

Abstract

Discoidin-domain receptors 1 and 2 (DDR1 and DDR2) are new potential targets for anti-inflammatory-drug discovery. A series of heterocycloalkynylbenzimides were designed and optimized to coinhibit DDR1 and DDR2. One of the most promising compounds, 5n, tightly bound to DDR1 and DDR2 proteins with Kd values of 7.9 and 8.0 nM; potently inhibited the kinases with IC50 values of 9.4 and 20.4 nM, respectively; and was significantly less potent for a panel of 403 wild-type kinases at 1.0 μM. DDR1- and DDR2-kinase inhibition by 5n was validated by Western-blotting analysis in primary human lung fibroblasts. The compound also dose-dependently inhibited lipopolysaccharide (LPS)-induced interleukin 6 (IL-6) release in vitro and exhibited promising in vivo anti-inflammatory effects in an LPS-induced-acute-lung-injury (ALI) mouse model. Compound 5n may serve as a lead compound for new anti-inflammatory drug discovery.
Introduction
Discoidin-domain receptors (DDRs), including DDR1 and DDR2, are nonintegrin collagen-receptor kinases with a unique extracellular domain homologous to that of the discoidin I protein of Dictyostelium discoideum.1−7 DDRs are involved in the regulation of cellular morphogenesis, differentiation, proliferation, adhesion, migration, and invasion.1−7 Collective evidence demonstrates that DDR1 and DDR2 are critical mediators of inflammatory-cytokine secretion.2,4,7 Dysregulation of the receptors has been implicated in a variety of inflammatory diseases, such as atherosclerosis, osteoarthritis, and organ fibrosis.1−7 For instance, collagen-induced activation of DDR1b markedly amplifies the production of interleukin 8 (IL-8), macrophage inflammatory protein 1α (MIP-1α), and monocyte-chemoattractant protein 1 (MCP-1) by macrophages during inflammatory responses.8 Renal cortical slices of DDR1-null mice showed a blunted response of chemokine secretion in response to lipopolysaccharide (LPS), which was accompanied by protection against LPS-induced mortality.9 A similar situation was also found in bleomycin-induced lung injury.10 Pharmacological inhibition of DDR1 by small molecules has been shown to reduce inflammatory cytokines and demonstrate promising therapeutic effects in mouse inflammation models.11,12 Activation of DDR2 was also reported to increase the production of cytokines such as IL-12, tumor-necrosis factor α (TNF-α), and interferon γ (INF-γ) by human dendritic cells13,14 and contribute significantly to inflammatory disorders. Collagen I mediated activation of DDR2 is critical for fibrogenesis and promotes resolution of lung inflammation.15 Silencing DDR2 expression was reported to decrease alcohol-induced liver injury and fibrosis in a model for early-stage alcoholic liver disease.16 Additionally, DDR2 can mediate hepatic-stellate-cell activation, proliferation, and migration during acute liver injury, highlighting the profibrotic activity of DDR2.17 Other studies also reported that activation of DDR2 by collagen I could induce the expression of DDR1 in primary human lung fibroblasts,18 indicating potential crosstalk between these two receptors. Therefore, dual inhibition of DDR1 and DDR2 might be a promising strategy for anti-inflammatory-drug discovery.
A number of DDR1 and DDR2 inhibitors have been reported to date (Figure 1). However, most of these molecules suffer from relatively low target specificity.19−21 For example, In addition to DDR1 and -2, inhibitors 1 and 2 also show strong inhibition of Abl, c-Kit, and cSrc. 4-[(4-Methylpiperazin-1-yl)methyl]-N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)benzamide (imatinib), 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoro-methyl)phenyl]-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]benzamide (nilotinib), and N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide (dasatinib) exhibit strong DDR1- and DDR2-inhibitory activities, but neither DDR1 nor DDR2 is their primary target.22 It is highly desirable to identify new selective DDR1 and -2 dual inhibitors for biological investigation and therapeutic development. Herein, we report the design and synthesis of heterocycloalkynylbenzimides as new selective DDR1 and -2 dual inhibitors with promising in vivo therapeutic effects in an LPS-induced-acute-lung-injury (ALI) mouse model.
Figure 1.

Chemical structures of the representative reported DDR1 and DDR2 inhibitors.
Chemistry
The designed DDR1 and -2 inhibitors were readily prepared using palladium-catalyzed Sonogashira coupling23 as the key steps (Scheme 1). Briefly, commercially available methyl 3-aminobenzoates (6) went through diazotization and iodization to yield intermediates 7, which were treated with ethynyltrimethylsilane under palladium catalysis to afford the Sonogashira-coupling products, deprotection of which produced the terminal alkynes (8). Intermediates 8 were reacted with 3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)aniline to produce intermediates 9 under basic conditions. Compounds 9 coupled with aromatic bromides under Sonogashira conditions to give compounds 5a–5l. Alternatively, intermediates 8 could also couple with aromatic bromides and then react with 3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)aniline under basic conditions to produce final products 5m–5r.
Scheme 1. Synthesis of Compounds 5a–5r.

Reagents and conditions: (a) (i) Concd H2SO4, sodium nitrite (NaNO2), H2O; (ii) potassium iodide (KI), H2O, 40–70% (two steps). (b) (i) Trimethylsilyl acetylene, CuI, bis(triphenylphosphine)palladium(II) chloride (PdCl2(PPh3)2), triethylamine (Et3N), acetonitrile (MeCN), 60 °C; (ii) K2CO3, MeOH, room temperature (rt), 88–92% (two steps). (c) t-BuOK, tetrahydrofuran (THF), −20 °C to rt, 92%. (d) Het-Br, CuI, PdCl2(PPh3)2, ethyl diisopropylamine (DIPEA), N,N-dimethylformamide (DMF), 80 °C, 40–85%. (e) CuI, PdCl2(PPh3)2, DIPEA, DMF, 80 °C, 64–91%. (f) 3-((4-Methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)aniline, t-BuOK, THF, −20 °C to rt, 74–87%.
Molecule Design
Compound 4 (7rh) is a newly discovered selective DDR1 inhibitor from our group (Figure 2A),24 which potently inhibits the kinase activity of DDR1 with an IC50 value of 9.7 nM. It also exhibited a modest suppressive effect on DDR2, with an IC50 value of 175 nM, but it was significantly less potent against the majority of a panel of 395 nonmutated kinases. In view of its promising target selectivity and outstanding pharmacokinetic (PK) properties,24 compound 4 was chosen as a starting lead compound for further structural modification to achieve selective dual inhibition of DDR1 and DDR2.
Figure 2.

Potential binding modes of new inhibitors with DDR1 and DDR2 proteins. (A) Molecular docking of 4 into DDR1 (PDB: 3ZOX). (B) Molecular docking of 4 into the DDR2 homology model. (C) Molecular docking of 5j into the DDR1 structure (PDB: 3ZOX). (D) Molecular docking of 5j into the DDR2 homology model. Regular hydrogen bonds are indicated by black dashed lines. The distances between two atoms are indicated by red dashed lines. The key residues are shown as yellow sticks.
DDR2 shares an approximate 57% sequence identity with DDR1 in its kinase domain (Figure S1).7 Thus, a homologous model of DDR2 was first generated on the basis of the DDR1 crystal structure (PDB code 3ZOS) to provide an initial structural basis for inhibitor optimization. It was shown that compound 4 could bind to the inactive configurations of DDR1 and DDR2 with similar type II binding modes (Figure 2A,B). The inhibitor was predicted to form four hydrogen bonds with the Met704, Glu672, and Asp784 residues of DDR1. Favorable van der Waals contacts could also be formed in the allosteric pocket. However, compound 4 failed to form a hydrogen-bond interaction with the corresponding Met95 of DDR2 because of its inappropriate orientation. Further investigation also suggested that potential stereohindrance between the pyrazolo[1,5-a]pyrimidin head of 4 and the Tyr94 and Met95 hinge residues might contribute to its significantly lower potency with DDR2 (Figure 2B). These preliminary computational analyses indicated that replacement of the pyrazolo[1,5-a]pyrimidine moiety with alternative hinge-binding heterocycles could be a feasible strategy to achieve dual inhibition against DDR1 and DDR2.
Results and Discussion
Compound 5a with a pyrimidine moiety was first designed and synthesized to exhibit similar inhibitory potencies against DDR1 and DDR2 to those of inhibitor 4. It was predicted that the introduction of a hydrogen-bond-donating group at the 2-position of 5a could potentially capture an additional hydrogen-bond interaction with the Met95 residue of the DDR2 protein to improve its kinase-inhibitory potency. Indeed, both the 2-aminopyrimidine (5b) and 2-(methylamino)pyrimidine (5c) derivatives displayed improved DDR2-inhibitory potencies with IC50 values of 45.3 and 83.1 nM, respectively (Table 1). The inhibitory activities against DDR1 were also improved by approximately 2-fold, with IC50 values of 3.9 and 5.0 nM, respectively. Not surprisingly, the introduction of a dimethylamino group at the 2-position almost totally abolished the inhibition of the DDR1 and DDR2 kinases by the resulting compound (5d). Although compounds 5b and 5c exhibited good inhibitory potencies against DDR1 and DDR2, they were almost equally potent against Abl1. The lack of target specificity makes these compounds less attractive for further investigation. Several five-member heterocycles were also utilized as potential hinge-binding moieties (Table 1). Although the introduction of 3-furan (5e), 3-thiophene (5g), and 2-furan (5f) significantly decreased the kinase-inhibitory potencies against DDR1 and DDR2, the 1-methyl-1H-imidazole (5h)- and 1,2-dimethyl-1H-imidazole (5i)-substituted derivatives exhibited strong inhibition against DDR1 and DDR2 kinases, with IC50 values of 16.0 and 94.2 nM (for 5h) and 7.9 and 34.1 nM (for 5i), respectively. Encouraged by the results of compounds 5h and 5i, bicyclic derivatives (5j–5l) were further designed and synthesized by utilizing a conformational-constraint strategy.25 It was shown that cyclization significantly improved their potencies against both DDR1 and DDR2. Compounds 5j–5l suppressed the kinase activity of DDR2 with IC50 values of 7.0, 13.1, and 10.4 nM, respectively. The compounds also displayed strong inhibition against DDR1, with IC50 values of 3.2, 3.9, and 6.2 nM, respectively. Thus, compound 5j represented one of the most potent dual inhibitors against DDR1 and DDR2 in these derivatives. Further computational study suggested that compound 5j bound to DDR1 and DDR2 with a DFG-out conformation (Figure 2C,D). The imidazo[1,2-a]pyrazine group could fit nicely into the hinge regions of DDR1 and DDR2 with hydrogen-bond interactions with Met704 and Met95, respectively (Figure 2D). Unfortunately, compound 5j also strongly inhibited the kinase activity of Abl1, with an IC50 value of 9.4 nM. Thus, further structural optimization was conducted with the aim to improve the inhibitor’s target selectivity.
