

Abstract
Protein-based conjugates are valuable constructs for a variety of applications. Conjugation of proteins to fluorophores is commonly used to study their cellular localization and the protein-protein interactions. Modification of therapeutic proteins with either polymers or cytotoxic moieties greatly enhances their pharmacokinetics and potency. To label a protein of interest, conventional direct chemical reaction with the side-chains of native amino acids often yields heterogeneously modified products. This renders their characterization complicated, requires difficult separation steps and may impact protein function. Although modification can also be achieved via the insertion of unnatural amino acids bearing bioorthogonal functional groups, these methods can have lower protein expression yields, limiting large scale production. As a site-specific modification method, enzymatic protein labelling is highly efficient and robust under mild reaction conditions. Significant progress has been made over the last five years in modifying proteins using enzymatic methods for numerous applications, including the creation of clinically relevant conjugates with polymers, cytotoxins or imaging agents, fluorescent or affinity probes to study complex protein interaction networks, and protein-linked materials for biosensing. This review summarizes developments in enzymatic protein labelling over the last five years for a panel of ten enzymes, including sortase A, subtiligase, microbial transglutaminase, farnesyltransferase, N-myristoyltransferase, phosphopantetheinyl transferases, tubulin tyrosin ligase, lipoic acid ligase, biotin ligase and formylglycine generating enzyme.
Graphic abstract:
1. Introduction
The ability to manipulate proteins, from the construction of protein-based conjugates to surface immobilization, has been central to recent rapid advances in the study of fundamental biology as well as in areas of the biotechnology and pharmaceutics. Labelling of proteins with fluorophores aids in identifying their cellular localization.1 Compared to the green fluorescent protein (GFP), the small size of these organic molecules makes them less likely to perturb the native structure and function of the protein being studied.2 Even when enzymatic labelling methods are employed that require incorporation of additional small peptide tags into the proteins of interest, the combined size of the tag and the fluorophore is still substantially smaller than the bulky GFP. Synthesis/semisynthesis of proteins with unambiguous post-translational modification states also helps to elucidate the roles of the individual modification, facilitating the study of protein-protein interactions (PPIs).3 In the field of biotechnology, the immobilization of proteins on solid surfaces has been extensively employed for the development of bio-sensors,4 recyclable catalysts,5 and protein microarrays.6 Biomaterials, including protein functionalized nanoparticles, hydrogels and liposomes have also been explored for various applications, including drug delivery systems7 and bio-responsive materials.8 As a major component of modern medicine, the use of protein-based therapeutics continues to grow.9 To achieve improved therapeutic outcomes, enormous efforts have been made in protein engineering. For example, conjugation of polyethylene glycol (PEG) polymer chains to small therapeutic polypeptides significantly improves their pharmacokinetic properties.10 To augment the efficacy of cancer treatments using antibodies alone, the incorporation of cytotoxic components, such as small molecule drugs11 and radioactive isotopes12 make antibody-based therapeutics more potent. Having an imaging agent in place of a toxic drug creates sensitive and specific targeted imaging probes for cancer diagnosis.13,151,172
Although protein conjugation can be achieved by installing cargos to proteins non-selectively by reacting with functional groups from the side chains of native amino acids, site-specific labelling provides homogeneously modified products. Precise control over the position of modification eliminates potential detrimental effects on the stability and/or function of the protein. For example, almost all PEGylated proteins are less active than their unmodified versions, in part due to blockage of the functional site by the bulky PEG polymer.10 In this case, site-specific incorporation of the PEG polymer offers a tremendous advantage over non-selective methods since an optimal position can be chosen to minimize disruption and retain more activity. Compared to early generations of antibody-drug conjugates (ADCs) with variable drug-to-antibody (DAR) ratios and positions of attachment, site-specific ADCs are expected to possess better therapeutic indicies, promoting the development of more efficacious therapeutic agents.14 To achieve site-specific protein modification, several strategies have been developed, including enzymatic labelling methods. In addition to being highly selective, enzymatic reactions usually take place under mild conditions with fast kinetics and high yield. Accordingly, significant progress has been made in applying enzymatic labelling methods for protein modification in the last decade. Numerous reviews on this topic have been published15–17 including one by Rashidian et al. in 2013 that summarized several enzymatic labelling strategies and their early applications.18 In this review, we focus on the developments that have occurred in the last five years (2013–2018). First, protein modification methods based on reactions occurring on either the native functional groups present in proteins or those inserted via nonsense suppression methods are described in Section 2. The bioorthogonal reactions that are widely used for protein bioconjugation are also included there. In section 3, a panel of 10 enzymes that have been widely explored will be discussed in detail with a focus on applications reported since 2013. These enzymes are organized into four categories, 1) peptidases: sortase A and subtiligase; 2) transferases: microbial transglutaminase, farnesyltransferase, N-myristoyltransferase and phosphopantetheinyl transferase; 3) ligases: tubulin tyrosine ligase, lipoic acid ligase and biotin ligase; 4) oxidoreductases: formylglycine generating enzyme. Finally, some concluding remarks and efforts to compare these different methods are presented. As this area continues to grow, we hope this review article will stimulate additional innovation and developments as well as provide some insights to facilitate the selection of the optimal enzyme to meet specific needs.
2. Chemistry on proteins
To label a protein of interest, both chemical and genetic methods have been developed to introduce modifications through either natural amino acid residues or synthetic analogues thereof.19,20 This has been achieved by exploiting the inherent reactivity of the functional groups present using direct chemical modification, insertion of an unnatural amino acid via nonsense suppression, or utilization of enzymatic labelling strategies. Due to the limited stability of biomolecules, the chemical reactions employed for protein modification must be biocompatible and proceed rapidly under mild conditions.
2.1. Direct chemical modification
Over the years, a wide variety of chemical reactions have been explored to specifically functionalize the side chains of certain amino acids through direct chemical modification (see Figure 1 for some commonly used reactions). In order to be compatible with sensitive proteins, these reactions generally require efficiency at ambient temperatures and aqueous conditions. Cysteine and lysine are the common targets for functionalization. Aromatic residues including tyrosine and tryptophan can also be modified successfully to create protein conjugates with a fluorophore, biotin or cytotoxic drugs.21 More recently, novel photocatalysis and redox-based strategies for the modification of tryptophan22 and methionine,23 respectively, have been reported. Although generally residue-specific, recent efforts have been focused on developing site-specific strategies to target a specific residue in the proteins. For detailed information of these developments, the reader is referred to other comprehensive reviews of this topic.20,24
Figure 1.

Selected direct chemical modifications of amino acids. (A) Cysteine modification via a) disulfide exchange, b) maleimide, or c) photo-catalysed thiol-ene couplings. (B) Lysine modification via coupling with d) isothiocyanate or activation with e) sulfonyl chloride or f) fluorine-substituted aromatic esters.
Among the 20 canonical amino acids, cysteine has served as the most convenient target owing to its highly nucleophilic sulfhydryl side chain group that offers a distinct reactive site in proteins.7 Modification can be made based on its ability to react via exchange reactions with disulfide-containing reagents, as well as its ability to undergo alkylation with suitable electrophiles including α-halocarbonyls (e.g. iodoacetamides) and Michael acceptors (e.g. maleimides).25 One of the most commonly used cysteine modifications is the reaction with maleimides, which has been applied to modify antibodies with fluorophores, cytotoxic drugs or PEG scaffolds.25 The frequent use of this reaction has resulted in the commercialization of a wide variety of maleimide derivatives, which in turn has stimulated additional applications. Examples of more recently developed approaches for cysteine alkylation involve photocatalysed reactions such as the irradiation of 3-(hydroxymethyl)-2-naphthol derivatives (NQMPs) to generate a Michael acceptor intermediate,26 or the use of alkene- and alkyne-functionalized reagents to initiate radical reactions for thiol-ene27 and thiol-yne28 coupling reactions, respectively. While great efforts have been made towards modification of native cysteines, the most critical disadvantage is that most methods lack site-specificity (when multiple cysteine residues are present), which results in heterogeneous product mixtures.20,24 Moreover, cysteines are relatively uncommon in protein sequences and often buried in within the protein structure when involved in disulfide bridges, making them difficult to access.29 Genetic incorporation of cysteine residues at sites of interest may disrupt protein structure and promote aggregation.30
Reactions with the primary amines present on the side chains of lysine residues has also been widely explored for protein modification.31,32 Owing to the strong ionic character of amines (usually present in its protonated form), lysines are frequently present on the surface of proteins and are thereby favourable targets especially for multi-site conjugations. Harder electrophiles, including NHS esters,33 isothiocyanates,34 and sulfonyl chlorides35 were initially used for lysine labelling. However, such reactions were found to display cross-reactivity with the protein N-terminus. To achieve more selective modification, kinetically controlled lysine modification using specially designed NHS-activated reagents was developed.36 Activated esters with fluoro-substituted aromatic leaving groups were also recently harnessed to specifically label an antibody fragment on a specific lysine residue present in a unique chemical environment.37 Despite potential chemoselectivity, the labelling efficiency is largely determined by the solvent accessibility and the chemical environment of the target lysine. Further improvement is needed.
2.2. Modification using unnatural amino acids
In addition to modifying native amino acids, the introduction of nonnatural amino acids into target proteins can also provide reactive handles for protein conjugation. These modified residues often bear bioorthogonal functional groups, which allow for more diverse and more specific subsequent conjugation reactions.38–41
To date, two unnatural amino acid (UAA)-incorporation methods have been described. One of them exploits the ability of the existing aminoacyl tRNA synthetase (aaRS) to recognize certain UAAs. In this scenario, the UAAs compete with the endogenous native amino acid substrates for the aaRS.42 Using this method, a collection of amino acids bearing bioorthogonal moieties including azidomethionine, homopropargylalanine, and homoallylglycine have been successfully incorporated into target proteins using methionyl-tRNA (MetRS).43–45 However, it should be noted that the MetRS will potentially replace all the methionine residues with the UAA analogue. As a result, a mixture of heterogeneously/multiply modified protein-conjugates will be obtained after modification, which is often not desirable.
A second, more selective approach, involves the reassignment of the stop codons, particularly the amber codon UAG, to insert a UAA.46 It was found that suppressor tRNAs found in some organisms cause the introduction of amino acids at UAG codons in lieu of stopping translation. Based on this discovery, directed evolution methods have been used to generate aaRS that specifically aminoacylate the UAA and then transfers it to an orthogonal tRNA.47 This aaRS-tRNA pair is then introduced into a host which expresses the target gene with the amber codon at a position of the protein sequence designated for modification. Such orthogonality provides a high degree of specificity without interference from endogenous natural amino acids and their complementary aaRS-tRNA pairs.
This technology has led to the development of a plethora of UAAs with diverse structures and functional groups (see Figure 2 for selected UAA structures). For instance, UAAs equipped with fluorescent reporters,48 photocross-linkers,49 or affinity handles50 have been reported. Functionalization of UAAs with reactive functional groups, which can participate in a wide range of bioorthogonal reactions, is also achievable, allowing conjugation of proteins to a broad range of cargos. However, several limitations still exist, including decreased expression yield of the desired full-length proteins compared to the wild-type, the production of truncated protein products that complicates purification, the compatibility of the introduced orthogonal aaRS-tRNA pair to the expression system and the availability of the required plasmids.51
Figure 2.

Structures of selected UAAs bearing fluorescent, cross-linker, affinity, or bioorthogonal handles.
2.3. Bioorthogonal reactions
Bioorthogonal functional groups refer to functionalities that are not typically present in biological systems, that can undergo reactions that do not occur with natural functionalities and are inert within the cellular environment. Incorporating these exogenous functional groups into a target protein, by the insertion of a UAA or enzymatic methods, enables selective modification to produce a homogenous product. In addition, to exploit their orthogonality, bioorthogonal reactions with high specificity have also been developed and successfully applied in a vast number of applications. In this section, several reactions that are commonly utilized for enzymatic protein labelling will be described. These include aldehyde/ketone condensations, azide-based click reactions and the tetrazine ligation. For a more comprehensive discussion of their mechanisms and applications, other reviews are readily accessible.52–54
Aldehydes and ketones are among the first functionalities that have been utilized for bioorthogonal protein labelling.55 Carbonyl groups can react with α-effect nucleophiles, such as alkoxyamines and hydrazides in aqueous solutions to form oximes and hydrazones (Scheme 1A). The reactions favour acidic conditions and their rates are rather slow at neutral pH. Initially, aniline was employed to accelerate the reactions under both acidic and neutral conditions.56 Catalysts with enhanced water solubility were later developed to further accelerate the reaction at neutral pH, including 5-methoxyanthranilic acid,57 m-phenylenediamine58 and p-phenylenediamine59. Electronic and acid/base effects also greatly influence the reaction rate at biologically relevant pH values, and carbonyl compounds with neighbouring acidic/basic groups can form hydrazones at elevated rates60. Although oximes are more stable than hydrozones, the C=N bond is still susceptible to hydrolysis61. To overcome this limitation, several alternative ligations strategies have been developed to form stable C-C linkages, such as the Pictet-Spengler ligation62/hydrazine-Pictet-Spengler ligation (Scheme 1B),63 and the trapped-Knoevenagel ligation,64 all of which have been applied to construct ADCs.
Scheme 1.

Selected bioorthogonal reactions based on aldehyde functionality. (A) Oxime/hydrazone ligation. (B) Pictet-Spengler ligations.
Aldehydes can be easily introduced into proteins of interest by formylglycine generating enzymes, which can convert a cysteine to an aldehyde-bearing formylglycine.65 Aldehyde and ketone containing substrate analogues for farnesyltransferase,66 N-myristoyltransferase,67 tubulin tyrosine ligase68 and lipoic acid ligase69 have also been developed to facilitate site-specific enzymatic protein labelling. It should be noted that while efficient for in vitro protein modification and cell surface labelling,70 the aldehyde/ketone-based reactions are less suited for in vivo applications due to the presence of endogenous carbonyl-containing compounds as well as the potential toxicity of the catalysts required for their efficient modification.
The azide group is essentially absent in biological systems and is generally inert to endogenous functionalities present in in the biological milieu, rendering it an ideal candidate for bioorthogonal reactions.71 Due to its small size, an azide group can be easily incorporated into various enzyme substrates for protein labelling. Introduction of the reactive partners for azides, including terminal alkynes and strained-alkynes, to make enzyme substrate analogues can also be achieved. Examples for different enzymes are discussed in detail in the following section. Three bioorthogonal reactions involving azides have been extensively explored, including the Staudinger ligation, the copper-catalysed azide-alkyne cycloaddition (CuAAC) and the strained-promoted azide-alkyne cycloaddition (SPAAC).
The Staudinger ligation (Scheme 2A), traceless-Staudinger ligation (Scheme 2B) and Staudinger-phosphite/phosphonite reactions exploit the reaction between azides and trivalent-phosphine reagents to form stable amide bonds.72 As one of the earliest developed bioorthogonal reactions, the Staudinger ligation has been successfully utilized in both in vitro and in vivo applications.73 However, the utility of these reactions is largely limited by their slow reaction rate and the oxidation propensity of the requisite phosphine reagents.52 CuAAC is a versatile and powerful tool for protein labelling. In the presence of Cu(I) as the catalyst, which is usually generated in situ from the reduction of Cu(II) by reducing agents, an azide and a terminal alkyne react to form a stable triazole product (Scheme 2C). 52,74 To stabilize the Cu(I) ion, a variety of chelating ligands have been synthesized and employed.75–79 Owing to its fast kinetics and excellent selectivity, the CuAAC reaction has been used for numerous in vitro protein modification applications. A major disadvantage of the CuAAC reaction arises from the cytotoxicity of the Cu(I) catalyst towards living cells, restricting its in vivo applications.80 To reduce toxicity, efforts have been made to decrease the copper concentration while maintaining reaction efficiency by optimizing azide structures81–83 and designing novel chelating ligands.77,80
Scheme 2.

Selected bioorthgonal reactions based on azide functionality. (A) The Staudinger ligation. (B) The traceless Staudinger ligation. (C) CuAAC reaction. (D) SPAAC reaction. DBCO is shown as an example for the strained alkyne compound.
SPAAC is also known as the copper-free click reaction. In the absence of a catalyst, alkynes can be activated via ring strain allowing them to react with azides directly (Scheme 2D), albeit at a much slower rate.74 Attempts to improve cycloaddition rates by optimizing the structure of strained alkynes have been limited by the inherent instability of these compounds under physiological conditions.54 Nevertheless, the SPAAC reaction has been widely exploited in bioconjugation with numerous accomplishments, especially for in vivo applications.84,85 The increasing number of commercially available strained-alkyne modified cargos has also facilitated its application. It should be noted that certain limitations are still present, including the hydrophobicity of the aromatic ring-fused strained alkynes and the potential for side-reactions with thiols from cellular proteins.86 Further tailoring and optimization of the CuAAC and SPAAC reactions will undoubtedly expand their utility for various biological applications.
The tetrazine ligation is the most rapid bioorthogonal reaction developed to date.52 The second order rate constant for the reaction between a tetrazine and its reactive partners ranges from 1 to 106 M−1s−1, depending on the structure of the strained alkene (Scheme 3A) or alkyne reagent (Scheme 3B) employed including norbornenes, cyclopropenes, trans-cyclo-octenes (TCO) and bicyclononynes.87 Tetrazines with different substituents also exhibit varied stability and reactivity towards strained alkenes.54,88 The capability to tune the reaction rate means that specific reagents can be chosen based on the needs dictated by the specific in vitro or in vivo application. In particular, the rapid kinetics are extremely useful for assembling radioisotope-labelled proteins in cases where very short half-life isotopes are employed.89 In addition, the extremely fast kinetics and selectivity has allowed the conjugation reaction to be performed in situ in live mice for tumour pre-targeting imaging applications.90 Importantly, the tetrazine ligation is orthogonal to the CuAAC and SPAAC reactions, enabling simultaneous dual labelling.91 Introduction of tetrazine moieties into a target protein has been reported using microbial transglutaminase with a tetrazine-amine substrate.92 As for the strained alkyne or alkyne compounds, a TCO-modified triglycine peptide substrate for sortase A has been reported for the construction of immuno-PET reagents.93 Lipoic acid analogues with TCO94 and norbornene95 were also utilized for protein fluorophore labelling in live cells. TCO-bearing analogues have also been developed for PFTase.96
Scheme 3.

Selected examples of tetrazine ligations. (A) Tetrazine ligation with TCO. (B) Tetrazine ligation with a strained alkyne
3. Enzymatic protein labelling strategies
As efficient catalysts for chemical reactions, enzymes have been widely applied in a variety of applications. Their high specificity, rapid reaction rates and ability to function under mild reaction conditions make them excellent choices for protein labelling purposes. Here, recent examples of work performed with enzymes from four classes including peptidases, transferases, ligases and oxidoreductases are described. In addition to those discussed below, other enzymes, including peroxidase,16 tyrosinase,97 and the enzymes involved in glycan synthesis,18 have also been employed on a more limited basis.
3.1. Peptidases
3.1.1. Sortase A.
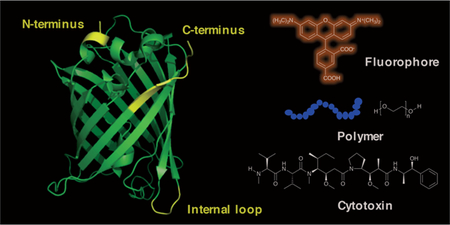
Sortase A (SrtA) from Staphylococcus aureus is a Ca2+-dependent transpeptidase.98 It recognizes a consensus sequence, LPXTG (known as a sortag) and cleaves the amide bond between the threonine and glycine residues, forming an acyl-enzyme-intermediate. Subsequent attack from the N-terminal amine of an oligoglycine-terminating peptide as the nucleophile yields a ligated product with a new peptide bond (Scheme 4A).99 SrtA has been extensively studied for site-specific labelling of peptides and proteins. Protocols have been published describing detailed procedures to label the N-terminus,100 C-terminus and the internal loops of proteins.101 Several enzyme variants, including an evolved penta-mutant with enhanced catalytic efficiency102 and a Ca2+-independent mutant103 as well as SrtA homologs from other bacterial sources with different recognition sequences have been reported to meet the requirements of various applications.104–107 Some of the early uses include protein lipid modification,108 cyclization,104 and cell-surface labelling,109 most of which have been summarized in several reviews.18,110,111 As an active area of research, an enormous range of applications have been reported since 2013, including but not limited to the semisynthesis of proteins with post-translational modifications,3,112 protein immobilization on solid surfaces,113–118 protein labelling on liposomes,119,120 virus-like particles121,122 and hydrogels123–126 as well as cell surface labelling127 and in vivo protein labelling.128–130 A few of these applications will be discussed in detail below.
Scheme 4.

