

Abstract
Human pluripotent stem cells (hPSCs) offer considerable potential for biomedical applications including drug screening and cell replacement therapies. Clinical translation of hPSCs requires large quantities of high quality cells, so scalable methods for cell culture are needed. However, current methods are limited by scalability, the use of animal-derived components, and/or low expansion rates. We recently reported a thermoresponsive 3D hydrogel for scalable hPSC expansion and differentiation into several defined lineages. This system would benefit from increased control over material properties to further tune hPSC behavior, and here we demonstrate a scalable 3D biomaterial with the capacity to tune both the chemical and mechanical properties to promote hPSC expansion under defined conditions. This 3D biomaterial, comprised of hyaluronic acid and poly(N-isopropolyacrylamide), has thermoresponsive properties that readily enable mixing with cells at low temperatures, physical encapsulation within the hydrogel upon elevation at 37°C, and cell recovery upon cooling and re-liquefaction. After optimization, the resulting biomaterial supports hPSC expansion over long cell culture periods while maintaining cell pluripotency. The capacity to modulate the mechanical and chemical properties of the hydrogel provides a new avenue to expand hPSCs for future therapeutic application.
Keywords: stem cell culture, thermoresponsive hydrogels, hyaluronic acid, poly(N-isopropylacrylamide)
Graphical Abstract
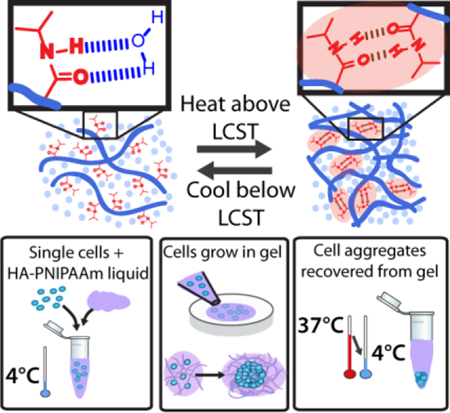
1. Introduction
Human pluripotent stem cells (hPSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), are highly promising for cell replacement therapies in regenerative medicine due to their capacities to self-renew and differentiate into all cell types of an adult organism.[1] hPSC-derived cells could substantially reduce the need for tissue and organ donation and, with additional cellular engineering, alleviate constraints of immune rejection.[2] hPSCs are also an important complement to animal studies in pharmacology and toxicology screens.[1i, 3] Because of these potential benefits, these cells are currently being researched for biomedical applications including cell replacement therapies, tissue engineering, and drug screening.
Despite this promise, biomedical translation of hPSCs is limited by the gap between cell culture at laboratory vs. clinical scales.[4] For example, replacement of cardiomyocytes in a single patient with a myocardial infarction would require at least 1×109 cells.[4c, 5] While research scale culture may achieve these numbers for animal studies or small trials, scalable platforms are necessary to achieve the >1015 cells entailed in treating the over 700,000 patients affected every year in the U.S.[6] Moreover, there are numerous other diseases or conditions including Type I diabetes, Parkinson’s disease, and Alzheimer’s disease that also have large patient populations and require substantial numbers of cells for treatment.[1e, 7] Furthermore, while methods to differentiate cells after expansion have substantially improved recently, still only a fraction of hPSCs develop into the desired lineage, and this efficiency must be accounted for when calculating the number of hPSCs needed. Finally, the low survival rates of hPSC-derived cells upon implantation necessitate even higher cell production scales.[4a, 4b]
The most widely used hPSC culture platforms rely on two-dimensional (2D) surfaces, which have been successfully harnessed for expansion and differentiation into a range of cell types and enabled translation into several clinical trials.[8] However, the majority of these systems require the use of animal tissue-derived surface coatings, such as Matrigel. This mouse tumor-derived mixture contains hundreds of proteins,[9] and as a result has limited reproducibility due to batch-to-batch variability, poses risks of pathogen transfer to hPSCs, and raises the possibility of immunogen transfer and subsequent immune responses in patients.[10] Importantly, 2D surfaces also necessitate exceedingly large culture surface areas (e.g. approximately 106 m2 for 1015 cells) and are thus not amenable to large-scale manufacturing for later stage clinical trials and commercialization.[8]
Three-dimensional (3D) culture systems are a promising and potentially necessary alternative to 2D culture platforms, as they potentially address the problem of scalability.[11] Several 3D systems have been established for hPSC culture, including cells adhered to microcarrier spheres, grown in suspension, or encapsulated in a soft material.[4c] Compared to 2D, 3D platforms offer microenvironments that more closely emulate in vivo conditions and as a result offer potential as a more efficient, robust, and predictable system for cell expansion and differentiation.[11d, 12] Recent work has also demonstrated the general importance of 3D environments on cell behavior, including differentiation and morphogenesis.[13] Moreover, encapsulation of hPSCs into a suitable material has been shown to improve cell yield relative to 2D platforms, prevent cell aggregation, and protect cells from shear stresses.[4c]
We recently reported a defined 3D hPSC culture system composed of poly(N-isopropylacrylamide) (PNIPAAm or PNIPAM) and poly(ethylene glycol) (PEG) for hPSC expansion, and the same system could be tuned for differentiation into midbrain dopaminergic neurons and oligodendrocyte precursors.[11b, 14] This PEG-PNIPAAm system substantially improved cell expansion levels, and additionally increased the proportion of the desired cell types, relative to 2D platforms. Importantly, the material is thermoresponsive, allowing for simple cell encapsulation and harvesting by switching the temperature. While promising, the initial PEG-PNIPAAm system offers limited control over the hydrogel’s mechanical and rheological properties, rendering homogenous cell encapsulation during scale-up difficult, and it is not amenable to conjugation of biochemical cues. A scalable system with greater control over the 3D microenvironment could significantly enhance the use of hPSCs for biomedical applications.
Here we develop a scalable, thermoresponsive biomaterial composed of hyaluronic acid (HA) and PNIPAAm (Figure 1). This synthesized biomimetic polymer enables controllable tuning of hydrogel mechanical properties, achieves a wide range of liquid-gel transition temperatures customizable to specific applications or culture conditions, and has the potential for conjugation of biochemical cues to tune the chemical properties. Furthermore, we show that this material provides a supportive microenvironment for hPSC expansion from both single cells and aggregates in a fully-defined, animal component-free environment that can readily be liquefied to recover the cells for further processing or growth (Figure 1b-d). Eliminating the need for cell-cell contact, such as aggregates of a very specific size range, at the start of each hPSC culture passage increases the potential for homogenous expansion compared to 2D culture.[15] Finally, this fully-defined material maintains hPSC pluripotency after multiple passages, illustrating the ability of engineered polymer systems to support stem cell expansion.
Figure 1.
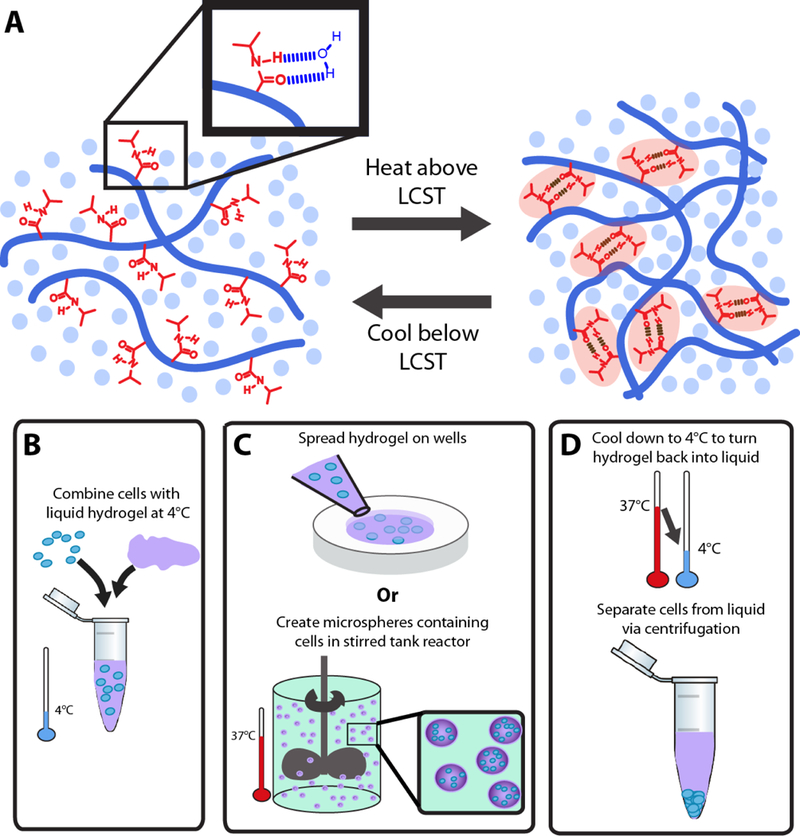
Schematic of HA-PNIPAAm showing PNIPAAm amide groups hydrogen bonding with water below the LCST and hydrogen bonding with each other above the LCST, thereby creating hydrophobic microdomains and converting the material to a physically crosslinked hydrogel (A). B-D illustrate the steps for cell encapsulation.
