

Abstract
The development of new catalysts for selective nitrene transfer is a continuing area of interest. In particular, the ability to control the chemoselectivity of intermolecular reactions in the presence of multiple reactive sites has been a long-standing challenge in the field. In this paper, we demonstrate examples of silver-catalyzed, nondirected, intermolecular nitrene transfer reactions that are both chemoselective and flexible for aziridination or C‒H insertion, depending on the choice of ligand. Experimental probes present a puzzling picture of the mechanistic details of the pathways mediated by [(tBu3tpy)AgOTf]2 and (tpa)AgOTf. Computational studies elucidate these subtleties and provide guidance for the future development of new catalysts exhibiting improved tunability in group transfer reactions.
Graphical Abstract
INTRODUCTION
Amines are important functional groups occurring in a diverse array of molecules with significant biological and therapeutic value. One effective strategy for streamlining the synthesis of amines involves the selective introduction of a new C‒N bond at a specific site in a hydrocarbon precursor. Diverse transition metals1–8 are known to catalyze the insertion of metal nitrenes into either a C‒H or C=C bond to yield amines or aziridines, respectively. Substrate control has traditionally been the primary factor dictating the chemoselectivity of the amination event; for example, intramolecular aminations of homoallylic sulfamates tend to yield the aziridination product using popular dinuclear Rh(II) tetracarboxylate catalysts.2 One notable exception is the dinuclear, chiral carboxamidate complex Rh2(S-nap)4, which can favor intramolecular C‒H bond insertion over aziridination.9 More recently, it has been shown that the chemoselectivity of nitrene transfer can be altered to favor C‒H insertion by changing the metal identity to Ru, Co, Mn, or Fe, which results in a change from a concerted to a stepwise nitrene transfer event.3,4b,5,8
Achieving catalyst control over the chemoselectivity of intermolecular nitrene transfer using a single metal is even more challenging, particularly when compared to metal-catalyzed carbene transfer reactions.10 A rare example of a Rh-based system selective for intermolecular nitrene insertion into allylic C‒H bonds employs Rh2((S)-nta)4 (nta = (S)-N-1,8-naphthoylalanine) in the presence of chiral sulfonimidamides;11 however, this behavior relies on a strong match between the chiral Rh catalyst and the optically pure nitrene precursor. In general, most dinuclear Rh catalysts that are successful for intramolecular nitrene transfer perform poorly in intermolecular reactions.12 Two major factors impeding the development of more modular and less expensive catalysts for nitrene transfer include a poor understanding of how reactivity differs in intra-versus intermolecular processes, as well as how the mechanism and selectivity of the nitrene transfer responds to modifications to the supporting ligands for a particular metal. A better understanding of these issues is necessary to spur further progress toward our ultimate goal of developing new, inexpensive catalysts for predictable, tunable, and intermolecular site-selective nitrene transfer reactions, a long-standing goal in the field.
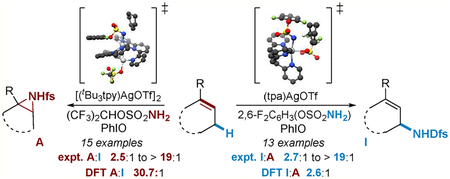
We have demonstrated that silver(I) catalysis offers unique opportunities to tune the chemo- and site-selectivity of intra-molecular nitrene transfer (Scheme 1A,B).13–15 Changes to the ligand, the AgOTf/ligand ratio, and the nitrene precursor all influence the coordination geometry and reactivity of the putative nitrene. For example, preference for aziridination or C‒H bond insertion in a homoallylic carbamate can be controlled by simply changing the ratio of 1,10-phenanthroline (phen) to AgOTf (Scheme 1A).13 Enforcing different coordination geometries at Ag(I) using diverse ligands, including 4,4′-di-tert-butyl-2,2′-bipyridine (tBubpy) and tris(2-pyridylmethyl)amine (tpa), enables site-selective amination of chemically distinct C‒H bonds (Scheme 1B).14,15 Expanding Ag(I) catalysis to tunable, intermolecular nitrene transfer reactions has been challenging; in this work, we describe initial experimental and computational studies to better understand these reactions (Scheme 1C).
Scheme 1.

Ag-Catalyzed Chemo- and Site-Selective Aminations
RESULTS AND DISCUSSION
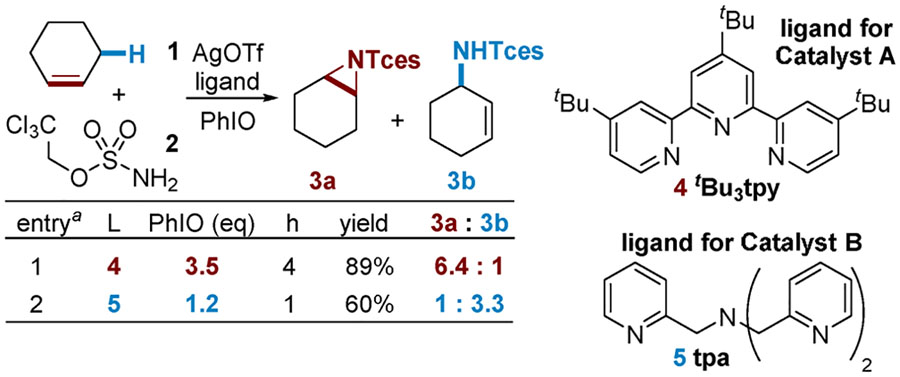
Initial Experimental Studies.
The reaction of cyclohexene (1), 2,2,2-trichloroethylsulfamate (TcesNH2, 2), and iodosobenzene (PhIO) with Ag catalysts supported by a variety of readily available N-donor ligands (see the Supporting Information for further details) was carried out (Table 1). We found that AgOTf ligated by tert-butylterpyridine 4 (entry 1) gave good selectivity for production of the aziridine 3a over the allylic amine 3b.7d–f X-ray crystallographic and DOSY NMR studies showed that the silver catalyst supported by 4 exists in both the solid and solution states as the dimer [(tBu3tpy)-AgOTf]2 (hereafter called catalyst A).7e,13 Interestingly, employing the tpa ligand 5 (entry 2) resulted in a preference for allylic C‒H amination to 3b over aziridination. A crystal structure of a silver(I) complex bearing a ligand similar to 5 has been reported, suggesting the formula (tpa)Ag(OTf) for catalyst B.16
Table 1.
Impact of the Ligand on Silver-Catalyzed Nitrene Transfer
|
NMR yield; Cl3CCH2Cl internal standard. Conditions: 5 equiv of 1, 1 equiv of 2, 10 mol % of AgOTf, 12 mol % of ligand, PhIO, 4 Å MS,CH2Cl2.
