

Abstract
Ancient DNA (aDNA) studies often rely on standard methods of mutation calling, optimized for high-quality contemporary DNA but not for excessive contamination, time- or environment-related damage of aDNA. In the absence of validated datasets and despite showing extreme sensitivity to aDNA quality, these methods have been used in many published studies, sometimes with additions of arbitrary filters or modifications, designed to overcome aDNA degradation and contamination problems. The general lack of best practices for aDNA mutation calling may lead to inaccurate results. To address these problems, we present ARIADNA (ARtificial Intelligence for Ancient DNA), a novel approach based on machine learning techniques, using specific aDNA characteristics as features to yield improved mutation calls. In our comparisons of variant callers across several ancient genomes, ARIADNA consistently detected higher-quality genome variants with fast runtimes, while reducing the false positive rate compared with other approaches.
Keywords: ancient DNA, genome variants, machine learning
1. Introduction
Prior to the development of next-generation sequencing (NGS) methods for aDNA sequencing, comparative analysis relied on the physical analysis of remains. The combination of increased quality from NGS technology as well as new methodology for reliable extraction and library preparation of ancient DNA (aDNA) samples has led to an influx of genomic studies of extinct species.1–6 Early genomic studies adopted mitochondrial genome sequence analysis representing a leap forward in evolutionary research. Until now, many such aDNA studies have been mostly limited to mitochondrial and small genome regions,7–11 given the problems with extraction of usable DNA in sufficient quantity.3,5,12–14 Recent advances have enabled amplifying enough nuclear DNA to allow for complete genome sequencing of ancient samples, although also leading to potentially compounding effects on coverage, quality, and contamination.3,4,12,15 Accuracy in these studies is especially important for comparative and evolutionary analysis against living species.1,6,12,16–20 Experimental and computational methods upstream of mutation calling were developed attempting to mitigate complications of aDNA sequencing including contamination from microbes and human handling, fragmentation, depurination, and deamination.3,12–15,19–29
Such methods often rely on detecting degradation of aDNA due to extensive exposure to the environment and the physical handling of samples over time,12,22,23 which is used to differentiate between sample and noise.4,21,24–26 Filtering out contamination of contemporary DNA from aDNA samples uses short read lengths, as aDNA is often highly fractured; thus long read lengths are comparatively rare and likely a contemporary contaminant.2,13,21,26,27 Other features can be used for reducing error in aDNA studies, such as substitutions arising from depurination events frequently occurring before strand breaks,24 or deamination events often found at the ends of fragments.24,25 Compensation for these nucleotide change events is frequently made by masking such substitutions if they occur towards the end of reads.2,16
Although these methods have been shown to decrease the bias caused by aDNA damage and contamination,3,4,21 there is yet to be a consensus method to address the issue of mutation calling. Typical approaches are often ad hoc extensions using existing algorithms, such as GATK.30 However, there is little similarity among these extensions in the filtering of read depth, quality, masking locations, or mapping characteristics.1,2,16–18,31 For example, recent publications on the sequencing of various woolly mammoth and ancient human whole genomes have all utilized differing methods in the quality control of sequencing reads despite working with similar datasets.31 Additionally, a study in Neandertal genomes has demonstrated that the use of GATK on even highly processed sequence data potentially yields inaccurate results.32
Due to the variation of quality and quantity of usable DNA in ancient samples and divergent methods to extract the maximum amount of information, there can be large discrepancies in aDNA findings and interpretations.2,12,16–18,31 Many of the currently employed variant calling algorithms are utilized with limited validation of results or proof of efficacy due to the constraints of aDNA sample availability.
Here, we introduce ARIADNA (ARtificial Intelligence for Ancient DNA), a novel approach for detecting single-nucleotide variants (SNVs) in aDNA samples. Given the lack of validated ground truth datasets for aDNA, it uses common and unique variants in multiple woolly mammoth genomes to train a predictive machine learning (ML) model. ARIADNA employs our fast GROM genome scanning engine33 to find all potential SNVs (PSNVs) found as deviations between sample and reference genomes and then utilizes a boosted regression tree algorithm for training and classification of potential mutation sites. The unique features of the corresponding sites are used by our algorithm to determine the difference between bona fide mutations in aDNA and noise due to aDNA degradation or contamination. We compared ARIADNA results on (i) woolly mammoth genomes with both the most commonly employed mutation caller, GATK, and a recently developed Bayesian model, AntCaller,34 (ii) the Altai Neandertal genome with GATK and AntCaller, as well as with SNV calls from two studies,1,32 and (iii) a simulated aDNA genome. Our comparisons demonstrate that ARIADNA provides the most accurate and comprehensive mutation call sets with stable nucleotide substitution frequencies and high call quality in these ancient genomes.
