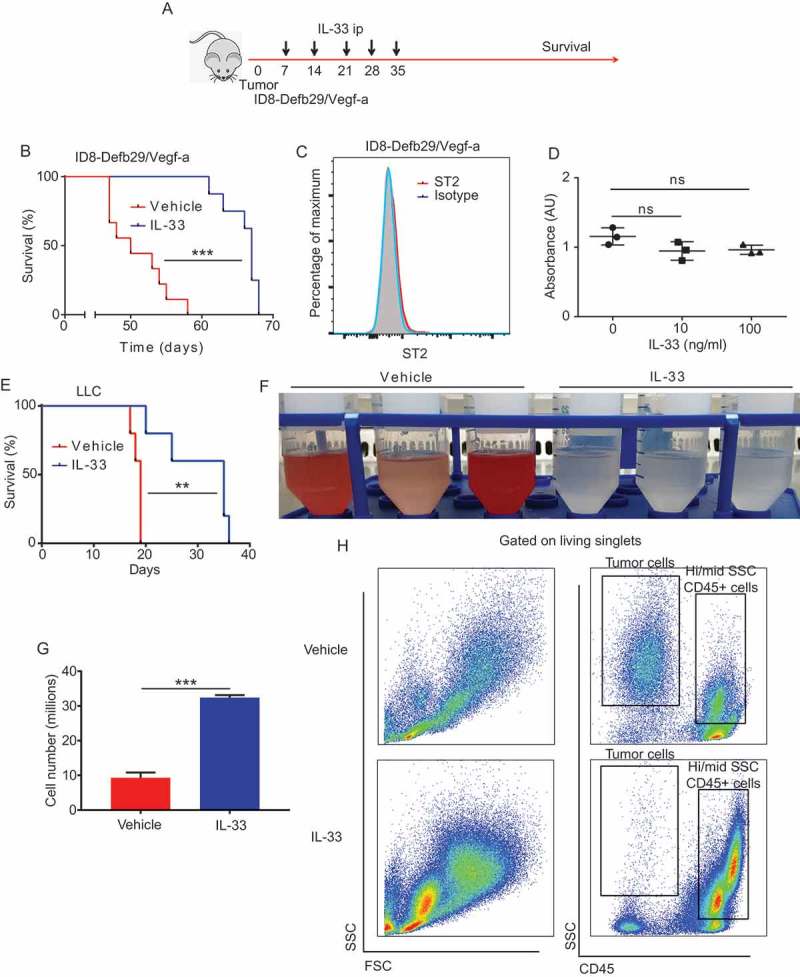
IL-33 delays ovarian cancer tumor progression.
(A) Schematic of IL-33 survival experiments (B) Survival plot of mice bearing intraperitoneal ID8-Defb29/Vegf-a syngeneic tumors treated intraperitoneally with IL-33 or PBS at days 7, 14, 21, 28 and 35 after tumor challenge (n = 9 per group, pooled from 2 independent experiments). (C) Expression flow cytometry of ST2 receptor by ID8-Defb29/Vegf-a (2 independent experiments) (D) ID8-Defb29/Vegf-a in vitro proliferation in the presence of IL-33 (n = 3 per group; 2 independent experiments) (E) Survival plot of mice bearing intraperitoneal Lewis lung carcinoma treated intraperitoneally with IL-33 or PBS at days 7, 8, 9, 10 and 11 after tumor challenge (n = 5 per group). (F) Peritoneal wash of mice bearing intraperitoneal ID8-Defb29/Vegf-a syngeneic tumors treated intraperitoneally with IL-33 or PBS at days 21 and 28, harvested at day 30 (n = 3mice per group; 3 independent experiments). (G) Cell count from peritoneal wash of mice bearing intraperitoneal ID8-Defb29/Vegf-a syngeneic tumors treated intraperitoneally with IL-33 or PBS at days 21 and 28, harvested at day 30 (> 3 independent experiments). (H) Representative flow cytometry plots of the ascites fluid (tumor microenvironment) of mice bearing intraperitoneal ID8-Defb29/Vegf-a syngeneic tumors treated intraperitoneally with mature IL-33 or PBS at days 21 and 28, harvested at day 30. (> 3 independent experiments). Log-Rank test, ANOVA, t-test. AU: arbitrary units, ns: not significant, **p < 0.01, ***p < 0.001.
To gain a better understanding of the effect of IL-33 on the peritoneal microenvironment we again challenged mice with ID8-Defb29/Vegf-a tumor and euthanized the mice after the third IL-33 administration to perform peritoneal lavage. Surprisingly, we found no accumulation of bloody ascites in the peritoneal cavity of the IL-33 treated mice (Figure 1f) suggesting decreased tumor burden. Correspondingly, in the IL-33 peritoneal lavage there is a substantial increase in the total number of CD45+ leukocytes (Figure 1g), especially intermediate and highly granular CD45+ cells and a decrease CD45- SSC hi tumor cells (Figure 1h).
These data support that local IL-33 administration delays peritoneal cancer progression through a tumor independent mechanism, and this survival is associated with decreased intraperitoneal tumor cells, as well as increased peritoneal leukocytes.
IL-33 promotes an allergic like infiltration of the peritoneal cavity
IL-33 is associated with the pathogenesis of allergy and asthma.12,13 We were curious if an allergic phenotype could be playing a role in the antitumor response observed. Accordingly, we analyzed the cytokine expression patterns from the peritoneal cells derived from IL-33 treated vs control mice. IL-33 treated mice exhibited higher levels of expression of IL-5 and IL-13, two classical cytokines of allergic and Th2 responses,14 when compared to controls (Figure 2a&b). However, in contrast to classical Th2 responses, there were no differences in IL-10 expression (Figure 2c). In accordance with an allergic response, we also found an increase in the IL-33 receptor, ST2 (Figure 2d)15 and a dramatic increase in the levels of Ym1(Figure 2e).16
Figure 2.