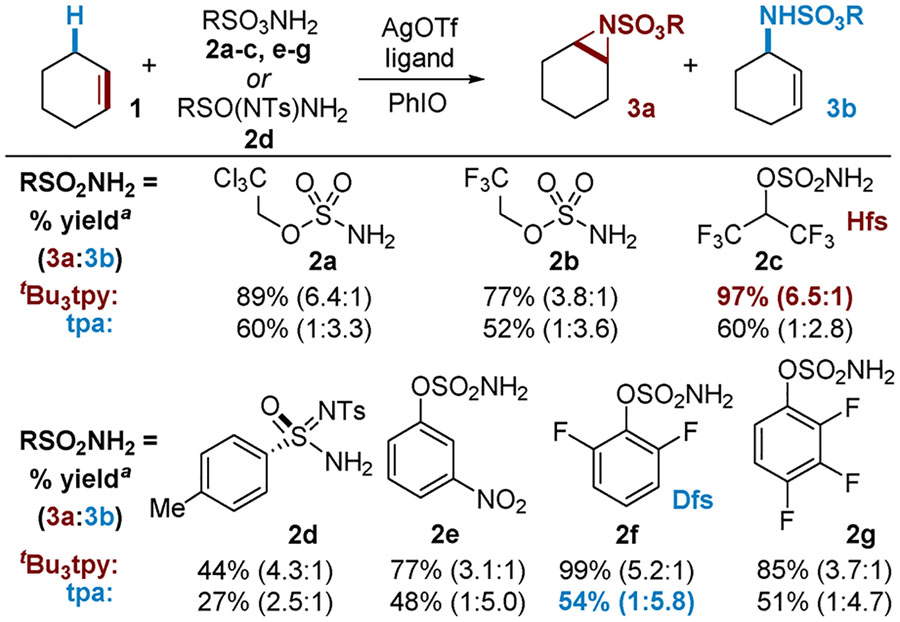
Having demonstrated the ability to control the chemo-selectivity of Ag-catalyzed amination by changing the nature of the ligand, the identity of the nitrene precursor was varied in an attempt to further improve selectivity (Table 2). While the 3a/3b selectivity for aziridination catalyzed by A was not improved with alkylsulfamate 2c as compared to 2a, the yield did increase to 97%. Other nitrene precursors resulted in either lower yields or decreased selectivity for the aziridine 3a. When B was employed, the C‒H amination product was favored to a greater extent employing arylsulfamates 2e–2g, with 2f displaying the highest 3b/3a ratio of 5.8:1. It is worth noting that in the majority of examples, the overall preference of A for aziridination and that of B for C‒H amination is independent of the identity of the nitrene precursor, supporting catalyst control of nitrene transfer. The one exception is the chiral sulfonimidamide 2d previously employed by Dauban in Rh-catalyzed nitrene transfer;11 in this case, aziridination was preferred using both A and B.
Table 2.
Influence of the Nitrene Precursor on Chemoselectivity
|
Total NMR yield; Cl3CCHCl2 as the internal standard. Conditions: 5 equiv of 1, 1 equiv of 2, 10 mol % of AgOTf, 12 mol % of ligand, 4 Å MS, CH2Cl2, rt. tBu3tpy catalyst A: 3.5 equiv PhIO, 4 h; tpa catalyst B:1.2 equiv of PhIO, 30 min.
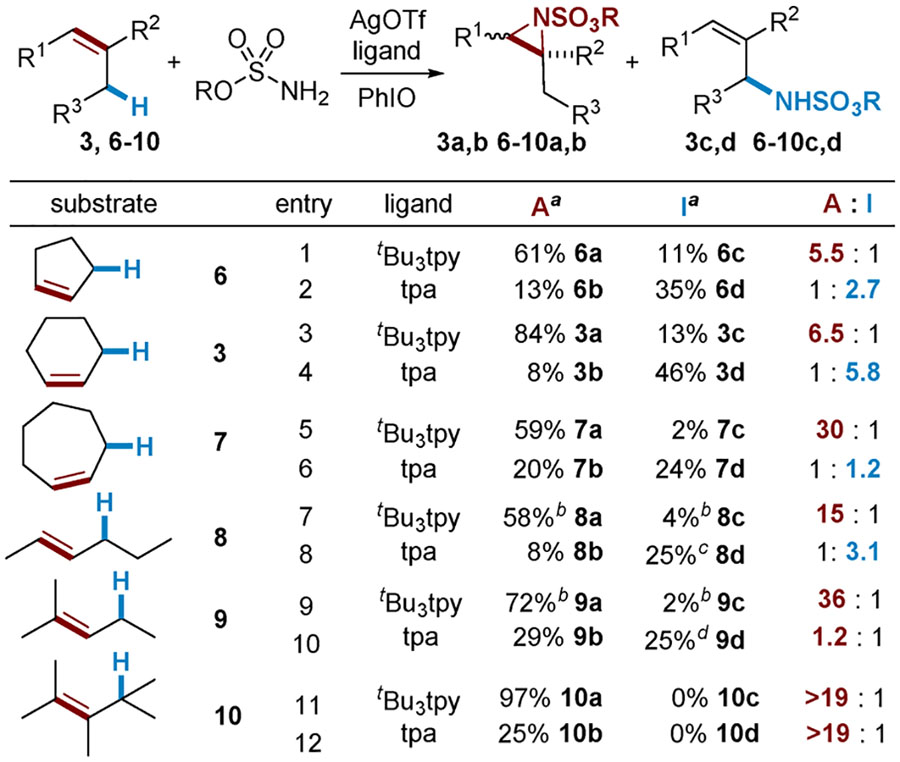
The scope of tunable nitrene transfer was briefly explored with selected substrates containing both C=C and C‒H functionalities (Table 3). Five- and six-membered rings (entries 1–4) give chemodivergent amination. However, as the size of the ring increases (entries 5 and 6), chemoselectivity for the aziridine product also increases with both catalysts A and B. The lesser selectivity for C‒H amination with B in this case may be attributed to poor stabilization of an allylic radical via conjugation with the double bond in the seven-membered ring due to competing transannular interactions, providing indirect evidence that B may operate via a stepwise nitrene transfer pathway. Reaction of trans-hex-2-ene 8 (entries 7 and 8) gave no isomerization in either aziridination or C‒H amination reactions but did show a rearranged product using B (entry 8), also indicative of a potential stepwise pathway. A is not overly sensitive to steric effects, giving excellent selectivity for aziridination of both tri- and tetrasubstituted alkenes 9 and 10 (entries 9 and 11). However, achieving selectivity for C‒H insertion in acyclic substrates employing B is challenging, as aziridination becomes competitive (entries 10 and 12). In order to rationalize these observations and achieve a better understanding of the reasons for the lack of chemocontrol, extensive computational studies were carried out as described later in this report.
Table 3.
Tunable, Catalyst-Controlled Amination*
|
A: HfsNH2 (0.25 mmol), 10 mol % of AgOTf, 12 mol % of tBu3tpy,3.5 equiv of PhIO, CH2Cl2, 4 Å MS, 4 h, rt. I: DfsNH2 (0.25 mmol), 10 mol % of AgOTf, 12 mol % of tpa, 1.2 equiv of PhIO, CH2Cl2, 4 Å MS, 1 h, rt.
Isolated yield.
NMR yield, Cl3CCH2Cl internal standard.
trans only, 6% rearranged.
3% rearranged.