2. Materials and methods
The backbone of our method consists of a ML algorithm tailored for aDNA mutation calling by utilizing unique features found in aDNA samples. We implement the use of boosted regression tree models35 with the available python library, scikit learn,36 building a succession of additive decision trees to best classify known data (training set). The algorithm assigns thresholds for feature values used within the trees from given data of true positive (TP) and false positive (FP) calls in the training set. Through a series of these trees, prediction deviance from known truth is continually reduced. Our feature set is generated using a modification of our comprehensive mutation caller GROM33 to act as a genome scanner and output feature information at potential mutation locations. These features include common measures such as read depth, SNV count, read and base quality as well as features unique to aDNA, such as distance from read end, C→T substitutions, and neighbouring mutation rates (Table 1). Once this series of trees is built from the training data, a model is constructed and classification of further data (known, for testing, or unknown, for implementation) can take place (Fig. 1). A hold back set of known mutations is used to test performance. The known values of the holdout set are not used in any way during training.
Table 1.
Features used in the ML classification algorithm
| Mutation probability | A count (low mapq) | A prior nucleotide | SNV base quality (high mapq) |
|---|---|---|---|
| Read depth (high mapq) | C count (low mapq) | T PRIOR NUCLEOTIDE | SNV base quality (high and low mapq) |
| Read depth (low mapq) | G count (low mapq) | C prior nucleotide | SNV mapping quality (high mapq) |
| Unmapped (forward) | T count (low mapq) | G prior nucleotide | SNV mapping quality (high and low mapq) |
| Unmapped (reverse) | A reference | A following nucleotide | SNV base quality read count (high mapq) |
| Soft-clipping read depth | T reference | T following nucleotide | SNV mapping quality read count (high mapq) |
| A count | C reference | C following nucleotide | SNV read count (high and low mapq) |
| C count | G reference | G following nucleotide | SNV position in read |
| G count | A SNV | A and soft-clipping | SNV forward strand |
| T count | T SNV | C and soft-clipping | |
| Repeat region | C SNV | G and soft-clipping | |
| Nearby SNV count | G SNV | T and soft-clipping |
Figure 1.

The design of the ML method for training and implementation.
We tested our method on four woolly mammoth samples M4, M25, Wrangel Island, and Oimyakon.16,17 These samples originated from two different studies, with the M4 and M25 samples being suspected of experiencing high levels of problems associated with aDNA sequencing.31 WGS fastq files for woolly mammoths M4 and M25 were downloaded from the Sequence Read Archive (SRA), http://www.ncbi.nlm.nih.gov/sra (project accession number: PRJNA281811). WGS fastq files for the Wrangel and Oimyakon woolly mammoths were downloaded from the European Nucleotide Archive (ENA), http://www.ebi.ac.uk/ena (accession number: ERP008929). WGS fastq files were mapped to the African Elephant reference genome loxAfr3, downloaded from UCSC (https://genome.ucsc.edu, http://hgdownload.soe.ucsc.edu/goldenPath/loxAfr3/bigZips/), using BWA MEM, version 0.7.4, with default parameters. Duplicates were removed using SAMtools,37 version 0.1.19. ARIADNA, GATK, and AntCaller were run on the resulting alignment files using default parameters. We limited analysis to supercontigs/scaffolds ≥1,000,000 bases. PSNVs were detected using GROM, customized to include output of additional features from Table 1.
Simulated aDNA was generated using Gargammel.38 Alignment data from scaffold_100 of the elephant Parvathy16 against the African Elephant reference genome was used to guide creation of the simulated aDNA sample. Variant sites to be used as a known validation were reported using SAMtools mpileup, before modification by Gargammel, and required a minimum variant allele frequency of 20%. Allele frequencies up to 70% were considered heterozygous and those greater than 70% homozygous. Six independent aDNA read simulations between 5× and 30× read coverage were constructed using Gargammel. The simulated aDNA reads were then aligned to the African Elephant reference genome loxAfr3 using BWA MEM as described above. ARIADNA, GATK, and AntCaller were run using default parameters. Comparison between the SAMtools output (pre-simulation) and the various caller outputs (post-aDNA simulation) were used to calculate the False Discovery Rate (FDR).
Additional testing was performed on the Altai Neandertal chromosome 1,1 using BAM files hosted at Max Planck Institute for Evolutionary Anthropology (http://cdna.eva.mpg.de/Neandertal/altai/). Calls and feature information were produced by GROM. The VCF files produced by GATK and snpAD from the respective publications1,32 were used for comparison. These were downloaded from the hosted Neandertal files at Max Planck Institute for Evolutionary Anthropology (for the 2014 dataset: http://cdna.eva.mpg.de/Neandertal/altai/AltaiNeandertal/VCF/, for the 2017 dataset: http://cdna.eva.mpg.de/Neandertal/Vindija/VCF/) to better observe mutation rates and nucleotide change frequencies. The GATK output was filtered, removing all listed “Low quality” calls from the vcf file before any comparisons were made. Further comparisons were made from 20 random genomes of the 1,000 Genomes Project;39 10 individuals from the European population group, and 10 individuals from the East Asian population group. These two groups are believed to be the contemporary populations that are most related to the Neandertals. Here mutation information was provided through the 1,000 Genomes Project VCF (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/).