Table 1. In Vitro Inhibitory Activities of Compounds 5a–5r against DDR1, DDR2, and Abl1a.


DDR1- and DDR2-inhibition experiments were performed using the LANCE ULTRA kinase assay according to the manufacturer’s instructions. Abl1-activity experiments were performed using the FRET-based Z′-Lyte assay according to the manufacturer’s instructions. The data are mean values from at least four independent experiments.
It has been previously demonstrated that the flag-methyl group was critical for Abl1 inhibitors to achieve potency against the kinase.26 Replacement of an original methyl with an ethyl group helped us to successfully identify a highly selective DDR1 inhibitor, 4.24 It was hypothesized that the Abl1 inhibition might be diminished by optimizing the flag-ethyl group in compound 5j. Compounds 5m–5p were consequently designed and synthesized on the basis of this hypothesis. It was shown that the flag-alkyl moiety indeed had a significant impact on the inhibitory potency against Abl1. When the ethyl group in 5j was replaced with a methyl substituent, the resulting compound, 5m, demonstrated 24-fold improved potency against Abl1, with an IC50 value of 0.4 nM, but the modification barely affected the DDR1 and DDR2 inhibition. Encouragingly, the isopropyl derivative (5n) exhibited significantly decreased Abl1-inhibitory potency, with an IC50 value of 494 nM, whereas it retained the strong inhibition against DDR1 and DDR2, with IC50 values of 9.4 and 20.4 nM, respectively. Compound 5n represented one of the most selective DDR1–DDR2 dual inhibitors over Abl1. The cyclic propyl compound, 5o, was less selective, whereas the tert-butyl derivative, 5p, had almost totally abolished DDR2 inhibition. The isopropyl-group-substituted compounds, 5q and 5r, also demonstrated obviously decreased Abl1 inhibition, but their potencies against DDR2 were also apparently lost. The relatively weak target-inhibitory activities made compounds 5q and 5r less attractive for further biological investigation.
To elucidate the details of the interaction of 5n with DDR1, we determined the X-ray cocrystal structure of their complex refined at 2.1 Å resolution (Figure 3 and Table S3). It was confirmed that 5n fits nicely into the ATP-binding site of DDR1 with a similar binding mode to that predicted by the docking model with 5j (Figure 2C). The imidazo[1,2-a]pyrazine moiety of 5n was observed to form an essential hydrogen bond with Met704 in the hinge region. Two additional hydrogen bonds were also formed between the amide and Glu672 and Asp784, whereas the flag isopropyl fitted nicely into a hydrophobic pocket formed by residues Val624, Ala653, Lys655, and Met699.
Figure 3.
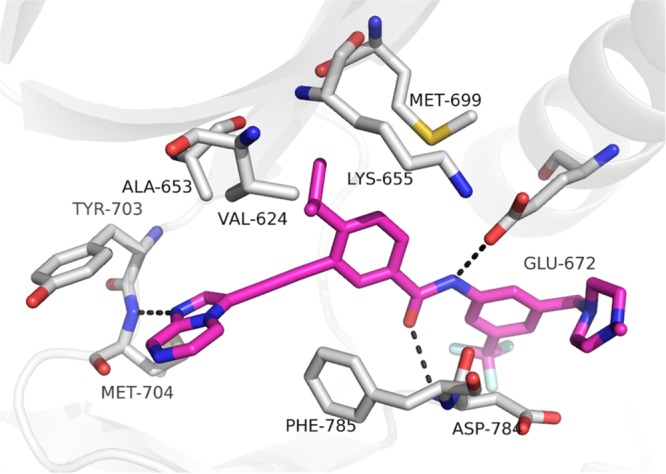
Cocrystal structure of 5n with DDR1 (PDB: 6GWR). Hydrogen bonds are indicated by dashed black lines. The key residues are shown as gray sticks. Compound 5n is shown as magenta sticks.
DDR1–DDR2 dual inhibition by 5n was validated by determining its binding affinities with the receptors (conducted by DiscoveRx).27 It was shown that 5n bound tightly to DDR1 and DDR2 with binding-constant (Kd) values of 7.9 and 8.0 nM, respectively. The target selectivity of 5n was further evaluated by conducting a kinase-selectivity-profiling study against a panel of 468 kinases (including 403 nonmutated kinases) at 1.0 μM, which is approximately 125-fold above its Kd values against the DDR1 and -2 targets. The results indicated that 5n exhibited good target selectivity with S(10) and S(1) scores of 0.032 and 0.017, respectively (Supporting Information).27 For instance, 5n showed a 100% competition rate (100% inhibition, ctrl = 0%) with DDR1 and DDR2 at 1.0 μM, whereas it only showed obvious binding with a minor portion of the kinases investigated. The major off-targets (inhibition > 90%, ctrl < 10%) included Abl1, ephrin type-A receptor 2 (EPHA2), EPHA7, EPHA8, ephrin type-B receptor 2 (EPHB2), lymphocyte-specific protein tyrosine kinase (LCK), serine–threonine kinase 10 (LOK), angiopoietin-1 receptor (TIE2), nerve growth-factor receptor A (TrkA), TrkB, and TrkC. The binding affinities (Kd) or kinase-inhibitory activities (IC50) of compound 5n against these off-targets were further determined by using DiscoveRx’s platform or in-house kinase assays (Table 2). It was shown that compound 5n was approximately 10–46-fold less potent against the majority of the off-target kinases. However, this compound seemed to be equally potent against the human-disease-related LOK, TrkB, and TrkC.28,29 The off-target inhibition of 5n on TrkB and TrkC was further proven by a LANCE ULTRA kinase assay, which exhibited IC50 values of 40 and 18 nM, respectively.
Table 2. Binding Affinities of Compound 5n against a Panel of Off-Target Kinasesa.
| kinase | Kd value or IC50 (nM) | kinase | Kd value or IC50 (nM) |
|---|---|---|---|
| Abl1 | 494b | LOK | 10c |
| EPHA2 | 260c | TIE2 | 370c |
| EPHA7 | 200c | TrkA | 100c |
| EPHA8 | 79c | TrkB | 11c (40)b |
| EPHB2 | 260c | TrkC | 9.3c (18)b |
| LCK | 180c |
Reported data are means from two independent experiments.
The kinase-inhibitory activities (IC50) were evaluated by using in-house kinase assays.
The binding affinities (Kd) were determined by using DiscoveRx’s platform.
The inhibitory effect of compound 5n on the activation of DDR1 and DDR2 was also investigated in primary human lung fibroblasts (Figure 4). The results clearly revealed that 5n dose-dependently inhibited the phosphorylation of DDR1 and DDR2, whereas it did not exhibit an obvious impact on the activation of c-Abl at concentrations of 50 and 100 nM.
Figure 4.

Effects of DDR1 and DDR2 inhibition by 5n on signaling in primary human lung fibroblasts. Lysates were probed for the indicated targets by Western-blot analysis. Primary human lung fibroblasts were treated with col I (50 μg/mL) and DMSO or different concentrations of 5n for 8 h. Cell lysates were harvested and subjected to immunoprecipitation or Western blotting. Activation of DDR2 was detected by immunoprecipitation. Protein lysates were also probed for p-DDR1, DDR1, p-c-Abl, c-Abl, and tubulin.
Given the critical function of DDR1 and DDR2 in the inflammatory process, we determined the potential anti-inflammatory effect of 5n by measuring its capability to suppress the LPS-induced release of a representative cytokine, IL-6. It was shown that compound 5n dose-dependently inhibited LPS-induced production of IL-6 in mouse primary peritoneal macrophages (MPMs) as evaluated by using an enzyme-linked immunosorbent assay (ELISA), supporting its promising in vitro anti-inflammatory activity (Figure 5).
Figure 5.

Compound 5n inhibited LPS-induced IL-6 release in a dose-dependent manner in MPMs. Each bar represents the means ± SE of 3–5 independent experiments. Statistical significance relative to the LPS group is indicated, **p < 0.01.
The therapeutic potential of 5n was further investigated in an LPS-induced-acute-lung-injury (ALI) mouse model.30 Compound 5n was orally administered at 20 or 40 mg/kg twice daily (BID) for 7 days prior to the administration of LPS (20 μL, 5 mg/kg) on the basis of its pharmacokinetics parameters (Table S4). Pretreatment with compound 5n reduced LPS-induced pulmonary edema as determined by the lung wet/dry (W/D) ratio (Figure 6A). The total protein concentrations in bronchial-alveolar-lavage fluid (BALF) had increased markedly after LPS administration compared with those of the control group (Figure 6B), and this was dose-dependently inhibited by 5n (Figure 6B). LPS instillation also resulted in significant pulmonary congestion, thickening of the alveolar wall, and interstitial edema (Figure 6C). These pathological changes induced by LPS were significantly reduced by treatment with 5n (Figure 6C). Moreover, the compound was well-tolerated and there was no animal deaths or obvious body-weight changes after the mice received 200 or 400 mg/kg administration of compound 5n (Figure S3).
Figure 6.

Compound 5n attenuated lung pathophysiological changes in LPS-challenged mice. (A) Lung W/D ratio. (B) Protein concentration in BALF. (C) H&E staining. Statistical significance relative to the LPS group is indicated, *p < 0.05, **p < 0.01.
Pro-inflammatory cytokines, which are secreted in the early phase of an inflammatory response, are critical in ALI. Thus, the levels of pro-inflammatory cytokines in BALF and serum were also determined. Compound 5n effectively decreased the levels of TNF-α and IL-6 both in BALF and serum (Figure 7A–D). Additionally, LPS-induced elevation of neutrophils and total cell numbers in BALF were also significantly reduced by treatment with 5n (Figure 7E,F). We further examined the effect of 5n on macrophage infiltration in lung tissue through CD68 immunohistochemical staining. As shown in Figure 7G, LPS induced a significant accumulation of macrophages in the lung, whereas there was no significant difference in the number of CD68-positive macrophages between the 5n-treated and control groups. Thus, we concluded that the administration of 5n resulted in significant therapeutic protection from LPS-induced pulmonary inflammation in vivo.
Figure 7.