IL-33 promotes an allergic like infiltration of the peritoneal cavity.
Mice were challenged with intraperitoneal ID8-Defb29/Vegf-a tumors and treated at days 7,14 and 21 with intraperitoneal IL-33 or PBS. Two days later we performed a peritoneal wash and analyzed the peritoneal cellular compartment. (A) Quantitative real time PCR showing relative quantification of IL-5, (B) IL-13 (product of IL-13 and GAPDH of each mouse shown in agarose gel), (C) IL-10, (D)ST2 and (E)Ym1 from IL-33 treated mice relative to PBS treated mice (n = 5 mice per group). t-test. ns: not significant, *p < 0.05, ***p < 0.001.
IL-33 promotes peritoneal CD4 t-cell and b cell activation and eosinophil recruitment
The IL-33 receptor ST2 is preferentially expressed on the surface of Th2 CD4 T-cells, which can induce their proliferation with unique cytokine production upon activation.17 Following IL-33 treatment the total peritoneal T-cell numbers were not different compared to control treated animals (Figure 3a). However, in the IL-33 treated mice, CD4 T-cells were overrepresented as the animals exhibited a significant decrease of CD8 T-cells locally (Figure 3b&c). In contrast there was an enhancement of the CD4 T cell effector function as these cells exhibited an activated phenotype as illustrated by increased levels of expression of CD44 and CD69 (Figure 3d) as well as an increased expression of the CD40L activation maker (Figure 3e). This activation was predominantly observed for the peritoneal IL-33 treated CD4+ T-cell group. CD4 activation likely results from a convergence of multiple factors elicited by IL-33, since IL-33 alone was able to only modestly enhance Th2 skewed CD4 T cell activation (but not naïve CD4) (Supplemental Figure 1a). Furthermore, IL-33 did not directly induce CD4 T cell proliferation (Supplemental Figure 1b).
Figure 3.
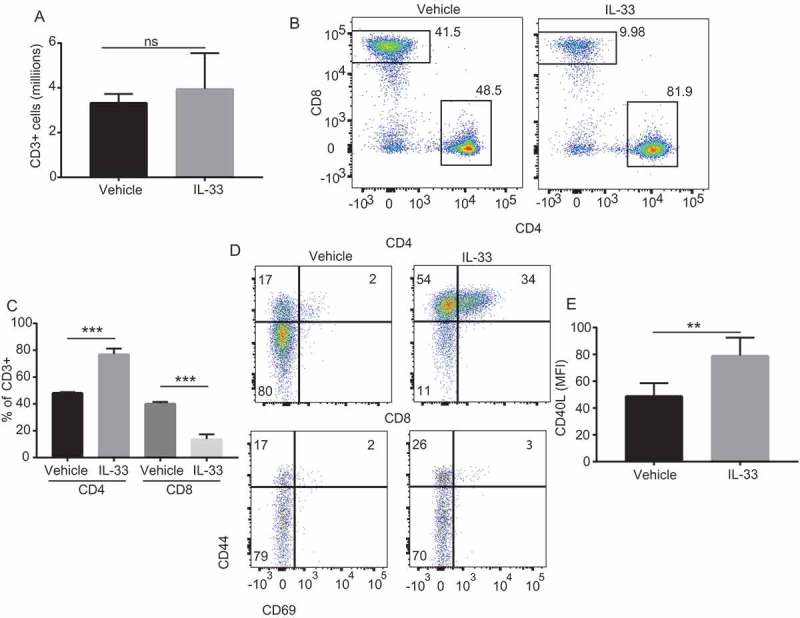
IL-33 promotes recruitment and activation of peritoneal CD4 T cells.
(A) Number of CD3+ cells in the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a treated with IL-33 or PBS (n = 5 mice per group, 2 independent experiments) (B) Representative flow cytometry plots and (C) percentage values of CD4+ and CD8+ T-cells in the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a treated with IL-33 or PBS (n = 5 mice per group, 2 independent experiments). (D) Representative flow cytometry plots showing CD44 and CD69 of CD4+ and CD8+ T-cells from the peritoneal cavity of ID8-Defb29/Vegf-a bearing mice treated with IL-33 or PBS (n = 5 mice per group, 2 independent experiments) (E) Mean fluorescence intensity of CD40L from CD4+ T-cells from of ID8-Defb29/Vegf-a bearing mice treated with IL-33 or PBS (n = 5 mice per group, 2 independent experiments). t-test and ANOVA. ns: not significant, **p < 0.01 ***p < 0.001.
We next analyzed the cytokine expression pattern of CD4 T-cells in the peritoneal cavity of IL-33 treated mice. We observed an allergic like response with higher expression of IL-5 and IL-13 than controls (Figure 4a-c).18,19 Consistent with a productive allergic response we found significantly higher activation of B-cells (Figure 4d) and increased levels of IgE (Figure 4e). B cell numbers were not different in IL-33 vs PBS treated mice (Suppl Figure 2a). In contrast to tumor-associated regulatory responses, B cells showed a dramatic decrease in IL-10 production compared to controls (Figure 4f).20 As in allergic responses we also found an intense eosinophil infiltration in the tumor microenvironment of IL-33 treated mice (Figure 4g).21 Taken together, these data suggest that direct peritoneal delivery of IL-33 can mediate an allergic-like CD4 T-cell activation and expansion, which mediates eosinophil recruitment and promotes uniquely B-cell activation with predominant class switching into IgE and decrease in production of IL-10.
Figure 4.

IL-33 promotes peritoneal CD4 T-cell and B cell activation and eosinophil recruitment.