To identify variants affecting essential genes, a list of essential human genes was downloaded from the Online GEne Essentiality database (http://ogee.medgenius.info/browse/). Only genes designated as “essential” were used in the analysis. Variants found in these genes were uploaded to the Ensembl Variant Effect Predictor (https://www.ensembl.org/vep) to categorize impact. The inbred region of the Neandertal genome analysed (chromosome 21: 17081807-35881807) was selected according to Prüfer et al.32
Given the lack of validated aDNA datasets, we used two simplifying assumptions for the development of our training and testing sets. First, we considered PSNVs shared between all woolly mammoth genomes to be TP locations (we did not take zygosity into account when designing our datasets). Conversely, potential mutation locations that only occurred in a single individual were deemed FP. We reasoned that these two sets will contain large numbers of primarily TP and FP, respectively, sufficient to train an effective classifier. The shared PSNVs that occurred in all woolly mammoth samples served as validation that the mutation did not occur as a result of contamination, degradation, or sequencing artefact, as opposed to unique PSNVs. Additionally, the use of woolly mammoth training samples for this study (as compared with Neandertal or ancient human) reduced the risk of misrepresented calls due to misalignment or contamination by closely related human samples when analysing the Altai Neandertal genome.13,40
3. Results and discussion
3.1 Mammoth genomes
We used GROM to scan the mammoth genomes for any evidence of difference with the reference genome. This yielded an average of one PSNV per 140 bp, i.e. between 18 million and 23 million locations per genome. Of these, 15 million PSNVs shared some evidence in all woolly mammoth genomes, while 6.6 million sites were unique to single woolly mammoth genomes. In the absence of validated SNV datasets, we used shared PSNVs as TPs and unique PSNVs as FPs (the remaining PSNVs were shared between 2 or 3 mammoths and not used for training). Although it is certain that some true mutations were picked up in the FP set, we reasoned that the effects of this mis-classification of events would be diminished due to the high frequency of real FPs among unique PSNVs. Additionally, the validation of TPs across four genomes should alleviate problems with mis-classification during application of the trained model. Mutation events shared between either two or three of the woolly mammoth samples were ignored in ARIADNA model training to eliminate excessive uncertainty.
We utilized two woolly mammoth genomes from separate studies for the purpose of training our ML model, a specimen from Wrangel Island and an M4 sample (a noisy and potentially contaminated candidate). One million shared and one million unique PSNVs from each of the two woolly mammoths in our training set were selected at random, resulting in four million training sites total. For the test set we used the two additional woolly mammoths from each study, Oimyakon and M25, and examined the results from the largest contig (contig_0). The data from these two genomes are not used in any way as part of the training set in order to avoid over-fitting or learning the unique characteristics of all available SNVs. In contig_0, the Oimyakon and M25 samples contained 799,849 and 960,816 PSNVs, respectively. Our algorithm utilized a feature set (Fig. 1) from the GROM genome scanner and the boosted regression tree ML module implemented using scikit learn.36 This gave our algorithm 45 different features to utilize (Table 1). The parameters of the boosted regression trees algorithm in scikit learn were set to 200 trees in the construction of the classifier, and a learning rate of 0.01.
ARIADNA identified 607,354 and 599,847 mutations in contig_0 of the Oimyakon and M25 woolly mammoth samples, respectively. Of these, 569,556 (Oimyakon) and 587,621 (M25) mutation sites are shared between all four woolly mammoths. Additional 32,050 (Oimyakon) and 87,130 (M25) mutation sites are shared with at least one other woolly mammoth. Only a small number of variants identified by ARIADNA in either woolly mammoth sample are unique, 5,748 (Oimyakon) and 12,226 (M25), making up 1.0% (Oimyakon) and 2.0% (M25) of all mutation calls.
To compare our results with other methods, we also employed the commonly used GATK HaplotypeCaller30 and the more recently developed AntCaller34 on all available woolly mammoth genomes. GATK made more calls than ARIADNA (as we anticipated), while AntCaller made the fewest number of calls, with both GATK and AntCaller having the high proportion of low-support calls (Figs 2 and 3, Tables 2 and 3). In the Oimyakon sample GATK made 825,955 calls, a 36% increase over our method and AntCaller made 497,718 calls, an 18% decrease compared with ARIADNA. In the M25 sample, 1,214,873 calls were made by GATK, more than twice as many as ARIADNA, while AntCaller made the fewest number of calls, 432,188 (Table 3). However, the most drastic increase is in the number of calls made by GATK that had no evidence of a variant in any other woolly mammoth. For Oimyakon this was 47,663 mutations and 280,049 mutations for M25 (Fig. 2C). This comprised 5.8% and 23.1% of the calls GATK made in their respective genomes (Fig. 2A), a striking discrepancy, suggesting that GATK over-predicted mutations at a very high rate in ancient genomes, especially in datasets with substantial noise. Such over-prediction by GATK has also been noted in a recent study on Neandertals by Prüfer et al.,32 despite the comparatively high quality of NGS data in the Neandertal. A similar behaviour was seen in AntCaller, where only 65,004, or 13% of the calls in Oimyakon were unique, but in M25, 102,820, 23.7% of the calls made were unique.
Figure 2.

Performance of ARIADNA, GATK, and AntCaller on the woolly mammoth genomes. (A) Proportion of shared (with at least one other sample) and unique (sample-specific) calls among all calls are plotted for ARIADNA (solid), GATK (check pattern), and AntCaller (diagonal pattern) on contig_0 of the woolly mammoth genomes, with total numbers of shared and unique calls plotted in (C). Spectra of nucleotide changes shown as proportions (B) and total numbers (D) are plotted for ARIADNA (solid), GATK (check pattern), and AntCaller (diagonal pattern) on contig_0 of the woolly mammoth genomes. Oimyakon sample (three leftmost bars on each x-axis position) is represented in blue and M25 (three rightmost bars on each x-axis position) are in red.
