

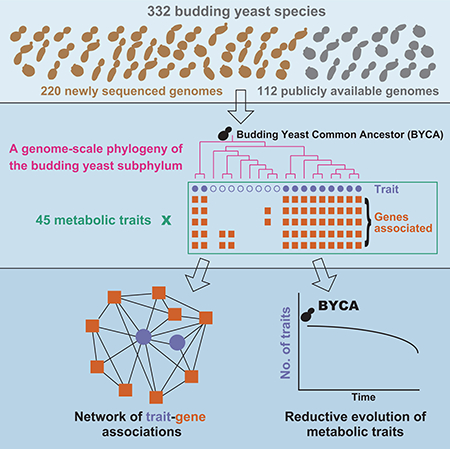
Summary
Budding yeasts (subphylum Saccharomycotina) are found in every biome and are as genetically diverse as plants or animals. To understand budding yeast evolution, we analyzed the genomes of 332 yeast species, including 220 newly sequenced ones, which represent nearly a third of all known budding yeast diversity. Here we establish a robust genus-level phylogeny comprised of 12 major clades, infer the timescale of diversification from the Devonian Period to the present, quantify horizontal gene transfer (HGT), and reconstruct the evolution of 45 metabolic traits and the metabolic toolkit of the Budding Yeast Common Ancestor (BYCA). We infer that BYCA was metabolically complex and chronicle the tempo and mode of genomic and phenotypic evolution across the subphylum, which is characterized by very low HGT levels and widespread losses of traits and the genes that control them. More generally, our results argue that reductive evolution is a major mode of evolutionary diversification.
In brief
An integrated phylogeny of over 300 budding yeast species encompasses the natural diversity and history of diversification of Saccharomycotina with insights into a metabolically-complex common ancestor and common reductive evolution leading to metabolic specialization.
Graphical Abstract
Introduction
Yeasts - unicellular fungi that lack fruiting bodies - have evolved multiple times across fungi (Stajich et al., 2009), but most known yeast species belong to the subphylum Saccharomycotina, hereafter referred to as budding yeasts or simply yeasts. This diverse group, whose genetic diversity is on par with the plant and animal lineages (Figure 1), includes the baker’s yeast and premier eukaryotic model system Saccharomyces cerevisiae (Peter et al., 2018), the common human commensal and opportunistic pathogen Candida albicans, and over 1,000 other known species with more continuing to be discovered (Dujon and Louis, 2017; Hittinger et al., 2015; Kurtzman et al., 2011).
Figure 1. Levels of evolutionary sequence divergence within the budding yeast subphylum are on par with levels observed in animals and plants.

The phylogenetic distance (in terms of amino acid substitutions / site) between iconic species in budding yeasts (Saccharomyces cerevisiae), animals (Homo sapiens), and plants (Arabidopsis thaliana) and other representative species in each lineage. For each lineage, phylogenetic distance was estimated from a concatenated ML tree inferred from analysis of 295 single-copy BUSCO genes. S. cerevisiae = Saccharomyces cerevisiae; S. paradoxus = Saccharomyces paradoxus; S. uvarum = Saccharomyces uvarum; C. glabrata = Candida glabrata; K. lactis = Kluyveromyces lactis; Wy. anomalus = Wickerhamomyces anomalus; D. hansenii = Debaryomyces hansenii; C. albicans = Candida albicans; B. bruxellensis = Brettanomyces bruxellensis; L. starkeyi = Lipomyces starkeyi; Y. lipolytica = Yarrowia lipolytica. Images representing taxa were drawn by hand, taken from PhyloPic (http://phylopic.org), or modified from Google Images. The data matrix and ML tree in Newick format used for calculating sequence divergence in each of the three lineages are provided in the Figshare depository.
Yeasts exhibit remarkably diverse heterotrophic metabolisms, which have allowed them to successfully partition nutrients and ecosystems to inhabit every continent and every major aquatic and terrestrial biome (Kurtzman et al., 2011; Opulente et al., 2018). Comparative genomic investigations have served as the launching pads into the evolution of budding yeast metabolism and ecological specialization in numerous clades, including the clade of S. cerevisiae and its close relatives (Hittinger, 2013), the genus Lachancea (Vakirlis et al., 2016), the C. albicans/Candida tropicalis clade (Butler et al., 2009), and the Candida glabrata/Nakaseomyces clade (Gabaldon et al., 2013). Although these investigations have shaped our understanding of budding yeast evolution, they have either focused on small slices of biodiversity or have been very broad investigations using collections of taxa skewed toward biomedically or industrially relevant yeasts (Gabaldón et al., 2013; Génolevures Consortium et al., 2009; Riley et al., 2016).
This sparse and sporadic sampling of genome sequences has forestalled efforts to examine the impact of different evolutionary processes or to identify major evolutionary trends across the entire subphylum. For example, several illustrative studies have shown how the presence of specific genes and pathways correlates with phenotypes, including clear cases of gains (Gonsalves et al., 2018; Hall and Dietrich, 2007) and losses (Hittinger et al., 2004; Riley et al., 2016; Slot and Rokas, 2010; Wolfe et al., 2015), but the frequency and generality of these observations remain unclear. Collectively, the use of a relatively small subset of distantly related budding yeast genomes has prevented the state-of-the-art quantification and statistical analyses that would be required to understand the tempo and mode of genomic and metabolic evolution across the subphylum.
To address this gap, we sequenced the genomes of 220 budding yeast species and coupled them with the published genomes of an additional 112 species to investigate the evolution of biodiversity of the subphylum Saccharomycotina. By interrogating genomic and metabolic trait variation, we reconstructed a robust genome-wide phylogeny, established the geological timeline of budding yeast diversification, quantified HGT in budding yeast genomes, and inferred the evolution of 45 metabolic traits and their underlying genetic toolkit from the ~400 million-year-old budding yeast common ancestor (BYCA) to the present.
Results and Discussion
332 yeast genomes spanning the diversity of the yeast subphylum
We sampled the genomes of 332 budding yeast species representing 79 / 92 genera across the subphylum Saccharomycotina (STAR Methods). Of the 332 yeast genomes, 112 were publicly available and previously published, 24 sequenced by RIKEN were publicly available (http://www.icm.riken.ip/cgi-bin/nbrp/nbrplist.cgi) but are described here for the first time, and 196 were newly sequenced by the Y1000+ Project (http://www.y1000plus.org) (Figures 2 and S1; Table S1). 195 / 196 newly sequenced Y1000+ Project genomes were from type strains (the other was from an authentic strain), as were at least 81 / 136 publicly available ones; in addition, 72 / 79 included genera were represented by their type species (Table S1).
Figure 2. Time-calibrated phylogeny of the budding yeast subphylum.

Divergence times were estimated using the autocorrelated clock model of rate variation across different lineages implemented in MCMCTree (clock=3), with a topology reconstructed from the concatenation-based maximum likelihood analysis of 2,408 amino acid orthologous groups (OGs) under a single LG+G4 model. The thirty-two internal branches that were not robustly recovered across our analyses are marked with circles. The 220 genomes published in this study are shown in bold. The bar plot next to each species indicates genomic quality assessed by a set of 1,759 BUSCO genes. “Complete” indicates the fraction of full-length BUSCO genes; “Fragmented” indicates the fraction of genes with a partial sequence; and “Missing” indicates the fraction of genes not found in the genome. Note that the CUG-Ser1 clade includes interspersed taxa from the families Debaryomycetaceae, Metschnikowiaceae, and Cephaloascaceae; the CUG-Ser2 clade includes the families Ascoideaceae and Saccharomycopsidaceae; the newly recovered CUG-Ala clade includes several taxa in need of reassignment; the Pichiaceae clade includes several taxa in need of reassignment; and the Dipodascaceae/Trichomonascaceae clade includes interspersed taxa mainly from these two families. MYA: million years ago. See also Figures S1, S2 and S3 and Tables S1 and S2.
Compared to the 136 publicly available genomes, the 196 newly sequenced genomes (Y1000+ Project genomes) had generally smaller average N50 values (new = 417.2 kb vs. public = 1,235.4 kb), but they had comparable degrees of genome assembly completeness (new = 91.3% vs. public = 94.0%), GC contents (new = 40.3% vs. public = 41.1%), and average numbers of predicted genes (new = 5,822.4 genes vs. public = 5,525.7 genes), suggesting that they are suitable for most evolutionary genomic analyses.
A robust phylogeny for the subphylum Saccharomycotina
To infer the budding yeast phylogeny, we assembled two full data matrices (2408OG data matrix and 1292BUSCO data matrix) and 7 additional ones (by subsampling subsets of genes in the 2408OG data matrix) and analyzed them using three phylogenetic strategies (concatenation under a single partition, concatenation under gene-based partitioning, and coalescence). These analyses produced a strongly supported and largely concordant phylogeny (Figure 2); only 32 / 331 yeast internodes were not recovered by all 27 phylogenies, with most instances of incongruence being due to whether the data matrix was analyzed by concatenation or coalescence (Figure S2).
Surprisingly, one of the resolved internodes concerned the placement of the CUG-Ser2 clade, which was equivocal in previous phylogenomic studies (Riley et al., 2016; Shen et al., 2016a, 2017). For example, the most recent phylogenomic analysis, in which the CUG-Ser2 clade was represented by a single taxon, showed that the clade’s placement was unduly influenced by inclusion of the DPMI (in S. cerevisieae) gene (Shen et al., 2017). However, our sampling of three additional representative taxa from the CUG-Ser2 clade eliminated the gene’s disproportionate influence and resolved this clade’s placement in the budding yeast phylogeny (Figure S3).
The new phylogeny divides the subphylum into 12 major clades (Figure 2), at least two and up to eight more than previously recognized (Dujon and Louis, 2017; Hittinger et al., 2015; Kurtzman and Boekhout, 2017; Kurtzman et al., 2011; Shen et al., 2016a). Several families are now well circumscribed (e.g., Pichiaceae), while other families are not reciprocally monophyletic (e.g., Dipodascaceae/Trichomonascaceae), leading us to consider them as major clades comprised of multiple known families. One major reorganization stems from the recognition that the split between the genus Sporopachydermia and other budding yeasts is very deep. Although previous authors had recognized affinities to the families Dipodascaceae and Trichomonascaceae, our analyses placed the genus Sporopachydermia on its own long branch with deep affinities to the family Alloascoideaceae in most analyses (Figure 2). A second major reorganization stems from the placement of a group of species that all translate the CUG codon as alanine (the CUG-Ala clade) (Krassowski et al., 2018; Riley et al., 2016) as a lineage that is separate from and sister to the Pichiaceae clade. In previous studies, some members of these two clades were placed together, while the taxonomic placements of taxa in the genera Nakazawaea, Pachysolen, and Peterozyma were still unclear (Kurtzman and Boekhout, 2017; Kurtzman et al., 2011). Our results suggest that the CUG-Ala clade represents a new, yet-to-be-described taxonomic family.
A timescale for budding yeast diversification
To estimate divergence times of the budding yeast subphylum, we employed Bayesian relaxed clock approach (Yang, 2007) with four generally accepted calibration points (STAR Methods) on the 2408OG data matrix. We estimated the origin of the subphylum at 317–523 (95% credibility interval; posterior mean date: 404) million years ago (MYA); the origin of the CUG-Ser1 clade, which contains the major opportunistic pathogen C. albicans, at 178–248 (210) MYA, the origin of the whole genome duplication (WGD) clade at 82–105 (93) MYA, and the divergence of S. cerevisiae and C. albicans from their sister species (Saccharomycesparadoxus and Candida dubliniensis) at 4.0–5.8 (4.9) MYA and 5.0–14.0 (8.7) MYA, respectively (Figure 2; Table S2).
Horizontal gene transfer (HGT) into budding yeasts
To identify the tempo and mode of HGT into the budding yeast gene lineage, we investigated all 1,538,912 genes that reside in the 8,792 contigs > 100 kb using a robust and conservative phylogeny-based approach (STAR Methods). We found that 878 genes in 186 yeast genomes were likely acquired via 365 distinct HGT events from non-fungal (mostly bacterial) sources (Figures 3A and S4; Table S3). Gene Ontology enrichment analysis of the 878 genes shows that most are associated with metabolism-related terms (Figure S5; Table S3). Furthermore, 616 / 878 genes (~70%) were significantly supported with the approximately unbiased (AU) test (Shimodaira, 2002), a value quite similar to the percentage supported by AU tests among previously described HGT-acquired genes (15 / 21, or ~71%; Table S3). Although precisely quantifying the fraction of HGT events is challenging, the approximately 0.04% (616 / 1,538,912) to 0.06% (878 / 1,538,912) of genes identified to have been putatively acquired via HGT suggests that the process is a very small contributor to overall yeast genome diversification.