Attenuation of lung inflammation by 5n in LPS-treated mice. (A) Amount of cytokine TNF-α in BALF. (B) Amount of cytokine TNF-α in serum. (C) Amount of cytokine IL-6 in BALF. (D) Amount of cytokine IL-6 in serum. (E) Amount of neutrophils in BALF. (F) Amount of total cells in BALF. (G) Immunohistochemical staining of CD68. Statistical significance relative to the LPS group is indicated, *p < 0.05, **p < 0.01.
To confirm the anti-inflammatory effects of 5n, we further evaluated the potency of the compound in terms of its inhibition of inflammatory-gene expression. As shown in Figure 8, LPS increased mRNA levels of pro-inflammatory cytokines TNF-α, IL-6, IL-1β, and IL-12 and adhesion molecules intercellular cell-adhesion molecule-1 (ICAM-1) and vascular cell-adhesion molecule 1 (VCAM-1; Figure 8A–F, respectively), whereas compound 5n treatment significantly abrogated LPS-induction of these inflammatory markers. The data collectively support that 5n exhibits potent therapeutic effects on ALI by down-regulating pro-inflammatory-cytokine expression.
Figure 8.

Effects of 5n on the expression of inflammatory genes in lung tissue. Levels of TNF-α (A), IL-6 (B), IL-1β (C), IL-12 (D), ICAM-1 (E), and VCAM-1 (F) as determined by an RT-qPCR assay. Statistical significance relative to the LPS group is indicated, *p < 0.05, **p < 0.01.
Conclusions
In summary, a series of heterocycloalkynylbenzimides were optimized to coinhibit both DDR1 and DDR2. One of the most promising candidates, 5n, tightly bound to the DDR1 and DDR2 proteins with Kd values of 7.9 and 8.0 nM and potently inhibited DDR1- and DDR2-kinase function with IC50 values of 9.4 and 20.4 nM, respectively, but it was obviously less potent against the majority of the 403 wild-type kinases at 1.0 μM. The compound exhibited promising anti-inflammatory effects in vitro and in vivo. To the best of our knowledge, this is the first in vivo investigation of selective DDR1 and DDR2 dual inhibitors as novel anti-inflammation agents.
Experimental Section
General Methods for Chemistry
All reagents and solvents were used as purchased from commercial sources without further purification. Flash chromatography was performed using 300-mesh silica gel. All reactions were monitored by TLC using silica-gel plates with fluorescence F254 and UV-light visualization. 1H NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz or a Bruker AV-500 spectrometer at 500 MHz. 13C NMR spectra were recorded on a Bruker AV-500 spectrometer at 125 MHz. Coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) relative to an internal standard (TMS). Low- and high-resolution of ESI-MS was recorded on an Agilent 1200 HPLC-MSD mass spectrometer and an Applied Biosystems Q-STAR Elite ESI-LC-MS/MS mass spectrometer, respectively. Purities of the final compounds, 5a–5r, were determined to be >95% by reverse-phase high-performance liquid chromatography (HPLC, Dionex Summit HPLC; Diamonsil C18 column, 5.0 μm, 4.6 × 250 mm, Dikma Technologies; PDA-100 photodiode-array detector; ASI-100 autoinjector; p-680A pump). A flow rate of 1.0 mL/min was used with a mobile phase of 90% MeOH in H2O with 0.1% modifier (ammonia, v/v).
4-Ethyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)-3-(pyrimidin-5-ylethynyl)benzamide (5a)
To a solution of 4-ethyl-3-ethynyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (487 mg, 1.13 mmol) in DMF (10 mL) were added 5-bromopyrimidine (200 mg, 1.26 mmol), DIPEA (0.4 mL, 2.26 mmol), PdCl2(PPh3)2 (80 mg, 0.11 mmol), and CuI (21 mg, 0.11 mmol). The mixture was filled with argon and stirred at 80 °C overnight. The mixture was poured into ice water, and the precipitate was collected and further purified by flash chromatography on silica gel to give the final compound, 5a (464 mg, 81% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1 H), 9.23 (s, 1 H), 9.07 (s, 2 H), 8.22 (d, J = 1.6 Hz, 1 H), 8.19 (s, 1 H), 8.02 (s, 1 H), 8.01 (dd, J = 8.0, 1.6 Hz, 1 H), 8.00 (d, J = 1.6 Hz, 1 H), 7.56 (d, J = 8 Hz, 1 H), 7.35 (s, 1 H), 3.54 (s, 2 H), 2.94 (q, J = 7.6 Hz, 2 H), 2.39 (br s, 4 H), 2.34 (br s, 4 H), 2.15 (s, 3 H), 1.28 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ165.1, 159.0, 157.4, 150.3, 141.2, 140.3, 132.7, 131.7, 129.7, 129.6 (q, J = 31.1 Hz), 129.0, 124.6 (q, J = 270.8 Hz), 124.2, 120.9, 120.4, 119.2, 115.5 (d, J = 3.6 Hz), 93.8, 87.2, 61.8, 55.1, 52.9, 46.1, 27.5, 14.9. HRMS (ESI) for C28H28F3N5O [M + H]+, calcd: 508.2319, found: 508.2316. Purity 99.5% (tR = 12.79 min).
3-((2-Aminopyrimidin-5-yl)ethynyl)-4-ethyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5b)
Compound 5b was prepared by following a similar procedure to that for 5a. Yield, 70%. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1 H), 8.47 (s, 2 H), 8.19 (s, 1 H), 8.12 (s, 1H), 8.01 (s, 1 H), 7.92 (d, J = 8.0 Hz, 1 H), 7.49 (d, J = 8.0 Hz, 1 H), 7.35 (s, 1 H), 7.19 (s, 2 H), 3.54 (s, 2 H), 2.89 (q, J = 7.2 Hz, 2 H), 2.40 (br s, 4 H), 2.33 (br s, 4 H), 2.15 (s, 3 H), 1.26 (t, J = 7.2 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.3, 162.5, 160.6, 149.5, 141.2, 140.4, 132.6, 131.0, 129.7 (q, J = 30.6 Hz), 128.8, 128.5, 124.6 (q, J = 271.5 Hz), 124.2, 122.3, 120.4, 115.5, 106.3, 90.1, 89.5, 61.9, 55.1, 53.0, 46.1, 27.5, 14.9. HRMS (ESI) for C28H29F3N6O [M + H]+, calcd: 523.2428, found: 523.2414. Purity 99.6% (tR = 12.05 min).
4-Ethyl-3-((2-(methylamino)pyrimidin-5-yl)ethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5c)
Compound 5c was prepared by following a similar procedure to that for 5a. Yield, 76%. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1 H), 8.53–8.48 (m, 2 H), 8.19 (s, 1 H), 8.12 (s, 1 H), 8.02 (s, 1 H), 7.93–7.91 (m, 1 H), 7.65–7.64 (m, 1 H), 7.50 (d, J = 8.0 Hz, 1 H), 7.35 (s, 1 H), 3.54 (s, 2 H), 2.90 (q, J = 7.6 Hz, 2 H), 2.85 (d, J = 4.8 Hz, 3 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.15 (s, 3 H), 1.26 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.3, 161.5, 160.4, 149.5, 141.1, 140.4, 132.6, 131.0, 129.7 (q, J = 31.4 Hz), 128.8, 128.5, 124.6 (q, J = 270.6 Hz), 124.2, 122.3, 120.4, 115.5, 105.8, 90.1, 89.6, 61.9, 55.1, 52.9, 46.1, 28.2, 27.5, 14.8. HRMS (ESI) for C29H31F3N6O [M + H]+, calcd: 537.2584, found: 537.2584. Purity 98.7% (tR = 14.44 min).
3-((2-(Dimethylamino)pyrimidin-5-yl)ethynyl)-4-ethyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5d)
Compound 5d was prepared by following a similar procedure to that for 5a. Yield, 77%. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1 H), 8.56 (s, 2 H), 8.19 (s, 1 H), 8.13 (s, 1 H), 8.01 (s, 1 H), 7.92 (d, J = 8.0 Hz, 1 H), 7.49 (d, J = 8.0 Hz, 1 H), 7.35 (s, 1 H), 3.54 (s, 2 H), 3.17 (s, 6 H), 2.89 (q, J = 7.2 Hz, 2 H), 2.40 (br s, 4 H), 2.33 (br s, 4 H), 2.15 (s, 3 H), 1.26 (t, J = 7.2 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.2, 160.3, 160.1, 149.5, 141.1, 140.4, 132.6, 130.9, 129.6 (q, J = 31.1 Hz), 128.8, 128.5, 124.6 (q, J = 270.8 Hz), 124.2, 122.2, 120.4, 115.5, 105.2, 90.3, 89.5, 61.9, 55.1, 52.9, 46.1, 37.1, 27.5, 14.8. HRMS (ESI) for C30H33F3N6O [M + H]+, calcd: 551.2741, found: 551.2733. Purity 98.5% (tR = 21.11 min).
4-Ethyl-3-(furan-3-ylethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5e)
Compound 5e was prepared by following a similar procedure to that for 5a. Yield, 50%. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1 H), 8.19–8.18 (m, 2 H), 8.13 (d, J = 1.6 Hz, 1 H), 8.01 (s, 1 H), 7.95–7.92 (m, 1 H), 7.81 (t, J = 1.6 Hz, 1 H), 7.50 (d, J = 8.0 Hz, 1 H), 7.35 (s, 1 H), 6.73 (d, J = 1.2 Hz, 1 H), 3.54 (s, 2 H), 2.87 (q, J = 7.6 Hz, 2 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.15 (s, 3 H), 1.26 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.2, 149.6, 146.9, 144.5, 141.1, 140.4, 132.6, 131.1, 129.7 (q, J = 31.3 Hz), 128.8, 128.7, 124.6 (q, J = 270.7 Hz), 124.1, 122.1, 120.3, 115.5, 112.7, 89.1, 85.4, 61.9, 55.0, 52.9, 46.0, 27.5, 14.8. HRMS (ESI) for C28H28F3N3O2 [M + H]+, calcd: 496.2206, found: 496.2204. Purity 98.6% (tR = 14.21 min).
4-Ethyl-3-(furan-2-ylethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5f)
Compound 5f was prepared by following a similar procedure to that for 5a. Yield, 46%. 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1 H), 8.18–8.17 (m, 2 H), 8.01 (s, 1 H), 7.98–7.96 (m, 1 H), 7.82 (d, J = 1.6 Hz, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.35 (s, 1 H), 6.96 (d, J = 3.6 Hz, 1 H), 6.65–6.64 (m, 1 H), 3.54 (s, 2 H), 2.87 (q, J = 7.6 Hz, 2 H), 2.40 (br s, 4 H), 2.33 (br s, 4 H), 2.15 (s, 3 H), 1.26 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.0, 149.7, 145.6, 141.1, 140.3, 136.1, 132.7, 131.1, 129.7 (q, J = 31.3 Hz), 129.3, 129.0, 124.6 (q, J = 270.8 Hz), 124.2, 121.0, 120.4, 116.7, 115.5, 112.1, 91.5, 83.8, 61.9, 55.0, 52.9, 46.1, 27.5, 14.8. HRMS (ESI) for C28H28F3N3O2 [M + H]+, calcd: 496.2206, found: 496.2198. Purity 99.6% (tR = 14.91 min).