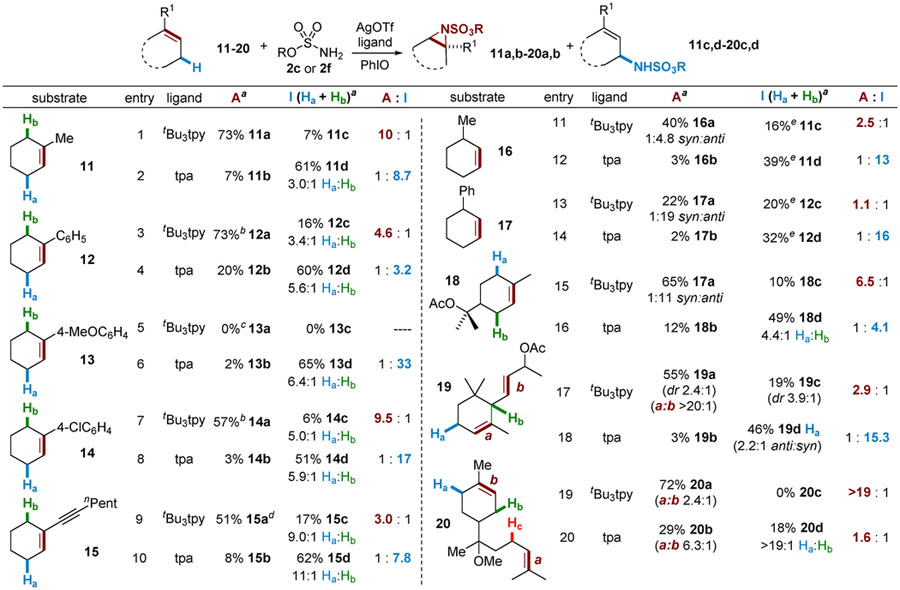
A series of substituted cyclohexenes of varying complexity were investigated further in tunable silver-catalyzed amination (Table 4). Addition of a Me group to the alkene of 11 increases selectivity for aziridination with A (entry 1) and improves the preference for C‒H amination with B; amination of the less hindered allylic C‒Ha bond is favored over C‒Hb by a ratio of3.0:1 (entry 2). Aryl-substituted alkenes 12 and 14 (entries 3 and 7) resulted in good selectivity using A, but competing epoxidation occurs with electron-rich 13 (entry 5). Excellent selectivity for C‒H amination is observed for 12–14 using B (entries 4, 6, and8). Catalyst B shows sensitivity to steric effects, as amination of the less congested Ha in 12–14 is preferred over the more hindered Hb by a ratio of approximately 6:1 in all three cases. Conjugation of an alkyne with the cyclohexene of 15 gives good chemoselectivity with both catalysts (entries 9 and 10); however, the aziridine is not stable to chromatography, undergoing rearrangement and hydrolysis to aldehyde 15a.17 Good site-selectivity for the amination of Ha over Hb was also noted using 15 (entry 10). Moving the Me group from the vinyl carbon of 11 to the allylic position in 16 (entries 11 and 12) decreases selectivity for aziridination with A. In contrast, the selectivity of C‒H amination was improved with B; however, allylic transposition occurs to give 11d and 16d. The addition of an allylic Ph group in 17 further decreases selectivity in aziridination (entry 13) but showed increased preference for insertion to 12d (entry 14). These experiments provide insights into the mechanistic details of Ag-catalyzed intermolecular nitrene transfer that will be further discussed in the context of our computational studies (vide infra).
Table 4.
Tunable Amination of C=C and C‒H Bonds*
|
A: 10 mol % of AgOTf, 12 mol % of tBu3tpy, HfsNH2 (0.25 mmol), 3.5 equiv of PhIO, CH2Cl2, 4 Å MS, 4 h, rt. I: 10 mol % of AgOTf, 12 mol % of tpa, DfsNH2 (0.25 mmol). 1.2 equiv of PhIO, CH2Cl2 4 Å MS, 1 h, rt.
Isolated yields.
Aziridine was opened with MeOH.
Formation of the epoxide as the major product.
56% NMR yield of aziridine, isolated as the aldehyde in 51% yield.
Transposition of the allylic bond was noted to give the trisubstituted alkene as the product. The ratio of expected to allylic product was approximately 1:1, while transposition was highly dominant for 17.
A series of natural-product-derived substrates 18–20, containing multiple alkene and allylic C‒H functionalities, were explored. Alkene 18 gives good selectivity for aziridination and a high 11:1 anti/syn ratio (entry 15). The sensitivity of B to steric effects is illustrated in entry 16, where activation of Ha is now preferred over Hb by a ratio of 4.4:1 (compare to entry 2). The α-ionone derivative 19 shows poorer selectivity for aziridination (entry 17), perhaps due to the increased steric demand of the more electron-rich alkene a. Both chemo- and site-selectivity for C‒H insertion (entry 18) are excellent. Finally, only aziridination of bisabolyl methyl ether 20 is observed with A (entry 19), with the exocyclic alkene preferred over the endo-cyclic alkene by a ratio of 2.4:1. The presence of two electron-rich alkenes in 20 results in poor selectivity for C‒H amination (entry 20); however, site-selectivity for the less hindered Ha is excellent, favoring Ha over Hb by a ratio of >19:1.
Mechanistic Studies.
Further experimental studies were carried out to better understand the mechanistic details of amination pathways promoted by A and B with both cyclic and acyclic substrates. In Scheme 2A, subjecting 11 to aziridination conditions in the absence and presence of TEMPO as a radical inhibitor showed a modest 10% decrease in yield, which is not a definitive indication that radical intermediates are involved. In contrast, subjecting 11 to C‒H amination conditions did result in a substantial decrease in yield in the presence of TEMPO, indicating the possibility of a radical intermediate. These results suggest that catalysts A and B operate under mechanistically distinct pathways. However, we note that the C‒H amination products of 16 and 17 employing either catalyst A or B contain a transposed double bond (Table 4, entries 11–14). This suggests both silver catalysts are capable of promoting stepwise nitrene transfer pathways involving radical intermediates. Surprisingly, TEMPO inhibition studies with 16 (Scheme 2B) show decreased yields of the insertion product but have little effect on the aziridination yield. Explaining these puzzling results is important for developing better Ag(I) catalysts and were addressed using computational studies (vide infra).
Scheme 2.

Influence of TEMPO on the Amination of 11 and 16
aA: HfsNH2 (0.25 mmol), 10 mol % of AgOTf, 12 mol % of tBu3tpy, 3.5 equiv of PhIO, CH2Cl2, 4 Å MS, 4 h, rt. bI: DfsNH2 (0.25 mmol), 10 mol % of AgOTf, 12 mol % of tpa, 1.2 equiv of PhIO, CH2Cl2, 4 Å MS, 1 h, rt.
Further mechanistic studies with acyclic alkenes (Schemes 3 and 4) reveal that catalyst A generates 21a exclusively as the cis isomer (Scheme 3), while B gives 21b as a 13:1 mixture of cis/trans isomers. Both catalysts favor aziridination, although the selectivity with B (2.9:1) is much lower than that observed with A (>20:1). Slight isomerization of the alkene from >20:1 cis/trans to 13:1 cis/trans in the C‒H amination product 21b occurs in both the absence and presence of TEMPO.
Scheme 3.

Influence of the Radical Inhibitor TEMPO on the Amination of Acyclic Alkene 21
aA: HfsNH2 (0.25 mmol), 10 mol % of AgOTf, 12 mol % of tBu3tpy,3.5 equiv of PhIO, CH2Cl2, 4 Å MS, 4 h, rt. bI: DfsNH2 (0.25 mmol), 10 mol % of AgOTf, 12 mol % of tpa, 1.2 equiv of PhIO, CH2Cl2, 4 Å MS, 1 h, rt.
Scheme 4.

Allylic Isomerization Studies
a5 equiv of 22, 1 equiv of HfsNH2, 10 mol % of AgOTf, 12 mol % of tBu3tpy, 3.5 equiv of PhIO, 0.05 M CH2Cl2, 4 Å MS, 4 h, rt. b5 equiv of 22, 1 equiv of DfsNH2, 10 mol % of AgOTf, 12 mol % of tpa,1.2 equiv of PhIO, 0.05 M CH2Cl2, 4 Å MS, 1 h, rt.
While the addition of TEMPO using catalyst B supported the presence of radical intermediates in the nitrene transfer pathway, the minimal isomerization of 21 (Scheme 3) with both A and B was unexpected. To shed more insight into this question, a sterically congested alkene 22 was subjected to both aziridination and C‒H amination conditions (Scheme 4). These experiments suggest the presence of radical intermediates in both pathways, as transposition of the alkene double bond was noted using both catalysts. The question was why catalyst A appeared in many cases to give concerted nitrene transfer, as opposed to the clear-cut evidence that B proceeded through a stepwise nitrene transfer pathway.
Computational Studies.