Figure 3. Very few budding yeast genes were acquired via horizontal gene transfer (HGT).

(A) Mapping of the 878 putative HGT-acquired genes on the 332-taxon phylogeny of budding yeasts (Figure 2). These 878 genes were acquired through 365 distinct HGT events, of which 230 appear to be species-specific and the other 135 involve two or more species. (B) Two independent HGT events provided osmotolerant budding yeasts the genetic machinery to produce the osmoprotectant glycine betaine. On the top, the biochemical pathway for the biosynthesis of glycine betaine from choline-O-sulfate is shown. Choline-O-sulfate is first converted by the action of choline sulfatase into choline; then, choline is converted by the action of choline oxidase into glycine betaine. On the bottom, the phytogenies for the choline sulfatase and choline oxidase genes are shown. Note that the sequences of the sister lineages to the W. domercqiae and W. versatilis choline sulfatase sequences are from Proteobacteria, whereas the sequences of the sister lineages to the W. domercqiae and W. versatilis choline oxidase sequences are from Actinobacteria. (C) Two independent events provided two different lineages of budding yeasts (the W/S clade and a clade of three species in the genus Kluyveromyces) the genetic machinery to metabolize the mitochondrial toxin propionate-3-nitronate (P3N). On the top, the biochemical pathway for the oxidation of P3N is shown, where P3N is converted into malonic semialdehyde via the action of nitronate monooxygenase. On the bottom, the phylogeny for the nitronate monooxygenase acquired by an ancestor of W. domercqiae and W. versatilis is shown. Note that the sequences of the sister lineages to the W. domercqiae and W. versatilis nitronate monooxygenase sequences are from Proteobacteria in the genus Pseudomonas, whereas the sequences of the sister lineages to the three Kluyveromyces nitronate monooxygenase sequences are from Proteobacteria in the genus Acinetobacter. The budding yeast species inferred to have been the recipients of horizontally transferred genes are shown in bold. Data matrices and phylogenies for all HGT-acquired genes are provided in the Figshare repository. See also Figures S4 and S5 and Table S3.
From the 878 HGT-acquired genes, 333 were present in the genomes of the six representative species from the W/S clade (Figure 3A; Table S3). In contrast, the genomes of 57 / 103 species in the CUG group, whose genetic codes deviate from the universal one, did not contain any HGT-acquired genes (Figure 3A). The fraction of HGT-acquired genes was significantly higher in the 226 yeast genomes that use the universal genetic code than in the 103 CUG group genomes (0.071% of 1,054,747 genes vs. 0.025% of 484,165 genes; Fisher’s exact test; P-value = 2.2e-16; which remained significant even after exclusion of the W/S clade species at 0.041% of 1,027,319 genes vs. 0.025% of 484,165 genes; Fisher’s exact test; P-value = 2.5e-05), supporting the hypothesis that altered genetic codes serve as barriers to HGT (Richards et al., 2011; Woese, 2000).
Finally, our dense genome sampling also facilitated the identification of cases involving the independent acquisition of genes participating in the same metabolic pathway into the same yeast lineage, as well as the independent acquisition of the same gene in two distantly related yeast lineages (Figures 3B and3C). Specifically, an ancestor of Wickerhamiella (Candida) versatilis and Wickerhamiella domercqiae, two highly osmotolerant species, acquired a choline oxidase associated with the production of the osmoprotectant betaine from choline; remarkably, that same ancestor appears to have also independently acquired a choline sulfatase involved in the conversion of choline-O-sulfate into choline (Figure 3B). W. versatilis and W. domercqiae also horizontally acquired a nitronate monooxygenase likely associated with resistance to propionate 3-nitronate (P3N; a plant and fungal toxin) from genus Pseudomonas bacteria (Figure 3C).
Evolution of metabolic traits across budding yeasts
To examine the evolution of budding yeast metabolism, we used Bayesian inference to estimate rates of gain and loss for a compilation of 45 discrete metabolic traits in 274 / 332 budding yeasts across the subphylum (Kurtzman et al., 2011; Opulente et al., 2018) (STAR Methods; Table S4). We found that 39 / 45 traits experienced higher rates of loss, whereas the remaining 6 experienced higher rates of gain (Table 1; Figure S6). These trends were statistically significant for 16 / 39 traits where the rate of loss was higher than the rate of gain and for 1 / 6 traits where the rate of gain was higher than the rate of loss (Table 1). As the metabolic trait data came from multiple sources and the strains tested sometimes differed from the strains sequenced, we also experimentally determined the growth on 13 / 45 metabolic traits and a control trait (glucose) (Table S4) for 328 / 332 strains of the budding yeast species whose genomes we analyzed. We found that the average consistency of the character states shared between the compilation of available metabolic trait data and our experimental measurements was 94% (Table S4), suggesting that our inferences are based on accurate data.
Table 1.
Rates of trait gain (q01), loss (q10), and posterior probabilities (PP) of ancestral states 0 and 1 of the Budding Yeast Common Ancestor (BYCA) for 45 metabolic traits.
| Trait | q01 | q10 | PP (0) | pp (1) | Trait | q01 | q10 | PP (0) | pp (1) |
|---|---|---|---|---|---|---|---|---|---|
| Glucose Fermentation | 1.16 | 0.37 | 0.96 | 0.04 | L-Arabinose* | 0.02 | 1.36 | 0.00 | 1.00 |
| Galactose Fermentation* | 0.31 | 1.48 | 0.87 | 0.13 | D-Arabinose | 0.18 | 2.27 | 0.99 | 0.01 |
| Sucrose Fermentation | 0.40 | 2.00 | 0.98 | 0.02 | D-Ribose | 0.17 | 0.96 | 0.99 | 0.01 |
| Maltose Fermentation* | 0.34 | 3.17 | 0.56 | 0.44 | Methanol | 0.03 | 0.27 | 1.00 | 0.00 |
| Lactose Fermentation* | 0.17 | 78.03 | 0.50 | 0.50 | Ethanol | 0.73 | 0.14 | 0.00 | 1.00 |
| Raffinose Fermentation* | 0.27 | 2.35 | 0.76 | 0.24 | Glycerol* | 1.95 | 0.23 | 0.40 | 0.60 |
| Trehalose Fermentation* | 0.38 | 1.55 | 0.00 | 1.00 | Erythritol* | 0.03 | 1.38 | 0.00 | 1.00 |
| Inulin* | 0.23 | 3.49 | 0.12 | 0.88 | Ribitol | 0.33 | 0.38 | 0.97 | 0.03 |
| Sucrose | 0.21 | 0.92 | 0.68 | 0.32 | Galactitol* | 0.13 | 1.85 | 0.09 | 0.91 |
| Raffinose* | 0.23 | 2.13 | 0.00 | 1.00 | D-Mannitol | 0.11 | 0.27 | 0.00 | 1.00 |
| Melibiose* | 0.06 | 2.89 | 0.00 | 1.00 | D-Glucitol | 0.04 | 0.23 | 0.00 | 1.00 |
| Galactose | 0.49 | 0.31 | 0.01 | 0.99 | myo-Inositol | 0.01 | 1.43 | 0.00 | 1.00 |
| Lactose* | 0.18 | 2.74 | 0.04 | 0.96 | DL-Lactate | 0.68 | 0.89 | 0.66 | 0.34 |
| Trehalose | 0.62 | 0.83 | 0.28 | 0.72 | Succinate | 0.35 | 0.20 | 0.00 | 1.00 |
| Maltose | 0.27 | 0.92 | 0.67 | 0.33 | Citrate | 0.46 | 0.85 | 0.08 | 0.92 |
| Melezitose | 0.43 | 1.22 | 0.83 | 0.17 | D-Gluconate | 0.75 | 0.97 | 0.58 | 0.42 |
| Methyl-a-D-glucoside | 0.24 | 1.53 | 0.00 | 1.00 | D-Glucosamine* | 0.04 | 1.35 | 0.01 | 0.99 |
| Soluble Starch* | 0.11 | 2.25 | 0.00 | 1.00 | N-Acetyl-D-glucosamine | 0.08 | 0.57 | 0.21 | 0.79 |
| Cellobiose | 0.24 | 0.75 | 0.17 | 0.83 | Hexadecane | 0.17 | 0.89 | 0.91 | 0.09 |
| Salicin | 0.10 | 0.76 | 0.20 | 0.80 | Nitrate* | 0.01 | 2.12 | 0.02 | 0.98 |
| L-Sorbose | 0.50 | 0.51 | 0.01 | 0.99 | Nitrite | 0.16 | 1.18 | 0.95 | 0.05 |
| L-Rhamnose* | 0.02 | 1.97 | 0.03 | 0.97 | 2-Keto-D-gluconate | 0.53 | 0.48 | 0.87 | 0.13 |
| D-Xylose | 0.12 | 0.38 | 0.00 | 1.00 |
Note: q01 (instantaneous transition rate of trait gain): the rate of change from state 0 (absent) to state 1 (present). q10 (instantaneous transition rate of trait loss): the rate of change from state 1 to state 0.
Asterisks indicate traits in which rates q01 and q10 are significantly different (assessed by Bayes Factors). PP (0) and PP (1) denote posterior probabilities of ancestral states 0 (absence) and 1 (presence) for the Budding Yeast Common Ancestor (BYCA), respectively. Every value in this table is derived from the largest peak of density of posterior distributions. See also Figure S6 and Table S4.
Metabolic trait reconstruction also allowed us to infer the ancestral state at each node in the budding yeast phylogeny (STAR Methods). For example, we inferred that BYCA was most likely a metabolically complex organism capable of assimilating 27 / 45 of the carbon- and nitrogen-containing compounds analyzed (average posterior probability of trait presence = 0.64; average posterior probability of trait absence = 0.36) (Figure 4A; Table 1). Using a stricter cutoff (posterior probability > 0.9), we found 21 traits that were most likely present in BYCA (e.g., nitrate, xylose, and galactose assimilation), and 8 that were most likely absent (e.g., glucose fermentation and methanol, ribose, and hexadecane assimilation) (Table 1).
Figure 4. Evolution of metabolic traits across the budding yeast subphylum.

(A) The number of traits per major clade (columns) is depicted in a scatterplot where each grey dot corresponds to a species. Red dots indicate representative species, and black dots represent the median number of traits across each family. On the right is the distribution across the subphylum Saccharomycotina in histogram form. The red line corresponds to the inferred number of metabolic traits present (i.e., posterior probability of trait presence > 0.5 in Table 1) in the BYCA (budding yeast common ancestor). The blue dashed and solid lines represent the 75th (25 traits), 50th (median; 20 traits), and 25th (12 traits) percentiles of the numbers of traits, respectively. Representative species names are written using a four-letter abbreviation as follows: Lsta: Lipomyces starkeyi; Ylip: Yarrowia lipomyces; Wver: Wickerhamiella versatilis; Stbo: Starmerella bombicola; Bbru: Brettanomyces bruxellensis; Calb: Candida albicans; Cyja: Cyberlindnera jadinii; Klac: Kluyveromyces lactis; Cgla: Candida glabrata; Scer: Saccharomyces cerevisiae. (B) Heatmap showing the fraction of species in each major clade (columns) that display a representative set of metabolic traits; values near white indicate major clades (whose species are) lacking the trait, and values near black indicate major clades with the trait. To the left of the heatmap, the presence (black) or absence (white) of a trait in BYCA (inferred from ancestral trait reconstruction) is shown. To the right of the heatmap, well-characterized genes whose distributions are significantly associated with each trait are shown. (C) Positive association network for genes and traits. Traits and genes are represented by squares and circles, respectively. Trait communities are represented by the following colors: magenta, Contains Galactose; purple, Modified Glucose, green, Respiratory; orange, Glucosides; and cyan, Sugar Alcohols & Pentose Phosphate Pathway (Opulente et al., 2018). Associations among gene and traits were calculated using Mutual Information (MI) analysis, and negative associations were detected using a Jaccard index (all values less than 0.25 were considered negative associations and excluded). Edges connecting genes to traits are colored gray and are represented by solid lines for associations that had a MI value greater than or equal to 0.15; for genes already appearing in the figure, dashed lines representing MI values between 0.10 and 0.15 were included. The inset includes genes associated with GPHN, which encodes gephyrin. See also Figure S6 and Tables S4 and S5.