2. Materials and Methods
2.1. Materials
Hyaluronic acid (HA) was purchased from LifeCore Biomedical. Divinyl sulfone, N-isopropylamine (NIPAAm) monomer, butyl methacrylate monomer, azoisobutyronitrile (AIBN), 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (DMP), dioxane, sodium borohydride (NaBH4), Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), ethylenediaminetetraacetic acid (EDTA), and triethanolamine (TEOA) were obtained from Sigma Aldrich. NIPAAm monomer was recrystallized in hexanes, and butyl methacrylate was purified through an alumina column before use. The hESC cell line H1 and hiPSC line TCTFs were used for cell culture and cellular analysis. TCTF cells, also known as 8FLVY6C2, are an iPSC-Triple Fusion-ZFN derived from fibroblasts.[16]
Antibodies:
Oct4 (Santa Cruz Biotechnology; 1:200), Nanog (Abcam; 1:250), αSMA (Abcam; 1:200), FOXA2/HNF3β (Millipore; 1:500), and β3Tub (Covance 1:500).
2.2. PNIPAAm-SH synthesis
PNIPAAm confers thermoresponsive behavior to a material (Figure 1a) [16] and was therefore utilized for our hydrogel. To conjugate the PNIPAAm to our HA-VS backbone we needed to generate PNIPAAm with chain end thiols that we could then react with the vinyl groups. Thiol terminated PNIPAAm was synthesized under reversible addition-fragmentation chain transfer (RAFT) polymerization using a cleavable chain transfer agent (CTA), 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (DMP), and azoisobutyronitrile (AIBN) as the initiator (Figure S1a).[17] NIPAAm monomer was dissolved in dioxane with AIBN and DMP and placed in a 60°C oil bath for 24 hours.[17] The resulting polymer was precipitated into hexane, filtered, and dried in a vacuum oven overnight. A GPC with RI and UV detector using THF as the solvent was used to analyze the molecular weight of the PNIPAAm-CTA polymers based on the dn/dc value for PNIPAAm. The CTA was cleaved to reveal a thiol group at the chain ends of the PNIPAAm polymer by reduction with NaBH4 [18]. The PNIPAAm-CTAs were dissolved in DI water in a 3-neck round bottom flask. NaBH4 (30-50 molar excess to polymer chains) was dissolved in 5 mL DI water before quickly adding to the PNIPAAm-CTA solution. The vessel was hooked up to N2 with an outlet valve to ensure gas buildup within the vessel did not occur and to maintain an oxygen-free atmosphere for the reaction. After 2 hours of reaction the PNIPAAm-CTA yellow solution was placed in dialysis tubing (MWCO 3400) and dialyzed against DI water with 0.1M NaCl for multiple days. 20 mM TCEP and 10 mM EDTA were then reacted with the PNIPAAm-SH solution for 2 hours to reduce any disulfide bonds that may have formed during purification. After disulfide reduction, the polymer solution was dialyzed against phosphate buffered saline (PBS) with 1% trifluoroacetic acid to remove any residual chemicals and to maintain free thiols. The PNIPAAm-SH solution was then centrifuged to separate any remaining solids or unremoved CTA and then lyophilized. 1H NMR spectrometry and GPC were used to analyze resulting PNIPAAm-SH polymers.
In order to control the thermoresponsive behavior of our hydrogel, PNIPAAm-SH variants containing butyl methacrylate were also generated. Butyl methacrylate monomer was included at specific molar ratios during the synthesis of PNIPAAm-CTA (i.e. 2.5, 3.5, 5, or 10 mol %) to produce random copolymers of P(NIPAAm-r-BMA-CTA). Subsequent purification and analysis was identical to PNIPAAm-SH synthesis.
2.3. HA functionalization with vinyl sulfone groups
HA was first reacted with divinyl sulfone (DVS) to provide vinyl sulfone groups for the conjugation of components with free thiol groups (Figure S1b). HA was dissolved in 0.1 M NaOH to deprotonate a portion of the hydroxyl groups and reacted with DVS at varying molar ratios [19]. Reactions were carried out with molar excess DVS to hydroxyl groups in order to prevent crosslinking. The solutions were constantly mixed and allowed to react for 20 minutes before quenching with 1 M HCl. The resulting HA functionalized with vinyl sulfone (HA-VS) solution was dialyzed against DI water for multiple days and freeze-dried. 1H nuclear magnetic resonance (NMR) spectrometry was utilized to determine the amount of vinyl sulfone groups attached to the HA polymer by comparing the peaks at ~2 ppm to those above ~6 ppm.
2.4. HA-PNIPAAm polymer synthesis and hydrogel preparation
Once PNIPAAm-SH and HA-VS were generated individually, they were reacted together and dissolved in aqueous solution to form a hydrogel (Figure S1c). HA-VS was dissolved in 0.3 M triethanolamine (TEOA) buffer pH 8.0 with 1 mM EDTA at 4°C. After fully dissolved, the PNIPAAm-SH was added to the solution at weight ratio 5:1 PNIPAAm-SH: HA-VS. The solutions were constantly mixed at 4°C for 4-16 hours before adding 20 mM TCEP and 10 mM EDTA for ~1 hour to break any disulfide bonds and allow more PNIPAAm-SHs to attach. If heparin, peptides or other biochemical cues are desired within the hydrogel system, these thiol-functionalized components can be added at this point. 10mM cysteine was added last to react with any free vinyl sulfone groups that had not been reacted yet. These HA-PNIPAAm solutions were dialyzed against PBS using MWCO 100 kDa dialysis tubing for multiple days and then lyophilized. 1H NMR spectrometry was used to analyze the relative ratios of HA peaks to PNIPAAm peaks of the final polymers. Polymers were analyzed by rheology to determine the LCST, storage modulus, and the viscosity of the hydrogel.
2.5. Rheology analysis
Hydrogels were made by dissolving the dry polymer in PBS or cell culture media at concentrations between 5-10 wt/vol%. An Anton-Paar MCR 301 rheometer with an 8 mm parallel plate was utilized. Amplitude sweeps, frequency sweeps, and temperature sweeps from 4°C to 50°C (at a rate of 1-2°C/minute), and temperature sweeps from 4°C to 37°C followed by a 30 minute hold at 37°C were analyzed to determine which polymers had desired properties for cell culture and expansion. A 25 mm parallel plate was utilized for viscosity measurements at 4°C.