The only previously reported, extensive computational study on Ag-catalyzed nitrene transfer explores aziridination using a trispyrazolyl borate complex (catalyst C).7a Aziridination was found to proceed through an initial C-N bond formation step, but a minimum energy crossing point (MECP) between the triplet and closed-shell singlet surface induces direct formation of the aziridine from the C-N bond formation transition state, resulting in retention of the stereochemistry of the alkene. The reactivity observed with our ligands on Ag(I) led to an initial hypothesis that catalyst A follows a single-step mechanism similar to that described by Pérez and co-workers, and catalyst B follows a stepwise pathway.7a However, our experimental probes of the mechanism (Schemes 2–4) yield puzzling results. Experiments in Scheme 2 on cyclic alkenes appear to support our hypothesis, as the use of TEMPO as a radical inhibitor did not impact the aziridination pathways involving catalyst A. The same is true for the acyclic alkene 21 in Scheme 3; however, the data in Scheme 4 suggest that even catalyst A is capable of promoting stepwise nitrene transfer, as allylic C=C double bond transposition is observed. To find a mechanistic explanation for these seemingly contradictory observations, as well as to investigate the reasons for the chemoselectivity differences between A and B, we decided to employ quantum chemical methods to study the reactivity of these two catalysts at the level of density functional theory (DFT). Here, B3LYP-D3 is used for the structures of critical points along the reaction coordinates, and we employ complete active space self-consistent field (CASSCF) for the electronic structure of the nitrene complexes. The validity of our choice of B3LYP-D3 is demonstrated by benchmarking various functionals against the crystal structure of catalyst A, and B3LYP-D3 was found to be one of the most robust functionals in reproducing the observed molecular geometry (see Table S4 for quantitative assessments of the functionals).
Structural Features of Nitrene Intermediates.
To begin our computational studies, the nitrene complex of B, (tpa)Ag(OTf) (NSO3R) B(N), was optimized with the nitrene ligand bound in the equatorial site (cis to the tertiary amine) and the OTf− ion bound in the axial site. This nitrene complex of catalyst B was chosen for computational studies of its reactivity due to the fact that the chemoselectivity of B is affected by the identity of the Ag salt employed, supporting the hypothesis that the anionic OTf ligand remains bound to B(N) during catalysis (see the Supporting Information for the discussion of other potential structures of B(N)). Catalyst A has been shown to exist as a dimer with one of the two OTf ions dissociated in its crystal structure, leaving two inequivalent Ag sites.7e AgN has a coordination sphere composed entirely of tBu3tpy N atoms, while AgO also has a coordinated OTf ion. To improve computational efficiency, the tert-butyl groups on the tBu3tpy ligand were replaced by H atoms [(tpy)2 Ag2OTF]+, , called A′). The nitrene complex of A′ was optimized with the nitrene ligand bound to the AgN, denoted as A′(N). This coordination isomer was chosen due to the fact that catalyst A displays minimal counteranion effects. The effect of the truncation on the structures of A and A(N) is also assessed in Figures S3 and S4 in the Supporting Information (see Tables S9, S20, and S22 for a quantitative comparison and A′(N) and A′(NO), nitrene binding to the AgO atom of catalyst A, between A and A′, and A(N) and A′(N), respectively). The optimized structures of B(N) and A′N are shown in Figure 1.
Figure 1.

Optimized structures of the Ag–nitrene intermediates investigated. A′(N) and B(N) are shown on the left (a) and right (b), respectively.
Electronic Features of Nitrene Intermediates.
The electronic structures of B(N) and A′(N) have been investigated both at the DFT and CASSCF levels of theory. For both B(N) and A′(N), the lowest-energy state was found by DFT to be a triplet state, in which a Ag(II) ion is ferromagnetically coupled to a doublet nitrene anion radical (Ag(II)↑−↑{NSO3R}–). The ferromagnetic alignment of spins in this complex is due to the orthogonality of the Ag–N σ* and N pπ magnetic orbitals. Analysis of the Kohn– Sham orbitals shows that the Ag–N bond is composed of a three-electron σ component and a three-electron π component, with an overall calculated Mayer bond order of 0.56 and 0.57 for the (antiferromagnetically coupled) open-shell (OS) singlet and triplet B(N), respectively, and 0.63 and 0.55 for OS singlet and triplet A′(N), respectively. The σ* orbital in both complexes is strongly covalent with ~20% polarization toward Ag, whereas the π* orbital is strongly polarized toward the nitrene N atom by 85–90%. The OS singlet excited state is low-lying, at 4.13 and8.27 kcal·mol−1 above the triplet ground state for B(N) and A′(N), respectively. The frontier orbitals are shown in Figure S5. Analysis of the CASSCF wave function indicates that there is only one configuration that contributes strongly to the electronic structure of both complexes in their triplet state. This con-figuration can be described as (σ)2(π′*)2(π″*)1(σ*)1. On the other hand, the OS singlet has multiconfigurational character with a major configuration (~70%) of (σ)2(π′*)2(π″*)2(σ*)0 and a minor configuration (~30%) of (σ)2(π′*)2(π″*)0(σ*)2. A second closed-shell (CS) singlet excited state, discussed previously in the literature,7a was located 7.23 and 12.1 kcal·mol−1 above the triplet ground state for B(N) and A′(N), respectively. This particular excited state, which could be described as either a Ag(I) ion bound to a singlet nitrene or a Ag(III)-imido complex, is not discussed due to its high energy.
We recently discussed the necessity for a metal-nitrene intermediate to have a pair of empty α and β molecular orbitals with N character in order to be able to undergo concerted nitrene transfer.18 Given this constraint, we would expect nitrene transfer from these silver complexes with triplet ground states (i.e., having two empty β molecular orbitals with N character) to occur via a stepwise mechanism. However, Pérez and co-workers found in their computational study that it is possible for intersystem crossing to occur directly following an initial single-electron transition state in such a way as to avoid radical-containing intermediate species.7a Thus, an examination of the nitrene transfer potential energy surface (PES) for both A′(N) and B(N) is warranted.
Reactivity Landscape for Nitrene Transfer.
PESs for Nitrene Transfer from B(N) to Substrate 1 Modeling Experimental Chemoselectivity.
Two distinct transition states (TSs) have been located as substrate 1 approaches B(N). The first involves partial formation of an N-H bond (C‒H bond amination (I), TStpa,I,1, the numerical subscript denotes the substrate identity) on either the triplet (3TS) or OS singlet (OS1TS) PES. The second TS involves partial formation of a C-N bond (aziridination (A), TStpa,A,1) on the triplet (3TS) PES. Both OS1TStpa,I,1 and 3TStpa,I,1 were found to be lower in energy than the 3TStpa,A,1 (10.1 and 10.2 vs 10.7 kcal·mol−1, respectively), consistent with the experimentally observed chemo-selectivity. After HAT is complete, intermediates (Int) were found on both the OS singlet (OS1Inttpa,I,1) and triplet (3Inttpa,I,1) PES (−19.2 and −19.7 kcal·mol−1, respectively), with a slight favoring of the triplet state. Radical rebound occurs without an energetic barrier after the complete formation of OS1Inttpa,I,1 on the OS singlet surface to give the aminated product and regenerate catalyst B (−71.4 kcal·mol−1). The complete reaction coordinate for both reactions catalyzed by B(N) is shown in Figure 2. The presence of an intermediate after hydrogen-atom abstraction on both the OS singlet and triplet PESs and the fact that the two intermediates are close in energy suggest that the 3Inttpa,I,1 could be long-lived enough to be trapped by radical inhibitors. This finding is consistent with our experimental observations that a radical quencher lowers the yield of the C‒H inserted product of B(N) in the reactions of 11 and 21 (Schemes 2 and 3). In contrast, substrate aziridination by B(N) occurs on the triplet PES via 3TStpa,A,1; no OS1TStpa,A,1 was located. Complete formation of the C–N bond gives 3Inttpa,A,1; a reorganization of the electronic structure is presumed to occur to give OS1Inttpa,A,1 (0.44 kcal·mol−1 in terms of vertical excitation energy), which then leads to the formation of the aziridine product, 1PCA,tpa,1. Similar to the case for C‒H insertion, the presence of 3Inttpa,A,1, along with the absence of a fast recombination OS1TStpa,A,1, suggests that trapping by radical inhibitors is possible (vide infra), a hypothesis that is consistent with our experimental observations.