4-Ethyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)-3-(thiophen-3-ylethynyl)benzamide (5g)
Compound 5g was prepared by following a similar procedure to that for 5a. Yield, 40%. 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1 H), 8.19 (s, 1 H), 8.15 (s, 1 H), 8.02 (s, 1 H), 7.94–7.93 (m, 2 H), 7.69–7.67 (m, 1 H), 7.50 (d, J = 8.0 Hz, 1 H), 7.35 (s, 1 H), 7.29 (d, J = 4.8 Hz, 1 H), 3.54 (s, 2 H), 2.90 (q, J = 7.6 Hz, 2 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.15 (s, 3 H), 1.27 (t, J = 7.2 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.2, 149.8, 141.1, 140.4, 132.6, 131.2, 130.4, 129.9, 129.7 (q, J = 31.3 Hz), 128.7, 127.5, 124.6 (q, J = 270.6 Hz), 124.2, 122.1, 121.5, 120.3, 115.5, 89.4, 86.9, 61.9, 55.1, 52.9, 46.1, 27.6, 14.8. HRMS (ESI) for C28H28F3N3OS [M + H]+, calcd: 512.1978, found: 512.1973. Purity 98.3% (tR = 16.21 min).
N-(4-Ethyl-3-((1-methyl-1H-imidazol-5-yl)ethynyl)phenyl)-3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)benzamide (5h)
Compound 5h was prepared by following a similar procedure to that for 5a. Yield, 46%. 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1 H), 8.19 (s, 1 H), 8.16 (s, 1 H) 8.01 (s, 1 H), 7.95 (d, J = 8.0 Hz, 1 H), 7.82 (s, 1 H), 7.52 (d, J = 8.0 Hz, 1 H), 7.37 (s, 1 H), 7.35 (s, 1 H), 3.75 (s, 3 H), 3.54 (s, 2 H), 2.90 (q, J = 7.2 Hz, 2 H), 2.40 (br s, 4 H), 2.33 (br s, 4 H), 2.15 (s, 3 H), 1.27 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.2, 149.4, 141.2, 140.3, 140.2, 134.6, 132.7, 130.9, 129.6 (q, J = 31.3 Hz), 128.9, 128.8, 124.6 (q, J = 270.7 Hz), 124.2, 121.7, 120.4, 115.5, 115.4, 94.2, 82.3, 61.8, 55.1, 52.9, 46.1, 32.2, 27.6, 14.9. HRMS (ESI) for C28H30F3N5O [M + H]+, calcd: 510.2475, found: 510.2471. Purity 99.4% (tR = 12.93 min).
N-(3-((1,2-Dimethyl-1H-imidazol-5-yl)ethynyl)-4-ethylphenyl)-3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)benzamide (5i)
Compound 5i was prepared by following a similar procedure to that for 5a. Yield, 48%. 1H NMR (400 MHz, DMSO-d6) δ 10.5 (s, 1 H), 8.19 (s, 1 H), 8.15 (d, J = 1.2 Hz, 1 H), 8.01 (s, 1 H), 7.95–7.92 (m, 1 H), 7.51 (d, J = 8.4 Hz, 1 H), 7.35 (s, 1 H), 7.24 (s, 1 H), 3.64 (s, 3 H), 3.54 (s, 2 H), 2.89 (q, J = 7.6 Hz, 2 H), 2.40–2.36 (m, 11 H), 2.15 (s, 3 H), 1.27 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.2, 149.1, 147.1, 141.1, 140.4, 133.0, 132.7, 130.8, 129.6 (q, J = 31.1 Hz), 128.9, 128.7, 124.6 (q, J = 270.7 Hz), 124.2, 121.8, 120.3, 115.5, 115.3, 94.0, 83.1, 61.9, 55.1, 52.9, 46.1, 31.2, 27.6, 14.9, 13.6. HRMS (ESI) for C29H32F3N5O [M + H]+, calcd: 524.2632, found: 524.2628. Purity 99.6% (tR = 13.87 min).
4-Ethyl-3-(imidazo[1,2-a]pyrazin-3-ylethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5j)
Compound 5j was prepared by following a similar procedure to that for 5a. Yield, 78%. 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1 H), 9.21 (d, J = 1.6 Hz, 1 H), 8.64 (dd, J = 4.8, 1.6 Hz, 1 H), 8.31 (d, J = 2.0 Hz, 1 H), 8.26 (s, 1 H), 8.19 (s, 1 H), 8.14 (d, J = 4.8 Hz, 1 H), 8.01–7.98 (m, 2 H), 7.56 (d, J = 8.4 Hz, 1 H), 7.35 (s, 1 H), 3.54 (s, 2 H), 2.98 (q, J = 7.6 Hz, 2 H), 2.39 (br s, 4 H), 2.33 (br s, 4 H), 2.14 (s, 3 H), 1.31 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.7, 150.1, 144.2, 141.7, 141.2, 140.8, 140.5, 133.3, 132.1, 131.8, 130.1 (q, J = 31.1 Hz), 129.9, 129.4, 125.1 (q, J = 270.6 Hz), 124.7, 121.6, 120.9, 119.7, 116.0, 109.9, 98.6, 80.2, 62.3, 55.6, 53.4, 46.6, 28.1, 15.4. HRMS (ESI) for C30H29F3N6O [M + H]+, calcd: 547.2428, found: 547.2425. Purity 99.8% (tR = 11.47 min).
4-Ethyl-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5k)
Compound 5k was prepared by following a similar procedure to that for 5a. Yield, 76%. 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1 H), 8.72 (dd, J = 4.4, 1.2 Hz, 1 H), 8.26 (dd, J = 9.2, 1.6 Hz, 1 H), 8.23 (m, 2 H), 8.20 (s, 1 H), 8.03 (s, 1 H), 7.99 (dd, J = 8.0, 2.0 Hz, 1 H), 7.56 (d, J = 8.0 Hz, 1 H), 7.39 (q, J = 3.6 Hz, 1 H), 7.36 (s, 1 H), 3.55 (s, 2 H), 2.99 (q, J = 7.2 Hz, 2 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.16 (s, 3 H), 1.31 (t, J = 7.2 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.1, 149.8, 145.4, 141.1, 140.3, 140.1, 138.5, 132.7, 131.0, 129.6 (q, J = 31.2 Hz), 129.2, 129.0, 126.5, 124.6 (q, J = 270.7 Hz), 124.2, 121.5, 120.4, 119.4, 115.5, 112.2, 96.6, 81.1, 61.9, 55.1, 52.9, 46.1, 27.6, 15.0. HRMS (ESI) for C30H29F3N6O [M + H]+, calcd: 547.2428, found: 547.2424. Purity 99.5% (tR = 23.41 min).
N-(4-Ethyl-3-(imidazo[1,2-a]pyridin-3-ylethynyl)phenyl)-3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)benzamide (5l)
Compound 5l was prepared by following a similar procedure to that for 5a. Yield, 83%. 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1 H), 8.59 (d, J = 6.4 Hz, 1 H), 8.28 (s, 1 H), 8.20 (s, 1 H), 8.07 (s, 1 H), 8.03 (s, 1 H), 7.97 (d, J = 8.0 Hz, 1 H), 7.75 (d, J = 8.8 Hz, 1 H), 7.55 (d, J = 8.0 Hz, 1 H), 7.46 (t, J = 7.2 Hz, 1 H), 7.36 (s, 1 H), 7.20 (t, J = 6.8 Hz, 1 H), 3.54 (s, 2 H), 2.98 (q, J = 7.6 Hz, 2 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.15 (s, 3 H), 1.32 (t, J = 7.6 Hz, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.3, 149.2, 145.8, 141.2, 140.3, 138.9, 132.8, 131.2, 129.6 (q, J = 31.0 Hz), 128.9, 127.0, 125.9, 124.6 (q, J = 270.6 Hz), 124.2, 121.7, 120.4, 1128.0, 115.5, 114.5, 108.0, 97.4, 81.2, 61.8, 55.1, 52.9, 46.1, 27.1, 14.9. HRMS (ESI) for C31H30F3N5O [M + H]+, calcd: 546.2475, found: 547.2465. Purity 99.6% (tR = 16.56 min).
3-(Imidazo[1,2-a]pyrazin-3-ylethynyl)-4-methyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5m)
Compound 5m was prepared by following a similar procedure to that for 9. Yield, 82%. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1 H), 9.22 (d, J = 1.2 Hz, 1 H), 8.68 (dd, J = 4.8, 1.6 Hz, 1 H), 8.32 (d, J = 1.6 Hz, 1 H), 8.28 (s, 1 H), 8.20 (s, 1 H), 8.14 (d, J = 4.4 Hz, 1 H), 8.03 (s, 1 H), 7.97 (dd, J = 8.0, 1.6 Hz, 1 H), 7.57 (d, J = 8.0 Hz, 1 H), 7.36 (s, 1 H), 3.55 (s, 2 H), 2.63 (s, 3 H), 2.40 (br s, 4 H), 2.35 (br s, 4 H), 2.16 (s, 3 H). 13C NMR (125 MHz, DMSO-d6) δ 165.6, 144.3, 144.1, 141.6, 141.2, 140.8, 140.4, 133.2, 131.7, 131.6, 130.9, 130.1 (d, J = 31.4 Hz), 129.6, 125.1 (d, J = 270.4 Hz), 124.7, 122.3, 120.9, 119.8, 116.0, 109.9, 98.9, 80.6, 62.3, 55.5, 53.4, 46.5, 21.4. HRMS (ESI) for C29H25f3N6O [M + H]+, calcd: 533.2271, found: 533.2275. Purity 95.4% (tR = 7.78 min).
