

Abstract
Metastasis is the cause of death in the majority (~90%) of malignant cancers. The oral potassium-sparing diuretic amiloride and its 5-substituted derivative 5-N,N-(hexamethylene)amiloride (HMA) reportedly show robust antitumor/ metastasis effects in multiple in vitro and animal models. These effects are likely due, at least in part, to inhibition of the urokinase plasminogen activator (uPA), a key protease determinant of cell invasiveness and metastasis. This study reports the discovery of 6-substituted HMA analogs that show nanomolar potency against uPA, high selectivity over related trypsin-like serine proteases, and minimal inhibitory effects against epithelial sodium channels (ENaC), the diuretic and antikaliuretic target of amiloride. Reductions in lung metastases were demonstrated for two analogs in a late-stage experimental mouse metastasis model, and one analog completely inhibited formation of liver metastases in an orthotopic xenograft mouse model of pancreatic cancer. The results support further evaluation of 6-substituted HMA derivatives as uPA-targeting anticancer drugs.
INTRODUCTION
The urokinase plasminogen activator (uPA) is a trypsin-like serine protease (TLSP) that has been implicated in the invasive spread and metastatic dissemination of transformed cells in several aggressive cancers, including triple-negative breast, pancreatic, gastric, colorectal, and prostate cancers.1–5 Clinically, uPA expression provides the highest level of evidence (level 1)6 for predicting poor patient prognosis in breast cancer and it is one of the strongest predictors of progression-free survival and overall survival.7–9
uPA binds to cell surfaces via a specific interaction with its cognate receptor urokinase plasminogen activator receptor (uPAR), which is expressed on cancer cells and the supporting stroma, often at the invasive front of tumors.10–12 uPAR-bound uPA activates colocalized plasminogen to yield the broad spectrum TLSP plasmin,13 a key mediator of directed pericellular proteolysis that activates multiple downstream proteases (e.g., matrix metalloproteinases and cathepsins), ultimately leading to degradation of the extracellular matrix and basement membrane components that confine primary tumors.3,14,15 The uPA-initiated proteolytic cascade is thus a key determinant of local invasion and metastatic spread via the lymphatic and circulatory systems.16,17
uPA attracted much interest in the 1990s and early 2000s as an anticancer target in academic and industry laboratories (reviewed by Ngo et al.18 and Tyndall et al.19). For example, efforts from Abbott Laboratories led to highly potent naphthamidine-based inhibitors, e.g., 1.20–23 X-ray cocrystal structures of these and other inhibitors uncovered a novel subsite, termed S1β, adjacent to the S1 binding pocket in uPA. Targeting of this site afforded potency gains in a variety of arylamidine-based inhibitors.20,24 The poor drug properties of arylamidines ultimately proved to be a major stumbling block, however, and compounds were not advanced to the clinic.25 Astex Therapeutics were able to repurpose the oral antiarrhythmic drug mexiletine into a potent, orally active non-amidine-containing uPA inhibitor 2,26 but no clinical studies have been reported to date.
3a and its oral hydroxyamidine prodrug 3b (Heidelberg Pharma AG; formerly WILEX AG) are, to the best of our knowledge, the only uPA inhibitors to have undergone clinical evaluation in cancer.27 Phase II studies of oral 3b in combination with gemcitabine showed a 17% increase in 1-year survival in patients with locally advanced, nonresectable pancreatic cancers compared to patients treated with gemcitabine alone.28 In a separate phase II study, patients with HER2-negative metastatic breast cancers who had received adjuvant chemotherapy following primary diagnosis and were subsequently treated with 3b in combination with capecitabine showed an increase in median progression-free survival (8.3 months) compared to patients receiving capecitabine alone (4.3 months).29
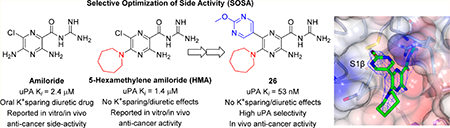
Amiloride 4 is an oral potassium-sparing diuretic that has been used for over 4 decades in the treatment of hypokalemia, hypertension, and cirrhosis.30,31 The diuretic and antikaliuretic properties of amiloride arise through inhibition of renal epithelial sodium channels (ENaCs).32,33 Several groups have reported that the compound also shows anticancer side-activities in multiple in vitro and rodent models (reviewed by Matthews et al.).16 In one study, high doses of amiloride (200 mg/kg in drinking water ad libitum) caused complete remission in a prostate xenograft mouse model.34 It has been suggested that the anticancer effects of amiloride observed at the high doses used in this and other animal studies likely arise, at least in part, from inhibition of uPA proteolytic activity (IC50 = 7 μM).35 Amiloride is unsuitable for use clinically as an anticancer drug, however, due to its low maximum daily dose ceiling (20 mg/day), where overdose risks hyperkalemia and associated cardiac events.36 Nevertheless, the demonstrable in vivo anticancer side-activities of amiloride, combined with excellent drug-like properties (inferred from decades of oral clinical use) and low micromolar potency against uPA, suggest it as an attractive starting scaffold for repurposing using the selective optimization of side-activity (SOSA) approach37 into a high potency uPA inhibitor for treating uPA-driven cancers.16 Moreover, the reduced basicity of the acyl guanidine group relative to arylamidines potentially offers a solution to the poor drug properties of earlier inhibitors.25 We propose that effective dosing of an analog suitably optimized for high uPA potency that lacks the diuretic and antikaliuretic effects of amiloride may safely and effectively recapitulate in humans the anticancer activities observed with high doses of amiloride in animals.
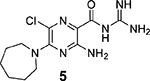
Our preliminary structure−activity exploration of amiloride analogs as uPA inhibitors focused on the effects of varying substituents at the 5-NH2 group.38 Relatively flat SAR trends were observed, with multiple analogs carrying diverse 5-substituents returning IC50 values in the range 1−50 μM. Large increases in potency relative to amiloride were not realized, with the most active compound (5-N,N′-dimethylethylenediamine amiloride) showing only a ~2-fold increase (IC50 = 3 μM). A key finding, however, was that 5-N,N-(hexamethylene)amiloride 5 (HMA, Figure 1) retained potency against uPA (IC50 = 6 μM).
Figure 1.

Structures of reported uPA inhibitors.
HMA has a long history of use as a pharmacological tool39 and, like amiloride, reportedly shows in vitro and in vivo anticancer activities.40–45 For example, HMA has been shown to delay primary tumor growth and increase thermosensitivity of SCK mammary carcinoma cells in a mouse allograft model,40,46 and it was recently shown to suppress sorafenib-resistant engraftment of FLT3-ITD+ acute myeloid leukemia primary cells in mice, with total suppression observed in combination with sorafenib.43 Importantly, alkyl substitution of the 5-NH2 group of amiloride (as in HMA) appears to reduce inhibitory effects against ENaCs.46,47 On the basis of this, HMA 5 was selected as the starting point for SOSA optimization.
There have been many structure−activity studies exploring the effects of substitution at various positions around the pyrazine core of amiloride,46 but there is a surprising lack of reports on analogs carrying variations at the 6-position of amiloride, HMA, or any other 5-substituted amilorides.48 This study reports the synthesis, uPA structure−activity relationships, and in vivo anticancer properties of HMA analogs carrying (hetero)aryl substituents at the 6-position.
RESULTS AND DISCUSSION
Chemistry.
The reported X-ray cocrystal structure of amiloride bound to uPA49 indicated that the 6-Cl group projects toward the S1β subsite, suggesting that other groups attached here might better occupy the site and increase potency. A diverse series of 6-(hetero)aryl substituted HMA analogs was produced from the common 5-N,N-(hexamethylene)amiloride methyl ester intermediate 7 (Scheme 1). Ester 7 was obtained via nucleophilic aromatic substitution of the commercially available 3-amino-5,6-dichloropyrazine-2-methyl ester 6 with hexamethyleneimine. The reported method for preparing 7,38 which involves heating 6 with hexamethyleneimine in DMF at 100 °C for 1 h, requires multiple column chromatography steps to obtain pure 7 (65% yield). We found that when the reaction was instead performed at reflux in 2-propanol in the presence of N,N-diisopropylethylamine (DIPEA), an excellent yield of 7 (86%) was obtained after only 2 h. The improved procedure could be performed on multigram scale in an open flask with the pure 7 crystallizing from the reaction mixture and isolated by simple filtration.
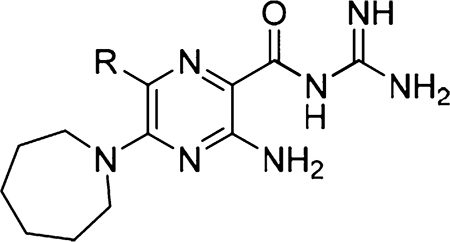
Scheme 1. Synthesis of 6-(Hetero)aryl HMA Analogs 9−32a.

aReagents and conditions: (a) (CH2)6NH, DIPEA, iPrOH, reflux, 2 h, 86%; (b) R-B(OH)2 or R-B(pin), Pd(PPh3)4, K2CO3, toluene/MeOH (4:1), reflux, 0.5−18 h; (c) guanidine (2 M in MeOH), DMF, rt, 18 h.
Direct attachment of (hetero)aryl substituents to the 6-position of 7 was achieved using standard Suzuki−Miyaura cross-coupling conditions with commercial arylboronic acids (or pinacol boronates), Pd(PPh3)4, and K2CO3 in toluene/MeOH (4:1) at reflux. A total of twenty-three 6-(hetero)aryl methyl esters 9a−32a were produced in yields ranging from 34 to 93%. The methyl esters were converted to their respective acylguanidines 9−32 in 2−92% by treatment with guanidine. Low guanidinylation yields were initially obtained using the reported method38 leading us to search for an improved procedure. Treating the methyl esters in DMF with a 2 M stock solution of guanidine in MeOH was found to give much higher yields, with most reactions proceeding to completion at room temperature and no major side products forming. The revised procedure was simple to carry out and allowed multiple guanidinylation reactions to be performed in parallel, and the 2 M guanidine solution could be stored under N2 in a refrigerator for up to 2 weeks.50
6-(2-(Alkylamino)pyrimidin-5-yl) HMA analogs 33−35 (Scheme 2) were obtained from 6-(2-chloropyrimidin-5-yl) methyl ester intermediate 8. Compound 8 was synthesized in 64% yield by Suzuki−Miyaura coupling of 7 with (2-chloropyrimidin-5-yl)boronic acid. Nucleophilic aromatic substitution of the 2-chloro group with the requisite amines yielded esters 33a−35a in 83−87% yields, which were converted to acyl guanidines 33−35 in 85−89% yields using the new guanidinylation procedure.
Scheme 2. Synthesis of 6-(2-(Alkylamino)pyrimidin-5-yl) Analogs 33−35a.

aReagents and conditions: (a) (2-chloropyrimidin-5-yl)boronic acid, Pd(PPh3)4, K2CO3, toluene/MeOH (4:1), reflux, 18 h, 64%; (b) (i) R-NH2, DIPEA, DMF, rt, 2 d or (ii) R-NH2, DIPEA, i-PrOH, reflux, 4 h, 83−87%; (c) guanidine (2 M in MeOH), DMF, rt, 18 h, 85−89%.
Previous reports describing napthamidine-based inhibitors20,21 identified that the 2-aminopyrimidine group shows favorable binding in the uPA S1β pocket. On the basis of this, 2-aminopyrimidinyl derivative 41 was synthesized using the route outlined in Scheme 3. The 3-NH2 group of methyl ester 7 was di-Boc protected using Boc-anhydride to give 36 in 85% yield. Halogen exchange from the 6-chloro group to 6-bromo in preparation for Buchwald−Hartwig amination was carried out over two steps. Hydrodechlorination using 1 atm of hydrogen in the presence of Pd/C and MgO was successful, although it was accompanied by loss of one Boc-group giving mono-Boc derivative 37 in 92% yield. Introduction of the 6-bromo substituent using N-bromosuccinimide (NBS) gave 38 (60% yield), and Buchwald−Hartwig amination of 38 with 2-aminopyrimidine using Pd2(dba)3/Xantphos afforded the 6-substituted pyrazine methyl ester 39 in 30% yield. Removal of the remaining Boc-group with trifluoroacetic acid (98% yield) followed by guanidinylation delivered 41 (35% yield).
Scheme 3. Synthesis of 2-Aminopyrimidinyl Derivative 41a.

aReagents and conditions: (a) (Boc)2O, CH2Cl2, DIPEA, DMAP, rt, 16 h, 85%; (b) Pd/C, H2 (1 atm), MgO, MeOH, rt, 48 h, 92%; (c) N-bromosuccinimide, CH2Cl2, rt, 5 h, 60%; (d) 2-aminopyrimidine, Pd2(dba)3/Xantphos, 1,4-dioxane, Cs2CO3, 100 °C, 18 h, 30%; (e) TFA, CH2Cl2, rt, 16 h, 98%; (f) guanidine (2 M in MeOH), DMF, rt, 35%.
Inhibition of uPA and Structure−Activity Relationships.
Inhibition of catalytically active, low molecular weight human uPA by 9−35 and 41 was determined using a fluorometric enzyme activity assay.51 Concentration−response curves for representative analogs 18 and 27 are provided in Figure 2, and calculated Ki values are presented in Table 1.
Figure 2.

Inhibition of low molecular weight human uPA by 18 (black) and 27 (red). Data points represent the mean ± SEM (n = 3) from a single representative experiment.
Table 1.
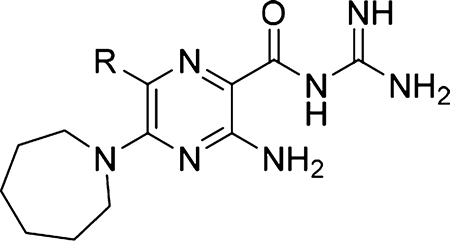
Inhibition of uPA by 6-Substituted HMA Analogsa
 | |||||
|---|---|---|---|---|---|
| Compound | R | uPA Ki (nM) [n] | Compound | R | uPA Ki (nM) [n] |
| 4 | amiloride | 2433 ± 192 [1] | 22 | 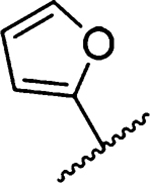 |
156 ±20 [2] |
| 5 |  |
1356 ± 183 [2] | 23 | 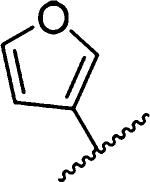 |
166 ±34 [2] |
| 9 |  |
5020 ± 1750 [1] | 24 |  |
1778 ± 249 [1] |
| 10 |  |
16441 ± 1311 [1] | 25 |  |
108 ±17 [2] |
| 11 |  |
4255 + 194 [1] | 26 | 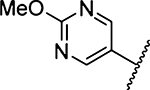 |
53 ± 5 [2] |
| 12 | 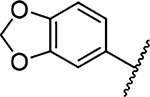 |
934 ± 151 [1] | 27 | 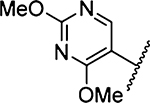 |
42 ± 2 [4] |
| 13 | 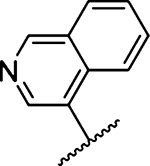 |
3539 ± 192 [2] | 28 | 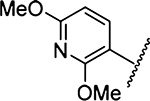 |
884 ± 14 [2] |
| 14 | 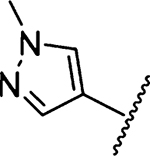 |
508 ± 9 [2] | 29 | 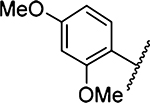 |
2198 ±53 [2] |
| 15 | 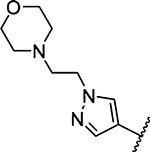 |
658 ± 66 [2] | 30 | 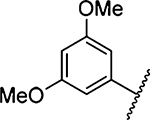 |
1319 ± 310 [1] |
| 16 | 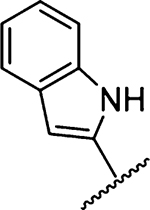 |
5670 ± 1026 [1] | 31 | 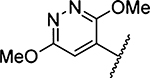 |
315 ±34 [2] |
| 17 | 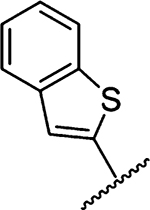 |
3308 ± 300 [1] | 32 | 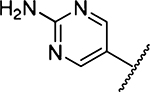 |
21 ±3 [2] |
| 18 | 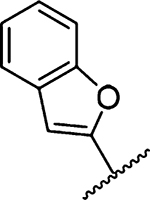 |
183 ±10 [4] | 33 | 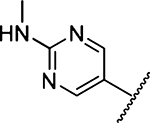 |
43 ± 4 [2] |
| 19 | 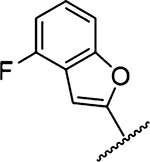 |
88 ± 33 [2] | 34 | 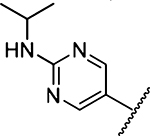 |
42 ± 4 [2] |
| 20 | 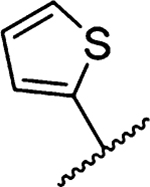 |
2417 ±251 [1] | 35 | 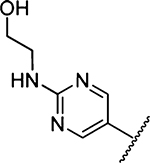 |
41 ± 2 [2] |
| 21 | 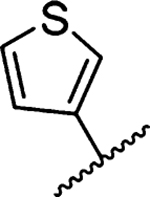 |
1027 ± 170 [1] | 41 | 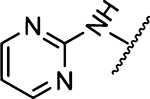 |
228 ± 22 [2] |
For compounds where n > 1, values represent the mean ± SEM from identical repeat assays conducted on different days. Ki values were calculated from experimentally determined IC50 values according to the methods of Cheng and Prussof.52
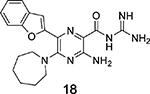
Amiloride 4 and HMA 5 returned Ki values of 2433 nM and 1356 nM, respectively, under the assay conditions. Replacing the 6-Cl group of HMA with a phenyl substituent 9 led to ~3.7-fold decrease in potency. Substitution of the phenyl ring at the para-position (10 and 11) did not improve potency, while conversion to the 3,4-methylenedioxy derivative 12 provided a slight increase relative to HMA (Ki = 934 nM). Appending an isoquinolyl group 13 reduced activity nearly 3-fold relative to HMA. N-Methylpyrazole derivative 14 showed a ~2.7-fold increase (Ki = 508 nM). Extension of the N-methyl group of 14 to N-ethylmorpholino 15 had only a minor effect (Ki = 658 nM). Surprisingly, while 2-indolyl 16 and 2-benzothiophenyl substituents 17 showed reduced activity, the single atom change to 2-benzofuranyl derivative 18 afforded a significant boost in potency (Ki = 183 nM), which increased 2-fold upon addition of a fluorine atom at the benzofuran 4-position (19, Ki = 88 nM). The preference for oxygen in the heteroaryl ring was also seen with the 2- and 3-furanyl derivatives 22 and 23, respectively, which both showed nM potency, whereas 2- and 3-thiophene derivatives 20 and 21 were micromolar inhibitors.
2-Fluoropyridine substitution (24 Ki = 1778 nM) afforded similar activity to HMA 5, while addition of a 5-pyrimidinyl group 25 (Ki = 108 nM) gave a ~12.6-fold increase. Addition of a methoxy group to the pyrimidine 2-position yielded the first sub-100 nM inhibitor 26 (Ki = 53 nM), and adding a second methoxy group to the pyrimidine 4-position 27 (Ki = 42 nM) gave a further increase. The favorable binding of the pyrimidine group was evident from the significant losses in potency observed with dimethoxypyridine 28 and dimethoxyphenyl derivatives 29 and 30. Loss of potency with the dimethoxypyridazine 31 was less dramatic (Ki = 315 nM). An additional 2-fold gain in potency was achieved upon addition of an amine to the pyrimidine 2-position, yielding the most potent inhibitor in the study (32 Ki = 21 nM). Further gains in potency were not obtained through N-alkyl substitution, with N-methyl 33, N-isopropyl 34, and N-hydroxyethyl 35 derivatives showing activity similar to 26. The 2-aminopyrimidine derivative 41 obtained using Buchwald−Hartwig chemistry showed lower potency (Ki = 228 nM).
X-ray Crystallography.
An X-ray cocrystal structure was obtained for HMA 5 bound to the catalytic domain of human uPA (Figure 3). The binding orientation and interactions observed in the HMA:uPA structure mirrored those previously observed with amiloride 4.49 The key salt bridge interaction was present between the acylguanidine of HMA and the Asp189 side chain carboxylate located at the base of the S1 binding pocket. H-bonds were present between the backbone carbonyl of Gly219 and the terminal NH and amide NH groups of the acyl guanidine. The second terminal guanidine-NH formed an H-bond to the side chain hydroxyl of Ser190, and the exocyclic NH2 group at C3 formed an H-bond to the side chain hydroxyl of Ser195. A bridging H-bond network was observed between a surface-bound water molecule, the acyl guanidine carbonyl oxygen, Ser190 side chain hydroxyl, and a guanidine NH group. A sulfate ion from the crystallization buffer was observed in the oxyanion hole of both structures.
Figure 3.