(A) Real-time quantitative-PCR of IL-5 expression in CD4+ T-cells sorted from the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a syngeneic tumors treated intraperitoneally with IL-33 or PBS (triplicates, pooled from 5 mice per group, 2 independent experiments). (B) Percentage of CD4+ T-cells expressing IL-5 in the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a tumors treated intraperitoneally with IL-33 or PBS (n = 5 per group). (C) Real-time quantitative-PCR of IL-13 expression in CD4+ T-cells sorted from the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a (triplicates, pooled from 5 mice per group (D) Levels of MHCII expressed in B-cells from the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a tumors treated intraperitoneally with IL-33 or PBS (n = 5 per group, 2 independent experiments). (E) Histogram showing levels of IgE in peritoneal fluid obtained by paracentesis from mice bearing intraperitoneal ID8-Defb29/Vegf-a syngeneic tumors treated intraperitoneally with IL-33 or PBS (pooled of 3 independent experiments with n = 3–5 mice per group each). (F) Real-time quantitative-PCR of IL-13 expression in B-cells sorted from the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a (n = 5 per group). (G) Histogram and flow cytometry plots showing the percentage of eosinophils the peritoneal cavity of mice bearing intraperitoneal ID8-Defb29/Vegf-a tumors treated intraperitoneally with IL-33 or PBS (n = 5 per group, 2 independent experiments). T-test. *p < 0.05 ***p < 0.001.
IL-33 promotes activation of peritoneal macrophages
Unlike other tumor locations, the peritoneal cavity is commonly exposed to gut breaching and ascending genital microorganisms, which makes it particularly sensitive to changes in innate immunity. Therefore, the peritoneal cavity has a resident population of ontogenically differentiated resident macrophages that serve as first line of defense.22 Allergic reactions are characterized by the presence of an activated macrophages, also described as wound-healing or M2a macrophages.23-25 Accordingly, we found that peritoneal macrophages presented this allergic like activated phenotype shown by an increased production of Ym-1 and IL-13 (Figure 5a&b). These macrophages also expressed the IL-33 receptor ST2 (Figure 5c), thus allowing for their direct IL-33 stimulation. These differed from the M2c regulatory macrophages or tumor-associated macrophages by a lack of expression of the regulatory cytokine IL-10 (Figure 5d). Interestingly, this phenotype was associated with decreased expression of CD40 (Figure 5e & Suppl. Figure 1c) and CD80 (Figure 5f & Suppl. Figure 1d) but an increase in CD86 (Figure 5g) compared to PBS treated mice. The differential regulation of CD80 and CD86 has been previously reported in asthmatic patients, suggesting an important role for CD86 after allergen challenge.26
Figure 5.
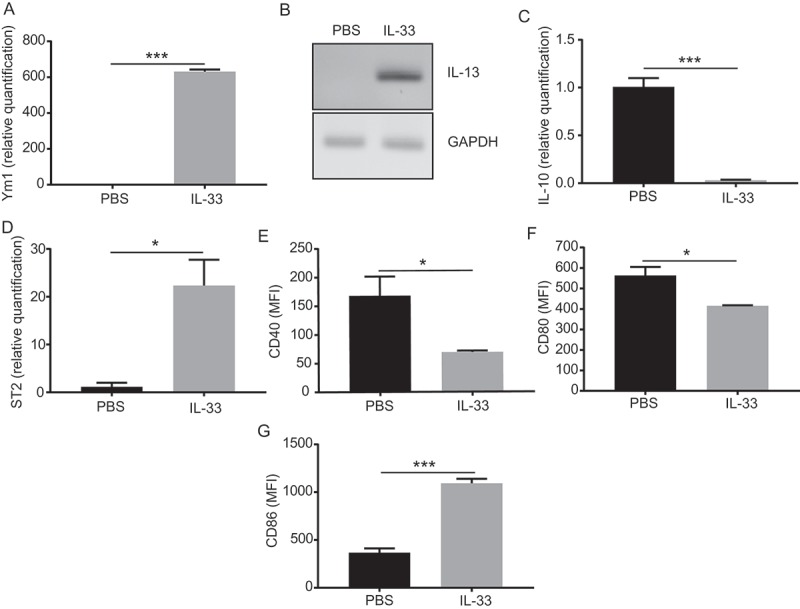
IL-33 promotes activation of peritoneal macrophages.
Mice were challenged with intraperitoneal ID8-Defb29/Vegf-a tumors and treated at days 7,14 and 21 with intraperitoneal IL-33 or PBS. Two days later we performed a peritoneal wash and performed peritoneal cell staining for flow cytometry and flow cytometry associated cell sorting of CD11b+F4/80+ peritoneal macrophages. (A) Quantitative real time PCR showing relative quantification of Ym1, (B) IL-13, (C) ST2 and (D) IL-10 from IL-33 treated mice relative to macrophages from PBS treated mice. Mean fluorescence intensity of macrophages stained for (E) CD40, (F) CD80 and (G) CD86 from IL-33 or PBS treated mice. (triplicates, pooled from 5 mice per group, 2 independent experiments, 2 independent experiments). t-test. *p < 0.05 ***p < 0.001
In summary the data support that the local inflammatory milieu mediated by IL-33 induces the activation of tumor-associated peritoneal macrophages similar to that occurring in allergic disease.
In these studies, maintenance of the allergic response appears important for the IL-33 mediated delay of ovarian tumor progression
To study the etiology of peritoneal tumor delay after treatment with IL-33, we investigated the tumoricidal activity of the cells derived from peritoneal washes of mice that had received IL-33 or PBS. Cells derived from the peritoneal cavity of IL-33 treated mice were able to kill ID8-Defb29/Vegf-a tumor cells upon coincubation (Figure 6a). To determine whether the activated macrophages or the newly recruited eosinophils were the responsible for this tumor cell lysis, we repeated the cytotoxicity assay with macrophages or eosinophils isolated from IL-33 treated mice or macrophages from PBS treated mice. To our surprise, eosinophils sorted from the peritoneal cavity of IL-33 treated mice were not able to lyse tumor cells (Figure 6b). However, IL-33 activated macrophages were able to lyse tumor cells directly in vitro (Figure 6b).
