

Abstract
Potassium channels that exhibit the property of inward rectification (Kir channels) are present in most cells. Cloning of the first Kir channel genes 25 years ago led to recognition that inward rectification is a consequence of voltage-dependent block by cytoplasmic polyamines, which are also ubiquitously present in animal cells. Upon cellular depolarization, these polycationic metabolites enter the Kir channel pore from the intracellular side, blocking the movement of K+ ions through the channel. As a consequence, high K+ conductance at rest can provide very stable negative resting potentials, but polyamine-mediated blockade at depolarized potentials ensures, for instance, the long plateau phase of the cardiac action potential, an essential feature for a stable cardiac rhythm. Despite much investigation of the polyamine block, where exactly polyamines get to within the Kir channel pore and how the steep voltage dependence arises remain unclear. This Minireview will summarize current understanding of the relevance and molecular mechanisms of polyamine block and offer some ideas to try to help resolve the fundamental issue of the voltage dependence of polyamine block.
Keywords: potassium channel, polyamine, spermidine, ion channel, protein structure
Physiology of inward rectifier potassium channels
Voltage-dependent changes in the conductance of K+, Na+, and Ca2+ channels underlie the electrical signals or action potentials that are essential to all excitable processes, and indeed to life itself (1). Physiologically, intracellular [K+] is ∼140 mm, whereas extracellular [K+] is only ∼4 mm. As a consequence, K+-selective conductances normally reverse at negative voltages and exhibit larger outward currents (at voltages positive to the reversal potential) than inward currents (at voltages negative to reversal) as illustrated in Fig. 1A. “Inward” or “anomalous” rectification refers to the phenomenon whereby K+ conductance is paradoxically reduced at positive potentials. It is a prominent feature of one major subfamily of K+ channels, the so-called “inward rectifier” (Kir) channels that are present in almost all cells (2). The functional role of Kir channels depends critically on the degree of inward rectification that they exhibit. Classical strong inward rectification, first described in skeletal muscle (3), is a property of a key current (IK1) in cardiac myocytes, as well as in glial cells and neurons in the central nervous system (4–7). Rectification of these channels is sufficiently strong that very little current flows at potentials positive to about −40 mV. In the heart, the high-potassium conductance at negative voltages ensures a very stable resting potential, but inward rectification results in suppression of conductance at depolarized potentials, allowing for the normally long plateau potentials that ensure a long refractory period and suppression of cardiac arrhythmias (8). In contrast to classical inward rectifiers, renal Kir channels (9) and ATP-sensitive K+ (KATP) channels, present in multiple cell types (10), display only “weak” rectification and allow substantial outward current to flow at positive potentials. Between the two extremes, potassium channels showing intermediate rectification properties, many of these channels being strongly dependent on ligand activation, often through G-proteins or other second messenger systems, are particularly prominent in the brain (2).
Figure 1.

A, key features of inward rectification. Under normal physiological conditions (high [K+] inside and low [K+] outside), electrodiffusive conductance through a K+-selective pore would result in large outward currents at positive potentials (top, dashed line). Inward rectification refers to decreasing currents at more positive voltages, with either a weak voltage dependence (top, yellow line) or strong voltage dependence (red line). The voltage dependence is assessed as the current relative to the unblocked or nonrectifying current (relative conductance, Grel) as a function of voltage (bottom). B, structure of Kir channels. Two opposing (a and c) subunits of Kir2.2, resolved at 2.8 Å (75) (Protein Data Bank code 5KUM) are visible, with the other two (b and d) subunits hidden, to show the ion-permeable pore of the channel. The pore is lined by distinct selectivity filter (SF), transmembrane inner cavity, and cytoplasmic pore regions. Residues that control polyamine-induced rectification in Kir2.1 are indicated (red), as well as residues that induce strong rectification when an acidic substitution is introduced in Kir6.2 (yellow). All residues are numbered according to Kir2.1 sequence. C, alignment of strong (Kir2.1 and Kir4.1) and weak (Kir1.1 and Kir6.2) Kir channel members. Representative members of Kir channel subfamilies are aligned between residues 100 and 320 (in Kir2.1). Note conservation of negative residues at the rectification controller in strong inward rectifier Kir2.1 and Kir4.1 and variable conservation of other key pore-lining negative residues.
Cloning of the first Kir channel genes in 1993 (11, 12) led to the elucidation of the structural components of each of the major types of inward rectifier channels, as well as the molecular basis of inward rectification. This Minireview represents a personal review of this 25-year effort, highlighting the remaining unknowns. For further insights, the reader is recommended to other detailed reviews (2, 13, 14).
Structure of inward rectifier channels
Over the last 15 years, high-resolution crystal structures of bacterial and eukaryotic Kir channels have revealed a highly conserved architecture: Kir channels are generated by tetrameric arrangements of identical or similar Kir subunits, each of which comprises a transmembrane domain (TMD)2 and a large cytoplasmic domain (Fig. 1B) (15–17). The TMD is conserved in overall structure throughout the potassium channel family (18) and includes two membrane-spanning α-helices (the outer and inner helices, termed M1 and M2, respectively) (19, 20), connected by an extracellular turret region, a short pore helix, and the ion selectivity filter. The selectivity filter (Fig. 1B, SF) at the outer end of the channel generates the narrowest part of the pore that contains four selective binding sites for K+ ions. Below the selectivity filter, the inner M2 helices line the “inner cavity” continuation of the pore. Below that, the cytoplasmic (Kir) domain, unique to Kir channels and consisting primarily of multiple β-sheets, lines a long extension of the pore (the cytoplasmic pore) below the transmembrane region through which permeant ions and blockers must pass (Fig. 1B). Although the cytoplasmic pore region is generally quite wide (Fig. 1B), and ions are likely to be fully hydrated within it, there is a narrowing at what is termed the “G-loop,” potentially an additional location of channel gating (21, 22).
Mechanism of inward rectification
Pore-block by internal cations
We have a clear picture of the physically distinct processes that underlie voltage-dependent transitions of the depolarization-activated Kv, Na+, and Ca2+ channels. Following activation, current declines, a process referred to as “inactivation,” which involves blocking of the pore by a positively charged cytoplasmic “ball” domain that gains access to its binding site after the channel has opened (23, 24). Parallels between the voltage-dependent inactivation of Kv channels and rectification of Kir channels first led Armstrong (25) to hypothesize that inward rectification might also arise from a fundamentally similar process, i.e. that inward rectification might result from a positively charged substance blocking the channel in a voltage-dependent manner, from the internal side of the membrane. Mg2+ and Na+ ions were subsequently shown to cause such effects in weak inward rectifier KATP channels (26, 27) and in cardiac inward rectifier channels (28), but a seemingly intrinsic and much more steeply voltage-dependent process was clearly a dominant cause of strong inward rectification in IK1 and other strong inward rectifier channels (29–31).