X-ray cocrystal structures of (a) amiloride 449 (PDB code 1F5L) and (b) HMA 5 (PDB code 5ZA7, 1.7 Å resolution) bound to uPA. Blue wire basket = 2mFo − DFC map contoured at 1.5σ.
X-ray cocrystal structures were similarly obtained for benzofuran (18, 19), furan (22, 23), pyrimidine 25, 2-methoxypyrimidine 26, 2,4-dimethoxypyrimidine 27, and N-methylpyrazole 14 analogs (Figures 4 and 5. X-ray refinement statistics are provided in Supporting Information, Table S1). Hbonding networks reminiscent of those seen in the HMA:uPA structure were observed in all complexes. The seven-membered hexamethylene ring appeared to sample multiple conformations, as evidenced by low electron density in all structures. Replacement of the 6-Cl group with (hetero)aryl substituents did not significantly alter the binding positions or orientation of the pyrazine core or acylguanidines relative to HMA 5. The 6-substituent occupied S1β in all cases, and sulfate ion occupied the oxyanion hole in some complexes.
Figure 4.

X-ray cocrystal structures of: (a) 22 (PDB code 5ZAE, 1.7 Å), (b) 23 (PDB code 5ZAF, 1.7 Å), (c) 18 (PDB code 5ZA9, 1.6 Å), and (d) 19 (PDB code 5ZC5, 1.9 Å) bound to uPA. Blue wire baskets represent 2mFo − DFC maps contoured at 1.5σ.
Figure 5.

X-ray cocrystal structures of (a) 25 (PDB code 5ZAG, 2.0 Å), (b) 26 (PDB code 5ZAH, 3.0 Å), (c) 27 (PDB code 5ZAJ, 1.7 Å), and (d) 14 (PDB code 5ZA8, 1.9 Å) bound to uPA. Blue wire baskets represent 2mFo − DFC maps contoured at 1.5σ.
The structure obtained with 2-furanyl analog 22 (Figure 4a) revealed that the ~8.7-fold increase in potency relative to HMA 5 may arise from new H-bonds formed between the furanyl O, a bridging water molecule, and the amide NH of Gly219. Favorable van der Waals contacts between the furan ring and Gly219, the Cys191-Cys220 disulfide, Ser146 backbone carbonyl, and the side chain Cβ and Cγ of Gln192 were also apparent. Changing to the 3-furanyl substituent 23 had little effect on uPA activity despite the furanyl oxygen no longer making the water-bridged H-bond interaction with Gly219 (Figure 4b). Flexibility of the Arg217 side chain was noted in this structure, where it had shifted toward S1β to participate in the Gly219-bridging water H-bond network. Relative to 22, the plane of the furan ring in 23 was tilted 11°, a pose that promoted van der Waals interactions with Cys220. Interactions with S1β residues Cys191-Cys220, Ser146, and the Gln192 side chain were preserved, and there appeared to be a dipolar interaction between the side chain carbonyl of Gln192 and the polarized furan 2-CH group.
Ring fusion to give benzofuran 18 led to a slight decrease in potency (Ki = 183 nM) relative to 22 despite this group extending further into S1β and almost completely occupying the subsite (Figure 4c). The plane of the benzofuran ring was essentially perpendicular to the pyrazine core, but the ring oxygen had flipped 180° relative to the furanyl oxygen of 22. This orientation allowed the benzofuran 7-CH group to fill a small, hydrophobic dimple formed by Lys143, Gln192, and Ser146. Additional hydrophobic contacts were seen between the benzofuran 3- and 4-position CH groups and Gly219. Similar to what was observed in the HMA 5 and 22 structures, the side chain of Arg217 adopted a conformation well away from S1β. The structure with 19 suggested that the 2-fold increase in potency obtained after fluorine substitution at the benzofuran 4-position (19 Ki = 88 nM) arose from favorable interactions between the 4-fluoro group and the side chain guanidine of Arg217 (possibly an H-bond), which had shifted back toward S1β in this complex. The interaction with Arg217 also appeared to cause a slight twisting about the benzofuran−pyrazine axis away from perpendicular, with the 78° dihedral angle in 18 reducing to 68° in 19.
The 5-pyrimidine substituent of 25 was found to partially occupy S1β (Figure 5a), making van der Waals interactions with the Cys191-Cys220 disulfide, Ser146, and Gln192 residues. A dipole interaction between the polarized 4-CH group of the pyrimidine and the Gln192 side chain carbonyl appeared to be present, along with an H-bond between the amide NH of Gly219 and the lone pair of electrons on a pyrimidine nitrogen. The polarized 6-CH on the pyrimidine appeared to interact with the Gly219 carbonyl group.
Addition of a methoxy group to the pyrimidine 2-position 26 led to a ~2-fold increase in potency, but it did not appear to make any specific interactions with the protein, instead orienting toward bulk solvent (Figure 5b). The side chain of Arg217 had flipped back toward S1β driven by a polar interaction with the pyrimidine nitrogen (possibly an H-bond).
Introduction of a second methoxy substituent onto the pyrimidine ring 27 resulted in a small increase in potency relative to 26. The pyrimidine and pyrazine rings were almost perpendicular (dihedral angle 80°), allowing for increased occupancy of S1β and projection of a methoxy group toward the small hydrophobic dimple formed by Lys143, Gln192, and Ser146 (Figure 5c). The second methoxy substituent was projected toward bulk solvent. In contrast to 25 and 26, the Arg217 side chain was positioned well away from S1β, apparently to avoid a steric clash with the pyrimidine 2-OCH3 group of 27.
The N-methylpyrazole substituent of 14 adopted a coplanar arrangement with the pyrazine reducing its penetration into S1β (Figure 5d). Interactions were observed between the pyrazole 3-CH and 5-CH groups and the backbone of Gly219 and the side chain of Gln192, respectively. The surface water molecule bound to Gly219 in the furanyl structures 22 and 23 was observed but did not appear to interact with the pyrazole.
Inhibition of Trypsin-like Serine Proteases.
HMA 5 and 6-substituted analogs 18, 19, and 26 were screened against a panel of trypsin-like serine proteases (TLSPs) using chromogenic assays (Table 2). In agreement with earlier reports showing that occupancy of uPA’s S1β subsite confers high selectivity over other TLSPs,20,24,53 each of the compounds showed 20−100+ fold selectivity for uPA.
Table 2.
uPA Selectivity versus Related Trypsin-like Serine Proteases of HMA 5 and 6-Substituted Analogs 18, 19, and 26a
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | ||||||||||
| Compound | R | uPA | tPA | Plasmin | Thrombin | Trypsin | Human plasma kallikrein | Factor Xa | Factor Xla | Activated protein C |
| 5 |  |
2.4 | >10 | >10 | >10 | >10 | >50 | >50 | >50 | >50 |
| 18 | 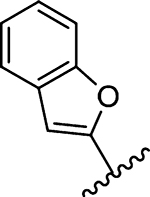 |
0.53 | >10 | >10 | >10 | >10 | 11.3 | >50 | 31.0 | >50 |
| 19 | 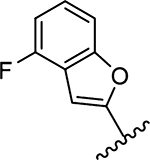 |
0.43 | >20 | >20 | >20 | N/D | 16.7 | >50 | 23.3 | >50 |
| 26 | 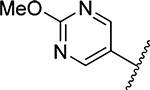 |
0.039 | >20 | >20 | >20 | N/D | >50 | >50 | >50 | >50 |
Values were obtained using chromogenic enzyme inhibition assays.38 N/D = not determined.
To further probe uPA selectivity, superfamily wide screening against 85 serine hydrolases was performed in situ using the recently described EnPlex activity-based protein profiling (ABPP) assay.54 In this approach, individual serine hydrolases are conjugated to differently colored Luminex beads and the ability of inhibitory compounds to outcompete a biotinylated serine-reactive/streptavidin-phycoerythrin probe at the active site of each enzyme provides a rapid and semiquantitative readout of enzyme inhibition. Results from the Enplex screen with amiloride 4, HMA 5, and 6-substituted analogs 18, 25, and 26 are shown in Figure 6.
Figure 6.

EnPlex54 screening of amiloride 4, HMA 5 and analogs 18, 25, and 26 for inhibition of 85 serine hydrolases. Gray boxes indicate wells in which no fluorescence was observed. The singular readouts suggesting activity of 4 and 26 against LIPE are artifacts. Black arrows indicate decreasing compound concentration from 33.3 μM to 10 nM.
Amiloride showed slight activity against only a few serine hydrolases (C1r, C1s, DDHD1, factor VIIa, and LACTB), consistent with its reported high selectivity for uPA.35 HMA 5 showed activity against the highly homologous S28 protease family members dipeptidyl peptidase 2 (DPP7) and prolylcarboxypeptidase (PRCP).55 Surprisingly, inhibitory activity against uPA was not observed for HMA under these assay conditions. 2-Benzofuranyl analog 18 showed the least selectivity among the tested compounds, inhibiting plasma kallikrein (KLKb1) and trypsin. 5-Pyrimidinyl analog 25 showed remarkable selectivity for uPA, with slight inhibition of trypsin being the only off-target TLSP activity observed. 2-Methoxypyrimidinyl analog 26 showed slight activity against C1s, plasma kallikrein, plasmin, prolylcarboxy peptidase, trypsin, and transmembrane protease serine 11D (TMPRSS11d).
Human Cell Cytotoxicity.
HMA 5 and representative analogs 18, 19, 25, and 26 were tested for cytotoxicity against four human cell lines (Table 3). HMA 5 showed toxicity in the low micromolar range against all cell lines, with HEK-293 embryonic kidney and HT-1080 fibrosarcoma cells being most sensitive. In general, the cell lines were more sensitive to 18, suggesting the benzofuran group may contribute to cytotoxic effects. MDA-MB-231 cells were ~3-fold more sensitive to the 4-fluorinated benzofuran 19, but the compound was less toxic than 18 toward the other three cell types. Cytotoxicity was not a general feature of 6-substituted HMA analogs since pyrimidine analogs 25 and 26 showed much lower activity against all cells. The lower log D7.4 of the pyrimidines (i.e., 2.8 and 3.4, respectively) relative to benzofuran 18 (i.e., log D7.4 = 5.1) and HMA (log D7.4 = 3.8) suggests lipophilicity may be a contributor to cytotoxicity.
Table 3.
Effect of HMA and Selected Analogs on Human Cell Viability
 | |||||
|---|---|---|---|---|---|
| Compound | R | MDA-MB-231 IC50 (μM) | HepG2 IC50 (μM) | HEK-293 IC50 (μM) | HT-1080 IC50 (μM) |
| 5 |  |
27.3 | 20.2 | 2.8 | 6.6 |
| 18 | 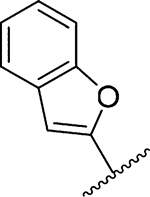 |
10.9 | 12.7 | 3.1 | 4.6 |
| 19 | 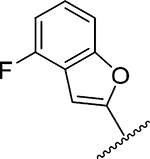 |
3.2 | 68.4 | 12.0 | 2.1 |
| 25 | 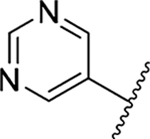 |
>100 | >100 | >100 | 37.1 |
| 26 | 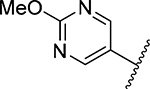 |
>100 | 57.3 | 74.9 | 44.8 |
Off-Target Activity against GPCRs.
Amiloride and related analogs are known to bind to G-protein-coupled receptors (GPCRs).56,57 For example, amiloride is a known antagonist of the adenosine 2A receptor (A2A).58–60 Recent work describing the effects of substitution at the amiloride 5-position on A2A antagonism identified analogs with increased potency, including HMA 5.61 In the current study, a representative selection of 6-substituted HMA analogs were evaluated alongside HMA 5 for A2A antagonism using a radiolabeled ligand displacement assay (Figure 7). In all cases, the presence of the 6-substituent diminished antagonist effects relative to HMA, with the majority of compounds showing poor A2A binding.
Figure 7.

Single concentration screening (10 μM) of HMA 5 and 6-substituted HMA analogs as antagonists of the human adenosine A2A receptor (hA2AAR-WT). Radioligand [3H]ZM241385 was present at 2.5 nM. Total binding (TB, 100%) of [3H]ZM241385 was determined in the absence of inhibitors. Nonspecific binding (NSB, 0%) was determined in the presence of 100 μM A2A agonist 5′-(N-ethylcarboxamido)adenosine (NECA). Data represent the mean ± SEM from at least three independent experiments conducted in duplicate. Methods were as described in ref 61.
Development of active site-directed uPA inhibitors has been complicated by the observation that compounds optimized for high affinity against human uPA often show reduced potency against the homologous mouse enzyme.62 This species selectivity can confound interpretation of data from mouse models as the decreased affinity for mouse uPA may lead to an underestimation of compound efficacy. To explore this issue in the 6-substituted HMA class, we measured the activities of 4, 5, 18, and 26 against mouse uPA (Table 4).
Table 4.
Activities of Compounds 4, 5, 18, and 26 against Human and Mouse uPA
| Compound | Human uPA Ki (nM)a | Mouse uPA Ki (nM)b | Human/Mouse Selectivity Ratio |
|---|---|---|---|
 |
2,433 | 2,313 | 0.95 |
 |
1,356 | 9,308 | 6.86 |
 |
183 | 1,802 | 9.85 |
 |
53 | 1,611 | 30.40 |
Experimental replicates were performed as described in Table 1.
Value is the average of two independent experiments.
Amiloride 4 was essentially equipotent against mouse and human uPA. Addition of the 5-hexamethylene ring in 5 decreased affinity for mouse uPA by almost 7-fold, while compounds 18 and 26 showed larger (10- to 30-fold) reductions. These findings align with reported trends for compounds bearing substituents that target uPA’s S1β subsite.62 The lower potency against mouse uPA was a key consideration in dose selection for in vivo studies with 18 and 26 (see below).
Diuretic and Antikaliuretic Effects.
The potassium-sparing activity of amiloride arises from inhibition of epithelial sodium channels (ENaCs) in the kidneys, leading to reduced urinary excretion of K+.32 In cases of overdose, the potassium-sparing effects can lead to hyperkalemia and cardiac arrhythmias.63 As such, removal of ENaC activity is essential for development of a 6-substituted HMA analog suitable for use in cancer. Early work from the Cragoe group at Merck established that alkyl substitution of the 5-NH2 group of amiloride reduces ENaC activity47 and showed that HMA produces >80-fold lower inhibition than amiloride in a deoxycorticosterone acetate (DOCA) induced K+ excretion model in adrenalectomized rats.47 6-Substituted HMA analogs were thus expected to show similarly low activity against ENaCs. To confirm this, a representative selection of analogs was tested against recombinant HEK-293 cells that overexpress the α, β, and γ subunits of human ENaCs (Figure 8). In line with expectations, amiloride 4 produced a strong effect (68% inhibition of ENaC at 10 μM), whereas HMA 5 showed greatly reduced activity and 6-substituted analogs 11, 18, 25, and 26 showed similar or lower inhibition than HMA.
Figure 8.

Inhibition of ENaC activity by amiloride 4, HMA 5 and 6-substituted analogs 11, 18, 25, and 26 in transfected HEK-293 cells. All compounds were present at 10 μM. Data represent the mean ± SD (n = 4).
Amiloride 4, HMA 5, and analogs 18 and 26 were next tested in a rat model of diuresis to confirm that loss of ENaC inhibitory potency corresponded to an absence of diuretic and antikaliuretic effects in vivo (Figure 9). With the exception of vehicle controls, all animals were dosed with 25 mg/kg acetazolamide to induce diuresis64 prior to receiving 1.5 mg/kg of amiloride 4, HMA 5, 18, or 26. Urine was collected over a 6 h period following dosing. Consistent with previous reports, amiloride 4 showed potent diuretic and antikaliuretic effects, increasing urine volume and sodium excretion by 34% and 159%, respectively, while decreasing potassium excretion by 92% relative to acetazolamide-treated controls (Na+/K+ excretion ratio = 63, p < 0.0001 for all measurements).64 Compound 18 did not significantly decrease K+ excretion, while HMA 5 and 2-methoxypyrimidine analog 26 showed a very slight potassium-sparing effect (24% reduction, p < 0.001). HMA 5, 18, and 26 did not alter Na+ excretion or urine volume, and the Na+/K+ excretion ratios for the three compounds were no different to acetazolamide treated controls (range = 2.44−2.68).
Figure 9.