Enzymatic labelling by SrtA. (A) Canonical C-terminal labelling catalysed by SrtA using oligoglycine substrates. (B) Labelling of the lysine (in a pilin domain) by SrtA using LPETG peptide substrates. (C) Protein labelling at the C-terminus by SrtA using primary amine or hydrazide-containing substrates. POI: protein of interest. Functionality/residues from the enzymatically added substrate are highlighted in red.
In recent years, SrtA has been applied to label full-length antibodies131,132 and their derivatives133–135 to create homogeneous conjugates. Grawunder and coworkers at NBE-Therapeutics AG disclosed the construction of homogeneous and site-specific counterparts of brentuximab vedotin (Adcetris) and trastuzumab emtansine (Kadcyla), using the evolved SrtA penta-mutant.131 The sortag was incorporated at the C-termini of both the heavy chain and light chain of the antibody. For the light chain modification, poor labelling was initially obtained due to the inaccessibility of the termini. The insertion of a short peptide spacer between the C-terminus and the sortag was shown to improve the labelling yield substantially. This strategy has also been widely adopted to enhance labelling of sterically hindered or buried protein termini. Cytotoxic drugs were appended to a penta-glycine moiety with the same linkers used in their corresponding commercial counterparts and subsequently conjugated to the antibodies by SrtA. The enzymatically generated brentuximab vedotin had a DAR (Drug to Antibody Ratio) of 3.2, indicating conjugation efficiency of 80%. When tested for tumour killing in vitro and in vivo, similar potencies were observed between the site-specifically conjugated and the chemically conjugated ADCs. Researchers from the same company later described the generation of ADCs bearing novel anthracycline-based cytotoxins by sortase-mediated antibody conjugation (SMAC) technology.132 They showed that these novel ADCs exhibited potencies exceeding those of Kadcyla and Adcetris, both of which are based on conventional tubulin-targeting payloads. This finding highlighted the importance of exploring alternative toxic payloads for the preparation of highly potent next-generation ADCs.
In addition to regular antibodies, SrtA has also been utilized to label a nanobody,93,136–139 a single-domain antibody (VHH) derived from alpacas and camels. Rashidian et al. used this strategy to label VHH for imaging and radio-diagnostic applications.93 They incorporated the sortag at the C-terminus of VHHs as this site is positioned away from the target-binding region. Labelling of two VHH constructs, DC8, an anti-Class II MHC nanobody and DC13, targeting CD11b, were evaluated. Dual-functionalized triglycine substrates comprised of two bioorthogonal handles were employed. One of the handles was used for the introduction of a fluorophore or 18F isotope while the other one was conjugated to a PEG moiety or a second VHH protein to tune the serum half-life or avidity of the conjugates. From PET imaging studies, the bivalent VHH homodimers were found to allow visualization of their targets more effectively than the monomers in vivo. Meanwhile, the PEGylated VHHs also displayed improved target staining. All the constructs exhibited high specificity. Apart from the imaging applications, Ploegh and coworkers also made structurally defined homogeneous VHH-drug conjugates.137 An anti-Class II MHC recognizing VHH7 was modified with a C-terminal sortag and reacted with triglycine peptides containing DM1 as the toxic payload using SrtA. A VHH7-NIR dye conjugate was also prepared to evaluate target binding, cellular internalization and in vivo localization of the VHH7. When the VHH7-DM1 was subjected to in vivo efficacy tests using highly invasive B cell lymphoma mouse xenografts, the drug-conjugate was shown to decrease tumour size significantly compared to the control group and to reduce metastatic spread.
In the area of creating protein-polymer conjugates, Gao and coworkers reported the in situ growth of a polymer chain directly from a target protein.140 In this study, a PEG-like polymer, POEGMA was polymerized from a therapeutically relevant protein, interferon alpha (IFN α), to form a site-specific and stoichiometrically modified IFN α-POEGMA conjugate.141 Briefly, IFN α was encoded with a C-terminal sortag and labelled with glycine substrates appended with an atom transfer radical polymerization (ATRP) initiator by SrtA. The resulting IFN α-Br acted as a macroinitiator. In the presence of OEGMA monomers under appropriate conditions, the polymer chain was grown from the IFN α protein, forming an IFN α-POEGMA conjugate with a high conversion of 90%. Importantly, when compared to PEGASYS, a commercial non-specifically PEGylated IFN α product, the IFN α-POEGMA exhibited superior pharmacokinetics and showed improved anticancer efficacy over PEGASYS, presumably due to decreased blockage of binding site using the site-specific conjugation strategy. As an alternative to PEGylation, this site-specific in situ growth technology is promising for the development of more effective next-generation protein therapeutics.
In addition to the use of canonical oligoglycine-containing peptides as substrates, SrtA was shown to have relaxed specificity for the amine nucleophiles.178,179 When positioned close to the active site of the SrtA, the non-protonated primary amine from a lysine residue can intercept the acyl-enzyme intermediate, forming an isopeptide bond (Scheme 4B).180,181 Chilkoti and coworkers took advantage of this reaction and used SrtA to site-specifically conjugate small molecules to lysine residues on proteins.182 They first examined the labelling reaction between a sortag-containing peptide and a pilin domain (PLN) peptide, which was derived from a natural bacterial protein that can form protein polymers via other related sortase enzymes. The PLN sequence contained a valine at the N-terminus and an internal lysine residue. Successful isopeptide ligation in the presence of SrtA was confirmed by MS. The authors then linked the pilin domain sequence to a fibronectin type III (Fn3) domain protein, an alternative target-binding protein. Fn3 was generated with C-terminal elastin-like polypeptides (ELP) to facilitate expression and purification. Several copies of the pilin domain were inserted between Fn3 and ELP to generate Fn3-PLN3-ELP. A biotin-appended sortag peptide was incubated with Fn3-PLN3-ELP overnight with SrtA. The conjugated product was detected by Western blotting against biotin while a control experiment with Fn3-ELP lacking the pilin domain and sortase did not yield modified proteins. The site of modification and the formation of the isopeptide bond was further confirmed by LC-MS/MS characterization of the trypsin-digested protein, revealing site-specific modification at the pilin lysine with no off-target labelling. Having established the isopeptide ligation with the Fn3 protein, the authors modified a monoclonal antibody with pilin domain insertion at the C-terminus of the heavy chain using a biotinylated sortag peptide. Approximately 1.8 biotins were incorporated per antibody, and the modified antibody retained its antigen binding capability, as validated by immunofluorescence microscopy. Thus, this isopeptide ligation can be employed to conjugate multiple cargos to a protein of interest at either internal or terminal sites, presenting a new bioconjugation technique for protein labelling. Optimization of the sequence and length of the pilin domain may further improve the reaction kinetics and ligation yield.
Not limited to the ε-amino groups of lysine residues, other primary amine-containing compounds have also been explored as substrates for SrtA (Scheme 4C).183,184 Cochran and coworkers reported the usage of a SrtA7M Hepta-mutant (SrtA7M)128 to create bioorthogonally tagged proteins of interest directly from E. coli using inexpensive, cell permeable and commercially available amine compounds.129 This SrtA mutant combined the mutations from the evolved enzyme with high catalytic activity and the mutations that abolished Ca2+ binding, yielding a Ca2+ independent SrtA variant with high reactivity. To label proteins directly in E. coli, SrtA7M was coexpressed with several target proteins engineered with C-terminal sortag respectively. Upon the addition of 3-azido-1-propanamine to the growth culture, they showed that three different target proteins were modified with the azide functional groups. Although endogenous bacterial proteins containing an LPXTG sortag were identified from the E. coli BL21(DE3) proteome using a RefSeq protein database search, SrtA7M-expressing cell lysate incubated with Cy3-DBCO exhibited minimal background labelling, suggesting the high specificity of the SrtA reaction. By using this technique, large quantities of labelled protein were obtained from cell lysate with one-step purification, greatly facilitating the synthetic process. Interestingly, hydrazine and its derivatives were also shown to be accepted by SrtA as nucleophilic substrates.185 Liu and coworkers first evaluated the SrtA-mediated hydrazinolysis of a model sortag peptide. A yield of greater than 95% was detected by HPLC while increasing the hydrazine concentration accelerated the reaction. The hydrazinolysis reaction was then applied to protein labelling. Several applications were illustrated including hydrazide-based protein semisynthesis as an alternative to native chemical ligation, installation of alkyne or azide functionalities that were further coupled to polymers to create PEGylated proteins, and construction of protein-fluorophore conjugates using a hydrazide-modified fluorophore. Protein synthesis using SrtA-mediated protein hydrazide ligation was also exemplified in recent work by Li and coworkers, where they synthesized p62 protein with various phosphorylation states and studied their interactions with K63 diubiquitin.112
These noncanonical nucleophile substrates can be used to complement the use of oligoglycine compounds, significantly broadening the substrate scope for SrtA. One limitation associated with the oligoglycine substrates is that the ligated product can also be recognized and cleaved by SrtA, resulted in decreased ligation yield. Through the use of these alternative substrates, the ligated protein lacks the glycine residue present in the sortag sequence, which helps to prevent hydrolysis and to improve conversion. However, further engineering of the SrtA to enhance specificity and reactivity towards the noncanonical substrates is highly desirable so that less SrtA and substrates can be used to achieve acceptable yields.
To assist the purification of labelled proteins, efforts have been made to develop one-pot labelling and purification techniques.186,187 As one example, Tsourkas and coworkers initially described the sortase-tag expressed protein ligation (STEPL) technique.188 A construct containing the protein of interest followed by the sortag sequence, a (GGS)5 flexible linker, SrtA and a His-tag was designed. The expressed protein fusion was absorbed onto Ni-NTA resin from the cell lysate. Upon the addition of Ca2+ and various oligoglycine peptides with different cargos, the target proteins were labelled and eluted, leaving the SrtA still attached to the resin (due to the His-tag). Both the concentrations of the Ca2+ and the oligoglycine peptides had an impact on the ligation yield and product purity, which needed to be optimized. Although successfully employed to label a HER2-recognizing affibody, the method was not generally applicable since some proteins expressed poorly once linked to SrtA and in some rare cases the fused SrtA became catalytically inactive. To overcome those limitations, the same group developed a proximity-based sortase-mediated ligation (PBSL) technique.189 In this system, the target protein was expressed as fusion to the C-terminal sortag, a (GGS)5 linker and a SpyTag peptide. The latter is a short 13-residue peptide that is covalently linked to its partner protein, SpyCatcher, upon binding. An additional construct where SrtA was fused to the N- or C-terminus of a His-tagged form of SpyCatcher was also prepared. The SrtA-SpyCatcher fusion was first immobilized on resin, which was then used to capture eGFP-LPETG-SpyTag from clarified cell lysates. The captured eGFP was modified and released from the resin by the addition of Ca2+ and triglycine peptides. Importantly, the cleaved SpyTag remained attached to the SpyCatcher on resin, making PBSL a traceless labelling technique. Peptide concentration and the reaction time were optimized to minimize product hydrolysis and achieve high ligation efficiency. When comparing the ligation yield between PBSL, STEPL and the conventional sortase reaction using an anti-CD3 ScFv protein, the STEPL failed to produce the target protein fusion with SrtA while PBSL yielded 2.5-fold more labelled protein than the traditional reaction in a much shorter time. A similar on-resin cleavage and ligation strategy was also reported by Cheng et al. to functionalize peptides.190 The peptide target with a C-terminal LPXTG sortag was synthesized on PEGA resins that are hydrophilic and permeable to macromolecules up to 35 kDa using standard solid phase peptide synthesis (SPPS). Diglycine compounds with various functional moieties, including biotin, lipid, PEG polymer and polypeptides, were utilized to successfully label and release the ligated peptide from the resin in one-pot using SrtA.
In summary, SrtA has been extensively applied for site-specific protein modification both in vitro and in vivo. Although the wild-type enzyme is limited by slow reaction kinetics and only moderate ligation yields, methods to improve the reaction have been developed and will continue to be explored. It should be noted that despite the improved catalytic efficacy, the evolved penta-mutant enzyme has a decreased affinity towards the nucleophilic substrate,102 and exhibited elevated hydrolysis in some cases.191 As mentioned above, further engineering of SrtA to improve reaction kinetics while suppressing hydrolysis should allow more efficient labelling using much lower substrate and enzyme concentrations.
3.1.2. Subtiligase.
Subtiligase is an engineered peptide ligase derived from subtilisin BPN’, a serine protease obtained from Bacillus amyloliquefaciens.192 It catalyses the ligation reaction of an acyl-donor peptide ester to the N-terminal α-amine of the acceptor peptide, forming a native peptide bond (Scheme 5). Compared to the parent subtilisin, the active site S221 was changed to cysteine (S221C) in subtiligase. This mutation shifted the catalytic mechanism to strongly favour aminolysis over hydrolysis.143 A second mutation of P225 to alanine (P225A) improved the aminolysis efficiency, due to the reduced steric crowding in the active site.143 The double mutant (S221C/P225A) of subtilisin BPN’ is known as subtiligase.193
Scheme 5.

Protein N-terminal labelling by Subtiligase using peptide ester substrates.
Capable of catalyzing the formation of a native peptide bond, subtiligase has been used for the synthesis/semisynthesis of large proteins. Notably, using synthetic peptide fragments as the building blocks, UAAs or residues equipped with post-translational modifications can be easily incorporated into the synthetic protein at any defined position. In an early example, Wells and coworkers performed the total synthesis of Ribonuclease A from 6 peptide fragments using subtiligase.194 Beyond the native protein, variants containing unnatural catalytic residues were also constructed, which helped to elucidate the catalytic mechanism of the enzyme. It should be noted that one major limitation associated with this total synthesis technique is that it is only applicable to proteins that can be refolded in vitro. As to protein semisynthesis, subtiligase is also able to carry out ligations with recombinant protein thioester fragments. As an alternative to the expressed protein ligation (EPL), which requires a cysteine at the ligation junction, careful selection of the conjugation site allowed enzyme-catalysed expressed protein ligation by subtiligase to be used to synthesize proteins with their precise wild-type primary sequence. In 2016, Cole and coworkers demonstrated that phosphorylated PTEN, a tumour suppressor lipid phosphatase, synthesized by subtiligase exhibited a more tightly closed conformation than that of a Y379C mutant generated from EPL, which was found to behave anomalously in cells.145
Since the enzyme can distinguish the N-terminal α-amine unambiguously over the lysine ε-amine, subtiligase has also been applied to study apoptosis by probing unblocked protein N-termini generated from proteolytic cleavage by caspases and caspase-like enzymes.195 A biotinylated peptide ester tag was designed by Wells and coworkers, which contained a biotin moiety for affinity enrichment and a TEV protease cleavage site to release the captured peptides from beads. To identify newly exposed N-termini in apoptotic cells, both Jurkat cells treated with or without etoposide, a topoisomerase II inhibitor, were analysed. Proteins in the cell lysate were labelled with the peptide ester tag by subtiligase. Following trypsin digestion, the biotin-labelled peptides were enriched using avidin immobilized beads and then subjected to LC-MS/MS characterization. The detected N-terminal sequence information was valuable in mapping the cleavage site of the caspase-like enzymes. Interestingly, the identified sequences in this study were generally not predicted by in vitro caspase substrate specificity studies, underlining the importance of cellular profiling over in vitro assays. The same subtiligase labelling technique was also utilized to monitor the regulation of post-translational modifications occurring at the protein N-terminus, such as N-α-acetylation.196
For protein labelling applications using subtiligase, it is straightforward to envision that peptide ester substrates containing bio-orthogonal functional groups can be ligated to the N-terminus of a protein target, enabling subsequent conjugation. The site-specificity relies on the ability of the enzyme to recognize only the N-terminal α-amine rather than a specific peptide recognition tag, which is characteristic of other enzymes, such as SrtA. In this scenario, a broad substrate specificity would be particularly useful so that a variety of proteins can be modified by subtiligase without the effort of genetic modification. Previous qualitative studies revealed that subtiligase exhibited relatively broad reactivity towards α-amine peptides with some sequence preference. The incomplete characterization made it difficult to predict whether a specific protein of interest would be modified by subtiligase or not. Very recently (2018), Weeks et al. employed an approach, called proteomic identification of ligation sites (PILS), to comprehensively characterize the N-terminal sequence specificity of subtiligase.144 To ensure full coverage of the sequence diversity, peptide libraries were generated from proteases digestions of the E. coli proteome. A similar biotin-containing peptide ester tag as described above was utilized to label substrate peptides, which were then captured on beads, released, and analysed by LC-MS/MS. It was found that the reactivity of subtiligase was mainly determined by the first two residues (P1’ and P2’) of the peptide substrates. Substantial sequence preferences were observed in both P1’ (small amino acids, methionine and arginine) and P2’ (aromatic and large hydrophobic residues) positions. To expand the substrate scope, subtiligase mutants were designed and generated to modify peptide sequences with disfavoured residues by the wild-type enzyme. For example, a Y217K mutant was found to improve the reactivity of sequences with an acidic P1’ residue while an F189R mutant increased modification efficiency towards peptides containing an acidic P2’ residue. A free-access web-based tool, α-Amine Ligation Profiling Informing N-terminal Modification Enzyme Selection (ALPINE), was established to help guide the selection of the optimal subtiligase variants for modification of proteins and peptides based on user-defined N-terminal sequences. As a proof of concept, an anti-GFP antibody was modified with an azide-containing peptide at the N-terminus of the heavy chain. Subsequent SPAAC reaction with a DBCO-modified cargo was employed to install either a biotin, a Cy3 fluorophore, an MMAE cytotoxin, a 30-mer oligonucleotide or a PEG polymer to the labelled antibody. Importantly, the Cy3 modified anti-GFP Fab was shown to retain its targeting-binding capability. In the cases where the N-terminus of the protein substrate was buried, multiple rounds of labelling by subtiligase or introduction of a short peptide extension were shown to substantially improve the conjugation efficacy.
Overall, subtiligase is a useful tool for protein labelling, whose utility has not been fully explored. The reaction is fast, site-selective and highly efficient. More importantly, the ability to directly modify the native N-terminal sequence of protein substrates eliminates the need for additional genetic manipulation. This feature combined with the web-based ALPINE tool greatly facilitates the labelling process, which can be readily adapted for a variety of applications.
3.2. Transferases
3.2.1. Microbial transglutaminase.
Transglutaminase catalyses amide bond formation between the γ-carboxamide group of a glutamine residue and the ε-amino group of a lysine (Scheme 6). Microbial transglutaminase (MTG), isolated from Streptomyces mobaraense, has been widely utilized in the food and textile industries to improve the texture and appearance of the products via protein cross-linking as well as for biotechnological applications to create various protein conjugates.97,197 Substrate specificity studies reveal that the surface accessibility of the glutamine or lysine residues and the backbone flexibility where they are located are crucial factors that dictate enzyme reactivity.198,199 Additionally, the activity of MTG is also affected by the surrounding residues close to the modification sites.200 Therefore, even though multiple glutamine or lysine residues are present on a protein surface, only a few of them can be modified by MTG.198 Concerning the acyl acceptor substrate, both lysine-containing peptides and primary amines with less steric hindrance are found to be recognized by the enzyme.201 Several peptide sequences containing the glutamine or lysine residue, referred to Q-tag (Scheme 6A) and K-tag (Scheme 6B) respectively, have been found to be efficient substrates for MTG and can be engineered into a protein of interest for site-specific modification.202–204 Concerning Q-tags, in addition to the classic and well-studied sequence of LLQG,205 a Q-tag denoted as 7M48 (WALQRPH),206 optimized from peptide sequences selected from phage-displayed libraries202 was shown to have improved reaction rates; that was confirmed by engineering 7M48 into maltose binding protein (MBP) and analysing the labelling using propargylamine as the small molecule substrate.147 Importantly, these tag sequences can be inserted at either the C-terminus,207 N-terminus208 or internal loops of the proteins of interest.209
Scheme 6.