Although the genetic bases for many of these metabolic traits remain unknown, the genes involved in a handful of metabolic pathways are well characterized, allowing us to not only reconstruct characterized genetic pathways, but to also use the co-occurrence of traits and genes across the budding yeast phylogeny to identify novel connections and functions. To this end, we used a mutual information-based approach to analyze the phylogenetic distribution of functional annotations of genes in yeast species, together with the metabolic trait values of those species (STAR Methods). We then overlaid our gene-to-trait scores onto a network of trait-to-trait ecological associations among 784 budding yeast species (Opulente et al., 2018). These analyses recovered multiple positive gene-to-trait associations for gene-trait pairs that are established in model systems, such as genes encoding beta-galactosidases (e.g., LAC4) with growth on lactose; alcohol oxidases (e.g., MOX) with growth on methanol; and the GAL1, GAL7, and GAL10 genes with growth on galactose (Figure 4B; Table S5) (Riley et al., 2016). These examples validate the utility of this analytical approach for elucidating the genetic underpinnings of metabolic trait variation.
As expected from our analyses showing a strong trend toward metabolic trait loss (Table 1) and previous studies on the losses of iconic genetic pathways (Hittinger et al., 2004; Riley et al., 2016; Slot and Rokas, 2010; Wolfe et al., 2015), the distributions of many of the genes involved in these metabolic processes (e.g., the genes required to assimilate D-glucosamine and A-acetyl-D-glucosamine and the genes required for L-rhamnose assimilation) were consistent with widespread losses, which included both ancient losses deep within major clades and more recent losses differentiating closely related taxa.
Complex interactions between metabolic pathways affect gene retention
By analyzing these gene and trait networks, we also identified genes whose associations with specific metabolic traits has heretofore received only limited attention in select budding yeasts. For example, although the model yeast S. cerevisiae cannot assimilate nitrate or nitrite (Kurtzman et al., 2011), we found that the genomes of 50 budding yeasts that could assimilate either of these nitrogen sources were generally predicted to encode nitrate/nitrite transporters (YNTs, 39/50), nitrate reductases (YNRs, 40/50), and nitrite reductases (YNIs, 47/50) (Figures 4C and5). These proteins are homologs of a pathway commonly deployed in filamentous fungi to import nitrate (YNTs), reduce nitrate into nitrite (YNRs), and reduce nitrite into ammonia (YNIs) for central metabolism (Slot and Hibbett, 2007), strongly suggesting that the same pathway is deployed across many budding yeasts (Perez et al., 1997). As with most other metabolic traits (Figure 4), nitrate assimilation genes were frequently lost, both anciently (e.g., in the CUG-Ser1 clade) and more recently (e.g., in the genus Cyberlindnera).
Figure 5. Interconnections and interdependence of nitrate assimilation, xanthine assimilation, and the molybdopterin cofactor (Moco) biosynthesis pathways across the budding yeast subphylum.

The concentric tracks on the periphery of the figure depict (from inner to outer) the phylogenetic distribution of: growth phenotype on media containing nitrate or nitrite as a sole nitrogen source (inner green circles) and genes encoding proteins involved in nitrate/nitrite assimilation (green squares); hypoxanthine/xanthine assimilation (orange squares); and Moco biosynthesis (blue squares). The underlying phylogeny and distribution of taxa is the same as in Figure 2, but species names have been omitted. The central diagram depicts the individual steps of nitrate assimilation (green, top), xanthine assimilation (orange, right), and Moco biosynthesis (blue, left) pathways, with proteins involved shown in bold: nitrate transporter (YNT), nitrate reductase (YNR), nitrite reductase (YNI), cyclic pyranopterin monophosphate synthase (MOCS1), molybdopterin synthase (MOCS2), gephyrin (GPHN), molybdopterin sulfurtransferase (MS), and xanthine dehydrogenase (XDH). Molecules in the pathways: guanosine triphosphate (GTP), cyclic pyranopterin monophosphate (cPMP), molybdopterin (MPT), adenylated molybdopterin (MPT-AMP), molybdenum cofactor (Moco), and thiomolybdenum cofactor (Moco-sulfide). Solid arrows indicate subsequent steps in each pathway; dashed lines indicate use of a specific cofactor by an enzyme. See also Table S6.
Our network-based approach also enabled us to use the phylogenetic distribution of the nitrate assimilation pathway to identify additional metabolic genes, pathways, and traits that co-varied but lacked obvious ecological connections. For example, we uncovered several significant associations with the gene encoding gephyrin (Figure 4C; Table S5), an enzyme involved in the biosynthesis of the molybdenum cofactor (Moco) used by the nitrate reductase enzyme (Schwarz and Mendel, 2006). When we examined secondary associations (genes associated with genes that are in turn associated with traits), we identified genes predicted to encode the full Moco biosynthesis pathway (Figures 4C inset and 5; Table S6).
Surprisingly, the phylogenetic distribution of various components of the Moco biosynthesis and nitrate assimilation pathways revealed that YNRs were never present in the absence of the Moco biosynthesis pathway, but the reverse was often the case (Figure 5). Furthermore, most of the organisms that harbored the Moco biosynthesis pathway but lacked the nitrate assimilation genes also contained the genes encoding xanthine dehydrogenases (XDHs) (Figure 5). In filamentous fungi, XDH is responsible for the assimilation of xanthine and hypoxanthine and requires a thiolated form of Moco, which is created during an additional processing step performed by a specialized Moco sulfurtransferase (MS) (Bittner et al., 2001). The phylogenetic distributions of the genes encoding XDHs and MSs were strongly correlated; of the 83 species with either gene, 78 (or 94%) were predicted to have both genes, illustrating how the underlying metabolic architecture of genetic pathways can lead to remarkable evolutionary linkages between traits that may seem unconnected at first.
Conclusion
Genome sequencing and analyses of 332 metabolically diverse species allowed us to infer that the last common ancestor of budding yeasts, BYCA, was metabolically complex, that very few genes were acquired via HGT, and that the observed metabolic diversity of extant budding yeasts was largely achieved through repeated, extensive losses of metabolic traits through reductions in their underlying genetic toolkit (Figure 6). Thus, the portrait of BYCA that emerges is similar to an archetypal member of its sister subphylum, Pezizomycotina, which contains iconic filamentous fungi, such as Aspergillus and Neurospora. These filamentous fungi have much larger genomes (~30–80 Mb) and gene sets (~10,000 - ~13,000 genes, including metabolic genes). Consequently, they exhibit a broader appetite and consume a wide array of nitrogen and carbon sources by deploying many genes not present in S. cerevisiae (Wisecaver et al., 2014).
Figure 6. The evolution of the budding yeast subphylum is characterized by lineage-variable HGT and widespread losses of genes and traits.

The x-axis depicts the posterior mean of the age of (representative) nodes in the budding yeast phylogeny, and the y-axis depicts the number of metabolic traits inferred to have been present at these nodes. The lines of different colors represent the evolutionary trajectories (in the spaces of time and metabolic traits) for 7 representative yeast taxa and their common ancestors (depicted by dots). For each ancestral node, metabolic traits were considered to be present when the posterior probability of ancestral state 1 (present) was > 0.5 for the node. The gray region is the 95% confidence interval for the number of metabolic traits present across budding yeast evolution based on ancestral trait reconstruction of the distribution of inferences for 45 metabolic traits across 274 budding yeasts. The inferred numbers of HGT-acquired genes are depicted by the circles of different sizes next to each taxon’s name. C. albicans = Candida albicans; L. starkeyi = Lipomyces starkeyi; Cy. jadinii = Cyberlindnera jadinii; B. bruxellensis = Brettanomyces bruxellensis; W. versatilis = Wickerhamiella versatilis; Y. lipolytica = Yarrowia lipolytica; S. cerevisiae = Saccharomyces cerevisiae; BYCA: Budding Yeast Common Ancestor; MYA: million years ago; Dev. = Devonian; Carb. = Carboniferous; Per. = Permian; Tri. =Triassic; Jur. = Jurassic; Cre. = Cretaceous; Pal. = Paleogene; and Neo. = Neogene. See also Figure S7.
How have budding yeasts survived and even thrived while undergoing this reductive evolution? Although the arc of budding yeast evolution bends strongly toward reduced genomes and metabolisms across history, among both extant and inferred ancestral yeasts, plenty of taxa have much broader metabolic capabilities than the mean (Figures 6 and S7). One attractive hypothesis, whose testing will require even denser taxon sampling, is that generalist lineages with less reduced genomes and more metabolic capabilities are more likely to produce new species and less likely to go extinct. Evolutionary dynamics favoring generalists over specialists could conceivably play out over geological time through some form of clade selection (Stanley, 1975; Williams, 1992), by drawing from the considerable genetic variation segregating within species (Hittinger et al., 2010; Peter et al., 2018), or both.
The observed pattern of widespread metabolic trait and gene losses complements the well-established losses or reductions of several flagship eukaryotic genomic and molecular features in many budding yeasts, such as introns, the molecular machinery for RNA interference, and H3K9me2/3 heterochromatin (Dujon and Louis, 2017). Evolution by loss is well documented for parasitic and symbiotic lineages (Dujon et al., 2004; Katinka et al., 2001; Spanu et al., 2010; Vogel and Moran, 2013; Wolfe et al., 1992) and in the aftermath of the whole genome duplication events where most second copies are lost (Soltis et al., 2015;
Wolfe et al., 2015). Nonetheless, the magnitude and pervasiveness of metabolic trait and gene loss across 400 million years of budding yeast evolution in organisms with widely divergent, free-living lifestyles provides unexpectedly broad support for the argument that reductive evolution is a major contributor to genome evolution (Albalat and Canestro, 2016; Wolf and Koonin, 2013), in general, and not simply associated with specific lifestyles and genomic events.
STAR ★ METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests may be directed to and will be fulfilled by the corresponding authors Chris Todd Hittinger (cthittinger@wisc.edu) and Antonis Rokas (antonis.rokas@vanderbilt. edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Taxon sampling
Detailed information about the strains and genomes of the 332 budding yeast species sampled and analyzed in this study can be found in Table S1. We did not include genomes associated with different strains from the same species or with known interspecies hybrids. Taxonomy, strain ID, and source information of 332 budding yeasts and the 11 outgroup taxa (9 representatives of the subphylum Pezizomycotina and 2 representatives of the subphylum Taphrinomycotina) used in this study are provided in Table S1. All sequenced strains have been publicly deposited in the NRRL, CBS, and/or JCM strain collections.
The percentage of newly sequenced 220 species (196 newly sequenced genomes from the Y1000+ Project and 24 publicly available but unpublished genomes from RIKEN) in each of the 12 major clades ranged from 41% to 100%, with an average of 78% (Figure S1). Specifically, these newly sequenced 220 species represent 63 different genera; 74 are representatives of 36 genera (e.g., Ambrosiozyma, Saturnispora, and Barnettozyma) whose genomes have not been previously sequenced; 111 are representatives of 25 genera (e.g., Ogataea, Kazachstania, Lipomyces, and Cyberlindnera) that previously had one or two publicly available genomes; and the remaining 35 are representatives of genera (e.g., Candida and Hanseniaspora) that already had 3 or more publicly available genomes. The sampled 332 budding yeasts use three different genetic codes: 229 follow the universal genetic code and decode the CUG codon as leucine (Figure 2; Table S1); 5 in the genera Nakazawaea, Pachysolen, and Peterozyma, decode CUG codon as alanine (CUG-Ala clade); 94 in the families Debaryomycetaceae, Metschnikowiaceae, and Cephaloascaceae encode CUG codon as serine (CUG-Ser1 clade); and 4 in the families Ascoideaceae and Saccharomycopsidaceae also decode CUG codon as serine (CUG-Ser2 clade).