Effects of amiloride 4, HMA 5, and 6-substituted analogs 18 and 26 on urine flow and excretion of Na+ and K+ in Sprague Dawley rats. Rats were dosed with 25 mg/kg acetazolamide prior to administering 1.5 mg/kg of test compound. Data represent the mean ± SEM (n = 8): (∗∗) p < 0.001, (∗∗∗∗) p < 0.0001 relative to acetazolamide control.
Experimental Lung Metastasis Model.
Two analogs were advanced to anticancer pilot studies to establish efficacy in vivo. Compounds 18 and 26 were selected as representative examples showing different potency and polarity (18 log D7.4 = 5.1, 26 log D7.4 = 3.4). In the first study, amiloride 4 and 6-substituted HMA analogs 18 and 26 were examined for antimetastasis effects in an experimental mouse model of late-stage lung metastasis. Luciferase-tagged HT-1080 human fibrosarcoma cells were used in the model after confirming cell surface expression of uPA/uPAR (Supporting Information Figure S1). Assays of lung homogenates following organ harvest at end point were used to quantitate lung tumor burden and response to treatment. Animals received 2.5 × 105 HT-1080 cells via lateral tail vein injection on day 0 and were weighed and scored for clinical signs (scoresheet Supporting Information Figure S2) over 21 days. Animals received 7.5 mg kg−1 day−1 of compound (or vehicle) via ip injection from day −1 to day 20. This dose was selected based on extensive in vivo pharmacokinetic profiling and represented a compromise between tolerability and attempting to demonstrate efficacy with compounds that show reduced potency against murine uPA.65
Amiloride 4 did not significantly inhibit metastasis in this model at the concentration tested. Benzofuranyl analog 18 decreased metastasis by 21% (p = 0.0297) relative to control, but there was no statistically significant difference between amiloride and 18 (Figure 10). 2-Methoxypyrimidine analog 26 significantly decreased lung metastasis (33%, p = 0.0007).
Figure 10.

Effects of amiloride 4, 18, and 26 in experimental mouse model of late-stage lung metastasis. All compounds were administered once daily at 7.5 mg kg−1 day−1 for 21 days. Lung metastasis was quantified by end point luciferase assay of lung homogenates. Data represent the mean ± SEM (18 and 26 n = 6; vehicle and 4 n = 4): (∗) p < 0.03; (∗∗∗) p < 0.001 relative to vehicle control, (∧) p < 0.03 relative to 4.
Orthotopic Model of Pancreatic Cancer.
The in vivo antitumor and antimetastasis effects of 18 and 26 were evaluated alongside the standard of care drug gemcitabine in an orthotopic xenograft mouse model of pancreatic cancer (Figure 11). Briefly, female athymic nude mice (BALBc nu/nu, 8−11 weeks of age) were anesthetized, an incision was made in the left flank, and the spleen and tail of the pancreas were externalized. A mixture of human pancreatic cancer cells (AsPC-1) 1 × 106 and human pancreatic stellate cells (hPSCs) 1 × 106 in 50 μL of PBS was injected into the tail of the pancreas. uPA expression was confirmed in both cell lines by Western blot analysis (Supporting Information Figure S3).66 The pancreas and spleen were then reinternalized, and the wound was closed. Seven days after cell implantation, mice were randomized to receive treatment with gemcitabine (75 mg/kg po twice weekly), 18, or 26 (10 mg/kg ip daily) for 28 days (n = 4 mice/group). The higher dose (relative to the lung metastasis model) was chosen to increase the likelihood of observing efficacy. In line with previous observations in this model, gemcitabine showed only modest inhibitory effects on the primary tumor.67 Measurement of tumor volume at end point revealed that 18 and 26 produced similar reductions to gemcitabine on the primary mass (Figure 11A). Notably, liver metastases (key site in this model) were reduced in mice treated with compounds 18 and 26 compared to those treated with gemcitabine, with no macroscopic metastatic lesions observed in any mice that received 26 (Figure 11B). The absence of liver metastases in all animals treated with 26 was confirmed by histology (Supporting Information Figure S4).
Figure 11.