Enzymatic protein labelling by MTG. (A) Labelling of target proteins containing Q-tag sequences using lysine or Primary amine Substractes. (B) Labelling of K-tag modified protein using a ZQG peptide dervatized with various cargos.
Construction of protein conjugates using MTG can be accomplished in one step using peptide or amine substrates appended to various cargos. Early examples included fluorophores,207,210 the PEG polymers,211 metal chelators,212 or oligonucleotides.204 Since 2013, two-step approaches consisting of initial protein modification with a bioorthogonal functional group, such as alkyne,147 azide,146,148 or tetrazine92 have been widely explored. Subsequent conjugation reactions are then performed to install the desired cargos onto the protein of interest. In addition, to avoid tedious purification steps and improve overall yield, one-pot conjugation combining the enzymatic protein labelling and bioorthogonal reactions has also been studied. In 2016, Pelletier and coworkers discovered that the addition of glutathione preserved the activity of MTG in the presence of copper.15 Since the enzymatic reactions catalysed by MTG occurred significantly faster than the CuAAC reaction, they were able to conjugate α-lactalbumin, bearing two reactive glutamines, to a Cy5 fluorophore by simply mixing MTG, the amine-substrates with either alkyne or azide, the clickable Cy5 and the CuAAC reagents. To follow up, they expanded the reaction scope to metal-free reactions and demonstrated that this one-pot strategy was also compatible with SPAAC and tetrazine ligation reactions.92
Due to the high demand for manufacturing homogeneous antibody conjugates, the utility of MTG for such purposes has been extensively explored. Early work by Schibli and coworkers evaluated the labelling of full-length antibodies by MTG using both amine and glutamine substrates.213 It was found that glycosylated antibodies were poorly modified by MTG while an unglycosylated antibody could be labelled efficiently with amine substrates. To explain the difference in reactivity, they identified the modification site to be Q295, which is spatially close to N297.212 As a conserved glycosylation site, N297 could mask the neighbouring Q295 upon glycosylation, rendering it inaccessible to MTG. Based on these findings, the authors developed a method to create antibody conjugates with uniform composition by first deglycosylating the antibodies using PNGase F to expose Q295. The resulting antibodies were then reacted with MTG to install functional moieties, including metal chelators for imaging purposes212 and cytotoxins for therapeutic applications.148 Since Q295 is preserved in all IgG subtypes, this method can potentially be applied to any given antibody without the need for genetic manipulation.
The integration of Q tags into antibody sequences has also been explored for site-specific modification. Through screening of multiple conjugation sites on an antibody, Rajpal and coworkers demonstrated that the modification location had an impact on the linker stability and the in vivo pharmacokinetics of the ADC, creating the opportunity to optimize the therapeutic index of ADCs by tuning the labelling site.214 Beyond these preliminary studies, in 2016, Strop et al. developed a Trop-2 targeted ADC using site-specific MTG-mediated conjugation and characterized its anti-tumour efficacy and toxicity comprehensively. An LLQGA tag was appended to the C-terminus of the antibody heavy chains and then linked to a lysine-vc-PAB-toxin moiety by MTG.215 The resulting ADC was highly efficacious in multiple mice xenografts bearing different solid tumours. The safety profile with nonhuman primates indicated on-target epithelial toxicities, which are fully recoverable. These preclinical efficacy and safety results supported the advanced clinical development of the ADC against multiple solid tumour types. Lastly, antibody labelling at native or engineered lysine residues has been reported more recently, allowing the construction of orthogonally dual-labelled antibodies.216,217
To streamline the process of antibody conjugation, on-bead modification methods were also developed. In 2016, Graziani and coworkers showed that antibodies immobilized on protein A beads could still be modified by MTG with high efficacy.218 This procedure replaced multiple purification steps by simple washing and could be easily adapted to a 96-well format, allowing rapid screening and optimization of the ADCs. In a related approach, Spycher et al. attached MTG onto glass microbeads.216 It was shown that the MTG on the solid support retained its catalytic activity to link different substrates to various proteins, including scFv and full-length antibodies. Notably, higher selectivity was observed for the immobilized MTG in that while two lysine residues on avidin could be modified by MTG in solution, only the one located in a highly-disordered region was labelled using the immobilized MTG. That difference was attributed to the decreased rotational flexibility of the immobilized MTG, which can be potentially beneficial to produce highly homogeneous protein conjugates.
Besides protein modification with small molecules, construction of protein-polymer conjugates using MTG has also been extensively investigated.211,219 PEGylation of small therapeutic proteins by transferring an amine-functionalized PEG onto intrinsic glutamine residues has been demonstrated. As noted earlier, accessibility and chain flexibility are important determinants of MTG reactivity. Therefore, it is possible to achieve site-selective modification on the endogenous glutamine residues without genetic engineering. Such examples included the PEGylation of human growth factor (hGH, Q40 and Q141),220 human interleukin-2 (hIL-2, Q74),221 granulocyte colony stimulating factor (GSCF, Q134),222 and more recently interferon alpha-2b (IFN α−2b, Q101).223 To further increase the selectivity of MTG, Pasut and coworkers explored the use of organic co-solvents, which was known to influence protein conformation and flexibility.224 It was found that highly selective conjugation could be achieved when appropriate co-solvents were used. For example, only Q141 was PEGylated when the enzymatic reaction with hGH was performed in 50% ethanol.225 Different from the widely explored modification on glutamine residues, MTG mediated PEGylation of lysine residues is relatively rare and has only been studied since 2016. In one case, a PEG polymer was linked to N-benzyloxycarbonyl-L-glutaminylglycine (ZQG), a well-studied glutamine-bearing dipeptide substrate for MTG, Similar to observations of glutamine labelling, selectivity was also achieved with several proteins. K41 on GSCF149 and K164 on IFN α−2b223 were found to be modified by MTG despite the presence of other lysine residues on the proteins.
Protein labelling with oligonucleotides via MTG has also been reported. In one of the earliest examples, Goto and coworkers incorporated ZQG onto the 5’-end of aminated DNA, forming ZQG-DNA that could be recognized by MTG.204 A K-tag (MKHKGS) was fused to two model proteins, bacterial alkaline phosphatase (AP) and eGFP at either N- or C-terminus. The AP-DNA conjugates were immobilized onto cDNA-displaying microplates via DNA hybridization. Subsequent addition of a fluorescent AP substrate yielded an intense fluorescence signal in the AP-DNA treated wells, indicating that the biological functions of both the DNA and the AP enzyme were retained. This concept was further expanded to use ZQG-dUTP as the building blocks to synthesize DNA/RNA–(enzyme)n conjugates, which can be applied for sensitive DNA/RNA detection.226,227 More recently, Takahara et al. designed NH2-dUTP that was easier to synthesize and could be added to the 3’-end of a DNA aptamer by terminal dexoynucleotidyl transferase (TdT).228 The NH2-clustered aptamer was crosslinked to Q-tag (FYPLQMRG) fused eGFP by MTG. Approximately 3 to 6 copies of eGFP were loaded onto each aptamer. Notably, purification of the conjugates from unreacted eGFP was readily achieved by the simple use of a centrifugal filter due to their large size. The conjugated aptamer retained its binding capability towards cell surface targets as confirmed by confocal imaging. Similarly, a polymer chain was also used to mount multiple proteins. The same group prepared a polymerizable methacrylate-ZQG monomer and polymerized it with acrylamide to form polymer chains.229 A K-tag (MRHKGS) was introduced onto the C-terminus of the protein substrate, in this case, a fusion protein containing a binding domain to the antibody Fc region and a chimeric alkaline phosphatase (IPP-PG). The polymer-enzyme conjugates were used in place of the secondary antibody-enzyme constructs for the detection of biomolecules via ELISA. Due to the clustering of proteins in one polymer chain, a maximum of 5-fold increase in signal detection was achieved when compared to that of the free protein, highlighting the advantageous signal amplification effect from multi-valency.
Overall, MTG is a versatile and promising tool for protein modification. Along with a number of demonstrated fruitful applications, directed evolution methods to engineer mTG for enhanced activity205 and transglutaminase from other bacterial sources that can be utilized for protein labelling have also been reported,230,231 facilitating future applications. The ability of MTG to label intrinsic residues on protein substrates greatly simplifies the production process. However, due to its relatively broad substrate specificity, it is difficult to predict whether a given protein of interest will be modified or not. Experiments are required to determine the extent of modification. Extra care must also be taken to identify potential protein-crosslink by-products and to evaluate whether modification of particular endogenous conjugation site is detrimental to protein function or stability.
3.2.2. Farnesyltransferase
Prenylation is a post-translational protein-lipid modification that is catalysed by farnesyltransferase (FTase). The enzyme recognizes a C-terminal CaaX motif and transfers the isoprenoid moiety from the natural substrate, farnesyl diphosphate (FPP), to the CaaX-box cysteine via a thioether linkage (Scheme 7); the recognition sequence is denoted as a CaaX box, where C is a cysteine, a represents small aliphatic amino acids, and X determines the substrate specificity towards different enzymes in the same family.232 Extensive studies investigating the enzyme specificity revealed that FTase is promiscuous towards both the peptide recognition sequence233–236 and the isoprenoid substrate.234,237,238 Interestingly, it was discovered very recently (2018) that in addition to the canonical CaaX box, some peptides and proteins bearing C-terminal CaaaX motif can also be accepted by FTase as substrates.239 This finding expanded the scope of prenylation within the proteome, implying more proteins may be potentially prenylated, whose cellular functions remain to be explored. For protein labelling applications, a number of isoprenoid analogues have been previously developed functionalized with various groups, including fluorophores,240–242 photoaffinity moieties,243–245 biotin,246 azide-,247–250 alkyne-,152,251,252 and aldehyde-functionalities66,253. It has been shown that these analogues can be incorporated into the target proteins bearing engineered C-terminal CaaX-box sequences. A variety of applications were demonstrated, ranging from cellular protein imaging,254 proteomic analysis,246,247,255 surface immobilization,151,249,256 and construction of protein conjugates.251,257 These early applications were summarized in the review by Rashidian et al.18 The examples discussed below focus on developments since 2013.
Scheme 7.

Protein labelling at C-terminus of a POI terminating in a CaaX-box sequence by FTase using isoprenoid analogues bearing bioorthogonal functional groups.
One of the common traits of the isoprenoid analogues described above is that they all contained only one functional moiety, such as a fluorophore/biotin or a reactive handle for subsequent conjugation. To enable protein dual-functional labelling, Distefano and coworkers designed an analogue containing both an alkyne and an aldehyde functional group.154 With these two handles, they were able to install a fluorophore as well as a PEG polymer into the proteins of interest. Since the CuAAC and oxime ligation are orthogonal to each other, both conjugation reactions can be performed simultaneously without cross interference. The designated probe is particularly useful for creating multi-functional protein conjugates, such as theranostic reagents and PEGylated protein-drug conjugates with improved pharmacokinetic properties.258 Beyond labelling individual proteins, this multifunctional substrate has been used to assemble nanoring structures bearing multiple copies of a model protein that can be internalized into cells. To expand the scope of compatible bioorthogonal reactions, Wollack et al. also developed a TCO-functionalized isoprenoid analogue and demonstrated that it could be efficiently transferred onto protein substrates by FTase.96 Subsequent reactions with tetrazines were also confirmed by MS. Since tetrazine ligation is much faster than the SPAAC reaction without the need of a metal catalyst, this strategy can be potentially useful for site-specific protein labelling inside living cells.
Although FTase is sufficiently promiscuous to process these bulkier isoprenoid analogues, their catalytic efficiencies are generally lower than that of the FPP. Aiming to address this limitation, Dozier et al. analysed how different FTase mutations within the active site could be used to better accommodate these larger analogues.259 Based on the crystal structures of FTase and previously reported literature,260 three key residues were identified that were in close contact with the third isoprenoid unit of FPP. Accordingly, three mutants were prepared, including W102A, Y154A, and Y205A. A panel of four analogues was examined, each containing an aryl aldehyde-, a TCO-, a coumarin- or a nitrobenzoxadiazole (NBD) group. It was found that the mutant enzymes catalysed the reactions more efficiently than the wild-type enzyme to a varying degree. Of particular note, the Y205A mutant exhibited a 300-fold increase in kcat/KM for the coumarin substrate, manifesting a catalytic efficiency comparable to that of the wild-type enzyme for the natural substrate, FPP. Overall this study established these mutant enzymes as useful tools for protein labelling. Based on those results, it should be feasible to further engineer FTase to create mutants with substrate specificities orthogonal to the wild-type enzyme, which can be used for simultaneous multi-protein labelling.261 In related work, it was shown that concurrent dual-protein labelling could be achieved using FTase and geranylgeranyltransferase-I (GGTase-I).155 GGTase-I has a larger active site than that of FTase. As a result, it catalyses the transfer of the 20-carbon isoprenoid unit from geranylgeranyl diphosphate (GGPP) to the protein substrates. Different specificities are also observed for the CaaX motif. Combining these features, Zhang et al. showed that by selecting appropriate CaaX sequences and isoprenoid substrates, orthogonal dual protein modification could be achieved. They utilized this strategy to install ketone- and alkyne-functionalities into two model proteins respectively and demonstrated that the subsequence conjugation using oxime ligation and CuAAC reactions could also be performed in one-pot without undesired side-products. Based on these results, it should be possible to expand this simultaneous labelling system to include engineered FTase enzymes for potential in vivo multi-protein modification applications.
To implement protein immobilization using FTase, Poulter and coworkers initially incorporated alkyne- and azide-containing isoprenoid analogues into GFP and GST engineered with a C-terminal CVIA sequence. The modified proteins were then oriented on a solid support using CuAAC and Staudinger ligation reactions.151 In a follow-up study, they further demonstrated that the regioselectively immobilized GST retained its enzymatic activity.262 In 2013, they constructed antibody arrays by immobilizing antibody binding proteins A, G and L onto glass slides using a similar strategy,157,263 enabling antibody capturing for direct and sandwich-type immunofluorescent antigen detection. Besides glass slides, proteins labelled with alkyne isoprenoid analogues can also be deposited onto the azide-modified self-assembled monolayers prepared on the gold surfaces using the CuAAC reaction, as illustrated by the Poulter264 and Maynard265 groups, respectively.
To capitalize on this highly site-specific protein modification strategy, Tretyakova and Distefano constructed structurally defined DNA-protein conjugates using FTase.156 An azide-containing isoprenoid analogue was incorporated into eGFP engineered with a C-terminal CVIA sequence and then linked to oligonucleotides via alkyne functional groups attached at the 5-position of thymidine using the CuAAC reaction. The resulting conjugates served as analogues of naturally occurring DNA-protein cross-links (DPCs) and were then used to investigate the effect of DPCs on the ability of human DNA polymerases to bypass these large lesions for the purpose of repair. It was demonstrated that lesions containing an entire protein blocked all human polymerases tested while DPCs formed with a short peptide could be bypassed, suggesting that proteolytic degradation to remove the replication block imposed by the DPCs occurs prior to DNA repair.
To explore the therapeutic potential of FTase-catalysed protein modification, Kim and coworkers utilized the enzymatic prenylation and oxime ligation to synthesize homogeneous protein-toxin conjugates for targeted therapy.153 In this study, EGFR-binding repebody proteins, engineered with a C-terminal CVIM sequence, were labelled with ketone-containing isoprenoid analogues by FTase and then conjugated to aminooxy-modified MMAF. Near quantitative labelling and subsequent conjugation were achieved. The resulting repebody-MMAF conjugates (RDCs) displayed much higher cytotoxicity to EGFR-positive cell lines compared to the free drug molecule while the toxic effects on EGFR-negative cells was negligible. A significant tumour regression response was observed when the RDCs were injected into xenograft mice bearing EGFR-positive tumours, emphasizing the therapeutic potential of the RDCs. In addition, this labelling strategy was developed by LegoChem Biosciences, Inc to establish ConjuALL™ platform technology,266 which utilized prenyltransferase to make site-specific antibody-drug conjugates, further demonstrating the potential of using FTase to construct site-specific protein conjugates with therapeutic significance.
Overall, the FTase labelling reaction is highly selective and efficient. Importantly, the sequence requirement of only four amino acid residues as the recognition sequence minimizes the potential perturbation of the target protein structure. It is especially well-suited for labelling of proteins whose C-terminus is far away from the site of action and when high homogeneity of the product is desired.
3.2.3. N-myristoytransferase.
N-myristoylation is an important protein-lipid modification in all eukaryotes, which can occur co-translationally or post-translationally.267,268 The enzyme N-myristoyltransferase (NMT) catalyses the transfer of myristate from myristoyl-CoA to the N-terminal glycine of protein substrates resulting in the formation of an amide linkage. The enzyme recognition sequence on the protein substrate is usually represented as GXXXS/T, where G is an absolute requirement and X can be a variety of amino acids.268,269 Comprehensive substrate specificity studies of yeast NMT conducted by Gordon and coworkers revealed that the enzyme has a relatively conserved myristoyl-CoA binding site, preferring analogues mimicking the structure of myristic acid concerning chain length and flexibility.158,270,271
To explore protein labelling using NMT, early work by Tate and coworkers utilized azide and alkyne-bearing CoA-myristic acid analogues to label both a synthetic model peptide and the Plasmodium falciparum ADP ribosylation factor 1 (pfARF1) protein, which is a natural substrate for NMT.160,272 Importantly, labelling of the protein substrate was performed in E. coli (Scheme 8), where the target protein was coexpressed with Candida albicans NMT. The bacterial culture was supplemented with either the azide or alkyne myristic acid analogues during protein expression. It should be noted that endogenous enzymatic activities in E. coli are critical to convert the analogues into their active CoA forms and to cleave the N-terminal methionine residue on the nascent proteins to expose the N-terminal glycine for enzyme recognition. A detailed protocol for the in vivo labelling procedure has been published by Heal et al.273 Building upon that early work, a variety of applications have been demonstrated since 2013, which are summarized below.
Scheme 8.

Protein labelling by NMT in E. coli. The plasmids expressing the POI and NMT are both transformed into E. coli. At the time of expression, myristic acid analogues are added to the culture medium, which are then converted to CoA modified substrates by endogenous enzymatic activities. Proteins are labelled in vivo at the N-terminus.
Not limited to the native protein substrates for NMT, Tirrel and coworkers investigated the possibility of labelling recombinant proteins with engineered N-myristoylation motifs.161 Two engineered GFP constructs, each carrying the N-terminal sequence from a natural myristoylated protein, were coexpressed with human NMT (hNMT) in E. coli. The azide-containing mystic acid analogue (12-ADA) was added to the culture during protein expression and the resulting cell lysate was collected and incubated with TAMRA-alkyne. Successful protein-fluorophore conjugation was confirmed using in-gel fluorescence imaging. Additionally, they were able to immobilize the myristoylated GFP onto a DBCO-functionalized solid surface using the SPAAC reaction directly from crude cell lysate. Utilizing the same labelling strategy, Tirrel and Ursem later modified an engineered Ca2+ binding protein, calmodulin (CaM), with azide functionality.162 The engineered and myristoylated CaM maintained its binding capability to Ca2+ and was functionally active. In addition to the construction of CaM-TAMRA conjugates from cell lysate, CaM-affinity resins were also prepared by immobilizing CaM onto DBCO-functionalized reins, which demonstrated superior performance in purifying CaM-binding proteins. These examples demonstrate the feasibility of labelling non-natural substrate proteins with engineered N-terminal motifs by NMT.
Taking advantage of the in situ myristoylation in engineered E. coli, Ho et al. recently developed a facile method for protein imaging in bacteria.159 A dual-plasmid system, one encoding hNMT and methionyl aminopeptidase and the other one for expressing the protein of interest, were transformed into E. coli. A total of four target proteins with polar and septal spatial distributions were studied. The azide-containing myristic acid analogue 12-ADA was supplied during protein expression. Fluorophore labelling experiments were first conducted in cell lysates where the bacterial cells were lysed and incubated with a BCN-modified BODIPY dye using the SPAAC reaction. Successful conjugation was confirmed by in-gel fluorescence analysis. Next, they performed protein conjugation in fixed cells by incubating the dye with fixed and permeabilized cells. Fluorescence was observed via confocal imaging for each target protein with its expected localization pattern. Finally, for protein labelling in live cells, cells were treated with the fluorophore reagent without fixation or permeabilization. The desired localization of each protein was again observed. A slightly more hydrophilic azide-containing myristic acid analogue with an oxygen substitution at the C6 position was also studied in the lysate and live cell labelling experiments. Similar results were achieved comparable to those obtained with 12-ADA, suggesting that the toolbox of fatty acid analogues can be expanded by tuning the structure of the acyl chain.
Since the acyl-chain of myristic acid is highly hydrophobic, myristoylation has also been exploited to construct lipid-peptide hybrid biomaterials. Chilkoti and coworkers fused the NMT recognition sequence to the elastin-like polypeptide (ELP).274 The ELP was coexpressed with yeast NMT in E. coli and was myristoylated in the presence of myristic acid. The purified lipid-modified ELP self-assembled into micelles with a hydrophobic core while the unmodified polypeptides remained as soluble unimers. The authors illustrated that the morphology of the self-assembled structures could be tuned by varying the length and composition of the ELP peptide. The presence of the hydrophobic core inside the micelles helped to promote physical encapsulation of hydrophobic drug molecules so that the micelles could serve as a drug delivery system. Although encapsulated doxorubicin (DOX) and paclitaxel showed decreased cytotoxicity in cell cultures compared to the free drugs, in vivo pharmacokinetics studies revealed a 6.5-fold higher serum half-life for the encapsulated DOX compared to the free drug. The simple generation of a self-assembled lipidated peptide-polymer drug delivery system directly from E. coli without additional labelling and purification steps greatly streamlines the production process. As a delivery system that functions through passive encapsulation, these micelles can be particularly useful for transporting drugs that lack reactive handles or in cases where chemical modification is detrimental to the bioactivity of the drug.
In summary, N-myristoyltransferase is a powerful tool for site-specific protein modification at the N-terminus. The ability to perform myristoylation in vivo in engineered E. coli without the need to purify the NMT enzyme markedly facilitates the modification process. Due to the lack of endogenous NMT in bacteria, in situ generation of modified myristoylated proteins can be especially useful for imaging cellular proteins and studying PPIs in live bacterial cells.
3.2.4. Phosphopantetheinyl transferases.
Phosphopantetheinylation is a process where a phosphopantetheine (Ppant) group from CoA is transferred to peptidyl (PCP) or acyl carrier protein (ACP) domains of several synthetases by phosphopantetheinyl transferases (PPTase).275 These domains can be fused to proteins of interest for site-specific labelling. A serine residue in a conserved sequence motif is modified through the formation of a phosphoester bond with the hydroxyl group, along with the concomitant release of ADP (Scheme 9). Surfactin phosphopantetheinyl transferase (Sfp) is the most utilized PPTase in enzymatic labelling strategies owing to its known promiscuity in recognizing a wide range of modified phosphopantetheine CoA thioesters and ability to transfer them onto PCPs.276 Similarly, acyl carrier protein synthase (AcpS), which recognizes an ACP domain, has also been used to label ACP-fused proteins with fluorescent tags277 or immobilize them onto resin supports.278 Although PCP and ACP have served well for labelling proteins as fusion constructs, they are relatively large (75 to 80 amino acids) compared to the tags employed in other enzymatic labelling approaches. To overcome that size limitation, Walsh and coworkers took advantage of phage display to screen for short peptide tags that can serve as replacements of PCP and ACP.163 From a genomic library derived from Bacillus subtilis, an 11-residue peptide denoted as ybbR (DSLEFIASKLA) was found to be the best substrate for Sfp-catalysed biotin-Ppant labelling. This tag can be incorporated onto the N- or C-terminus of proteins of interest or inserted into internal flexible loops, enabling efficient labelling in a single step (Scheme 9B). Later, the same group developed peptide tags S6 (GDSLSWLLRLLN) and A1 (GDSLDMLEWSLM) that manifested higher efficiency and specificity for Sfp- and AcpS-catalysed protein labelling, respectively.279 The orthogonality between these two peptide tags towards their corresponding PPTase allowed sequential labelling of two different target proteins performed in vitro or on cell surfaces. Furthermore, NMR-based experiments on these short peptide tags identified six key amino acids critical for interaction with the enzyme.280 That allowed the design of an 8-residue peptide, A4 (DSLDMLEW) containing 5 of the 6 key residues that displayed comparable efficiency to that of A1 in AcpS-mediated protein labelling.
Scheme 9.