Yeast growth
Most yeast strains were obtained from the USDA Agricultural Research Service (ARS) NRRL Culture Collection in Peoria, Illinois, USA. Strains from all yeast species were initially plated from freezer stock on yeast extract peptone dextrose (YPD) plates and grown for single colonies. For all carbon and nitrogen testing, we set up 5 replicates on separate days using different colonies for each yeast species. Yeast strains from each species were inoculated into YPD and grown for 48 hours at room temperature. After 48 hours of growth, we randomized and arrayed species into a 96-well deep well plate for storage and future use. Using the same cultures, we also inoculated species into a minimal-based starvation medium containing 0.1% glucose, 5g/L ammonium sulfate, and 1.7g/L Yeast Nitrogen Base (w/o amino acids, ammonium sulfate, or carbon) and grew them overnight. After 24 hours of growth, we transferred the yeast species into carbon or nitrogen treatment plates for phenotyping.
METHOD DETAILS
Genome sequencing and assembly
For each of the 196 newly sequenced species from the Y1000+ Project (Table S1), genomic DNA (gDNA) was isolated using a modified phenol:chloroform extraction method that used a second round of phenol:chloroform to remove additional proteins, sonicated, and ligated to Illumina sequencing adaptors as previously described (Hittinger et al., 2010). The paired-end libraries were submitted for paired end sequencing (2 × 250 base pairs) on an Illumina HiSeq 2500 instrument.
Paired-end Illumina DNA sequence reads were used to generate whole genome assemblies using the meta-assembler pipeline iWGS v1.1 (Zhou et al., 2016). We first preprocessed the raw sequenced reads by trimming of adapters and low-quality bases with Trimmomatic v0.33 (Bolger et al., 2014) and Lighter v1.1.1 (Song et al., 2014). Next, we identified the optimal k-mer length for each genome’s assembly using KmerGenie v1.6982 (Chikhi and Medvedev, 2014). We then used the processed sequence reads as input into six different de novo assembly tools: ABYSS v1.5.2 (Simpson et al., 2009), DISCOVAR r51885 (Weisenfeld et al., 2014), MASURCA v2.3.2 (Zimin et al., 2013), SGA v0.10.13 (Simpson and Durbin, 2012), SOAPdenovo2 v2.04 (Luo et al., 2012), and SPADES v3.7.0 (Bankevich et al., 2012). The resulting genome assemblies were assessed for quality with QUAST v4.4 (Gurevich et al., 2013); as the “best” assembly for each genome we chose the one with the highest genome size and N50 value (i.e., the contig or scaffold value above which 50% of the total length of the sequence assembly can be found).
For the genomes of the 24 species sequenced by RIKEN (Table S1), a paired-end library with an approximate insert size of 240 bp was prepared from the genomic DNA using the TruSeq DNA PCR-free Library Preparation Kit (Illumina, Inc., San Diego, CA, USA) according to the manufacturer’s protocols. A mate-pair library with an approximate insert size of 3 kb was also prepared from DNA using the Nextera Mate Pair Sample Preparation Kit (Illumina, Inc.) with some modifications (Park et al., 2013). Both paired-end and mate-pair libraries were sequenced on a HiSeq 2500 (Illumina, Inc.) to generate 151-base paired-end reads. Mate-pair libraries derived from the DNA of Hyphopichia homilentoma JCM 1507, Candida sorboxylosa JCM 1536, Candida intermedia JCM 1607, Wickerhamia fluorescens JCM 1821, Cyberlindnera fabianii JCM 3601, Saccharomycopsis malanga JCM 7620, Candida carpophila JCM 9396, Candida succiphila JCM 9445, Wickerhamiella domercqiae JCM 9478, Sporopachydermia quercuum JCM 9486, Starmerella bombicola JCM 9596, Candida boidinii JCM 9604, Nakazawaeapeltata JCM 9829, Scheffersomyces lignosus JCM 9837, and Ambrosiozyma kashinagacola JCM 15019 were sequenced on a MiSeq (Illumina, Inc.) to generate 309-base paired-end reads. All mate-pair reads were processed using NextClip software, v0.8 (Leggett et al., 2014) to trim the adapter sequences. The estimated sequencing depths ranged from 76* for Yarrowia deformans JCM 1694 to 281* for Wickerhamiella versatilis JCM 5958. The ALLPATHS-LG software version 51828 (for Cyberlindnera fabianii JCM 3601, Wickerhamiella domercqiae JCM 9478, and Starmerella bombicola JCM 9596), version 52155 (for Hyphopichia homilentoma JCM 1507, Candida sorboxylosa JCM 1536, Candida intermedia JCM 1607, Priceomyces haplophilus JCM 1635, Yarrowia deformans JCM 1694, Wickerhamia fluorescens JCM 1821, Ambrosiozyma monospora JCM 7599, Saccharomycopsis malanga JCM 7620, Candida carpophila JCM 9396, Candida succiphila JCM 9445, Sporopachydermia quercuum JCM 9486, Candida boidinii JCM 9604, Nakazawaea peltata JCM 9829, Scheffersomyces lignosus JCM 9837, Yarrowia keelungensis JCM 14894, and Ambrosiozyma kashinagacola JCM 15019), and version 52488 (for Wickerhamiella versatilis JCM 5958, Ascoidea asiatica JCM 7603, Alloascoidea hylecoeti JCM 7604, Ogataea methanolica JCM 10240, and Millerozyma acaciae JCM 10732) were used to assemble the reads into scaffolds using the default parameters (Gnerre et al., 2011). Small contigs (<1 kb) were not included in the final genome assemblies. The sequence library generation and sequencing were performed at the Genome Network Analysis Support Facility, RIKEN Center for Life Science Technologies (Yokohama, Japan).
Assessment of genome assemblies
The qualities of the genome assemblies of the 196 newly sequenced (Y1000+ Project genomes) and 136 publicly available (RIKEN plus previously published genomes) budding yeast species were assessed by quantifying their completeness based on the expected gene content of the Benchmarking Universal Single-Copy Orthologs (BUSCO), version 2.0.1 (Waterhouse et al., 2018), as described previously (Shen et al., 2016a). We used a set of 1,759 BUSCO genes inferred to be saccharomyceta-specific (“saccharomyceta” is an informal taxonomic rank used by many databases, including NCBI: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Info&id=716545) and single-copy in at least 90% of 60 genomes in the OrthoDB Version 9 database (Zdobnov et al., 2017) to evaluate the completeness of the 332 genome assemblies. Briefly, for each BUSCO gene, its consensus orthologous protein sequence among the 60 reference genomes was used as query in a tBLASTn search against each genome to identify up to three putative genomic regions, and the gene structure of each putative genomic region was predicted by AUGUSTUS v 3.2.2 (Stanke and Waack, 2003). Next, the sequences of these predicted genes were aligned to the HMM-profile of the BUSCO gene, and the ones with alignment bit-scores higher than a pre-set cutoff (90% of the lowest bit-score among the 60 reference genomes) were kept. If only one predicted gene from a genome was retained and its aligned sequence length in the HMM-profile alignment was > 95% of the aligned sequence lengths of genes in the 60 reference genomes, the BUSCO gene was classified to be present in the genome examined as single-copy, “full-length”. If two or more predicted genes from a genome were retained and their aligned sequence lengths in the HMM-profile alignments were > 95% of the aligned sequence lengths of genes in the 60 reference genomes, the BUSCO gene was classified as duplicated, “full-length”. If one or more predicted genes from a genome were retained but their aligned sequence lengths in the HMM-profile alignment were < 95% of the aligned sequence lengths of genes in the 60 reference genomes, the BUSCO gene was classified as “fragmented”. If no predicted gene from a genome was retained, the BUSCO gene was classified as “missing”. For each genome, we then calculated the fractions of single-copy (full-length) genes, duplicated (full-length) genes, fragmented genes, and missing genes, which in turn provided us with a measure of the completeness of gene content in each of the 332 genomes (Shen et al., 2016a).
Genome annotation
With the exception of Saccharomyces cerevisiae and Candida albicans, whose genome annotations are of exceptionally high quality, for consistency we annotated all the other 330 budding yeast genomes analyzed in our study using the MAKER genome annotation pipeline v2.31.8 (Holt and Yandell, 2011). Genome annotation using MAKER occurs in an iterative manner and relies on multiple inputs, some of which were universal to all genomes (e.g., homology evidence), whereas the others were species-specific (e.g., parameters for ab initio gene predictors). The procedure described below was followed for the annotation of all 330 genomes.
The homology evidence used in our genome annotation consists of fungal protein sequences in the SwissProt database (release 2016_11;
ftp://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2016_11/knowledgebase/uniprot_sprot-only2016_11.tar.gz) and annotated protein sequences of select yeast species from Mycocosm (Grigoriev et al., 2014), a web portal developed by the US Department of Energy Joint Genome Institute for fungal genomic analyses. Three ab initio gene predictors were used with the MAKER pipeline, including GeneMark-ES v4.32 (Ter-Hovhannisyan et al., 2008), SNAP v2013–11-29 (Korf, 2004), and AUGUSTUS v 3.2.2 (Stanke and Waack, 2003), each of which was trained for each individual genome. For GeneMark-ES, repeats in the genome sequence were first soft-masked using RepeatMasker v4.0.6 (http://www.repeatmasker.org) with the library Repbase library release-20160829 and the “-species” parameter set to “saccharomycotina” for all genomes. GeneMark-ES was then trained on the masked genome sequence using the self-training option (“--ES”) and the branch model algorithm (“--fugus”), which is optimal for fungal genome annotation. On the other hand, the training of both SNAP and AUGUSTUS requires pre-existing gene models as training data. Therefore, we carried out an initial MAKER analysis where gene annotations were generated directly from homology evidence (the “protein2genome” option in the “maker_opts.ctl” control file was set to 1) without using any ab initio gene predictors. The resulting gene annotations supported by homology evidence (“keep_preds” set to 0) were then used to train SNAP and AUGUSTUS.
Once all three ab initio gene predictors were trained, they were used together with homology evidence to conduct a first round of full MAKER analysis; it should be noted that, here, the homology evidence was only used to inform gene predictions (“protein2genome” set to 0). Resulting gene models supported by homology evidence (“keep_preds” set to 0) were used to re-train SNAP and AUGUSTUS. A second round of MAKER analysis was conducted using the newly trained SNAP and AUGUSTUS parameters, and once again the resulting gene models with homology supports were used to re-train SNAP and AUGUSTUS. Finally, a third round of MAKER analysis was performed using the new SNAP and AUGUSTUS parameters. All resulting gene models were reported (“keep_preds” set to 1), and these comprise the final set of annotations for the genome.
Thirty-nine out of the 136 published genomes analyzed in our study have previously been annotated. To evaluate the quality of our genome annotations, we performed direct comparisons between our annotations and existing ones for the same species. Genome annotations of 39 budding yeast species were downloaded from their respective sources and compared to the corresponding annotations generated in our study using Cuffcompare v2.2.1 (Trapnell et al., 2012). Using the existing annotations as the reference, our annotations achieved high levels of specificity (ranging from 92% to 99.2%, with an average of 97.3%) and sensitivity (ranging from 85.3% to 99.3%, with an average of 94.9%) at the base pair level, as well as contained few missing exons (the fraction of exons missing ranged from 2.7% to 15%, with an average of 4.8%) and genes (the fraction of genes missing ranged from 1.1% to 11.9%, with an average of 3.9%) across the 39 budding yeast species. Note that all of these calculations assume that the previously published annotations contain no errors, which might inflate estimates of inaccuracies in our annotations.
Identification of potential hybrid species
Although we excluded the genomes of known hybrid species from our study, the possibility exists that some of the 332 budding yeast genomes are the products of hybridization (Hittinger et al., 2015). To test whether this was the case, for each of the 332 budding yeast genomes, we examined the genome-wide distribution of Ks (number of synonymous substitutions per synonymous site) values determined from pairs of orthologous genes identified in the species of interest and its closest relative in the budding yeast phylogeny depicted in Figure 2 (following Ortiz-Merino et al. 2017). To calculate the Ks distribution for a species of interest, for each gene in the species of interest we identified its closest homolog in the genome of their closest relative using BlastP with an e-value cut off of 10’10. The protein sequences of the gene pairs were then aligned using MAFFT, version 7.299 (Katoh and Standley, 2013), with options “--genafpair --maxiterate 1000”. DNA/codon alignments were then generated by threading the DNA sequence onto the protein alignment using Pal2Nal (Suyama et al., 2006). Using the DNA/codon alignments, Ks was calculated using the LWL85m method implemented in the YN00 module of paml4 (Yang, 2007).