(A) Effects of gemcitabine (Gem), 18, and 26 on end point primary tumor volume in an orthotopic xenograft pancreatic cancer model. Mice received oral gemcitabine 75 mg/kg twice weekly or daily intraperitoneal injections of 18 or 26 (10 mg/kg) for 28 days. Data represent the mean ± SEM (n = 4 per group). (B) Number of mice with histologically confirmed liver metastases after receiving gemcitabine, 18, and 26 (n = 4 per group): (∗) p = 0.0285 vs vehicle, χ2 test.
CONCLUSIONS
This study reveals that 6-substituted HMA derivatives are potent inhibitors of human uPA with potential for development into a new class of anticancer drugs. Access to a diverse series of 6-(hetero)aryl analogs was afforded via a simple two step Suzuki−Miyaura/acyl guanidinylation approach using commercial boronic acids/esters and a common 6-chloropyrazine intermediate 7. The key intermediate was easily synthesized on multigram scale in a single step from commercial reagents. Buchwald−Hartwig chemistry was established on a related 6-bromopyrazine scaffold to access 6-aminopyrimidine HMA derivative 41. Given the general reactivity of halopyrazines in transition-metal-catalyzed cross-coupling reactions,68 it is likely that a great many other substituents could be appended at the 6-position of HMA using this chemistry. Screening of such a library against proteins that reportedly bind to HMA (e.g., P2X7,69 NHE1,70 HIV-1 Vpu,71 influenza A M2,72 GnRHR,73 GABA-A ρ−1,74 ASIC1a,75 and GIRK)76 could help to identify more potent and selective probes for these targets, and possibly therapeutic leads.
Leading compounds from the study showed uPA inhibitory potencies more than 10-fold higher than HMA, with X-ray crystallography revealing that the gains arose from interactions formed between the appended 6-substituents and uPA’s S1β pocket. High target selectivity was demonstrated across the serine hydrolase superfamily, and selected analogs showed no activity against ENaCs in vitro or K+-sparing/diuretic effects in vivo. Analogs carrying 2-benzofuranyl and 5-pyrimidinyl substituents emerged as distinct lead classes with differing physicochemical properties and human cell cytotoxicity profiles. Examples from each class were found to inhibit formation of lung metastases in a mouse experimental metastasis model, and 2-methoxypyrimidine analog 26 completely inhibited formation of liver macrometastases in a mouse pancreatic cancer model. In summary, this work reports the successful repurposing of the diuretic amiloride into potent uPA inhibitors with attractive pharmacological properties for further evaluation as a novel class as anticancer agents for treating uPA-driven malignancies.
EXPERIMENTAL SECTION
Chemistry: General.
All solvents and chemicals were purchased and used without further purification except iPrOH, which was distilled from BaO and stored over 4 Å molecular sieves under Ar. Progress of reactions was monitored using Merck silica gel 60 F254 TLC plates with EtOAc/pet. spirit mixtures as eluent. Compound spots on TLC plates were visualized under UV light at 254 nm. Column chromatography was performed using Merck silica gel 60 (230−400 mesh). Melting points (mp) were measured in open capillaries using a Griffin analog melting point apparatus (Thermo-Fisher Scientific, Waltham, MA, USA) and are uncorrected. 1H and 13C NMR spectra were recorded using a Varian Inova 500 or Varian Premium Shielded 500 (499.74 MHz for 1H; 125.66 MHz for 13C) in deuterated solvents. Chemical shifts are reported in parts per million (δ ppm) relative to TMS for 1H or the solvent peak for 13C. High-resolution mass spectrometry (HRMS) analysis was performed using a Waters XEVO QToF mass spectrometer (Waters Corporation, Milford, MA, USA) with leucine encephalin as internal standard. Purity of all final compounds was confirmed at >95% using high performance liquid chromatography (HPLC). HPLC analyses were performed using a Shimadzu CLASS-VP LC10 analytical HPLC system (Shimadzu Corporation, Kyoto, Japan) with detection at 254 nm. Gradient elutions were performed using mixtures of solvent 100% H2O/0.1% TFA and 100% CH3CN/0.1% TFA on a Waters VisionHT 3 μm C18 column (150 mm × 4.6 mm) at a flow rate of 0.5 mL.min−1. Column temperature was not controlled. Only one previous 6-substituted HMA analog (6-phenyl HMA) has been reported, but no characterization or uPA inhibition data were described.77
Chemistry: Experimental Procedures and Compound Characterization. Synthesis of Methyl 3-Amino-6-chloro-5-hexamethyleneimine-2-pyrazinecarboxylate (7).
To a suspension of methyl 3-amino-5,6-dichloro-2-pyrazinecarboxylate (5.55 g, 25.0 mmol) in 2-propanol (50 mL) was added hexamethyleneimine (2.73 g, 3.10 mL, 27.5 mmol). DIPEA (3.55 g, 4.79 mL, 27.5 mmol) was added, and the reaction mixture was heated at reflux for 2 h. The reaction mixture was allowed to cool to room temperature, which caused the product to crystallize. The solid product was collected by vacuum filtration and washed with cold 2-propanol (2 × 10 mL) and then diethyl ether (10 mL). After drying, the pure product was obtained as pink crystals (6.1 g, 86%). Mp 109−111 °C. 1H NMR (CDCl3): δ 6.13 (br s, 2H), 3.89 (s, 3H), 3.82 (t, J = 6.0 Hz, 4H), 1.84 (s, 4H), 1.58 (s, 4H). 13C NMR (CDCl3): δ 166.4, 153.7, 153.1, 120.3, 111.3, 52.0, 51.1, 28.4, 26.7. MS (ESI): m/z 285 (M + H)+, 307 (M + Na)+. HRMS (ESI+) Anal. For C12H17N4NaO2+: calcd mass 307.0932, found 307.0938.
General Method A: Suzuki−Miyaura Coupling Reactions.
Methyl 3-amino-6-chloro-5-hexamethyleneimine-2-pyrazine-carboxylate 7 (1 equiv) was combined with K2CO3 (10 equiv), the appropriate boronic acid (1.5 equiv), and Pd(PPh3)4 (5 mol %) in a two-neck round-bottom flask. The flask was connected to a condenser and purged with nitrogen. A 4:1 dry toluene/MeOH mixture (60 mL) was added via syringe, and the reaction mixture was heated at reflux for 0.5−18 h. The mixture was allowed to cool to room temperature and filtered through Celite (10 × 3 cm, eluting with 3 × 20 mL of EtOAc). The filtrate was evaporated to dryness and the residue purified by silica gel flash column chromatography using EtOAc/pet. spirit.
Methyl 3-Amino-5-(azepan-1-yl)-6-(4-(methylthio)-phenyl)-pyrazine-2-carboxylate (10a).
General method A using 4-(methylthio)phenylboronic acid (93 mg, 0.545 mmol) afforded 10a as a yellow solid; yield 77% (104 mg, 0.279 mmol). Mp 132−134 °C. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 6.20 (bs, 2H), 3.88 (s, 3H), 3.36 (t, J = 5.5 Hz, 4H,), 2.48 (s, 3H), 1.62 (s, 4H), 1.43 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.4, 154.8, 153.4, 137.8, 137.5, 131.2, 128.3, 126.8, 112.5, 51.9, 51.0, 28.0, 27.1, 16.1. HRESI-MS m/z (M + H+) Anal. for C19H25N4O2S: calcd mass, 373.1698. Found: 373.1688. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 36.1 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(4-(trifluoromethyl)-phenyl)-pyrazine-2-carboxylate (11a).
General method A using 4-(trifluoromethyl)phenylboronic acid (570 mg, 3.0 mmol) afforded 11a as a white solid; yield 64% (505 mg, 1.28 mmol). Mp 158−160 °C. 1H NMR (500 MHz, CDCl3): δ 7.63 (s, 4H), 6.13 (br s, 2H, NH2), 3.89 (s, 3H), 3.34 (s, 4H), 1.65 (s, 4H), 1.45 (s, 4H). 13C NMR (126 MHz, CDCl3): δ 167.4, 154.9, 153.7, 144.6, 129.9, 129.3, 128.2, 125.6, 125.1, 113.2, 52.1, 51.3, 28.1, 27.3. HRESI-MS m/z (M + Na+) Anal. For C19H21F3N4NaO2: calcd mass 417.1526. Found 417.1514. Anal.-HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 29.3 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(benzo[d][1,3]dioxol-5-yl)pyrazine-2-carboxylate (12a).
General method A using benzo-[d][1,3]dioxol-5-ylboronic acid (85 mg, 0.515 mmol) afforded 12a as a yellow solid; yield 51% (66 mg, 0.178 mmol). Mp 156−158 °C. 1H NMR (500 MHz, CDCl3) δ 7.02 (s, 1H), 6.93 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 5.96 (s, 2H), 3.88 (s, 4H), 3.38 (s, 4H), 1.63 (s, 4H), 1.44 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.6, 154.9, 153.5, 147.9, 147.1, 134.9, 131.5, 121.7, 112.2, 108.7, 108.3, 101.1, 51.9, 51.0, 28.1, 27.1. HRESI-MS m/z (M + H+) Anal. for C19H23N4O4: calcd mass 371.1719. Found 371.1734. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 31.8 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(isoquinolin-4-yl)-pyrazine-2-carboxylate (13a).
General method A using isoquinolin-4-ylboronic acid (271 mg, 1.06 mmol) afforded 13a as a brown solid; yield 58% (156 mg, 0.413 mmol). Mp 140−142 °C. 1H NMR (500 MHz, CDCl3) δ 9.23 (s, 1H), 8.58 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 3.86 (s, 3H), 3.21 (t, J = 5.5 Hz, 4H), 1.47 (s, 4H), 1.38 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.3, 155.8, 154.0, 152.1, 143.0, 134.3, 131.8, 130.8, 128.4, 128.0, 127.4, 126.7, 124.5, 112.9, 52.0, 50.4, 27.8, 26.8. HRESI-MS m/z (M + Na+) 400, Anal. for C21H24N5NaO2: calcd mass 400.1749. Found 400.1747. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 22.3 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(1-methyl-1H-pyrazol-4-yl)pyrazine-2-carboxylate (14a).
General method A using 1methyl-1-H-pyrazol-4-ylboronic acid (110 mg, 0.526 mmol) afforded 14a as a yellow solid; yield 77% (89 mg, 0.269 mmol). Mp 134−136 °C. 1H NMR (500 MHz, CDCl3) δ 7.64 (s, 1H), 7.57 (s, 1H), 6.17 (bs, 2H), 3.91 (s, 3H), 3.89 (s, 3H), 3.47 (t, J = 6 Hz, 4H), 1.66 (s, 4H), 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.4, 155.5, 153.4, 138.7, 129.1, 125.3, 122.6, 112.4, 52.1, 51.2, 39.0, 28.0, 27.2. HRESI-MS m/z (M + H+) 331, Anal. for C16H23N6O2: calcd mass 331.1882. Found 331.1894. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 25.2 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(1-(2-morpholinoethyl)-1H-pyrazol-4-yl)pyrazine-2-carboxylate (15a).
General method A using 1-(2-morpholinoethyl)-1H-pyrazol-4-ylboronic acid (165 mg, 0.536 mmol) afforded 15a as a yellow solid; yield 80% (119 mg, 0.277 mmol). Mp 132−134 °C. 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 1H), 7.61 (s, 1H), 6.24 (bs, 2H), 4.25 (s, 2H), 3.89 (s, 3H), 3.69 (s, 4H), 3.46 (s, 4H), 2.84 (s, 2H), 2.50 (s, 4H), 1.65 (s, 4H), 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.2, 155.5, 153.3, 138.6, 128.7, 125.2, 122.1 112.4, 66.9, 58.2, 53.7, 51.9, 51.1, 49.6, 27.9 27.0. HRESI-MS m/z (M + H+), Anal. for C22H36N7O3: calcd mass 430.2567. Found 430.2553. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 21.6 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(1H-indol-2-yl)-pyrazine-2-carboxylate (16a).
General method A using 1H-indol-2-ylboronic acid (278 mg, 1.73 mmol) afforded 16a as an orange solid; yield 34% (87.5 mg, 0.239 mmol). Mp 152−154 °C. 1H NMR (500 MHz, CDCl3) δ 9.14 (s, 1H), 7.57 (d, 1H), 7.37 (d, 1H, J = 7.7 Hz), 7.16 (t, 1H, J = 7.0 Hz), 7.08 (t, 1H, J = 7.0 Hz), 6.52 (s, 1H), 3.93 (s, 3H), 3.58 (t, J = 5.75 Hz, 4H), 1.70 (s,4H), 1.50 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.2, 155.1, 153.2, 136.0, 135.7, 129.0, 124.5, 122.4, 120.7, 119.90, 112.70, 111.0, 102.0, 52.1, 51.7, 28.1, 27.5. HRESI-MS m/z (M + H+) 388, Anal. for C20H24N5O2: calcd mass 388.1730. Found 388.1731. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 29.8 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(benzo[b]thiophen-2-yl)-pyrazine-2-carboxylate (17a).
General method A using benzo[b]-thiophen-2-ylboronic acid (534 mg, 3.0 mmol) afforded 17a as a yellow solid; yield 78% (600 mg). Mp 146−148 °C. 1H NMR (500 MHz, CDCl3): δ 1.48 (s, 4H), 1.69 (s, 4H), 3.51 (s, 4H), 3.91 (s, 3H), 6.13 (br s, 2H, NH2), 7.22 (s, 1H), 7.21 (m, 2H), 7.71 (d, J = 6.9 Hz, 1H), 7.80 (d, J = 7.0 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 167.3, 155.3, 153.6, 143.3, 140.2, 140.1, 125.4, 124.5, 124.4, 123.6, 122.3, 122.0, 113.2, 52.3, 51.3, 28.2, 27.4. HRESI-MS m/z (M + K+) Anal. For C20H22KN4O3 calcd mass 405.1394. Found 405.1361. Anal.-HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 31.9 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(benzofuran-2-yl)-pyrazine-2-carboxylate (18a).
General method A using benzofuran-2-ylboronic acid (486 mg, 3.0 mmol) afforded 18a as a yellow solid; yield 57% (420 mg). Mp 121−123 °C. 1H NMR (500 MHz, CDCl3): δ 1.46 (s, 4H), 1.66 (s, 4H), 3.45 (t, J = 5.8 Hz, 4H), 3.90 (s, 3H), 6.13 (br s, 2H, NH2), 6.93 (s, 1H), 7.21 (t, J = 7.3 Hz, 1H), 7.25 (t, J = 7.2 Hz, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.56 (d, J = 7.5 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 167.2, 164.9, 154.8, 154.6, 129.2, 124.4, 123.1, 121.4, 121.2, 112.9, 111.5, 105.1, 52.2, 50.2, 28.2, 27.3. HRESI-MS m/z (M + H+) Anal. for C20H23N4O2: calcd mass 367.1831. Found. 367.1770. Anal.-HPLC (70:30 H2O/0.1% CH3CN/0.1% TFA → 100% CH3CN/ 0.1% TFA over 30 min, tR = 29.5 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(4-fluorobenzofuran-2yl)-pyrazine-2-carboxylate (19a).
General method A using 4-fluorobenzofuran-2-ylboronic acid (95 mg, 0.531 mmol) afforded 19a as a yellow solid; yield 58% (80 mg, 0.210 mmol). Mp 108−110 °C. 1H NMR (126 MHz, CDCl3): 7.27 (d, 1H), 7.20 (m, 1H), 6.99 (s, 1H), 6.93 (t, 1H, J = 8.5 Hz), 3.91 (s, 3H), 3.46 (s, 4H), 1.69 (s, 4H), 1.49 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.0, 156.9, 156.2, 154.7, 154.0, 138.3, 124.6, 120.5, 118.0, 115.9, 110.4, 108.6, 107.6, 52.2, 50.0, 28.0, 27.1. HRESI-MS m/z (M + Na+) Anal. for C20H21FN6NaO4: calcd mass 407.1495. Found 407.1504. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 21.2 min).
Methyl-3-amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(thiophen-2-yl)-pyrazine-2-carboxamide (20a).
General method A using thiophen-2-ylboronic acid (70 mg, 0.549 mmol) afforded 20a as a light yellow solid; yield 85% (99 mg, 0.299 mmol). Mp 118−120 °C. 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 5 Hz, 1H), 7.03 (d, J = 3 Hz, 1H), 6.98 (m, 1H), 3.89 (s, 3H), 3.43 (t, J = 5.7 Hz, 4H), 1.67 (s, 4H), 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.3, 155.2, 153.5, 142.8, 126.9, 126.0, 125.8, 125.5, 112.7, 52.1, 51.1, 28.2, 27.3. HRESI-MS m/z (M + Na+) Anal. for C16H20N4NaO2: calcd mass 355.1205. Found 355.1193. Anal. HPLC (100 H2O/0.1% TFA →100% CH3CN/ 0.1% TFA over 30 min, tR = 28.8 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(thiophen-3-yl)-pyrazine-2-carboxylate (21a).
General method A using thiophen-3-ylboronic acid (75 mg, 0.584 mmol) afforded 21a as a white solid; yield 85% (100 mg, 0.302 mmol). Mp 118−120 °C. 1H NMR (500 MHz, CDCl3) δ 7.38 (s, 1H), 7.29 (m, 1H), 7.25 (d, J = 5 Hz, 1H), 3.88 (s, 3H), 3.38 (t, J = 5.8 Hz, 4H), 1.63 (s, 4H), 1.44 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.4, 155.3, 153.5, 141.3, 128.1, 127.9, 125.4, 122.8, 112.2, 52.0, 50.9, 28.0, 27.1. HRESI-MS m/z (M + H+) Anal. For C16H21N4NaO2S: calcd mass 355.1205. Found 355.1197. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 28.3 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(furan-2-yl)-pyrazine-2-carboxylate (22a).
General method A using furan-2-ylboronic acid (59 mg, 0.526 mmol) afforded 22a as an off white solid; yield 98% (111 mg, 0.352 mmol). Mp 116−118 °C. 1H NMR (500 MHz, CDCl3): δ 7.41 (s, 1H), 6.54 (d, J = 2.3 Hz, 1H), 6.45 (s, 1H), 6.13 (br s, 2H, NH2), 3.89 (s, 3H), 3.37 (t, J = 5.5 Hz, 4H), 1.66 (s, 4H), 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3): δ 167.1, 154.8, 154.0, 152.2, 141.4, 122.0, 112.1, 111.6, 108.6, 52.0, 50.0, 28.0, 26.9. HRESI-MS m/z (M + Na+) Anal. for C16H21N4NaO3: calcd mass 339.1418. Found 339.1433. Anal.-HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 27.1 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(furan-3-yl)-pyrazine-2-carboxylate (23a).
General method A using furan-3-ylboronic acid (107 mg, 0.389 mmol) afforded 23a as a yellow solid; yield 93% (114 mg, 0.360 mmol). Mp 122−124 °C. 1H NMR (500 MHz, CDCl3) δ 7.64 (s, 1H), 7.41 (s, 1H), 6.61 (s, 1H), 6.20 (bs, 2H), 3.89 (s, 3H), 3.48 (t, J = 5 Hz, 4H), 1.66 (s, 4H), 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.3, 155.6, 153.6, 142.6, 140.6, 125.8, 124.8, 112.6, 111.2, 52.0, 51.0, 28.0, 27.1. HRESI-MS m/z (M + H+) Anal. for C16H21N4O3: calcd mass 316.1614. Found 316.1604. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 30.9 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2-fluoropyridin-3-yl)-pyrazine-2-carboxylate (24a).
General method A using 2-fluoropyridin-3-ylboronic acid (75 mg, 0.529 mmol) afforded 24a as a brown solid; yield 76% (92 mg, 0.266 mmol). Mp 156−158 °C. 1H NMR (500 MHz, CDCl3) δ 8.17 (d, J = 4.3 Hz, 1H), 8.06 (m, 1H), 7.29 (t, J = 5.5 Hz, 1H), 3.89 (s, 3H), 3.36 (s, J = 5.7 Hz, 4H), 1.63 (s, 4H), 1.46 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.2, 161.3, 155.2, 154.1, 146.8, 141.1, 124.5, 123.8, 122.2, 113.3, 52.3, 50.3, 28.1, 27.3. HRESI-MS m/z (M + Na+) Anal. for C17H20FN5NaO2: calcd mass 368.1499. Found 368.1489. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 26.3 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(pyrimidin-5-yl)-pyrazine-2-carboxylate (25a).
General method A using pyrimidin-5-ylboronic acid (279 mg, 2.25 mmol) afforded 25a as a yellow solid; yield 91% (450 mg). Mp 176−178 °C. 1H NMR (500 MHz, CDCl3): δ 9.13 (s, 1H), 8.89 (s, 2H), 6.13 (br s, 2H, NH2), 3.91 (s, 3H), 3.52 (t, J = 6.0 Hz, 4H), 1.68 (s, 4H), 1.48 (s, 4H). 13C NMR (126 MHz, CDCl3): δ 167.1, 157.1, 155.7, 155.3, 153.9, 134.9, 124.5, 114.5, 52.3, 51.5, 28.0, 27.4. HRESI-MS m/z (M + Na+) Anal. for C16H20N6NaO2: calcd mass 351.1545. Found. 351.1535. Anal.-HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 23.4 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2-methoxypyrimidin-5yl)-pyrazine-2-carboxylate (26a).
General method A using 2-methoxypyrimidin-5-ylboronic acid (79 mg, 0.513 mmol) afforded 26a as a yellow solid; yield 88% (111 mg, 0.310 mmol). Mp 168−170 °C. 1H NMR (500 MHz, CDCl3): 8.66 (s, 2H), 6.13 (br s, 2H, NH2), 4.05 (s, 3H), 3.90 (s, 3H), 3.38 (t, J = 5.5 Hz, 4H), 1.68 (s, 4H), δ 1.48 (s, 4H). 13C NMR (126 MHz, CDCl3): δ 166.4, 164.7, 158.3, 155.3, 153.8, 128.8, 125.2, 113.9, 55.2, 52.2, 51.4, 28.0, 27.3. HRESI-MS m/z (M + H+) Anal. for C17H22N6O3: calcd mass 359.1832. Found 359.1848. Anal.-HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tr = 28.0 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2,4-dimethoxypyrimidin-5-yl)-pyrazine-2-carboxylate (27a).