PPTase-catalysed reactions for site-specific enzymatic labelling. (A) Proteins fused with PCP or ACP as recognition domains for modification of CoA derivatives. (B) Shorter tags (ybbR, S6, A1, and A4) can be inserted within exposed loops of the POI for internal modification.
Since 2014, the use of shorter peptide tags in the PPTase-catalysed protein labelling method has led to the efficient preparation of numerous protein conjugates for biomaterials, therapeutic, and imaging applications. The cargo of interest can be directly coupled with CoA and transferred to target proteins in one-step without the need for secondary conjugation reactions. Thus, direct attachment of fluorophores, affinity handles, drugs or macromolecules to proteins of interest is possible. For example, Gaub and coworkers designed Protein-DNA chimeras through Sfp-catalysed conjugation of ybbR-tagged GFP using CoA-modified DNA for single molecule analysis, enabling direct assembly of proteins via a Single-Molecule Cut & Paste technique (SMC&P).165 In SMC&P, the cantilever of an atomic force microscope (AFM) was used to transfer protein-DNA hybrids from a disordered state in the depot site and rearrange them at the target site. The DNA in the protein conjugates serves as an anchor for the cantilever tip.281 This technique allows for understanding complex networks of proteins that are spatially arranged on a platform. The covalent linkage between target proteins and DNA made from the Sfp-mediated approach improved protein-based SMC&P analyses. As a precise and efficient method, the utility of this protein-DNA coupling technique can be extended to the fields of nanobiotechnology and protein engineering. In the field of ADC construction, insertion of the peptide tags, ybbR and S6, into exposed loops of an antibody was explored to prepare a homogenous product.164 The cytotoxin auristatin was coupled to CoA through maleimide chemistry and loaded onto trastuzumab via Sfp-mediated reaction. Interestingly, it was found that conjugation on the CH1 domain resulted in favourable pharmacokinetics and minimal drug loss while attachment on the CH2 domain generally produced thermally unstable conjugates that resulted in rapid clearance. When various regions in CH1 were evaluated as modification sites, the resulting ADCs displayed similar in vitro potencies. In addition to being highly potent and selective in vitro, substantial tumour regression was also observed in a mouse xenograft tumour model. The versatility of this approach provides precise control of the site and stoichiometry of drug attachment in ADCs.
Protein modification with fluorescent reporters or affinity handles have been essential to study PPIs. Handel and coworkers fused an S6 tag to the C-terminus of several chemokines, followed by conjugation to fluorophore-containing CoA derivatives.282 Previous experiments using maleimide chemistry to prepare chemokine-fluorophores via genetically introduced cysteine residues led to protein aggregation and thus low yields.283 In contrast, PPTase-mediated labelling of three chemokines (CXCL12, CCL2, and CCL21) provided conjugates in high yields and allowed their interaction with the respective receptors to be studied using flow cytometry and cellular imaging. Catteneo and coworkers incorporated different PPTase recognition tags into neurotrophin and its receptors.284 Using a mutagenesis strategy, successful insertions of the A4 tag into nerve growth factor (NGF), as well as A1 and S6 into either tropomyosin kinase receptor A (TrkA) or p75 neurotrophin (P75NTR) receptor were achieved. It was shown that the purified NGF could be biotinylated with CoA-biotin catalysed by AcpS and remain functional. To image the two different receptors on the surface of a living cell, a sequential dual-color staining procedure was performed. Cells transfected with the two orthogonally-tagged receptors were first treated with CoA-biotin and AcpS, followed by incubation with streptavidin-quantum dot S-QDot 525. Then a second botinylation using Sfp was performed followed by incubation with SQDot 655. Distinct colors were observed under confocal imaging, indicating successful labelling. The ability to dually label two cell surface proteins could be potentially applied to study protein-receptor interactions involved in signal transduction pathways.
The combination of PPTase-based labelling with other enzymatic labelling methods has allowed for the development of FRET-based reporters. Schwarzer and coworkers combined PPTase- and SrtA-mediated labelling techniques to simultaneously incorporate GST and mCherry into a synthetic peptide bearing a fluorescein (FRET pair to mCherry).285 In another dual labelling strategy, Geierstanger and coworkers genetically encoded a C-terminal S6 tag and an internal pyroline-carboxy-lysine (Pcl) residue, in IgE to allow conjugation to different fluorophores for FRET study via enzymatic Sfp labelling and chemical oxime-ligation reaction, respectively.286 As a UAA known to be specifically reactive with a wide range of 2-aminobenzaldehyde-functionalized reagents,287 Pcl was genetically encoded in place of a surface-exposed proline. Based on the crystal structure of IgE, this site is suitable for FRET-based studies as it is close to the C-terminus in the open state (apo) but moves away upon binding the specific IgE receptor (holo state).286 After purification, the protein was dually labelled with Alexa Fluor 488 (donor)-CoA and 2-aminobenzaldyhe-Alexa Fluor 594 (acceptor). Efficient labelling with no cross-reactivity between the enzymatic and chemical modifications was achieved in a one-pot manner under near physiological conditions. The probe acted as a good FRET biosensor for evaluating IgE binding with its high-affinity receptor FcεRI, a key event involved in the allergic response cascade. A decrease in FRET efficiency was observed that correlated with increased intra- and inter-chain distances during the binding event in vitro. Since the Pcl and S6 tag modified proteins of interest can be expressed in mammalian cells, this method could be useful for site-specific dual labelling of complex proteins that are resistant to recombinant expression in bacterial hosts.
The development of PPTase-mediated protein labelling in concert with the discovery of shortened enzyme recognition tags has greatly facilitated the preparation of functional protein conjugates. In contrast to other enzymatic labelling strategies that are restricted to either termini of a protein, labelling by PPTase is more versatile with regards to the modification site within the protein sequence. However, limitations on solubility and cell permeability of the synthetic CoA analogues may compromise its application in in vivo studies. Chemoenzymatic approaches have been explored in site-specific protein labelling of PCP- and ACP-fused proteins in vivo using smaller synthetic substrate analogues.288,289 In this approach, the analogues are synthesized as D-pantetheine derivatives, which are then metabolically converted to CoA-analogues inside the host, catalysed by three promiscuous enzymes involved in the CoA biosynthetic pathway: pantothenate kinase (CoAA), phosphopantetheine adenylyltransferase (CoAD), and dephospho-CoA kinase (CoAE).290 Recently, an Sfp mutant was employed that could accept substrate analogues without the need for CoA phosphorylation by CoAE, which is the problematic step in the CoA biosynthetic pathway.291 In vivo labelling with PPTases through chemoenzymatic methods has thus far been limited to proteins fused with large PCP and ACP domains. Extending this method to labelling proteins inserted with the short peptide tags has yet to be explored but should prove fruitful.
3.3. Ligase
3.3.1. Tubulin tyrosine ligase
In eukaryotic cells, tubulin tyrosine ligase (TTL) catalyses the ATP-dependent addition of a tyrosine residue to the C-terminus of α-tubulin forming a native peptide bond (Scheme 10A).292 Together with an uncharacterized carboxypeptidase, the C-terminus of α-tubulin is subjected to a post-translational process where a tyrosine residue can be reversibly added and removed, which is important in regulating microtubule function.293 TTL suppression has been reported to be associated with progression of neuroblastoma,294 and even in normal cells, TTL has been shown to play a vital role in controlling neuronal organization.295
Scheme 10.

TTL-mediated attachment of tyrosine analogues (A) and functionalized glycines (B) to the POI engineered with a C-terminal Tub-tag.
Soon after the discovery of this enzyme, it was found that TTL-mediated labelling of α-tubulin is not limited to the native tyrosine substrates, but can be extended to functionalized tyrosine analogues, which prompted biotechnological applications of TTL. 3-Fluorotyrosine was one of the first tyrosine-derivatives found to be accepted by the enzyme.296 Enzymatic incorporation of a fluorine into the target protein allowed the use of 19F NMR to study the PPIs that occur near the C-terminal region of α-tubulin. While studying the effects of nitrotyrosination on tubulin, Arce and coworkers found that nitrotyrosine, which is naturally present in cells, could be reversibly incorporated into tubulin by TTL and removed by the carboxypeptidase without detrimental effects to cells.297 Years after this report, Bane and coworkers were able to extend the range of tyrosine analogues for protein labelling on live cells.298 By measuring the degree of inhibition of TTL-mediated [3H]-L-tyrosine incorporation into tubulin, 3-formyltyrosine and 3-azidotyrosine were identified as acceptable substrates with efficiencies lower than the native tyrosine but comparable to that of 3-nitrotyrosine. Since the use of azides often involves the CuAAC reaction which requires the use of copper that may have negative effects on tubulin assembly, initial efforts focused on 3-formyltyrosine as a model system. When reacted with a synthetic coumarin-hydrazine, the hydrazone formation reaction with 3-formyltyrosine was completed within 120 min with a second-order rate constant of 53 M−1min-1. Interestingly, hydrazone formation through a covalent linkage led to a red shift of both the absorption and emission maxima of the coumarin, as well as an increased quantum yield, which allowed the conjugates to be detected even in the presence of unreacted fluorophore. This unique bathochromic shift was useful in later cell labelling studies since it obviated the need for medium exchange (to remove unreacted probe) after incubating cells with fluorophore-probes. Importantly, fluorophore labelling using this method did not hinder polymerization of tubulin and was found to be a suitable method for live cell imaging. Incubation of coumarin hydrazine with CHO cells grown in 3-formyltyrosine containing medium led to strongly fluorescing cells. Western blotting analysis confirmed that the probe was selectively linked to α-tubulin.
Recently, to expand the scope of TTL-catalysed labelling of proteins other than tubulin, Leonhardt and coworkers set out to find a sequence that can be efficiently recognized by the enzyme (Tub-tag).68 A 14-residue Tub-tag peptide which mimics the C-terminal sequence of tubulin was initially discovered to be an acceptable substrate using 3-L-formyltyrosine or 3-N3-L-tyrosine and a recombinant TTL. For protein labelling studies, two types of GFP-specific nanobodies (GBP1 and GBP4), GFP, and ubiquitin were prepared, all containing the Tub-tag sequence at the C-terminus. Importantly, the insertion of Tub-tag did not affect the fluorescence of GFP, and the engineered nanobodies retained their binding ability, indicating minimal perturbation of the protein structure and function. A wide range of bioorthogonal reactions were explored exploiting the introduced azide or aldehyde moiety, including SPACC, Staudinger ligation, and hydrazone- and oxime-forming reactions. The two-step modification method was also shown to be highly selective even when the labelling was performed in E. coli lysate. To demonstrate the utility of TTL-mediated ligation towards enrichment and isolation of target proteins from crude lysate, GBP1-biotin was prepared by sequential incubation with 3-N3-L-tyrosine and DBCO-biotin, followed by immobilization on streptavidin-coated magnetic beads. Western blotting was used to confirm specific GFP pulldown from HEK cell lysates. Applicability for immunostaining was highlighted using GBP1 labelled with Alexa Fluor 594 (GBP1-Alexa594) via TTL-mediated ligation and oxime formation. HeLa cells expressing GFP-LamininB1 that localizes in the inner membrane of the nucleus were fixed, permeabilized and stained with GBP1-Alexa594. Super-resolution microscopy showed colocalization of the GFP and Alexa Fluor 594 signals, indicating successful binding of TTL-modified nanobodies to GFP in the intracellular environment.
More recently, Hackenberger and coworkers explored the substrate scope of TTL. Exploiting the plasticity of the TTL active site, they were able to achieve one step protein labelling with biotin and a fluorescent coumarin using the wild-type enzyme without any necessary mutation of the active site.167 Initial labelling efficiencies tested on a carboxyfluorescein-labelled C-terminal Tub-tag peptide showed that while ortho-substituted tyrosine derivatives become fully ligated, substrates with para-substituents including an azide or alkyne are not well-recognized by the TTL. For hydrophobic amino acids, labelling was found to be especially efficient for those bearing aromatic rings. Importantly, the carboxylic acid group of the substrate amino acid was found to be essential for enzyme recognition. Based on these observations, the TTL activity towards functionalized amino acids was tested (Scheme 10B). Fluorescent molecules including a coumarin-functionalized glycine and β-(1-azulenyl)-L-alanine were found to be acceptable substrates. A biotinylated tyrosine that contained an ethylene glycol linker to provide flexibility and liberation from the spatial constraints of the active site was also found to be successfully ligated. To understand this substrate preference of TTL, two types of computational studies were carried out. Both studies led to a conclusion that interactions in the active site of enzyme stabilized ortho-substituted tyrosines due to spatial conditions as well as hydrophobic molecules with ring structures due to π-interactions. A solvent-exposed channel extending out of the active site was also observed, which was consistent with the experimental result where labelling was accomplished with the biotin-functionalized tyrosine analogue. Based on this insight, one-step fluorescent labelling was performed on various functional proteins engineered with a Tub-tag at their C-termini. Incubation of cells expressing GFP-fusion proteins with GBP-coumarin showed colocalized signals of GFP and coumarin, demonstrating the utility of the one-step labelling method for cellular imaging. Additionally, annexin V, an apoptosis marker protein, was labelled with coumarin through the one-step process and showed comparable results for visualizing apoptotic cells to those obtained using commercial probes. This demonstrated the potential of this method to improve the performance of currently available probes, which are partially limited by their product heterogeneity that results from the non-specific modification process used for their preparation.
In summary, TTL has been applied to site-specifically modify proteins with various functionalities, allowing a broad range of bioorthogonal reactions to be selected for conjugation to additional cargos. Interest in TTL is starting to re-emerge due to the recent findings that the wild-type enzyme itself can accept a broad range of substrates.167 Understanding the mechanism that governs the substrate-enzyme interactions has enabled one-step TTL-mediated labelling using functional probes of the protein of interest and has made TTL a powerful tool. Combination with enzyme engineering is expected to allow labelling of an even broader range of substrates to facilitate novel applications.
3.3.2. Lipoic acid ligase
Lipoic acid ligase (LplA) recognizes a specific LplA acceptor peptide (LAP) and catalyses the attachment of a lipoate moiety to a lysine residue in LAP through an ATP-dependent reaction (Scheme 11), which was first recognized by Koike and coworkers.299 LplA was later found to be capable of recognizing lipoate analogues,300,301 which prompted studies focused on using this enzyme for site-specific labelling. Members of the Ting lab initially showed that azide-containing analogues could be successfully incorporated into the LAP peptide using LplA. Fusion of the LAP peptide to cell-surface proteins, followed by labelling with LplA and subsequent conjugation with cyclooctyne-probes allowed selective protein imaging of live cells.168 While initial studies required a 22 amino acid LAP peptide for LplA recognition, further studies employing yeast surface display selection led to the discovery of a 13 residue LAP, GFEIDKVWYDLDA, that has higher catalytic efficiency and reduced capacity for protein structure disruption, which therefore has been used as the standard LAP since then.302
Scheme 11.