In known hybrid species, such as Saccharomyces pastorianus and Zygosaccharomyces parabailii (Dunn and Sherlock, 2008; Ortiz-Merino et al., 2017), the genome-wide distribution of Ks values is bimodal, reflecting the fact that some genes in the hybrid genome are most closely related to one parental species and some to the other more divergent parent. In contrast, non-hybrid species have a unimodal Ks distribution, reflecting the fact that genes originated from a single parental species. We visually inspected the resulting Ks distributions and found that the newly sequenced genomes of Citeromyces siamensis and Martiniozyma abiesophila had bimodal distributions of Ks similar to those of S. pastorianus and Z. parabailii (Dunn and Sherlock, 2008; Ortiz-Merino et al., 2017), suggesting that Ci. siamensis and Ma. abiesophila are of potentially hybrid origin. Consistent with this hypothesis, Ci. siamensis and Ma. abiesophila also had the highest numbers of predicted genes (12,786 and 12,589, respectively), the highest numbers of gene duplicates (24.1% and 23.6%, respectively), and the highest genome sizes (24.8 Mb and 14.5 Mb, respectively) in our data set (Table S1). Both hybrid taxa were not associated with any of 32 incongruent intemodes (Figure 2).
Phylogenomic data matrix construction
We generated two complete data matrices (2408OG data matrix and 1292BUSCO data matrix) from the genomes of 332 budding yeasts and 11 outgroups, as well as 7 additional data matrices by subsampling subsets of the orthologous groups of genes (OGs) in the 2408OG data matrix.
a) 2408OG data matrix
The 2408OG orthologous group data matrix was constructed based on a 5-step workflow. In step 1, we used all protein sequences of the 2,012,541 genes present in the 332 budding yeasts and 11 outgroups to perform an all-versus-all search using BLASTP with an e-value cutoff of 10-10. We then used the BLASTP results to cluster homologous protein sequences using the Markov Cluster (MCL) algorithm implemented in OrthoMCL, version 2.0 (Li et al., 2003); we adopted the widely used inflation parameter of 1.5 for two reasons. First, this inflation parameter value was found to be the optimal one in a previous evaluation of the effects of different inflation parameter values on orthology assignment in the budding yeasts (Salichos and Rokas, 2011). Second, our examination of a range of different inflation parameter values (from 1.2 to 2.0, with a step of 0.1) showed that all values led to the generation of nearly sets of clusters of homologous genes. OrthoMCL clustering resulted in the identification of 171,715 singleton clusters that contain a single protein and 61,763 clusters that contain two or more proteins. Plotting of the distributions of the lengths of genomic contigs that contain the 171,715 singleton clusters and 61,763 clusters with two or more proteins showed that the two distributions were very similar. Retaining only those clusters with gene occupancy ≥ 50%, that is those clusters that were present in at least half (≥ 172) of the 343 genomes (332 budding yeasts and 11 outgroups), resulted in the identification of 4,036 putative OGs.
In step 2, we inspected these 4,036 putative OGs for the presence of two or more sequences (i.e., paralogous sequences) from a taxon. For each putative OG, we first aligned its protein sequences using the program MAFFT, version 7.299 (Katoh and Standley, 2013), with the parameters “--auto” and “--maxiterate 1000” and removed columns whose site occupancy was less than 0.01 from the resultant alignment using the program Phyutility, version 2.2.6 (Smith and Dunn, 2008), with the parameters “-aa” and “-clean 0.01”. We then used each trimmed OG alignment to reconstruct a quick but “approximate” maximum likelihood (ML) tree using the program FastTree, version 2.1.9 (Price et al., 2010), with the LG model of amino acid substitutions (Le and Gascuel, 2008), a discrete gamma approximation with 20 categories (-gamma), 4 rounds of minimum-evolution subtree-prune-regraft moves (-spr 4), and the more exhaustive ML nearest-neighbor interchange option enabled (-mlacc 2 -slownni). Whenever there were 2 or more protein sequences from a specific taxon in a given OG, we identified the best (i.e., putatively orthologous) one by using a tree-based method (maximally inclusive subtree) implemented in PhyloTreePruner, version 1.0 with a minimum internal support value of 0.95 (Kocot et al., 2013). This resulted in the retention of 2,908 OGs with gene occupancy > 50%.
In step 3, we performed multiple sequence alignment for each of the 2,908 OGs using the E-INS-i strategy (--genafpair --maxiterate 1000) as implemented by the program MAFFT, version 7.299 (Katoh and Standley, 2013), and excluded ambiguously aligned regions using trimAl v1.4 with the “gappyout” option on (Capella-Gutierrez et al., 2009). We then examined all the resulting alignments and removed protein sequences whose lengths were shorter than 50% the length of the trimmed multiple sequence alignment length of the OG to which they belonged. We also removed OGs whose total trimmed multiple sequence alignment length was <167 amino acid sites. These filters resulted in the retention of 2,424 OGs, each of which had > 50% gene occupancy and > 167 amino acid site alignment length.
In step 4, to minimize the inclusion of potentially spurious sequences, we inferred an ML phylogram for each OG using IQ-TREE 1.5.1 (Nguyen et al., 2015) with an automatic detection for the best-fitting model of amino acid evolution and then used the ML phylogram to identify and to remove all protein sequences that resulted in terminal branch lengths that were at least 20-times longer than the median of all terminal branch lengths across the phylogram. This step led to the removal of 421 potentially spurious sequences from 292 OGs; the remaining 2,132 OGs did not contain any spurious sequences.
In the final step (step 5), we redid mutiple sequence alignment and trimming for those 292 OGs that we removed as spurious sequences as part of step 4. From the 292 OGs, 276 OGs retained ≥ 50% gene occupancy and ≥ 167 amino acid site alignment length and, thus, were kept; the remaining 16 were discarded.
Retention of the 2,132 OGs from step 4 and the 276 OGs from step 5 yielded a final data matrix consisting of 2,408 orthologous groups (OGs) (1,162,805 amino acid sites) of genes from the 332 budding yeast taxa and 11 outgroups.
b) 1292BUSCO data matrix
As BUSCO genes are a set of reliable markers for phylogenomic inference of diverse lineages (Waterhouse et al., 2018), including the budding yeasts (Shen et al., 2016a), we used 1,757 / 1,759 single-copy, full-length BUSCO genes from 332 budding yeasts and 11 outgroups to construct a data matrix (the EOG09344D43 and EOG09344ST8 BUSCO genes were excluded because we were unable to consistently recover them from our genomes). The number of BUSCO genes whose protein sequences are all present in the same orthologous group (OG) identified by OrthoMCL with the inflation value of 1.5 is 1,650 (~94% out of 1,757). Multiple sequence alignment; trimming of ambiguously aligned regions; removal of short, spurious, or paralogous sequences; and filtering for ≥ 50% gene occupancy and ≥ 167 amino acid site alignment length were done in the same way as described above for the 2408OG data matrix. Application of these filters resulted in a data matrix of 1,292 BUSCO genes (527,069 amino acid sites), each of which had ≥ 50% gene occupancy and ≥ 167 amino acid site alignment length.
c) 7 additional subsampled data matrices
To explore the stability of phylogenetic relationships among the 332 budding yeasts, we constructed 7 additional data matrices by subsampling subsets of OGs from the 2408OG data matrix as follows:
1. OG2BUSCO data matrix: This data matrix was constructed by retaining only those OGs that were present in both the 2408OG data matrix and the 1292BUSCO data matrix. Overlapping OGs between the two data matrices were determined by BLASTP (e-value cutoff of 10−10). Briefly, a query 2408OG sequence was considered to be overlapping to a subject 1292BUSCO sequence only if they were from the same taxon and were at least 95% similar at the protein sequence level. OGs between the two data matrices were considered overlapping only if all their sequences were overlapping, resulting in a data matrix of 1,081 OGs (545,300 amino acid sites), each of which had ≥ 50% gene occupancy and ≥ 167 amino acid site alignment length.
2. OG2PHYLOME data matrix: This data matrix was constructed by retaining only those OGs that were present in both the 2408OG data matrix and the phylomeDB, version 4 (Huerta-Cepas et al., 2014). First, 1,838 orthologs from the S. cerevisiae phylome (P21) containing at least 11 taxa were selected. The S. cerevisiae phylome was curated using 21 budding yeast genomes (8 are representatives from the CUG-Ser1 clade; 12 are representatives from the Saccharomycetaceae; 1 is a representative from the Dipodascaceae/Trichomonascaceae clade). Overlap between the two sets of OGs was determined in the same way as described above for the OG2BUSCO data matrix, resulting in a data matrix of 819 OGs (317,158 amino acid sites), each of which had ≥ 50% gene occupancy and ≥ 167 amino acid site alignment length.
3. Top500_ABS data matrix: This data matrix was constructed by retaining the 500 OGs (from the 2408OG data matrix) with the highest average bootstrap support (ABS) value of all internal branches on the ML gene tree (Shen et al., 2016b) and contains 472,241 amino acid sites.
4. Top500_completeness data matrix: This data matrix was constructed by retaining the 500 OGs (from the 2408OG data matrix) with the highest gene occupancy (Shen et al., 2016b) and contains 295,429 amino acid sites.
5. Top500_ informativeness data matrix: This data matrix was constructed by retaining the 500 OGs (from the 2408OG data matrix) with the highest information content and contains 174,183 amino acid sites. Information content was calculated based on quartet-mapping (Nieselt-Struwe and von Haeseler, 2001) implemented in MARE, version 0.1.2 (http://mare.zfmk.de), with default settings.
6. Top500_length data matrix: This data matrix was constructed by retaining the 500 OGs (from the 2408OG data matrix) with the longest trimmed multiple sequence alignment lengths (Shen et al., 2016b), and contains 494,658 amino acid sites.
7. Top500_treeness2RCFV data matrix: This data matrix was constructed by retaining the 500 OGs (from the 2408OG data matrix) with the highest ratio of treeness to relative composition frequency variability (RCFV) (Phillips and Penny, 2003; Shen et al., 2016b), and contains 328,534 amino acid sites. Treeness is defined as the ratio (sum of lengths of all internal branches) / (total tree length) (Phillips and Penny, 2003; Shen et al., 2016b). RCFV (Phillips and Penny, 2003) is defined as:
Where s is the number of the character states (here the s is 20 for amino acids) and t is the number of taxa in a given trimmed OG alignment. Fy is the frequency of state i for the yth taxon, and is the average frequency of state i across t taxa.
Phylogenetic analyses
For each of these 9 data matrices, we inferred individual gene trees, as well as three estimates of the species phylogeny; two species phylogeny estimates were obtained by concatenation (concatenation under a single partition and concatenation under gene-based partitioning) (Rokas et al., 2003) and one by coalescence (Mirarab and Warnow, 2015).
Individual gene tree inference:
Individual gene trees were reconstructed using maximum likelihood (ML) analysis. For each gene, we conducted 10 independent tree searches (5 used starting trees inferred by parsimony and the other 5 used random starting trees) to obtain the best-scoring ML tree using RAxML, multithread version 8.2.3 (Stamatakis, 2014), under the best-fitting model of amino acid substitution selected by the IQ-TREE program (option -m TEST -mrate G4) with the Bayesian information criterion (BIC).
Concatenation-based ML species tree inference:
Our recent evaluation of 19 empirical phylogenomic data matrices showed that IQ-TREE and RAxML/ExaML typically recovered the ML trees with the highest-observed likelihood scores (Zhou et al., 2018). Moreover, our pilot concatenation-based ML tree inferences under a single “LG (Le and Gascuel, 2008) +G4 (Yang, 1996)” model of amino acid substitutions using the multi-threaded version of IQ-TREE v1.5.1 (Nguyen et al., 2015) and the MPI parallel version of ExaML v3.0.17 (Kozlov et al., 2015) for the 2408OG and 1292BUSCO data matrices, showed that IQ-TREE and ExaML produced topologically identical ML trees with very similar likelihood scores, but IQ-TREE did so faster (IQ-TREE: ~3,000 CPU hours / single tree search; ExaML: ~4,200 CPU hours / single tree search). Therefore, we adopted the program IQ-TREE to infer concatenation-based ML trees under a single partition (LG+G4) and gene-based partitions (i.e., model parameters were unlinked across genes with the -q option in IQ-TREE), respectively. Branch support for each internode in the ML tree was evaluated with 100 rapid bootstrapping replicates using RAxML, hybrid version 8.2.3 (Stamatakis, 2014) with the CAT model with 25 categories instead of G4 model.