Polyamines as the cause of steeply voltage-dependent “intrinsic” rectification
The first members of the Kir channel subfamily were cloned in 1993, Kir1.1 (12) and Kir2.1 (32). This was rapidly followed by cloning of multiple additional representatives of ultimately all seven Kir subfamilies (2). The availability of cloned Kir channels in the Kir2 subfamily (which encodes the classical cardiac inward rectifier (33, 34)) permitted high-level expression in endogenous system, and facilitated the discovery of polyamines as the agents of strong inward rectification (35–37). In macro-patch experiments on strong inward rectifier channels expressed in Xenopus oocytes, we first showed that rectification gradually disappears after patch excision into divalent ion-free solutions (35), as earlier reported in cardiac IK1 channels (38). Strikingly, however, strong rectification was restored by placing excised membrane patches close to the surface of the oocyte, suggesting that soluble intrinsic factors released from the oocyte might be the cause of the rectification. Size- and charge-exclusion chromatography (35) indicated that the active agents were small polycationic organic amines, and HPLC confirmed that the naturally occurring polyamines (spermine, spermidine, and putrescine) are indeed released from the oocyte (35) (subsequently demonstrated to be through connexin hemi-channels (39)). Critically, application of micromolar levels of these polyamines to inside-out membrane patches is sufficient to restore all the essential features of classical inward rectification (Fig. 2A) (35, 36, 40, 41). The voltage dependence of the spermine and spermidine block is markedly steeper than Mg2+ block (35, 36, 40, 41), which explains why classical “intrinsic” inward rectification is much steeper than that predicted by Mg2+ block alone. The same essential nature of polyamine-induced rectification has since been demonstrated in all the Kir2 (34, 42–44), Kir3 (45), and Kir4 (46, 47) subfamily members that exhibit strong rectification physiologically. Members of the Kir1 and Kir6 subfamilies exhibit only shallow inward rectification, shown to be a consequence of only weakly voltage-dependent and low potency (millimolar sensitivity) block by Mg2+ and polyamines (27, 37, 48, 49).
Figure 2.
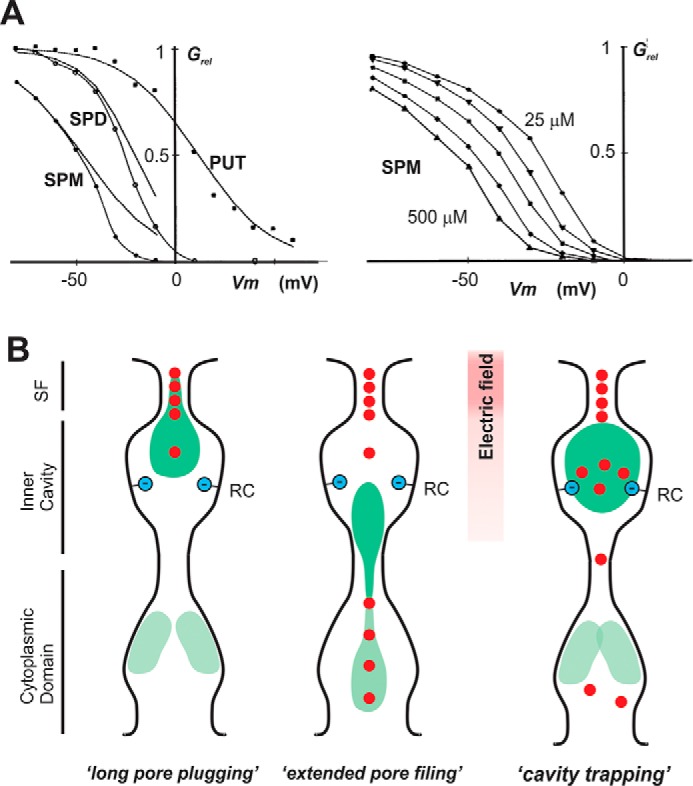
A, characteristics of polyamine-dependent rectification (redrawn from Ref. 38). Left, Grel versus voltage plots for Kir2.3 currents in the presence of 250 μm spermine4+ (SPM), spermidine3+ (SPD), or putrescine2+ (PUT). Note that both potency and steepness of the voltage dependence increase as the polyamine size and charge increase. Right, Grel versus voltage plots for Kir2.3 currents in the presence of increasing spermine concentrations (25, 50, 100, 150, and 250 μm). The Grel-Vm curves shift in parallel. Note that there is both a shallow and steep component to the voltage dependence for spermidine and spermine. B, left, “long pore plugging” model of polyamine-dependent rectification. Shallow rectification is assumed to arise from movement of the polyamine into the cytoplasmic cavity (pale site), up to the entrance to the inner cavity, potentially carrying some polyamine charge into the electric field. Steep rectification is assumed to arise from movement of the polyamine deep into the pore, between the rectification controller and the selectivity filter (dark site), with substantial polymine charge moving across the field and displacing K+ ions from the filter in the process. Center, “extended pore-filing” model of polyamine-dependent rectification. Shallow rectification is assumed to arise from movement of the polyamine into the cytoplasmic cavity (pale site), in the process “pushing” on a “chain” of K+ ions that extends through the selectivity filter. Steep rectification is assumed to arise from forward movement of the polyamine toward the rectification controller (dark site), moving up to four or five K+ ions through the selectivity filter in the process. Right, “cavity-trapping” model for polyamine-dependent rectification. We propose a new hybrid model that may reconcile objections to earlier models. Shallow rectification is again assumed to arise from movement of the polyamine into the cytoplasmic cavity (pale site), potentially displacing K+ ions into the inner cavity. The charge associated with block, determining how steep rectification will be, is determined by the net charge on the wall of the inner cavity. With four negative charges at the rectification controller, there will be four K+ counterions present. Steep rectification is then assumed to arise from movement of the polyamine into the inner cavity, through a narrow single filing region at the entrance, in the process displacing these K+ ions into and through the selectivity filter.
In search of the voltage dependence of polyamine action: The appeal of the “long pore plugging” concept
The steepness of the voltage dependence of polyamine block increases roughly in direct equivalence to the charge on the polyamine itself, from +2 (putrescine2+) to +3 (spermidine3+) to +4 (spermine4+) (35, 36), although it can be even higher. External potassium ions relieve polyamine-dependent rectification by increasing the apparent polyamine off-rate (43), as expected for a channel blocker that interacts with permeant ions within the pore. As a linear molecule, spermine is very long (almost 20 Å long), compared with a K+ ion, but of similar diameter (∼3 Å). It was an obvious possibility that, in blocking Kir channels, spermine lies along the pore axis, binding at multiple sites that would otherwise be occupied by K+ ions. Our original conception was that the polyamine would enter deeply into the pore, entering what we now recognize as the selectivity filter, which is otherwise occupied by two or more K+ ions (18, 50), thereby achieving the necessary voltage dependence by moving essentially all four spermine charges through the electric field–what we termed “long-pore plugging” (Fig. 2B).
Close inspection revealed that spermine block of Kir2 subfamily channels also includes a shallow voltage-dependent component at more negative voltages (Fig. 2A), which suggested a second blocking component, perhaps within the cytoplasmic pore (Fig. 2B) (40, 51). We originally proposed two concentration-dependent binding reactions (i.e. two polyamines independently entering the channel pore), and a voltage-dependent transition deep within the electric field to account for the steep voltage-dependent component of spermine-induced rectification (40).
In this earliest conception, based on the original pore-blocking model of Woodhull (52), the voltage-dependent block results from the movement of the charged blocker itself into the electric field; interactions of the blocking particle with permeant ions are ignored. If the channel was blocked by only one spermine molecule, but the entering spermine molecule had to sweep out permeant ions to reach its binding site, excess charge movement could result, as first pointed out by Ruppersberg et al. (53), and hence the voltage dependence of the block at a selectivity filter site could be underestimated.