Table 2.
Variant detection rate in basepairs per SNV (sum of scaffold lengths divided by the total number of SNVs called) by different callers in two woolly mammoth genomes
| GATK | AntCaller | ARIADNA | |
|---|---|---|---|
| Oimyakon woolly mammoth | 157 | 157 | 214 |
| M25 woolly mammoth | 107 | 300 | 216 |
Table 3.
High and low evidence calls made by different algorithms
| M25 woolly mammoth | All calls | High evidence calls | Low evidence calls | % of low evidence calls |
|---|---|---|---|---|
| GATK | 1,214,873 | 951,754 | 263,119 | 21.66 |
| AntCaller | 432,188 | 385,310 | 46,878 | 10.85 |
| ARIADNA | 599,847 | 596,199 | 3,648 | 0.61 |
| Altai Neandertal | ||||
| snpAD | 216,469 | 206,433 | 10,036 | 4.64 |
| GATK | 379,115 | 334,491 | 44,624 | 11.77 |
| ARIADNA | 272,990 | 270,943 | 2,047 | 0.75 |
Low-evidence calls are defined as having either <30% of reads supporting each call or originating from regions with <1/3 of the median read coverage.
Another indication of over-prediction can be observed in the large difference in the counts of nucleotide change type between ARIADNA and GATK for the two mammoth genomes. ARIADNA call sets were robust; we observed very little change in counts or proportion of nucleotide substitution types in either Oimyakon or M25 mammoths (Fig. 2B and D). Conversely, there were large discrepancies in the GATK predictions (>2.5-fold) in such counts between the woolly mammoth genomes (Fig. 2D).
Further, we found that in the Oimyakon samples, nearly 99% of the mutations identified using ARIADNA were shared in at least one of the woolly mammoth samples, compared with 94% called by GATK and 87% by AntCaller. This was even more starkly contrasted in the noisier M25 dataset, where 98% of variants ARIADNA detected were common in all mammoths, unlike 77% for GATK and 76% for AntCaller. More surprisingly, in this noisy dataset the use of GATK resulted in 23% of total calls being unique to M25. When analysed with GATK and AntCaller, there was a large increase in rate for unique calls between Oimyakon and M25; from less than 6% to 23% using GATK and from 13% to 23% using AntCaller (Fig. 2A). The difference in the rate of unique calls between Oimyakon and M25 using ARIADNA methods was only 1.1% (Fig. 2A). Such robustness in predicted mutation rate strongly suggests that ARIADNA likely produced a more reliable call set in aDNA than that of either GATK or AntCaller, with the latter two producing the highest and the lowest variant counts in the noisy M25 (Tables 2 and 3).
Finally, following an approach used to establish the high noise levels in the mammoth NGS data,29 we tested the quality of calls produced by GATK, AntCaller, and ARIADNA by comparing the share of reads supporting called SNVs in 20,000 randomly sampled calls of Oimyakon and M25 samples. In both cases GATK and AntCaller produced more calls with biased heterozygous read support and from lower coverage regions than ARIADNA (Fig. 3). This difference was noticeable in the Oimyakon sample (Fig. 3A, C and E), but it was quite substantial in the noisy M25 sample, where ARIADNA SNVs showed much higher read support versus those of GATK or AntCaller (Fig. 3B, D and F). We found that in the M25 mammoth, nearly 22% and 12% of the calls made by GATK and AntCaller, respectively, were of lower quality, i.e. had either <30% of reads supporting the call or originated from regions covered by <1/3 of the median number of reads (Table 3). In contrast, ARIADNA was able to filter out many heterozygous SNVs with biased read support in noisy aDNA data.
Figure 3.

Improvements in calling woolly mammoth variants with ARIADNA. A total of 20,000 randomly sampled calls from the Oimyakon (A, C, E) and M25 (B, D, F) mammoth genomes, binned by read depth (x-axis) and by share of supporting reads (SSR, y-axis), are shown for AntCaller (A, B), GATK (C, D), and ARIADNA (E, F). The size of each coloured point corresponds to the count of calls that were sampled with the given read depth/SSR combination. SSR y-axis ranges are coloured as blue [0.9–1.0], green [0.6–0.8], yellow [0.3–0.5], and red [0–0.2], to provide visual representation of large proportions of SNVs with low read evidence for SNV detected by AntCaller (A, B) and GATK (C, D), and much stronger read support for ARIADNA calls (E, F).