2.6. Cell culture
The HA-PNIPAAm copolymer was sterilized in ethanol followed by evaporation of the ethanol in a sterile environment. Once dry, the sterile HA-PNIPAAm copolymer was dissolved in cold Essential 8 (E8) media with ROCK inhibitor (ROCKi). Encapsulating cells within these hydrogels from 2D culture on Matrigel (Corning) or continuously from hydrogel to hydrogel followed a previous protocol we developed (Figure 1b-d).[11b] Passaging cells from 2D culture on Matrigel was performed by first enzymatically treating the cells on Matrigel with accutase (Life Technologies) for 5 minutes at 37°C to release the cells. The solution was rinsed over the plate multiple times to collect all the cells and then placed on ice. Mechanical disruption of any remaining aggregates using both a 1 mL pipette and a 200 μL pipette ensured single cells in the population. The solution was centrifuged for 3 minutes at 1.4 rpm and the cells were then combined at a density of 1×106 cells in 100 μL cold hydrogel solution and then pipetted onto tissue culture plates. The plates were placed in an incubator at 37°C for approximately 10 minutes before adding E8 media with 10 μM Rho kinase (ROCK) inhibitor (Y-27632, Selleckchem). After 5 days, ice cold PBS was added to the wells to reliquify the hydrogel. The cell solution was centrifuged for 3 minutes at 1.0 rpm before accutase was mixed with the cell pellet and placed in a water bath at 37°C for 10-12 minutes. Same protocol is followed at this point after the accutase step from Matrigel. When cells were plated as small aggregates, the cells were grown in Mebiol (CosmoBio) for 1-2 days before using cold PBS and centrifugation to release from hydrogel; no accutase was used to break up the small aggregates.
2.7. Antibody staining
In order to investigate pluripotency marker expression in cells cultured in the HA-PNIPAAm hydrogels, hPSC aggregates were recovered from the hydrogels by using cold PBS and centrifugation. Aggregates were plated onto Matrigel-coated plates for 48 hours before fixing with 4 w/v% paraformaldehyde (PFA) at room temperature for 15 minutes, permeabilized with 0.25% Triton X-100 for 15 minutes and blocked with 5 v/v% serum plus primary antibodies at 4°C for 16 hours. After multiple washing steps, secondary antibodies in 2 w/v% BSA were added and incubated at 4°C for 4 hours. Cells were washed again multiple times before imaging. The percentage of Oct4+ or Nanog+ nuclei was quantified using CellProfiler software.
2.8. Embryoid body differentiation
In order to analyze the differentiation capacity into all three germ layers, hPSC aggregates were recovered from hydrogels via cold PBS and centrifugation and plated onto 0.1% gelatin-coated plates (as opposed to growing on low-adhesion plates to form the EBs) and grown in 20 v/v% fetal bovine serum (FBS) in DMEM/F12 media (Life Technologies) plus 10 μM β-mercaptoethanol, 1 v/v% Glutamax (Life Technologies), 1 v/v% minimum essential medium non-essential amino acids (Invitrogen), and 1 v/v% Penn/Strep. After 25-30 days, the cells were fixed and stained as above.
2.9. Teratoma analysis
All procedures in animals followed established NIH guidelines for animal care and use and were approved by the UC Berkeley Animal Care and Use Committee (ACUC), the Committee for Laboratory and Environmental Biosafety (CLEB), and the Stem Cell Research Oversight committee (SCRO). 6-week old female NOD/SCID mice (Jackson) were subcutaneously injected in the hind flank with a 100 μl suspension of 500,000 TCTF cells that had been cultured in HA-PNIPAAm for 5 passages. Eight weeks after injection, animals were sacrificed and tumors harvested. Tumors were fixed, and parts were sectioned and prepared for Haemotoxylin and Eosin (H&E) as previously described (Fritz et al., Molecular therapy, 2015).[20] H&E staining was performed on a Shandon Varistain automated stainer (ThermoFisher). Additionally, other parts of the tumor were dehydrated in 30 (w/v) % sucrose, embedded in OCT, and cryo-sectioned using a freezing microtome. Tumor sections were mounted on slides and stained for immunohistochemistry. Images for immunohistochemistry and histology were taken on a Zeiss AxioObserver fluorescence microscope.
2.10. Statistical Analysis
Analysis of variance (ANOVA) single factor analysis was performed on experiments with more than two experimental groups. Standard deviation was the measure of uncertainty in all the materials characterization and pluripotency staining data. Standard error was the measure of uncertainty in the cell culture data. Error bars indicate standard deviation or standard error of three replicates per data point (n=3). All statistical analysis and graphing were performed on Microsoft Excel.
3. Results
3.1. Polymer design and synthesis
We sought to develop a fully-defined, thermoresponsive biomaterial with tunable mechanical and chemical properties for hPSC expansion. Our design contains a hyaluronic acid (HA) backbone functionalized with PNIPAAm chains to achieve thermoresponsive behavior (Figure S1). HA was chosen as the hydrophilic block for its abundant hydroxyl and carboxyl groups that can enable functionalization with polymers (e.g. PNIPAAm), peptides, or proteins. Additionally, HA is a major structural component in the extracellular matrix and has been found to influence proliferation, nutrient diffusion, and differentiation.[21] As the initial step, grafting PNIPAAm as the hydrophobic block, 1MDa HA was functionalized with vinyl sulfone groups and analyzed via 1H NMR (Figure S1b). The area under the curve for the HA peak at 2 ppm was compared to the area under the curves for the vinyl sulfone peaks at 6-7 ppm (Figure S2). This analysis indicated vinyl sulfone modification at averages of 28%, 48%, and 72% of the total number of HA disaccharides (HA-VS) with 1.5, 2.5, and 3.5 times the molar excess of divinyl sulfone to hydroxyl groups, respectively (Figure S2c). The result confirmed that increasing the molar excess divinyl sulfone reacted to HA results in higher functionalization.
To create a thermoresponsive hydrogel, thiol-terminated PNIPAAm was synthesized and clicked to the pendant vinyl groups on HA-VS. PNIPAAm was polymerized via RAFT polymerization to yield a low polydispersity polymer, followed by a reduction reaction, to generate a terminal thiol (PNIPAAm-SH) (Figure S1a, Figure S3a). Gel permeation chromotography (using refractive index detectors and a polystyrene standard in tetrahydrofuran solvent) determined the molecular weight of the PNIPAAm-SH polymers to be 18.2 kg/mol with a polydispersity of 1.03 (Figure S3b). The PNIPAAm-SH polymers were conjugated to the HA-VS via thiol-ene click chemistry, resulting in a brush polymer comprised of a hydrophilic HA backbone and hydrophobic PNIPAAm side chains (Figure S1c).[22] Conjugation of the PNIPAAm-SH to HA-VS was confirmed by the presence of large peaks corresponding to PNIPAAm via 1H NMR (Figure S4a).
3.2. Polymer design optimization
An optimal hydrogel should maintain stem cell pluripotency, support cell proliferation, and allow for easy pipetting, plating, and cell recovery. Specifically, several design criteria that impact cell culture were defined. Material storage modulus (or stiffness) has been reported to affect stem cell proliferation and differentiation and thus was designated as an important variable for optimizing cell expansion.[13b, 13c] The hydrogels were designed to maintain a storage modulus between 0.5-4 kPa at 37°C based on our previous 3D hydrogels that supported hPSC culture.[11b] Additionally, the hydrogels need to maintain mechanical stability to facilitate hPSC growth in suspension. Therefore, the polymer solution was designed to transition from a liquid to a hydrogel below 25°C, i.e. the lower critical solution temperature (LCST), in order to prevent the gel from dissolving or fracturing during cell culture maintenance and passage. Specifically, the LCST is the critical temperature below which the polymer mixture is soluble in aqueous solution and above which the PNIPAAm polymer blocks microphase separate to result in a hydrogel (Figure 1a).
To generate hydrogels with a storage modulus between 0.5- 4 kPa at 37°C and an LCST at or below 25°C, we identified several critical input variables in our polymer synthesis and characterized their effects on the hydrogel. We first reacted a single batch of the 18.2 kg/mol PNIPAAm-SH with HA at different molecule weights, and VS modifications of 28%, 48%, and 72%. The resulting polymer was then dissolved in aqueous solution at different concentrations and analyzed by rheology to determine the storage modulus at 37°C and LCST.