In search of the voltage dependence of polyamine action: Insights from channel structures
The Kir channel permeation pathway has now been elucidated in exquisite detail (Fig. 1B), and many mutations that affect polyamine blocking have been identified (Fig. 1A). Aspartate 172, located in the M2 region of Kir2.1, was the first residue implicated in the classical inward rectification of these channels. Subsequent mutational analyses showed that this residue is a major determinant of both polyamine-blocking affinity and voltage dependence (35, 37, 51, 54). Neutralization of this residue in Kir4.1 reduces apparent affinity for both Mg2+ and spermine by 5 orders of magnitude (37). Conversely, introduction of a negative charge at the equivalent site induces strong rectification in otherwise only weakly rectifying Kir1.1 and Kir6.2 channels (48, 49); hence, this residue has been termed the “rectification controller.” However, the rectification controller does not exert an “all-or-none” effect. Some Kir channels that exhibit relatively strong rectification lack an acidic residue at this position (e.g. Kir3.2), and rectification is weakened but not lost in Kir2.1[D172N] mutant channels (54–56). Importantly, strong rectification can be induced in both Kir6.2 and Kir2.1 not just when acidic residues are introduced at the rectification controller residue but at any pore-facing location in the inner cavity (Fig. 1B) (56, 57). It is thus generally agreed that polyamines must directly interact with or sense the charge at this site, but as discussed below, it is controversial whether this is as far as the spermine gets or whether this residue determines how much further the spermine travels in the blocking process. Compelling support for the idea that the spermine actually reaches beyond the rectification controller is provided by experiments showing that spermine occupancy of the deep site can protect against methyl thiosulfhydryl modification of cysteine residues introduced one helix turn above, but not one helix turn below, the rectification controller (58, 59), as well as in silico docking experiments that indicate stable spermine binding at the top of the inner cavity and entrance to the selectivity filter in Kir channel model structures (58).
Multiple other residues in pore-lining regions of the Kir domain have also been shown to be important for polyamine block (Fig. 1B), including two (Glu-224 and Glu-299 in Kir2.1) that create a ring of negative charge near the top of the cytoplasmic pore (Fig. 1B) (19, 20). When these residues are neutralized, the rate of block is slowed (56, 60, 61), and the apparent affinity of the deepest site is reduced (51, 56, 60, 62, 63). In addition, two more acidic pore-lining residues (Asp-255 and Asp-259), which form a second ring of charge closer to the cytoplasmic end of the channel (21), as well as a neighboring aromatic residue (Phe-254 in Kir2.1) (64), also contribute to polyamine block (Fig. 1B). At least for this lower negative charge ring, the primary effect of neutralizing these residues is on the rates of polyamine entry and exit to deeper sites within the pore (64–66), rather than on affinity, but still it seems clear that multiple potential locations exist where spermine could reside as part of the deep blocking process. Within the narrow confines of the channel pore, it is likely that bulk electrostatics will not apply and that significant long-range electrostatic effects are possible (67), but these cytoplasmic pore-lining charges are likely to be too far from the Asp-172 site for a single bound spermine ion to directly contact both (Fig. 1B). As discussed below, these structural features of rectification have led to a second model of inward rectification, which we will term the “extended pore filing” model, to explain the uniquely steep voltage-dependent block that is the essential feature of strong inward rectification.
In search of the voltage dependence of polyamine action: The appeal of the “extended pore filing” concept
Polyamine analogs of different lengths and valences highlight the fact that movement of charged polyamines through the transmembrane field cannot solely account for the steeply voltage-dependent rectification of Kir2.x channels. Diamino alkanes, with hydrocarbon length from 4 (putrescine, 1,4-diaminobutane) up to 12, all cause voltage-dependent rectification (68). Putrescine blocks with an effective valence of +2, but as the hydrocarbon chain length increases, the effective valence of channel block increases up to the same steep voltage dependence as the spermine block (i.e. equivalent to +4–5 elementary charges) in 1,12-diaminododecane, which has a similar length to spermine (68). Because these diamines all carry only two charges, blocker migration across the electric field clearly cannot then fully account for the steep voltage dependence that is observed. Observations such as these add to the argument that at least some of the voltage dependence of the polyamine block must arise from displacement of potassium ions by the migrating polyamine as it approaches a binding site deep within the Kir pore, “pushing” charge carried by K+ ions through the pore, and that shorter blockers might not migrate as deeply into the channel pore as longer blockers, thereby displacing fewer ions as they reach their binding site (56, 68).
Interestingly, despite their considerably higher charge, extended polyamine analogs with up to 10 amines do not exhibit any steeper voltage dependence than spermine (59, 69). This is also consistent with the notion that the effective valence associated with polyamine block does not depend on movement of the entire blocker through the transmembrane field, but rather the coupled displacement of permeant K+ ions as the blocker migrates to its maximal depth within the pore. In addition, modification of the terminal amines of spermine or other polyamine analogs typically results in weakening of blocker affinity but not of the effective charge associated with the block (58, 70).
The recognition that the Kir cytoplasmic domain extends the Kir channel pore into the cytoplasm raised the possibility that there could be an extended chain of permeant ions connecting the selectivity filter potassium ions through the inner cavity ion to additional ions in the cytoplasmic pore (15). If so, polyamines entering the cytoplasmic pore could “push” the chain, resulting in ions at the head moving through the filter and carrying the necessary charge associated with block across the electric field, without a need for the polyamine itself to enter the electric field. As originally envisioned (15, 56), such coupled movement of polyamines and potassium ions through the pore requires that that the ions move in single file. As subsequent mutagenesis studies indeed implicated charged residues closer to the cytoplasmic side of the channel in controlling the voltage dependence of rectification, Lu and co-workers (56, 64, 66, 70, 71) have championed this “extended pore filing” model (Fig. 2B), proposing that, even in its deepest site, spermine is located below the rectification controller, outside the electric field, with essentially all of the voltage dependence of rectification resulting from spermine “pushing” the chain of K+ ions forward, and in the process moving four or more K+ ions across the filter and through the electric field.
In search of a unifying model for inward rectification: The contradictory issues
The two models described above are based on distinct conceptions of the nature of the charge movement associated with polyamine block and location of the polyamine within the Kir channel pore. They can each account for specific features as described above, but there are experimental findings that are not readily accounted for by either. The “long pore plugging” model, in assuming that steep voltage dependence arises from the polyamine reaching the selectivity filter, cannot easily account for the finding that charges far from this location (i.e. Glu-224) can affect steady-state spermine affinity. Moreover, as discussed below, no crystal structures have demonstrated spermine binding at the top of the inner cavity or in the selectivity filter of Kir channels. Conversely, the “extended pore filing” concept requires single ion filing to begin at the very bottom of the cytoplasmic pore. Xu et al. (64) noted that at the level of Phe-254, the pore is relatively narrow, potentially providing a “gasket” through which spermine can enter in single file, but this has not been tested further. More importantly, spermine can be trapped inside Kir channels that are closed at or near the M2 helix bundle crossing (72), and strong rectification can be induced in Kir6.2 by introduction of a negative charge right at the entrance to the selectivity filter (57), neither of which is trivially consistent with spermine residing between the cytoplasmic pore and the inner cavity.