General method A using 2,4-dimethoxypyrimidin-5-ylboronic acid (79 mg, 0.350 mmol) afforded 27a as a yellow solid; yield 91% (124 mg, 0.319 mmol). Mp 150−152 °C. 1H NMR (500 MHz, CDCl3) δ 8.36 (s, 1H), 4.03 (s, 3H), 3.94 (s, 3H), 3.87 (s, 3H), 3.39 (s, 4H), 1.60 (s, 4H,), 1.46 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 168.3, 167.2, 164.8, 158.1, 155.3, 154.1, 123.8, 117.1, 112.6, 55.0, 54.1, 52.0, 50.0, 28.0, 27.0. HRESI-MS m/z (M + H+) Anal. for C18H25N6O4 calcd mass 389.1937. Found389.1929. Anal.-HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 27.1 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2,6-dimethoxypyridin3-yl)-pyrazine-2-carboxylate (28a).
General method A using 2,6-dimethoxypyridin-3-ylboronic acid (200 mg, 0.703 mmol) afforded 28a as a yellow solid; yield 42% (115 mg, 0.297 mmol). Mp 134−136 °C. 1H NMR (500 MHz, CDCl3) δ 7.66 (d, J = 8.0 Hz, 1H), 6.38 (d, J = 8.0 Hz, 1H), 3.92 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.45 (bs, 2H), 3.31 (bs, 2H), 1.57 (s, 4H), 1.44 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.5, 162.8, 159.7, 155.3, 153.9, 141.5, 127.6, 116.2, 112.2, 101.3, 53.7, 53.5, 51.9, 50.1, 28.1, 27.1. HRESI-MS m/z (M + H+), Anal. for C19H25N5O4: calcd mass 388.1985. Found 388.1983. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 31.6 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2,4-dimethoxyphenyl)-pyrazine-2-carboxylate (29a).
General method A using 2,4-dimethoxyphenylboronic acid (128 mg, 0.702 mmol) afforded 29a as a yellow solid; yield 53% (143 mg, 0.371 mmol). Mp 106−108 °C. 1H NMR (500 MHz, CDCl3): δ 7.33 (d, J = 8.3 Hz, 1H), 6.54 (dd, J = 8.3 Hz, J = 2.1 Hz 1H), 6.42 (d, J = 2.1 Hz, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.70 (s, 3H), 3.44 (s, 2H), 3.26 (s, 2H), 1.58 (s, 2H), 1.53 (s, 2H), 1.46 (s, 2H), 1.39 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 166.0, 160.9, 158.2, 155.4, 154.0, 131.1, 129.2, 123.7, 111.8, 105.1, 98.7, 55.7, 55.7, 52.0, 50.6, 27.8, 26.9. HRESI-MS m/z (M + Na+), Anal. For C20H27N4NaO4: calcd mass 409.1852. Found 409.1866. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 28.6 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(3,5-dimethoxyphenyl)-pyrazine-2-carboxylate (30a).
General method A using 3,5-dimethoxyphenylboronic acid (128 mg, 0.702 mmol) afforded 30a as a yellow solid; 62% (166 mg, 0.430 mmol). Mp 96−98 °C. 1H NMR (500 MHz, CDCl3) δ 6.64 (s, 2H), 6.40 (s, 1H), 3.88 (s, 3H), 3.81 (s, 6H), 3.38 (t, J = 6.0 Hz, 4H), 1.64 (s, 4H), 1.44 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.5, 161.0, 154.7, 153.7, 142.8, 131.6, 112.0, 106.3, 100.0, 55.6, 52.0, 50.9, 28.1, 27.1. HRESI-MS m/z (M + Na+), Anal. for C20H26N4NaO4: calcd mass 409.1852. Found 409.1867. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 30.0 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(3,6-dimethoxypyridazin-4-yl)pyrazine-2-carboxylate (31a).
General method A using 3,6-dimethoxypyridazin-4-ylboronic acid (97 mg, 0.526 mmol) afforded 31a as a yellow solid; yield 68% (94 mg, 0.242 mmol). Mp 152−154 °C. 1H NMR (500 MHz, CDCl3) δ 7.17 (s, 1H), 4.08 (s, 3H), 4.00 (s, 3H), 3.88 (s, 3H), 3.43 (s, 2H), 3.29 (s, 2H), 1.63 (s, 4H), 1.44 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 167.00, 162.8, 159.6, 154.7, 154.1, 135.1, 122.9, 119.4, 113.1, 54.8, 54.6, 52.1, 49.7, 28.0, 27.2. HRESI-MS m/z (M + Na+), Anal. for C18H24N6NaO4: calcd mass 411.1757. Found 411.1762. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 21.2 min).
Methyl 3-Amino-6-(2-aminopyrimidin-5-yl)-5-(azepan-1-yl)-pyrazine-2-carboxylate (32a).
General method A using 2-aminopyrimidin-5-ylboronic acid (208 mg, 1.5 mmol) afforded 32a as a yellow solid; yield 83% (285 mg). Mp 216−218 °C. 1H NMR (500 MHz, DMSO-d6): δ 8.27 (s, 2H), 7.04 (br s, 2H, NH2), 6.76 (br s, 2H, NH2), 3.74 (s, 3H), 3.41 (t, J = 5.9 Hz, 4H), 1.59 (s, 4H), 1.41 (s, 4H). 13C NMR (126 MHz, DMSO-d6): δ 27.0, 27.9, 51.0, 51.9, 112.3, 123.9, 126.5, 154.2, 155.1, 157.4, 163.0, 167.3, 163.0, 157.4, 155.1, 154.2, 126.5, 123.9, 112.3, 51.9, 51.0, 27.9, 27.0. HRESI-MS m/z (M + H+), Anal. for C16H22N7O2: calcd mass 344.1835. Found 344.1833. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 14.2 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2-chloropyrimidin-5-yl)-pyrazine-2-carboxylate (8).
General method A using (2-chloropyrimidin-5-yl)boronic acid (475 mg, 3 mmol) afforded 8 as a yellow solid (460 mg, 64%). Mp 168−170 °C. 1H NMR (CDCl3): δ 8.76 (s, 2H), 6.13 (br s, 2H, NH2), 3.91 (s, 3H), 3.35 (t, J = 5.8 Hz, 4H), 1.71 (s, 4H), 1.49 (s, 4H). 13C NMR (CDCl3): δ 27.3, 27.8, 51.4, 52.2, 114.6, 122.8, 133.3, 153.7, 155.0, 158.0, 159.2, 166.8. MS (ESI): m/z 363 (M + H)+, 385 (M + H)+. HRESI-MS m/z (M + H+), Anal. for C16H20ClN6O2: calcd mass 363.1336. Found 363.1347. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 17.6 min).
General Method B. Synthesis of Methyl 3-Amino-5-(azepan1-yl)-6-(2-(substituted amino)pyrimidin-5-yl)pyrazine-2-carboxylates 33a and 34a.
To a solution of methyl 3-amino-5-(azepan-1-yl)-6-(2-chloropyrimidin-5-yl)pyrazine-2-carboxylate 8 (0.25 mmol) in DMF (2 mL) at room temperature was added the appropriate amine (0.75 mmol). DIPEA (0.75 mmol) was then added, and the reaction mixture was stirred at room temperature for 2 days. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic layer was washed with brine, dried (MgSO4), and evaporated. The residue was purified by silica gel flash column chromatography using EtOAc/pet. spirit.
Methyl 3-Amino-5-(azepan-1-yl)-6-(2-(methylamino)-pyrimidin-5-yl)pyrazine-2-carboxylate (33a).
General method B using 8 (91 mg, 0.25 mmol) and methylamine (2 M in MeOH, 0.375 mL, 0.75 mmol) gave 33a as a yellow solid; yield 87% (78.0 mg). Mp 158−160 °C. 1H NMR (500 MHz, CDCl3): δ 8.45 (s, 2H), 6.22 (br s, 2H, NH2), 5.28 (q, J = 4.9 Hz, 1H, NH), 3.90 (s, 3H), 3.42 (t, J = 5.9 Hz, 4H), 3.04 (d, J = 5.1 Hz, 3H), 1.67 (s, 4H), δ 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3): δ 167.4, 162.0, 157.2, 155.3, 153.7, 126.9, 124.2, 113.3, 52.3, 51.3, 28.8, 28.0, 27.3. HRESI-MS m/z (M + H+), Anal. for C17H24N7O2 358.1991, found 358.2001. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 14.8 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2-(isopropylamino)-pyrimidin-5-yl)pyrazine-2-carboxylate (34a).
General method B using 8 (91 mg, 0.25 mmol) and isopropylamine (45 mg, 0.75 mmol) gave 34a as a yellow solid; yield 83% (80 mg). Mp 155−157 °C. 1H NMR (500 MHz, CDCl3): δ 8.43 (s, 2H), 6.25 (br s, 2H), 5.10 (d, J = 7.8 Hz, 1H), 4.16 (septet, J = 6.8 Hz, 1H), 3.89 (s, 3H), 3.43 (t, J = 5.9 Hz, 4H), 1.67 (s, 4H), 1.47 (s, 4H), 1.25 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 167.2, 157.0, 155.1, 153.5, 126.8, 123.8, 113.1, 52.0, 51.1, 43.0, 27.8, 27.1, 22.9. HRESI-MS m/z (M + H+), Anal. for for C19H28N7O2 386.2304, found 386.2318. Anal. HPLC (100 H2O/ 0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 16.1 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(2-(2-hydroxyethyl)-amino)pyrimidin-5-yl)pyrazine-2-carboxylate (35a).
To a solution of methyl 3-amino-5-(azepan-1-yl)-6-(2-chloropyrimidin-5-yl)-pyrazine-2-carboxylate 8 (91 mg, 0.25 mmol) in 2-propanol (5 mL) was added ethanolamine (16.8 mg, 0.017 mL, 0.275 mmol). DIPEA (35.5 mg, 0.048 mL, 0.275 mmol) was then added, and the reaction mixture was heated at reflux for 2 h. After cooling to room temperature, additional equivalents of ethanolamine (0.275 mmol) and DIPEA (0.275 mmol) were added, and the reaction mixture was heated for a further 2 h. The mixture was allowed to cool to room temperature overnight, which caused the product to crystallize. The solid product was collected by vacuum filtration and washed with cold 2-propanol (2 × 2 mL) and diethyl ether (2 mL). After drying, the pure product was obtained as light yellow crystals (82 mg, 84%). Mp 164−166 °C 1H NMR (500 MHz, CDCl3): δ, 8.44 (s, 2H), 6.23 (br s, 2H), 5.64 (t, J = 5.5 Hz, 1H), 3.90 (s, 3H), 3.85 (t, J = 4.4 Hz, 2H), 3.62 (q, J = 4.9 Hz, 2H), 3.42 (t, J = 5.6 Hz, 4H), 1.68 (s, 4H), 1.48 (s, 4H). 13C NMR (126 MHz, CDCl3): δ 167.1, 161.7, 156.9, 155.1, 153.5, 126.2, 124.6, 113.3, 63.6, 52.1, 51.1, 44.9, 27.8, 27.1. HRESI-MS m/z (M + H+), Anal. For C18H26N7O3 388.2097, found 388.2088. Anal. HPLC (100 H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 14.4 min).
General Method C. Synthesis of Acyl Guanidines Using the Reported Method.
38 Free guanidine was generated via neutralization of guanidine·HCl with Na+ iPrO− in iPrOH to create a 2-fold molar excess relative to the 6-substituted pyrazinoyl methyl ester. A stock solution of iPrO− was prepared by adding Na (typically 30−70 mg) in anhydrous iPrOH under N2 and heated at 70 °C for 30 min. From this stock solution an amount of iPrO− was removed via syringe and made up to 10 mL in anhydrous iPrOH. To this solution was added a stoichiometric amount of guanidine hydrochloride, and the mixture was stirred at 70 °C for 30 min. This solution was gravity filtered onto the neat 6-substituted pyrazinoyl methyl ester. Once filtration was complete, the mixture was heated to reflux and allowed to react for 1−6 h. The reactions were monitored by MS and TLC and terminated by removal of solvent under reduced pressure. Final products were isolated by rp-HPLC. All compounds were isolated as TFA salts and exchanged to their respective HCl salts by stirring with quaternary ammonium Cl− anion exchange resin in MeOH prior to biological evaluation.
General Method D. Improved Synthesis of Acyl Guanidines.
Dry CH3OH (50 mL) was slowly added with stirring to small pieces of sodium (2.3 g, 0.1 mol) cooled on ice under nitrogen. After complete dissolution of the sodium, guanidine hydrochloride (9.55 g, 0.1 mol) was added and the resulting mixture was stirred at room temperature for 1.5 h. Filtration of the white precipitate gave a 2 M solution of free guanidine in methanol, which was collected in a sealed dry flask under nitrogen and stored in a refrigerator.
To a suspension of the requisite methyl ester (1 equiv, 0.2−0.5 mmol) in DMF (5 mL) was added 2 M guanidine in methanol (10 equiv, 2−5 mmol), and the resulting mixture was stirred at room temperature overnight. Brine (20 mL) was added and the mixture extracted with EtOAc (3 × 30 mL). The organic layer was washed with 10% NaCl (2 × 30 mL), dried (MgSO4), and concentrated under reduced pressure. The crude mixture was dissolved in 5 mL of 30% CH3CN/70% H2O and 0.1% acetic acid and loaded onto a reverse phase silica gel chromatography column. Isocratic elution was performed with CH3CN/H2O mixtures containing 0.1% acetic acid. The pooled fractions containing pure compound by TLC analysis were concentrated under reduced pressure and neutralized with solid NaHCO3. The aqueous mixture was extracted with EtOAc (3 × 30 mL), and the combined organic layer was washed with brine, dried (MgSO4), and evaporated to give the free acylguanidine.
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(4-(methylthio)phenyl)pyrazine-2-carboxamide (10).
General method C using 10a (70 mg, 0.214 mmol) gave 10 as a yellow solid; yield 5% (6 mg, 0.015 mmol). Mp 152−154 °C. 1H NMR (500 MHz, CDCl3) δ 10.31 (s, 1H), 9.44 (s, 2H), 8.47 (s, 2H), 7.44 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 3.40 (t, J = 5.5 Hz, 4H) 1.60 (s, 4H), 1.46 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.0, 156.6, 155.7, 153.6, 139.0, 136.4, 131.2, 128.1, 126.6, 111.0, 51.5, 27.9, 27.1, 15.5. HRESIMS m/z (M + H+) Anal. for C19H26N7OS: calcd mass 400.1920. Found 400.1935. Anal. HPLC. (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 28.4 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(4-(trifluoromethyl)phenyl)pyrazine-2-carboxamide (11).
General method D using 11a (394 mg, 1.0 mmol) gave 11 as a yellow solid; yield 92% (385 mg). Mp 172−175 °C. 1H NMR (CD3OD): δ 7.68 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 3.38 (t, J = 5.0 Hz, 4H), 1.65 (s, 4H), 1.46 (s, 4H). 13C NMR (CD3OD): δ 167.9, 160.2, 155.8, 155.0, 145.6, 130.1, 130.0 (d, J = 32 Hz), 129.2, 125.8 (d, J = 2.7 Hz), 124.3 (d, J = 170), 112.9, 51.6, 28.6, 27.6. HRESI-MS m/z (M + H+) Anal. for C19H23F3N7O: calcd mass 422.1916. Found 422.1911. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 27.9 min).
3-Amino-5-(azepan-1-yl)-6-(benzo[d][1,3]dioxol-5-yl)-N-carbamimidoylpyrazine-2-carboxamide (12).
General method C using 12a (54 mg, 0.146 mmol) gave 12 as a yellow oil; yield 9% (6 mg, 0.015 mmol). Mp 154−156 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.71 (s, 2H), 8.27 (s, 2H), 7.31 (s, 1H), 6.96 (m, 2H), 6.08 (s, 2H), 3.40 (t, J = 5.75 Hz, 4H), 1.60 (s, 2H), 1.39 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.2, 155.2, 154.8, 153.6, 147.26, 146.7, 133.4, 130.3, 121.4, 112.7, 108.5, 108.0, 101.2, 50.4, 27.2, 26.1. HRESI-MS m/z (M + H+) Anal. for C19H24N7O3: calcd mass 398.1941. Found 398.1929. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 26.4 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(isoquinolin-4-yl)pyrazine-2-carboxamide (13).
General method C using 13a (156 mg, 0.413 mmol) gave 13 as an orange solid; yield 15% (32 mg, 0.079 mmol). Mp 158−160 °C. 1H NMR (500 MHz, CD3OD) δ 10.47 (s, exchange with solvent), 9.57 (bs, 1H), 9.19 (s, exchange with solvent), 9.09 (s, exchange with solvent) 8.69 (bs, 1H), 8.40 (d, J = 8.2 Hz, 1H), 8.01 (t, J = 7.5 Hz, 1H), 7.91 (t, J = 7.5 Hz, 1H), 7.81 (d, J = 8.2 Hz, 1H), 3.37 (s, 2H), 3.23 (s, 2H), 1.53 (s, 4H), 1.44 (s, 4H). 13C NMR (126 MHz, CD3OD) δ 167.5, 157.7, 156.9, 156.4, 149.7, 137.6, 136.9, 136.9, 136.8, 131.6, 131.4, 129.0, 125.7, 124.7, 113.0, 51.6, 29.0, 27.8. HRESI-MS m/z (M + H+) Anal. for C20H24N7OS: calcd mass 410.1763. Found 410.1758. Anal. HPLC (70:30 H2O/0.1% TFA/ CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 22.7 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(1-methyl-1H-pyrazol-4-yl)pyrazine-2-carboxamide (14).
General method C using 14a (69 mg, 0.207 mmol) gave 14 as a yellow oil; yield 38% (32 mg, 0.193 mmol). Mp 150−152 °C. 1H NMR (500 MHz, CDCl3) δ 10.49 (s, 1H), 8.83 (s, 2H), 8.52 (s, 2H), 8.00 (s, 1H), 7.57 (s, 1H), 5.49 (bs, 2H), 3.93 (s, 3H), 3.50 (t, J = 5.75 Hz, 4H), 1.69 (s, 4H), 1.49 (s, 4H).13C NMR (126 MHz, CDCl3) δ 166.6, 156.4, 156.1, 153.4, 137.4, 130.7, 125.1, 121.7, 111.1, 51.5, 38.7, 27.9, 27.3. HRESI-MS m/z (M + H+) Anal. for C16H24N9O: calcd mass 358.2104. Found 358.2118. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 21.0 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(1-(2-morpholinoethyl)-1H-pyrazol-4-yl)pyrazine-2-carboxamide (15).
General method C using 15a (99 mg, 0.229 mmol) gave 15 as an orange oil; yield 20% (22 mg, 0.048 mmol). Mp 157−159 °C. 1H NMR (500 MHz, CDCl3) δ 10.48 (s, 1H), 8.86 (s, 2H), 8.65 (s, 2H), 7.89 (s, 1H), 7.56 (s, 1H), 4.67 (s, 2H), 3.93 (s, 4H), 3.69 (s, 2H,), 3.47 (s, 4H), 3.42 (bs, 2H), 3.05 (bs, 2H,) 1.67 (s, 4H), 1.49 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.6, 156.4, 156.1, 153.6, 140.0, 130.6, 124.1, 122.9, 111.3, 63.8, 57.2, 53.3, 51.4, 46.4, 27.9, 27.2. HRESI-MS m/z (M + H+) Anal. for C21H33N10O2: calcd mass 457.2788. Found 457.2796. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 19.0 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(1H-indol-2-yl)pyrazine-2-carboxamide (16).
General method C using 16a (76 mg, 0.209 mmol) gave 16 as an orange solid; yield 31% (29 mg, 0.074 mmol). Mp 152−154 °C. 1H NMR (500 MHz, CDCl3) δ 11.09 (s, 1H), 10.82 (s, 1H), 8.82 (s, 2H), 7.92 (s, 2H), 7.55 (d, 1H, J = 7.7 Hz), 7.41 (d, 1H, J = 7.7 Hz), 7.12 (t, 1H, J = 7 Hz), 7.03 (t, 1H), 6.44 (s, 1H), 3.64 (t, J = 5.0 Hz, 4H), 1.73 (s, 4H), 1.50 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.7, 156.2, 155.7, 153.0, 136.6, 134.9, 128.3, 125.7, 122.7, 120.5, 119.8, 111.7, 110.8, 102.3, 52.1, 27.9, 27.6. HRESI-MS m/z (M + H+) Anal. for C20H28N7O3: calcd mass 393.2151. Found 393.2159. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 24.4 min).
3-Amino-5-(azepan-1-yl)-6-(benzo[b]thiophen-2-yl)-N-carbamimidoylpyrazine-2-carboxamide (17).
General method D using 17a (383 mg, 1.0 mmol) gave 17 as a yellow solid; yield 78% (320 mg). Mp 220−224 °C. 1H NMR (500 MHz, CDCl3) δ 10.48 (s, 1H), 8.92 (s, 2H), 8.76 (s, 2H), 7.60 (d, J = 7.5 Hz, 1H), 7.43 (d, J = 7.5 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 7.20 (s, 1H), 3.50 (t, J = 5.75 Hz, 4H), 1.69 (s, 4H), 1.49 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 168.2. 164.9, 160.7, 156.5, 155.4, 144.1, 141.3, 141.2, 125.8, 125.5, 124.7, 123.5, 123.0, 113.5, 52.0, 29.2, 28.1. HRESI-MS m/z (M + H+) C20H24N7OS: calcd mass 410.1763. Found 410.1758. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 27.5 min).
3-Amino-5-(azepan-1-yl)-6-(benzofuran-2-yl)-N-carbamimidoylpyrazine-2-carboxamide (18).
General method D using 18a (366 mg, 1.0 mmol) gave 18 as a yellow solid; yield 86% (338 mg). Mp 130−132 °C; 1H NMR (CD3OD): δ 7.64 (d, J = 7.5 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 7.05 (s, 1H), 3.51 (t, J = 5.0 Hz, 4H), 1.71 (s, 4H), 1.54 (s, 4H). 13C NMR (CD3OD): δ 167.4, 164.3, 154.6, 154.5, 154.4, 154.1, 129.7, 124.9, 123.7, 121.6, 121.1, 112.4, 111.5, 105.6, 50.3, 28.6, 27.3. HRESI-MS m/z (M + H+) Anal. for C20H24N7O2: calcd mass 394.1991. Found 394.1986. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 27.5 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(4-fluoro-benzofuran-2-yl)pyrazine-2-carboxamide (19).
General method C using 19a (54 mg, 0.139 mmol) gave 19 as an orange solid; yield 26% (15 mg, 0.036 mmol). Mp 132−136 °C. 1H NMR (500 MHz, CD3OD) δ 10.47 (s, exchange with solvent), 8.98 (s, exchange with solvent), 8.33 (s, exchange with solvent), 7.36 (t, J = 8.7 Hz, 1H), 7.32 (q, J = 5.5 Hz, 1H), 7.22 (s, 1H), 7.02 (t, J = 8.7 Hz, 1H), 3.44 (t, J = 5.8 Hz, 4H) 1.72 (s, 4H), 1.51 (s, 4H). 13C NMR (126 MHz, CD3OD) δ 167.3, 158.2, 157.4, 157.0, 156.9, 156.2, 156.2, 155.7, 126.6, 121.2, 112.2, 109.8, 108.7, 102.4, 51.1, 29.0, 27.9. HRESI-MS m/z (M + H+) Anal. for C20H22FN7O2,: calcd mass 412.1897. Found 412.1909. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 25.6 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(thiophen-2-yl)pyrazine-2-carboxamide (20).