HA-VS with 28% and 48% vinyl sulfone functionalization formed uniform hydrogels at 37°C and a liquid polymer solution at 4°C. HA-PNIPAAm polymers generated from 72% HA-VS also formed hydrogels at 37°C but were too viscous at 4°C, making them difficult to homogenously mix with cells (Figure S5). For subsequent work, 48% HA-VS was used instead of 28% HA-VS to leave residual vinyl groups that can be used to functionalize additional components, such as peptides for bio-functionalization (Figure S2).
The effects of HA molecular weight on the storage modulus and the LCST were determined by reacting different molecular weight HA-VS with a single batch of PNIPAAm-SH. Increasing molecular weights of HA should increase polymer entanglement in the gel phase, thereby increasing the storage modulus. Keeping the percent vinyl sulfone functionalization of HA at 48%, HA molecular weights of 200 kDa, 700 kDa, and 1 MDa were reacted to PNIPAAm-SH. The resulting polymers were dialyzed for several days to remove unreacted PNIPAAm-SH, lyophilized, and dissolved in PBS at 2.5, 5, 7.5, and 10 wt/vol% (w/v%). When dissolved at 7.5 w/v%, these polymers formed hydrogels that exhibited storage moduli in the range of 1-3 kPa at 37°C (Figure 2a). These data represent 3 independently synthesized HA-VS-PNIPAAm hydrogels generated from the same batches of HA-VS and PNIPAAm-SH. In addition, the LCST decreased from ~34°C to ~20°C as the molecular weight increased from 200 kDa to 1 MDa. Subsequent HA-PNIPAAm polymers were generated using 1 MDa HA-VS, as it was within the desirable storage modulus range and had the lowest LCST.
Figure 2.
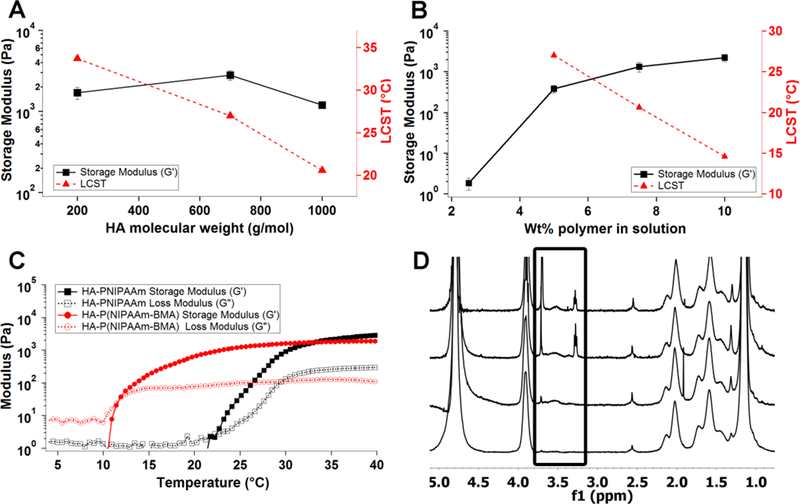
Rheology data for HA-PNIPAAm hydrogels and 1H NMR spectra of HA-PNIPAAm with PEG conjugation. A-B: Storage modulus at 37°C (black squares) and LCST (red triangles) as a function of HA molecular weight (A) and polymer concentration in solution (B). All error bars represent storage modulus averaged over 30 minutes at a constant temperature of 37°C, and concentrations are 7.5 w/v% unless otherwise noted. Error bars represent the standard deviation (n=3). Panel A gels were synthesized with 48% vinyl functionalized HA-VS. Panel B shows the same 1 MDa ~48% HA-PNIPAAm gel at different concentrations. C. Storage modulus (G’) and loss modulus (G”) as a function of temperature for an HA-PNIPAAm polymer (black) and HA-P(NIPAAm-r-BMA) with 2.5% butyl methacrylate (red). D. 1H NMR spectra of HA-PNIPAAm with PEG conjugation, where the PEG peaks are visible around 3.3 and 3.7 ppm. From top to bottom: HA-PNIPAAm with 10 kDa PEG, HA-PNIPAAm with 5 kDa PEG, HA-PNIPAAm with 2 kDa PEG, and HA-PNIPAAm.
In addition to changing the molecular weight of the polymer, we can also increase the polymer concentration to increase storage modulus. A higher concentration reduces the average distance between polymer chains, thereby increasing the average number of hydrogen bonds between PNIPAAm chains in the gelled state. As a result, the storage modulus should increase, and the LCST should decrease. HA-PNIPAAm copolymer was dissolved in PBS at concentrations of 2.5, 5, 7.5, and 10 wt/vol% (w/v%). Polymer concentrations less than 5 w/v% resulted in hydrogels below the desired storage modulus range at 37 °C (< 500 Pa) (Figure 2b). Polymer solutions resuspended at 7.5 and 10 w/v% both generated hydrogels with storage moduli averaging 1 kPa (Figure 2b). Within this range, as concentration increased, the storage modulus increased and the LCST decreased (Figure 2b). In contrast, polymers dissolved at concentrations greater than 10 w/v% did not fully dissolve at 4 °C or would dehydrate and shrink in size when plated at 37 °C, yielding a polymer rich phase and a separate water phase (Figure S5).
To determine whether the LCST of the hydrogels could be lowered even further than the 20°C achieved with 7.5 w/v% 1 MDa HA-PNIPAAm, butyl methacrylate was randomly polymerized with the N-isopropylacrylamide resulting in a random copolymer P(NIPAAm-r-BMA) and subsequently attached to the HA-VS (Figure S6). Incorporation of butyl methacrylate groups increased the hydrophobicity of the PNIPAAm block and ultimately lowered the temperature at which a hydrogel was formed. The LCST of HA-PNIPAAm generated using 48% vinyl modified HA-VS, 1 MDa HA, and 7.5 w/v% polymer concentration was approximately 20°C. Incorporation of 2.5 mol% BMA decreased the LCST to 11°C while maintaining storage moduli between 800 Pa and 2 kPa (Figure 2c). In contrast, incorporation of 3.5-10 mol% BMA did not result in continuous single-phase gel networks at 37°C but instead led to phase separation upon heating, likely due to the higher hydrophobicity of the polymers (Figure S5). As a result, either 0 or 2.5% BMA was used in all subsequent studies. The addition of BMA may facilitate applications where an LCST below 20°C is necessary.
Finally, to demonstrate the polymer could be chemically functionalized, for example for the future incorporation of proteins or peptides, we attached thiol-terminated polyethylene glycol (PEG) chains of 2 kDa, 5 kDa, and 10 kDa to the HA-PNIPAAm homopolymer. PEG was used as a proof of principle because it is a large polymer that is easily detected by 1H NMR. From 1H NMR, following conjugation and extensive dialysis, we noted an increasing size of the PEG peak with increasing PEG length in comparison to the HA-PNIPAAm peaks, indicating attachment to the copolymer (Figure 2d). Approximately 5%, 18%, and 12% of the HA disaccharide was modified with 2 kDa, 5 kDa, and 10 kDa PEG, respectively, based on 1H NMR where the area under the curve for the PEG peak at 3.2 ppm was normalized to the area under the curve for the N-acetyl proton for HA at 2 ppm. While an approximation since the HA is heavily modified with PNIPAAm and may have overlapping PNIPAAm peaks at 2 ppm, this relative comparison is consistent with the expected results in that the percent conjugation is roughly constant for the three experimental groups.