Some resolution of the above issues may ultimately be provided by high-resolution structures that unambiguously identify the location of polyamines within the Kir channel pore. However, despite the availability of multiple Kir structures, blocker-bound structures have remained rather elusive. In 2010, Clarke et al. (73) first reported KirBac3.1 structures with spermine bound in the cytoplasmic vestibule or in the G-loop/bundle crossing region, partially buried within subunit interfaces. Clarke et al. (73) also reported an axial spermine occupying the permeation pathway at residue Leu-124, corresponding to the rectification controller residue Asp-172 in Kir2.1. However, electrophysiological or other functional studies of the polyamine block of KirBac3.1 are lacking, and given that the rectification controller is a neutral residue in KirBac3.1, it seems unlikely that this channel will actually show strong rectification (74). Hence, the ultimate relevance of these prokaryotic channel structures to strong inward rectification in eukaryotic Kir channels is not entirely clear. Unfortunately, subsequent high-resolution crystal structures of eukaryotic Kir2.2 and Kir3.2, which are both bona fide strong inward rectifiers, in numerous states and with multiple ligands bound (19, 20, 22, 75), have singularly failed to show bound polyamines. In our own laboratory, we have obtained sub-3 Å resolution structures of Kir2.2 in the presence of even very high spermine concentrations, but we still have not observed unambiguous spermine densities. Why not? Polyamines also cause inward rectification of AMPA/kainate receptors (102), and recent cryoEM structures of AMPA receptors (76) directly demonstrate binding sites for spermine and related spider toxin and adamantine derivatives. In this case, the blockers reside very deeply within the pore, essentially filling the selectivity filter itself, providing a ready explanation for the steep voltage dependence of block by “long pore plugging” in these channels, with a combination of blocker entry into the filter and displacement of permeant ions.
In search of a unifying model for inward rectification: A potential solution?
One poorly explained aspect of inward rectification just might hold a clue to the explanation. Spermine and other polyamines also block cyclic nucleotide-gated (CNG) channels, structurally related in the transmembrane region to Kir channels but lacking the Kir domain. CNG channel block exhibits a very prominent U-shaped voltage dependence, in which spermine block is steep and potent, as in Kir channels, but is completely relieved at more positive voltages, resulting in an N-shaped current-voltage relationship (77). This is most reasonably explained as resulting from a voltage-dependent permeation, or “punch-through,” of the polyamine through the channel. Despite the very high potency and voltage dependence of polyamine block of Kir channels, many also exhibit what is clearly an incomplete block at the most positive voltages. This has usually also been interpreted as resulting from a finite rate of permeation of the polyamine through the channel, requiring access to and through the selectivity filter (44, 65). However, unlike CNG channels, the incomplete block of Kir channels does not typically manifest as a voltage-dependent relief of block, i.e. as increasing conductance at increasing positive voltages, but rather there is a nonzero plateau in the conductance–voltage relationship in the presence of polyamines (47, 62, 78). The electric field may extend through the inner cavity to the cytoplasmic pore, but is almost certainly most concentrated at the selectivity filter (79), and therefore such a “standing” conductance, rather than a voltage-dependent relief of block (as seen in CNG channels), is not the predicted outcome of a “punch-through” mechanism involving polyamines traversing the electric field and permeating the channel.
Lu and co-workers (64, 66) have suggested that shorter blockers may not efficiently occlude the channel pore, particularly at shallow sites, and thereby some “slippage” or “bypass” of permeating ions may contribute to the smaller observed effective valence of block by shorter polyamines. Although this idea muddies the original “extended pore filing” concept by implying nonsingle-filing in the cytoplasmic pore, it does suggest an appealing potential explanation of Kir channel rectification that may resolve the above issues. We now propose a hybrid model, which we will term the “cavity-trapping” model (Fig. 2B). The essential features of the model are that steeply voltage-dependent polyamine block does require entry of the polyamine into the inner cavity of the channel, and it is only from this location that spermine displaces multiple resident potassium ions across the selectivity filter to achieve the relevant voltage dependence of block. The model requires the following: 1) that there be single filing of ions (including polyamines) into the inner cavity of Kir channels, and 2) that it is the inner cavity that contains the necessary number of K+ ions to achieve high-charge movement across the filter. We suggest that this will be essentially the same number of soluble cations as there are negative charges on the cavity surface; hence, there should be at least four K+ ions in the inner cavity with four acidic charges at the rectification controller, but perhaps not even one with neutral rectification controller residues. At first glance, these two premises of the model run counter to two long-held assumptions: 1) that when open and conducting, the entrance to the inner cavity should be wide, and 2) that the inner cavity may contain just one, centrally located, potassium ion. However, there is little or no evidence to support either of these assumptions.
First, in all crystallized Kir channel structures, the pore is narrow at the bundle crossing region, such that K+ ions would be excluded sterically from passing through. Even when charges introduced at this location generate constitutively open channels (80), or when all recognized gating requirements are met by mutation and phosphatidylinositol 4,5-bisphosphate binding (75), crystal structures are still so narrow as to require single ion filing at the bottom of the TMD.
Second, exactly how many cations are present in the inner cavity is a largely unaddressed issue. Although the inner cavity volume is such that even a single K+ ion within it will be at an effective concentration of ∼1 m, far higher than the bulk solution, if the “rectification controller” residues are all charged, then four counter charges must also be present. There are multiple lines of evidence that the acidic rectification controller residues must be charged. (i) Asparagine or glutamine side chains cannot substitute for aspartate or glutamate (81). (ii) The valence and potency of spermine-dependent rectification are dependent on the exact number of acidic charges at the rectification controller (82, 83). (iii) The voltage dependence of rectification is halved if only two acidic residues are present (e.g. in dimeric construct channels (57)) or if four acidic residues are present, but two positively charged moieties are also introduced into the inner cavity above the rectification controller by cysteine modification (84). (iv) methyl thiosulfhydryl triethylamine (MTSET+) modification of the inner cavity below the rectification controller causes kinetic trapping of spermine but no reduction of voltage dependence (84). With compelling evidence that spermine4+ enters the inner cavity, it is reasonable that, when absent, four other positive charges, i.e. four K+ ions, must be present.
The “cavity-trapping” model could provide a ready explanation for the failure to identify bound polyamines in the channel. First, because the model depends only on the net negativity in the inner cavity to determine the steepness of rectification, it can explain insensitivity of rectification to the exact location of the “rectification controller” charges within the inner cavity (57). Because it assumes that spermine in the inner cavity replaces mobile potassium ions, the polyamine need not occupy any single position. In addition, the model does not require that the polyamine gain access to the selectivity filter itself, and hence, even in a maximally blocked channel, K+ ions may be present in the filter. In these features, the model encompasses many of those in the “extended pore-filing” model but restricts the action to the inner cavity, explaining why mutations below this level, or even introduction of positive charge within the inner cavity but below the rectification controller (84), act primarily on polyamine access and not affinity. The model can also provide a ready explanation for “standing” conductance (i.e. incomplete block) in the maximally blocked channel: if the polyamine fails to replace all K+ ions in the inner cavity, i.e. if the inner cavity negativity is enough to hold more than four K+ ions or if shorter polyamines do not provide sufficient (i.e. +4) charges, then there may still be residual K+ ions within the cavity in addition to the polyamines, and a fixed “slippage” conductance of K+ ions past the polyamine into and through the filter could result. For very long diamines, even though K+ ions would be required to follow the spermine into the cavity to maintain charge balance, the steric bulk of the molecule might be sufficient to force a similar number of K+ ions to leave the cavity as when spermine enters. In contrast, for polyamine analogs that contain a bulky headgroup, the polyamine tail may gain access to the inner cavity, displacing K+ ions and causing inward rectification, but the head may block the entrance, thereby obviating any “slippage” conductance, as seen for instance in philanthotoxin block of Kir2 (44) or Kir4 channels (47).