Figure 2.

Reaction coordinate of nitrene transfer from B(N) to 1 to give either the C‒H amination (brown) or aziridine product (brown). R = reactant, TS = transition state, Int = intermediate, and PC = product complex. Subscript I = C‒H insertion and A = aziridination; superscript OS1 and 3 denote open-shell singlet and triplet spin states, respectively. Solid lines denote the triplet PES, and dashed lines denote the open-shell singlet PES. Arrows and the size of the arrows represent α or β spin populations for the electrons on Ag (silver), N (brown), and organic substrate (black). Black arrows inside the ring indicate that electron density is delocalized over the allylic radical and arrows outside the ring represent electron density localized on the carbon proximal to the arrow. Numbers shown are Gibbs free energies; numbers enclosed in parentheses are enthalpies, both taking into account the solvent (CH2Cl2). Note: the energy for the blue dashed line enclosed by quotation marks was obtained from converging the single point energy at the OS singlet state based on the geometry of the solid blue line below it.
PESs for Nitrene Transfer from A′(N) to Substrate 1 Modeling Experimental Chemoselectivity.
Four distinct TSs are located as 1 approaches A′(N). Partial formation of the N–H bond (TStpy,I,1) or C–N bond (TStpy,A,1) occurs on either the triplet or OS singlet PES. 3TStpy,I,1 and 3TStpy,A,1 are lower in energy than OS1TStpy,I,1 and OS1TStpy,A,1 by 5.60 and 7.62 kcal·mol–1, respectively. 3TStpy,A,1 was found to be lower in energy than 3TStpy,I,1 (9.38 vs 11.4 kcal·mol–1), in agreement with the experimental selectivity. Both 3TStpy,I,1 and 3TStpy,A,1 lead to the formation of triplet state intermediates, 3Inttpy,I,1 and 3Inttpy,A,1 (−16.0 and −8.32 kcal·mol−1, respectively). However, these intermediates do not evolve to form products on the triplet surface. Instead, it was found that OS1TStpy,I,1 and OS1TStpy,A,1 lead to the direct formation of product complexes, 1PCtpy,I,1 and 1PCtpy,A,1, respectively (−81.7 and −62.5 kcal· mol−1, respectively) without accessing an intermediate. The complete reaction coordinate for both reactions by A′(N) is shown in Figure 3. The experimental observation that A is resistant to inhibition by TEMPO (Schemes 2 and 3) suggests that direct access of the OS singlet PES after 3TStpy (the crosses in Figure 3) to yield products is the major reaction pathway (vide infra).
Figure 3.

Reaction coordinate of nitrene transfer from A′(N) to 1 to give either C‒H insertion (blue) or aziridine products (brown). R = reactant, TS = transition state, Int = intermediate, and PC = product complex. Subscript I = C‒H insertion and A = aziridination and superscript OS1 and 3 denote open-shell singlet and triplet spin states, respectively. Solid lines denote the triplet PES, and dashed lines denote the open-shell singlet PES. Arrows and the size of the arrows represent α or β spin populations for the electrons on Ag (silver), N (blue), and organic substrate (black). Black arrows inside the ring represent electron density delocalized over the allylic radical, and arrows outside the ring represent electron density localized on the carbon proximal to the arrow. Numbers shown are Gibbs free energies, and numbers enclosed in parentheses are enthalpies, both taking into account the solvent (CH2Cl2).
The shape of the PES and the experimentally observed tolerance of the aziridination pathway toward radical inhibition are strongly reminiscent of the system described by Pérez and co-workers for Ag complexes supported by tripodal ligands.7a These resemblances prompted us to perform a comparison between B(N), A′(N), and the Pérez (TpMe2)Ag(NTs) (C(N);TpMe2 = hydro(3,5-dimethyl-1-pyrazolylborate)) system in order to achieve a deeper understanding of reactivity in our silver-catalyzed intermolecular nitrene transfer reactions.
Differential Mechanisms for Nitrene Transfer from B(N), A′(N), and C(N) to Organic Substrates.
Results for the reaction coordinates of nitrene transfer from B(N) and A′(N) to substrate 1 (vide supra) contrast with those recently reported for Ag-nitrene intermediates supported by tripodal Tp ligands.7a,19 In previously reported examples, only a triplet and CS singlet state were located, with the triplet favored in energy by 7.6 kcal·mol−1.7a The large spin population of 1.32 on the N atom, along with the inability to locate an OS singlet state, led to the description of C(N) as a Ag(I) complex with a coordinated triplet nitrene ligand. In contrast, we located OS singlet states for B(N) and A′(N) with spin populations (−0.78 on N and 0.78 on (tpa) Ag for B(N) and −0.74 on N and 0.74 on (tpy)2Ag2 for A′(N)), which prompt us to describe these silver-nitrene complexes as Ag(II) ions coupled to nitrene anion radicals, despite the fact that spin densities on the triplet states are similar to that of C(N) (1.26 on N and 0.74 on (tpa)Ag for B(N) and 1.27 on N and 0.64 on (tpy)2Ag2 for A′(N)).
Unlike either A′(N) or C(N), B(N) performs nitrene transfer via a mechanism that generates Inttpa on both the triplet and OS singlet PESs. This allows radical inhibition of the reaction, in contrast to the tolerance toward TEMPO exhibited by A′(N) and C(N) (Scheme 5). Importantly, the divergence in mechanism between these three catalysts occurs after the initial transition state. Therefore, the differences in the mechanism for nitrene transfer from B(N), A′(N), and C(N) are not likely due to their different ground state electronic structures. Instead, the nature of the transition states needs to be examined along with the thermodynamic driving forces en route to product formation.
Scheme 5.

Comparisons among A′(N), B(N), and C(N)
Nature of the C‒H Amination Transition States from A′(N), B(N), and C(N).
It is useful to examine the general geometrical features of nitrene transfer transition states. For simplicity, we will analyze in detail only the C‒H amination transition states. In concerted insertions of nitrenes into C‒H bonds, as is seen in Rh2-catalyzed reactions, the transition state involves simultaneous (though asynchronous) C-N and N-H bond formation and C‒H cleavage.18 These three interactions take place within a triangular, three-membered ring transition state (left, Scheme 6). Stepwise reactions proceed first through a hydrogen atom transfer (HAT) transition state leading to radical intermediates. The transition state involves a linear N⋯H⋯C geometry (right, Scheme 6). These geometries are electronically dictated: the three-membered ring transition state requires a stabilizing pair of electrons and therefore an empty N orbital (or a pair of α/β holes with N characters to accept the electrons of the C‒H bond). The linear N⋯H⋯C structures result from three-center/three-electron or three-center/four-electron bonds and are seen for the more electron-rich systems. All silver-nitrene complexes are electron rich and will therefore use HAT transition states for C‒H amination (and corresponding single-electron transition states for aziridination). The major question of this work, however, is to explain when the HAT transition state leads to the formation of radical intermediates versus when it is followed by a barrierless rebound.
Scheme 6.

Diagrams of Concerted and HAT Transition State Geometries for C‒H Amination, Including the Features of Stepwise Transition States vs Those Leading to Barrierless Rebound
Geometric details of the C‒H amination transition states of catalysts A′, B, and C are given in Table 5 (a more complete set of data for catalyst A′ and B is given in Tables S23, S24 and Tables S27 and S28, respectively, in the Supporting Information). Since catalyst A performs C‒H amination via a barrierless recombination pathway and B via a stepwise pathway, it is useful to compare their geometric differences. Both catalysts feature typical HAT transition states with N⋯H⋯C angles very close to linearity. However, the lowest-energy transition state (triplet) for catalyst A′ also clearly shows signs of Ag–N bond cleavage since its Ag–N bond length is increased by 0.05 Å from the geometry of A′(N). The Ag–N bond length for B is unchanged (0.01 Å). The observation that transition states leading to barrierless recombination involve Ag–N bond cleavage is also true of aziridination transition states. Catalysts A and C undergo aziridination via barrierless rebound, and their Ag–N bond distances are elongated by 0.06 and 0.04 Å, respectively, in their transition state geometries.