3-(Imidazo[1,2-a]pyrazin-3-ylethynyl)-4-isopropyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5n)
Compound 5n was prepared by following a similar procedure to that for 9. Yield, 80%. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1 H), 9.22 (s, 1 H), 8.64 (d, J = 4.0 Hz, 1 H), 8.32 (s, 1 H), 8.28 (s, 1 H), 8.20 (s, 1 H), 8.15 (d, J = 4.0 Hz, 1 H), 8.02 (s, 2 H), 7.62 (d, J = 8.0 Hz, 1 H), 7.36 (s, 1 H), 3.61–3.56 (m, 1 H), 3.54 (s, 2 H), 2.39 (br s, 4 H), 2.33 (br s, 4 H), 2.15 (s, 3 H), 1.34 (d, J = 6.8 Hz, 6 H). 13C NMR (125 MHz, DMSO-d6) δ 165.2, 153.8, 143.7, 141.2, 140.7, 140.3, 140.0, 132.8, 131.8, 131.3, 129.6 (q, J = 31.0 Hz), 129.5, 126.1, 124.6 (q, J = 270.9 Hz), 124.2, 120.7, 120.4, 119.3, 115.5, 109.4, 98.1, 79.9, 61.8, 55.1, 52.9, 46.1, 32.0, 23.2. HRMS (ESI) for C31H31F3N6O [M + H]+, calcd: 561.2584, found: 561.2580. Purity 99.4% (tR = 14.87 min).
4-Cyclopropyl-3-(imidazo[1,2-a]pyrazin-3-ylethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5o)
Compound 5o was prepared by following a similar procedure to that for 9. Yield, 85%. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1 H), 9.21 (d, J = 1.2 Hz, 1 H), 8.68–8.66 (m, 1 H), 8.31 (d, J = 1.6 Hz, 1 H), 8.28 (s, 1 H), 8.19 (s, 1 H), 8.14 (d, J = 4.8 Hz, 1 H), 8.02 (s, 1 H), 7.96–7.94 (m, 1 H), 7.35 (s, 1 H), 7.15 (d, J = 8.4 Hz, 1 H), 3.54 (s, 2 H), 2.56–2.53 (m, 1 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.15 (s, 3 H), 1.23–1.17 (m, 2 H), 0.93–0.89 (m, 2 H). 13C NMR (125 MHz, DMSO-d6) δ 165.6, 150.2, 144.1, 141.6, 141.2, 140.8, 140.5, 132.5, 131.9, 131.7, 130.1 (q, J = 31.1 Hz), 129.9, 125.1 (q, J = 270.8 Hz), 124.7, 124.5, 122.2, 120.9, 119.9, 116.0, 110.0, 99.1, 80.3, 62.3, 55.6, 53.4, 46.6, 15.0, 11.1. HRMS (ESI) for C31H29F3N6O [M + H]+, calcd: 559.2428, found: 559.2427. Purity 98.2% (tR = 14.51 min).
4-(tert-Butyl)-3-(imidazo[1,2-a]pyrazin-3-ylethynyl)-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5p)
Compound 5p was prepared by following a similar procedure to that for 9. Yield, 85%. 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1 H), 9.23 (d, J = 1.2 Hz, 1 H), 8.66–8.65 (m, 1 H), 8.36 (d, J = 2.0 Hz, 1 H), 8.27 (s, 1 H), 8.21 (s, 1 H), 8.16 (d, J = 4.4 Hz, 1 H), 8.03 (s, 1 H), 8.01–7.98 (s, 1 H), 7.66 (d, J = 8.8 Hz, 1 H), 7.37 (s, 1 H), 3.56 (s, 2 H), 2.42 (br s, 4 H), 2.34 (br s, 4 H), 2.16 (s, 3 H), 1.61 (s, 9 H). 13C NMR (125 MHz, DMSO-d6) δ 165.6, 155.4, 144.3, 141.7, 141.2, 140.8, 140.3, 135.0, 133.3, 131.8, 130.2 (q, J = 31.4 Hz), 129.7, 127.1, 125.1 (q, J = 270.9 Hz), 124.7, 121.0, 120.7, 119.8, 116.0, 110.1, 101.3, 82.5, 62.3, 55.6, 53.4, 46.6, 36.8, 30.7. HRMS (ESI) for C32H33F3N6O [M + H]+, calcd: 575.2741, found: 575.2735. Purity 99.1% (tR = 15.84 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-isopropyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5q)
Compound 5q was prepared by following a similar procedure to that for 9. Yield, 81%. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1 H), 8.73–8.71 (m, 1 H), 8.27–8.19 (m, 4 H), 8.02–8.00 (m, 2 H), 7.61 (d, J = 8.4 Hz, 1 H), 7.40–7.35 (m, 2 H), 3.66–3.59 (m, 1 H), 3.56 (s, 2 H), 2.40 (br s, 4 H), 2.33 (br s, 4 H), 2.15 (s, 3 H), 1.33 (d, J = 6.8 Hz, 6 H). 13C NMR (125 MHz, DMSO-d6) δ 165.6, 154.5, 146.0, 141.7, 140.8, 140.5, 139.1, 133.2, 131.7, 130.1 (d, J = 31.4 Hz), 129.9, 127.0, 126.7, 125.1 (q, J = 270.9 Hz), 124.7, 121.6, 120.9, 120.0, 116.0, 112.7, 97.1, 81.8, 62.3, 55.6, 53.4, 46.6, 32.3, 23.6. HRMS (ESI) for C31H31F3N6O [M + H]+, calcd: 561.2584, found: 561.2579. Purity 99.1% (tR = 28.98 min).
3-(Imidazo[1,2-a]pyridin-3-ylethynyl)-4-isopropyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (5r)
Compound 5r was prepared by following a similar procedure to that for 9. Yield, 80%. 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1 H), 8.58 (d, J = 6.8 Hz, 1 H), 8.28 (d, J = 2.0 Hz, 1 H), 8.20 (s, 1 H), 8.07 (s, 1 H), 8.02 (s, 1 H), 8.00–7.98 (m, 1 H), 7.75 (d, J = 9.2 Hz, 1 H), 7.60 (d, J = 8.4 Hz, 1 H), 7.48–7.44 (m, 1 H), 7.36 (s, 1 H), 7.22–7.19 (m, 1 H), 3.61–3.56 (m, 1 H), 3.55 (s, 2 H), 2.40 (br s, 4 H), 2.34 (br s, 4 H), 2.16 (s, 3 H), 1.35 (d, J = 6.8 Hz, 6 H). 13C NMR (125 MHz, DMSO-d6) δ 165.8, 153.9, 146.3, 141.7, 140.8, 139.4, 133.3, 131.9, 130.1 (q, J = 31.4 Hz), 129.6, 127.5, 126.5, 126.4, 125.1 (q, J = 270.7 Hz), 124.7, 121.8, 120.9, 118.5, 116.0, 115.0, 108.5, 97.9, 81.9, 62.3, 55.6, 53.4, 46.6, 32.5, 23.6. HRMS (ESI) for C32H32F3N5O [M + H]+, calcd: 560.2632, found: 560.2628. Purity 99.6% (tR = 17.89 min).
Methyl 4-Ethyl-3-iodobenzoate (7a)24
To a suspension of methyl 3-amino-4-ethylbenzoate (10 g, 55.8 mmol) in water (100 mL) was added concd H2SO4 (10 mL) at 0 °C, and then a solution of NaNO2 (4.6 g, 67 mmol) in water (50 mL) was added dropwise. After the mixture has been stirred at 0 °C for 2 h, a solution of KI (10.2 g, 61.4 mmol) in water (50 mL) was added dropwise, and the mixture was warmed to rt slowly. The reaction was quenched with a saturated Na2S2O3 solution and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, concentrated in vacuo, and further purified by flash chromatography on silica gel to give the title compound, 7a (11.3 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 1.6 Hz, 1 H), 7.92–7.90 (m, 1 H), 7.24 (d, J = 2.8 Hz, 1 H), 3.87 (s, 3 H), 2.75 (q, J = 7.6 Hz, 2 H), 1.19 (t, J = 7.6 Hz, 3 H).
Methyl 3-Iodo-4-methylbenzoate (7m)24
Compound 7m was prepared by following a similar procedure to that for 7a. Yield, 68%. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1 H), 7.89 (dd, J = 8.0, 1.2 Hz, 1 H), 7.29 (d, J = 8.0 Hz, 1 H), 3.90 (s, 3 H), 2.47 (s, 3 H).
Methyl 3-Iodo-4-isopropylbenzoate (7n)
Compound 7n was prepared by following a similar procedure to that for 7a. Yield, 50%. 1H NMR (400 MHz, DMSO-d6) δ 8.33 (d, J = 2.0 Hz, 1 H), 7.93 (dd, J = 8.0, 2.0 Hz, 1 H), 7.47 (d, J = 8.0 Hz, 1 H), 3.84 (s, 3 H), 3.20–3.10 (m, 1 H), 1.20 (d, J = 6.8 Hz, 6 H). MS (ESI) m/z 305 [M + H]+.
Methyl 4-Cyclopropyl-3-iodobenzoate (7o)
Compound 7o was prepared by following a similar procedure to that for 7a. Yield, 63%. 1H NMR (400 MHz, DMSO-d6) δ 8.32 (d, J = 2.0 Hz, 1 H), 7.86–7.83 (m, 1 H), 7.05 (d, J = 8.0 Hz, 1 H), 3.83 (s, 3 H), 2.09–2.03 (m, 1 H), 1.11–1.06 (m, 2 H), 0.76–0.72 (m, 2 H). MS (ESI) m/z 303 [M + H]+.
Methyl 4-(tert-Butyl)-3-iodobenzoate (7p)
Compound 7p was prepared by following a similar procedure to that for 7a. Yield, 40%. 1H NMR (400 MHz, DMSO-d6) δ 8.47 (d, J = 1.2 Hz, 1 H), 7.91–7.89 (m, 1 H), 7.59 (d, J = 8.4 Hz, 1 H), 3.84 (s, 3 H), 1.51 (s, 9 H). MS (ESI) m/z 319 [M + H]+.
Methyl 4-Ethyl-3-ethynylbenzoate (8a)24
To a solution of methyl 4-ethyl-3-iodobenzoate (11.3 g, 39 mmol) in CH3CN (100 mL) were added PdCl2(PPh3)2 (548 mg, 0.78 mmol), CuI (149 mg, 0.78 mmol), and Et3N (16 mL, 117 mmol). The mixture was filled with Ar and stirred at 60 °C overnight. The reaction mixture was filtered through a pad of Celite and concentrated under vacuum. The residue was redissolved in MeOH (100 mL), and K2CO3 (16 g, 117 mmol) was added. The mixture was added at rt for 1 h. The reaction mixture was filtered through a pad of Celite and concentrated under vacuum. The resulting residue was purified by a silica-gel column to give the title compound, 8a (6.6 g, 90% yield). 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.95–7.92 (m, 1 H), 7.29–7.26 (m, 1 H), 3.90 (s, 3 H), 3.28 (s, 1 H), 2.86 (q, J = 7.6 Hz, 2 H), 1.25 (t, J = 7.6 Hz, 3 H).