Physiological and pathological relevance of polyamine-dependent rectification
Cellular polyamine levels and Kir channel rectification
Polyamines have been of interest as cellular metabolites since they were first discovered by van Leeuwenhoek in the 17th century. Acting as stabilizers for DNA structure (85), they are essential for normal and neoplastic cell growth and remain of great interest in cancer biology (86), as well as in autoimmune diseases (87), but their involvement in many cellular processes may be related to their ion channel-blocking effects. Only nanomolar to micromolar concentrations of free polyamines are needed to reproduce the degree of potassium channel inward rectification seen in intact cells (41, 88, 89), and induction of inward rectification may be the most potent physiological property of polyamines. Although cellular polyamines are typically highly buffered, total cellular polyamine concentrations (10–10,000 μm) (90) are sufficient to cause very strong rectification of Kir channels. Given the steep voltage dependence of polyamine block, it is likely that free cytoplasmic concentrations are always high enough to affect the degree of channel block somewhere within the membrane potential range traversed during an action potential (−70 to +30 mV) and that alterations of polyamine levels will therefore alter excitability. Fakler et al. (41) demonstrated that inclusion of ATP in the whole-cell patch-clamp pipette could relieve inward rectification of Kir2.1 channels, consistent with chelation of free polyamines. Bianchi et al. (91) demonstrated relief of inward rectification of endogenous currents in RBL-1 cells after treatment with an inhibitor of the polyamine synthetic enzyme S-adenosylmethionine decarboxylase. Such treatment resulted in increased cellular putrescine and decreased spermidine and spermine levels, with a shallowing of the I-V relationship and significantly increased outward Kir currents. We observed similar effects in Xenopus oocytes expressing Kir2.1 channels (92) and utilizing a Chinese hamster ovary cell line that is deficient in ornithine decarboxylase activity and requires putrescine in the medium for normal cell growth (93). We also demonstrated changes in rectification of expressed Kir2 (Kir2.3) channels, as a result of polyamine depletion (92). In these cells, removal of putrescine from the medium leads to a gradual decline in intracellular levels of putrescine, then spermidine, and finally spermine, and these changes correlate with alterations in channel-blocking kinetics predicted by excised-patch experiments with varying polyamine concentrations. The effects of altered polyamine levels on inward rectification and excitability in intact tissues are largely unexplored, but in the hearts of mice overexpressing ornithine decarboxylase (94), IK1 density was reduced but, surprisingly, the voltage dependence of rectification was essentially unchanged, despite putrescine and cadaverine levels being highly elevated (35-fold) and spermidine increased by 3.6-fold (although spermine was essentially unchanged). IK1 currents were also studied in myocytes from Gyro mice in which spermine synthase is disrupted, leading to a complete loss of spermine. In this case, IK1 current densities were not altered, but the steepness of rectification was reduced (94), demonstrating the role of spermine in controlling rectification. Intracellular dialysis of myocytes with putrescine, spermidine, or spermine caused reduction, no change, and an increase of the steepness of rectification, respectively (94). Taken together with kinetic analysis of IK1 activation, these results are consistent with spermine being a major rectifying factor at potentials positive to the potassium reversal potential (EK), spermidine dominating at potentials around and negative to EK, and putrescine being of lesser significance in rectification in the mouse heart.
Clinical relevance of inward rectification
Gyro mice demonstrate that even when spermine synthase is completely absent, the homeostatic maintenance of other polyamines ensures that even though rectification is affected (94), it is never likely to be fundamentally absent. Whether changes in inward rectification play any significant role in the pathologies resulting from altered polyamine metabolism, such as Snyder-Robinson syndrome, in which spermine synthase is disrupted, (95) remains unclear. Conversely, a consequence of altering rectification by genetic mutation of the Kir channel has been demonstrated (96). Although humans bearing Kir2.1 loss-of-function mutations suffer from Anderson-Tawil syndrome (97), characterized in part by long QT intervals on the electrocardiogram, heterozygous neutralization of the Kir2.1 “rectification controller” (mutation D172N) in two affected individuals resulted in a gain-of-function relief of rectification and increased Kir2.1 potassium current during the action potential (96). This prematurely shortens the action potential, leading to short QT syndrome (SQT3) (8). This predicts a steeper steady-state restitution curve for SQT3 patients, which may predispose them to reentrant arrhythmias (8).
It remains an unexploited possibility that pharmacological manipulation of polyamine levels in specific tissues might be an approach to treatment of disorders of excitability by altering voltage dependence of potassium channel activity. An enormous number of polyamine derivatives have been developed (98, 99), but pharmacological approaches to modulate strong inward rectifier potassium channels, in general, and to exploit polyamine-dependent rectification, in particular, have not been very forthcoming. However, a few polyamine analogs with well-understood actions in other channels may have additional specific effects in Kir channels related to polyamine block. Memantine is a blocker of glutamate receptor channels but also suppresses Kir current amplitude in apparent competition with spermine, causing cell depolarization (100). The antiarrhythmic drug propafenone blocks human cardiac Kir2 channels, apparently by decreasing the negative electrostatic charge sensed by polyamines within the cytoplasmic pore (101). Propafenone-induced block was markedly increased at lower intracellular [K+] and decreased at more positive voltages, resulting in effective reduction of inward rectification, consistent with propafenone binding within the cytoplasmic domain in such a way that it decreased the net negative charge sensed by K+ ions and polyamines.
Perspective and prospects
Expression cloning of the first inward rectifier channel genes 25 years ago led to elucidation of the structural basis of classical anomalous rectifiers, G-protein–activated potassium channels, and ATP-sensitive potassium channels. High-level expression of these cloned channels permitted the molecular basis of inward rectification–voltage-dependent block by polyamines-to be substantially defined. Despite that, a completely satisfying structural basis of polyamine–Kir channel interaction has been elusive. Although it is currently no more than a hypothesis, we hope that the new “cavity-trapping” model we propose will spur efforts to test it and that these efforts may help solve the long-standing question of where the fundamentally steep voltage dependence of inward rectification arises. From a physiological and clinical perspective, the possibility that alterations in polyamine levels during altered physiological or pathophysiological conditions might affect cellular excitability by altering Kir channel rectification is also still underexplored. Although short QT syndrome has been shown to result from a mutation that decreases the cardiac Kir2.1-dependent IK1 channel rectification, pharmacological exploitation of polyamine-dependent rectification is also still an unattained goal.
This work was supported by National Institutes of Health Grant HL140024 (to C. G. N). This article is part of a series on “Polyamines,” written in honor of Dr. Herbert Tabor's 100th birthday. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TMD
- transmembrane domain
- CNG
- cyclic nucleotide-gated
- EK
- potassium reversal potential.