Table 5.
Geometrical Features of Optimized Transition States
| C–C amination transition states | |||
|---|---|---|---|
| catalyst | spin statea | ∠N···H···C (deg) | Δd(Ag−N)b (Å) |
| A′ | singlet | 171 | 0.03 |
| triplet | 172 | 0.05 | |
| B | singlet | 173 | −0.03 |
| triplet | 176 | 0.01 | |
| aziridination transition states | |||
| catalyst | spin statea | Δd(N–C)c (Å) | Δd(Ag–N)b (Å) |
| A′ | singlet | 0.627 | 0.01 |
| triplet | 0.736 | 0.06 | |
| B | triplet | 0.626 | 0.03 |
| C | singlet | 0.335 | −0.06 |
| triplet | 0.625 | 0.04 | |
The singlet spin states are all optimized open-shell singlets.
Δd(Ag–N) is defined as the difference in Ag–N(nitrene) bond distance between the transition state geometry and the reactant complex geometry.
Δd(N–C) is defined as the difference between the two N⋯C distances in the transition state.
This geometrical observation is also supported by an energetic analysis. First, we compare ΔH⧧ of the singlet and triplet transition states. In contrast to B(N), which has a similar enthalpic driving force at both OS1TStpa,I,1 and 3TStpa,I,1 (), OS1TStpy,1 for A′(N) is much less exothermic than 3TStpy,1 ( for TStpy,A,1 and for TStpy,I,1). Since the for the formation of the first C–N bond in aziridination and the N–H bond in C‒H insertion should be very similar on both PESs, the difference in between 3TStpy and OS1TStpy must result mainly from the breaking of the Ag–N bond during OS1TStpy. Comparing the Ag–N bond lengths in B(N) and A′(N), they differ by ~0.07 Å, a difference that may be attributed to partial chelating character of the nitrene ligand in A′(N), which contains a weak Ag⋯O interaction of 2.7 Å, reminiscent of the quasi-bidentate nitrene coordination mode in C(N). This implies that the bond between Ag and N is stronger in the OS singlet A′(N) than in the triplet A′(N); thus, it takes more energy to break the Ag–N bond during OS1TStpy to form the bond with the incoming atom from the substrate (Δ(Ag–N)TS‑R = +0.062 Å for OS1TStpy,A,1 and Δ(Ag–N)TS‑R = +0.077 Å for OS1TStpy,I,1 versus Δ(Ag–N)TS‑R = –0.01 Å for OS1TStpa,I). Once past OS1TStpy, the Ag–N bond has elongated enough to form 1PCtpy in a barrierless radical recombination step. The transient process leading to the formation of 1PCtpy means that the A′(N)–substrate complex must cross from 3TStpy to 1PCtpy through an MECP (where dashed and solid lines cross in Figures 2 and 3). Through this analysis, we observe that nitrene complexes with a semichelating coordination mode to Ag lead to nitrene transfer transition states having significant Ag–N bond cleavage character, which results in barrierless recombination and an absence of radical intermediates.
Interplay of Enthalpy and Entropy of Activation on the Differential Chemoselectivity of Nitrene Transfer from B(N) and A′(N) to Substrate 1
The differential chemoselectivity of nitrene transfer from B(N) and A′(N) to substrate 1 is satisfactorily reproduced by DFT studies of the reaction coordinates on both the triplet and OS singlet PESs using OS1TStpa,1,I or using 3TStpa,1,I vs ; vs ; see below for the origin of the approximately equal signs). The agreement with experiment justifies a further analysis of thermodynamic parameters, namely, enthalpy, entropy, and Gibbs free energy of activation/formation. Activation of the C=C bond on the triplet PES is the most exothermic step among all TSs for both B(N) and A′(N) (Table 6). This is sensible, as breaking the C=C π-bond in an alkene is less energetically costly than breaking the C‒H σ-bond at an allylic position.20 However, when the entropy of activation is taken into account, B(N) suffers more than A′(N) for both C–N bond formation and HAT. The fact that B(N) is more negatively impacted by ΔS A⧧ is due to steric hindrance around the nitrene site, which restricts the flexibility of the incoming substrate, as can be seen from space-filling models of B(N) and A′(N) (Figure 4). This larger negative entropic contribution of 3TStpa,1, in general, and of 3TStpa,A,1 compared to 3TStpa,I,1, is the major thermodynamic factor in the differential chemoselectivity of B(N) and A′(N). Hence, the chemoselectivity of A′(N) toward aziridination of 1 is enthalpy-driven, whereas that of B(N) toward amination of 1 is entropy-driven.
Table 6.
Enthalpy (kcal·mol−1), Entropy (cal·mol−1·K−1), and Gibbs Free Energy (kcal·mol−1) of Activation for Nitrene Transfer from B(N) to 1 and 21 on Either the Triplet or OS Singlet Surface
| TS | substrate | ΔS‡ | ||
|---|---|---|---|---|
| OS1TStpa,I | 1 | −4.30 | −48.3 | 10.1 |
| 3TStpa,I | 1 | −4.53 | −49.5 | 10.2 |
| 3TStpa,A | 1 | −4.75 | −51.7 | 10.7 |
| 3TStpy,I | 1 | −3.00 | −48.3 | 11.0 |
| 3TStpy,A | 1 | −5.58 | −50.2 | 9.38 |
| OS1TStpa,I | 21 | −4.89 | −57.0 | 12.1 |
| 3TStpa,I | 21 | −0.51 | −47.5 | 13.7 |
| 3TStpa,A | 21 | −4.69 | −53.4 | 11.2 |
Figure 4.

Space-filling models of B(N) (a) viewed from the top of the nitrene and (b) viewed from the side. Space-filling models of A′(N) (c) viewed from the top of the nitrene and (d) viewed from the side. Color code: silver, gray; sulfur, yellow; fluorine, green; oxygen, red; nitrogen, blue; carbon, black; and hydrogen, white.
Fine-Tuning the Chemoselectivity of Nitrene Transfer from B(N) to Cyclic versus Acyclic Alkenes.
To further examine our hypothesis that the interplay of ΔH⧧ and ΔS⧧ fine-tunes the chemoselectivity of Ag–nitrene complexes, the reaction coordinate for nitrene transfer from B(N) to acyclic substrate 21 was studied. Replacing cyclic alkenes by acyclic alkenes (i.e., 1 by 21) changes the experimental chemoselectivity of B(N). Correspondingly, we found that the lowest-energy TS is the aziridination transition state 3TStpa,A,21, which is 0.89 kcal·mol−1 more stable than the lowest-energy C‒H amination transition states TStpa,I,21, OS1TStpa,I,21. This gives kA/kI = 4.52 ≈ PA/PI, assuming that the product-determining steps are the rate-determining steps. Note that there are only two internal secondary allylic C‒H bonds in 21, compared to four accessible C=C sites composed of C2-re, C2-si, C3-re, and C3-si. This could potentially increase the PA/PI to 9.04. The calculated product ratios match the experimental product ratio of 2.9:1 well and, more importantly, the observed chemoselectivity. To understand the influence of ΔH⧧ and ΔS⧧ on the change of chemoselectivity from 1 to 21, their contributions to the TStpa are summarized in Table 6. is more favorable for HAT than C–N bond formation for 21 as compared to 1 because the migrating allylic C‒H bond can adopt a nearly coplanar alignment with the π orbitals of the C=C bond to allow some degree of resonance stabilization as the C‒H bond starts to break. This stabilization is less prominent in 1 due to the constraint imposed by the cyclic structure (Figure 5). However, the staggered conformation of 21 forces the propyl substituent to impose steric hindrance on B(N), a feature that is absent in 1. The reason for this is the constraint imposed by the cyclic structure of 1 that forces C4 to stay in a pseudo-gauche conformation with respect to the C=C bond (Figure 6). This steric effect reflects heavily on ΔS⧧, hence making 3TStpa,A,21 more energetically accessible than OS1TStpa,I,21.