Coalescence-based species tree inference:
For each data matrix, we used the set of individual ML gene trees (see section on individual gene tree inference above) to infer the coalescence-based phylogeny with ASTRAL-II, version 4.10.2 (Mirarab and Wamow, 2015). This is a summary species tree method that aims to account for individual gene tree heterogeneity due to incomplete lineage sorting (ILS). The reliability of each internal branch in the coalescence-based species tree was evaluated using the local posterior probability (LPP) measure.
Quantification of incongruence:
For each of 27 species phylogenies inferred from the 9 data matrices (2 original data matrices + 7 subsampled data matrices) under the three different approaches (two concatenation-based and one coalescence-based), we used internode certainty (ICA) to quantify the degree of incongruence for every internode by considering all prevalent conflicting bipartitions among individual ML gene trees (Salichos and Rokas, 2013). The (partial) internode certainty (ICA) values were calculated as implemented in RAxML, multithread version 8.2.3 (option -f i).
Molecular dating
We used the Bayesian method MCMCTree in the paml4.9e package (Yang, 2007) to estimate divergence times among the 332 budding yeasts using the 2408OG data matrix. The input tree was derived from the concatenation-based ML analysis under a single LG+G4 model (Figure 2). Since budding yeasts lack a reliable fossil record, we adopted four well-estimated ranges of divergence from four internodes of the budding yeast phylogeny as our calibrations (Kumar et al., 2017; Marcet-Houben and Gabaldon, 2015). These were: the Saccharomyces cerevisiae -Saccharomyces uvarum split (lower bound: 14.3 MYA - upper bound: 17.94 MYA), the Saccharomyces cerevisiae - Kluyveromyces lactis split (103 MYA - 126 MYA), the Saccharomyces cerevisiae - Candida albicans split (161 MYA - 447 MYA), and the origin of the subphylum Saccharomycotina (304 MYA −590 MYA).
Because Bayesian molecular dating is computationally intractable for data matrices that contain hundreds of species and thousands of genes (Irisarri et al., 2017), such as the 2408OG one, we created 50 replicate data matrices, each of which comprised of a different, randomly chosen subset of 100 OGs, to infer the budding yeast timetree (inference of each of the replicates took ~500 CPU hours). For each replicate, we first estimated branch lengths under a single LG+G4 model with codeml in the paml4.9 e package (Yang, 2007) and obtained a rough mean of the overall mutation rate. Next, we applied the approximate likelihood method (dos Reis and Yang, 2011) to estimate the gradient vector and Hessian matrix with Taylor expansion (option usedata=3). Last, we assigned (a) the gamma-Dirichlet prior for the overall substitution rate (option rgene_gamma) as G(1, 12.5), with a mean of 0.08 (meaning 8×10−10 amino acid substitutions per site per year), (b) the gamma-Dirichlet prior for the rate-drift parameter (option sigma2 gamma) as G(1, 10), and (c) the parameters for the birth-death sampling process with birth and death rates X=p=1 and sampling fraction p=0. We employed the autocorrelated-rate model (option clock=3) to account for the rate variation across different lineages and used soft bounds (left and right tail probabilities equal 0.025) to set minimum and maximum values for the four calibration splits mentioned above. The MCMC run was first run for 100,000 iterations as burn-in, then sampled every 500 iterations until a total of 3,000 samples was collected. Lastly, the divergence time estimate for each internal branch was calculated as the average across the timetrees produced by the 50 replicates.
In addition to the Bayesian MCMCTree method, we also used the non-Bayesian RelTime method, as implemented in the command line version of MEGA7 (Kumar et al., 2016). As RelTime is computationally much less demanding than MCMCTree (Mello et al., 2017), we conducted divergence time estimation using the complete 2408OG data matrix. We used the same ML tree and calibrations as we did for the MCMCTree analysis (see above). Comparison of divergence time estimates between RelTime and MCMCTree revealed that they were broadly consistent (Pearson’s correlation coefficient r = 0.87, P-value < 2.2e-16; average time deviation = ~ 19.5%), which is in agreement with a previously published comparison of the two methods based on analyses of 8 empirical phylogenomic data matrices (Mello et al., 2017). Furthermore, both our study and that of Mello and colleagues (Mello et al., 2017) found that the RelTime estimates were generally older than MCMCTree estimates for the deep internodes of the budding yeast phylogeny (e.g., for the internodes between 12 major clades) and were generally younger than the MCMCTree estimates for shallower internodes (e.g., within the families Pichiaceae, Saccharomycodaceae, Saccharomycetaceae, Phaffomycetaceae, the CUG-Ala clade, and the CUG-Ser1 clade).
Horizontal gene transfer (HGT) analyses
Identification of HGT events:
To detect genes in budding yeast genomes that may have been horizontally acquired from non-fungal organisms, we employed a robust and conservative phylogeny-based approach (Husnik and McCutcheon, 2017; Marcet-Houben and Gabaldon, 2010; Richards et al., 2011; Wisecaver et al., 2016). Briefly, for a given budding yeast gene, we inferred it to have been acquired by HGT if there was substantial topological disagreement between the gene tree and its associated species tree and the budding yeast gene sequence was robustly nested within the donor lineage in the gene tree.
To avoid spurious results due to the presence of small genomic fragments of contaminant organisms in our genome assemblies (Schonknecht et al., 2014), we limited our analyses to those genes that resided in genomic contigs or scaffolds that were ≥ 100 kb. This filter resulted in the analysis of 1,538,912 predicted genes (out of a total of 1,892,694 genes) in 329 yeast genomes. The remaining three genomes (Botryozyma nematodophila, Blastobotrys nivea, and Citeromyces siamensis) did not contain genomic contigs that were ≥ 100 kb).
For each gene, we evaluated whether it had been horizontally acquired using a two-step workflow.
In step 1, we first carried out a BLASTP search against a custom database (nr+) consisting of the NCBI non-redundant (nr) protein database (last accessed January 20, 2017) and all predicted protein sequences from 329 yeasts genomes, with an e-value cutoff of 10’10. We next used custom Perl scripts to: (a) assign taxonomic information to each BLAST hit rating with the dump files downloaded from the NCBI Taxonomy database (nodes.dmp, merged.dmp, and names.dmp; ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomv). and then (b) parse the BLAST hits. based on their taxonomic information. into three different lineages (RECIPIENT: Saccharomycotina; GROUP: Fungi; OUTGROUP: non-fungal) so as to obtain three values: bbhO (BLAST bitscore of the best hit in OUTGROUP lineage). bbhG (bitscore of the best hit in GROUP lineage but not in RECIPIENT lineage). and maxB (bitscore of the query to itself).
Using this information. we next calculated: (a) the Alien Index (AI) value (Wisecaver et al.. 2016). which is a normalized difference of bitscore between the best hit in OUTGROUP lineage and the best hit in GROUP lineage but not in RECIPIENT lineage: and (b) the percentage of species from OUTGROUP lineage (outg_pct) in the list of the top 300 hits that have different taxonomic species names (so that we avoid over-representation of multiple strains of the same species) (Marcet-Houben and Gabaldon. 2010). From the 1.538.912 genes analyzed. we found 1.806 genes with AI value ≥ 0.1 and outg_pct ≥ 90%.
In step 2. we retrieved the 300 most similar homologs that have different taxonomic species names from the nr+ database (see above). aligned them by the MAFFT. version 7.299 (Katoh and Standley, 2013), with “--auto” option, and trimmed ambiguously aligned regions using trimAl v1.4 (Capella-Gutierrez et al., 2009) with “-automated1” option. We then used the resulting alignment to infer the ML tree using IQ-TREE 1.5.1 (Nguyen et al., 2015) with its best-fitting model of amino acid evolution and 1000 ultrafast bootstrapping replicates (Minh et al., 2013). Lastly, we rooted each ML tree at the midpoint using the ape and phangorn R packages and visualized it using the command version of iTOL v3 (Letunic and Bork, 2016). After manually inspecting all 1,806 ML trees, we identified 878 genes in 186 / 329 budding yeast genomes whose phylogenies indicated they were putatively acquired via HGT. A summary table file that contains species name, gene name, genomic contig ID, genomic contig length, HGT status, and Gene Ontology (GO) term for each of 1,538,912 genes in 329 yeast genomes that we examined has been deposited on the Figshare data repository.
Analysis of HGT-acquired genes:
For each of 878 genes inferred to have been horizontally transferred into budding yeast genomes, we used the gene sequence and taxon names of their closest relatives on the ML tree to infer the gene name, gene function, and likely donor lineage of the HGT-acquired gene (Table S3). To examine the biological processes, cellular components, and molecular functions that these 878 HGT-acquired genes are associated with, we conducted gene ontology (GO) enrichment analysis using topGO 2.28.0 (Alexa and Rahnenfuhrer, 2016). We found that these genes were significantly enriched in metabolism-related terms, such as metabolic process, oxidation-reduction process, carbohydrate metabolic process (terms in Biological process), beta-galactosidase complex, integral component of membrane (terms in Cellular component), acetyltransferase activity, and catalytic activity (terms in Molecular function) (Figure S5). In the list of 878 putative HGT-acquired genes (Table S3), the largest number of horizontally acquired genes, 169, was found in the genome of W. versatilis, one of the six representative species from the W/S clade (Figure 3A). Transcriptome data in NCBI (SRA accession numbers: SRR5942408, SRR5942407, SRR5942426, SRR5942425, SRR5942428, SRR5942427, SRR5942422, SRR5942421) showed that at least 91 of its 169 horizontally acquired genes had expression values > 5 FPKM (Fragments Per Kilobase per Million mapped fragments), suggesting that most are likely functional. Gene Ontology (GO) enrichment analysis of 169 HGT-acquired genes in the genome of W. versatilis show they are significantly enriched in GO term functions and processes, such as oxidation-reduction process, metabolic process, catalytic activity, cofactor binding, and oxidoreductase activity.
Examination of the 878 HGT-acquired gene phylogenies showed that they stem from 365 distinct HGT events, 230 species-specific ones and 135 that involve two or more species (Figure S4; Table S3). The average age of these 365 HGT events was 66.3 MYA (95% credibility interval: 56.6–76.0), the average protein sequence identity between the HGT-acquired yeast gene and the most closely related non-fungal donor gene was 58%, and the average percentage of descendent species that retained a given HGT-acquired gene was 40.7%. These results suggest that most HGT events tend to affect one or a few species, they are relatively ancient or divergent, and are frequently lost.
Finally, the examination of the genomic locations of the 878 HGT-acquired genes showed that 77 genes were physically linked in 13 contiguous clusters of 3 or more genes each. Of these 13 horizontally acquired gene clusters, 4 were shared by several budding yeast species, while the other 9 were species-specific.
Robustness of HGT inference:
Inference of HGT can be noise-prone (Husnik and McCutcheon, 2017; Martin, 2018; Roger, 2018). To gauge the robustness of inference of HGT-acquired genes, we performed five additional sets of analyses.
First, we compared the list of 878 putative HGT-acquired genes to the list of 30 previously identified instances of HGT in budding yeasts. We found that 21 / 30 (70%) of these previously detected instances of HGT were included in our set of 878 HGT-acquired genes, corroborating the sensitivity and conservativeness of our high-throughput approach for identifying HGT (Table S3).
Second, to gauge whether setting the values of AI value ≥ 0.1 and outg_pct ≥ 90% in step 1 was too strict, we also examined the top 1,806 candidates with the highest AI scores for outg_pct ≥ 10%. Examination of the overlap between the set of 1,806 genes recovered with AI value ≥ 0.1 and outg_pct ≥ 90% and the set of the top 1,806 genes recovered with outg_pct ≥ 10% showed that that 605 / 878 strongly supported cases of HGT were present in both sets.
Third, we examined diverse properties of contigs containing HGT-acquired genes and compared to those of contigs lacking HGT-acquired genes. Examination of the distribution of sequence lengths of the 480 genomic contigs that contain 878 HGT-acquired genes, alongside the 8,312 genomic contigs that do not contain any HGT-acquired genes, showed that contigs containing the HGT-acquired genes were typically longer than contigs that lack them. We further used different cutoff values of contig length (from ⩾ 100 kb to ⩾ 1,500 kb with steps of 100 kb) to examine the percentage of HGT-acquired genes over total number of gene examined and found that it slightly increased with contig size.