General method C using 20a (99 mg, 0.298 mmol) gave 20 as a yellow oil; yield 2% (2 mg, 0.006 mmol). Mp 136−138 °C. 1H NMR (500 MHz, CDCl3) δ 10.10 (s, 1H), 10.04 (bs, 2H), 8.08 (bs, 2H), 7.26 (d, J = 5 Hz, 1H), 7.03 (d, J = 3 Hz, 1H), 3.47 (t, J = 5.75 Hz, 4H), 1.68 (m, 4H), 1.49 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 167.0, 156.3, 156.1, 153.4, 139.9, 128.4, 128.2, 125.2, 124.5, 111.0, 51.0, 27.8, 27.1. HRESI-MS m/z (M + H+) Anal. for C16H21N7O: calcd mass 360.1607. Found 360.1610. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 24.2 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(thiophen-3-yl)pyrazine-2-carboxamide (21).
General method C using 21a (103 mg, 0.309 mmol) gave 21 as an orange oil; yield 17% (24 mg, 0.067 mmol). Mp 138−140 °C. 1H NMR (500 MHz, CDCl3) δ 10.31 (s, 1H), 8.62 (bs, 2H), 8.29, (bs, 2H), 7.78 (m, 3H), 7.64 (dd, J = 8.5, 1.5 Hz, 1H,), 7.44 (m, 2H), 6.23 (bs, 2H), 3.38 (t, J = 5.75, 4H), 1.59 (s, 4H), 1.42 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.7, 156.2, 156.0, 153.6, 140.1, 128.4, 127.8, 125.9, 123.6, 110.7, 51.3, 27.9, 27.1. HRESIMS m/z (M + H+) Anal. for C16H21N7O: calcd mass 360.1607. Found 360.1611. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 25.2 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(furan-2-yl)-pyrazine-2-carboxamide (22).
General method D using 22a (316 mg, 1.0 mmol) gave 22 as a yellow solid; yield 91% (310 mg). Mp 143−146 °C. 1H NMR (CD3OD): δ 7.50 (s, 1H), 6.55 (d, J = 3.0 Hz, 1H), 6.50 (s, 1H), 3.37 (t, J = 6.0 Hz, 4H), 1.64 (s, 4H), 1.47 (s, 4H). 13C NMR (CD3OD): δ 168.1, 160.7, 156.3, 156.0, 153.4, 142.8, 122.4, 112.6, 112.4, 109.9, 50.9, 29.2, 27.8. HRESI-MS m/z (M + H+) Anal. for C16H22N7O2: calcd mass 344.1835. Found 344.1830. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 24.3 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(furan-3-yl)-pyrazine-2-carboxamide (23).
General method C using 23a (90 mg, 0.285 mmol) gave 23 as a yellow solid; yield 35% (41 mg, 0.119 mmol). Mp 146−148 °C. 1H NMR (500 MHz, CDCl3) δ 10.42 (s, 1H), 8.54 (s, 4H), 7.63 (s, 1H), 7.42 (s, 1H), 6.63 (s, 1H), 5.35 (bs, 2H), 3.52 (t, J = 5.75 Hz, 4H), 1.68 (s, 4H), 1.49 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.4, 156.3, 155.8, 153.6, 143.2, 141.1, 125.3, 124.9, 110.8, 110.7, 51.4, 27.9, 27.1. HRESI-MS m/z (M + H+) Anal. for C16H22N7O2: calcd mass 344.1835. Found 344.1837. Anal. HPLC (70:30 H2O/0.1% TFA/ CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 25.5 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2-fluoropyridin-3-yl)pyrazine-2-carboxamide (24).
General method C using 24a (100 mg, 0.290 mmol) gave 24 as a yellow solid; yield 13% (18 mg, 0.048 mmol). Mp 160−162 °C. 1H NMR (500 MHz, CDCl3) δ 10.42 (s, 1H), 8.59 (bs, 4H), 8.17 (m, 2H), 7.29 (t, J = 5.4 Hz) 5.13 (bs, 2H), 3.38 (s, 4H), 1.64 (s, 4H), 1.47 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.5, 160.7, 158.8, 156.0, 155.9, 154.0, 146.9, 141.4, 124.2, 123.4, 122.5, 111.8, 50.4, 27.9, 27.2. HRESI-MS m/z (M + H+) Anal. for C17H22FN8O: calcd mass 373.1901. Found 373.1916. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 22.2 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(pyrimidin-5-yl)pyrazine-2-carboxamide (25).
General method D using 25a (328 mg, 1.0 mmol) gave 25 as a yellow solid; yield 87% (305 mg). Mp 188− 190 °C. 1H NMR (CD3OD): δ 8.98 (s, 1H), 8.97 (s, 2H), 3.35 (t, J = 5.5 Hz, 4H), 1.67 (s, 4H), 1.47 (s, 4H). 13C NMR (CD3OD): δ 162.2, 156.5, 156.3, 155.7, 155.5, 154.0, 153.5, 135.2, 122.2, 51.0, 28.0, 27.1. HRESI-MS m/z (M + H+). Anal. for C16H22N9O: calcd mass 356.1947. Found 356.1942. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 20.1 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2-methoxypyrimidin-5-yl)pyrazine-2-carboxamide (26).
General method D using 26a (358 mg, 1.0 mmol) gave 26 as a yellow solid; yield 90% (345 mg). Mp 148−150 °C. 1H NMR (CD3OD): δ 8.77 (s, 2H), 4.08 (s, 3H), 3.46 (t, J = 5.5 Hz, 4H), 1.72 (s, 4H), 1.54 (s, 4H). 13C NMR (CD3OD): δ 166.7, 164.1, 163.4, 158.1, 154.8, 153.6, 128.6, 124.2, 112.8, 54.3, 27.7, 26.7. HRESI-MS m/z (M + H+) Anal. for C17H24N9O2: calcd mass 386.2053. Found 386.2048. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 20.7 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2,4-dimethoxypyrimidin-5-yl)pyrazine-2-carboxamide (27).
General method C using 27a (104 mg, 0.267 mmol) gave 27 as a yellow solid; yield 54% (65 mg, 0.156 mmol). Mp 184−186 °C. 1H NMR (CD3OD) δ 10.76 (s, 1H), 9.27 (s, 2H), 8.51 (s, 2H), 8.48 (s, 2H), 4.08 (s, 3H), 3.35 (t, J = 5.75 Hz, 4H), 1.66 (s, 4H), 1.46 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 166.6, 164.0, 157.8, 156.1, 153.9, 127.8, 124.7, 112.5, 55.6, 51.6, 27.8, 27.1. HRESI-MS m/z (M + H+) Anal. for C18H26N9O3: calcd mass 416.2159. Found 416.2158. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 21.4 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2,6-dimethoxypyridin-3-yl)pyrazine-2-carboxamide (28).
General method C using 28a (98 mg, 0.253 mmol) gave 28 as a yellow solid; yield 13% (15 mg, 0.036 mmol). Mp 152−154 °C. 1H NMR (500 MHz, CDCl3) δ 10.28 (s, 1H), 8.77 (s, 2H), 8.14 (s, 2H), 7.61 (d, J = 8.0 Hz), 6.39 (d, J = 8.0 Hz, 1H), 3.94 (s, 3H), 3.87 (s, 3H), 3.52 (s, 2H), 3.35 (s, 2H), 1.60 (s, 4H), 1.51 (s, 2H), 1.44 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 166.0, 163.2, 159.2, 155.4, 153.5, 141.1, 128.0, 114.6, 110.2, 101.3, 53.8, 53.5, 50.4, 27.7, 26.81. HRESI-MS m/z (M + H+) Anal. for C19H27N8O3: calcd mass 415.2206. Found 415.2191. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 26.1 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2,4-dimethoxyphenyl)pyrazine-2-carboxamide (29).
General method C using 29a (123 mg, 0.317 mmol) gave 29 as a yellow solid; yield 8% (12 mg, 0.029 mmol). Mp 156−158 °C. 1H NMR (500 MHz, CDCl3) δ 10.19 (s, 1H), 9.10 (bs, 2H), 7.79 (bs, 2H), 7.23 (d, J = 8.1 Hz), 6.58 (d, J = 8.1 Hz), 6.48 (s, 1H) 3.86 (s, 3H), 3.74 (s, 3H), 3.51 (s, 2H), 3.36 (s, 2H), 1.59 (s, 4H), 1.50 (s, 2H), 1.45 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 166.0, 161.5, 157.8, 155.7, 155.4, 153.8, 130.6, 129.6, 122.1, 109.8, 105.0, 98.7, 55.7, 55.6, 50.6, 27.8, 26.9. HRESI-MS m/z (M + H+) Anal. for C20H28N7O3: calcd mass 414.2254. Found 414.2269. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 26.7 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(3,5-dimethoxyphenyl)pyrazine-2-carboxamide (30).
General method C using 30a (147 mg, 0.381 mmol) gave 30 as a yellow solid; yield 26% (44 mg, 0.116 mmol). Mp 138−140 °C. 1H NMR (500 MHz, CDCl3) δ 10.75 (s, 1H), 8.81 (bs, 2H), 8.60 (bs, 2H), 6.73 (d, J = 2.2 Hz), 6.58 (t, J = 2.2 Hz, 1H), 3.84 (s, 6H), 3.38 (s, 2H), 1.64 (s, 4H), 1.45 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 167.0, 160.1, 156.4, 155.6, 153.8, 141.6, 132.9, 110.9, 106.5, 100.3, 55.9, 51.0, 28.0, 27.1. HRESI-MS m/z (M + H+) Anal. for C20H28N7O3: calcd mass 414.2254. Found 414.2244. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 25.8 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(3,6-dimethoxypyridazin-4-yl)pyrazine-2-carboxamide (31).
General method C using 31a (94 mg, 0.243 mmol) gave 31 as a yellow solid; yield 17% (21 mg, 0.051 mmol). Mp 202−204 °C. 1H NMR (500 MHz, CD3OD) δ 10.38 (s, exchange with solvent), 8.77 (s, exchange with solvent), 8.21 (s, exchange with solvent), 7.35 (s, 1H), 4.06 (s, 3H), 3.99 (s, 3H), 3.35 (s, 4H), 1.66 (s, 4H), 1.55 (s, 2H), 1.45 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 167.3, 164.1, 160.9, 156.9, 156.8, 156.1, 136.3, 124.0, 121.4, 112.2, 55.4, 55.3, 51.0, 28.9, 27.9. HRESI-MS m/z (M + H+) Anal. for C18H26N9O3: calcd mass 416.2159. Found 416.2150. Anal. HPLC (70:30 H2O/0.1% TFA/CH3CN/0.1% TFA → 00% CH3CN/0.1% TFA over 30 min, tR = 19.0 min).
3-Amino-6-(2-aminopyrimidin-5-yl)-5-(azepan-1-yl)-N-carbamimidoylpyrazine-2-carboxamide (32).
General method D using 32a (86 mg, 0.25 mmol) gave 32 as a yellow solid; yield 78% (72 mg). Mp 225−228; 1H NMR (DMSO-d6): δ 1.41 (s, 4H), 1.60 (s, 4H), 3.38 (t, J = 5.9 Hz, 4H), 6.73 (br s, 2H), 7.25 (br s, 1H), 8.40 (s, 2H). 13C NMR (DMSO-d6): δ 167.5, 162.9, 159.1, 157.5, 154.9, 153.7, 126.0, 123.6, 112.9, 51.1, 28.0, 27.0. HRESI-MS m/z (M + H+), Anal. for C16H23N10O3: calcd mass 371.2056. Found 371.2067. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 10.2 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2-(methylamino)pyrimidin-5-yl)pyrazine-2-carboxamide (33).
General method D using 33a (39 mg, 0.11 mmol) gave 33 as a yellow solid; yield 85% (36 mg). Mp 168−170; 1H NMR (CD3OD): δ 8.47 (s, 2H), 3.48 (t, J = 5.6 Hz, 4H), 2.97 (s, 3H), 1.69 (s, 4H), 1.51 (s, 4H). 13C NMR (CD3OD): δ 28.0, 28.5, 29.0, 52.0, 113.5, 124.4, 127.3, 155.4, 156.7, 158.3, 158.5, 162.9, 168.3, 162.9, 158.5, 158.3, 156.7, 155.4, 127.3, 124.4, 113.5, 52.0, 29.0, 28.5, 28.0. HRESI-MS m/z (M + H+) Anal. for C17H25N10O: calcd mass 385.2213. Found 385.2195. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 14.8 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2-(isopropylamino)pyrimidin-5-yl)pyrazine-2-carboxamide (34).
General method D using 34a (35 mg, 0.09 mmol) gave 34 as a yellow solid; yield 89% (32 mg). Mp 172−175 °C. 1H NMR (CD3OD): δ 8.46 (s, 2H), 4.14 (septet, J = 6.8 Hz, 1H), 3.47 (t, J = 5.6 Hz, 4H), 1.69 (s, 4H), 1.51 (s, 4H), 1.25 (d, J = 6.5 Hz, 6H). 13C NMR (CD3OD): δ 21.6, 26.8, 27.8, 42.8, 50.9, 112.3, 123.2, 126.2, 154.2, 155.5, 157.1, 158.8, 160.6, 167.1, 160.6, 158.8, 157.1, 155.5, 154.2, 126.2, 123.3, 112.3, 50.9, 42.8, 27.8, 26.8, 21.6. HRESI-MS m/z (M + H+) Anal. for C19H29N10O: calcd mass 413.2526. Found 413.2525. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 16.1 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(2-((2-hydroxyethyl)amino)pyrimidin-5-yl)pyrazine-2-carboxamide (35).
General method D using 35a (50 mg, 0.13 mmol) gave 35 as a Yellow solid: yield 86% (46 mg). Mp 188−191 °C. 1H NMR (CD3OD): δ 8.48 (s, 2H), 3.72 (t, J = 5.6 Hz, 2H),), 3.54 (t, J = 5.6 Hz, 2H), 3.45 (t, J = 5.8 Hz, 4H), 1.68 (s, 4H), 1.50 (s, 4H). 13C NMR (CD3OD): δ 167.1, 162.3, 160.8, 158.2, 156.3, 154.9, 126.5, 124.7, 117.5, 61.7, 51.9, 44.6, 28.9, 27.8.. HRESI-MS m/z (M + H+) Anal. for C18H27N10O2: calcd mass 415.2318. Found 415.2324. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 14.4 min).
Methyl 3-((Di-tert-butyloxycarbonyl)amino)-5-(azepan-1-yl)-6-chloro-2-pyrazinecarboxylate (36).
To a stirred solution of 7 (2.85 g, 10 mmol) and DMAP (61 mg, 0.5 mmol) in 1,2dichloroethane (40 mL) was added a solution of di-tert-butyl dicarbonate (6.57 g, 30 mmol) in 1,2-dichloroethane (20 mL) at rt. The reaction mixture was heated at 70 °C for 18 h. After cooling to room temperature, additional equivalents of di-tert-butyl dicarbonate (3.28 g, 15 mmol) and DIPEA (10 mL) were added and the reaction mixture was heated for a further 18 h. Solvents were evaporated and the residue was partitioned between EtOAc (100 mL) and water (100 mL). The aqueous layer was extracted with EtOAc (3 × 75 mL), and the combined organic layer was washed with brine, dried (MgSO4), and concentrated. The residue was purified by silica gel flash column chromatography using EtOAc/pet. spirit (10−15%) to give 36 (4.10 g, 85%) as a white solid. Mp 116−118 °C. 1H NMR (CDCl3): δ 3.89 (s, 3H), 3.88 (t, J = 6.1 Hz, 4H), 1.55 (s, 4H), 1.84 (s, 4H), 1.41 (s, 18H). 13C NMR (CDCl3): δ 163.0, 152.3, 150.4, 145.4, 128.6, 126.1, 83.3, 52.8, 51.6, 28.1, 28.1, 26.7. MS (ESI): m/z 507 (M + Na)+. HRESI-MS m/z (M + H+) Anal. for C22H33ClN4O6Na: calcd mass 507.1986. Found 507.2000. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min).
Methyl 5-(Azepan-1-yl)-3-((tert-butoxycarbonyl)amino)-pyrazine-2-carboxylate (37).
To a mixture of chloropyrazine 36 (3.30 g, 6.8 mmol), Pd/C (476 mg, 10 mol %), and MgO (408 mg, 10.2 mmol) was added dry MeOH (50 mL). The flask was charged with H2 (1 atm), and the reaction mixture was stirred at rt for 2 days. The solvent was evaporated and the residue redissolved in EtOAc and filtered through Celite, eluting with EtOAc. The organic layer was washed with brine, dried (MgSO4), and evaporated to give the crude mono Boc-protected product 37 as a white solid (2.22 g, 92%). Crude product showed sufficient purity and was used in the next step with further purification. Mp 97−100 °C. 1H NMR (400 MHz, CDCl3) δ 10.28 (s, 1H), 7.68 (s, 1H), 3.94 (s, 3H), 3.62 (br s, 4H), 1.58 (br s, 4H), 1.53 (s, 9H), 1.84 (br s, 4H). 13C NMR (CDCl3): δ 167.3, 153.8, 150.6, 150.3, 122.4, 113.5, 80.7, 52.3, 47.9, 28.3, 27.4, 26.6. HRESI-MS m/z (M + H+) Anal. for C17H27N4O4: 351.2032. Found 351.2032. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min).
Methyl 5-(Azepan-1-yl)-6-bromo-3-((tert-butoxycarbonyl)-amino)pyrazine-2-carboxylate (38).
To a solution of pyrazine methyl ester 37 (2.10 g, 6.0 mmol) in CH2Cl2 (90 mL) was added N-bromosuccinimide (1.18 g, 6.6 mmol), and the reaction mixture was stirred at rt for 5 h. The mixture was treated with water (50 mL) and stirred for 10 min. The organic layer was separated, washed with sodium bicarbonate solution (2 × 50 mL) and brine (50 mL), dried (MgSO4), and concentrated. The residue was purified by silica gel flash column chromatography using EtOAc/pet. spirit (10−15%) to give 38 (1.56 g, 60%) as a white solid. Mp 114−115 °C. 1H NMR (CDCl3): δ 10.04 (s, 1H), 3.95 (t, J = 6.0 Hz, 4H), 3.92 (s, 3H), 1.89 (br s, 4H), 1.60 (t, J = 3.1 Hz, 4H), 1.52 (s, 9H). 13C NMR (CDCl3): δ 166.3, 153.0, 150.0, 147.7, 114.8, 111.9, 81.0, 52.5, 51.4, 28.2, 28.2, 26.8. MS (ESI): m/z 429 (M + H)+. HRESI-MS m/z (M + Na+) Anal. for C17H25BrN4O4Na: calcd mass 451.0957. Found 451.0959. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min).
Methyl 5-(Azepan-1-yl)-3-((tert-butoxycarbonyl)amino)-6-(pyrimidin-2-ylamino)pyrazine-2-carboxylate (39).
A resealable Schlenk tube was charged with bromopyrazine 38 (429 mg, 1.0 mmol), 2-aminopyrimidine (135 mg, 1.4 mmol), Pd2(dba)3 (10 mg, 0.011 mmol, 1 mol % Pd), Xantphos (13 mg, 0.022 mmol), Cs2CO3 (426 mg, 1.4 mmol), and degassed dioxane (5 mL). The Schlenk tube was capped and carefully subjected to three cycles of evacuation−backfilling with N2. It was then sealed and the reaction mixture heated at 100 °C for 20 h. The mixture was cooled to rt, diluted with THF (20 mL), filtered through Celite, and concentrated. The residue was purified by silica gel flash column chromatography using EtOAc/pet. spirit (50−60%) to give 39 (130 mg, 30%) as a white solid. Mp 168−170 °C. 1H NMR (CDCl3): δ 10.16 (s, 1H), 8.35 (d, J = 4.8 Hz, 2H), 7.23 (s, 1H), 6.71 (t, J = 4.8 Hz, 1H), 3.90 (s, 3H), 3.77 (t, J = 6.0 Hz, 4H), 1.73 (br s, 4H), 1.53 (s, 9H), 1.48 (br s, 4H). 13C NMR (CDCl3): δ 166.8, 161.3, 158.3, 152.3, 150.2, 147.9, 126.6, 113.2, 111.5, 80.5, 52.4, 50.5, 28.3, 28.0, 26.8. MS (ESI): m/z 444 (M + H)+. HRESI-MS m/z (M + H+) Anal. for C21H30N7O4: calcd mass 444.2359. Found 444.2374. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min).
Methyl 3-Amino-5-(azepan-1-yl)-6-(pyrimidin-2-ylamino)-pyrazine-2-carboxylate (40).
To a solution of Boc-protected pyrazine 39 (60 mg, 0.135 mmol) in CH2Cl2 (4 mL) at 0 °C was added TFA (0.1 mL), and the mixture was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue partitioned between CH2Cl2 (20 mL) and saturated NaHCO3 solution (20 mL). The organic layer was dried (MgSO4) and concentrated and the residue purified by silica gel flash column chromatography using EtOAc/pet. spirit (60−70%) to give 40 (45 mg, 98%) as a gray solid. Mp 194−196 °C. 1H NMR (CDCl3): δ 8.34 (d, J = 4.8 Hz, 2H), 7.04 (s, 1H), 6.67 (t, J = 4.8 Hz, 1H), 6.20 (br s, 2H, NH2), 3.87 (s, 3H), 3.67 (t, J = 6.0 Hz, 4H), 1.69 (br s, 4H), 1.48 (t, J = 3.2 Hz, 4H). 13C NMR (CDCl3): δ 166.8, 161.8, 158.4, 154.2, 153.5, 124.3, 112.8, 109.4, 52.0, 50.6, 50.3, 28.2, 26.7. HRESI-MS m/z (M + H+) Anal. for C16H22N7O2: calcd mass 344.1835. Found 344.1848. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min).
3-Amino-5-(azepan-1-yl)-N-carbamimidoyl-6-(pyrimidin-2-ylamino)pyrazine-2-carboxamide (41).
General method D using 40 (35 mg, 0.1 mmol) gave 41 (30 mg, 81%) as a yellow solid. Mp 210−212 °C. 1H NMR (CD3OD): δ 8.25 (d, J = 4.9 Hz, 2H), 6.70 (t, J = 4.9 Hz, 1H), 3.70 (t, J = 6.0 Hz, 4H), 1.63 (br s, 4H), 1.43 (t, J = 3.1 Hz, 4H). 13C NMR (CD3OD): δ 165.7, 162.2, 158.3, 155.6, 154.9, 154.8, 124.1, 112.3, 108.0, 50.4, 27.8, 26.2. MS (ESI): m/z 371 (M + H)+. HRESI-MS m/z (M + H+) Anal. for C16H23N10O: calcd mass 371.2056. Found 371.2057. Anal. HPLC (100% H2O/0.1% TFA → 100% CH3CN/0.1% TFA over 30 min, tR = 14.3 min).
Cell Culture Conditions.
ATCC certified cell lines were used for all experiments. Cells were cultured at 37 °C, 95% humidity, and 5% (v/v) CO2 in a ThermoScientific Heracell 150i CO2 incubator (ThermoScientific) and maintained in DMEM/Hi glucose culture medium supplemented with 10% heat-inactivated FCS. Adherent cells were passaged via the addition of trypsin/0.05% EDTA solution (GIBCO), followed by centrifugation and resuspension in fresh media using a 5 mL Luerlock syringe fitted with an 18-gauge needle (Terumo, Tokyo, Japan). All cell lines were routinely tested for the presence of mycoplasma infection.
Fluorometric uPA Activity Assay.
Urokinase from human kidney cells (catalog no. U4010, Sigma-Aldrich, St. Louis, MO, USA) was diluted to 900 nM, aliquoted, and stored at –80 °C until use in experiments (typically 4−5 plates assayed on the same day). Thawed enzyme stocks were maintained at –20 °C between during assays using a Nalgene Labtop Cooler Jr (catalog no. 5115–0012, Thermo-Fisher Scientific, Waltham, MA, USA). A 5 mM stock solution of urokinase fluorescent substrate III (Z-Gly-Gly-Arg-AMC, Calbiochem catalog no. 672159, Merck Millipore, MA, USA, human uPA Km = 400 μM) was prepared in dH2O/10% DMSO and aliquoted with storage at –20 °C. Assay buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 0.5 mM EDTA, 0.01% v/v Tween-20) was prepared and used within 7 days with storage at 4 °C. On the day of assay, compound stocks (20−50 mM in anhydrous DMSO) were serially diluted in assay buffer in a clear Greiner CELLSTAR 96-well plate on ice (Greiner Bio-One GmBH, Kremsmünster, Austria) to give each dilution at 2× the final assayed concentration. In a separate black Greiner CELLSTAR 96-well plate (Greiner Bio-One GmBH, Kremsmünster, Austria), residual volumes of assay buffer were added via multichannel pipet to compound fluorescence blanks and inhibited enzyme control wells, as appropriate. Next, 90 μL of dilute fluor (250 μM final) was added to all wells, followed by 100 μL of 2× compound dilution to appropriate wells. Immediately prior to assay, 900 nM uPA stock was diluted to 15 nM and 10 μL of this dilution was added to all relevant wells, initiating the reaction. Reaction progress was monitored using a POLARStar OMEGA fluorescence plate reader (fluorescence intensity mode, top-optic, ex 355 nm, em 450 nm, 20 flashes per well, double orbital shaking for 3 s prior to every read, cycle time 60 s, 45−60 cycles, incubation at 37 °C, BMG-Labtech, Ortenberg, Germany). Changes in fluorescence over a 15 min period from the linear portion of the reaction progress curve were used to determine IC50 values by fitting log-transformed data to a sigmoidal dose−response curve in GraphPad Prism v7.0 (GraphPad Software, La Jolla, CA, USA). Experiments with Mouse uPA (active mouse urokinase, HMW, Molecular Innovations Inc., MI, USA) were performed as above with a final enzyme concentration of 20 nM (Km = 580 μM). Ki values were calculated according to the methods of Cheng and Prussof.52