References
- 1. Hille B. (1992) Ionic Channels of Excitable Membranes, Sinauer Associates, Sunderland, MA [Google Scholar]
- 2. Nichols C. G., and Lopatin A. N. (1997) Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191 10.1146/annurev.physiol.59.1.171 [DOI] [PubMed] [Google Scholar]
- 3. Katz B. (1949) Les constantes electriques de la membrane du muscle. Arch. Sci. Physiol. 2, 285–299 [Google Scholar]
- 4. Nakajima Y., Nakajima S., and Inoue M. (1988) Pertussis toxin-insensitive G protein mediates substance P-induced inhibition of potassium channels in brain neurons. Proc. Natl. Acad. Sci. U.S.A. 85, 3643–3647 10.1073/pnas.85.10.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hestrin S. (1987) The properties and function of inward rectification in rod photoreceptors of the tiger salamander. J. Physiol. 390, 319–333 10.1113/jphysiol.1987.sp016703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Newman E. A. (1993) Inward-rectifying potassium channels in retinal glial (Muller) cells. J. Neurosci. 13, 3333–3345 10.1523/JNEUROSCI.13-08-03333.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brismar T., and Collins V. P. (1989) Inward rectifying potassium channels in human malignant glioma cells. Brain Res. 480, 249–258 10.1016/0006-8993(89)90190-X [DOI] [PubMed] [Google Scholar]
- 8. Nichols C. G., Makhina E. N., Pearson W. L., Sha Q., and Lopatin A. N. (1996) Inward rectification and implications for cardiac excitability. Circ. Res. 78, 1–7 10.1161/01.RES.78.1.1 [DOI] [PubMed] [Google Scholar]
- 9. Giebisch G., Hunter M., and Kawahara K. (1990) Apical potassium channels in Amphiuma diluting segment: effect of barium. J. Physiol. 420, 313–323 10.1113/jphysiol.1990.sp017914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 471–476 10.1038/nature04711 [DOI] [PubMed] [Google Scholar]
- 11. Kubo Y., Reuveny E., Slesinger P. A., Jan Y. N., and Jan L. Y. (1993) Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature 364, 802–806 10.1038/364802a0 [DOI] [PubMed] [Google Scholar]
- 12. Ho K., Nichols C. G., Lederer W. J., Lytton J., Vassilev P. M., Kanazirska M. V., and Hebert S. C. (1993) Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362, 31–38 10.1038/362031a0 [DOI] [PubMed] [Google Scholar]
- 13. Baronas V. A., and Kurata H. T. (2014) Inward rectifiers and their regulation by endogenous polyamines. Front. Physiol. 5, 325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu Z. (2004) Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 66, 103–129 10.1146/annurev.physiol.66.032102.150822 [DOI] [PubMed] [Google Scholar]
- 15. Nishida M., and MacKinnon R. (2002) Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111, 957–965 10.1016/S0092-8674(02)01227-8 [DOI] [PubMed] [Google Scholar]
- 16. Kuo A., Gulbis J. M., Antcliff J. F., Rahman T., Lowe E. D., Zimmer J., Cuthbertson J., Ashcroft F. M., Ezaki T., and Doyle D. A. (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300, 1922–1926 10.1126/science.1085028 [DOI] [PubMed] [Google Scholar]
- 17. Nishida M., Cadene M., Chait B. T., and MacKinnon R. (2007) Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26, 4005–4015 10.1038/sj.emboj.7601828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., and MacKinnon R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 10.1126/science.280.5360.69 [DOI] [PubMed] [Google Scholar]
- 19. Tao X., Avalos J. L., Chen J., and MacKinnon R. (2009) Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326, 1668–1674 10.1126/science.1180310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hansen S. B., Tao X., and MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 10.1038/nature10370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pegan S., Arrabit C., Zhou W., Kwiatkowski W., Collins A., Slesinger P. A., and Choe S. (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8, 279–287 10.1038/nn1411 [DOI] [PubMed] [Google Scholar]
- 22. Whorton M. R., and MacKinnon R. (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208 10.1016/j.cell.2011.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoshi T., Zagotta W. N., and Aldrich R. W. (1990) Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538 10.1126/science.2122519 [DOI] [PubMed] [Google Scholar]
- 24. Zhou M., Morais-Cabral J. H., Mann S., and MacKinnon R. (2001) Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 411, 657–661 10.1038/35079500 [DOI] [PubMed] [Google Scholar]
- 25. Armstrong C. M. (1969) Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J. Gen. Physiol. 54, 553–575 10.1085/jgp.54.5.553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ciani S., and Ribalet B. (1988) Ion permeation and rectification in ATP-sensitive channels from insulin-secreting cells (RINm5F): effects of K+, Na+ and Mg2+. J. Membr. Biol. 103, 171–180 10.1007/BF01870947 [DOI] [PubMed] [Google Scholar]
- 27. Nichols C. G., Ho K., and Hebert S. (1994) Mg2+-dependent inward rectification of ROMK1 potassium channels expressed in Xenopus oocytes. J. Physiol. 476, 399–409 10.1113/jphysiol.1994.sp020141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vandenberg C. A. (1987) Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc. Natl. Acad. Sci. U.S.A. 84, 2560–2564 10.1073/pnas.84.8.2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuda H., Saigusa A., and Irisawa H. (1987) Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature 325, 156–159 10.1038/325156a0 [DOI] [PubMed] [Google Scholar]
- 30. Matsuda H., Matsuura H., and Noma A. (1989) Triple-barrel structure of inwardly rectifying K+ channels revealed by Cs+ and Rb+ block in guinea-pig heart cells. J. Physiol. 413, 139–157 10.1113/jphysiol.1989.sp017646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oliva C., Cohen I. S., and Pennefather P. (1990) The mechanism of rectification of iK1 in canine Purkinje myocytes. J. Gen. Physiol. 96, 299–318 10.1085/jgp.96.2.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kubo Y., Baldwin T. J., Jan Y. N., and Jan L. Y. (1993) Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature 362, 127–133 10.1038/362127a0 [DOI] [PubMed] [Google Scholar]
- 33. Panama B. K., McLerie M., and Lopatin A. N. (2007) Heterogeneity of IK1 in the mouse heart. Am. J. Physiol. Heart Circ. Physiol. 293, H3558–H3567 10.1152/ajpheart.00419.2007 [DOI] [PubMed] [Google Scholar]
- 34. Panama B. K., and Lopatin A. N. (2006) Differential polyamine sensitivity in inwardly rectifying Kir2 potassium channels. J. Physiol. 571, 287–302 10.1113/jphysiol.2005.097741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopatin A. N., Makhina E. N., and Nichols C. G. (1994) Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372, 366–369 10.1038/372366a0 [DOI] [PubMed] [Google Scholar]
- 36. Ficker E., Taglialatela M., Wible B. A., Henley C. M., and Brown A. M. (1994) Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science 266, 1068–1072 10.1126/science.7973666 [DOI] [PubMed] [Google Scholar]
- 37. Fakler B., Brändle U., Bond C., Glowatzki E., König C., Adelman J. P., Zenner H. P., and Ruppersberg J. P. (1994) A structural determinant of differential sensitivity of cloned inward rectifier K+ channels to intracellular spermine. FEBS Lett. 356, 199–203 10.1016/0014-5793(94)01258-X [DOI] [PubMed] [Google Scholar]