Fourth, we compared the percentages of HGT-acquired genes in the 220 newly sequenced genomes and in the 112 publicly available genomes and found them to be similar. In contrast, the percentage of HGT-acquired genes in the 164 genomes with the highest N50 values was significantly higher than the percentage in the 165 genomes the lowest N50 values.
Finally, to test whether the maximum likelihood (ML) gene tree (i.e., the tree with the highest ML score, which shows that the horizontally acquired yeast gene was nested within a clade of non-fungal donor genes) was statistically different from a constrained ML tree in which all fungal and yeast genes were forced to be monophyletic, we applied the approximately unbiased (AU) (Shimodaira, 2002) test in the software package CONSEL (Shimodaira and Hasegawa, 2001), version 0.20. We found that 616 out of 878 putative HGT-acquired genes are significantly supported with the AU test (AU test; p-values <0.05) (Table S3).
Analyses of trait evolution
In this study, we reconstructed the evolution of 45 discrete metabolic traits in 274 budding yeasts representing the 12 major clades. All metabolic trait data were obtained from The Yeasts: A Taxonomic Study and pertain to budding yeasts’ abilities to grow on different substrates (Kurtzman et al., 2011).
For each discrete metabolic trait, we scored each taxon for its ability to grow (hereafter scored as “1”), not grow (hereafter scored as “0”), show variation in growth / absence of growth across different strains (hereafter scored as “v”), or lack of information / missing data (hereafter scored as “n”) (Table S4).
To conduct analyses of trait evolution and ancestral state inference, we used Bayes MultiState module in the BayesTraits, version 3 (Pagel et al., 2004), because of its ability to apply reversible jump MCMC (rjMCMC) to optimize model uncertainty with different parameters. To infer ancestral states and the rates of trait gain and loss, BayesTraits takes as input the species phylogeny, the trait states, and a model of trait evolution. For the input species phylogeny, we used the ML tree with branch lengths inferred from the 2408OG data matrix using a single LG+G4 model (Figure 2), which was then pruned to keep the 274 budding yeast species for which there were metabolic trait data.
For each trait, v and n values were recoded to be “01” (this means that the character’s state can be either “0” or “1”) and “-” in the BayesTraits analyses, respectively. We calculated the rate of gain (q01: rate of change from 0 to 1) and the rate of loss (q10: rate of change from 1 to 0) across the budding yeast phylogeny. To test whether the rates of trait gain and loss were significantly different or not, we compared estimates of marginal likelihoods under a model of trait evolution in which the rates of trait gain and loss were unconstrained against those obtained under a model in which the rates of gain and loss were constrained to be equal. We determined whether the two models were significantly different using log Bayes Factors (lnBF): 2(log marginal likelihood [unconstrained] -log marginal likelihood [constrained]); the rates of gain and loss for a given trait were considered to be significantly different when lnBF > 2.
For each metabolic trait, we also inferred the posterior probability of each of the two character states (0 and 1) at the root (i.e., the budding yeast common ancestor [BYCA]) and at each of 272 internodes across the phylogeny. Each analysis was run for 10 million generations, sampling parameters every 4,000 generations until 2,000 samples were collected. The stepping stone sampler was used to estimate marginal likelihoods, sampling 200 stones in which each stone was run 1,000 generations.
Finally, we plotted the kernel density of the posterior distribution for the rates of trait gain and loss, as well as posterior probabilities (PP) of states 0 and 1 at each intemode, and identified the largest peak values from their densities. To visualize the ancestral state at each internode across the budding yeast phylogeny, we used the pie char function in iTOL v3 (Letunic and Bork, 2016).
Validation of metabolic trait data
As the metabolic trait data in The Yeasts: A Taxonomic Study (Kurtzman et al., 2011) comes from multiple sources, contains missing data, and the strains tested sometimes differ from the sequenced strains, we also experimentally determined the growth for 328 / 332 strains of the budding yeast species whose genomes we used on 13 / 45 discrete metabolic traits, 11 carbon-based and 2 nitrogen-based (Table S4). As a control, we also experimentally determined yeast species growth on glucose, a substrate on which all sampled strains are known to grow (Kurtzman et al., 2011).
Carbon treatment plates contained a minimal media base with ammonium sulfate and one of twelve carbon sources at a 2% concentration (Kurtzman et al., 2011). The 12 carbon sources tested in our analyses included glucose, sucrose, raffinose, galactose, lactose, maltose, cellobiose, L-rhamnose, D-xylose, glycerol, D-glucosamine, and V-acetyl-D-glucosamine. Nitrogen treatment plates contained a minimal media base with either potassium nitrate or sodium nitrite as the nitrogen source and 2% glucose (Kurtzman et al., 2011). Nitrogen treatment plates required two rounds of growth to accurately measure growth of species; therefore, after a week of growth, we pinned the yeast species into a second round of the respective nitrogen source. After a week of growth, we visually scored yeast species for growth on each carbon or nitrogen source. We then compared growth across replicates for each species and treatment; a species was determined to grow on a carbon source if it showed growth ≥ 50% of the time across replicates. Our results show that the average consistency of the character states shared between the compilation of available metabolic trait data and our experimental measurements was 94% (Table S4), suggesting that our inferences are based on accurate data.
Trait and gene association network analyses
Calculations of positive associations among carbon and nitrogen assimilation traits were obtained from a recently published study (Opulente et al., 2018). A positive association network was created in the R package igraph v. 1.0.1 (Csardi and Nepusz, 2006), and trait communities in these networks were determined through the Clauset-Newman-Moore algorithm (fast.greedy community), an approach that determines communities by maximizing the total network’s modularity.
The input data matrix consisted of species-specific presence and absence data for functional annotations of genes, together with the species-specific qualitative information about their capabilities to grow in the conditions used in the ancestral trait reconstruction analyses. The functional annotations were Kyoto Encyclopedia of Genes and Genomes (KEGG) database entries or annotations (Kanehisa et al., 2016b) obtained through GhostKOALA (Kanehisa et al., 2016a). Mutual information coefficients were calculated for each pairwise combination of KEGG annotation and trait using the R packages infotheo, minet (Meyer, 2008; Meyer et al., 2008), and WCGNA (Langfelder and Horvath, 2008), and the resulting association network was trimmed with the ARACNE algorithm (Margolin et al., 2006). Strong associations were defined by a cutoff of 0.15 nats or higher, and weak associations were considered between 0.10 and 0.15 nats. The positive or negative character of the associations was determined by calculating the Jaccard index for each pairwise combination, with a cutoff at 0.25.
Data tracks shown on Figure 5 were subject to the following adjustments: (1) the nitrite growth track contained exclusively data from validation experiments performed in this study (Table S4), due to limited data in the literature (Table S4); (2) for the nitrate growth track, positive growth was imputed in cases of disagreement between the validation and literature; and (3) due to partial annotation redundancy for specific steps of the Moco biosynthesis pathway, MOCS1 and MOCS2 data tracks were collated from multiple annotations, with MOCS1 activity being considered present if either the K03637, K03639, or K20967 KEGG annotation was found in a genome and with MOCS2 activity being considered present if either the K03635, K03636, or K21232 KEGG annotation was found in a genome.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses and quantification approaches associated with genome sequencing, accuracy and completeness of assembly, accuracy and completeness of annotation, phylogenetic data matrix reconstruction, horizontal gene transfer (HGT) inference, and metabolic trait evolution can be found in the relevant sections of the Method Details.
The Fisher’s exact tests were conducted in R version 3.4.0.
DATA AND SOFTWARE AVAILABILITY
Raw sequence read and genome assembly data for all 220 new genomes have been deposited at DDBJ/ENA/GenBank and their accession numbers are provided in Table S1. All budding yeast genome assemblies and annotations, phylogenetic data matrices, multiple sequence alignments, phylogenetic trees, the metabolic trait data matrix, trait ancestral character state reconstructions, additional figures and tables, R codes, and the custom Perl scripts used in this study are available on the Figshare data repository (10.6084/m9.figshare.5854692; https://figshare.com/articles/Tempo_and_mode_of_genome_evolution_in_the_budding_yeast_subphylum/5854692). All sequenced strains have been publicly deposited in the NRRL, CBS, and/or JCM strain collections.Supplemental Excel titles and legends
Supplementary Material
Highlights.
332 genomes, including 220 newly sequenced, covering ~ 1/3 of known budding yeasts
Genome-scale inference of robust phylogeny and timetree of budding yeast subphylum
Reconstruction of 45 metabolic traits infers complex budding yeast common ancestor
Reductive evolution of traits and genes is a major mode of evolutionary diversification
Acknowledgments
We dedicate this manuscript to the memory of the late Cletus P. Kurtzman, a trailblazer of budding yeast taxonomy and systematics. We thank members of the Rokas and Hittinger labs, in particular Abbigail L. LaBella, for feedback and discussions on the project, Mario dos Reis, Qiqing Tao, and Sudhir Kumar for helpful suggestions with divergence time estimation analyses, and the yeast taxonomy community for depositing sequenced strains into culture collections. This work was supported in part by the National Science Foundation (DEB-1442113 to AR, DEB-1442148 to CTH and CPK, IOS-1401682 to JHW, and DGE-1256259 to QKL) and the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02–07ER64494 and DE-SC0018409 to Timothy J. Donohue). CTH is a Pew Scholar in the Biomedical Sciences and Vilas Faculty Early Career Investigator, supported by the Pew Charitable Trusts and Vilas Trust Estate, respectively. AR is supported by a Guggenheim fellowship, JLS by Vanderbilt’s Biological Sciences graduate program, XZ in part by the National Key Project for Basic Research of China (973 Program, No. 2015CB150600), DTD by a NHGRI training grant to the Genomic Sciences Training Program (5T32HG002760), and QKL by a National Institutes of Health Predoctoral Training Program (5T32GM007133). DL was supported by CONICET (PIP 392), FONCyT (PICT 2542), and Universidad Nacional del Comahue (B199). CAR was supported by the Conselho Nacional de Desenvolvimento Cíentifico e Tecnológico (CNPq-Brazil) and Fundaçáo de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG). This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, the Center for High-Throughput Computing at UW-Madison, Lucigen Corporation, the CIPRES Science Gateway, and the UW Biotechnology Center DNA Sequencing Facility. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albalat R, and Canestro C (2016). Evolution by gene loss. Nat. Rev. Genet 17, 379–391. [DOI] [PubMed] [Google Scholar]
- Alexa A, and Rahnenfuhrer J (2016). Gene set enrichment analysis with topGO. Http://Www.Bioconductor.Org/.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner F, Oreb M, and Mendel RR (2001). ABA3 is a molybdenum cofactor sulfurase required for activation of aldehyde oxidase and xanthine dehydrogenase in Arabidopsis thaliana. J. Biol. Chem 276, 40381–40384. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Rasmussen MD, Lin MF, Santos MAS, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, et al. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, and Gabaldon T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi R, and Medvedev P (2014). Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37. [DOI] [PubMed] [Google Scholar]
- Csárdi G, and Nepusz T (2006). The igraph software package for complex network research. InterJournal Complex Syst. 1695, 1–9. [Google Scholar]
- Dujon BA, and Louis EJ (2017). Genome Diversity and Evolution in the Budding Yeasts (Saccharomycotina). Genetics 206, 717–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, de Montigny J, Marck C, Neuveglise C, Talla E, et al. (2004). Genome evolution in yeasts. Nature 430, 35–44. [DOI] [PubMed] [Google Scholar]