Figure 6.
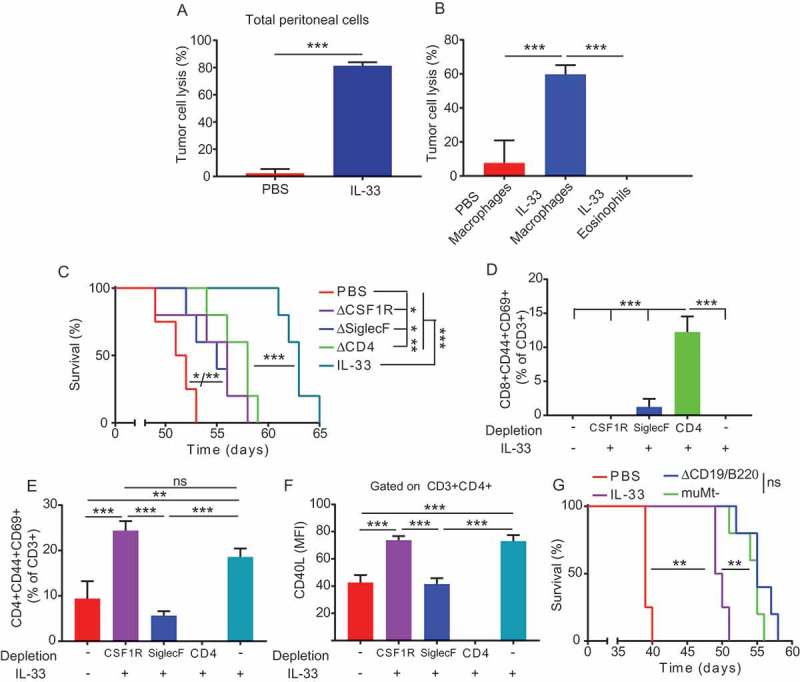
Maintenance of the Th2 response is necessary for the IL-33 mediated delay tumor progression in ovarian cancer.
(A) Cytotoxicity of peritoneal cells from IL-33 or PBS treated ID8-Defb29/Vegf-a bearing mice measured by luciferase absorbance after co-culture of 18 hours with ID8-Defb29/Vegf-a-luciferase tumor cells (triplicates, 2 independent experiments). (B) Cytotoxicity of macrophages or eosinophils sorted from the peritoneal cavity of IL-33 or PBS treated ID8-Defb29/Vegf-a bearing mice measured by luciferase absorbance after co-culture of 18 hours with ID8-Defb29/Vegf-a-luciferase tumor cells in the presence of 100ng/ml of IL-33 (triplicates, 2 independent experiments) (C) Survival plot of mice bearing intraperitoneal ID8-Defb29/Vegf-a tumors treated intraperitoneally with mature IL-33 or PBS at days 7, 14, 21, 28 and 35 after tumor challenge with anti-mouse CSF1R, Siglec-F or CD4 antibodies or irrelevant IgG (n = 5 per group). (D) Percentage of activated (CD44+CD69+) CD8+ T-cells in the peritoneal cavity of the different groups shown in (D). (E) Percentage of activated (CD44+CD69+) CD4+ T-cells in the peritoneal cavity of the different groups shown in (D). (F) CD40L expression of CD4+ T-cells in the peritoneal cavity of the different groups shown in (D) measured as mean fluorescent intensity. (G) Survival plot of mice bearing intraperitoneal ID8-Defb29/Vegf-a tumors treated intraperitoneally with mature IL-33 or PBS at days 7, 14, 21, 28 and 35 after tumor challenge with or without B cell depletion or B cell KO (muMt-) (n = 5 per group).t-test, ANOVA, Log Rank. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the relative contribution of peritoneal macrophages, eosinophils or CD4 T-cells in mediating in vivo anti-tumor effects of IL-33, we treated ID8-Defb29/Vegf-a tumor bearing mice with IL-33 while depleting macrophages, eosinophils or CD4+ T-cells. Depletion of any of these cell types resulted in a negative impact on the IL-33 anti-tumor effects, but none of these individually fully abrogated the overall effect (Figure 6c). Depletion of CD4+ T-cells was associated with a marked increase in activated CD8 T-cells in the tumor microenvironment (Figure 6d). This increase in activated CD8+ T-cells was unable to exert the anti-tumor effect that occurs with the predominant presence of CD4+ T-cells in the presence of IL-33. The relevance of activated CD4 T-cells was further supported by the results of eosinophil depletion. Eosinophil depletion showed a lack in activation of CD4 T-cells comparable to the mice receiving only PBS (Figure 6e), with them also harboring lower levels of CD40L (Figure 6f) and a restoration of the CD4:CD8 ratio to that of PBS treated mice (Supplemental Figure 3a), which suggests that eosinophils are required for optimal activation of intratumoral CD4 T-cells. As in CD4 depletion, eosinophil depletion resulted in decreased survival advantage of IL-33 therapy (Figure 6c).
On the other hand, macrophage depletion did not show any difference in proportions of T cells when compared with IL-33 treatment alone, suggesting that the absence of direct anti-tumor activity elicited by the macrophages is impairing the IL-33 effectiveness in the case of their depletion (Figure 6d-f and Supplemental Figure 3a). Additionally, depletion of both CD4 T cells and eosinophils did not alter the upregulation of CD86 and ST2 or the cytotoxic ability of peritoneal macrophages upon IL-33 treatment (Supplemental Figure 3b-d).
Together the data support that IL-33-induced increase in survival requires the presence of activated CD4 T-cells, the recruitment of eosinophils in the tumor microenvironment with the activation of local peritoneal macrophages in order to obtain the full survival benefit.