LplA-mediated protein labelling using lipoate analogues bearing various bioorthogonal functional groups to a POI containing an engineered C-terminal LplA acceptor peptide.
Since 2013, the scope of lipoate analogues employed has been significantly broadened, leading to a much wider range of compatible biorthogonal reactions that can be used for LplA-mediated labelling. Wombacher and coworkers were able to attach p-iodophenyl derivatives to peptides or proteins bearing the LAP motif using LplA, followed by palladium catalysed Sonogashira cross-coupling between the labelled aryl halide moiety and an alkyne-probe to prepare site-specific protein-fluorophore conjugates.170 Since these analogues are bulkier than the native lipoate substrate, four mutant LplA constructs were also prepared with an enlarged binding pocket. In accordance with previous reports, mutation of W37I was found to be the most crucial mutation for the enzyme to accept lipoate analogues. Using the LplA W37V mutant, a two-step labelling was performed on dihydrofolate reductase from E. coli (eDHFR) which contains an N-terminal LAP to create a fluorescently labelled form of DHFR. However, non-specific labelling of the alkyne-probe on halide-free DHFR was also observed, which was likely due to palladium catalysed thiol-yne coupling. Although this could be prevented by blocking all free thiols with excess N-ethylmaleimide, development of new palladium catalysts with higher selectivity is desirable. More recently, a series of norbornene-bearing lipoate analogues was also developed, which can be used in tetrazine ligation reactions.95 A small library of analogues was prepared to select the most efficient substrate. Interestingly, overall conformational flexibility was found to be as important as the length of the derivatives in determining enzyme reactivity. Using the LplA W37V mutant enzyme, tetrazine-fluorescein was successfully conjugated to the norbornene-labelled eDHFR containing an N-terminal LAP-tag, in purified form or when diluted into cell lysate. Live-cell labelling was also achieved with HEK293T cells that were transiently transfected with pDisplay-LAP2-CFP-TM fusion protein. After expression, the construct localized to the plasma membrane exposing the extracellular LAP-tag and was then modified by LplA W37V mutant. Incubation with tetrazine-TAMRA showed colocalization of the cyan fluorescent protein (CFP) signal and TAMRA fluorescence with minimal background, highlighting the site-specific advantages of LplA-mediated labelling and the copper-free conjugation conditions of tetrazine ligation. More recently, an analogue containing 18F was also developed, enabling one-step modification of an Fab fragment, modified by LAP-insertion, with radio-isotopes that can be potentially useful for diagnostic imaging.303
Recently, in addition to modifications of protein termini, labelling by LplA has been developed to functionalize internal sites, even simultaneously at multiple sites.169 In the first report of exploiting LplA for labelling internal-sites by inserting LAP into loop regions, a series of GFP constructs was prepared. When the expression level of different constructs was compared, it was found that the LAP-containing GFPs were expressed at an extent comparable to that of the wild-type GFP, as evaluated by the normalized intrinsic fluorescence of GFP in crude E. coli lysate. More importantly, LAP-constructs showed higher expression than GFP containing two azide-functionalized UAA residues (AzF-GFP). The superior expression levels observed underscore the advantages of protein labelling using enzymatic methods over the incorporation of UAAs, especially when modification of multiple sites is desired. After incorporating 10-azidodecanoic acid into GFP constructs using LplAW37V, subsequent PEGylation was performed by SPAAC using DBCO-PEG (Mw 5 kDa), whose conversion yield, and product homogeneity all exceeded that of non-specifically PEGylated wild-type GFP. The utility of the method was further demonstrated by glycosylation, fatty acid modification, and surface immobilization of GFP. Interestingly, a unique uniform orientation was obtained, which was different from those labelled at the termini.169 This opens up the possibility of creating protein chips that require particular orientations of proteins, which are not accessible by labelling through either terminus.
McNaughton and coworkers recently applied LplA-mediated labelling for activation of immunotherapeutics by enzymatically linking an antibody recruiting domain (ARD) to a cell binding domain (CBD).69 An aldehyde-bearing lipoate analogue was incorporated into a HER2 binding nanobody (denoted as 5F7) that contains an N-terminal LAP using LplAW37I. Dinitrophenyl (DNP)-functionalized hydrazine was then conjugated to form a chimeric construct where the DNP acts as an ARD, and the 5F7 a CBD. Successful target binding was confirmed by confocal imaging with SK-BR-3 cells that overexpress HER2. To test the therapeutic capability of DNP-5F7, SK-BR-3 cells were incubated with DNP-5F7, anti-DNP antibodies, and peripheral blood mononuclear cells (PMBCs), which induced cytotoxicity when they interact with antibodies. Cell death was shown to be selectively induced on HER2 expressing cells with good potency (EC50 60 nM).
In summary, a variety of applications have been achieved using LplA-mediated enzymatic protein labelling followed by subsequent bioorthogonal reactions. Particularly, recent developments that allow site-specific labelling of internal regions of a target protein opens up new possibilities for situations where N- or C-terminal labelling is less desirable.
3.3.3. Biotin ligase
Biotin ligase catalyses the conjugation of biotin derivatives onto proteins in an ATP-dependent manner. In their seminal report, Ting and coworkers used an E. coli biotin ligase (BirA) to label biotin-derivatives to target proteins through the lysine residue of an acceptor peptide (AP), which consists of 15 amino acids (GLNDIFEAQKIEWHE), appended onto the N- or C-terminus of a protein (Scheme 12A).171 By exploiting a ketone moiety in a conjugated biotin analogue, they were able to attach a photoactive hydrazide reagent to cyan fluorescent protein-AP using oxime chemistry. Protein labelling on the surface of live cells was also shown, where the biotinylated target proteins were visualized using streptavidin-Alexa Fluor 568. In a subsequent report, Ting and coworkers expanded the range of tolerable substrates by screening various biotin analogues against biotin ligases from nine different microorganisms.172 Pyrococcus horikoshii biotin ligase (PhBL) was found to promote the ligation of azide- and propargyl-functionalized biotin analogues when evaluated on a purified domain of an endogenous biotin acceptor protein (p67). Using the Staudinger ligation, azide-p67 was conjugated to a phosphine-FLAG peptide. The ability to functionalize target proteins with analogues bearing bioorthogonal moieties highlights the utility of biotin ligase for the construction of protein conjugates.
Scheme 12.

Biotin ligase mediated-protein labelling. (A) Protein labelling using biotin ligase with biotin analogues that contain bioorthogonal functional groups. B. Biotin ligase from S. tokodaii forms a complex with its biotin-modified protein substrate bearing the BCCP domain.
A unique advantage of biotin incorporation is that it can spontaneously associate with streptavidin with extremely high affinity, which allows conjugation to be performed under protein- and cell-compatible conditions.304 Taking advantage of this association, the biotin ligase labelling technique has been applied to study PPIs. In work performed by Ting and coworkers, two protein constructs were created, one target protein fused to BirA, and the other one modified with an enzyme recognizable AP. The interaction between these two proteins was gauged by measuring the extent of biotinylation on the AP-protein using a streptavidin-probe.305 As expected, a PPI between a well-studied protein pair consisting of FKGP and FRB, which dimerizes in the presence of rapamycin, was detected by incubation of FKGP-AP and FRB-BirA with biotin and rapamycin, followed by streptavidin blotting. However, due to the high concentrations of FKGP and FRB in live cells that nearly reach the KM of AP (25 μM) for BirA, a PPI was detected independent of rapamycin in live HEK cells. Therefore, the AP was truncated at the C-terminal end to reduce its affinity with BirA so that it would only be modified when present in close contact with the enzyme. This strategy led to a successful detection of rapamycin-dependent biotinylation. Evaluation of a phosphorylation-dependent PPI between Cdc25C phosphatase and 14–3-3ε phosphoserine/threonine binding protein was performed to demonstrate the general utility of the method. Incubating mCherry-AP-Cdc25C constructs with BirA-14–3-3ε in the presence of biotin, followed by the addition of streptavidin-Alexa Fluor 647 showed colocalized signals of the two fluorophores, indicating successful detection of the PPI. Mutation of Cdc25C that blocked phosphate binding led to the disappearance of the Alexa Fluor 647 signal, confirming that the visualized PPI was indeed a result of Cdc25C and 14–3-3ε interaction.
The biotinylation by BirA is a two-step process, in which biotin and ATP first form a biotinoyl-5’-AMP intermediate, which remains inside the enzyme active site until it reacts with the lysine residue of the AP. By mutating BirA so that it releases the highly reactive biotinoyl-AMP species into its surroundings, Burke and coworkers were able to induce proximity-dependent biotinylation of lysine residues on proteins neighbouring the BirA-fused target protein, enabling PPI identification in the native cellular environment.306 This technique was denoted as BiolD. To illustrate its applicability, a genetic fusion of the mutant BirA to lamin-A protein was created, which allowed identification of proteins in close contact with lamin-A in live HEK cells. More recently, Roux and coworkers improved the proximity-identification method by developing a smaller enzyme that led to more selective and efficient labelling of neighbouring proteins (BiolD2 approach).175 As the smallest biotin ligase known to date, the enzyme from Aquifex aeolicus (Aabiotin ligase) was employed. To make it usable for proximity-dependent biotinylation, the enzyme was humanized and mutated within the biotin catalytic domain (R40G) to release the reactive intermediate. The engineered Aabiotin ligase mutant was superior to the previously employed BirA enzyme, as it could be used under a wider range of temperatures and detect proteins that were otherwise difficult to identify before. Additionally, it required less biotin substrate due to its higher efficiency. It was also shown that the “biotinylation range” (the distance between the BirA-fused target protein and the protein which is biotinylated via lysine modification) could be increased by inserting a flexible peptide linker between the protein being studied and the fused biotin ligase.
Site-specific protein labelling has been demonstrated using biotin ligases functionalized with various probes, which is distinctive from other enzymatic labelling methods in that the enzyme itself becomes covalently attached to the target of interest (Scheme 12B). Hayashi and coworkers developed a labelling method that involved biotin ligase from Sulfolobus tokodaii (Stbiotin ligase), which forms a stable complex with the biotinylated form of biotin carboxyl carrier protein (BCCP). The Stbiotin ligase first catalyses the biotinylation reaction of BCCP and then associates with the resulting product. Exploiting this unique property, Hayashi and coworkers used Stbiotin ligase for fluorescence imaging of cells.173 Fluorophore-labelled biotin ligase was prepared by mutating R152 to a cysteine, followed by reaction with a maleimide-functionalized fluorescein or DyLight549. In the presence of biotin, this modified Stbiotin ligase was then attached to the bradykinin B2 receptor (B2R), a membrane protein expressed in HEK293 cells, that had a truncated form of BCCP (69 residues) appended onto the N-terminus. Visualization of the cell surface due to fluorescently labelled BR2 was observed by confocal microscopy. To demonstrate the utility of the method for labelling within cells, BR2 that contained a C-terminal BCCP was coexpressed with GFP-Stbiotin ligase in HEK293 cells. The GFP signal was observed on the plasma membrane (on the luminal side) upon biotinylation, which indicated successful labelling.
Recently, the complex formation of Stbiotin ligase and BCCP was utilized to facilitate oriented immobilization of antibodies onto the surface of a sensor chip.174 A sequential procedure was used, starting with immobilization of Stbiotin onto a gold surface. To make the BCCP-interaction site accessible, N151 and R152 located on the opposite side of biotin ligase were replaced with cysteine, which was be adsorbed onto the surface via thiol-gold interactions. Next, either one or two copies of the synthetic IgG-binding domains from protein A, Z-domain, was fused to the N-terminus of BCCP and was immobilized onto the gold surface upon biotinylation. The Stbiotin ligase-BCCP complex positioned between the Z-domain and the surface acted as a rigid spacer to promote antibody capture. The efficiency of the prepared sensor chips to capture antibodies was evaluated by monitoring changes in frequency from the sensor readout as anti-GFP antibodies were introduced followed by GFP. It was shown that the frequency change caused by GFP rose from specific antigen-antibody interactions, which demonstrated successful capture of anti-GFP antibodies by the immobilized Z-domain. Along with a dose-dependent response to GFP, the designed sensor showed high sensitivity that allowed detection of concentrations as low as 0.1 nM GFP, which was impossible to achieve when Z-domains were directly attached to the surface via cysteines.
Overall, some features of biotin ligase are unique compared to other enzymatic labelling systems. Instead of only serving as a catalyst, the enzyme can be more involved in the reaction. For example, it can be fused to the protein of interest or form a complex with its biotinylated protein substrate. While most enzymes focus on improving the substrate specificity, the studies described above have often involved mutating biotin ligase to render it more promiscuous so that labelling is not sequence-specific but distance-dependent for proximity sensing. Given these unique features, it is likely that more applications will be reported in the future.
3.4. Oxidoreductases
Formylglycine generating enzyme.
Formylglycine generating enzymes (FGEs) catalyse the post-translational modification of type 1 sulfatases by oxidation and subsequent hydrolysis of the thiol moiety of cysteine within the CXPXR motif to an aldehyde-bearing formylglycine (FGly) (Scheme 13). The enzyme was first identified by Figura and coworkers when studying diseases related to sulfatase deficiency.307 Subsequently, Bertozzi and coworkers exploited FGE for site-specific protein modification.65 In their pioneering work, two types of aldehyde tags were tested on three different recombinant proteins expressed in E. coli. One of the tags was a full-length sulfatase motif that consisted of 13 amino acids (LCTPSRGSLFTGR) and the other a shorter tag (LCTPSR) which contained only the core conserved residues. Aldehyde formation on the cysteine residue inside the tag was shown to occur regardless of whether the tag was present at either the C- or N-terminus of the protein substrate. Subsequently, site-specific protein modification was performed by reacting an aminooxy-functionalized affinity tag, fluorophore or PEG with the formylglycine residue, demonstrating the generality of the FGE-mediated labelling technique. In a follow-up study, peptide library screening was conducted to identify new aldehyde tag sequences that would minimize any perturbation of the target protein structure.308 Noncanonical sequences including LCTASR and LCTASA were found to be recognized to a similar degree compared to the canonical sequence of LCTPSR by Mycobacterium tuberculosis FGE (MtFGE) in vitro and by endogenous FGE-like activities in E. coli. Soon after, Bertozzi and coworkers expanded their methodology of protein labelling in prokaryotic cells to mammalian cells,177 which was an important improvement since many therapeutic proteins are expressed in mammalian cells due to the need for them to be post-translationally modified. They showed that an IgG Fc fragment expressed in CHO cells showed conversion of cysteine to formylglycine by endogenous FGE, with efficiencies ranging from 25–67% by having a 13-(LCTPSRAALLTGR) or 6-residue tag (LCTPSR) appended at either the N- or C-terminus. The efficiencies were significantly improved by coexpressing human FGE along with the engineered Fc proteins, indicating that insufficient levels of endogenous FGE relative to the overexpressed Fc proteins was a limiting factor. Multiple proteins bearing the aldehyde tag were shown to be modified selectively using this method. Overall, in the early development stages of FGE-mediated labelling, broadening the range of aldehyde tags, exploring different protein expression and labelling systems, testing various protein species, and confirming the selectivity of the subsequent conjugation reactions with the inserted aldehyde moiety were the major focuses, laying a solid foundation for later biotechnological development.
Scheme 13.