3.3. Synthesized hydrogels support hPSC culture
After generating polymers with the storage moduli between 0.5-4 kPa at 37°C and an LCST below 25°C, HA-PNIPAAm and HA-P(NIPAAm-r-BMA) polymers were analyzed for cell culture. Specifically, 1 MDa HA-PNIPAAm hydrogels were dissolved at concentrations above 10 w/v%, resulting in storage module of 4 kPa at 37°C. Cells were mixed with these polymer solutions at 4°C followed by gelation and thus cell encapsulation at 37°C. The hydrogel was incubated for 10 minutes at 37°C before adding cell culture media warmed to 37°C.While the hPSCs remained viable, the hydrogel did not support hPSC proliferation, as they never grew beyond single cells potentially due to the significant hydrogel shrinkage that constrained the cells above 10 w/v% (Figure S5). Hydrogels generated from 1 MDa HA-PNIPAAm and 1 MDa HA-P(NIPAAm-r-BMA) at either 7.5 or 10 w/v% exhibited storage moduli of approximately 1 kPa at 37°C and supported hPSC viability and proliferation (Figure 3). A representative temperature sweep for these polymers is shown in Figure 3a. At 4°C the viscosities for these polymers were lower than 0.1 Pa·sec at shear rates of 10-100 Hz (Figure S4b). Even at low shear rates of 0.1-1 Hz, the viscosity was still only 0.2-0.4 Pa·sec, which is amenable for gentle mixing of cells with the cooled hydrogel to form a homogenous encapsulation of hPSCs (Figure 3b, Figure S4b).
Figure 3.
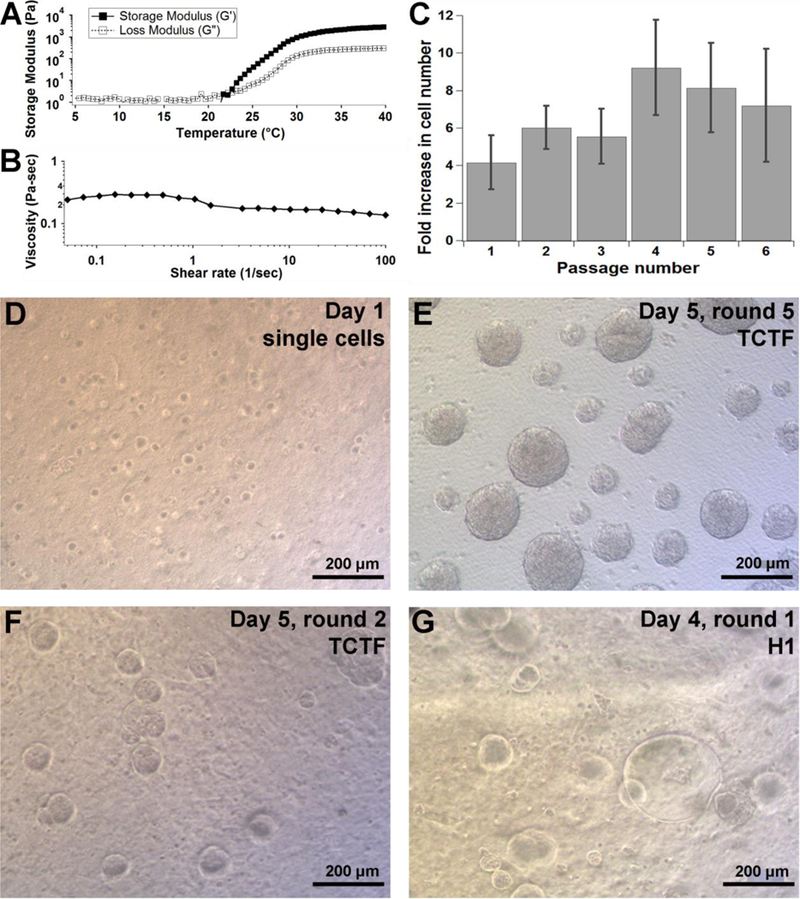
A. Rheology plot showing typical behavior of hydrogels that worked well for cell culture B. Viscosity plot of the hydrogel at 4°C C. TCTF cell expansion rates up to 6 continuous passages, at 5 days per passage. Error bars represent standard error (n=3).Based on ANOVA single factor analysis, p-value = 0.447, indicating no statistical difference between all 6 passages. D-G. Pictures of cell growth for different cell lines in HA-PNIPAAm and HA-P(NIPAAm-r-BMA) gels. D. Day 1 TCTF single cells. E. Day 5 in the fifth passage of the TCTF line plated as single cells in HA-PNIPAAm gels. F. Day 5 TCTF cells plated as single cells for 2nd passage in HA-P(NIPAAm-r-BMA) gel. G. Day 4 H1 cells plated as single cells for 1st passage in HA-P(NIPAAm-r-BMA) gel. Scale bars are all 200μm.
To demonstrate the potential of HA-PNIPAAm for cell culture, the hiPSC line TCTF and the hESC line H1 were grown in the hydrogels under fully defined conditions. Cells were cultured in Essential 8 (E8) medium supplemented with 10 μM Rho kinase (ROCK) inhibitor (Y-27632). A single cell suspension of TCTF hPSCs were encapsulated in the HA-PNIPAAm hydrogel, grown for 5 days per passage, and maintained overall for 6 consecutive passages. Expansion rates for TCTF cells are shown in Figure 3. The TCTF cell line expanded between 4-12 fold after 5 days in the material and remained proliferative over the 6 passage experiment (Figure 3c). As a proof of principle of its applicability to other hPSC lines, H1 hESCs were grown in these hydrogels from single cells, resulting in an approximate 4-9 fold expansion rate over 5 days (Figure S7). TCTF iPSCs grown from single cells through multiple passages maintained consistently-sized morphologically round aggregates each day. Figure 3d-g shows images of single cells and aggregates grown from single cells within the HA-PNIPAAm homopolymer and HA-P(NIPAAm-r-BMA) hydrogels. Each image represents an independent synthesis repeat of the hydrogel. Importantly, this is the first HA-based hydrogel to successfully culture single cells through multiple passages, and is only the second thermoreversible hydrogel capable of hPSC single-cell culture [11b].
3.4. Synthesized hydrogels maintain hPSC pluripotency after long periods of cell culture
To assess hPSC pluripotency after culture within HA-PNIPAAm hydrogels, antibody staining for pluripotency markers Nanog and Oct4 (also known as POU5F1) was performed. Greater than 80% of TCTF cells from passages 4-6 expressed Nanog and Oct4 (Figure 4). To functionally assess pluripotency in vitro, embryoid bodies (EB) were generated from TCTF cells after they were passaged 5 times in the gel and grown on gelatin-coated plates for 25-30 days before fixing and staining for three germ layers. The mesoderm layer ultimately generates muscle, bone, connective tissue, and blood; the endoderm layer creates the gastrointestinal tract and lung; and the ectoderm generates skin and the central nervous system. EB differentiation showed cells stained positive for smooth muscle actin that suggests mesoderm lineage commitment, HNFβ/FoxA2 for endoderm lineage, and Tuj1 for ectoderm lineage in the HA-PNIPAAm hydrogels (Figure 4c).
Figure 4.