B cell depletion increases IL-33 anti-tumor efficacy
To further study the effect of activated peritoneal B cells in the IL-33 treated mice, we treated ID8-Defb29/Vegf-a tumor bearing mice with IL-33 while depleting B-cells or in B cell deficient mice (muMt-). Interestingly, lack of B cells during IL-33 treatment resulted in an increased survival when compared to the IL-33 treatment alone (Figure 6g). This suggests that allergic phenotype B cells induced by IL-33 results detrimental for the in vivo anti-tumor effect of IL-33.
Discusion
Here we describe that the cytokine IL-33 is able to extend survival in metastatic ovarian cancer by driving an allergic like local immune microenvironment. Local IP administration of IL-33 into ovarian tumor-bearing mice drove the recruitment and activation of allergic like CD4+ T-cells in the peritoneal cavity, recruitment of eosinophils and secretion of IgE. This inflammatory milieu resulted in driving a phenotypically novel population of peritoneal macrophages capable of direct tumor cell killing. These changes in the ovarian tumor microenvironment were able to promote a long increase in survival in ovarian-cancer bearing mice.
IL-33 has been described as a key initiator of acute local inflammation and tissue-repair.27 IL-33 is present in the cell nucleus under normal conditions and only released from cells after injury or necrosis. After its release, IL-33 is inactivated by proteolytic cleavage28 or oxidation.29 IL-33 is predominantly expressed in epithelial and endothelial cells. High constitutive release generates an allergic inflammation with eosinophilia.30 Increased secretion of IL-33 is a characteristic finding in the pathogenesis of allergy12,31 and asthma.13,32,33 In our ovarian cancer tumor model the local increase in IL-33 resulted in a IL5+IL-13+ skewing of CD4+ T-cells and recruitment and activation of eosinophils, similar to allergic disease. This allergic response differed from a classical regulatory Th2 response in that there was a general downregulation of IL-10. As determined by our depletion experiments, this induced allergic reaction was able to delay tumor progression more effectively than the infiltration by activated CD8+ T-cells which we observed interestingly following CD4+ T-cell depletion. Although ovarian cancer can be immunogenic, it is generally protected by a suppressive tumor microenvironment that prevents priming of tumor-specific T-cells and suppresses the direct effect of anti-tumor CTLs.34-36 This environment renders difficult tumor effective resolution by a Th1 response based on CD8+ T-cells through mechanisms of T cell anergy and exhaustion.37,38 An allergic-like antitumor response has the advantage of having its effectors in the innate immune compartment, not requiring specific priming or generation of CTL immunity, suggesting a unique way to exploit IL-33 in the treatment of peritoneal tumors.
The peritoneal cavity is a privileged site for IL-33 treatment as it is contained and it possesses an ontogenically differentiated lineage of tissue-resident macrophages.39,40 This macrophage population represents the first line of defense against microbes breaching the intestine and ascendant gynecological infections.22 As has been reported for other sites,41-43 we observed that the peritoneal administration of IL-33 was able to promote an allergic like macrophage activation in the peritoneal tumor-associated macrophages. As reported previously for CD40 activated CD86+ activated macrophages in the microenvironment of pancreatic cancer,44 or activated peritoneal macrophages were able to promote tumor cytotoxicity in vitro and delay tumor progression in vivo. Additionally, IL-33 activated macrophages significantly decreased the expression of IL-10, an immunosuppressive cytokine that is normally high in tumor-associated ascites and associated with unfavorable prognoses.45-47 Another peculiarity of the peritoneal immune environment is the presence of B-1 cells, which have been shown to be activated directly by IL-33 resulting in an increased attraction of monocytes-macrophages.48,49 The role of B cells in tumor progression and anti-tumor immunity is not fully elucidated. Previous studies have found that B cell depletion results in an increased tumor growth,50 although it has also been shown, as in our case, to enhance survival in combination with checkpoint inhibitors.51
In conclusion, we show that intraperitoneal administration of IL-33 represents a promising location for treatment against peritoneally confined metastatic cancers. The survival advantage conferred by this therapy does not depend on classic anti-tumor Th1 response but on an allergic like-tissue remodeling response elicited by CD4+ T-cells, eosinophils and macrophages, with less reliant on effector CD8 T cell immunity. Further study of this unique cytokine in local delivery for tumor therapy appears important.
Methods
Animals and cell lines
C57BL/6 and B6.129S2-Ighmtm1Cgn/J (muMt-) mice were purchased from The Jackson or Charles River Laboratory. Animal experiments were approved by the Institutional Animal Care and Use Committee at the Wistar Institute.
Parental ID8 cells were provided by Katherine Roby (Department of Anatomy and Cell Biology, University of Kansas Medical Center, Kansas City, KS) and retrovirally transduced to express Defb29 and Vegf-a.52 We generated ID8-Defb29/Vegf-a intraperitoneal tumors as described previously.53 We generated ID8-Defb29/Vegf-a-luciferase by lentivirally transducing them to express firefly luciferase with a puromycin resistance selection gene.
Mice were treated with 1 μg of murine IL-33 (Peprotech and eBioscience) dissolved in PBS or PBS alone once per week.
Flow cytometry
We used a BD LSRII flow cytometer or BD FACSAria cell sorter (BD Biosciences).
Anti-mouse antibodies used were directly fluorochrome conjugated. We used: anti-ST2 (DIH9), CD3e (17A2), CD4 (RM4-5), CD8b (YTS156.7.7), CD45 (30-F11), CD44 (IM7), CD69 (H1.2F3), IL-5 (TRFK5), CD40L (MR1), B220 (RA36B2), CD11b (M1/70), MHCII (M5/114), F4/80 (BM8), Cd80 (16-10A1), CD86 (GL-1) (all from BioLegend) and Siglec-F (1RNM44N, ThermoFisher). Live/dead exclusion was done with Zombie Yellow (BioLegend).
Cell proliferation assays
We plated 1,000 ID8-Defb29/Vegf-a cells on a 96 well plate and added 10 or 100ng/ml of IL-33 diluted in PBS or PBS alone. Three days later we performed MTS assays according to the manufacturer’s instructions (Cell Titer96 AQueous One Solution Cell Proliferation Assay, Promega).