FGE catalysed conversion of a cysteine to an aldehyde-bearing formylglycine residue. Labelling occurs in E. coli by coexpression of the tag-fused POI and FGE. The aldehyde functional group serves as a reactive handle for Subsequent Comjugation.
Site-specific protein glycosylation is important for understanding the role of glycosylation in many key biological processes and developing protein-based therapeutics. Bertozzi and coworkers were able to apply FGE-mediated labelling for site-specific protein glycosylation, by inserting an aldehyde tag into the target protein, followed by oxime formation with complex aminooxy-glycans that were synthesized through a novel synthetic route.309 A total of three complex glycans were synthesized. To label these complex glycans, a formylglycine-containing human growth hormone (hGH) was produced in E. coli by coexpression with MtFGE. Slightly increasing the acidity of the reaction buffer was found to improve the oxime ligation yield to allow more efficient coupling of the glycans to the target proteins without noticeable detrimental effects on either reactant. Since glycosylation sites in most proteins are located at internal positions instead of at the termini, Bertozzi and coworkers explored the labelling by FGE on proteins engineered with an internal enzyme recognition motif.310 Three recognition motifs were evaluated and incorporated into an Fc fragment. Comparing the expression levels of each construct in CHO cells and subsequent conjugation efficiencies with aminooxy-Alexa Fluor 488, the CTPSR motif was selected as it minimized effects on protein structure and had the highest modification level (76%). After optimizing the reaction conditions using O-benzylhydroxylamine, near quantitative oxime conjugation was obtained.
Over time, improved conjugation chemistry for aldehyde moieties has been developed, thereby increasing the power of FGE-mediated labelling. Oxime and hydrazone chemistries are the most commonly used methods for aldehyde-bearing proteins. However, the resulting C=N linkage favours acidic reaction conditions and is prone to hydrolysis, which limits the utility of these reactions to create conjugates intended to be used in biological systems that require long-term stability at physiological temperatures. Bertozzi and coworkers recently developed a modified Pictet-Spengler reaction (Scheme 1B), which provides ligation products with improved stability over conventional aldehyde-involving linkages.62 In a canonical Pictet-Spengler reaction, a tryptamine derivative reacts with an aldehyde to form a stable C-C bond. However, the reaction rates were very slow under protein-compatible conditions. To overcome this limitation, the aliphatic amine of tryptamine was replaced with a methylated-aminooxy moiety and was moved to the 2-position of indole to expose the more nucleophilic 3-position for subsequent reaction. The modified reaction, denoted as the Pictet-Spengler ligation, manifests rates that are 4–5 orders of magnitude faster than the conventional method in aqueous media at pH 4.5. The ligated product was shown to be stable after 5 days of incubation under aqueous conditions at pH 4.5 or 5.0. In contrast, the majority of oxime-linked reagents hydrolysed within 2 days. An anti-HER2 human IgG conjugated to Alexa Fluor 488 (AF488-α-HER2) was then prepared by combining FGE-mediated labelling and Pictet-Spengler ligation, resulted in fluorophore-labelled antibodies with preserved affinity for HER2, illustrating the applicability of this chemistry for modifying functional biomolecules.
In a subsequent report, Agarwal et. al introduced the hydrazino-Pictet-Spengler (HIPS) ligation, which was a further improved version of the Pictet-Spengler reaction, displaying fast reaction rates even under neutral conditions with long term hydrolytic stability.63 In this method, an alkylhydrazine-functionalized indole was used as the nucleophile that reacted with an aldehyde, which allowed rapid reaction rates at near neutral pH (pH 6.0). This new reaction was also utilized to construct antibody-fluorophore conjugates. Notably, when incubated in human plasma, the HIPS-conjugated protein showed no hydrolysis as verified by monitoring the loss of Alexa Fluor 488 fluorescence, while conjugates employing oxime-linkages deteriorated in a single day. More recently, a new carbonyl ligation method termed the trapped-Knoevengael ligation with faster reaction rates at pH 7 was introduced by Kudirka et al., further expanding the reaction scope that can be coupled to the FGE labelling technique.64
The combination of FGE modification and HIPS chemistry was used to study the effect of DAR and the conjugation site on the in vivo efficacy and pharmacokinetics of ADCs.176 Several modification sites were first evaluated, where three of them (the light chain, CH1 domain, and heavy chain C-terminus) were identified to minimize immunogenicity and aggregation. Benchmarking the clinically approved drug T-DMI, the antibody trastuzumab was site-specifically labelled with the cytotoxin DM1 at the three chosen sites using a combination of FGE modification and HIPs chemistry. Interestingly, while the fluorophore conjugates were shown to be stable, the DM1 conjugates were shown to lose their payload over time, to a varying degree depending on the conjugation site. This difference in stability was attributed to the labile ester bond that connected the DM1 payload to the aldehyde-reactive indole. Different efficacy as well as stability was observed when the conjugates were evaluated in tumour-bearing mouse xenografts. These results indicated that the distinct microenvironments at each site have unique influences on the performance of the corresponding ADC. Importantly, mice treated with the site-specifically created ADCs showed better overall improvements in mortality, body weight loss, liver toxicity and tissue damage in comparison with control constructs prepared by conventional lysine chemistry, emphasizing the value of controlled conjugation methods and linker chemistry on improving ADC effectiveness.
More recently, Rabuka and coworkers showed potential to increase DAR even higher by introducing a tandem Knoevenagel condesation and Michael addition strategy that allows conjugation of two payloads per site.311 In this method, a formylglycine-containing antibody is treated with pyrazolone-functionalized maytansine, where Knoevenagel condensation of the first pyrazolone-nucleophile occurs to generate a reactive enone that can undergo Michael addition with a second pyrazolone. Even with hydrophobic payloads in close proximity, Rabuka was able to achieve high efficacy and phamacokinetics by controlling the conjugation sites.
As a powerful protein labelling tool, FGE-mediated protein modification has been developed as a commercial platform (SMARTag™ technology) by Redwood Bioscience/Catalent for the construction of site-specific ADCs.312 The fact that labelling can be performed by coexpressing the tag-fused proteins of interest with FGE obviates the need for additional incubation steps which makes the method convenient while the plethora of bioorthogonal reactions that have been developed for aldehyde conjugation provides a versatile repertoire of chemistries.
Conclusions
Overall, a wide variety of protein conjugates have been created using enzymatic labelling methods. These conjugates have been applied in a diverse array of applications, including the study of protein sub-cellular localization and PPIs, surface functionalization for biochip and bio-responsive materials development, and the construction of next-generation protein therapeutics with enhanced pharmacokinetics and potency. Table 1 summarizes some of the key characteristics for the enzymatic modification methods discussed in this review and their applications. Amongst all these enzymatic methods, which collectively act as a powerful tool box, there is no one perfect solution without limitations. Understanding their respective strength and disadvantages can help to select the optimal technique to meet specific requirements. To label a given target protein, it is necessary to consider the effect of the engineered tag sequence and the location of the modification on the structure and function of the protein. Reaction kinetics also play an important role when limited substrate is supplied as well as when the target protein is sensitive to reaction conditions during long incubation times.
Table 1.
Compatible bioorthogonal functional groups with different enzymatic labelling methods and their applications
| Enzyme | Tag sequence | Labelling site | substrates | KM* μM | kcat* min−1 | kcat/KM* min−1 μM−1 | Bioorthogonal functional groups | Applications |
|---|---|---|---|---|---|---|---|---|
| Sortase A | LPXTG | C-terminus, internal sites | G(n) peptides, primary amines, hydrazides | 140 | 90 | 0.64102 | Azide,129 terminal alkyne,129 TCO,93 BCN,133 DBCO,139 | One-step labelling with cargos, conjugation to small molecules,93 polymers141 and polypeptides,142 protein semisynthesis,3 surface immobilization,113 in vivo labelling,130 cell-surface labelling127 |
| (GGG peptide) | ||||||||
| G(n) | N-terminus | LPXTG peptides | 7600 | 90 | 0.012102 | |||
| Subtiligase | - | N-terminal -NH2 | Peptide esters | 620 | 1260 | 2143 | Azide144 | One-step labelling with cargos, conjugation to small molecules,144 protein semisynthesis145 |
| (s-AAPF-glc-PG-amide peptide) | ||||||||
| Microbial transglutaminase | Q tag (LLQG) | Any site | Lysine peptides, Primary amines | - | - | - | Azide,146 terminal alkyne,147 DBCO,92 tetrazine,92 triphosphine92 | One-step labelling with cargos, conjugation to small molecules,148 polymers149 and polypeptides,150, surface immobilization147 |
| K tag (MKHKGS) | Glutamine peptides | 52660 | 2128 | 0.04151 | ||||
| (Z-QG) | ||||||||
| Farnesyltransferase | CaaX | C-terminus | Isoprenoid analogues | 1.71 | 31 | 1866 | Azide,151 terminal alkyne,152 TCO,96 aldehyde,66 ketone,153 | Conjugation to small molecules,154 polymers,66 polypeptides155 and oligonucleotides,156 surface immobilization157 |
| (FPP) | ||||||||
| N-myristoyltransferase | GNEASYPL | N-terminus | Myristic acid analogues | 5159 | - | - | Azide,159 terminal alkyne,160 ketone67 | Conjugation to small molecules,161 surface immobilization,162 in vivo labelling159 |
| (CoA-myristate) | ||||||||
| Phosphopantetheinyl transferase | GDSLSWLLRLLN | Any site | CoA derivatives | 60.8 | 14.7 | 0.242164 | - | One-step labelling with cargos, conjugation to small molecules164 and oligonucleotides,165 surface immobilization,163 cell-surface labelling166 |
| (CoA-biotin with ybbR tag by Sfp) | ||||||||
| Tubulin tyrosine ligase | Tub tag (VDSVEGEGEEEGEE) | C-terminal -COOH | Tyrosine analogues | - | - | - | Terminal alkyne,167 azide,68 halides,167 aldehyde,167 nitro167 | Conjugation to small molecules68,167 |
| Functionalized-glycine | ||||||||
| Lipoic acid ligase | GFEIDKVWYDLDA | Any site | Lipoic acid analogues | 4.5 | 15 | 3.3169 | Azide,169 terminal alkyne,168, norbornene,95 aldehyde,69 iodine,170 TCO,94 | Conjugation to small molecules 69 and polymers,169 surface immobilization,169 cell-surface labelling168 |
| (lipoic acid with E2p domain) | ||||||||
| Biotin ligase | GLNDIFEAQKIEWHE | N- or C-terminus | Biotin analogues | 0.3 | 18 | 60172 | Ketone,171 azide,172 terminal alkyne172 | Conjugation to small molecules,173 surface immobilization,174 cell-surface labelling,171 proximity labeling175 |
| (biotin with BCCP domain by BirA) | ||||||||
| Formylglycine generating enzyme | CXPXR | N- or C-terminus | - | - | - | - | Aldehyde65 | Conjugation to small molecules176 and polymers,65 cell-surface labelling,177 in vivo labelling177 |
The kinetic parameters are values reported for selected “small molecule” substrates from the “substrates” column. In particular, the kinetic parameters for protein substrates with different tag sequences are not provided.
In addition, protein conjugation to a desired cargo can be achieved in either one step or two steps using different enzymes. For the one-step approach, the desired cargo is first incorporated into an enzyme substrate and then transferred to the protein of interest by the enzyme in a single step. Examples of this include the preparation of protein-polymer conjugates using MTG or ADCs by SrtA, MTG and PPTase. For smaller functional moieties such as the coumarin fluorophore and biotin, one-step labelling can also be achieved with FTase, TTL and LplA. In the latter situation, some of the analogues were recognized by the wild-type enzyme, while others required an engineered protein to accommodate these bulkier substrate analogues. In some cases with larger substrates, the labelling yield may vary in a case dependent manner. Optimization of the reaction conditions is needed to achieve the best outcome.
In contrast to the one-step approach, the stepwise conjugation method is applicable to all the enzymes discussed above. The protein of interest is first labelled with a substrate analogue bearing a bioorthogonal functional group by the enzyme. Depending on the functional groups installed, subsequent reaction with their complementary reactive partner appended to cargo yields the desired protein conjugates. The functional groups that have been incorporated into substrate analogues for each enzyme are summarized in Table 1. Small modifications, such as the azide functionality, are compatible with most enzymes while the bulkier TCO or DBCO functional groups can only be employed by those with a larger substrate binding site. Using this modular approach, a diverse range of cargos can be incorporated into the target protein. More importantly, due to the high utility of the bioorthogonal reactions, a large collection of derivatized cargos are commercially available, making the conjugation process easy to access. However, the step-wise procedure typically requires an additional purification step, which leads to the loss of material. The final conjugation product may also contain multiple unreacted protein species that further complicates the purification procedure.
In addition to the intrinsic properties related to the specific enzymes, purification processes also affect the efficiency of protein conjugate production. For SrtA, NMT, PPTase and FGE, protein modification can be achieved in E. coli by coexpression of the target protein and the enzyme. The modified protein can be directly purified from the cell lysate eliminating the effort to prepare the enzyme and the target protein separately by chromatographic methods. It should be noted that for SrtA, a high concentration of the primary amine substrates is often needed to achieve acceptable labelling yields. Taking advantage of the easily separated microbeads, surface immobilized MTG has been developed to facilitate the removal of the catalyst from the desired protein conjugate. A capture and release strategy based on aldehyde reactivity and FTase labelling was also reported so that only the conjugated protein is eluted from the capture beads without the need to separate the unlabelled protein by chromatographic methods. Several one-pot procedures enabling site-specific modification and purification of the protein conjugates using SrtA have also been demonstrated, further streamlining the production process.
Significant progress in enzymatic protein modification has been made over the last five years. With the ever-growing demand for protein-based conjugates, the utility of enzymatic labelling methods will continue to be explored and expanded. The ability to combine different enzymatic methods also opens up numerous possibilities to create multi-functional protein constructs. Furthermore, future applications will undoubtedly benefit from the development of new substrate analogues, the capability of enzyme engineering and the discovery of novel bioorthogonal reactions. As the area grows, it is likely that novel applications and new ideas will continue to emerge.
Acknowledgements
This work was supported in part by the National Institutes of Health (GM084152) and by the National Science Foundation (CHE-1308655)
Biography
Yi Zhang received her BS degree in Chemistry in 2012 from Wuhan University, China. She is currently a PhD student in Professor Mark Distefano’s group in the Chemistry Department, University of Minnesota. Her research interests include applying enzymatic protein modification and conjugation methods for biotechnological and therapeutic applications.
Keun-Young “Kevin” Park, was born in Seoul, South Korea, and grew up in Seoul and Ithaca, New York. He received his B.S and M.S degree in Chemistry from Sungkyunkwan University, South Korea each in 2010 and 2012 with Professor Young-Uk Kwon. He served in the Republic of Korea Air Force as a lieutenant in charge of disaster management. He is currently pursuing a Ph.D. degree in Chemistry with Professor Mark. D. Distefano at the University of Minnesota. His research interests include enzymatic protein modification for pharmaceutical applications such as achievement of enhanced pharmacokinetics from conjugation of polymers.
Kiall Francis Suazo was born and raised in Malaybalay City, Philippines. He received both his B.S. (2009) and M.S. (2014) degrees in Chemistry from the University of the Philippines Diliman. Upon moving to the University of Minnesota in 2014, he joined the Distefano group to pursue his Ph. D. in Chemistry in the field of chemical biology. His current research revolves on chemical proteomic profiling of prenylated proteins involved in diseases, as well as biochemical assays and techniques to study protein prenylation.
Mark Distefano was born in Baton Rouge, LA, and grew up in California and Paris, France. He received his B.A. degree in Chemistry and Biochemistry from the University of California at Berkeley in 1985 and his Ph.D. degree from Massachusetts Institute of Technology in 1989, where he worked with Professor Christopher T. Walsh. He was a postdoctoral fellow in the laboratory of Peter B. Dervan at California Institute of Technology. He is currently Distinguished McKnight Professor of Chemistry and Medicinal Chemistry at the University of Minnesota. His current research is focused on understanding protein lipid modication including protein prenylation.
Footnotes
Conflicts of interest
There are no conflicts to declare
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Charron G, Li MM, MacDonald MR and Hang HC, Proc. Natl. Acad. Sci. U S A, 2013, 110, 11085–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jensen EC, Anat. Rec, 2012, 295, 2031–2036. [DOI] [PubMed] [Google Scholar]
- 3.Staus DP, Wingler LM, Choi M, Pani B, Manglik A, Kruse AC and Lefkowitz RJ, Proc. Natl. Acad. Sci. U S A, 2018, 115, 3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibraheem A and Campbell RE, Curr. Opin. Chem. Biol, 2010, 14, 30–36. [DOI] [PubMed] [Google Scholar]
- 5.Datta S, Christena LR and Rajaram YR, 3 Biotech, 2013, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sutandy FXR, Qian J, Chen C-S and Zhu H, Curr. Protoc. Protein Sci, 2013, 72, 27.21.21–27.21.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M and Thanou M, Pharmacol. Res, 2010, 62, 90–99. [DOI] [PubMed] [Google Scholar]
- 8.Ulijn RV, Bibi N, Jayawarna V, Thornton PD, Todd SJ, Mart RJ, Smith AM and Gough JE, Mater. Today, 2007, 10, 40–48. [Google Scholar]
- 9.Lagasse HA, Alexaki A, Simhadri VL, Katagiri NH, Jankowski W, Sauna ZE and Kimchi-Sarfaty C, F1000Res, 2017, 6, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dozier JK and Distefano MD, Int. J. Mol. Sci, 2015, 16, 25831–25864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beck A, Goetsch L, Dumontet C and Corvaia N, Nat. Rev. Drug Discov, 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- 12.Larson SM, Carrasquillo JA, Cheung NK and Press OW, Nat. Rev. Cancer, 2015, 15, 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Dongen GA, Visser GW, Lub-de Hooge MN, de Vries EG and Perk LR, Oncologist, 2007, 12, 1379–1389. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher D, Hackenberger CP, Leonhardt H and Helma J, J. Clin. Immunol, 2016, 36 Suppl 1, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braun AC, Gutmann M, Luhmann T and Meinel L, J. Controlled Release, 2018, 273, 68–85. [DOI] [PubMed] [Google Scholar]
- 16.Walper SA, Turner KB and Medintz IL, in Chemoselective and Bioorthogonal Ligation Reactions, ed. Algar WR, Dawson PE, Medintz IL, 2017, DOI: 10.1002/9783527683451.ch7.
- 17.Lotze J, Reinhardt U, Seitz O and Beck-Sickinger AG, Mol. Biosyst, 2016, 12, 1731–1745. [DOI] [PubMed] [Google Scholar]
- 18.Rashidian M, Dozier JK and Distefano MD, Bioconjugate Chem, 2013, 24, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephanopoulos N and Francis MB, Nat. Chem. Biol, 2011, 7, 876. [DOI] [PubMed] [Google Scholar]