- Dunn B, and Sherlock G (2008). Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 18, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, Martin T, Marcet-Houben M, Durrens P, Bolotin-Fukuhara M, Lespinet O, Arnaise S, Boisnard S, Aguileta G, Atanasova R, et al. (2013). Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium Génolevures, Souciet J-L, Dujon B, Gaillardin C, Johnston M, Baret PV, Cliften P, Sherman DJ, Weissenbach J, Westhof E, et al. (2009). Comparative genomics of protoploid Saccharomycetaceae. Genome Res. 19, 1696–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 108, 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonqalves C, Wisecaver JH, Kominek J, Oom MS, Leandro MJ, Shen X-X, Opulente DA, Zhou X, Peris D, Kurtzman CP, et al. (2018). Evidence for loss and reacquisition of alcoholic fermentation in a fructophilic yeast lineage. Elife 7, e33034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, et al. (2014). MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, and Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, and Dietrich FS (2007). The Reacquisition of Biotin Prototrophy in Saccharomyces cerevisiae Involved Horizontal Gene Transfer, Gene Duplication and Gene Clustering. Genetics 177, 2293–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT (2013). Saccharomyces diversity and evolution: a budding model genus. Trends Genet. 29, 309–317. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, and Carroll SB (2004). Parallel inactivation of multiple GAL pathway genes and ecological diversification in yeasts. Proc. Natl. Acad. Sci. USA 101, 14144–14149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Gonsalves P, Sampaio JP, Dover J, Johnston M, and Rokas A (2010). Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature 464, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Bai F-Y, Boekhout T, Gonsalves P, Jeffries TW, Kominek J, Lachance M-A, Libkind D, Rosa CA, et al. (2015). Genomics and the making of yeast biodiversity. Curr. Opin. Genet. Dev 35, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, and Yandell M (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Capella-Gutierrez S, Pryszcz LP, Marcet-Houben M, and Gabaldon T (2014). PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 42, D897–D902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnik F, and McCutcheon JP (2017). Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol 16, 67–79. [DOI] [PubMed] [Google Scholar]
- Irisarri I, Baurain D, Brinkmann H, Delsuc F, Sire J-Y, Kupfer A, Petersen J, Jarek M, Meyer A, Vences M, et al. (2017). Phylotranscriptomic consolidation of the jawed vertebrate timetree. Nat. Ecol. Evol 1, 1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, and Morishima K (2016a). BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol 428, 726–731. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, and Tanabe M (2016b). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katinka MD, Duprat S, Cornillot E, Metenier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, et al. (2001). Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414, 450–453. [DOI] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot KM, Citarella MR, Moroz LL, and Halanych KM (2013). PhyloTreePruner: A Phylogenetic Tree-Based Approach for Selection of Orthologous Sequences for Phylogenomics. Evol. Bioinform. Online 9, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I (2004). Gene finding in novel genomes. BMC Bioinformatics 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AM, Aberer AJ, and Stamatakis A (2015). ExaML version 3: a tool for phylogenomic analyses on supercomputers. Bioinformatics 31, 2577–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassowski T, Coughlan AY, Shen X-X, Zhou X, Kominek J, Opulente DA, Riley R, Grigoriev IV, Maheshwari N, Shields DC, et al. (2018). Evolutionary instability of CUG-Leu in the genetic code of budding yeasts. Nat. Commun 9, 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, and Hedges SB (2017). TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol 34, 1812–1819. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP, and Boekhout T (2017). Yeasts as Distinct Life Forms of Fungi In Yeasts in Natural Ecosystems: Ecology, Buzzini P, Lachance M-A, and Yurkov A, eds. (Cham: Springer International Publishing; ), pp. 1–37. [Google Scholar]
- Kurtzman CP, Fell JW, and Boekhout T (2011). The Yeasts: A Taxonomic Study (Elsevier Science; ). [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, and Gascuel O (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol 25, 1307–1320. [DOI] [PubMed] [Google Scholar]
- Leggett RM, Clavijo BJ, Clissold L, Clark MD, and Caccamo M (2014). NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, and Roos DS (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Houben M, and Gabaldon T (2010). Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 26, 5–8. [DOI] [PubMed] [Google Scholar]
- Marcet-Houben M, and Gabaldon T (2015). Beyond the Whole-Genome Duplication: Phylogenetic Evidence for an Ancient Interspecies Hybridization in the Baker’s Yeast Lineage. PLoS Biol. 13, e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera R, and Califano A (2006). ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinformatics 7, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WF (2018). Eukaryote lateral gene transfer is Lamarckian. Nat. Ecol. Evol 2, 754–754. [DOI] [PubMed] [Google Scholar]
- Mello B, Tao Q, Tamura K, and Kumar S (2017). Fast and Accurate Estimates of Divergence Times from Big Data. Mol. Biol. Evol 34, 45–50. [DOI] [PubMed] [Google Scholar]
- Meyer PE (2008). Information-Theoretic Variable Selection and Network Inference from Microarray Data. Universite Libre de Bruxelles. [Google Scholar]
- Meyer PE, Lafitte F, and Bontempi G (2008). minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics 9, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Nguyen MAT, and von Haeseler A (2013). Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol 30, 1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, and Warnow T (2015). ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31, i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, and Minh BQ (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieselt-Struwe K, and von Haeseler A (2001). Quartet-mapping, a generalization of the likelihood-mapping procedure. Mol. Biol. Evol 18, 1204–1219. [DOI] [PubMed] [Google Scholar]
- Opulente DA, Rollinson EJ, Bernick-Roehr C, Hulfachor AB, Rokas A, Kurtzman CP, and Hittinger CT (2018). Factors driving metabolic diversity in the budding yeast subphylum. BMC Biol. 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Merino RA, Kuanyshev N, Braun-Galleani S, Byrne KP, Porro D, Branduardi P, and Wolfe KH (2017). Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol. 15, e2002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel M, Meade A, and Barker D (2004). Bayesian estimation of ancestral character states on phylogenies. Syst. Biol 53, 673–884. [DOI] [PubMed] [Google Scholar]
- Pérez MD, Gonzalez C, Avila J, Brito N, and Siverio JM (1997). The YNT1 gene encoding the nitrate transporter in the yeast Hansenula polymorpha is clustered with genes YNI1 and YNR1 encoding nitrite reductase and nitrate reductase, and its disruption causes inability to grow in nitrate. Biochem. J 321 (Pt 2), 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergstrom A, Sigwalt A, Barre B, Freel K, Llored A, et al. (2018). Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, and Penny D (2003). The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenet. Evol 28, 171–185. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2 - Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, and Yang Z (2011). Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol 28, 2161–2172. [DOI] [PubMed] [Google Scholar]
- Richards TA, Leonard G, Soanes DM, and Talbot NJ (2011). Gene transfer into the fungi. Fungal Biol. Rev 25, 98–110. [Google Scholar]
- Riley R, Haridas S, Wolfe KH, Lopes MR, Hittinger CT, Goker M, Salamov AA, Wisecaver JH, Long TM, Calvey CH, et al. (2016). Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. USA 113, 9882–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger AJ (2018). Reply to “Eukaryote lateral gene transfer is Lamarckian.” Nat. Ecol. Evol 2, 755–755. [DOI] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, and Carroll SB (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804. [DOI] [PubMed] [Google Scholar]
- Salichos L, and Rokas A (2011). Evaluating ortholog prediction algorithms in a yeast model clade. PLoS One 6, e18755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salichos L, and Rokas A (2013). Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497, 327–331. [DOI] [PubMed] [Google Scholar]
- Schonknecht G, Weber APM, and Lercher MJ (2014). Horizontal gene acquisitions by eukaryotes as drivers of adaptive evolution. BioEssays 36, 9–20. [DOI] [PubMed] [Google Scholar]
- Schwarz G, and Mendel RR (2006). Molybdenum cofactor biosynthesis and molybdenum enzymes. Annu. Rev. Plant Biol 57, 623–647. [DOI] [PubMed] [Google Scholar]
- Shen X-X, Zhou X, Kominek J, Kurtzman CP, Hittinger CT, and Rokas A (2016a). Reconstructing the Backbone of the Saccharomycotina Yeast Phylogeny Using Genome-Scale Data. G3 (Bethesda) 6, 3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Salichos L, and Rokas A (2016b). A Genome-Scale Investigation of How Sequence, Function, and Tree-Based Gene Properties Influence Phylogenetic Inference. Genome Biol. Evol 8, 2565–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Hittinger CT, and Rokas A (2017). Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol 1, 0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H (2002). An approximately unbiased test of phylogenetic tree selection. Syst. Biol 51, 492–508. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, and Hasegawa M (2001). CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247. [DOI] [PubMed] [Google Scholar]
- Simpson JT, and Durbin R (2012). Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, and Birol I (2009). ABySS: A parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JC, and Hibbett DS (2007). Horizontal Transfer of a Nitrate Assimilation Gene Cluster and Ecological Transitions in Fungi: A Phylogenetic Study. PLoS One 2, e1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JC, and Rokas A (2010). Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. Proc. Natl. Acad. Sci. USA 107, 10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, and Dunn CW (2008). Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24, 715–716. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Marchant DB, Van de Peer Y, and Soltis DE (2015). Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev 35, 119–125. [DOI] [PubMed] [Google Scholar]
- Song L, Florea L, and Langmead B (2014). Lighter: fast and memory-efficient sequencing error correction without counting. Genome Biol. 15, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanu PD, Abbott JC, Amselem J, Burgis TA, Soanes DM, Stuber K, Loren van Themaat EV, Brown JKM, Butcher SA, Gurr SJ, et al. (2010). Genome Expansion and Gene Loss in Powdery Mildew Fungi Reveal Tradeoffs in Extreme Parasitism. Science 330, 1543–1546. [DOI] [PubMed] [Google Scholar]
- Stajich J, Berbee ML, Blackwell M, Hibbett DS, James TY, Spatafora JW, and Taylor JW (2009). The Fungi. Curr. Biol 19, R840–R845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, and Waack S (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 Suppl 2, ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stanley SM (1975). A theory of evolution above the species level. Proc. Natl. Acad. Sci. USA 72, 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, and Bork P (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, and Borodovsky M (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis N, Sarilar V, Drillon G, Fleiss A, Agier N, Meyniel J-P, Blanpain L, Carbone A, Devillers H, Dubois K, et al. (2016). Reconstruction of ancestral chromosome architecture and gene repertoire reveals principles of genome evolution in a model yeast genus. Genome Res. 26, 918–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KJ, and Moran NA (2013). Functional and Evolutionary Analysis of the Genome of an Obligate Fungal Symbiont. Genome Biol. Evol 5, 891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, and Zdobnov EM (2018). BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol 35, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Yin S, Sharpe T, Lau B, Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, et al. (2014). Comprehensive variation discovery in single human genomes. Nat. Genet 46, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC (1992). Natural Selection: Domains, Levels, and Challenges (New York: Oxford University Press; ). [Google Scholar]
- Wisecaver JH, Slot JC, and Rokas A (2014). The Evolution of Fungal Metabolic Pathways. PLoS Genet. 10, e1004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver JH, Alexander WG, King SB, Hittinger CT, and Rokas A (2016). Dynamic Evolution of Nitric Oxide Detoxifying Flavohemoglobins, a Family of Single-Protein Metabolic Modules in Bacteria and Eukaryotes. Mol. Biol. Evol 33, 19791987. [DOI] [PubMed] [Google Scholar]
- Woese CR (2000). Interpreting the universal phylogenetic tree. Proc. Natl. Acad. Sci. USA 97, 8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, and Koonin EV (2013). Genome reduction as the dominant mode of evolution. Bioessays 35, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Morden CW, and Palmer JD (1992). Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proc. Natl. Acad. Sci. USA 89, 10648–10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Armisén D, Proux-Wera E, ÓhÉigeartaigh SS, Azam H, Gordon JL, and Byrne KP (2015). Clade- and species-specific features of genome evolution in the Saccharomycetaceae. FEMS Yeast Res. 15, fov035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z (1996). Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol 11, 367–372. [DOI] [PubMed] [Google Scholar]
- Yang Z (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Tegenfeldt F, Kuznetsov D, Waterhouse RM, Simao FA, Ioannidis P, Seppey M, Loetscher A, and Kriventseva EV (2017). OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 45, D744–D749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Peris D, Kominek J, Kurtzman CP, Hittinger CT, and Rokas A (2016). in silico Whole Genome Sequencer & Analyzer (iWGS): A Computational Pipeline to Guide the Design and Analysis of de novo Genome Sequencing Studies. G3 (Bethesda) 6, 3655–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Shen X-X, Hittinger CT, and Rokas A (2018). Evaluating Fast Maximum Likelihood-Based Phylogenetic Programs Using Empirical Phylogenomic Data Sets. Mol. Biol. Evol 35, 486–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Marqais G, Puiu D, Roberts M, Salzberg SL, and Yorke JA (2013). The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.