For T cell proliferation we stained Th2 skewed CD4 T cells with Cell Trace CFSE (Invitrogen) and cocultured them with IL-33 (250ng/ml) or ConA (Invitrogen) and measured proliferation by flow cytometry after 72h.
Ige quantification
We obtained peritoneal fluid from the mice by paracentesis, removed the cellular component by centrifugation and used the liquid component to perform an IgE quantification ELISA following manufacturer’s instructions (Biolegend).
Quantitative real-time PCR
Sorted cell RNA was isolated by mechanical disruption and extracted using RNeasy kits (QIAGEN) according to manufacturer’s instruction. RNA was reverse transcribed using High Capacity Reverse Transcription kits (Applied-Biosystems). Quantification of the following genes was performed on the 7900 Fast Real Time PCR system (Applied Biosystem) using SYBR green reagents and the following primers ST2 (Forward: 5ʹ-GACATCAGCCAAGAAGTGAGAG-3ʹ; and Reverse: 5ʹ-AATCCTCCATACAACCACACAA-3ʹ), IL-5 (Forward: 5ʹ-CTCCAATGCATAGCTGGTGAT-3ʹ; and Reverse: 5ʹ-GAGATTCCCATGAGCACAGT-3ʹ), Ym1 (Forward: 5ʹ-TCACAGGTCTGGCAATTCTTCTG-3ʹ; and Reverse: 5ʹ-ACTCCCTTCTATTGGCCTGTCC-3ʹ), CD40 (Forward: 5ʹ- GACCTCCAAGTTCTTATCCTCAC-3ʹ; and Reverse: 5ʹ- CACTGATACCGTCTGTCATCC-3ʹ), CD80 (Forward: 5ʹ- TTGCCAGTAGATTCGGTCTTC-3ʹ; and Reverse: 5ʹ- TTGTGCTGCTGATTCGTCTT-3ʹ), IL-13 (Forward: 5ʹ- GTCCACACTCCATACCATGC-3ʹ; and Reverse: 5ʹ- GATCTGTGTCTCTCCCTCTGA-3ʹ) IL-10 (Forward: 5ʹ- ATGGCCTTGTAGACACCTTG-3ʹ; and Reverse: 5ʹ- GTCATCGATTTCTCCCCTGTG-3ʹ). mRNA expression was normalized by GAPDH levels (primers Forward: 5ʹ-CCTGCACCACCAACTGCTTA-3ʹ; and Reverse: 5ʹ-AGTGATGGCATGGACTGTGGT-3ʹ). The average of three independent analyses for gene and sample was calculated using the ΔΔ threshold cycle (Ct) method and was normalized to the endogenous reference control gene GAPDH.
Th2 skewing
We harvested splenocytes from C57Bl6 mice and performed CD4 T cell isolation using EasySep™ Mouse CD4 + T Cell Isolation Kit (Stemcell). We plate 1 million CD4 T-cells per well in 1ml of RPMI 10% FBS with 5ug/ml ConA, 20U/ml IL-2 and 50ng/ml IL-4. Cytokines were refreshed on day 3. We harvested the cells on day 6 to perform the experiments.
Cytotoxicity assay
We plated 10,000 target tumor cells in flat bottom 96 well plate. Before plating the effector cells, we washed away the tumor conditioned media and added fresh media and the appropriate number of effector cells per well (50,000 of sorted macrophages or eosinophils or 150,000 of total peritoneal cells in 200 μL) and 100ug/ml of IL-33. Following 18 hours we washed the wells with PBS and determined cytotoxicity using the Luciferase Assay (Promega) according to the manufacturer’s instructions. Cytotoxicity was calculated as (maximum viability control – individual well)/(maximum viability control – maximum death control)*100 as a percentage.
Macrophage, eosinophil, CD4 and b cell depletion
We depleted macrophages by intraperitoneally injecting 400μg of anti-mouse CSF1R antibody (AFS98, BioXcell) three times a week. Eosinophils were depleted by intraperitoneally injecting 15μg of anti-mouse Siglec-F (clone 238047; R&D Systems) three times a week. We depleted CD4+ cells by intraperitoneally injecting 400μg of anti-mouse CD4 (GK1.4, BioXcell) three times a week. Depletion was initiated 3 days prior to and continued throughout IL-33 treatment. As isotype control we used 400μg of rat IgG2a anti-trinitrophenol (2A3, BioXcell). We depleted B cells by intraperitoneally injecting 300μg of anti-mouse CD19 (1D3, BioXcell) and 300μg of anti-mouse B220 (RA3.3A1/6.1, BioXcell).54
Statistics
Differences between the means of experimental groups were calculated using a two-tailed unpaired Student’s t test or one-way ANOVA where more than two quantitative variables were measured. Error bars represent standard error of the mean. Survival rates were compared using the log-rank test. All statistical analyses were done using Graph Pad Prism 7.0. A p-value < 0.05 was considered statistically significant.
Funding Statement
This work was supported by the HHS | NIH | National Cancer Institute (NCI) [P50CA174523];Basser Foundation;W.W. Smith Family Trust;HHS | NIH | National Cancer Institute (NCI) [P30 CA010815];
Acknowlegdements
This work was supported by a Penn/Wistar Institute NIH SPORE (P50CA174523 to D.B.W.), the Wistar National Cancer Institute Cancer Center (P30 CA010815), the W.W. Smith Family Trust (to D.B.W.) and funding from the Basser Foundation (to D.B.W.). E.K.D was supported by F32 CA213795. We would like to thank the Wistar Flow Cytometry Facility and Animal Facility for their technical assistance.
Disclosure of interest
D.B. Weiner receives a commercial research grant from Inovio Pharmaceuticals, has received speakers bureau honoraria from Inovio Pharmaceuticals, GeneOne, AstraZeneca, has ownership interest (including patents) in Inovio Pharmaceuticals and is a consultant/advisory board member for Inovio Pharmacueticals. The other authors report no conflict of interests.