Figure 5.

Structure of OS1TStpa,I,1 (a) and OS1TStpa,I,21 (b). The allylic group, nitrene N atom, and the Ag atom are shown in spheres for emphasis. Insets: Neumann projections of substrate looking down along the C2–C3 bond (a) or C3–C4 bond (b).
Figure 6.

(a) Overlay of 3Inttpa,A,21 (mostly in beige) and 3Inttpa,A,1 (mostly in blue) shown on the left. Ag, the nitrene N, the pyridyl group closest to substrates, and substrates are shown in spheres for clarity.(b) Overlay of 3Inttpa,I,21 (mostly in beige) and 3Inttpa,I,1 (mostly in blue) shown on the right. Ag, the aryl sulfamate, and substrates are shown in spheres for clarity.
C=C cis/trans Isomerization of Acyclic Alkene 21 during Nitrene Transfer from B(N).
Radical intermediates are found on both triplet and OS singlet surfaces for B(N), with 3Inttpa,I lower in energy by only 0.6 kcal·mol−1. However, 3Inttpa,I cannot lead to the formation of products without first accessing OS1Inttpa,I via a minimum energy crossing point (where crosses are located in Figure 3) to allow the conservation of spin symmetry. The presence of an MECP after the initial formation of 3Inttpa,I suggests that this species will be relatively long-lived. In turn, this increased lifetime allows cis/trans isomerization of the allylic C=C bond via C–C rotation of the diradical 3Inttpa,I species to occur. This also explains why TEMPO inhibition is observed (Scheme 3). Similar arguments apply to Inttpa,A. To further support this hypothesis, the trans isomer of Inttpa,21 () was considered. Indeed, and were located and found to be more stable than their cis isomers (−7.02 kcal·mol−1 for and −3.55 kcal·mol−1 for ). This result implies that a pre-equilibrium between the two isomers occurs before radical recombination to yield the trans isomer as the major product. However, the distribution of different isomers also depends on the kinetic barrier of the isomerization, which, in turn, depends on the specific steric constraints of the substrate and the degree of C=C bond character in Inttpa,21, that is, the C=C bond is completely broken in Inttpa,A,1 to give a carbon radical at C3 while the C=C bond is half broken in Inttpa,I,1 to stabilize the carbon radical at C4 after HAT through resonance (Figure 7). This can be seen structurally in Figure 7a where the methyl group on C2 and the propyl group on C3 are no longer coplanar in 3Inttpa,A,21 and .
Figure 7.

(a) Overlay of 3Inttpa,21,A (mostly in beige) and (mostly in blue) shown on the left. The rest of the molecule except Ag, nitrene N, the pyridyl group closed to 21, and substrate 21 is omitted for clarity. (b) Overlay of 3Inttpa,I,21 (mostly in beige) and (mostly in blue) shown on the right, the rest of the molecule except Ag, the sulfamate, and substrate 21 is omitted for clarity.
Timing of Intersystem Crossing and C=C Bond Transposition.
MECPs are required to be present after the transition states in the reaction trajectories from both A′(N) and B(N). Yet, the two Ag–nitrene complexes display dissimilar reactivity. As discussed above, for catalyst B, the MECP must occur after intermediate formation, and this feature “delays” the formation of products, allowing a byproduct shunt to occur through bimolecular collision of Inttpa with TEMPO. On the other hand, for catalyst A, the MECP must occur before the formation of intermediates because no radical byproducts are observed, with the exception of C=C bond transposition products seen with substrate 16. Thus, the potential energy surface for this substrate was examined more closely. For this substrate, as the allylic C‒H bond on 16 breaks in the (triplet) transition state, electronic reorganization occurs almost instantaneously, as compared to the nuclear movement to form the C–N-bond-giving products. The HAT nature of the transition state yields an allylic radical with spin population on both C1 and C3. Bond formation between C1 and N occurs immediately on the OS singlet PES to give 16d. The driving force favoring C1 over C3, which is mostly steric, is due to ΔS⧧ (vide infra), hence giving the thermodynamic product. Indeed, 16d is formed directly (with no intermediates) as the product after OS1TStpy, consistent with experimental observations (Table 1 and Scheme 2). Thus, although C=C double bond transposition in this case was initially suggestive of a stepwise mechanism with radical intermediates, we find computationally that this transposition can occur via a barrierless recombination and does not necessitate a long-lived diradical intermediate. This reactivity is therefore consistent with a HAT mechanism with barrierless rebound from A′(N).
CONCLUSIONS
In conclusion, we have reported the first examples of simple silver catalysts capable of tunable, nondirected, and intermolecular chemoselective amination. We have found catalyst systems that demonstrate complementary chemoselectivities in alkenes containing sites for both aziridination and C‒H amination in nitrene transfer, largely independent of substrate identities. Computational studies showed that the tunable chemoselectivity between tpa and tBu3tpy ligands is a result of the steric profile around the Ag–nitrene intermediates. The means to tune the steric bulk around the nitrene site without changing the electronic structure of the silver–nitrene intermediates, such as the size of the bound counteranion, could potentially afford an additional tunability within the same ligand system. Furthermore, two distinct nitrene transfer mechanisms are observed in the computational results, the first involving a very late transition state from catalyst A, followed by a barrierless recombination step that preserves stereochemistry and the second having a very early transition state from catalyst B to yield radical intermediates. The major distinction between these two mechanisms appears to be the extent to which the Ag–N bond breaks during the HAT transition state. Future studies are focused on the development of increasingly selective amination catalysts using a broader array of supporting ligands and counteranions. The ability to correlate the coordination geometry of the catalyst with both reaction mechanism pathways and selectivity are goals currently under investigation.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded through an NSF-CAREER Award 1254397 and the Wisconsin Alumni Research Foundation to J.M.S. The NMR facilities at UW—Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389–01). The National Magnetic Resonance Facility at Madison is supported by the NIH (P41GM103399, S10RR08438, S10RR029220) and the NSF (BIR-0214394). J.F.B. thanks the Center for Selective C‒H Functionalization supported by the National Science Foundation (CHE01205646). The computational facility at Madison is supported in part by National Science Foundation Grant CHE-0840494 and at the UW—Madison Center For High Throughput Computing (CHTC) in the Department of Computer Sciences. The CHTC is supported by UW—Madison, the Advanced Computing Initiative, the Wisconsin Alumni Research Foundation, the Wisconsin Institutes for Discovery, and the National Science Foundation, and is an active member of the Open Science Grid, which is supported by the National Science Foundation and the U.S. Department of Energy’s Office of Science. U.S. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07981.
Experimental procedures and characterization for all new compounds (PDF)
NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews on nitrene transfer, see:Müller P; Fruit C Chem. Rev 2003, 103, 2905.Collet F; Lescot C; Dauban P Chem. Soc. Rev 2011, 40, 1926.Díaz-Requejo MM; Pérez PJ Chem. Rev 2008, 108, 3379.Collet F; Dodd R; Dauban P Chem. Commun 2009, 5061.