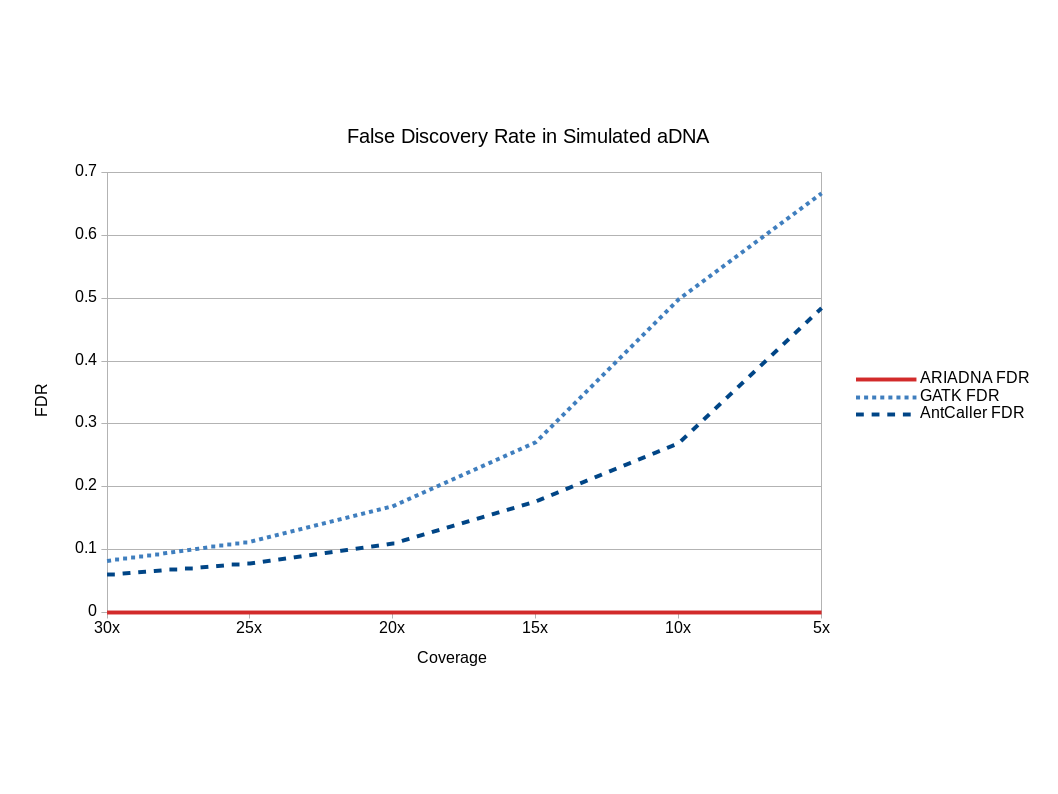
We also tested all three algorithms on six simulated datasets (changing read coverage from 5× to 30×) generated from the genome data of the closest relative of mammoths, an Asian elephant16 using Gargammel.38 Over-prediction by both GATK and AntCaller was observed in simulated datasets across all generated coverages (Supplementary Fig. S1). The false discovery rate (FDR) of both GATK and AntCaller increased dramatically with decreasing read coverage, reaching or exceeding half of the total predictions at 5× coverage. The FDR of ARIADNA remained consistently low (some 500-fold lower than for the other tools, the highest being 0.001 for 5× coverage). Thus, ARIADNA outperforms both GATK and AntCaller not only in empirical datasets but also in simulated ones.
3.2 Neandertal genome
We then tested if our model could be applied to other genomes. We used the same ML decision tree that was constructed with the woolly mammoth training set to analyse the Altai Neandertal. As a first benchmark, we used GATK calls on Altai Neandertal. As a second benchmark, we used the call set produced on that genome by Prüfer et al. using snpAD.32 All three methods utilized the identical bam files produced by Prüfer et al. We then compared call sets of GATK, snpAD, and ARIADNA to the SNVs in two 1,000 Genomes Project populations (European and East Asian).
Compared with these benchmarks of 379,115 GATK calls and 216,469 snpAD calls, ARIADNA made 272,990 calls, much closer to the number of mutations found in the two modern populations, 279,007 (European) and 283,776 (East Asian). This observation for all nucleotide changes also held true for each nucleotide change type, where ARIADNA consistently produced call sets that were most similar to the European and East Asian populations (Fig. 4).
Figure 4.

Performance of GATK, snpAD, and ARIADNA on the Altai Neandertal genome (chromosome 1). Spectra of nucleotide changes in variant call sets are plotted for GATK (dark blue, Altai Neandertal analysis of 2014, ref. 1), snpAD (light blue, Altai Neandertal analysis of 2017, ref. 32), European individuals (dark grey, EUR), East Asian individuals (light grey, EAS) and ARIADNA (red). (A) Share and (B) total number of specific substitution calls.
Our results are consistent with the earlier observations32 that GATK, being sensitive to aDNA noise and degradation, tends to over-predict the aDNA mutations, producing more variants than any other methodology. On the other hand, the approach utilizing snpAD32 seems to overcompensate in stringency and therefore to under-predict SNVs, reducing the amount of variation to less than what is otherwise found in the modern human population. In contrast, ARIADNA reported nucleotide substitutions with almost the same relative frequencies as other callers (Fig. 4A) but with absolute numbers being much closer to the modern human variation (Fig. 4B).