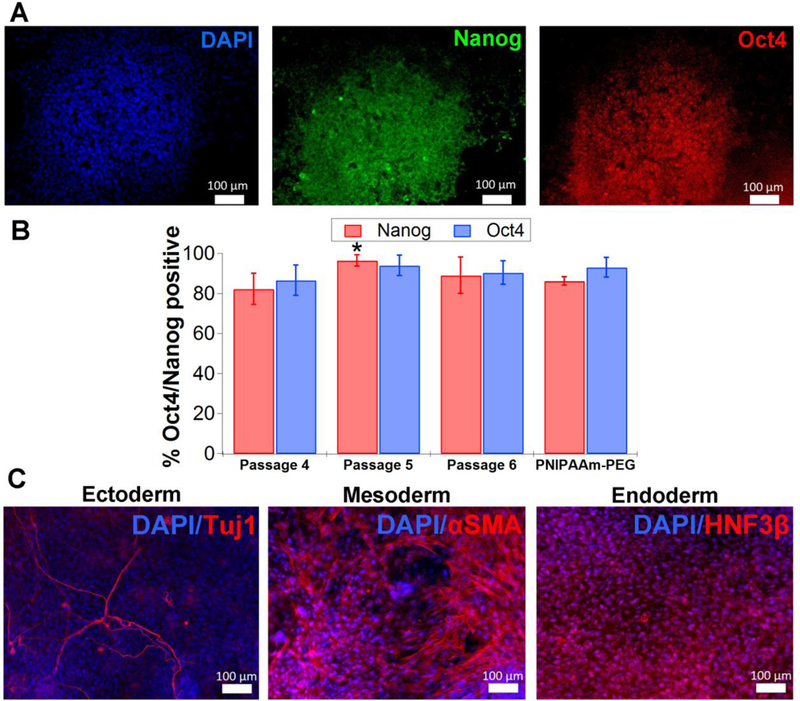
hPSCs remain pluripotent after propagation within gels. A. Antibody staining for pluripotency markers Nanog and Oct4 for TCTF iPSCs (after round 4). B. TCTF iPSCs have high percentages (>80%) of Oct4 and Nanog positive staining after 4, 5, and 6 passages as single cells within gels. At least 1,000 cells were analyzed for each condition. Error bars represent standard deviation (n=3 wells). Based on a two tailed t-test, there is no statistical significance between Oct4 and Nanog staining of cells cultured in HA-PNIPAAm when compared to PEG-PNIPAAm except for a slight difference in Nanog staining at the intermediate passage 5. * indicates p< 0.05. C. TCTF iPSCs remain pluripotent after multiple rounds cultured in hydrogels (round 3 shown here) by analysis of embryoid body differentiation. Aggregates were recovered from the hydrogels and plated on gelatin plates for ~25-30 days before performing antibody staining for all three germ layers (Tuj1 for ectoderm, αSMA for mesoderm, and HNF3β for endoderm). All scale bars are 100 μm.
To further analyze pluripotency, a teratoma assay was conducted. NOD/SCID mice were injected with ~5 × 105 TCTF cells that had been cultured in HA-PNIPAAm for 5 consecutive passages as single cell suspensions. Eight weeks after injection, tumors formed and were sectioned for immunohistochemistry and Haemotoxylin and Eosin (H&E) staining. Immunohistochemistry showed expression of smooth muscle actin, HNFβ, and Tuj1 (Figure 5b). H&E staining also confirmed the presence of mesodermal, endodermal, and ectodermal structures (Figure 5c). Together, these results demonstrate that the synthesized HA-PNIPAAm hydrogel maintained pluripotency of hPSCs after extended periods of in vitro culture.
Figure 5.
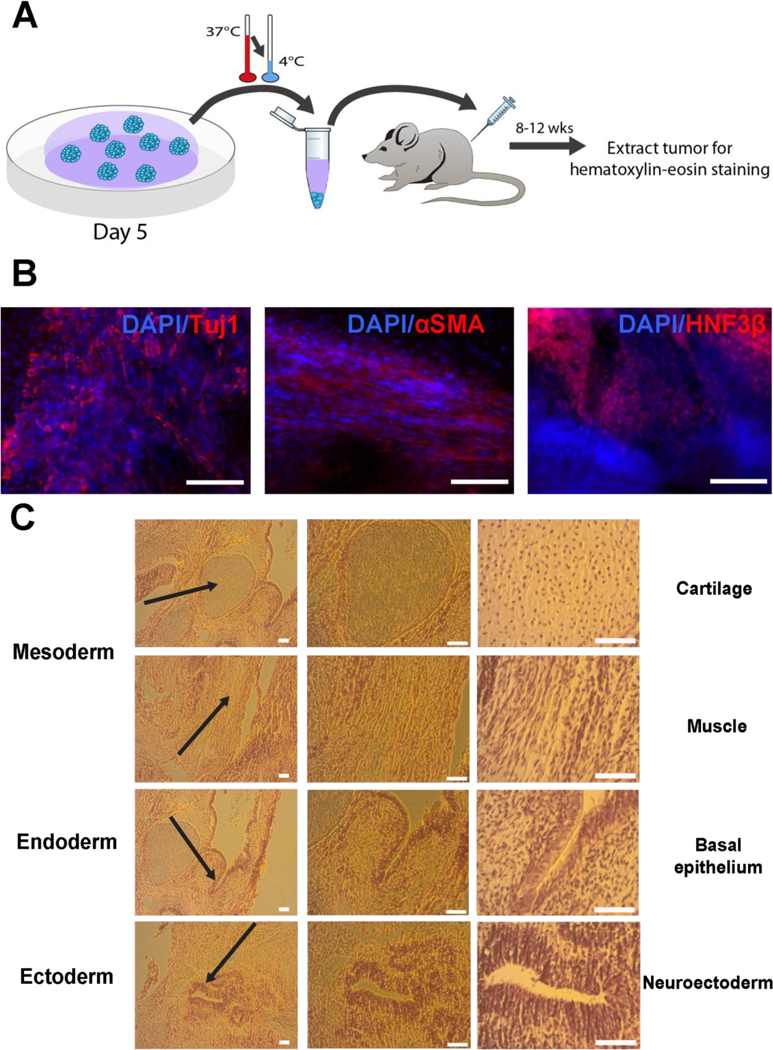
Teratoma analysis for TCTFs generated following 5 continuous passages in the hydrogels. A. Schematic of cell injection after 5 days of growth in 5th passage to NOD/SCID mice. B. TCTF iPSCs cultured after 5 continuous passages in gels stained positive for all three germ layers in teratomas formed in mice. C. TCTF iPSCs show all three germ layers via H&E staining. All scale bars are 100 μm.
4. Discussion
The biomedical and clinical use of hPSCs will require large quantities of cells, and hydrogels offer a scalable, defined approach to hPSC expansion and offer the potential to enhance cell quality and yield relative to commonly used 2D systems.[11] We synthesized HA-PNIPAAm polymers to develop a 3D, thermoreversible system with tunable mechanical and biochemical properties for scalable and defined cell culture, then applied the system for hPSC expansion. hPSCs proliferated starting from single cell suspension in the HA-PNIPAAm and expanded 4-10 fold after 5 days of culture in the material. Culture as single cells is extremely challenging in 2D, leading to low viability.[15b] As a result, cells are typically passaged as small mechanically and/or enzymatically dissociated clusters, which can lead to passage to passage variability. Cells were then readily recovered from the hydrogel by decreasing the temperature below the material’s LCST. After 5 consecutive passages in HA-PNIPAAm, hPSCs remained pluripotent, with over 80% of cells staining positive for Oct4 and Nanog, and were capable of generating the 3 germ layers in an in vitro EB and an in vivo teratoma assay. We show we can control the stiffness and LCST of the hydrogel by varying the molecular weight of HA used, the concentration of polymer in solution, and the incorporation of hydrophobic groups into the PNIPAAm chains. We also show that HA-PNIPAAm has sites available for conjugation of additional polymers or potentially biochemical cues, which can be added to improve expansion or direct cell differentiation.
Previous studies have shown PNIPAAm can be conjugated to hydrophilic polymers such as HA or PEG for tissue engineering, implantation, or drug delivery applications.[16, 23] These hydrogels had LCSTs ranging from 30°C-34°C, making them ideal for cell injection or drug release into a 37°C body. This study is the first demonstration of a system to use a HA hydrogel to successfully culture hPSCs from single cell suspension, tuned to a considerably lower LCST, and only the second thermoreversible system.[11b]
Stem cell microenvironments provide complex signals that can be physical and biochemical in nature.[8, 24] While emulating and simplifying these native microenvironments to in vitro culture systems, we must maintain control over key cues to support pluripotency or to direct cells towards certain lineages. For example, microenvironment stiffness affects stem cell fate. Mesenchymal stem cells differentiate into bone, muscle, or neurons on progressively softer substrates in 2D[13c] and also show a trend in 3D hydrogels with more fat cells generated in soft hydrogels compared to more bone cells in stiffer hydrogels.[25] Neural stem cells also respond to substrate stiffness, with stiffer substrates (>1 kPa) generating more astrocytes and softer substrates (<1 kPa) producing more neurons in 2D.[13b] In 3D alginate gels, a similar trend is seen with greater proliferation and neuronal differentiation with hydrogels >1 kPa storage modulus.[25] These effects have been primarily studied in 2D, and are beginning to be investigated in 3D. With our increased understanding of the importance of cell microenvironments, this HA-PNIPAAm system may offer some advantages for scale up and differentiation of hPSCs. Additionally, we show that material properties such as stiffness can be modulated and tuned by varying the synthesis conditions and reagents used to generate the polymer. For our application, we focused on HA-PNIPAAm hydrogels with storage moduli of ~1 kPa as they maintained their hydrogel shape and form during media changes while also promoting hPSC survival and proliferation.