- 20.Krall N, da Cruz FP, Boutureira O and Bernardes GJL, Nat. Chem, 2015, 8, 103. [DOI] [PubMed] [Google Scholar]
- 21.Seki Y, Ishiyama T, Sasaki D, Abe J, Sohma Y, Oisaki K and Kanai M, J. Am. Chem. Soc, 2016, 138, 10798–10801. [DOI] [PubMed] [Google Scholar]
- 22.Yu Y, Zhang LK, Buevich AV, Li G, Tang H, Vachal P, Colletti SL and Shi ZC, J. Am. Chem. Soc, 2018, 140, 6797–6800. [DOI] [PubMed] [Google Scholar]
- 23.Lin S, Yang X, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD and Chang CJ, Science, 2017, 355, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spicer CD and Davis BG, Nat. Commun, 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- 25.Gunnoo SB and Annemieke M, ChemBioChem, 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 26.Arumugam S, Guo J, Mbua NE, Friscourt F, Lin N, Nekongo E, Boons GJ and Popik VV, Chem. Sci, 2014, 5, 1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valkevich EM, Guenette RG, Sanchez NA, Chen YC, Ge Y and Strieter ER, J. Am. Chem. Soc, 2012, 134, 6916–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conte ML, Staderini S, Marra A, Sanchez-Navarro M, Davis BG and Dondoni A, Chem. Commun, 2011, 47, 11086–11088. [DOI] [PubMed] [Google Scholar]
- 29.Chalker JM, Bernardes GJL, Lin YA and Davis BG, Chem. Asian J., 2009, 4, 630–640. [DOI] [PubMed] [Google Scholar]
- 30.Koniev O and Wagner A, Chem. Soc. Rev, 2015, 44, 5495–5551. [DOI] [PubMed] [Google Scholar]
- 31.Sletten EM and Bertozzi CR, Angew. Chem. Int. Ed, 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosen CB and Francis MB, Nat. Chem. Biol, 2017, 13, 697. [DOI] [PubMed] [Google Scholar]
- 33.Kalkhof S and Sinz A, Anal. Bioanal. Chem, 2008, 392, 305–312. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura T, Kawai Y, Kitamoto N, Osawa T and Kato Y, Chem. Res. Toxicol, 2009, 22, 536–542. [DOI] [PubMed] [Google Scholar]
- 35.Patel MK, Vijayakrishnan B, Koeppe JR, Chalker JM, Doores KJ and Davis BG, Chem. Commun, 2010, 46, 9119–9121. [DOI] [PubMed] [Google Scholar]
- 36.Chen X, Muthoosamy K, Pfisterer A, Neumann B and Weil T, Bioconjugate Chem, 2012, 23, 500–508. [DOI] [PubMed] [Google Scholar]
- 37.Pham GH, Ou W, Bursulaya B, DiDonato M, Herath A, Jin Y, Hao X, Loren J, Spraggon G, Brock A, Uno T, Geierstanger BH and Cellitti SE, ChemBioChem, 2018, 19, 799–804. [DOI] [PubMed] [Google Scholar]
- 38.Hendrickson TL, de Crécy-Lagard V and Schimmel P, Annu. Rev. Biochem, 2004, 73, 147–176. [DOI] [PubMed] [Google Scholar]
- 39.Liu CC and Schultz PG, Annu. Rev. Biochem, 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 40.Heinz N, FEBS Lett, 2012, 586, 2057–2064. [DOI] [PubMed] [Google Scholar]
- 41.Lang K and Chin JW, Chem. Rev, 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- 42.de Graaf AJ, Kooijman M, Hennink WE and Mastrobattista E, Bioconjugate Chem, 2009, 20, 1281–1295. [DOI] [PubMed] [Google Scholar]
- 43.Link AJ and Tirrell DA, J. Am. Chem. Soc, 2003, 125, 11164–11165. [DOI] [PubMed] [Google Scholar]
- 44.Dieterich DC, Link AJ, Graumann J, Tirrell DA and Schuman EM, Proc. Natl. Acad. Sci. U S A, 2006, 103, 9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song W, Wang Y, Yu Z, Vera CIR, Qu J and Lin Q, ACS Chem. Biol, 2010, 5, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wals K and Ovaa H, Front. Chem, 2014, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L and Schultz PG, Chem. Biol, 2001, 8, 883–890. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Xie J and Schultz PG, J. Am. Chem. Soc, 2006, 128, 8738–8739. [DOI] [PubMed] [Google Scholar]
- 49.Ai HW, Shen W, Sagi A, Chen PR and Schultz PG, ChemBioChem, 2011, 12, 1854–1857. [DOI] [PubMed] [Google Scholar]
- 50.Gallivan JP, Lester HA and Dougherty DA, Chem. Biol, 1997, 4, 739–749. [DOI] [PubMed] [Google Scholar]
- 51.Kim CH, Axup JY and Schultz PG, Curr. Opin. Chem. Biol, 2013, 17, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lang K and Chin JW, ACS Chem. Biol, 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- 53.McKay CS and Finn MG, Chem. Biol, 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen X and Wu YW, Org. Biomol. Chem, 2016, 14, 5417–5439. [DOI] [PubMed] [Google Scholar]
- 55.Prescher JA and Bertozzi CR, Nat. Chem. Biol, 2005, 1, 13. [DOI] [PubMed] [Google Scholar]
- 56.Dirksen A, Hackeng TM and Dawson PE, Angew. Chem. Int. Ed., 2006, 45, 7581–7584. [DOI] [PubMed] [Google Scholar]
- 57.Crisalli P and Kool ET, J. Org. Chem, 2013, 78, 1184–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rashidian M, Mahmoodi MM, Shah R, Dozier JK, Wagner CR and Distefano MD, Bioconjugate Chem, 2013, 24, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wendeler M, Grinberg L, Wang X, Dawson PE and Baca M, Bioconjugate Chem, 2014, 25, 93–101. [DOI] [PubMed] [Google Scholar]
- 60.Kool ET, Park DH and Crisalli P, J. Am. Chem. Soc, 2013, 135, 17663–17666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalia J and Raines RT, Angew. Chem. Int. Ed, 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agarwal P, van der Weijden J, Sletten EM, Rabuka D and Bertozzi CR, Proc. Natl. Acad. Sci. U S A, 2013, 110, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC and Rabuka D, Bioconjugate Chem, 2013, 24, 846–851. [DOI] [PubMed] [Google Scholar]
- 64.Kudirka R, Barfield RM, McFarland J, Albers AE, de Hart GW, Drake PM, Holder PG, Banas S, Jones LC, Garofalo AW and Rabuka D, Chem. Biol, 2015, 22, 293–298. [DOI] [PubMed] [Google Scholar]
- 65.Carrico IS, Carlson BL and Bertozzi CR, Nat. Chem. Biol, 2007, 3, 321–322. [DOI] [PubMed] [Google Scholar]
- 66.Rashidian M, Song JM, Pricer RE and Distefano MD, J. Am. Chem. Soc, 2012, 134, 8455–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao H, Sun W, Song Z, Yu Y, Wang L, Chen X and Zhang Q, Chembiochem, 2017, 18, 324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schumacher D, Helma J, Mann FA, Pichler G, Natale F, Krause E, Cardoso MC, Hackenberger CP and Leonhardt H, Angew. Chem. Int. Ed, 2015, 54, 13787–13791. [DOI] [PubMed] [Google Scholar]
- 69.Gray MA, Tao RN, DePorter SM, Spiegel DA and McNaughton BR, Chembiochem, 2016, 17, 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zeng Y, Ramya TN, Dirksen A, Dawson PE and Paulson JC, Nat. Methods, 2009, 6, 207–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Debets MF, van der Doelen CW, Rutjes FP and van Delft FL, Chembiochem, 2010, 11, 1168–1184. [DOI] [PubMed] [Google Scholar]
- 72.Nieto-García O, Jaffee MB, Mühlberg M and Hackenberger CPR, in Chemoselective and Bioorthogonal Ligation Reactions, ed. Algar WR, Dawson PE, Medintz L, 2017, DOI: 10.1002/9783527683451.ch4.
- 73.Wang ZPA, Tian CL and Zheng JS, RSC Adv, 2015, 5, 107192–107199. [Google Scholar]
- 74.Hein JE, in In Chemoselective and Bioorthogonal Ligation Reactions, ed. Algar WR, Dawson PE, Medintz IL, 2017, DOI: 10.1002/9783527683451.ch2.
- 75.Chan TR, Hilgraf R, Sharpless KB and Fokin VV, Org. Lett, 2004, 6, 2853–2855. [DOI] [PubMed] [Google Scholar]
- 76.Hong V, Presolski SI, Ma C and Finn MG, Angew. Chem. Int. Ed, 2009, 48, 9879–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.del Amo DS, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL and Wu P, J. Am. Chem. Soc, 2010, 132, 16893–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Presolski SI, Hong V, Cho SH and Finn MG, J. Am. Chem. Soc, 2010, 132, 14570–14576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y and Wu P, Angew. Chem. Int. Ed., 2011, 50, 8051–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kennedy DC, McKay CS, Legault MC, Danielson DC, Blake JA, Pegoraro AF, Stolow A, Mester Z and Pezacki JP, J. Am. Chem. Soc, 2011, 133, 17993–18001. [DOI] [PubMed] [Google Scholar]
- 81.Kuang GC, Michaels HA, Simmons JT, Clark RJ and Zhu L, J. Org. Chem, 2010, 75, 6540–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR and Ting AY, Angew. Chem. Int. Ed, 2012, 51, 5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiang H, Zheng T, Lopez-Aguilar A, Feng L, Kopp F, Marlow FL and Wu P, Bioconjugate Chem, 2014, 25, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A and Bertozzi CR, Proc. Natl. Acad. Sci. U S A, 2010, 107, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dehnert KW, Baskin JM, Laughlin ST, Beahm BJ, Naidu NN, Amacher SL and Bertozzi CR, Chembiochem, 2012, 13, 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Geel R, Pruijn GJ, van Delft FL and Boelens WC, Bioconjugate Chem, 2012, 23, 392–398. [DOI] [PubMed] [Google Scholar]
- 87.Oliveira BL, Guo Z and Bernardes GJL, Chem. Soc. Rev, 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- 88.Karver MR, Weissleder R and Hilderbrand SA, Bioconjugate Chem, 2011, 22, 2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wades TJ, Wong EH, Weisman GR and Anderson CJ, Chem. Rev, 2010, 110, 2858–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rossin R, Lappchen T, van den Bosch SM, Laforest R and Robillard MS, J. Nucl. Med, 2013, 54, 1989–1995. [DOI] [PubMed] [Google Scholar]
- 91.Patterson DM and Prescher JA, Curr. Opin. Chem. Biol, 2015, 28, 141–149. [DOI] [PubMed] [Google Scholar]
- 92.Rachel NM, Toulouse JL and Pelletier JN, Bioconjugate Chem, 2017, 28, 2518–2523. [DOI] [PubMed] [Google Scholar]
- 93.Rashidian M, Wang L, Edens JG, Jacobsen JT, Hossain I, Wang Q, Victora GD, Vasdev N, Ploegh H and Liang SH, Angew. Chem. Int. Ed, 2016, 55, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu DS, Tangpeerachaikul A, Selvaraj R, Taylor MT, Fox JM and Ting AY, J. Am. Chem. Soc, 2012, 134, 792–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Best M, Degen A, Baalmann M, Schmidt TT and Wombacher R, ChemBioChem, 2015, 16, 1158–1162. [DOI] [PubMed] [Google Scholar]
- 96.Wollack JW, Monson BJ, Dozier JK, Dalluge JJ, Poss K, Hilderbrand SA and Distefano MD, Chem. Biol. Drug. Des, 2014, 84, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Milczek EM, Chem. Rev, 2018, 118, 119–141. [DOI] [PubMed] [Google Scholar]
- 98.Ilangovan U, Ton-That H, Iwahara J, Schneewind O and Clubb RT, Proc. Natl. Acad. Sci. U S A, 2001, 98, 6056–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jacobitz AW, Kattke MD, Wereszczynski J and Clubb RT, Adv. Protein Chem. Struct. Biol, 2017, 109, 223–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Theile CS, Witte MD, Blom AE, Kundrat L, Ploegh HL and Guimaraes CP, Nat. Protoc, 2013, 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE and Ploegh HL, Nat. Protoc, 2013, 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen I, Dorr BM and Liu DR, Proc. Natl. Acad. Sci. U S A, 2011, 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hirakawa H, Ishikawa S and Nagamune T, Biotechnol. Bioeng, 2012, 109, 2955–2961. [DOI] [PubMed] [Google Scholar]
- 104.Popp MW, Dougan SK, Chuang TY, Spooner E and Ploegh HL, Proc. Natl. Acad. Sci. U S A, 2011, 108, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matsumoto T, Takase R, Tanaka T, Fukuda H and Kondo A, Biotechnol. J, 2012, 7, 642–648. [DOI] [PubMed] [Google Scholar]
- 106.Das S, Pawale VS, Dadireddy V, Singh AK, Ramakumar S and Roy RP, J. Biol. Chem, 2017, 292, 7244–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nikghalb KD, Horvath NM, Prelesnik JL, Banks OGB, Filipov PA, Row RD, Roark TJ and Antos JM, Chembiochem, 2018, 19, 185–195. [DOI] [PubMed] [Google Scholar]
- 108.Antos JM, Miller GM, Grotenbreg GM and Ploegh HL, J. Am. Chem. Soc, 2008, 130, 16338–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Popp MW, Antos JM, Grotenbreg GM, Spooner E and Ploegh HL, Nat. Chem. Biol, 2007, 3, 707–708. [DOI] [PubMed] [Google Scholar]
- 110.Popp MW and Ploegh HL, Angew. Chem. Int. Ed, 2011, 50, 5024–5032. [DOI] [PubMed] [Google Scholar]
- 111.Ritzefeld M, Chemistry, 2014, 20, 8516–8529. [DOI] [PubMed] [Google Scholar]
- 112.Tan XL, Pan M, Zheng Y, Gao S, Liang LJ and Li YM, Chem. Sci, 2017, 8, 6881–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Uth C, Zielonka S, Horner S, Rasche N, Plog A, Orelma H, Avrutina O, Zhang K and Kolmar H, Angew. Chem. Int. Ed, 2014, 53, 12618–12623. [DOI] [PubMed] [Google Scholar]
- 114.Le RK, Raeeszadeh-Sarmazdeh M, Boder ET and Frymier PD, Langmuir, 2015, 31, 1180–1188. [DOI] [PubMed] [Google Scholar]
- 115.Qafari SM, Ahmadian G and Mohammadi M, RSC Adv, 2017, 7, 56006–56015. [Google Scholar]
- 116.Garg S, Singaraju GS, Yengkhom S and Rakshit S, Bioconjugate Chem, 2018, 29, 1714–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zou Z, Mate DM, Rubsam K, Jakob F and Schwaneberg U, ACS Comb. Sci, 2018, 20, 203–211. [DOI] [PubMed] [Google Scholar]
- 118.Ham HO, Qu Z, Haller CA, Dorr BM, Dai E, Kim W, Liu DR and Chaikof EL, Nat. Commun, 2016, 7, 11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tabata A, Ohkuba Y, Anyoji N, Hojo K, Tomoyasu T, Tatematsu Y, Ohkura K and Nagamune H, Anticancer Res, 2015, 35, 4411–4418. [PubMed] [Google Scholar]
- 120.Silvius JR and Leventis R, Bioconjugate Chem, 2017, 28, 1271–1282. [DOI] [PubMed] [Google Scholar]
- 121.Schoonen L, Pille J, Borrmann A, Nolte RJ and van Hest JC, Bioconjugate Chem, 2015, 26, 2429–2434. [DOI] [PubMed] [Google Scholar]
- 122.Patterson D, Schwarz B, Avera J, Western B, Hicks M, Krugler P, Terra M, Uchida M, McCoy K and Douglas T, Bioconjugate Chem, 2017, 28, 2114–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cambria E, Renggli K, Ahrens CC, Cook CD, Kroll C, Krueger AT, Imperiali B and Griffith LG, Biomacromolecules, 2015, 16, 2316–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arkenberg MR and Lin CC, Biomater. Sci, 2017, 5, 2231–2240. [DOI] [PubMed] [Google Scholar]
- 125.Gau E, Mate DM, Zou Z, Oppermann A, Töpel A, Jakob F, Wöll D, Schwaneberg U and Pich A, Biomacromolecules, 2017, 18, 2789–2798. [DOI] [PubMed] [Google Scholar]
- 126.Matsumoto T, Isogawa Y, Tanaka T and Kondo A, Biosens. Bioelectron, 2018, 99, 56–61. [DOI] [PubMed] [Google Scholar]
- 127.Pasqual G, Chudnovskiy A, Tas JMJ, Agudelo M, Schweitzer LD, Cui A, Hacohen N and Victora GD, Nature, 2018, 553, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hirakawa H, Ishikawa S and Nagamune T, Biotechnol. J, 2015, 10, 1487–1492. [DOI] [PubMed] [Google Scholar]
- 129.Glasgow JE, Salit ML and Cochran JR, J. Am. Chem. Soc, 2016, 138, 7496–7499. [DOI] [PubMed] [Google Scholar]
- 130.Wu Q, Ploegh HL and Truttmann MC, ACS Chem. Biol, 2017, 12, 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beerli RR, Hell T, Merkel AS and Grawunder U, PLoS One, 2015, 10, e0131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stefan N, Gebleux R, Waldmeier L, Hell T, Escher M, Wolter FI, Grawunder U and Beerli RR, Mol. Cancer Ther, 2017, 16, 879–892. [DOI] [PubMed] [Google Scholar]
- 133.Alt K, Paterson BM, Westein E, Rudd SE, Poniger SS, Jagdale S, Ardipradja K, Connell TU, Krippner GY, Nair AK, Wang X, Tochon-Danguy HJ, Donnelly PS, Peter K and Hagemeyer CE, Angew. Chem. Int. Ed, 2015, 54, 7515–7519. [DOI] [PubMed] [Google Scholar]
- 134.McCluskey AJ and Collier RJ, Mol. Cancer Ther, 2013, 12, 2273–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Greineder CF, Villa CH, Walsh LR, Kiseleva RY, Hood ED, Khoshnejad M, Warden-Rothman R, Tsourkas A and Muzykantov VR, Bioconjugate Chem, 2018, 29, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rashidian M, Keliher EJ, Bilate AM, Duarte JN, Wojtkiewicz GR, Jacobsen JT, Cragnolini J, Swee LK, Victora GD, Weissleder R and Ploegh HL, Proc. Natl. Acad. Sci. U S A, 2015, 112, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fang T, Duarte JN, Ling J, Li Z, Guzman JS and Ploegh HL, Angew. Chem. Int. Ed, 2016, 55, 2416–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Massa S, Vikani N, Betti C, Ballet S, Vanderhaegen S, Steyaert J, Descamps B, Vanhove C, Bunschoten A, van Leeuwen FW, Hernot S, Caveliers V, Lahoutte T, Muyldermans S, Xavier C and Devoogdt N, Contrast Media Mol. Imaging, 2016, 11, 328–339. [DOI] [PubMed] [Google Scholar]
- 139.van Lith SA, van Duijnhoven SM, Navis AC, Leenders WP, Dolk E, Wennink JW, van Nostrum CF and van Hest JC, Bioconjugate Chem, 2017, 28, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pang Y, Liu J, Qi Y, Li X and Chilkoti A, Angew. Chem. Int. Ed, 2016, 55, 10296–10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hu J, Wang G, Zhao W, Liu X, Zhang L and Gao W, Biomaterials, 2016, 96, 84–92. [DOI] [PubMed] [Google Scholar]
- 142.Witte MD, Cragnolini JJ, Dougan SK, Yoder NC, Popp MW and Ploegh HL, Proc. Natl. Acad. Sci. U S A, 2012, 109, 11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abrahmsen L, Tom J, Burnier J, Butcher KA, Kossiakoff A and Wells JA, Biochemistry, 1991, 30, 4151–4159. [DOI] [PubMed] [Google Scholar]
- 144.Weeks AM and Wells JA, Nat. Chem. Biol, 2018, 14, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Henager SH, Chu N, Chen Z, Bolduc D, Dempsey DR, Hwang Y, Wells JA and Cole PA, Nat. Methods, 2016, 13, 925–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rachel NM and Pelletier JN, Chem. Commun, 2016, 52, 2541–2544. [DOI] [PubMed] [Google Scholar]
- 147.Oteng-Pabi SK, Pardin C, Stoica M and Keillor JW, Chem. Commun, 2014, 50, 6604–6606. [DOI] [PubMed] [Google Scholar]
- 148.Dennler P, Chiotellis A, Fischer E, Bregeon D, Belmant C, Gauthier L, Lhospice F, Romagne F and Schibli R, Bioconjugate Chem, 2014, 25, 569–578. [DOI] [PubMed] [Google Scholar]
- 149.Mero A, Grigoletto A, Maso K, Yoshioka H, Rosato A and Pasut G, Polym. Chem, 2016, 7, 6545–6553. [Google Scholar]
- 150.Tanaka T, Kamiya N and Nagamune T, FEBS Lett, 2005, 579, 2092–2096. [DOI] [PubMed] [Google Scholar]
- 151.Yang MT, Chang CH, Wang JM, Wu TK, Wang YK, Chang CY and Li TT, J. Biol. Chem, 2011, 286, 7301–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gauchet C, Labadie GR and Poulter CD, J. Am. Chem. Soc, 2006, 128, 9274–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hosokawa A, Wollack JW, Zhang Z, Chen L, Barany G and Distefano MD, Int. J. Pept. Res. Ther, 2007, 13, 345–354. [Google Scholar]
- 154.Lee JJ, Choi HJ, Yun M, Kang Y, Jung JE, Ryu Y, Kim TY, Cha YJ, Cho HS, Min JJ, Chung CW and Kim HS, Angew. Chem. Int. Ed, 2015, 54, 12020–12024. [DOI] [PubMed] [Google Scholar]
- 155.Rashidian M, Kumarapperuma SC, Gabrielse K, Fegan A, Wagner CR and Distefano MD, J. Am. Chem. Soc, 2013, 135, 16388–16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang Y, Blanden MJ, Sudheer C, Gangopadhyay SA, Rashidian M, Hougland JL and Distefano MD, Bioconjugate Chem, 2015, 26, 2542–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yeo JE, Wickramaratne S, Khatwani S, Wang YC, Vervacke J, Distefano MD and Tretyakova NY, ACS Chem. Biol, 2014, 9, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Seo JS, Lee S and Poulter CD, J. Am. Chem. Soc, 2013, 135, 8973–8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kishore NS, Lu TB, Knoll LJ, Katoh A, Rudnick DA, Mehta PP, Devadas B, Huhn M, Atwood JL, Adams SP, Gokel GW and Gordon JI, J. Biol. Chem, 1991, 266, 8835–8855. [PubMed] [Google Scholar]
- 160.Ho SH and Tirrell DA, J. Am. Chem. Soc, 2016, 138, 15098–15101. [DOI] [PubMed] [Google Scholar]
- 161.Heal WP, Wickramasinghe SR, Leatherbarrow RJ and Tate EW, Org. Biomol. Chem, 2008, 6, 2308–2315. [DOI] [PubMed] [Google Scholar]
- 162.Kulkarni C, Kinzer-Ursem TL and Tirrell DA, Chembiochem, 2013, 14, 1958–1962. [DOI] [PubMed] [Google Scholar]
- 163.Kulkarni C, Lo M, Fraseur JG, Tirrell DA and Kinzer-Ursem TL, Bioconjugate Chem, 2015, 26, 2153–2160. [DOI] [PubMed] [Google Scholar]
- 164.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R and Walsh CT, Proc. Natl. Acad. Sci. U S A, 2005, 102, 15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Grünewald J, Klock HE, Cellitti SE, Bursulaya B, McMullan D, Jones DH, Chiu HP, Wang X, Patterson P, Zhou H, Vance J, Nigoghossian E, Tong H, Daniel D, Mallet W, Ou W, Uno T, Brock A, Lesley SA and Geierstanger BH, Bioconjugate Chem, 2015, 26, 2554–2562. [DOI] [PubMed] [Google Scholar]
- 166.Pippig DA, Baumann F, Strackharn M, Aschenbrenner D and Gaub HE, ACS Nano, 2014, 8, 6551–6555. [DOI] [PubMed] [Google Scholar]
- 167.Sunbul M, Yen M, Zou Y and Yin J, Chem. Commun, 2008, 0, 5927–5929. [DOI] [PubMed] [Google Scholar]
- 168.Schumacher D, Lemke O, Helma J, Gerszonowicz L, Waller V, Stoschek T, Durkin PM, Budisa N, Leonhardt H, Keller BG and Hackenberger CPR, Chem. Sci, 2017, 8, 3471–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fernández-Suárez M, Baruah H, Martínez-Hernández L, Xie KT, Baskin JM, Bertozzi CR and Ting AY, Nat. Biotechnol, 2007, 25, 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Plaks JG, Falatach R, Kastantin M, Berberich JA and Kaar JL, Bioconjugate Chem, 2015, 26, 1104–1112. [DOI] [PubMed] [Google Scholar]
- 171.Hauke S, Best M, Schmidt TT, Baalmann M, Krause A and Wombacher R, Bioconjugate Chem, 2014, 25, 1632–1637. [DOI] [PubMed] [Google Scholar]