Methyl 3-Ethynyl-4-methylbenzoate (8m)24
Compound 8m was prepared by following a similar procedure to that for 8a. Yield, 92%. 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 1.2 Hz, 1 H), 7.85 (dd, J = 8.0, 1.6 Hz, 1 H), 7.44 (d, J = 8.4 Hz, 1 H), 4.49 (s, 1 H), 3.84 (s, 3 H), 2.44 (s, 3 H).
Methyl 3-Ethynyl-4-isopropylbenzoate (8n)
Compound 8n was prepared by following a similar procedure to that for 8a. Yield, 89%. 1H NMR (400 MHz, DMSO-d6) δ 7.94–7.91 (m, 2 H), 7.51 (d, J = 8.0 Hz, 1 H), 4.49 (s, 1 H), 3.84 (s, 3 H), 3.45–3.39 (m, 1 H), 1.22 (d, J = 6.8 Hz, 6 H). MS (ESI) m/z 203 [M + H]+.
Methyl 4-Cyclopropyl-3-ethynylbenzoate (8o)
Compound 8o was prepared by following a similar procedure to that for 8a. Yield, 88%. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1 H), 7.82 (d, J = 8.0 Hz, 1 H), 6.99 (d, J = 8.4 Hz, 1 H), 4.49 (s, 1 H), 3.84 (s, 3 H), 2.42–2.35 (m, 1 H), 1.13–1.11 (m, 2 H), 0.83–0.81 (m, 2 H). MS (ESI) m/z 201 [M + H]+.
Methyl 4-(tert-Butyl)-3-ethynylbenzoate (8p)
Compound 8p was prepared by following a similar procedure to that for 8a. Yield, 88%. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (m, 1 H), 7.88 (d, J = 8.0 Hz, 1 H), 7.54 (d, J = 8.4 Hz, 1 H), 4.64 (s, 1 H), 3.84 (s, 3 H), 1.48 (s, 9 H). MS (ESI) m/z 217 [M + H]+.
4-Ethyl-3-ethynyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide (9)
To a solution of methyl 4-ethyl-3-ethynylbenzoate (6.6 g, 35.1 mmol) and 3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)aniline (9.1 g, 33.3 mmol) in THF (80 mL) was added t-BuOK (5.9 g, 52.6 mmol) at −20 °C. The mixture was warmed to rt slowly. After completion of the reaction, the mixture was poured into ice water and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, concentrated in vacuo, and purified by flash chromatography on silica gel to give the title compound, 9 (13.1 g, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1 H), 8.17 (s, 1 H), 8.10 (d, J = 2.0 Hz, 1 H), 8.00 (s, 1 H), 7.95–7.93 (m, 1 H), 7.48 (d, J = 8.4 Hz, 1 H), 7.35 (s, 1 H), 4.50 (s, 1 H), 3.54 (s, 2 H), 2.83 (q, J = 7.6 Hz, 2 H), 2.40–2.33 (m, 8 H), 2.16 (s, 3 H), 1.22 (d, J = 7.2 Hz, 3 H). MS (ESI) m/z 430 [M + H]+.
Methyl 3-(Imidazo[1,2-a]pyrazin-3-ylethynyl)-4-methylbenzoate (10m)
Compound 10m was prepared by following a similar procedure to that for 5a. Yield, 91%. 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1 H), 8.72 (d, J = 4.4 Hz, 1 H), 8.27 (s, 1 H), 8.21 (s, 1 H), 8.12 (d, J = 4.4 Hz, 1 H), 7.92 (dd, J = 8.0, 1.2 Hz, 1 H), 7.54 (d, J = 8.0 Hz, 1 H), 3.88 (s, 3 H), 2.60 (s, 3 H). MS (ESI) m/z 292 [M + H]+.
Methyl 3-(Imidazo[1,2-a]pyrazin-3-ylethynyl)-4-isopropylbenzoate (10n)
Compound 10n was prepared by following a similar procedure to that for 5a. Yield, 68%. 1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1 H), 8.26 (d, J = 4.4 Hz, 1 H), 8.24 (s, 1 H), 8.07 (d, J = 4.4 Hz, 1 H), 8.05–8.02 (m, 2 H), 7.44 (d, J = 8.0 Hz, 1 H), 3.94 (s, 3 H), 3.63–3.52 (m, 1 H), 1.36 (d, J = 6.4 Hz, 6 H). MS (ESI) m/z 320 [M + H]+.
Methyl 4-Cyclopropyl-3-(imidazo[1,2-a]pyrazin-3-ylethynyl)benzoate (10o)
Compound 10o was prepared by following a similar procedure to that for 5a. Yield, 82%. 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1 H), 8.70 (d, J = 4.0 Hz, 1 H), 8.26 (s, 1 H), 8.19 (s, 1 H), 8.11 (d, J = 4.0 Hz, 1 H), 7.88 (d, J = 8.4 Hz, 1 H), 7.10 (d, J = 8.4 Hz, 1 H), 3.86 (s, 3 H), 1.21–1.19 (m, 2 H), 0.88–0.85 (m, 2 H). MS (ESI) m/z 318 [M + H]+.
Methyl 4-(tert-Butyl)-3-(imidazo[1,2-a]pyrazin-3-ylethynyl)benzoate (10p)
Compound 10p was prepared by following a similar procedure to that for 5a. Yield, 64%. 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1 H), 8.65 (d, J = 4.0 Hz, 1 H), 8.26–8.25 (m, 2 H), 8.15 (s, 1 H), 7.96–7.94 (m, 1 H), 7.63 (d, J = 8.4 Hz, 1 H), 3.89 (s, 3 H), 1.57 (s, 9 H). MS (ESI) m/z 334 [M + H]+.
Methyl 3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-isopropylbenzoate (10q)
Compound 10q was prepared by following a similar procedure to that for 5a. Yield, 73%. 1H NMR (400 MHz, CDCl3) δ 8.49–8.47 (m, 1 H), 8.27 (d, J = 2.0 Hz, 1 H), 8.05–8.02 (m, 2 H), 8.00–7.98 (m, 1 H), 7.40 (d, J = 8.0 Hz, 1 H), 7.15–7.11 (m, 1 H), 3.92 (s, 3 H), 3.71–3.64 (m, 1 H), 1.35 (d, J = 6.8 Hz, 6 H). MS (ESI) m/z 320 [M + H]+.
Methyl 3-(Imidazo[1,2-a]pyridin-3-ylethynyl)-4-isopropylbenzoate (10r)
Compound 10r was prepared by following a similar procedure to that for 5a. Yield, 71%. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1 H), 8.17 (s, 1 H), 7.95 (d, J = 8.4 Hz, 1 H), 7.76 (br s, 1 H), 7.57 (d, J = 8.0 Hz, 1 H), 7.50–7.43 (m, 2 H), 7.19 (t, J = 6.8 Hz, 1 H), 3.87 (s, 3 H), 3.58–3.51 (m, 1 H), 1.32 (t, J = 7.2 Hz, 6 H). MS (ESI) m/z 319 [M + H]+.
Cells and Treatment
MPMs were prepared and cultured from C57BL/6 mice using the method described in our previous paper.30 MPMs were incubated in DMEM media (Gibco) supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37 °C with 5% CO2. Compounds were added into cell-cultural medium in DMSO solution with the final concentration of DMSO being 0.1‰.
Reagents
LPS was purchased from Sigma Chemical Company. Mouse TNF-α and IL-6 ELISA kits were purchased from eBiosscience. Anti-CD68 was from Santa Cruz Biotechnology. Trizol-reagent and primers were purchased from Invitrogen.
In Vitro Kinase Assay
The functional assays of the compounds’ effects on the kinase activity of Abl were determined using the FRET-based Z′-Lyte assay system according to the manufacturer’s instructions (Invitrogen). Tyrosine-2 peptide was used as the Abl substrate. The reactions were carried out in 384-well plates in a 10 μL reaction volume with the appropriate amounts of kinases in 50 mM HEPES (pH 7.5), 10 mM MgCl2, 1.0 mM EGTA, and 0.01% Brij-35. The reactions were incubated 1 h at room temperature in the presence of 2.0 μM substrate with 10 mM ATP and in the presence of various concentrations of the compounds. The development reagent was then added for a further 2 h room-temperature incubation, which was followed by the addition of the stop solution. The fluorescence-signal ratio of 445 nm (coumarin)/520 nm (fluorescein) was examined on an EnVision Multilabel Reader (PerkinElmer, Inc.).
The effects of the compounds on the kinases DDR1 and DDR2 were assessed by using a LanthaScreen Eu kinase-activity assay (Invitrogen). Kinase reactions are performed in a 10 μL volume in low-volume 384-well plates. The reaction buffer consists of 50 mM HEPES (pH 7.5), 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA, and the concentration of Fluorescein-poly-GAT substrate (Invitrogen) in the assay is 100 nM. Kinase reactions were initiated with the addition of 100 nM ATP in the presence of serial dilutions of the compounds. The reactions were allowed to proceed for 1 h at room temperature before a 10 μL preparation of EDTA (20 mM) and Eu-labeled antibody (4 nM) in TR-FRET dilution buffer was added. The final concentration of antibody in each assay well was 2 nM, and the final concentration of EDTA was 10 mM. The plate was allowed to incubate at room temperature for 1 h more before the TR-FRET-emission ratios of 665/340 nm were acquired on a PerkinElmer EnVision multilabel reader (PerkinElmer, Inc.). Data analysis and curve fitting were performed using GraphPad Prism4 software.
Active-Site-Dependent-Competition-Binding Assay: KINOME-Scan Screening
The binding affinity of 5n with DDR1 was analyzed by a KINOME-scan system, and the analysis was conducted by Ambit Bioscience. Briefly, kinases were tagged with DNA. The ligands were biotinylated and immobilized to streptavidin-coated beads. The binding reactions were assembled by incubating DNA-tagged kinases, immobilized ligands, and the test compounds in binding reactions (20% SeaBlock, 0.17× PBS, 0.05% tween-20, 6 mM DTT) for 1.0 h at room temperature. The affinity beads were washed with washing buffer (1× PBS, 0.05% Tween-20) first and then elution buffer (1× PBS, 0.05% Tween 20, 0.5 μM nonbiotinylated affinity ligands). The kinase concentration in the eluate was determined by quantitative PCR of the DNA tagged to the kinase. The ability of the test compound to bind to the kinase was evaluated with the following formula:
The negative control was DMSO (100% ctrl) and the positive control was a control compound (0% ctrl).