- (2).For selected examples of Rh-catalyzed nitrene transfers, see:Roizen JL; Harvey ME; Du Bois J Acc. Chem. Res 2012, 45, 911.Zalatan DN; Du Bois J Top. Curr. Chem 2009, 292, 347.Dequirez G; Pons V; Dauban P Angew. Chem., Int. Ed 2012, 51, 7384.Espino CG; Du Bois J Angew. Chem., Int. Ed 2001, 40, 598.Fiori KW; Du Bois JJ Am. Chem. Soc 2007, 129, 562.Collet F; Lescot C; Liang CG; Dauban P Dalton Trans. 2010, 39, 10401.Espino CG; Fiori KW; Kim M; Du Bois JJ Am. Chem. Soc 2004, 126, 15378.Zalatan DN; Du Bois JJ Am. Chem. Soc 2008, 130, 9220.Liang C; Robert-Peillard F; Fruit C; Muller P; Dodd RH; Dauban P Angew. Chem., Int. Ed 2006, 45, 4641.Lescot C; Darses B; Collet F; Retailleau P; Dauban PJ Org. Chem 2012, 77, 7232.Lebel H; Spitz C; Leogane O; Trudel C; Parmentier M Org. Lett 2011, 13, 5460.
- (3).Harvey ME; Musaev DG; Du Bois JJ Am. Chem. Soc 2011, 133, 17207. [DOI] [PubMed] [Google Scholar]
- (4).For selected references of Fe-catalyzed nitrene transfer, see:Halfen JA Curr. Org. Chem 2005, 9, 657.Paradine SM; White MC J. Am. Chem. Soc 2012, 134, 2036.Cramer SA; Jenkins DM J. Am. Chem. Soc 2011, 133, 19342.Hennessy ET; Liu RY; Iovan DA; Duncan RA; Betley TA Chem. Sci 2014, 5, 1526.Fantauzzi S; Caselli A; Gallo E Dalton Trans. 2009, 5434.Nakanishi M; Salit A; Bolm C Adv. Synth. Catal 2008, 350, 1835.
- (5).For selected examples of Co-catalyzed nitrene transfer, see:Lu HJ; Subbarayan V; Tao JR; Zhang XP Organometallics 2010, 29, 389.Lu H; Zhang XP Chem. Soc. Rev 2011, 40, 1899.Ruppel JV; Kamble RM; Zhang XP Org. Lett 2007, 9, 4889.
- (6).Cu-catalyzed nitrene transfer:Bagchi V; Paraskevopoulou P; Das P; Chi L; Wang Q; Choudhury A; Mathieson JS; Cronin L; Pardue DB; Cundari TR; Mitrikas G; Sanakis Y; Stavropoulos PJ Am. Chem. Soc 2014, 136, 11362.Duran F; Leman L; Ghini A; Burton G; Dauban P; Dodd RH Org. Lett 2002, 4, 2481.Lebel H; Lectard S; Parmentier M Org. Lett 2007, 9, 4797.Dauban P;Saniére L; Tarrade A; Dodd RH J. Am. Chem. Soc 2001, 123, 7707.Srivastava RS; Tarver NR; Nicholas KM J. Am. Chem. Soc 2007, 129, 15250.Barman DN; Nicholas KM Eur. J. Org. Chem 2011, 2011, 908.Fructos MR; Trofimenko S; Díaz-Requejo MM; Pérez PJ J. Am. Chem. Soc 2006, 128, 11784.
- (7).For selected recent examples describing Ag-catalyzed nitrene transfer, see:Maestre L; Sameera WMC; Díaz-Requejo MM; Maseras F; Pérez PJ J. Am. Chem. Soc 2013, 135, 1338.Li Z; Capretto DA; Rahaman R; He C Angew. Chem., Int. Ed 2007, 46, 5184.Gómez-Emeterio BP; Urbano J; Díaz-Requejo MM; Pérez PJ Organometallics 2008, 27, 4126.Li ZG; He C Eur. J. Org. Chem 2006, 2006, 4313.Cui Y; He CJ Am. Chem. Soc 2003, 125, 16202.Cui Y; He C Angew. Chem., Int. Ed 2004, 43, 4210.Llaveria J; Beltrán A; Sameera WMC; Locati A; Díaz-Requejo MM; Matheu MI; Castillón S; Maseras F; Pérez PJ J. Am. Chem. Soc 2014, 136, 5342.Yang M; Su B; Wang Y; Chen K; Jiang X; Zhang Y-F; Zhang X-S; Chen G; Cheng Y; Cao Z; Guo Q-Y; Wang L; Shi Z-J Nat. Commun 2014, 5, 4707.Zheng Q-Z; Jiao N Chem. Soc. Rev 2016, 45, 4590.
- (8).Paradine SM; Griffin JR; Zhao J; Petronico AL; Miller SM; White MC Nat. Chem 2015, 7, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zalatan DN; Du Bois JJ Am. Chem. Soc 2008, 130, 9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For selected references, see:Padwa A; Austin DJ; Price AT; Semones MA; Doyle MP; Protopopova MN; Winchester WR; Tran AJ Am. Chem. Soc 1993, 115, 8669.Doyle MP; Phillips IM Tetrahedron Lett. 2001, 42, 3155.Davies HML; Beckwith RE J. Chem. Rev 2003, 103, 2861.Doyle MP; Duffy R; Ratnikov M; Zhou L Chem. Rev 2010, 110, 704.Davies HML; Dick AR Top. Curr. Chem 2009, 292, 303.
- (11) (a).Liang C; Collet F; Robert-Peillard F; Muller P; Dodd RH; Dauban PJ Am. Chem. Soc 2008, 130, 343. [DOI] [PubMed] [Google Scholar]; (b) Lescot C; Darses B; Collet F; Retailleau P; Dauban PJ Org. Chem 2012, 77, 7232. [DOI] [PubMed] [Google Scholar]
- (12) (a).Progress in Inorganic Chemistry; Karlin KD, Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2014; Vol. 58, pp 225–302. [Google Scholar]; (b) Kornecki KP; Berry JF Chem. - Eur. J 2011, 17, 5827. [DOI] [PubMed] [Google Scholar]; (c) Kornecki KP; Berry JF Eur. J. Inorg. Chem 2012, 2012, 562. [Google Scholar]; (d) Kornecki KP; Berry JF Chem. Commun 2012, 48, 12097. [DOI] [PubMed] [Google Scholar]
- (13).Rigoli JW; Weatherly CD; Alderson JM; Vo BT; Schomaker JM J. Am. Chem. Soc 2013, 135, 17238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Alderson JM; Phelps AM; Scamp RJ; Dolan NS; Schomaker JM J. Am. Chem. Soc 2014, 136, 16720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Scamp RJ; Jirak JG; Dolan NS; Guzei IA; Schomaker JM Org. Lett 2016, 18, 3014–3017. [DOI] [PubMed] [Google Scholar]
- (16).Williams NJ; Gan W; Reibenspies JH; Hancock RD Inorg. Chem 2009, 48, 1407. [DOI] [PubMed] [Google Scholar]
- (17) (a).Li DR; Xia WJ; Tu YQ; Zhang FM; Shi L Chem. Commun 2003, 798. [PubMed] [Google Scholar]; (b) Li X; Wu B; Zhao XZ; Jia YX; Tu YQ; Li DR Synlett 2003, 5, 623. [Google Scholar]
- (18).Varela-Álvarez A; Yang T; Jennings H; Kornecki KP; Macmillan SN; Lancaster KM; Mack JBC; Du Bois J; Berry JF; Musaev DG J. Am. Chem. Soc 2016, 138, 2327. [DOI] [PubMed] [Google Scholar]
- (19).Besora M; Braga AC; Sameera WMC; Urbano J; Fructos M; Pérez PJ; Maseras FJ Organomet. Chem 2015, 784, 2. [Google Scholar]
- (20) (a).Blanksby SJ; Ellison GB Acc. Chem. Res 2003, 36, 255. [DOI] [PubMed] [Google Scholar]; (b) Luo Y-R Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.