Author contributions
A.P.P. designed and performed most experiments and co-wrote the manuscript; K.K.P. and E.K.D. provided intellectual and technical support; N.S. and D.V. contributed to the design of in vivo experiments and performed in vitro experiments; E.R. and K.W. performed mouse and in vitro experiments; U.Z. performed Q-PCR experiments and provided insight; D.B.W, J.R.C.G. and K.M. oversaw and designed the study and experiments, analyzed data, and co-wrote the manuscript.
Supplemental data
Supplemental data for this article can be access on the publisher’s here.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Maringe C, Walters S, Butler J, Coleman MP, Hacker N, Hanna L, Mosgaard BJ, Nordin A, Rosen B, Engholm G, et al. Stage at diagnosis and ovarian cancer survival: evidence from the International Cancer Benchmarking Partnership. Gynecol Oncol. 2012;127:75–82. doi: 10.1016/j.ygyno.2012.06.033. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA, Gynecologic Oncology G. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 4.Jaaback K, Johnson N, Lawrie TA. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database Syst Rev. 2016;CD005340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pujade-Lauraine E, Guastalla JP, Colombo N, Devillier P, Francois E, Fumoleau P, Monnier A, Nooy M, Mignot L, Bugat R, et al. Intraperitoneal recombinant interferon gamma in ovarian cancer patients with residual disease at second-look laparotomy. J Clin Oncol. 1996;14:343–350. doi: 10.1200/JCO.1996.14.2.343. [DOI] [PubMed] [Google Scholar]
- 6.Edwards RP, Gooding W, Lembersky BC, Colonello K, Hammond R, Paradise C, Kowal CD, Kunschner AJ, Baldisseri M, Kirkwood JM, et al. Comparison of toxicity and survival following intraperitoneal recombinant interleukin-2 for persistent ovarian cancer after platinum: twenty-four-hour versus 7-day infusion. J Clin Oncol. 1997;15:3399–3407. doi: 10.1200/JCO.1997.15.11.3399. [DOI] [PubMed] [Google Scholar]
- 7.Berek JS, Markman M, Stonebraker B, Lentz SS, Adelson MD, DeGeest K, Moore D. Intraperitoneal interferon-alpha in residual ovarian carcinoma: a phase II gynecologic oncology group study. Gynecol Oncol. 1999;75:10–14. doi: 10.1006/gyno.1999.5532. [DOI] [PubMed] [Google Scholar]
- 8.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Villarreal DO, Wise MC, Walters JN, Reuschel EL, Choi MJ, Obeng-Adjei N, Yan J, Morrow MP, Weiner DB. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res. 2014;74:1789–1800. doi: 10.1158/0008-5472.CAN-13-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez D, Ye C, Geng Z, Chen S, Fan J, Qin L, Long A, Wang L, Zhang Z, Zhang Y, et al. Exogenous IL-33 Restores Dendritic Cell Activation and Maturation in Established Cancer. J Immunol. 2017;198:1365–1375. doi: 10.4049/jimmunol.1501399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Z, Wang J, Wientjes MG, Au JL. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010;6:1625–1641. doi: 10.2217/fon.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinds DA, McMahon G, Kiefer AK, Do CB, Eriksson N, Evans DM, St Pourcain B, Ring SM, Mountain JL, Francke U, et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet. 2013;45:907–911. doi: 10.1038/ng.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson W, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piehler D, Eschke M, Schulze B, Protschka M, Muller U, Grahnert A, Richter T, Heyen L, Kohler G, Brombacher F, et al. The IL-33 receptor (ST2) regulates early IL-13 production in fungus-induced allergic airway inflammation. Mucosal Immunol. 2016;9:937–949. doi: 10.1038/mi.2015.106. [DOI] [PubMed] [Google Scholar]
- 15.Savinko T, Karisola P, Lehtimaki S, Lappetelainen AM, Haapakoski R, Wolff H, Lauerma A, Alenius H. ST2 regulates allergic airway inflammation and T-cell polarization in epicutaneously sensitized mice. J Invest Dermatol. 2013;133:2522–2529. doi: 10.1038/jid.2013.195. [DOI] [PubMed] [Google Scholar]
- 16.Welch JS, Escoubet-Lozach L, Sykes DB, Liddiard K, Greaves DR, Glass CK. TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J Biol Chem. 2002;277:42821–42829. doi: 10.1074/jbc.M205873200. [DOI] [PubMed] [Google Scholar]
- 17.Meisel C, Bonhagen K, Lohning M, Coyle AJ, Gutierrez-Ramos JC, Radbruch A, Kamradt T. Regulation and function of T1/ST2 expression on CD4+ T cells: induction of type 2 cytokine production by T1/ST2 cross-linking. J Immunol. 2001;166:3143–3150. [DOI] [PubMed] [Google Scholar]
- 18.Ngoc PL, Gold DR, Tzianabos AO, Weiss ST, Celedon JC. Cytokines, allergy, and asthma. Curr Opin Allergy Clin Immunol. 2005;5:161–166. [DOI] [PubMed] [Google Scholar]
- 19.Till S, Durham S, Dickason R, Huston D, Bungre J, Walker S, Robinson D, Kay AB, Corrigan C. IL-13 production by allergen-stimulated T cells is increased in allergic disease and associated with IL-5 but not IFN-gamma expression. Immunology. 1997;91:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. [DOI] [PubMed] [Google Scholar]
- 21.Judd LM, Heine RG, Menheniott TR, Buzzelli J, O'Brien-Simpson N, Pavlic D, O'Connor L, Al Gazali K, Hamilton O, Scurr M, et al. Elevated IL-33 expression is associated with pediatric eosinophilic esophagitis, and exogenous IL-33 promotes eosinophilic esophagitis development in mice. Am J Physiol Gastrointest Liver Physiol. 2016;310:G13–25. doi: 10.1152/ajpgi.00290.2015. [DOI] [PubMed] [Google Scholar]