Immunoprecipitation and Western-Blot Analysis
Primary human lung fibroblasts were cultured in Medium199 (M199, Sigma-Aldrich) containing 10% fetal bovine serum (FBS) and maintained at 37 °C in a humidified incubator with 5% CO2 and 95% air. Cells were cultured in 100 mm tissue-culture dishes in complete media (M199 with 10% FBS) until they reached a high density (∼80% confluence). Then, cells were starved for 4 h in M199 with 1% FBS. After that, cells were cultured in 5 mL of complete media with 50 μg/mL collagen and a concentration of 5n for 24 h. Collagen and collagen with DMSO were added as controls. Cells were lysed, supernatants were recovered by centrifugation at 13 000 rpm, protein concentrations were measured, and equal amounts of total protein were separated by SDS-PAGE. For immunoprecipitation, lysates were precleared with protein A/G beads (Thermo Fisher Scientific). We used 200 μg of cellular protein in 1 mL of lysis buffer per immunoprecipitation reaction. To each sample, 1 μg of DDR2 antibody (Cell Signaling #12133) was added with 50 μL of protein A/G bead slurry; each sample was then allowed to rotate overnight at 4 °C on a nutator. Immunoprecipitated complexes were washed twice in lysis buffer, boiled in sample buffer, and subjected to SDS-PAGE. Proteins were transferred to PVDF membranes (Bio-Rad), which was followed by blocking for 1 h in 5% bovine serum albumin in TBS-T. Membranes were incubated overnight at 4 °C with primary antibody: phospho-DDR1 (Tyr792, Cell Signaling #11994), DDR1 (Santa Cruz SC-532), DDR2 (Cell Signaling #12133), phospho-tyrosine (Millipore, 4G10), c-Abl (Santa Cruz sc-23), p-c-Abl (Santa Cruz sc-293130), or tubulin-α (Biorad, MCA77D800). Membranes were incubated with the corresponding HRP-conjugated secondary antibody (Pierce Biotechnologies) for 1 h. Specific bands were detected using the enhanced-chemiluminescence reagent (ECL, PerkinElmer Life Sciences) on autoradiographic film.
Crystallization and Structure Determination
The kinase domain of human DDR1 (Uniprot Q08345, residues 601–913) was expressed as an N-terminal 6×His fusion in Sf9 cells and purified by nickel-affinity chromatography, followed by tag cleavage with TEV protease and then size-exclusion chromatography on an S200 column (GE Healthcare). Protein at 13.6 mg/mL in 50 mM HEPES, 300 mM NaCl, 0.5 mM TCEP, and 2% DMSO was incubated with 1 mM compound 5n for 4 h on ice and then filtered to 0.22 μm. Sitting drops (150 nL) were set up, with the highest-resolution crystals being obtained from a 1:2 ratio of protein to mother liquor (10% ethylene glycol, 0.2 M sodium sulfate, 24% PEG3350, 0.1 M bis-tris-propane; pH 7.1). Crystals were cryoprotected in mother liquor supplemented with 20% ethylene glycol and vitrified in liquid nitrogen. Diffraction was carried out at the Diamond Light Source beamline I03 at 100 K. Data were indexed and integrated using XDS31,32 and scaled using AIMLESS.33 Phases were identified using molecular replacement in PHASER.34 Structures were built using PHENIX.AUTOBUILD35 and then refined and modified using PHENIX.REFINE36 and COOT.35 The refined structure was validated with MolProbity,36 and the atomic-coordinate files were deposited in the Protein DataBank with Autodep.37
Determination of Pharmacokinetic Parameters in Rats
Male SD rats weighing 180–220 g (Southern China Medical University) were utilized for the studies. The protocol was approved by the Animal Care and Use Committee, GIBH. Animals were given standard animal chow and water ad libitum in a climate-controlled room (23 ± 1 °C, 30–70% relative humidity, a minimum of 10 exchanges of room air per hour, and a 12 h light–dark cycle) for 1 week prior to experiments. The compound was dissolved in a solution containing 2% DMSO, 4% ethanol, 4% castor oil, and 90% ddH2O. The pharmacokinetic properties of the SD rats (male) were determined following iv and oral administration. Animals were randomly distributed into two experimental groups (n = 3). The oral groups were given 25 mg/kg by gastric gavage. The other group was dosed by injection into the tail vein (5 mg/kg). After single administration, whole blood samples (100–200 μL) were obtained from the orbital venous plexus at the following time points after dosing: 5, 10, and 30 min and 1, 2, 3, 4, 6, 8, 11, and 24 h (po) or 2, 10, and 30 min and 1, 2, 3, 4, 6, 8, 11, and 24 h (iv). Whole blood samples were collected in heparinized tubes. The plasma fraction was immediately separated by centrifugation (8000 rpm, 6 min, 4 °C) and stored at −20 °C until LC-MS analysis. The rats were humanely euthanasia by carbon dioxide 24 h after the experiment without pain. The pharmacokinetics parameters were calculated by analyzing the compound concentrations in plasma samples using the pharmacokinetic software DAS.2.0.
Animals
Male C57BL/6 mice (6–8 weeks age) were obtained from the Animal Center of Wenzhou Medical University. Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals. All animal experimental procedures were approved by the Wenzhou Medical University Animal Policy and Welfare Committee.
Acute-Toxicity Assay
Male C57BL/6 mice weighing 18–20 g were randomly divided into three groups (n = 5 per group). Mice were gavage-administered 200 μL of the drug (at 200 or 400 mg/kg in physiological saline). Mice in the control group received 200 μL of physiological saline. After drug administration, body-weight changes were recorded for 7 days.
LPS-Induced ALI
Male C57BL/6 mice were randomly divided into four groups, designated CON (control, 8 mice, given only the vehicle of 0.9% saline), LPS (8 mice, given 5 mg/kg LPS alone), LPS+20 mg/kg BID 5n (8 mice, given both 20 mg/kg compound 5n and 5 mg/kg LPS), and LPS+40 mg/kg BID 5n (8 mice, given both 40 mg/kg compound 5n and 5 mg/kg LPS). Prior to intratracheal injection of LPS, the mice were treated orally two times per day with 5n at dosages of 20 and 40 mg/kg continuously for 1 week. Mice were then euthanized with ketamine 6 h after LPS induction. Blood was collected; the chest cavity of each animal was carefully opened, and BALF and lung tissues were collected.
BALF Analysis
The collected BALF was centrifuged at 1000 rpm for 10 min at 4 °C, the supernatant was used for protein-concentration detection and subsequent cytokine determinations. The precipitation was resuspended using 50 μL of physiological saline. The total number of cells in the BALF was detected by a cell-counting instrument. The number of neutrophils in the BALF was examined using Wright–Gimesa stain.
Determination of TNF-α and IL-6
The levels of pro-inflammatory cytokines TNF-α and IL-6 were determined in cell culture, BALF, and serum with an ELISA kit according to the manufacturer’s instructions. The total amount of the inflammatory factor in the culture media was normalized to the total protein quantity of the viable cell pellets.
Real-Time Quantitative PCR (RT-qPCR)
Lung tissues were homogenized in TRIZOL reagent for the extraction of RNA according to each manufacturer’s protocol. Both reverse-transcription and quantitative PCR were carried out using a two-step M-MLV Platinum SYBR Green qPCR SuperMix-UDG kit (Invitrogen). An Eppendorf Mastercycler ep realplex detection system (Eppendorf) was used for RT-qPCR analysis. Primers for the genes encoding TNF-α, IL-6, IL-1β, IL-12, ICAM-1, VCAM-1, and β-actin were obtained from Invitrogen. The amount of each gene was determined and normalized to the amount of β-actin.
Lung Wet/Dry Weight Ratio
The right upper lobe of the lung was excised. After removal of the excess water on the tissue surface, the wet weight was recorded. The sample was then dried at 60 °C for 48 h until there was no more weight change to record the dry weight. The wet-weight/dry-weight ratio (W/D) was calculated and used as an index of lung edema.
Pulmonary Histopathology and Immunohistochemistry Analysis
The right lower lobe of the lung was excised and fixed with 4% formalin. The lung tissues were embedded with paraffin, sliced to 5 μm sections, and stained with hematoxylin and eosin (HE). Mouse-lung histopathology images were acquired using a microscope (Nikon Model Eclipse 80i, Nikon). The immunohistochemistry analysis was performed following the anti-CD68-antibody-staining protocol.
Statistical Analysis
All in vitro experiments were assayed in triplicate. Data are expressed as means ± SD. All statistical analyses were performed using GraphPad Pro Prism 6.0 (GraphPad). Student’s t test was employed to analyze the differences between sets of data. A p value <0.05 was considered statistically significant.
Computational Study
All the procedures were performed in Maestro 11.2 (version 11.2, Schrödinger, LLC). The 3D structure of DDR2 has not been determined to date, although many homologous structures with high sequence identity have been reported. We chose the crystal structure of human DDR1 (PDB: 3ZOS), which shares 57% sequence identity with DDR2, as a template to generate a homology model for the active form of DDR2. The homology model of DDR2 was built by Prime Homology Modeling, and all the parameters were the defaults.
The DDR2 protein was processed using the Protein Preparation Wizard workflow in Maestro 9.4 (version 11.2, Schrödinger, LLC) to add bond orders and hydrogens. All hetatm residues and crystal water molecules beyond 5 Å from the het group were removed. Compounds 5a, 5b, 5i, and 5j were built by in the LigPrep module using the OPLS-2005 force field. The Glide module was used as the docking program. The grid-enclosing box was placed on the centroid of the 0LI, which was extracted from the crystal structure of DDR1. The standard-precision (SP) approach of Glide was adopted to dock compounds 5a, 5b, 5i, and 5j to DDR2 with the default parameters.
Acknowledgments
The authors appreciate the financial support from the National Natural Science Foundation of China (21572230, 81673285, and 81425021), Guangdong Province (2014TQ01R341, 2015A030306042, 2015A030312014, and 2016A050502041 and the Nanyue-Baijie Award), and Jinan University. We thank Diamond Light Source for beamtime (proposal mx15433), as well as the staff of beamline I03 for assistance with data collection. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA; ULTRA-DD grant no. 115766), Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome (106169/ZZ14/Z).