- 38. Matsuda H. (1988) Open-state substructure of inwardly rectifying potassium channels revealed by magnesium block in guinea-pig heart cells. J. Physiol. 397, 237–258 10.1113/jphysiol.1988.sp016998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Enkvetchakul D., Ebihara L., and Nichols C. G. (2003) Polyamine flux in Xenopus oocytes through hemi-gap junctional channels. J. Physiol. 553, 95–100 10.1113/jphysiol.2003.047910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lopatin A. N., Makhina E. N., and Nichols C. G. (1995) The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J. Gen. Physiol. 106, 923–955 10.1085/jgp.106.5.923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fakler B., Brändle U., Glowatzki E., Weidemann S., Zenner H. P., and Ruppersberg J. P. (1995) Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell 80, 149–154 10.1016/0092-8674(95)90459-X [DOI] [PubMed] [Google Scholar]
- 42. Shieh R. C., John S. A., Lee J. K., and Weiss J. N. (1996) Inward rectification of the IRK1 channel expressed in Xenopus oocytes: effects of intracellular pH reveal an intrinsic gating mechanism. J. Physiol. 494, 363–376 10.1113/jphysiol.1996.sp021498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lopatin A. N., and Nichols C. G. (1996) [K+] dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J. Gen. Physiol. 108, 105–113 10.1085/jgp.108.2.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo D., and Lu Z. (2000) Mechanism of IRK1 channel block by intracellular polyamines. J. Gen. Physiol. 115, 799–814 10.1085/jgp.115.6.799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Makary S. M., Claydon T. W., Enkvetchakul D., Nichols C. G., and Boyett M. R. (2005) A difference in inward rectification and polyamine block and permeation between the Kir2.1 and Kir3.1/Kir3.4 K+ channels. J. Physiol. 568, 749–766 10.1113/jphysiol.2005.085746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fakler B., Bond C. T., Adelman J. P., and Ruppersberg J. P. (1996) Heterooligomeric assembly of inward-rectifier K+ channels from subunits of different subfamilies: Kir2.1 (IRK1) and Kir4.1 (BIR10). Pflugers Arch. 433, 77–83 10.1007/s004240050251 [DOI] [PubMed] [Google Scholar]
- 47. Kucheryavykh Y. V., Pearson W. L., Kurata H. T., Eaton M. J., Skatchkov S. N., and Nichols C. G. (2007) Polyamine permeation and rectification of Kir4.1 channels. Channels 1, 172–178 10.4161/chan.4389 [DOI] [PubMed] [Google Scholar]
- 48. Shyng S., Ferrigni T., and Nichols C. G. (1997) Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J. Gen. Physiol. 110, 141–153 10.1085/jgp.110.2.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu Z., and MacKinnon R. (1994) Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 371, 243–246 10.1038/371243a0 [DOI] [PubMed] [Google Scholar]
- 50. Zhou Y., Morais-Cabral J. H., Kaufman A., and MacKinnon R. (2001) Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 414, 43–48 10.1038/35102009 [DOI] [PubMed] [Google Scholar]
- 51. Yang J., Jan Y. N., and Jan L. Y. (1995) Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron 14, 1047–1054 10.1016/0896-6273(95)90343-7 [DOI] [PubMed] [Google Scholar]
- 52. Woodhull A. M. (1973) Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61, 687–708 10.1085/jgp.61.6.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ruppersberg P. J., Kitzing E. V., and Schoepfer R. (1994) The mechanism of magnesium block of NMDA receptors. Neurosciences 6, 87–96 10.1006/smns.1994.1012 [DOI] [Google Scholar]
- 54. Wible B. A., Taglialatela M., Ficker E., and Brown A. M. (1994) Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature 371, 246–249 10.1038/371246a0 [DOI] [PubMed] [Google Scholar]
- 55. Yi B. A., Lin Y. F., Jan Y. N., and Jan L. Y. (2001) Yeast screen for constitutively active mutant G protein-activated potassium channels. Neuron 29, 657–667 10.1016/S0896-6273(01)00241-0 [DOI] [PubMed] [Google Scholar]
- 56. Guo D., Ramu Y., Klem A. M., and Lu Z. (2003) Mechanism of rectification in inward-rectifier K+ channels. J. Gen. Physiol. 121, 261–275 10.1085/jgp.200208771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kurata H. T., Phillips L. R., Rose T., Loussouarn G., Herlitze S., Fritzenschaft H., Enkvetchakul D., Nichols C. G., and Baukrowitz T. (2004) Molecular basis of inward rectification: polyamine interaction sites located by combined channel and ligand mutagenesis. J. Gen. Physiol. 124, 541–554 10.1085/jgp.200409159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kurata H. T., Diraviyam K., Marton L. J., and Nichols C. G. (2008) Blocker protection by short spermine analogs: refined mapping of the spermine binding site in a Kir channel. Biophys. J. 95, 3827–3839 10.1529/biophysj.108.133256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kurata H. T., Marton L. J., and Nichols C. G. (2006) The polyamine binding site in inward rectifier K+ channels. J. Gen. Physiol. 127, 467–480 10.1085/jgp.200509467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taglialatela M., Ficker E., Wible B. A., and Brown A. M. (1995) C-terminus determinants for Mg2+ and polyamine block of the inward rectifier K+ channel IRK1. EMBO J. 14, 5532–5541 10.1002/j.1460-2075.1995.tb00240.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fujiwara Y., and Kubo Y. (2006) Functional roles of charged amino acid residues on the wall of the cytoplasmic pore of Kir2.1. J. Gen. Physiol. 127, 401–419 10.1085/jgp.200509434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kubo Y., and Murata Y. (2001) Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J. Physiol. 531, 645–660 10.1111/j.1469-7793.2001.0645h.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xie L. H., John S. A., and Weiss J. N. (2002) Spermine block of the strong inward rectifier potassium channel Kir2.1: dual roles of surface charge screening and pore block. J. Gen. Physiol. 120, 53–66 10.1085/jgp.20028576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu Y., Shin H. G., Szép S., and Lu Z. (2009) Physical determinants of strong voltage sensitivity of K+ channel block. Nat. Struct. Mol. Biol. 16, 1252–1258 10.1038/nsmb.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kurata H. T., Cheng W. W., Arrabit C., Slesinger P. A., and Nichols C. G. (2007) The role of the cytoplasmic pore in inward rectification of Kir2.1 channels. J. Gen. Physiol. 130, 145–155 10.1085/jgp.200709742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shin H. G., Xu Y., and Lu Z. (2005) Evidence for sequential ion-binding loci along the inner pore of the IRK1 inward-rectifier K+ channel. J. Gen. Physiol. 126, 123–135 10.1085/jgp.200509296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Robertson J. L., Palmer L. G., and Roux B. (2008) Long-pore electrostatics in inward-rectifier potassium channels. J. Gen. Physiol. 132, 613–632 10.1085/jgp.200810068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pearson W. L., and Nichols C. G. (1998) Block of the Kir2.1 channel pore by alkylamine analogues of endogenous polyamines. J. Gen. Physiol. 112, 351–363 10.1085/jgp.112.3.351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Loussouarn G., Marton L. J., and Nichols C. G. (2005) Molecular basis of inward rectification: structural features of the blocker defined by extended polyamine analogs. Mol. Pharmacol. 68, 298–304 [DOI] [PubMed] [Google Scholar]