When the project was initiated there was a large parameter space to explore, and we identified key parameters and conditions for generating thermoresponsive hydrogels. Hydrogels were initially designed to maintain a storage modulus between 0.5-4 kPa at 37°C based on our previous 3D hydrogels that supported hPSC culture.[11b] In order to achieve a storage modulus within this 0.5-4 Pka range, we recommend adjustments to the following parameters: polymer concentration in solution, % vinyl sulfone modification on HA-VS, and ratio of PNIPAAm-SH reacted with HA-VS. As seen in Figure 2b, lowering the concentration of polymer in solution is a simple means to create softer gels without changing the reactants used to synthesize the polymer, but the caveat is that the LCST also decreases. Alternatively, a higher %vinyl sulfone modification of HA can be used to provide additional sites for PNIPAAm-SH attachment, thereby increasing the storage modulus. Softer gels can conversely be achieved by attaching less PNIPAAm-SH to the HA-VS backbone. In this work, we reacted PNIPAAm-SH to HA-VS at a weight ratio of 5:1, which based on molar ratios is 175 molar excess PNIPAAm-SH reacted to the vinyl sulfone groups on HA-VS.
HA-PNIPAAm polymers were designed with an LCST at or below room temperature to allow for extended cell culture without significant hydrogel loss over time. Increasing the molecular weight of HA as well as the concentration of polymer in solution decreased the LCST. Future characterization could reveal that the former could result from more PNIPAAm polymers attached to each longer HA chain and/or more restricted freedom of motion for all of the chains, allowing the PNIPAAm polymers to microphase separate more readily. A similar effect with increasing polymer concentration minimizes the number of water molecules interacting with PNIPAAm, thereby decreasing the LCST. In addition, we investigated whether the LCST could be lowered below 25°C by incorporating a hydrophobic group such as butyl methacrylate during the PNIPAAm-CTA synthesis, which lowers the temperature at which the PNIPAAm forms hydrophobic microdomains and generates a hydrogel. The LCST of PNIPAAm is ~32°C, and introduction of butyl methacrylate decreased the LCST to 11°C. These HA-P(NIPAAm-r-BMA) hydrogels supported hPSC proliferation (Figure 3f-g) and could be utilized for future cell culture experiments requiring handling below 25°C.
In addition to stiffness, biochemical cues influence stem cell growth or differentiation. Soluble recombinant proteins used for stem cell expansion and differentiation are a dominant cost in cell culture, and conjugation of these signals to materials has the potential to reduce cell-mediated endocytosis and can increase the local concentration and potency of the peptide. [27] Specifically, the attachment of heparin would allow for capture of peptides or proteins through noncovalent interactions. There are several proteins and peptides with heparin binding domains including basic fibroblast growth factor, which is often used for pluripotent or neural stem cell propagation and in some cases differentiation. Additionally, adhesive peptides including RGD and others can be considered in order to broaden this hydrogel for applications in stem cell differentiation.[28] In functionalizing the HA-VS to attach PNIPAAm, we retained residual vinyl sulfone functionalities that could be used to attach additional materials or in the future biochemical cues. As a proof of concept, we conjugated thiol terminated PEGs of varying molecular weights.
5. Conclusions
We demonstrated a fully synthetic and tunable HA-PNIPAAm polymer system can maintain high hPSC viability and pluripotency over several rounds of cell culture passage. We achieved successful hPSC growth from single cells for more than 5 continuous passages with pluripotent cells. The replacement of animal derived materials with a fully synthetic polymer and animal-component free media significantly reduces risks of contamination and immunogenicity.[8] In addition, a fully synthetic system can minimize the effects of lot-to-lot variability seen with animal-derived materials.
HA hydrogels that are thermoreversible and tunable could be utilized to address a wide array of needs. The mechanical properties of the hydrogels could be tailored to suit specific types of stem cells and the environments that support their cell growth, for instance potentially softer for neural stem cell culture or stiffer for mesenchymal stem cell culture. In addition, the material could be modified for scalable, defined differentiation protocols to produce specific cell types desired for certain human therapeutics. There are also a multitude of biochemical cues that could be incorporated into this system to aid in differentiation or maintenance of cell lines. Ultimately it could be possible to expand cells within the HA-PNIPAAm hydrogels, incorporate biochemical cues for specific cell differentiation, and then inject into the body for cell implantation therapies. Transitioning into production using bioreactors would greatly expand the potential of these hydrogels for scalable production of stem cells.
Supplementary Material
Acknowledgements
We would like to thank the generosities of Dr. Nitash Balsara for use of his laboratory for polymer synthesis, Dr. Sanjay Kumar for use of his rheometer, Dr. Joseph Wu for the TCTF cell line, and Dr. Lin He for access to an H&E stainer. In addition, this work was funded by CIRM Grants DISC2-08982 and RT3-07800 and NIH grants R01 NS074831 and R01 NS083856.
This work reports a thermoresponsive hyaluronic acid and poly(N-isopropylacrylamide) based hydrogel that supports human pluripotent stem cell culture from single cells. The material can be chemically and mechanically tuned, and provides a fully defined and scalable method for stem cell expansion. Human pluripotent stem cells grown within the gel maintain viability and pluripotency.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References:
- [1].a) Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM, Science 1998, 282, 1145; [DOI] [PubMed] [Google Scholar]; b) Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S, Cell 2007, 131, 861; [DOI] [PubMed] [Google Scholar]; c) Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, Wagner R, Lee GO, Antosiewicz-Bourget J, Teng JM, Thomson JA, Nature methods 2011, 8, 424; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schulz TC, Young HY, Agulnick AD, Babin MJ, Baetge EE, Bang AG, Bhoumik A, Cepa I, Cesario RM, Haakmeester C, Kadoya K, Kelly JR, Kerr J, Martinson LA, McLean AB, Moorman MA, Payne JK, Richardson M, Ross KG, Sherrer ES, Song X, Wilson AZ, Brandon EP, Green CE, Kroon EJ, Kelly OG, D’Amour KA, Robins AJ, PloS one 2012, 7, e37004; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lindvall O, Kokaia Z, Martinez-Serrano A, Nature medicine 2004, 10 Suppl, S42; [DOI] [PubMed] [Google Scholar]; f) Badylak SF, Taylor D, Uygun K, Annual review of biomedical engineering 2011, 13, 27; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) McNeish J, Nature reviews. Drug discovery 2004, 3, 70; [DOI] [PubMed] [Google Scholar]; h) Jensen J, Hyllner J, Bjorquist P, Journal of cellular physiology 2009, 219, 513; [DOI] [PubMed] [Google Scholar]; i) Zuba-Surma EK, Wojakowski W, Madeja Z, Ratajczak MZ, Current pharmaceutical design 2012, 18, 2644. [DOI] [PubMed] [Google Scholar]