GATK made the greatest number of Neandertal SNV calls not made in any of the other algorithms tested here, 77,902, far ahead of snpAD, 3,374, or ARIADNA, 1,810. This was an expected result due to the reports of GATK being excessively sensitive, including noise-driven calls in its predictions.32 Neither ARIADNA nor snpAD made any common calls that were not identified by GATK. Similar to the behaviour in the woolly mammoth datasets, GATK had the highest proportion of calls made with lower coverage and lower read support, nearly 12% (Table 3), further indicating its sensitivity to aDNA noise, degradation, or contamination. Surprisingly, despite the least number of total calls made by snpAD, 4.64% of them also had low read coverage and low numbers of supporting reads, while ARIADNA produced the least number of such low-quality calls, 0.75% (Table 3).
To further compare the quality of calls made by ARIADNA, snpAD, and GATK, we analysed calls that were unique for each algorithm in the Altai Neandertal genome in essential human genes41 and catalogued potential effects using VEP42 (Table 4, shared calls not shown). GATK made by far the most unique calls in such genes, including the greatest number of missense mutations, stop loss, and stop gains. ARIADNA made the lowest number of unique calls in essential genes, followed by snpAD, and neither of the two methods produced unique missense, stop loss, or stop gain variants.
Table 4.
VEP-reported effects of algorithm-specific variants in essential genes of Altai Neandertal
| Algorithm-specific calls |
|||
|---|---|---|---|
| ARIADNA | snpAD | GATK | |
| Stop lost | 0 | 0 | 3 |
| Missense variant | 0 | 0 | 101 |
| Stop gained | 0 | 0 | 8 |
| Upstream gene variant | 2 | 7 | 1,056 |
| Non-coding transcript variant | 2 | 11 | 1,091 |
| Splice acceptor variant | 0 | 0 | 3 |
| 3 prime UTR variant | 0 | 0 | 171 |
| Incomplete terminal codon variant | 0 | 0 | 1 |
| Synonymous variant | 0 | 0 | 48 |
| Non-coding transcript exon variant | 1 | 0 | 205 |
| Regulatory region variant | 3 | 1 | 273 |
| 5 prime UTR variant | 1 | 0 | 22 |
| Splice region variant | 0 | 0 | 17 |
| Coding sequence variant | 0 | 0 | 1 |
| Stop retained variant | 0 | 0 | 1 |
| Intron variant | 8 | 17 | 2,772 |
| Downstream gene variant | 0 | 7 | 1,106 |
| Splice donor variant | 0 | 0 | 1 |
| NMD transcript variant | 7 | 9 | 1,063 |
| TF binding site variant | 0 | 0 | 6 |
Calls made by all three algorithms are not included in the counts.
And finally, we evaluated the performance of snpAD, AntCaller, and ARIADNA in a large inbred region of chromosome 21 of the Altai Neandertal, where GATK has been shown to make an excessive number of heterozygous (and thus likely erroneous) calls, compared with snpAD.32 Showing further improvement in call quality (Table 5), ARIADNA made fewer heterozygous calls than snpAD or AntCaller inside of the inbred region, both in total number and proportionally, despite making a greater number of calls than either snpAD or AntCaller overall. Additionally, outside of this inbred region, ARIADNA identified a greater number of heterozygous calls, both in total count and proportionally, than snpAD, and close to that of AntCaller.
Table 5.
Homozygous and heterozygous calls made within and outside of an inbred region of chromosome 21 of the Altai Neandertal
| Calls by algorithm |
|||
|---|---|---|---|
| ARIADNA | snpAD | AntCaller | |
| Total calls on chromosome 21 | 59,545 | 41,955 | 53,978 |
| calls outside of inbred region | |||
| heterozygous | 6,550 | 3,489 | 6,701 |
| homozygous | 23,071 | 16,618 | 19,581 |
| Calls inside of inbred region | |||
| Heterozygous | 424 | 701 | 2,215 |
| Homozygous | 29,500 | 21,147 | 25,481 |
Taken together, these comparisons demonstrate that ARIADNA trained on woolly mammoth genomes generated higher-quality SNV call sets in the Neandertal genome, when judged using both technical (read support and read depth) and biological criteria (fewest calls made in essential human genes and fewest heterozygous calls in inbred regions, with overall variation closest to other human samples). The ARIADNA model is intended for re-use and is available from Open Science Framework, https://osf.io/5bph4/.
A combination of GROM and ARIADNA is also much faster than GATK and AntCaller (snpAD has not been released and was not available for testing). In a direct comparison,33 GROM was 12–25 times faster than GATK on a single thread and more than 70 times faster on 24 threads. In our tests, AntCaller was 10–20 times slower than GROM on a single thread, somewhat faster than GATK, as in earlier AntCaller comparisons with GATK.34 Using the output from GROM, ARIADNA classifier run took between 5.5 min (Oimyakon genome) and 14.5 min (Neandertal genome) on a single thread, a significant speedup compared with >60 h of GATK runtime (all timings were performed on an Intel Xeon E5-2690 v3 processor, 2.60 GHz, with 24 threads and 128 GB RAM). A one-time ARIADNA training run took 4 h to generate a model, thus it can be easily scaled as appropriate validated datasets become available.
4. Conclusion
ARIADNA utilizes a comprehensive feature set, incorporating several features that are often used in ad hoc methods (Table 1). This includes identifying the position of the SNV within reads, base quality and mapping quality, nucleotide mismatch counts, and nucleotide change. We have also included novel features, such as accounting for nearby SNVs, adjacent nucleotides, and repeat regions, to better define difficult mapping regions or potential mutation hot-spots. The incorporation of several features as well as a decision tree ML model allows a dynamic level of filtering to compensate for changing NGS quality and read availability that is difficult to do with more static algorithms.