Chromogenic Trypsin-like Serine Protease (TLSP) Activity Assays.
For experiments using uPA (catalog no. U4010, Sigma-Aldrich, St. Louis, MO, USA), trypsin, tPA, thrombin, plasmin, and factor Xa, concentrated stocks were prepared in deionized H2O and stored at −80 °C until thawed for use. Thawed enzyme stocks were maintained at –20 °C between assays using a Nalgene Labtop Cooler Jr (catalog no. 5115–0012, Thermo-Fisher Scientific, Waltham, MA, USA). A 312.5 μM stock solution of S-2288 chromogenic substrate (HD-Ile-L-Pro-L-Arg-p-nitoraniline, Chromogenix, MA, USA, uPA Km = 200 μM, trypsin Km = 28 μM, tPA Km = 300 μM, thrombin Km = 3 μM, plasmin Km = 9 mM) was prepared in deionized H2O and aliquoted with storage at −20 °C. Assay buffer (10 mM HEPES, 150 mM NaCl, 0.01% v/v Tween-20, pH 7.4) was prepared and used within 7 days with storage at 4 °C. On the day of assay, compound stocks (20−50 mM in DMSO) were serially diluted in assay buffer in a clear Greiner CELLSTAR 96-well plate on ice (Greiner Bio-One GmBH, Kremsmünster, Austria) to give each dilution at 10× the final assay concentration. In a separate Greiner CELLSTAR 96-well plate (Greiner Bio-One GmBH, Kremsmünster, Austria) residual volumes of assay buffer were added via multichannel pipet to compound fluorescence blanks and inhibited enzyme control wells, as appropriate. Next, 80 μL of dilute substrate (250 μM final) was added to all wells, followed by 10 μL of 10× compound dilution to appropriate wells. Immediately prior to assay, concentrated enzyme stocks were diluted to 100 nM and 10 μL of this dilution was added to all relevant wells, initiating the reaction (final enzyme concentration = 10 nM). DMSO was present at a final concentration of 1% v/v. Change in absorbance over time at 405 nm was measured at 37 °C using a Molecular Devices SpectraMax Plus 384-well plate reader (Molecular Devices LLC, Sunnyvale, CA, USA). IC50 values were determined by plotting percentage of residual activity (V0) versus log drug concentration and fitted to sigmoidal dose response curves using GraphPad Prism v7.0 (GraphPad Software, La Jolla, CA, USA). The following chromogenic substrates and final enzyme concentrations were used for experiments with other TLSPs. S-2366, L-pyroGlu-L-Pro-L-Arg-p-nitroaniline, Chromogenix, MA, USA, [human plasma kallikrein] = 23.5 nM, Km = 430 μM, [factor XIa] = 2 nM, Km = 400 μM, [activated protein C] = 10 nM, Km = 200 μM.
Crystallization and X-ray Data Collection.
Expression and purification of recombinant human uPA protease domain were performed as previously described.78 Crystals were grown by the sitting drop vapor diffusion method and were obtained by equilibrating against a reservoir solution containing 50 mM sodium citrate (pH 4.6) and 2.0 M ammonium sulfate supplemented with 5% PEG400 at room temperature for a period of 3 days. For uPA:inhibitor cocomplexes at pH 7.4, the crystals were soaked for 1 week in new soaking buffer (40% PEG4000, 100 mM Tris-HCl, pH 7.4) containing 100 μM compound. Prior to X-ray data collection, crystals were soaked in a cryoprotectant solution containing 20% glycerol and snap-frozen in liquid N2. X-ray diffraction data of the crystals were collected at the BL17U beamline, Shanghai Synchrotron Radiation Facility (SSRF). The diffraction data were indexed and integrated using the HKL2000 program package.79 Structures were solved by molecular replacement80 using the uPA structure (PDB code 4DVA)81 as the search model. The electron density for the compound was clearly visible in the uPA active sites and was modeled based on the Fo − Fc difference map. The structures were refined using the CCP4 program package79 and manually adjusted using the molecular graphics program COOT82 iteratively until convergence. Solvent molecules were added using a Fo − Fc Fourier difference map at 2.5σ in the final refinement step. Statistics for data collection and final model refinement are summarized in the Supporting Information Table S1.
EnPlex Assays.
Enplex superfamily wide serine hydrolase activity screening was performed as described in ref 54.
Cytotoxicity Assays.
Subconfluent cells (~70−80% confluence) were harvested and dissociated into a single cell suspension. Cells were seeded at a density of 5000 cells/well (final volume 90 μL) into a Greiner CELLSTAR 96-well plate (Greiner Bio-One GmBH, Kremsmünster, Austria) via multichannel pipet and incubated for 18h prior to the addition of compounds. Compounds were serially diluted from 20 to 50 mM DMSO stocks in culture media in a separate 96-well plate under sterile conditions to give 10× stocks (10 nM to 1 mM) on the day of compound addition. 10 μL of the 10× compound/media stocks or matched vehicle/media solutions (for vehicle controls; final vehicle concentration was equal to 0.2%) were transferred via multichannel pipet into the assay plate containing 90 μL of cell suspension via multichannel pipet to give the treated assay plate at the final 1× drug concentrations (1 nM to 100 μM, n = 4 wells at each concentration). Drug blanks or vehicle media blanks were included as appropriate at each concentration to correct for intrinsic color of the compounds and the phenol red containing media. 200 μL media was added to outside wells to decrease evaporation but was not used in the assay itself. Compound-treated plates were incubated for 48 h prior to development. After 48 h, plates were removed from the incubator and 20 μL of CellTiter 96 Aqueous One solution cell proliferation assay (catalog no. G3581, Promega Corporation, Fitchburg, WI, USA) was added to each well. Plates were incubated for a further 2 h prior to reading at 490 nm using a SpectraMax Plus 384-well plate reader (Molecular Devices LLC, Sunnyvale, CA, USA). IC50 values (defined as the concentration of each treatment causing 50% inhibition of maximal cell viability) were determined using a logarithmic sigmoidal dose−response curve using the variable slope parameter to determine IC50 using GraphPad Prism v7.0 (GraphPad Software, La Jolla, CA, USA).
ENaC Inhibition Assays.
HEK-293 cells were cultured in Dulbecco’s modified Eagle medium/nutrient mixture F-12 (D-MEM/F-12) supplemented with 10% fetal bovine serum, 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, and appropriate selection antibiotics. Semiconfluent cells were transfected with cDNA for ENaC α, β, and γ subunits (genes SCNN1A, SCNN1B, and SCNN1G) prior to experimentation. Cells were plated at a density of 20 000 cells/well in a 384-well black wall, flat clear bottom microtiter plates (type: BD Biocoat poly-D-lysine multiwell cell culture plate) and incubated at 37 °C overnight. Following manufacturer instructions, cells were loaded with 20 μL of membrane potential dye solution for 30 min at 37 °C from the FLIPR membrane potential red dye fluorescence assay kit (Molecular Devices, Sunnyvale, CA, USA). This assay uses fluorometric detection of changes in membrane potential following modulation of transmembrane ion channel/exchanger activity, i.e., antagonism of stimulated ENaC activity in this case. Drug, vehicle, and control solutions were prepared from frozen stocks in DMSO on the day of experimentation through dilution into HEPES-buffered physiological salt solution (NaCl 137 mM, KCl 4 mM, CaCl2 1.8 mM, MgCl2 1 mM, HEPES 10 mM, glucose 10 mM, DMSO 0.3% v/v, pH 7.4) in a 384-well microtiter plate ready for addition to the assay plate by the FLIPRTETRA instrument. All analogs were tested at 8 concentrations (n = 4 wells at each concentration). Positive control cells were stimulated with 1 μM S-3969 (ENaC agonist, n = 4 wells).83 Negative control cells were first stimulated with 1 μM S-3969 and then treated with 30 μM benzamil (potent ENaC antagonist, n = 4 wells). Following dye loading, cells were preincubated with 5 μL of drug, vehicle, or control solutions for 5 min. Next 5 μL of S-3939 solution (final [S-3969] = 1 μM) was added to stimulate ENaC activity. All data were normalized to agonist-treated positive and antagonist-treated negative controls such that the agonist-treated signal corresponded to 100% activity and antagonist-treated signal corresponded to 0% activity. Normalized data were used to determine IC50 values for each analog using the log[inhibitor]-normalized response variable slope algorithm in GraphPad Prism v7.0 (GraphPad Software, La Jolla, CA, USA). These experiments were performed under contract by Charles River Laboratories, Cleveland, OH, USA.
Rat Diuresis and Kaliuresis Model.
Sixty-four male 21 week old Sprague-Dawley rats were randomized into 8 groups of 8 animals. All test animals received one intraperitoneal (ip) injection of either vehicle or 25 mg/kg acetazolamide followed by one intravenous (iv) injection of vehicle, 1.5 mg/kg amiloride or 1.5 mg/kg amiloride derivative. Acetazolamide was administered at an injection volume of 1 mL/kg and concentration of 25 mg/mL. Amiloride, HMA, 18, and 26 were administered at an injection volume of 1 mL/kg and concentration of 1.5 mg/mL. Monosodium acetazolamide was formulated in 0.9% saline. Amiloride and HMA were formulated in 50 mM acetate buffer (pH 5.5)/5% v/v DMSO, and 18 and 26 were formulated in 50 mM acetate buffer (pH 5.5)/5% v/v DMSO/0.5% v/v Tween-80. Immediately following dosing, all animals were placed into metabolic cages and total urine was collected over 6 h. A control group receiving ip and iv administrations of vehicle was also included for comparison. Urine Na+ and K+ levels were quantitated using a Konelab 20 XTi biochemistry analyzer (LLQ = 20 mM). This experiment was approved by the Austin Health Animal Ethics Committee (A2016/05420) and was conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, seventh edition, 2004. Statistical analyses were performed using a one-way ANOVA with Dunnett’s post hoc test in GraphPad Prism v7.0 (GraphPad Software, La Jolla, CA, USA).
Mouse Lung Metastasis Model.
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Wollongong (AE1708). Male and female NOD.Cg-Prkdc<scid>-IL2rg<tm1Wjl>SzJAusb mice (Australian BioResources, Moss Vale, NSW, Australia) at 7−8 weeks of age were randomized into treatment groups (n = 4−6/treatment group). Treatments commenced 1 day prior to tail vein administration of 2.5 × 105 luciferase-tagged HT-1080 human fibrosarcoma cells (BioWare Brite HT-1080-Red-Fluc) in an injection volume of 100 μL of PBS [except sham control (n = 1)]. Daily ip injections continued to day 20 with 7.5 mg/kg amiloride, 18, 26, or vehicle (50 mM acetate buffer, pH 5.5, 7.5% v/v DMSO, 0.75% Tween80). Animals were weighed and assessed for clinical signs daily. Humane end points of >15% acute weight loss (from previous 7 day maximum individual weight) or >20% chronic weight loss (from all-time individual maximum weight) or a body condition score >3 was employed. On day 21, all remaining animals were euthanized via slow CO2 asphyxiation, weighed, necropsied, and had their lungs removed. Lungs were snap frozen in liquid N2 and stored at –80 °C until homogenization. Lungs were homogenized on ice using a PRO Scientific Bio-Gen PRO200 homogenizer in cell lysis buffer (150 mM NaCl, 10 mM Tris base, 1% Triton X100 v/v, pH 8.0) and centrifuged twice at 4 °C and 1000 rpm for 15 min to remove cell debris. Homogenates were prepared to approximately 50 mg/mL protein based on individual wet lung weights. Luciferase activity was quantified according to the manufacturer’s instructions using the Molecular Diagnostics SpectraMax Glo Steady-Luc report assay kit. Luciferase assays were conducted in clear-bottomed white walled 96-well plates (Nunc) using a BMG Labtech PolarSTAR multimode plate reader. Data were analyzed using GraphPad Prism v7.0, and statistical significance was determined using the one-way ANOVA with Dunnett’s post hoc test.
Orthotopic Model of Pancreatic Cancer.
The orthotopic model has been described in detail previously.67,84 In brief, 6- to 8-week-old female athymic nude mice (BALB/c nu/nu) were anesthetized and an incision was made in the left flank, followed by exteriorization of the spleen and tail of the pancreas. A mixture of human PC cells (AsPC-1) 1 × 106 + human PSCs (hPSCs) 1 × 106 in 50 μL of PBS was injected into the tail of the pancreas. AsPC-1 cells were chosen for their high implantation rate (100% in our hands) and high reproducibility of tumor growth rates and metastasis. After intrapancreatic injection, organs were reinternalized and the abdomen was surgically closed. Mice receiving treatment were randomized into subgroups (n = 4 mice per group) on postsurgical day 7. These mice were treated as follows for 4 weeks: gemcitabine (75 mg/kg BIW oral gavage); 18 or 26 (10 mg/kg/day ip or vehicle (daily ip, BIW oral gavage). Compounds were formulated for ip injection in 50 mM sodium acetate buffer (pH 5.5)/ 10% (v/v) DMSO/1% (w/v/) Kolliphor HS-15 and filtered through 0.22 μm PVDF syringe-driven filters under sterile conditions. Pancreatic tumor growth was monitored by palpation. Mice were killed at 4 weeks after first ip injection. Tumors were resected and assessed for size and volume by two separate observers (tumor volume was calculated to two decimal points, according to an established formula (1/2(length × breadth × width) using digital Vernier calipers (Intech Tools, Thomas Town, VIC, Australia).84 The abdominal cavity, mesentery, spleen, liver, and lungs were examined and scored according to the presence or absence of visible macrometastatic nodules. These organs were then excised and fixed in 10% neutral buffered formalin overnight and processed using the Leica Peloris dual retort tissue processor (Germany). Histological staining was performed on 4 μm sections deparaffinised in xylene and rehydrated using graded ethanol washes. Haematoxylin and eosin staining were performed on a Leica autostainer XL.
This experiment was approved by the University of New South Wales Animal Care and Ethics Committee (Approval Number 15/106A) and was conducted in accordance with ARRIVE Guidelines.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Australian National Health and Medical Research Council (NHMRC, Project Grant APP1100432). The authors gratefully acknowledge Dr. Arnault Massink, Mrs Corine van Veen and Prof Ad P. Ijzerman, Leiden Academic Centre for Drug Research, Leiden University, for A2A activity screening. D.C.J. acknowledges funding from an NIH T32 GM115327-Tan training grant. D.A.B and D.C.J. acknowledge funding from a MSKCC Core Grant P30 (Grant CA008748).
ABBREVIATIONS USED
- ABPP
activity-based protein profiling
- ASIC1a
acid-sensing ion channel 1a
- C1r
complement C1r subcomponent
- C1s
complement component 1s
- DDHD1
phosphatidic acid-preferring phospholipase A1
- DIPEA
diisopropylethylamine
- DOCA
deoxycorticosterone acetate
- DPP7
dipeptidyl peptidase 2
- ENaC
epithelial sodium channel
- FLT3-ITD+
Fms-like tyrosine kinase internal tandem duplication positive
- GABA-A
γ-aminobutyric acidA receptor
- Gem
gemcitabine
- GIRK
G-protein-coupled inwardly rectifying potassium channel
- GnRHR
gonadotropin-releasing hormone receptor
- GPCR
G-protein-coupled receptor
- hA2AAR-WT
human adenosine A2A receptor
- HER2
human epidermal growth factor receptor 2
- HIV-1
human immunodeficiency virus 1
- HMA
5-(N,N-hexamethylene)amiloride
- Hpsc
human pancreatic stellate cells
- KLKb1
plasma kallikrein
- LACTB
serine β-lactamase-like protein
- NECA
5′-(N-ethylcarboxamido)adenosine
- NHE1
sodium hydrogen exchanger isoform 1
- NSB
non-specific binding
- P2X7
P2X purinoreceptor
- PRCP
prolylcar-boxypeptidase
- SAR
structure−activity relationship
- SOSA
selective optimization of side activity
- TB
total binding
- TLSP
trypsin-like serine protease
- TMPRSS11d
trypsin and transmembrane protease serine 11D
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00838.
X-ray crystallography refinement data and statistics, HT-1080 uPA and uPAR expression data, experimental late-stage metastasis model clinical score sheet (PDF) Molecular formula strings of all compounds (CSV)
REFERENCES
- (1).Ulisse S; Baldini E; Sorrenti S; D’Armiento M The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9 (1), 32–71. [DOI] [PubMed] [Google Scholar]
- (2).Brungs D; Chen J; Aghmesheh M; Vine KL; Becker TM; Carolan MG; Ranson M The urokinase plasminogen activation system in gastroesophageal cancer: A systematic review and meta-analysis. Oncotarget 2017, 8 (14), 23099–23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Croucher DR; Saunders DN; Lobov S; Ranson M Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat. Rev. Cancer 2008, 8 (7), 535–545. [DOI] [PubMed] [Google Scholar]
- (4).Harris NLE; Vennin C; Conway JRW; Vine KL; Pinese M; Cowley MJ; Shearer RF; Lucas MC; Herrmann D; Allam AH; Pajic M; Morton JP; Australian Pancreatic Cancer Genome Initiative; Biankin AV; Ranson M; Timpson P; Saunders DN SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene 2017, 36 (30), 4288–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yang JL; Seetoo D; Wang Y; Ranson M; Berney CR; Ham JM; Russell PJ; Crowe PJ Urokinase-type plasminogen activator and its receptor in colorectal cancer: independent prognostic factors of metastasis and cancer-specific survival and potential therapeutic targets. Int. J. Cancer 2000, 89 (5), 431–439. [DOI] [PubMed] [Google Scholar]