- [2].a) Bianco P, Robey PG, Nature 2001, 414, 118; [DOI] [PubMed] [Google Scholar]; b) Eberli D, Atala A, Method Enzymol 2006, 420, 287; [DOI] [PubMed] [Google Scholar]; c) Fisher MB, Mauck RL, Tissue engineering. Part B, Reviews 2013, 19, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Harrison RH, St-Pierre JP, Stevens MM, Tissue Eng Part B-Re 2014, 20, 1; [DOI] [PubMed] [Google Scholar]; e) Brown PT, Handorf AM, Jeon WB, Li WJ, Current pharmaceutical design 2013, 19, 3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Sommer CA, Mostoslavsky G, Journal of cellular physiology 2013, 228, 267; [DOI] [PubMed] [Google Scholar]; b) Ebert AD, Svendsen CN, Nature reviews. Drug discovery 2010, 9, 367; [DOI] [PubMed] [Google Scholar]; c) Ebert AD, Yu J, Rose FF Jr., Mattis VB, Lorson CL, Thomson JA, Svendsen CN, Nature 2009, 457, 277; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L, Nature 2009, 461, 402; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA, Proc Natl Acad Sci U S A 2009, 106, 15768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L, Nature 2011, 480, 547; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Laflamme MA, Murry CE, Nature biotechnology 2005, 23, 845; [DOI] [PubMed] [Google Scholar]; c) Serra M, Brito C, Correia C, Alves PM, Trends in biotechnology 2012, 30, 350. [DOI] [PubMed] [Google Scholar]
- [5].Jing D, Parikh A, Canty JM Jr., Tzanakakis ES, Tissue engineering. Part B, Reviews 2008, 14, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American C Heart Association Statistics, S. Stroke Statistics, Circulation 2017, 135, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Lock LT, Tzanakakis ES, Tissue Eng 2007, 13, 1399; [DOI] [PubMed] [Google Scholar]; b) Lunn JS, Sakowski SA, Hur J, Feldman EL, Annals of neurology 2011, 70, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Villa-Diaz LG, Ross AM, Lahann J, Krebsbach PH, Stem cells 2013, 31, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Hughes C, Radan L, Chang WY, Stanford WL, Betts DH, Postovit LM, Lajoie GA, Molecular & cellular proteomics : MCP 2012, 11, 1924; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hughes CS, Postovit LM, Lajoie GA, Proteomics 2010, 10, 1886; [DOI] [PubMed] [Google Scholar]; c) Hakala H, Rajala K, Ojala M, Panula S, Areva S, Kellomaki M, Suuronen R, Skottman H, Tissue engineering. Part A 2009, 15, 1775. [DOI] [PubMed] [Google Scholar]
- [10].van der Valk J, Brunner D, De Smet K, Fex Svenningsen A, Honegger P, Knudsen LE, Lindl T, Noraberg J, Price A, Scarino ML, Gstraunthaler G, Toxicology in vitro : an international journal published in association with BIBRA 2010, 24, 1053. [DOI] [PubMed] [Google Scholar]
- [11].a) Fan Y, Zhang F, Tzanakakis ES, ACS Biomaterials Science & Engineering 2016, DOI: 10.1021/acsbiomaterials.6b00253; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lei YG, Schaffer DV, P Natl Acad Sci USA 2013, 110, E5039; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu Y, Fox V, Lei Y, Hu B, Joo KI, Wang P, Journal of biomedical materials research. Part B, Applied biomaterials 2014, 102, 1101; [DOI] [PubMed] [Google Scholar]; d) Serra M, Correia C, Malpique R, Brito C, Jensen J, Bjorquist P, Carrondo MJ, Alves PM, PloS one 2011, 6, e23212; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Siti-Ismail N, Bishop AE, Polak JM, Mantalaris A, Biomaterials 2008, 29, 3946. [DOI] [PubMed] [Google Scholar]
- [12].a) Tibbitt MW, Anseth KS, Biotechnology and bioengineering 2009, 103, 655; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu J, Marchant RE, Expert review of medical devices 2011, 8, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Kumar S, Nature materials 2014, 13, 918; [DOI] [PubMed] [Google Scholar]; b) Saha K, Keung AJ, Irwin EF, Li Y, Little L, Schaffer DV, Healy KE, Biophysical journal 2008, 95, 4426; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677; [DOI] [PubMed] [Google Scholar]; d) Evans ND, Minelli C, Gentleman E, LaPointe V, Patankar SN, Kallivretaki M, Chen X, Roberts CJ, Stevens MM, European cells & materials 2009, 18, 1; [DOI] [PubMed] [Google Scholar]; e) Marklein RA, Burdick JA, Advanced materials 2010, 22, 175; [DOI] [PubMed] [Google Scholar]; f) Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordonez-Moran P, Clevers H, Lutolf MP, Nature 2016, 539, 560; [DOI] [PubMed] [Google Scholar]; g) Ranga A, Girgin M, Meinhardt A, Eberle D, Caiazzo M, Tanaka EM, Lutolf MP, Proc Natl Acad Sci U S A 2016, 113, E6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Adil MM, Rodrigues GM, Kulkarni RU, Rao AT, Chernavsky NE, Miller EW, Schaffer DV, Scientific reports 2017, 7, 40573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, Sasai Y, Nature biotechnology 2007, 25, 681; [DOI] [PubMed] [Google Scholar]; b) Xu Y, Zhu X, Hahm HS, Wei W, Hao E, Hayek A, Ding S, Proc Natl Acad Sci U S A 2010, 107, 8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC, Cell stem cell 2013, 12, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Tan H, Ramirez CM, Miljkovic N, Li H, Rubin JP, Marra KG, Biomaterials 2009, 30, 6844; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ohya S, Nakayama Y, Matsuda T, Biomacromolecules 2001, 2, 856; [DOI] [PubMed] [Google Scholar]; c) Ohya S, Sonoda H, Nakayama Y, Matsuda T, Biomaterials 2005, 26, 655; [DOI] [PubMed] [Google Scholar]; d) Peroglio M, Grad S, Mortisen D, Sprecher CM, Illien-Junger S, Alini M, Eglin D, European spine journal : official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society 2012, 21 Suppl 6, S839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Convertine AJ, Ayres N, Scales CW, Lowe AB, McCormick CL, Biomacromolecules 2004, 5, 1177; [DOI] [PubMed] [Google Scholar]; b) Ganachaud F, Monteiro MJ, Gilbert RG, Dourges MA, Thang SH, Rizzardo E, Macromolecules 2000, 33, 6738. [Google Scholar]
- [19].a) Scales CW, Convertine AJ, McCormick CL, Biomacromolecules 2006, 7, 1389; [DOI] [PubMed] [Google Scholar]; b) Moad G, Rizzardo E, Thang SH, Polym Int 2011, 60, 9. [Google Scholar]
- [20].Yu Y, Chau Y, Biomacromolecules 2012, 13, 937. [DOI] [PubMed] [Google Scholar]
- [21].Fritz AL, Adil MM, Mao SR, Schaffer DV, Molecular therapy : the journal of the American Society of Gene Therapy 2015, 23, 952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Toole BP, Nature reviews. Cancer 2004, 4, 528. [DOI] [PubMed] [Google Scholar]
- [23].Hoyle CE, Lowe AB, Bowman CN, Chemical Society reviews 2010, 39, 1355. [DOI] [PubMed] [Google Scholar]
- [24].a) Mortisen D, Peroglio M, Alini M, Eglin D, Biomacromolecules 2010, 11, 1261; [DOI] [PubMed] [Google Scholar]; b) Alexander A, Khan Ajazuddin, J., Saraf S, Saraf S, European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V 2014, 88, 575. [DOI] [PubMed] [Google Scholar]
- [25].a) Keung AJ, Kumar S, Schaffer DV, Annual review of cell and developmental biology 2010, 26, 533; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prestwich GD, Organogenesis 2008, 4, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ, Nature materials 2010, 9, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Kuhl PR, Griffith-Cima LG, Nature medicine 1996, 2, 1022; [DOI] [PubMed] [Google Scholar]; b) Alberti K, Davey RE, Onishi K, George S, Salchert K, Seib FP, Bornhauser M, Pompe T, Nagy A, Werner C, Zandstra PW, Nature methods 2008, 5, 645; [DOI] [PubMed] [Google Scholar]; c) Mahadik BP, Pedron Haba S, Skertich LJ, Harley BA, Biomaterials 2015, 67, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meng Y, Eshghi S, Li YJ, Schmidt R, Schaffer DV, Healy KE, FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2010, 24, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.