In summary, ARIADNA yielded consistent proportions of shared and unique mutations in the two woolly mammoth datasets compared with GATK and AntCaller. The frequency of nucleotide substitutions was also more stable using ARIADNA on the two woolly mammoth genomes than that of either GATK or AntCaller. And ARIADNA showed some 500-fold lower FDR compared with the other two algorithms when tested on six simulated aDNA datasets based on the genome of Asian elephant, the close relative of woolly mammoth.
Utilizing modern human variation from the 1,000 Genomes Project to compare results in the Altai Neandertal, we also found that the SNV calls made by ARIADNA were more consistent and potentially more relevant than calls of either GATK or snpAD. In the essential genes, ARIADNA made fewer Neandertal variant calls than either snpAD or GATK, and within an inbred region of the Altai Neandertal, ARIADNA made the lowest number of heterozygous calls.
This testing suggests that the approach we used for ARIADNA is superior for variant detection in ancient genome samples and has the capability to build models that can be utilized with very fast runtimes for improved variant finding across a range of species and read coverages.
5. Data availability
Whole genome sequencing fasta files for the woolly mammoths M4 and M25 and for the elephant Parvathy are available from the Sequence Read Archive (SRA), http://www.ncbi.nlm.nih.gov/sra (project accession number: PRJNA281811).
Whole genome sequencing fasta files for the Wrangel and Oimyakon woolly mammoths are available from the European Nucleotide Archive (ENA), http://www.ebi.ac.uk/ena (accession number: ERP008929).
Whole genome sequencing fasta files were mapped to the African reference genome loxAfr3, is available from UCSC (https://genome.ucsc.edu, http://hgdownload.soe.ucsc.edu/goldenPath/loxAfr3/bigZips/).
BAM files for the Altai Neandertal are hosted at Max Planck Institute for Evolutionary Anthropology (http://cdna.eva.mpg.de/Neandertal/altai/).
Vcf files utilizing GATK from Prüfer et al. 20141 are hosted at Max Planck Institute for Evolutionary Anthropology (http://cdna.eva.mpg.de/Neandertal/altai/AltaiNeandertal/VCF/).
Vcf files utilizing snpAD from Prüfer et al. 201732 are hosted at Max Planck Institute for Evolutionary Anthropology (http://cdna.eva.mpg.de/Neandertal/Vindija/VCF/).
1,000 Genomes variant information is available from the 1,000 Genomes ftp (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/).
The ARIADNA model and associated wrapper are available on Open Science Framework at https://osf.io/5bph4/.
Conflict of interest
The authors declare that they have no competing financial interests.
Funding
Work in A.G.’s laboratary is supported by the grants from the National Science Foundation (DBI-1458202), National Institutes of Health (R15CA220059) and New Jersey Health Foundation.
Supplementary Material
{kind=link}
References
- 1. Prüfer K., Racimo F., Patterson N., et al. 2014, The complete genome sequence of a Neanderthal from the Altai Mountains, Nature, 505, 43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parks M., Lambert D.. 2015, Impacts of low coverage depths and post-mortem DNA damage on variant calling: a simulation study, BMC Genomics, 16, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rizzi E., Lari M., Gigli E., De Bellis G., Caramelli D.. 2012, Ancient DNA studies: new perspectives on old samples, Genet. Sel. Evol., 44, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gansauge M., Meyer M.. 2014, Selective enrichment of damaged DNA molecules for ancient genome sequencing, Genome Res., 24, 1543–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prüfer K., Stenzel U., Hofreiter M., Pääbo S., Kelso J., Green R.. 2010, Computational challenges in the analysis of ancient DNA, Genome Biol., 11, R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith S., Kawash J., Karaiskos S., Biluck I., Grigoriev A.. 2017, Evolutionary adaptation revealed by comparative genome analysis of woolly mammoths and elephants, DNA Res., 24, 359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krings M., Geisert H., Schmitz R., Krainitzki H., Pääbo S.. 1999, DNA sequence of the mitochondrial hypervariable region II from the Neandertal type specimen, Proc. Natl. Acad. Sci. USA, 96, 5581–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noro M., Masuda R., Dubrovo I., Yoshida M., Kato M.. 1998, Molecular phylogenetic inference of the woolly mammoth Mammuthus primigenius, based on complete sequences of mitochondrial cytochrome b and 12S ribosomal RNA genes, J. Mol. Evol., 46, 314–26. [DOI] [PubMed] [Google Scholar]
- 9. Krause J., Dear P., Pollack J., et al. 2006, Multiplex amplification of the mammoth mitochondrial genome and the evolution of Elephantidae, Nature, 439, 724–7. [DOI] [PubMed] [Google Scholar]
- 10. Green R., Krause J., Ptak S., et al. 2006, Analysis of one million base pairs of Neanderthal DNA, Nature, 444, 330–6. [DOI] [PubMed] [Google Scholar]
- 11. Krings M., Stone A., Schmitz R., Krainitzki H., Stoneking M., Pääbo S.. 1997, Neandertal DNA sequences and the origin of modern humans, Cell, 90, 19–30. [DOI] [PubMed] [Google Scholar]