- 172.Chapman‐Smith A, Mulhern TD, Whelan F, Cronan JE Jr. and Wallace JC, Protein Sci, 2001, 10, 2608–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chen I, Howarth M, Lin W and Ting AY, Nat. Methods, 2005, 2, 99. [DOI] [PubMed] [Google Scholar]
- 174.Slavoff SA, Chen I, Choi YA and Ting AY, J. Am. Chem. Soc, 2008, 130, 1160–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sueda S, Yoneda S and Hayashi H, ChemBioChem, 2011, 12, 1367–1375. [DOI] [PubMed] [Google Scholar]
- 176.Miyao H, Ikeda Y, Shiraishi A, Kawakami Y and Sueda S, Anal. Biochem, 2015, 484, 113–121. [DOI] [PubMed] [Google Scholar]
- 177.Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ and Zheng Y, Mol. Biol. Cell, 2016, 27, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, de Hart GW, Garofalo AW, Holder P, Jones LC, Kudirka R, McFarland J, Zmolek W and Rabuka D, Bioconjugate Chem, 2014, 25, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J and Bertozzi CR, Proc. Natl. Acad. Sci. U S A, 2009, 106, 3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Parthasarathy R, Subramanian S and Boder ET, Bioconjugate Chem, 2007, 18, 469–476. [DOI] [PubMed] [Google Scholar]
- 181.Samantaray S, Marathe U, Dasgupta S, Nandicoori VK and Roy RP, J. Am. Chem. Soc, 2008, 130, 2132–2133. [DOI] [PubMed] [Google Scholar]
- 182.Mohlmann S, Mahlert C, Greven S, Scholz P and Harrenga A, Chembiochem, 2011, 12, 1774–1780. [DOI] [PubMed] [Google Scholar]
- 183.Dasgupta S, Samantaray S, Sahal D and Roy RP, J. Biol. Chem, 2011, 286, 23996–24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bellucci JJ, Bhattacharyya J and Chilkoti A, Angew. Chem. Int. Ed, 2015, 54, 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ito T, Sadamoto R, Naruchi K, Togame H, Takemoto H, Kondo H and Nishimura S, Biochemistry, 2010, 49, 2604–2614. [DOI] [PubMed] [Google Scholar]
- 186.Baer S, Nigro J, Madej MP, Nisbet RM, Suryadinata R, Coia G, Hong LP, Adams TE, Williams CC and Nuttall SD, Org. Biomol. Chem, 2014, 12, 2675–2685. [DOI] [PubMed] [Google Scholar]
- 187.Li YM, Li YT, Pan M, Kong XQ, Huang YC, Hong ZY and Liu L, Angew. Chem. Int. Ed, 2014, 53, 2198–2202. [DOI] [PubMed] [Google Scholar]
- 188.Bellucci JJ, Amiram M, Bhattacharyya J, McCafferty D and Chilkoti A, Angew. Chem. Int. Ed., 2013, 52, 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Policarpo RL, Kang H, Liao X, Rabideau AE, Simon MD and Pentelute BL, Angew. Chem. Int. Ed, 2014, 53, 9203–9208. [DOI] [PubMed] [Google Scholar]
- 190.Warden-Rothman R, Caturegli I, Popik V and Tsourkas A, Anal. Chem, 2013, 85, 11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wang HH, Altun B, Nwe K and Tsourkas A, Angew. Chem. Int. Ed, 2017, 56, 5349–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng X, Zhu T, Hong H, Zhou Z and Wu Z, Org. Chem. Front, 2017, 4, 2058–2062. [Google Scholar]
- 193.Heck T, Pham PH, Yerlikaya A, Thöny-Meyer L and Richter M, Catal. Sci. Technol, 2014, 4, 2946–2956. [Google Scholar]
- 194.Wells JA and Estell DA, Trends Biochem. Sci, 1988, 13, 291–297. [DOI] [PubMed] [Google Scholar]
- 195.Chang TK, Jackson DY, Burnier JP and Wells JA, Proc. Natl. Acad. Sci. U S A, 1994, 91, 12544–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jackson DY, Burnier J, Quan C, Stanley M, Tom J and Wells JA, Science, 1994, 266, 243–247. [DOI] [PubMed] [Google Scholar]
- 197.Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL and Wells JA, Cell, 2008, 134, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW, Sharfi H, Zhai B, Rodriguez-Mias R, Luithardt H, Cantley LC, Daley GQ, Asara JM, Gygi SP, Wagner G, Liu CF and Yuan J, Cell, 2011, 146, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mariniello L, Porta R, Sorrentino A, Giosafatto CV, Rossi Marquez G, Esposito M and Di Pierro P, Amino Acids, 2014, 46, 767–776. [DOI] [PubMed] [Google Scholar]
- 200.Spolaore B, Raboni S, Ramos Molina A, Satwekar A, Damiano N and Fontana A, Biochemistry, 2012, 51, 8679–8689. [DOI] [PubMed] [Google Scholar]
- 201.Rachel NM and Pelletier JN, Biomolecules, 2013, 3, 870–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Malesevic M, Migge A, Hertel TC and Pietzsch M, Chembiochem, 2015, 16, 1169–1174. [DOI] [PubMed] [Google Scholar]
- 203.Gundersen MT, Keillor JW and Pelletier JN, Appl. Microbiol. Biotechnol, 2014, 98, 219–230. [DOI] [PubMed] [Google Scholar]
- 204.Sugimura Y, Yokoyama K, Nio N, Maki M and Hitomi K, Arch. Biochem. Biophys, 2008, 477, 379–383. [DOI] [PubMed] [Google Scholar]
- 205.Lee JH, Song C, Kim DH, Park IH, Lee SG, Lee YS and Kim BG, Biotechnol. Bioeng, 2013, 110, 353–362. [DOI] [PubMed] [Google Scholar]
- 206.Tominaga J, Kemori Y, Tanaka Y, Maruyama T, Kamiya N and Goto M, Chem. Commun., 2007, 401–403. [DOI] [PubMed] [Google Scholar]
- 207.Deweid L, Neureiter L, Englert S, Schneider H, Deweid J, Yanakieva D, Sturm J, Bitsch S, Christmann A, Avrutina O, Fuchsbauer H-L and Kolmar H, Chem. Eur. J, DOI: 10.1002/chem.201803485. [DOI] [PubMed] [Google Scholar]
- 208.Oteng-Pabi SK and Keillor JW, Anal. Biochem, 2013, 441, 169–173. [DOI] [PubMed] [Google Scholar]
- 209.Taki M, Shiota M and Taira K, Protein Eng , Des. Sel., 2004, 17, 119–126. [DOI] [PubMed] [Google Scholar]
- 210.Tominaga J, Kamiya N, Doi S, Ichinose H, Maruyama T and Goto M, Biomacromolecules, 2005, 6, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 211.Mori Y, Goto M and Kamiya N, Biochem. Biophys. Res. Commun, 2011, 410, 829–833. [DOI] [PubMed] [Google Scholar]
- 212.Kamiya N, Abe H, Goto M, Tsuji Y and Jikuya H, Org. Biomol. Chem, 2009, 7, 3407–3412. [DOI] [PubMed] [Google Scholar]
- 213.Fontana A, Spolaore B, Mero A and Veronese FM, Adv. Drug Deliv. Rev, 2008, 60, 13–28. [DOI] [PubMed] [Google Scholar]
- 214.Jeger S, Zimmermann K, Blanc A, Grunberg J, Honer M, Hunziker P, Struthers H and Schibli R, Angew. Chem. Int. Ed, 2010, 49, 9995–9997. [DOI] [PubMed] [Google Scholar]
- 215.Mindt TL, Jungi V, Wyss S, Friedli A, Pla G, Novak-Hofer I, Grünberg J and Schibli R, Bioconjugate Chem, 2008, 19, 271–278. [DOI] [PubMed] [Google Scholar]
- 216.Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, Ho WH, Farias S, Casas MG, Abdiche Y, Zhou D, Chandrasekaran R, Samain C, Loo C, Rossi A, Rickert M, Krimm S, Wong T, Chin SM, Yu J, Dilley J, Chaparro-Riggers J, Filzen GF, O’Donnell CJ, Wang F, Myers JS, Pons J, Shelton DL and Rajpal A, Chem. Biol, 2013, 20, 161–167. [DOI] [PubMed] [Google Scholar]
- 217.Strop P, Tran TT, Dorywalska M, Delaria K, Dushin R, Wong OK, Ho WH, Zhou D, Wu A, Kraynov E, Aschenbrenner L, Han B, O’Donnell CJ, Pons J, Rajpal A, Shelton DL and Liu SH, Mol. Cancer Ther, 2016, 15, 2698–2708. [DOI] [PubMed] [Google Scholar]
- 218.Spycher PR, Amann CA, Wehrmüller JE, Hurwitz DR, Kreis O, Messmer D, Ritler A, Küchler A, Blanc A, Béhé M, Walde P and Schibli R, Chembiochem, 2017, 18, 1923–1927. [DOI] [PubMed] [Google Scholar]
- 219.Spidel JL, Vaessen B, Albone EF, Cheng X, Verdi A and Kline JB, Bioconjugate Chem, 2017, 28, 2471–2484. [DOI] [PubMed] [Google Scholar]
- 220.Puthenveetil S, Musto S, Loganzo F, Tumey LN, O’Donnell CJ and Graziani E, Bioconjugate Chem, 2016, 27, 1030–1039. [DOI] [PubMed] [Google Scholar]
- 221.Maso K, Grigoletto A and Pasut G, Adv. Protein Chem. Struct. Biol., 2018, 112, 123–142. [DOI] [PubMed] [Google Scholar]
- 222.Mero A, Spolaore B, Veronese FM and Fontana A, Bioconjugate Chem, 2009, 20, 384–389. [DOI] [PubMed] [Google Scholar]
- 223.Sato H, Hayashi E, Yamada N, Yatagai M and Takahara Y, Bioconjugate Chem, 2001, 12, 701–710. [DOI] [PubMed] [Google Scholar]
- 224.Scaramuzza S, Tonon G, Olianas A, Messana I, Schrepfer R, Orsini G and Caliceti P, J. Controlled Release, 2012, 164, 355–363. [DOI] [PubMed] [Google Scholar]
- 225.Spolaore B, Raboni S, Satwekar AA, Grigoletto A, Mero A, Montagner IM, Rosato A, Pasut G and Fontana A, Bioconjugate Chem, 2016, 27, 2695–2706. [DOI] [PubMed] [Google Scholar]
- 226.Mero A, Schiavon M, Veronese FM and Pasut G, J. Controlled Release, 2011, 154, 27–34. [DOI] [PubMed] [Google Scholar]
- 227.da Silva Freitas D, Mero A and Pasut G, Bioconjugate Chem, 2013, 24, 456–463. [DOI] [PubMed] [Google Scholar]
- 228.Kitaoka M, Tsuruda Y, Tanaka Y, Goto M, Mitsumori M, Hayashi K, Hiraishi Y, Miyawaki K, Noji S and Kamiya N, Chem. Eur. J, 2011, 17, 5387–5392. [DOI] [PubMed] [Google Scholar]
- 229.Kitaoka M, Mitsumori M, Hayashi K, Hiraishi Y, Yoshinaga H, Nakano K, Miyawaki K, Noji S, Goto M and Kamiya N, Anal. Chem, 2012, 84, 5885–5891. [DOI] [PubMed] [Google Scholar]
- 230.Takahara M, Wakabayashi R, Minamihata K, Goto M and Kamiya N, Bioconjugate Chem, 2017, 28, 2954–2961. [DOI] [PubMed] [Google Scholar]
- 231.Wakabayashi R, Yahiro K, Hayashi K, Goto M and Kamiya N, Biomacromolecules, 2017, 18, 422–430. [DOI] [PubMed] [Google Scholar]
- 232.Steffen W, Ko FC, Patel J, Lyamichev V, Albert TJ, Benz J, Rudolph MG, Bergmann F, Streidl T, Kratzsch P, Boenitz-Dulat M, Oelschlaegel T and Schraeml M, J. Biol. Chem, 2017, 292, 15622–15635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Oteng-Pabi SK, Clouthier CM and Keillor JW, PLoS ONE, 2018, 13, e0197956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z and Lin H, Chem. Rev, 2018, 118, 919–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hartman HL, Hicks KA and Fierke CA, Biochemistry, 2005, 44, 15314–15324. [DOI] [PubMed] [Google Scholar]
- 236.Krzysiak AJ, Scott SA, Hicks KA, Fierke CA and Gibbs RA, Bioorg. Med. Chem. Lett, 2007, 17, 5548–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hougland JL, Lamphear CL, Scott SA, Gibbs RA and Fierke CA, Biochemistry, 2009, 48, 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang YC, Dozier JK, Beese LS and Distefano MD, ACS Chem. Biol, 2014, 9, 1726–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Reigard SA, Zahn TJ, Haworth KB, Hicks KA, Fierke CA and Gibbs RA, Biochemistry, 2005, 44, 11214–11223. [DOI] [PubMed] [Google Scholar]
- 240.Labadie GR, Viswanathan R and Poulter CD, J. Org. Chem, 2007, 72, 9291–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Blanden MJ, Suazo KF, Hildebrandt ER, Hardgrove DS, Patel M, Saunders WP, Distefano MD, Schmidt WK and Hougland JL, J. Biol. Chem, 2018, 293, 2770–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Owen DJ, Alexandrov K, Rostkova E, Scheidig AJ, Goody RS and Waldmann H, Angew. Chem. Int. Ed, 1999, 38, 509–512. [DOI] [PubMed] [Google Scholar]
- 243.Chen APC, Chen YH, Liu HP, Li YC, Chen CT and Liang PH, J. Am. Chem. Soc, 2002, 124, 15217–15224. [DOI] [PubMed] [Google Scholar]
- 244.Dursina B, Reents R, Delon C, Wu Y, Kulharia M, Thutewohl M, Veligodsky A, Kalinin A, Evstifeev V, Ciobanu D, Szedlacsek SE, Waldmann H, Goody RS and Alexandrov K, J. Am. Chem. Soc, 2006, 128, 2822–2835. [DOI] [PubMed] [Google Scholar]
- 245.Chehade KAH, Kiegiel K, Isaacs RJ, Pickett JS, Bowers KE, Fierke CA, Andres DA and Spielmann HP, J. Am. Chem. Soc, 2002, 124, 8206–8219. [DOI] [PubMed] [Google Scholar]
- 246.Turek TC, Gaon I and Distefano MD, J. Org. Chem, 2001, 66, 3253–3264. [DOI] [PubMed] [Google Scholar]
- 247.Turek-Etienne TC, Strickland CL and Distefano MD, Biochemistry, 2003, 42. [DOI] [PubMed] [Google Scholar]
- 248.Nguyen UT, Guo Z, Delon C, Wu Y, Deraeve C, Fränzel B, Bon RS, Blankenfeldt W, Goody RS, Waldmann H, Wolters D and Alexandrov K, Nat. Chem. Biol, 2009, 5, 227–235. [DOI] [PubMed] [Google Scholar]
- 249.Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, Jaunbergs J, Weinbaum C, Tamanoi F, Falck J and Zhao Y, Proc. Natl. Acad. Sci. U S A, 2004, 101, 12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Rose MW, Rose ND, Boggs J, Lenevich S, Xu J, Barany G and Distefano MD, J. Pept. Res, 2005, 65, 529–537. [DOI] [PubMed] [Google Scholar]
- 251.Duckworth BP, Xu J, Taton TA, Guo A and Distefano MD, Bioconjugate Chem, 2006, 17, 967–974. [DOI] [PubMed] [Google Scholar]
- 252.Nguyen UT, Cramer J, Gomis J, Reents R, Gutierrez-Rodriguez M, Goody RS, Alexandrov K and Waldmann H, Chembiochem, 2007, 8, 408–423. [DOI] [PubMed] [Google Scholar]
- 253.Duckworth BP, Chen Y, Wollack JW, Sham Y, Mueller JD, Taton TA and Distefano MD, Angew. Chem. Int. Ed, 2007, 46, 8819–8822. [DOI] [PubMed] [Google Scholar]
- 254.Das D, Tnimov Z, Nguyen UT, Thimmaiah G, Lo H, Abankwa D, Wu Y, Goody RS, Waldmann H and Alexandrov K, Chembiochem, 2012, 13, 674–683. [DOI] [PubMed] [Google Scholar]
- 255.Rashidian M, Dozier JK, Lenevich S and Distefano MD, Chem. Commun, 2010, 46, 8998–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kim M, Kleckley TS, Wiemer AJ, Holstein SA, Hohl RJ and Wiemer DF, J. Org. Chem, 2004, 69, 8186–8193. [DOI] [PubMed] [Google Scholar]
- 257.DeGraw AJ, Palsuledesai C, Ochocki JD, Dozier JK, Lenevich S, Rashidian M and Distefano MD, Chem. Biol. Drug. Des, 2010, 76, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Weinrich D, Lin PC, Jonkheijm P, Nguyen UT, Schroder H, Niemeyer CM, Alexandrov K, Goody R and Waldmann H, Angew. Chem. Int. Ed, 2010, 49, 1252–1257. [DOI] [PubMed] [Google Scholar]
- 259.Khatwani SL, Kang JS, Mullen DG, Hast MA, Beese LS, Distefano MD and Taton TA, Bioorg. Med. Chem, 2012, 20, 4532–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Distefano MD and Rashidian M, U. S Pat, 9878045, 2018. [Google Scholar]
- 261.Dozier JK, Khatwani SL, Wollack JW, Wang YC, Schmidt-Dannert C and Distefano MD, Bioconjugate Chem, 2014, 25, 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Pickett JS, Bowers KE, Hartman HL, Fu HW, Embry AC, Casey PJ and Fierke CA, Biochemistry, 2003, 42, 9741–9748. [DOI] [PubMed] [Google Scholar]
- 263.Hougland JL, Gangopadhyay SA and Fierke CA, J. Biol. Chem, 2012, 287, 38090–38100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Viswanathan R, Labadie GR and Poulter CD, Bioconjugate Chem, 2013, 24, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Seo JS and Poulter CD, Langmuir, 2014, 30, 6629–6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Choi SR, Seo JS, Bohaty RFand Poulter CD, Bioconjugate Chem, 2014, 25, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tolstyka ZP, Richardson W, Bat E, Stevens CJ, Parra DP, Dozier JK, Distefano MD, Dunn B and Maynard HD, Chembiochem, 2013, 14, 2464–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kim Y, Park T, Woo S, Lee H, Kim S, Cho J, Jung D, Kim Y, Kwon H and Oh K, U. S. Pat, 13/466,875, 2012. [Google Scholar]
- 269.Giglione C, Fieulaine S and Meinnel T, Biochimie, 2015, 114, 134–146. [DOI] [PubMed] [Google Scholar]
- 270.Martin DD, Beauchamp E and Berthiaume LG, Biochimie, 2011, 93, 18–31. [DOI] [PubMed] [Google Scholar]
- 271.Towler DA, Adams SP, Eubanks SR, Towery DS, Jackson-Machelski E, Glaser L and Gordon JI, Proc. Natl. Acad. Sci. U S A, 1987, 84, 2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Devadas B, Lu T, Katoh A, Kishore NS, Wade AC, Mehta PP, Rudnick DA, Bryant ML, Adams SP, Li Q, Gokel GW and Gordon JI, J. Biol. Chem, 1992, 267, 7224–7239. [PubMed] [Google Scholar]
- 273.Lu T, Li Q, Katoh A, Hernandez J, Duffin K, Jackson-Machelski E, Knoll LJ, Gokel GW and Gordon JI, J. Biol. Chem, 1994, 269, 5346–5357. [PubMed] [Google Scholar]
- 274.Heal WP, Wickramasinghe SR, Bowyer PW, Holder AA, Smith DF, Leatherbarrow RJ and Tate EW, Chem. Commun, 2008, 480–482. [DOI] [PubMed] [Google Scholar]
- 275.Heal WP, Wright MH, Thinon E and Tate EW, Nat. Protoc, 2011, 7, 105–117. [DOI] [PubMed] [Google Scholar]
- 276.Luginbuhl KM, Mozhdehi D, Dzuricky M, Yousefpour P, Huang FC, Mayne NR, Buehne KL and Chilkoti A, Angew. Chem. Int. Ed, 2017, 56, 13979–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Beld J, Sonnenschein EC, Vickery CR, Noel JP and Burkart MD, Nat. Prod. Rep, 2014, 31, 61–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Yin J, Liu F, Li X and Walsh CT, J. Am. Chem. Soc, 2004, 126, 7754–7755. [DOI] [PubMed] [Google Scholar]
- 279.Kosa NM, Haushalter RW, Smith AR and Burkart MD, Nat. Methods, 2012, 9, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Rothmann M, Kosa NM and Burkart MD, RSC adv, 2014, 4, 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Zhou Z, Cironi P, Lin AJ, Xu Y, Hrvatin S, Golan DE, Silver PA, Walsh CT and Yin J, ACS Chem. Biol, 2007, 2, 337–346. [DOI] [PubMed] [Google Scholar]
- 282.Zhou Z, Koglin A, Wang Y, McMahon AP and Walsh CT, J. Am. Chem. Soc, 2008, 130, 9925–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Strackharn M, Pippig DA, Meyer P, Stahl SW and Gaub HE, J. Am. Chem. Soc, 2012, 134, 15193–15196. [DOI] [PubMed] [Google Scholar]
- 284.Kawamura T, Stephens B, Qin L, Yin X, Dores MR, Smith TH, Grimsey N, Abagyan R, Trejo J, Kufareva I, Fuster MM, Salanga CL and Handel TM, PLoS ONE, 2014, 9, e81454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Allen SJ, Hamel DJ and Handel TM, Cytokine, 2011, 55, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Marchetti L, De Nadai T, Bonsignore F, Calvello M, Signore G, Viegi A, Beltram F, Luin S and Cattaneo A, PLoS ONE, 2014, 9, e113708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Schmohl L and Schwarzer D, J. Pept. Sci, 2014, 20, 145–151. [DOI] [PubMed] [Google Scholar]
- 288.Grünewald J, Jones David H, Brock A, Chiu HP, Bursulaya B, Ng K, Vo T, Patterson P, Uno T, Hunt J, Spraggon G and Geierstanger BH, ChemBioChem, 2014, 15, 1787–1791. [DOI] [PubMed] [Google Scholar]
- 289.Ou W, Uno T, Chiu H-P, Grünewald J, Cellitti SE, Crossgrove T, Hao X, Fan Q, Quinn LL, Patterson P, Okach L, Jones DH, Lesley SA, Brock A and Geierstanger BH, Proc. Natl. Acad. Sci. U S A, 2011, 108, 10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Clarke KM, Mercer AC, La Clair JJ and Burkart MD, J. Am. Chem. Soc, 2005, 127, 11234–11235. [DOI] [PubMed] [Google Scholar]
- 291.Worthington AS and Burkart MD, Org. Biomol. Chem, 2006, 4, 44–46. [DOI] [PubMed] [Google Scholar]
- 292.Nazi I, Koteva KP and Wright GD, Anal. Biochem, 2004, 324, 100–105. [DOI] [PubMed] [Google Scholar]
- 293.Kittilä T and Cryle MJ, Biochem. Cell Biol, 2017, 96, 372–379. [DOI] [PubMed] [Google Scholar]
- 294.Ersfeld K, Wehland J, Plessmann U, Dodemont H, Gerke V and Weber K, J. Cell Biol, 1993, 120, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Janke C, Cell Biol J., 2014, 206, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kato C, Miyazaki K, Nakagawa A, Ohira M, Nakamura Y, Ozaki T, Imai T and Nakagawara A, Int. J. Cancer, 2004, 112, 365–375. [DOI] [PubMed] [Google Scholar]
- 297.Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, Vernet M, Schweitzer A, Saoudi Y, Pointu H, Bosc C, Salin PA, Job D and Wehland J, Proc. Natl. Acad. Sci. U S A, 2005, 102, 7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Monasterio O, Nova E, López-Brauet A and Lagos R, FEBS Letters, 1995, 374, 165–168. [DOI] [PubMed] [Google Scholar]
- 299.Bisig CG, Purro Silvia A, Contín María A, Barra Héctor S and Arce Carlos A, Eur. J. Biochem, 2002, 269, 5037–5045. [DOI] [PubMed] [Google Scholar]
- 300.Banerjee A, Panosian TD, Mukherjee K, Ravindra R, Gal S, Sackett DL and Bane S, ACS Chem. Biol, 2010, 5, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Reed LJ, Leach FR and Koike M, J. Biol. Chem, 1958, 232, 123–142. [PubMed] [Google Scholar]
- 302.Brookfield DE, Green J, Ali ST, Machado RS and Guest JR, FEBS Lett, 1991, 295, 13–16. [DOI] [PubMed] [Google Scholar]
- 303.Morris TW, Reed KE and Cronan JE Jr., J. Biol. Chem, 1994, 269, 16091–16100. [PubMed] [Google Scholar]
- 304.Puthenveetil S, Liu DS, White KA, Thompson S and Ting AY, J. Am. Chem. Soc, 2009, 131, 16430–16438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Drake CR, Sevillano N, Truillet C, Craik CS, VanBrocklin HF and Evans MJ, ACS Chem. Biol, 2016, 11, 1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Liu W, Samanta SK, Smith BD and Isaacs L, Chem. Soc. Rev, 2017, 46, 2391–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Fernández-Suárez M, Chen TS and Ting AY, J. Am. Chem. Soc, 2008, 130, 9251–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Roux KJ, Kim DI, Raida M and Burke B, J. Cell Biol, 2012, 196, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M and von Figura K, Cell, 2003, 113, 435–444. [DOI] [PubMed] [Google Scholar]
- 310.Rush JS and Bertozzi CR, J. Am. Chem. Soc, 2008, 130, 12240–12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Hudak JE, Yu HH and Bertozzi CR, J. Am. Chem. Soc, 2011, 133, 16127–16135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Smith EL, Giddens JP, Iavarone AT, Godula K, Wang LX and Bertozzi CR, Bioconjugate Chem, 2014, 25, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Kudirka RA, Barfield RM, McFarland JM, Drake PM, Carlson A, Bañas S, Zmolek W, Garofalo AW and Rabuka D, ACS Med. Chem. Lett, 2016, 7, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Catalent, https://www.catalent.com/index.php/content/view/full/2527, (accessed June 2018).