- (6).Harris L; Fritsche H; Mennel R; Norton L; Ravdin P; Taube S; Somerfield MR; Hayes DF; Bast RC Jr. American society of clinical oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol 2007, 25 (33), 5287–5312. [DOI] [PubMed] [Google Scholar]
- (7).Look MP; van Putten WL; Duffy MJ; Harbeck N; Christensen IJ; Thomssen C; Kates R; Spyratos F; Ferno M; Eppenberger-Castori S; Sweep CG; Ulm K; Peyrat JP; Martin PM; Magdelenat H; Brunner N; Duggan C; Lisboa BW; Bendahl PO; Quillien V; Daver A; Ricolleau G; Meijer-van Gelder ME; Manders P; Fiets WE; Blankenstein MA; Broet P; Romain S; Daxenbichler G; Windbichler G; Cufer T; Borstnar S; Kueng W; Beex LV; Klijn JG; O’Higgins N; Eppenberger U; Janicke F; Schmitt M; Foekens JA Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J. Natl. Cancer. Inst 2002, 94 (2), 116–128. [DOI] [PubMed] [Google Scholar]
- (8).Foekens JA; Peters HA; Look MP; Portengen H; Schmitt M; Kramer MD; Brunner N; Janicke F; Meijer-van Gelder ME; Henzen-Logmans SC; van Putten WL; Klijn JG The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer. Res 2000, 60 (3), 636–643. [PubMed] [Google Scholar]
- (9).Duffy MJ; O’Grady P; Devaney D; O’Siorain L; Fennelly JJ; Lijnen HJ Urokinase-plasminogen activator, a marker for aggressive breast carcinomas. Preliminary report. Cancer 1988, 62 (3), 531–533. [DOI] [PubMed] [Google Scholar]
- (10).Estreicher A; Muhlhauser J; Carpentier JL; Orci L; Vassalli JD The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J. Cell Biol 1990, 111 (2), 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lund IK; Illemann M; Thurison T; Christensen IJ; Hoyer-Hansen G uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets 2011, 12 (12), 1744–1760. [DOI] [PubMed] [Google Scholar]
- (12).Dohn LH; Illemann M; Høyer-Hansen G; Christensen IJ; Hostmark J; Litlekalsoy J; von der Maase H; Pappot H; Laerum OD Urokinase-type plasminogen activator receptor (uPAR) expression is associated with T-stage and survival in urothelial carcinoma of the bladder. Urol. Oncol 2015, 33 (4), 165.e15–165.e24. [DOI] [PubMed] [Google Scholar]
- (13).Ploug M; Ellis V Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett. 1994, 349 (2), 163–168. [DOI] [PubMed] [Google Scholar]
- (14).Duffy MJ; Duggan C The urokinase plasminogen activator system: a rich source of tumour markers for the individualised management of patients with cancer. Clin. Biochem 2004, 37 (7), 541–548. [DOI] [PubMed] [Google Scholar]
- (15).Rowe RG; Weiss SJ Navigating ECM barriers at the invasive front: the cancer cell-stroma interface. Annu. Rev. Cell Dev. Biol 2009, 25, 567–595. [DOI] [PubMed] [Google Scholar]
- (16).Matthews H; Ranson M; Kelso MJ Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: an orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer 2011, 129 (9), 2051–2061. [DOI] [PubMed] [Google Scholar]
- (17).Ranson M; Andronicos NM Plasminogen binding and cancer: promises and pitfalls. Front. Biosci., Landmark Ed 2003, 8, s294–304. [DOI] [PubMed] [Google Scholar]
- (18).Ngo JCK; Jiang L; Lin Z; Yuan C; Chen Z; Zhang X; Yu H; Wang J; Lin L; Huang M Structural basis for therapeutic intervention of uPA/uPAR system. Curr. Drug Targets 2011, 12 (12), 1729–1743. [DOI] [PubMed] [Google Scholar]
- (19).Tyndall JD; Kelso MJ; Clingan P; Ranson M Peptides and small molecules targeting the plasminogen activation system: towards prophylactic anti-metastasis drugs for breast cancer. Recent Pat. Anti-Cancer Drug Discovery 2008, 3 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- (20).Nienaber VL; Davidson D; Edalji R; Giranda VL; Klinghofer V; Henkin J; Magdalinos P; Mantei R; Merrick S; Severin JM; Smith RA; Stewart K; Walter K; Wang J; Wendt M; Weitzberg M; Zhao X; Rockway T Structure-directed discovery of potent non-peptidic inhibitors of human urokinase that access a novel binding subsite. Structure 2000, 8 (5), 553–563. [DOI] [PubMed] [Google Scholar]
- (21).Wendt MD; Geyer A; McClellan WJ; Rockway TW; Weitzberg M; Zhao X; Mantei R; Stewart K; Nienaber V; Klinghofer V; Giranda VL Interaction with the S1 beta-pocket of urokinase: 8-heterocycle substituted and 6,8-disubstituted 2-naphthamidine urokinase inhibitors. Bioorg. Med. Chem. Lett 2004, 14 (12), 3063–3068. [DOI] [PubMed] [Google Scholar]
- (22).Wendt MD; Rockway TW; Geyer A; McClellan W; Weitzberg M; Zhao X; Mantei R; Nienaber VL; Stewart K; Klinghofer V; Giranda VL Identification of novel binding interactions in the development of potent, selective 2-naphthamidine inhibitors of urokinase. Synthesis, structural analysis, and SAR of N-phenyl amide 6-substitution. J. Med. Chem 2004, 47 (2), 303–324. [DOI] [PubMed] [Google Scholar]
- (23).Bruncko M; McClellan WJ; Wendt MD; Sauer DR; Geyer A; Dalton CR; Kaminski MA; Weitzberg M; Gong J; Dellaria JF; Mantei R; Zhao X; Nienaber VL; Stewart K; Klinghofer V; Bouska J; Rockway TW; Giranda VL N-aphtha-midine urokinase plasminogen activator inhibitors with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett 2005, 15 (1), 93–98. [DOI] [PubMed] [Google Scholar]
- (24).Towle MJ; Lee A; Maduakor EC; Schwartz CE; Bridges AJ; Littlefield BA Inhibition of urokinase by 4-substituted benzo[b]thiophene-2-carboxamidines: an important new class of selective synthetic urokinase inhibitor. Cancer Res. 1993, 53 (11), 2553–2559. [PubMed] [Google Scholar]
- (25).Rockway TW; Giranda VL Inhibitors of the proteolytic activity of urokinase type plasminogen activator. Curr. Pharm. Des 2003, 9 (19), 1483–1498. [DOI] [PubMed] [Google Scholar]
- (26).Frederickson M; Callaghan O; Chessari G; Congreve M; Cowan SR; Matthews JE; McMenamin R; Smith DM; Vinkovic M; Wallis NG Fragment-based discovery of mexiletine derivatives as orally bioavailable inhibitors of urokinase-type plasminogen activator. J. Med. Chem 2008, 51 (2), 183–186. [DOI] [PubMed] [Google Scholar]
- (27).Sturzebecher J; Vieweg H; Steinmetzer T; Schweinitz A; Stubbs MT; Renatus M; Wikstrom P 3-Amidinophenylalanine-based inhibitors of urokinase. Bioorg. Med. Chem. Lett 1999, 9 (21), 3147–3152. [DOI] [PubMed] [Google Scholar]
- (28).Heinemann V; Ebert MP; Laubender RP; Bevan P; Mala C; Boeck S Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br. J. Cancer 2013, 108 (4), 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Positive Phase II data for MESUPRON in breast cancer. WILEX Half-Yearly Financial Report 2012; WILEX AG: Munich, Germany, 2012. [Google Scholar]
- (30).Spinar J; Spinarova L; Vitovec J Diuretics in monotherapy and in combination with other diuretics and nondiuretics in the treatment of hypertension. Vnitr. Lek 2013, 59 (6), 486–494. [PubMed] [Google Scholar]
- (31).Lant AF; Smith AJ; Wilson GM Clinical evaluation of amiloride, a potassium-sparing diuretic. Clin. Pharmacol. Ther 1969, 10 (1), 50–63. [DOI] [PubMed] [Google Scholar]
- (32).Kleyman TR; Cragoe EJ Jr. The mechanism of action of amiloride. Semin. Nephrol 1988, 8 (3), 242–248. [PubMed] [Google Scholar]
- (33).Palmer BF Regulation of potassium homeostasis. Clin. J. Am. Soc. Nephrol 2015, 10 (6), 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jankun J; Keck RW; Skrzypczak-Jankun E; Swiercz R Inhibitors of urokinase reduce size of prostate cancer xenografts in severe combined immunodeficient mice. Cancer Res. 1997, 57 (4), 559–563. [PubMed] [Google Scholar]
- (35).Vassalli JD; Belin D Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett. 1987, 214 (1), 187–191. [DOI] [PubMed] [Google Scholar]
- (36).Perazella MA Drug-induced hyperkalemia: old culprits and new offenders. Am. J. Med 2000, 109 (4), 307–314. [DOI] [PubMed] [Google Scholar]
- (37).Wermuth CG Selective optimization of side activities: another way for drug discovery. J. Med. Chem 2004, 47 (6), 1303–1314. [DOI] [PubMed] [Google Scholar]
- (38).Matthews H; Ranson M; Tyndall JD; Kelso MJ Synthesis and preliminary evaluation of amiloride analogs as inhibitors of the urokinase-type plasminogen activator (uPA). Bioorg. Med. Chem. Lett 2011, 21 (22), 6760–6766. [DOI] [PubMed] [Google Scholar]
- (39).Kleyman TR; Cragoe EJ Jr. Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol 1988, 105 (1), 1–21. [DOI] [PubMed] [Google Scholar]
- (40).Song CW; Lyons JC; Makepeace CM; Griffin RJ; Cragoe EJ Jr. Effects of HMA, an analog of amiloride, on the thermosensitivity of tumors in vivo. Int. J. Radiat. Oncol., Biol., Phys 1994, 30 (1), 133–139. [DOI] [PubMed] [Google Scholar]
- (41).Luo J; Tannock IF Inhibition of the regulation of intracellular pH: potential of 5-(N,N-hexamethylene) amiloride in tumour-selective therapy. Br. J. Cancer 1994, 70 (4), 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rich IN; Worthington-White D; Garden OA; Musk P Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na(+)/H(+) exchanger. Blood 2000, 95 (4), 1427–1434. [PubMed] [Google Scholar]
- (43).Man CH; Lam SS; Sun MK; Chow HC; Gill H; Kwong YL; Leung AY A novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AML. Blood 2014, 123 (16), 2530–2539. [DOI] [PubMed] [Google Scholar]
- (44).Ye JH; Gao J; Wu YN; Hu YJ; Zhang CP; Xu TL Identification of acid-sensing ion channels in adenoid cystic carcinomas. Biochem. Biophys. Res. Commun 2007, 355 (4), 986–992. [DOI] [PubMed] [Google Scholar]
- (45).Rowson-Hodel AR; Berg AL; Wald JH; Hatakeyama J; VanderVorst K; Curiel DA; Leon LJ; Sweeney C; Carraway KL Hexamethylene amiloride engages a novel reactive oxygen species- and lysosome-dependent programmed necrotic mechanism to selectively target breast cancer cells. Cancer Lett. 2016, 375 (1), 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kleyman TR; Cragoe EJ Jr. Cation transport probes: the amiloride series. Methods Enzymol. 1990, 191, 739–755. [DOI] [PubMed] [Google Scholar]
- (47).Cragoe EJ; Woltersdorf OW; Bicking JB; Kwong SF; Jones JH Pyrazine Diuretics. II. N-Amidino-3-amino-5-substituted 6Halopyrazinecarboxamides. J. Med. Chem 1967, 10 (1), 66–75. [DOI] [PubMed] [Google Scholar]
- (48).Patwardhan NN; Ganser LR; Kapral GJ; Eubanks CS; Lee J; Sathyamoorthy B; Al-Hashimi HM; Hargrove AE Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. MedChemComm 2017, 8 (5), 1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zeslawska E; Schweinitz A; Karcher A; Sondermann P; Sperl S; Sturzebecher J; Jacob U Crystals of the urokinase type plasminogen activator variant beta(c)-uPAin complex with small molecule inhibitors open the way towards structure-based drug design. J. Mol. Biol 2000, 301 (2), 465–475. [DOI] [PubMed] [Google Scholar]
- (50).Lee S; Yi KY; Hwang SK; Lee BH; Yoo S.-e.; Lee K (5-Arylfuran-2-ylcarbonyl)guanidines as cardioprotectives through the inhibition of Na+/H+ exchanger isoform-1. J. Med. Chem 2005, 48 (8), 2882–2891. [DOI] [PubMed] [Google Scholar]
- (51).Cochran BJ; Gunawardhana LP; Vine KL; Lee JA; Lobov S; Ranson M The CD-loop of PAI-2 (SERPINB2) is redundant in the targeting, inhibition and clearance of cell surface uPA activity. BMC Biotechnol. 2009, 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Yung-Chi C; Prusoff WH Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22 (23), 3099–3108. [DOI] [PubMed] [Google Scholar]
- (53).Nienaber V; Wang J; Davidson D; Henkin J Re-engineering of human urokinase provides a system for structure-based drug design at high resolution and reveals a novel structural subsite. J. Biol. Chem 2000, 275 (10), 7239–7248. [DOI] [PubMed] [Google Scholar]
- (54).Bachovchin DA; Koblan LW; Wu W; Liu Y; Li Y; Zhao P; Woznica I; Shu Y; Lai JH; Poplawski SE; Kiritsy CP; Healey SE; DiMare M; Sanford DG; Munford RS; Bachovchin WW; Golub TR A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nat. Chem. Biol 2014, 10 (8), 656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Soisson SM; Patel SB; Abeywickrema PD; Byrne NJ; Diehl RE; Hall DL; Ford RE; Reid JC; Rickert KW; Shipman JM; Sharma S; Lumb KJ Structural definition and substrate specificity of the S28 protease family: the crystal structure of human prolylcarboxypeptidase. BMC Struct. Biol 2010, 10 (16), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Soudijn W; Van Wijngaarden I; Ijzermann AP Allosteric modulation of G protein-coupled receptors: perspectives and recent developments. Drug Discovery Today 2004, 9 (17), 752–758. [DOI] [PubMed] [Google Scholar]
- (57).Gao ZG; Ijzerman AP Allosteric modulation of A(2A) adenosine receptors by amiloride analogues and sodium ions. Biochem. Pharmacol 2000, 60 (5), 669–676. [DOI] [PubMed] [Google Scholar]
- (58).Garritsen A; Beukers MW; Ijzerman AP; Soudijn W Recognition of adenosine receptors by amiloride and its analogues. Nucleosides Nucleotides 1991, 10 (5), 1107–1111. [Google Scholar]
- (59).Garritsen A; Ijzerman AP; Tulp M. M. Th.; Cragoe EJ Jr.; Soudijn W Receptor binding profiles of amiloride analogues provide no evidence for a link between receptors and the Na+/H+ exchanger, but indicate a common structure on receptor proteins. J. Recept. Res 1991, 11 (6), 891–907. [DOI] [PubMed] [Google Scholar]
- (60).de Lera Ruiz M; Lim Y-H; Zheng J Adenosine A2A receptor as a drug discovery target. J. Med. Chem 2014, 57 (9), 3623–3650. [DOI] [PubMed] [Google Scholar]
- (61).Massink A; Louvel J; Adlere I; van Veen C; Huisman BJ; Dijksteel GS; Guo D; Lenselink EB; Buckley BJ; Matthews H; Ranson M; Kelso M; Ijzerman AP 5′-Substituted amiloride derivatives as allosteric modulators binding in the sodium ion pocket of the adenosine A2A receptor. J. Med. Chem 2016, 59 (10), 4769–4777. [DOI] [PubMed] [Google Scholar]
- (62).Klinghofer V; Stewart K; McGonigal T; Smith R; Sarthy A; Nienaber V; Butler C; Dorwin S; Richardson P; Weitzberg M; Wendt M; Rockway T; Zhao X; Hulkower KI; Giranda VL Species specificity of amidine-based urokinase inhibitors. Biochemistry 2001, 40 (31), 9125–9131. [DOI] [PubMed] [Google Scholar]
- (63).McNay JL; Oran E Possible predisposition of diabetic patients to hyperkalemia following administration of potassium-retaining diuretic, amiloride (MK 870). Metab., Clin. Exp 1970, 19 (1), 58–70. [DOI] [PubMed] [Google Scholar]
- (64).Baer JE; Jones CB; Spitzer SA; Russo HF The potassium-sparing and natriuretic activity of N-amidino-3,5-diamino-6-chloropyrazinecarboxamide hydro-chloride dihydrate (amiloride hydrochloride). J. Pharmacol. Exp. Ther 1967, 157 (2), 472–485. [PubMed] [Google Scholar]
- (65). Comprehensive pharmacokinetic profiling of compounds 18 and 26 will be reported elsewhere.
- (66).Uhland K; Siphos B; Arkona C; Schuster M; Petri B; Steinmetzer P; Mueller F; Schweinitz A; Steinmetzer T; Van De Locht A Use of IHC and newly designed matriptase inhibitors to elucidate the role of matriptase in pancreatic ductal adenocarcinoma. Int. J. Oncol 2009, 35 (2), 347–357. [PubMed] [Google Scholar]
- (67).Pothula SP; Xu Z; Goldstein D; Biankin AV; Pirola RC; Wilson JS; Apte MV Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. Br. J. Cancer 2016, 114 (3), 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Nikishkin NI; Huskens J; Verboom W Transition metal-catalyzed functionalization of pyrazines. Org. Biomol. Chem 2013, 11 (22), 3583–3602. [DOI] [PubMed] [Google Scholar]
- (69).Chessell IP; Michel AD; Humphrey PP Properties of the pore-forming P2 × 7 purinoceptor in mouse NTW8 microglial cells. Br. J. Pharmacol 1997, 121 (7), 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Amith SR; Wilkinson JM; Fliegel L KR-33028, a potent inhibitor of the Na(+)/H(+) exchanger NHE1, suppresses metastatic potential of triple-negative breast cancer cells. Biochem. Pharmacol 2016, 118, 31–39. [DOI] [PubMed] [Google Scholar]
- (71).Rosenberg MR; Weaver LM; Casarotto MG Probing interactions of Vpu from HIV-1 with amiloride-based compounds. Biochim. Biophys. Acta, Biomembr 2016, 1858 (4), 733–739. [DOI] [PubMed] [Google Scholar]
- (72).Jalily PH; Eldstrom J; Miller SC; Kwan DC; Tai SS-H; Chou D; Niikura M; Tietjen I; Fedida D Mechanisms of action of novel influenza A/M2 viroporin inhibitors derived from hexamethylene amiloride. Mol. Pharmacol 2016, 90 (2), 80–95. [DOI] [PubMed] [Google Scholar]
- (73).Heitman LH; Ye K; Oosterom J; Ijzerman AP Amiloride derivatives and a nonpeptidic antagonist bind at two distinct allosteric sites in the human gonadotropin-releasing hormone receptor. Mol. Pharmacol 2008, 73 (6), 1808–1815. [DOI] [PubMed] [Google Scholar]
- (74).Snell HD; Gonzales EB 5-(N, N-Hexamethylene) amiloride is a GABA-A ρ1 receptor positive allosteric modulator. Channels 2016, 10 (6), 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Leng TD; Si HF; Li J; Yang T; Zhu M; Wang B; Simon RP; Xiong ZG Amiloride analogs as ASIC1a inhibitors. CNS Neurosci. Ther 2016, 22 (6), 468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Walsh KB Targeting GIRK channels for the development of new therapeutic agents. Front. Pharmacol 2011, 2, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Cox G; Ewart G; Gage P A Method of Modulating Ion Channel Functional Activity. PCT Int. Appl WO2000021538 A1, 2000. [Google Scholar]
- (78).Zhao G; Yuan C; Wind T; Huang Z; Andreasen PA; Huang M Structural basis of specificity of a peptidyl urokinase inhibitor, upain-1. J. Struct. Biol 2007, 160 (1), 1–10. [DOI] [PubMed] [Google Scholar]
- (79).Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (80).The CCP4 suite: Programs for protein crystallography. Collaborative Computational Project, Number 4. Acta Crystallogr., Sect. D: Biol. Crystallogr 1994, 50, 760–763, [DOI] [PubMed] [Google Scholar]
- (81).Jiang L; Botkjaer KA; Andersen LM; Yuan C; Andreasen PA; Huang M Rezymogenation of active urokinase induced by an inhibitory antibody. Biochem. J 2013, 449 (1), 161–166. [DOI] [PubMed] [Google Scholar]
- (82).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (83).Lu M; Echeverri F; Kalabat D; Laita B; Dahan DS; Smith RD; Xu H; Staszewski L; Yamamoto J; Ling J; Hwang N; Kimmich R; Li P; Patron E; Keung W; Patron A; Moyer BD Small molecule activator of the human epithelial sodium channel. J. Biol. Chem 2008, 283 (18), 11981–11994. [DOI] [PubMed] [Google Scholar]
- (84).Xu Z; Vonlaufen A; Phillips PA; Fiala-Beer E; Zhang X; Yang L; Biankin AV; Goldstein D; Pirola RC; Wilson JS; Apte MV Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol 2010, 177 (5), 2585–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.