- 12. Rasmussen M., Li Y., Lindgreen S., et al. 2010, Ancient human genome sequence of an extinct Palaeo-Eskimo, Nature, 463, 757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Green R., Briggs A., Krause J., et al. 2009, The Neandertal genome and ancient DNA authenticity, EMBO J., 28, 2494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Willerslev E., Cooper A.. 2005, Ancient DNA, Proc. Biol. Sci., 272, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malmström H., Storå J., Dalén L., Holmlund G., Götherström A.. 2005, Extensive human DNA contamination in extracts from ancient dog bones and teeth, Mol. Biol. Evol., 22, 2040–7. [DOI] [PubMed] [Google Scholar]
- 16. Lynch V., Bedoya-Reina O., Ratan A., et al. 2015, Elephantid genomes reveal the molecular bases of woolly mammoth adaptations to the Arctic, Cell Rep., 12, 217–28. [DOI] [PubMed] [Google Scholar]
- 17. Palkopoulou E., Mallick S., Skoglund P., et al. 2015, Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth, Curr. Biol., 25, 1395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lazaridis I., Patterson N., Mittnik A., et al. 2014, Ancient human genomes suggest three ancestral populations for present-day Europeans, Nature, 513, 409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schuenemann V., Peltzer A., Welte B., et al. 2017, Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods, Nat. Commun., 8, 15694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mathieson I., Lazaridis I., Rohland N., et al. 2015, Genome-wide patterns of selection in 230 ancient Eurasians, Nature, 528, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sawyer S., Krause J., Guschanski K., Savolainen V., Pääbo S.. 2012, Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA, PLoS One, 7, e34131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Noonan J., Hofreiter M., Smith D., et al. 2005, Genomic sequencing of Pleistocene cave bears, Science, 309, 597–9. [DOI] [PubMed] [Google Scholar]
- 23. Höss M., Dilling A., Currant A., Pääbo S.. 1996, Molecular phylogeny of the extinct ground sloth Mylodon darwinii, Proc. Natl. Acad. Sci. USA, 93, 181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Briggs A., Stenzel U., Johnson P., et al. 2007, Patterns of damage in genomic DNA sequences from a Neandertal, Proc. Natl. Acad. Sci. USA, 104, 14616–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brotherton P., Endicott P., Sanchez J., et al. 2007, Novel high-resolution characterization of ancient DNA reveals C> U-type base modification events as the sole cause of post mortem miscoding lesions, Nucleic Acids Res., 35, 5717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pääbo S. 1989, Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification, Proc. Natl. Acad. Sci. USA, 86, 1939–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pääbo S., Poinar H., Serre D., et al. 2004, Genetic analyses from ancient DNA, Annu. Rev. Genet., 38, 645–79. [DOI] [PubMed] [Google Scholar]
- 28. Hofreiter M., Serre D., Poinar H., Kuch M., Pääbo S.. 2001, Ancient DNA, Nat. Rev. Genet., 2, 353–9. [DOI] [PubMed] [Google Scholar]
- 29. Morozova I., Flegontov P., Mikheyev A. S., et al. 2016, Toward high-resolution population genomics using archaeological samples, DNA Res., 23, 295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DePristo M., Banks E., Poplin R., et al. 2011, A framework for variation discovery and genotyping using next-generation DNA sequencing data, Nat. Genet., 43, 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogers R., Slatkin M.. 2017, Excess of genomic defects in a woolly mammoth on Wrangel island, PLoS Genet., 13, e1006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prüfer K., de Filippo C., Grote S., et al. 2017, A high-coverage Neandertal genome from Vindija Cave in Croatia, Science, 358, 655–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith S., Kawash J., Grigoriev A.. 2017, Lightning-fast genome variant detection with GROM, Gigascience, 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou B., Wen S., Wang L., Jin L., Li H., Zhang H.. 2017, AntCaller: an accurate variant caller incorporating ancient DNA damage, Mol. Genet. Genomics, 292, 1419–30. [DOI] [PubMed] [Google Scholar]
- 35. Friedman J. 2001, Greedy function approximation: a gradient boosting machine, Ann. Stat., 29, 1189–232. [Google Scholar]
- 36. Pedregosa F., Varoquaux G., Gramfort A., et al. 2011, Scikit-learn: machine learning in Python, J. Mach. Learn. Res., 12, 2825–30. [Google Scholar]
- 37. Li H., Handsaker B., Wysoker A., et al. 2009, The sequence alignment/map format and SAMtools, Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Renaud G., Hanghøj K., Willerslev E., Orlando L.. 2017, gargammel: a sequence simulator for ancient DNA, Bioinformatics, 33, 577–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. 1000 Genomes Project Consortium. 2015, A global reference for human genetic variation, Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wall J., Kim S.. 2007, Inconsistencies in Neanderthal genomic DNA sequences, PLoS Genet., 3, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen W., Minguez P., Lercher M., Bork P.. 2012, OGEE: an online gene essentiality database, Nucleic Acids Res., 40, D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McLaren W., Gil L., Hunt S., et al. 2016, The ensembl variant effect predictor, Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.