- 70. Shin H. G., and Lu Z. (2005) Mechanism of the voltage sensitivity of IRK1 inward-rectifier K+ channel block by the polyamine spermine. J. Gen. Physiol. 125, 413–426 10.1085/jgp.200409242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Guo D., and Lu Z. (2003) Interaction mechanisms between polyamines and IRK1 inward rectifier K+ channels. J. Gen. Physiol. 122, 485–500 10.1085/jgp.200308890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Phillips L. R., and Nichols C. G. (2003) Ligand-induced closure of inward rectifier Kir6.2 channels traps spermine in the pore. J. Gen. Physiol. 122, 795–804 10.1085/jgp.200308953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clarke O. B., Caputo A. T., Hill A. P., Vandenberg J. I., Smith B. J., and Gulbis J. M. (2010) Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell 141, 1018–1029 10.1016/j.cell.2010.05.003 [DOI] [PubMed] [Google Scholar]
- 74. Cheng W. W., Enkvetchakul D., and Nichols C. G. (2009) KirBac1.1: it's an inward rectifying potassium channel. J. Gen. Physiol. 133, 295–305 10.1085/jgp.200810125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee S. J., Ren F., Zangerl-Plessl E. M., Heyman S., Stary-Weinzinger A., Yuan P., and Nichols C. G. (2016) Structural basis of control of inward rectifier Kir2 channel gating by bulk anionic phospholipids. J. Gen. Physiol. 148, 227–237 10.1085/jgp.201611616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Twomey E. C., Yelshanskaya M. V., Vassilevski A. A., and Sobolevsky A. I. (2018) Mechanisms of channel block in calcium-permeable AMPA receptors. Neuron 5;99(5):956–968.e4 10.1016/j.neuron.2018.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu Z., and Ding L. (1999) Blockade of a retinal cGMP-gated channel by polyamines. J. Gen. Physiol. 113, 35–43 10.1085/jgp.113.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guo D., and Lu Z. (2000) Pore block versus intrinsic gating in the mechanism of inward rectification in strongly rectifying IRK1 channels. J. Gen. Physiol. 116, 561–568 10.1085/jgp.116.4.561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., and MacKinnon R. (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417, 515–522 10.1038/417515a [DOI] [PubMed] [Google Scholar]
- 80. Bavro V. N., De Zorzi R., Schmidt M. R., Muniz J. R., Zubcevic L., Sansom M. S., Venien-Bryan C., and Tucker S. J. (2012) Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat. Struct. Mol. Biol. 19, 158–163 10.1038/nsmb.2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Abrams C. J., Davies N. W., Shelton P. A., and Stanfield P. R. (1996) The role of a single aspartate residue in ionic selectivity and block of a murine inward rectifier K+ channel Kir2.1. J. Physiol. 493, 643–649 10.1113/jphysiol.1996.sp021411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Glowatzki E., Fakler G., Brändle U., Rexhausen U., Zenner H. P., Ruppersberg J. P., and Fakler B. (1995) Subunit-dependent assembly of inward-rectifier K+ channels. Proc. Biol. Sci. 261, 251–261 10.1098/rspb.1995.0145 [DOI] [PubMed] [Google Scholar]
- 83. Shyng S., and Nichols C. G. (1997) Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110, 655–664 10.1085/jgp.110.6.655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kurata H. T., Zhu E. A., and Nichols C. G. (2010) Locale and chemistry of spermine binding in the archetypal inward rectifier Kir2.1. J. Gen. Physiol. 135, 495–508 10.1085/jgp.200910253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tabor C. W., and Tabor H. (1984) Polyamines. Annu. Rev. Biochem. 53, 749–790 10.1146/annurev.bi.53.070184.003533 [DOI] [PubMed] [Google Scholar]
- 86. Casero R. A. Jr., Murray Stewart T., and Pegg A. E. (2018) Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat. Rev. Cancer 18, 681–695 10.1038/s41568-018-0050-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hesterberg R. S., Cleveland J. L., and Epling-Burnette P. K. (2018) Role of polyamines in immune cell functions. Med. Sci. (Basel) March 8;6(1). pii: E22 10.3390/medsci6010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bowie D., and Mayer M. L. (1995) Neuron 15, 453–462 10.1016/0896-6273(95)90049-7 [DOI] [PubMed] [Google Scholar]
- 89. Feuerstein B. G., Szöllösi J., Basu H. S., and Marton L. J. (1992) α-Difluoromethylornithine alters calcium signaling in platelet-derived growth factor-stimulated A172 brain tumor cells in culture. Cancer Res. 52, 6782–6789 [PubMed] [Google Scholar]
- 90. Seiler N. (1994) in The Neuropharmacology of Polyamines (Carter C., ed) pp. 1–36, Harcourt Brace, New York [Google Scholar]
- 91. Bianchi L., Roy M. L., Taglialatela M., Lundgren D. W., Brown A. M., and Ficker E. (1996) Regulation by spermine of native inward rectifier K+ channels in RBL-1 cells. J. Biol. Chem. 271, 6114–6121 10.1074/jbc.271.11.6114 [DOI] [PubMed] [Google Scholar]
- 92. Shyng S. L., Sha Q., Ferrigni T., Lopatin A. N., and Nichols C. G. (1996) Depletion of intracellular polyamines relieves inward rectification of potassium channels. Proc. Natl. Acad. Sci. U.S.A. 93, 12014–12019 10.1073/pnas.93.21.12014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Steglich C., and Scheffler I. E. (1982) An ornithine decarboxylase-deficient mutant of Chinese hamster ovary cells. J. Biol. Chem. 257, 4603–4609 [PubMed] [Google Scholar]
- 94. Lopatin A. N., Shantz L. M., Mackintosh C. A., Nichols C. G., and Pegg A. E. (2000) Modulation of potassium channels in the hearts of transgenic and mutant mice with altered polyamine biosynthesis. J. Mol. Cell. Cardiol. 32, 2007–2024 10.1006/jmcc.2000.1232 [DOI] [PubMed] [Google Scholar]
- 95. Pegg A. E. (2016) Functions of polyamines in mammals. J. Biol. Chem. 291, 14904–14912 10.1074/jbc.R116.731661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Priori S. G., Pandit S. V., Rivolta I., Berenfeld O., Ronchetti E., Dhamoon A., Napolitano C., Anumonwo J., di Barletta M. R., Gudapakkam S., Bosi G., Stramba-Badiale M., and Jalife J. (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 96, 800–807 10.1161/01.RES.0000162101.76263.8c [DOI] [PubMed] [Google Scholar]
- 97. Tristani-Firouzi M., and Etheridge S. P. (2010) Kir 2.1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch. 460, 289–294 10.1007/s00424-010-0820-6 [DOI] [PubMed] [Google Scholar]
- 98. Zhu Q., Huang Y., Marton L. J., Woster P. M., Davidson N. E., and Casero R. A. Jr. (2012) Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 42, 887–898 10.1007/s00726-011-1004-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Huang Y., Marton L. J., Woster P. M., and Casero R. A. (2009) Polyamine analogues targeting epigenetic gene regulation. Essays Biochem. 46, 95–110 10.1042/bse0460007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tsai K. L., Chang H. F., and Wu S. N. (2013) The inhibition of inwardly rectifying K+ channels by memantine in macrophages and microglial cells. Cell. Physiol. Biochem. 31, 938–951 10.1159/000350112 [DOI] [PubMed] [Google Scholar]
- 101. Amorós I., Dolz-Gaitón P., Gómez R., Matamoros M., Barana A., de la Fuente M. G., Núñez M., Pérez-Hernández M., Moraleda I., Gálvez E., Iriepa I., Tamargo J., Caballero R., and Delpón E. (2013) Propafenone blocks human cardiac Kir2.x channels by decreasing the negative electrostatic charge in the cytoplasmic pore. Biochem. Pharmacol. 86, 267–278 10.1016/j.bcp.2013.04.023 [DOI] [PubMed] [Google Scholar]
- 102. Bowie D. (2018) Polyamine-mediated channel block of ionotropic glutamate receptors and its regulation by auxiliary proteins. J. Biol. Chem. 293, 18789–18802 10.1074/jbc.TM118.003794 [DOI] [PMC free article] [PubMed] [Google Scholar]