- 22.Melichar B, Freedman RS. Immunology of the peritoneal cavity: relevance for host-tumor relation. Int J Gynecol Cancer. 2002;12:3–17. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Z, Zhu L. Update on the role of alternatively activated macrophages in asthma. J Asthma Allergy. 2016;9:101–107. doi: 10.2147/JAA.S104508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byers DE, Holtzman MJ. Alternatively activated macrophages and airway disease. Chest. 2011;140:768–774. doi: 10.1378/chest.10-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, Pitman N, Mirchandani A, Rana B, van Rooijen N, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 26.Balbo P, Silvestri M, Rossi GA, Crimi E, Burastero SE. Differential role of CD80 and CD86 on alveolar macrophages in the presentation of allergen to T lymphocytes in asthma. Clin Exp Allergy. 2001;31:625–636. [DOI] [PubMed] [Google Scholar]
- 27.Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17:122–131. doi: 10.1038/ni.3370. [DOI] [PubMed] [Google Scholar]
- 28.Roy A, Ganesh G, Sippola H, Bolin S, Sawesi O, Dagalv A, Schlenner SM, Feyerabend T, Rodewald HR, Kjellen L, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem. 2014;289:237–250. doi: 10.1074/jbc.M112.435156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, Rees DG, Overed-Sayer CL, Woods J, Bond NJ, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6:8327. doi: 10.1038/ncomms9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, Cote-Sierra J. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. 2014;55:33–41. doi: 10.1016/j.jaut.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Nabe T, Wakamori H, Yano C, Nishiguchi A, Yuasa R, Kido H, Tomiyama Y, Tomoda A, Kida H, Takiguchi A, et al. Production of interleukin (IL)-33 in the lungs during multiple antigen challenge-induced airway inflammation in mice, and its modulation by a glucocorticoid. Eur J Pharmacol. 2015;757:34–41. doi: 10.1016/j.ejphar.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 32.Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, Martin JG, Hamid Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 33.Bahrami Mahneh S, Movahedi M, Aryan Z, Bahar MA, Rezaei A, Sadr M, Rezaei N, Universal Scientific E, Research N. Serum IL-33 Is Elevated in Children with Asthma and Is Associated with Disease Severity. Int Arch Allergy Immunol. 2015;168:193–196. doi: 10.1159/000442413. [DOI] [PubMed] [Google Scholar]
- 34.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 35.Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, Ahonen CL, Conejo-Garcia JR. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 2009;69:7329–7337. doi: 10.1158/0008-5472.CAN-09-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 2012;209:495–506. doi: 10.1084/jem.20111413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y, Zha Y, Gajewski TF. Molecular regulation of T-cell anergy. EMBO Rep. 2008;9:50–55. doi: 10.1038/sj.embor.7401138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. [DOI] [PubMed] [Google Scholar]
- 39.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15:731–744. doi: 10.1038/nri3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joshi AD, Oak SR, Hartigan AJ, Finn WG, Kunkel SL, Duffy KE, Das A, Hogaboam CM, et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol. 2010;11:52. doi: 10.1186/1471-2172-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He R, Yin H, Yuan B, Liu T, Luo L, Huang P, Dai L, Zeng K. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Mol Immunol. 2017;90:42–49. doi: 10.1016/j.molimm.2017.06.249. [DOI] [PubMed] [Google Scholar]
- 43.Lee JS, Seppanen E, Patel J, Rodero MP, Khosrotehrani K. ST2 receptor invalidation maintains wound inflammation, delays healing and increases fibrosis. Exp Dermatol. 2016;25:71–74. doi: 10.1111/exd.12833. [DOI] [PubMed] [Google Scholar]
- 44.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu L, Deng Z, Peng Y, Han L, Liu J, Wang L, Li B, Zhao J, Jiao S, Wei H. Ascites-derived IL-6 and IL-10 synergistically expand CD14(+)HLA-DR(-/low) myeloid-derived suppressor cells in ovarian cancer patients. Oncotarget. 2017;8:76843–76856. doi: 10.18632/oncotarget.20164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loercher AE, Nash MA, Kavanagh JJ, Platsoucas CD, Freedman RS. Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J Immunol. 1999;163:6251–6260. [PubMed] [Google Scholar]
- 47.Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood. 2006;107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed A, Koma MK. Interleukin-33 Triggers B1 Cell Expansion and Its Release of Monocyte/Macrophage Chemoattractants and Growth Factors. Scand J Immunol. 2015;82:118–124. doi: 10.1111/sji.12312. [DOI] [PubMed] [Google Scholar]
- 49.Komai-Koma M, Gilchrist DS, McKenzie AN, Goodyear CS, Xu D, Fy L. IL-33 activates B1 cells and exacerbates contact sensitivity. J Immunol. 2011;186:2584–2591. doi: 10.4049/jimmunol.1002103. [DOI] [PubMed] [Google Scholar]
- 50.DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–4016. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dai M, Wei H, Yip YY, Feng Q, He K, Popov V, Hellstrom I, Hellstrom KE. Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation. J Immunother. 2013;36:248–257. doi: 10.1097/CJI.0b013e3182943549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 53.Perales-Puchalt A, Svoronos N, Rutkowski MR, Allegrezza MJ, Tesone AJ, Payne KK, Wickramasinghe J, Nguyen JM, O’Brien SW, Gumireddy K, et al. Follicle-Stimulating Hormone Receptor Is Expressed by Most Ovarian Cancer Subtypes and Is a Safe and Effective Immunotherapeutic Target. Clin Cancer Res. 2015;521:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, Davidson MG, Kenkel JA, Segal E, Pusapati GV, et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. 2015;521:99–104. doi: 10.1038/nature14424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.