Glossary
Abbreviations Used
- LPS
lipopolysaccharide
- IL-6
interleukin 6
- MIP-1α
macrophage inflammatory protein 1α
- MPMs
mouse primary peritoneal macrophages
- ALI
mouse acute lung injure
- DDRs
discoidin-domain receptors
- RTKs
transmembrane-receptor tyrosine kinases
- MCP-1
monocyte-chemoattractant protein 1
- TNF-α
tumor-necrosis factor α
- INF-γ
interferon γ
- IC50
half-maximal (50%) inhibitory concentration of a substance
- ATP
adenosine triphosphate
- Kd
binding constant
- Abl
Abelson-murine-leukemia viral oncogene
- EPHA7
ephrin type-A receptor 7
- EPHB2
ephrin type-B receptor 2
- LCK
lymphocyte-specific protein tyrosine kinase
- LOK
serine–threonine kinase 10
- TIE2
angiopoietin-1 receptor
- TrkA
nerve growth-factor receptor A
- ELISA
enzyme-linked immunosorbent assay
- BID
twice daily
- W/D
wet/dry
- BALF
bronchial-alveolar-lavage fluid
- ICAM-1
intercellular cell-adhesion molecule 1
- VCAM-1
vascular cell-adhesion molecule 1
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01045.
Results of sequence alignment of DDR1 and DDR2, selectivity-profiling study of compound 5n, crystallization and structure determination of DDR1–5n, PK profiles of 5n, in vivo acute study of 5n, 1H and 13C NMR spectra of compounds 5a–5r, and HPLC traces of the representative compounds (PDF)
Compound-characterization checklist (XLS)
Molecular-formula strings (CSV)
Author Contributions
■ Z.W., Y.Z., and D.M.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Valiathan R. R.; Marco M.; Leitinger B.; Kleer C. G.; Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancerprogression. Cancer Metastasis Rev. 2012, 31, 295–321. 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger B. Discoidin domain receptor functions in physiologicaland pathological conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. 10.1016/B978-0-12-800180-6.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai L. K.; Luczynski M. T.; Huang P. H. Discoidin domainreceptors: a proteomic portrait. Cell. Mol. Life Sci. 2014, 71, 3269–3279. 10.1007/s00018-014-1616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borza C. M.; Pozzi A. Discoidin domain receptors in disease. Matrix Biol. 2014, 34, 185–192. 10.1016/j.matbio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothiwale S.; Borza C. M.; Lowe E. W. Jr.; Pozzi A.; Meiler J. Meiler, J.Discoidin domain receptor 1 (DDR1) kinase as target for structurebased drug discovery. Drug Discovery Today 2015, 20, 255–261. 10.1016/j.drudis.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju G. X.; Hu Y. B.; Du M. R.; Jiang J. L. Discoidin domainreceptors (DDRs): potential implications in atherosclerosis. Eur. J. Pharmacol. 2015, 751, 28–33. 10.1016/j.ejphar.2015.01.033. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lu X.; Ren X.; Ding K. Small molecule discoidindomain receptor kinase inhibitors and potential medical applications. J. Med. Chem. 2015, 58, 3287–3301. 10.1021/jm5012319. [DOI] [PubMed] [Google Scholar]
- Matsuyama W.; Wang L.; Farrar W. L.; Faure M.; Yoshimura T. Activation of discoidin domain receptor 1 isoform b with collagen up-regulates chemokine production in human macrophages: role of p38 mitogen-activated protein kinase and NF-kappa B. J. Immunol. 2004, 172, 2332–2340. 10.4049/jimmunol.172.4.2332. [DOI] [PubMed] [Google Scholar]
- Flamant M.; Placier S.; Rodenas A.; Curat C. A.; Vogel W. F.; Chatziantoniou C.; Dussaule J. C. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J. Am. Soc. Nephrol. 2006, 17, 3374–3381. 10.1681/ASN.2006060677. [DOI] [PubMed] [Google Scholar]
- Avivi-Green C.; Singal M.; Vogel W. F. Discoidin domain receptor 1-deficient mice are resistant to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 420–427. 10.1164/rccm.200603-333OC. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Bian H.; Bartual S. G.; Du W.; Luo J.; Zhao H.; Zhang S.; Mo C.; Zhou Y.; Xu Y.; Tu Z.; Ren X.; Lu X.; Brekken R. A.; Yao L.; Bullock A. N.; Su J.; Ding K. Structure-based design of tetrahydroisoquinoline-7-carboxamides as selective discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2016, 59, 5911–5916. 10.1021/acs.jmedchem.6b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Zhang Y.; Bartual S. G.; Luo J.; Xu T.; Du W.; Xun Q.; Tu Z.; Brekken R. A.; Ren X.; Bullock A. N.; Liang G.; Lu X.; Ding K. Tetrahydroisoquinoline-7-carboxamide derivatives as new selective discoidin domain receptor 1 (DDR1) inhibitors. ACS Med. Chem. Lett. 2017, 8, 327–332. 10.1021/acsmedchemlett.6b00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudel B.; Yoon D. S.; Lee J. H.; Lee Y. M.; Kim D. K. Collagen I enhances functional activities of human monocyte-derived dendritic cells via discoidin domain receptor 2. Cell. Immunol. 2012, 278, 95–102. 10.1016/j.cellimm.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Poudel B.; Ki H. H.; Lee Y. M.; Kim D. K. Induction of IL-12 production by the activation of discoidin domain receptor 2 via NF-kappaB and JNK pathway. Biochem. Biophys. Res. Commun. 2013, 434, 584–588. 10.1016/j.bbrc.2013.03.118. [DOI] [PubMed] [Google Scholar]
- Yang J.; Wheeler S. E.; Velikoff M.; Kleaveland K. R.; Lafemina M. J.; Frank J. A.; Chapman H. A.; Christensen P. J.; Kim K. K. Activated alveolar epithelial cells initiate fibrosis through secretion of mesenchymal proteins. Am. J. Pathol. 2013, 183, 1559–1570. 10.1016/j.ajpath.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z.; Liu H.; Sun X.; Guo R.; Cui R.; Ma X.; Yan M. RNA interference against discoidin domain receptor 2 ameliorates alcoholic liver disease in rats. PLoS One 2013, 8, e55860 10.1371/journal.pone.0055860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaso E.; Ikeda K.; Eng F. J.; Xu L.; Wang L. H.; Lin H. C.; Friedman S. L. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J. Clin. Invest. 2001, 108, 1369–1378. 10.1172/JCI200112373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz P. A.; Jarai G. Collagen I induces discoidin domain receptor (DDR) 1 expression through DDR2 and a JAK2-ERK1/2-mediated mechanism in primary human lung fibroblasts. J. Biol. Chem. 2011, 286, 12912–12923. 10.1074/jbc.M110.143693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters A.; Nguyen H. D.; Phan T.; Simard J. R.; Grutter C.; Engel J.; Rauh D. Identification of type II and III DDR2 inhibitors. J. Med. Chem. 2014, 57, 4252–4262. 10.1021/jm500167q. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Berdini V.; Buck I. M.; Carr M. E.; Cleasby A.; Coyle J. E.; Curry J. E.; Day J. E.; Day P. J.; Hearn K.; Iqbal A.; Lee L. Y.; Martins V.; Mortenson P. N.; Munck J. M.; Page L. W.; Patel S.; Roomans S.; Smith K.; Tamanini E.; Saxty G. Fragment-based discovery of potent and selective DDR1/2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 798. 10.1021/acsmedchemlett.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai H.; Tan L.; Beauchamp E. M.; Hatcher J. M.; Liu Q.; Meyerson M.; Gray N. S.; Hammerman P. S. Characterization of DDR2 inhibitors for the treatment of DDR2 mutated non-small cell lung cancer. ACS Chem. Biol. 2015, 10, 2687–2696. 10.1021/acschembio.5b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E.; Waters B.; Spiegel K.; Alnadaf T.; Manley P. W.; Buchdunger E.; Walker C.; Jarai G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. 10.1016/j.ejphar.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Sonogashira K. Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653, 46–49. 10.1016/S0022-328X(02)01158-0. [DOI] [Google Scholar]
- Gao M.; Duan L.; Luo J.; Zhang L.; Lu X.; Zhang Y.; Zhang Z.; Tu Z.; Xu Y.; Ren X.; Ding K. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamidesas novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2013, 56, 3281–3295. 10.1021/jm301824k. [DOI] [PubMed] [Google Scholar]
- Chang S.; Zhang L.; Xu S.; Luo J.; Lu X.; Zhang Z.; Xu T.; Liu Y.; Tu Z.; Xu Y.; Ren X.; Geng M.; Ding J.; Pei D.; Ding K. Design, synthesis, and biological evaluation of novel conformationallyconstrained inhibitors targeting epidermal growth factorreceptor threonine790 →methionine790 mutant. J. Med. Chem. 2012, 55, 2711–2723. 10.1021/jm201591k. [DOI] [PubMed] [Google Scholar]
- Li Y.; Shen M.; Zhang Z.; Luo J.; Pan X.; Lu X.; Long H.; Wen D.; Zhang F.; Leng F.; Li Y.; Tu Z.; Ren X.; Ding K. Design, synthesis, and biological evaluation of 3-(1H-1,2,3-triazol-1-yl)benzamide derivatives as potent pan Bcr-Abl inhibitors including the threonine(315)-->isoleucine(315) mutant. J. Med. Chem. 2012, 55, 10033–10046. 10.1021/jm301188x. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Belkina N. V.; Liu Y.; Hao J. J.; Karasuyama H.; Shaw S. LOK is a major ERM kinase in resting lymphocytes and regulates cytoskeletal rearrangement through ERM phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4707–4712. 10.1073/pnas.0805963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahira T.; Ueda N.; Kurihara M.; Matsushima S.; Ohmori H.; Fujii K.; Bhawal U. K.; Yamamoto K.; Kirita T.; Kuniyasu H. Tropomyosin receptor kinases B and C are tumor progressive and metastatic marker in colorectal carcinoma. Hum. Pathol. 2013, 44, 1098–1106. 10.1016/j.humpath.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Chen G.; Zhang Y.; Liu X.; Fang Q.; Wang Z.; Fu L.; Liu Z.; Wang Y.; Zhao Y.; Li X.; Liang G. Discovery of a new inhibitor of myeloid differentiation 2 from cinnamamide derivatives with antiinflammatory activity in sepsis and acute lung injury. J. Med. Chem. 2016, 59, 2436–2451. 10.1021/acs.jmedchem.5b01574. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H. R. Processing diffraction data with MOSFLM. Evolv. Methods Macro. Crystallogr. 2007, 245, 41–51. 10.1007/978-1-4020-6316-9_4. [DOI] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Guranovic V.; Dutta S.; Feng Z.; Berman H. M.; Westbrook J. D. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1833–1839. 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.