

Abstract
Bacterial pathogens subvert host cells by manipulating cellular pathways for survival and replication; in turn, host cells respond to the invading pathogen through cascading changes in gene expression. Deciphering these complex temporal and spatial dynamics to identify novel bacterial virulence factors or host response pathways is crucial for improved diagnostics and therapeutics. Dual RNA sequencing (dRNA-Seq) has recently been developed to simultaneously capture host and bacterial transcriptomes from an infected cell. This approach builds on the high sensitivity and resolution of RNA sequencing technology and is applicable to any bacteria that interact with eukaryotic cells, encompassing parasitic, commensal or mutualistic lifestyles. Several laboratory protocols have been presented that outline the collection, extraction and sequencing of total RNA for dRNA-Seq experiments, but there is relatively little guidance available for the detailed bioinformatic analyses required. This protocol outlines a typical dRNA-Seq experiment, based on a Chlamydia trachomatis-infected host cell, with a detailed description of the necessary bioinformatic analyses with currently available software tools.
Keywords: dRNA-Seq, Chlamydia, bioinformatics, host–pathogen, sequencing
Introduction
Background
On infection or other interactions, bacteria and their host eukaryotic cells engage in a complex interplay, as they negotiate their respective survival and defense strategies. Unraveling these coordinated regulatory interactions, virulence mechanisms and innate responses is key for our understanding of pathogenesis, disease and the development of therapeutics [1]. Traditional transcriptomic approaches such as microarrays have typically focused on either the prokaryotic or eukaryotic organism to investigate the host–bacteria interaction network [2]. However, this approach cannot decipher reciprocal changes in gene expression that contribute to the global infection system. Instead, an integrated approach is required that acknowledges both interaction partners, i.e. both bacteria and host, from the same biological sample. Owing to the increasing affordability and resolution of next-generation sequencing, this is now achievable via dual RNA sequencing (dRNA-Seq) [1].
RNA sequencing (RNA-Seq) was developed for the study of transcriptomes based on the massively parallel sequencing of RNA [3]. In a typical experiment, total mRNA from a sample is subjected to high-throughput next-generation sequencing and mapped to a reference genome to deduce the structure and/or expression state of each transcript [4]. Gene expression changes can be accurately measured between samples with high coverage and sensitivity, while alternative splicing analyses can be applied to identify novel isoforms and transcripts, RNA editing and allele-specific expression [5]. The high sensitivity and dynamic range of RNA-Seq has expanded our capability for whole-transcriptome analysis and enabled new insight into the functional elements of the genome [6].
dRNA-Seq extends these capabilities to two (or potentially more) interacting organisms, allowing the simultaneous monitoring of gene expression changes without disturbing the complex interactions that define host–bacteria infection dynamics. We applied dRNA-Seq to map host and bacteria transcriptomes from Chlamydia-infected host epithelial cells, which highlighted a dramatic early response to infection and numerous altered pathways within the host cell [1]. dRNA-Seq has since been successfully used to study host–bacteria interactions for Salmonella enterica [7], Azospirillum brasilense [8], Mycobacterium tuberculosis [9], Haemophilus influenzae [10], Yersinia pseudotuberculosis [11, 12], Streptococcus pneumoniae [13] and Actinobacillus pleuropnemoniae [14]. Published dRNA-Seq laboratory protocols have focused on the generation of sequencing libraries from ribosomal RNA (rRNA)-depleted samples [15, 16]. Here, we provide a detailed protocol for the critical bioinformatic analysis of dRNA-Seq data.
Advantages and limitations
Complementary DNA (cDNA) microarrays first enabled large-scale transcriptome analyses, allowing the expression pattern of tens of thousands of known genes to be measured. Drawbacks include (1) a high background signal [17], (2) cross-hybridization between genes of similar sequence, (3) the limit of expression-level detection to the 1000-fold range, compared with the actual cellular 1 000 000-fold range [18], (4) restriction of analysis to known or predicted mRNAs [19] and (5) the inability to detect novel transcripts [18]. Some of these were overcome with tiling arrays to measure antisense RNA expression and other noncoding RNA (ncRNA) transcripts, but the large size of eukaryotic genomes makes this inordinately costly [20]. Tag-based sequencing does enable the enumeration of individual transcripts, but this method requires existing gene structure information, can only sample a small region of a transcript and is incapable of capturing diverse classes of RNA and its isoforms.
RNA-Seq provides a wider dynamic range, higher technical reproducibility and a better estimate of absolute expression levels with lower background noise [21–23], and has become the primary method to examine transcriptomes. By allowing an unbiased determination of gene expression, high-resolution data on potentially transcribed regions upstream and downstream of the annotated coding region and posttranslational rearrangements such as splicing and different RNA isoforms can be reported [24]. As a result, RNA-Seq improves genome annotation and identifies new open reading frames, transcription start sites, the 5′ and 3′ untranslated regions of known genes and ncRNAs such as microRNA (miRNA), promoter-associated RNA and antisense 3′ termini-associated RNA [25]. dRNA-Seq can report these data for two (or potentially more) organisms from the same sample while providing powerful insight into novel interaction dynamics. For example, gene expression changes in one organism can be correlated with the responses of the other to capture crucial events that signify the dynamic mechanisms of host adaption and the progression of infection [1, 4, 7, 10, 26].
Despite these advantages, dRNA-Seq remains technically challenging. Up to 98% of the total RNA is rRNA [27]. Bacterial mRNA levels are typically low compared with the host, especially during early infection periods, often requiring mRNA depletion and/or enrichment approaches for cost-effective sequencing. Additionally, the quantity of mRNA detected by RNA-Seq is often a poor indicator for protein abundance because of mRNA instability and turnover [28, 29]. The wide range of expression levels can result in nonuniform coverage where only a few reads can be captured for genes subject to lower expression levels, while short isoforms and repeat sequences derived from the same gene may result in assembly ambiguities. These ambiguities are compounded when using de novo methods for genomes that are partially or fully unsequenced [21] but can be avoided when assembling reads to a reference genome. Transcript length bias can distort the identification of differentially expressed genes (DEGs) in favor of longer transcripts [30] but can be standardized with appropriate normalization techniques. Despite these challenges, dRNA-Seq is a powerful, economical, sensitive and species-independent platform for investigating the gene expression dynamics of host–bacteria interactions [4].
Overview of the technique
This protocol provides a detailed bioinformatics analysis pipeline for a typical dRNA-Seq host–bacteria analysis. We describe an experiment based on human epithelial carcinoma (HeLa) cells (host) infected with Chlamydia trachomatis (bacteria), which is a well-defined host–bacteria system; Chlamydia is an obligate intracellular bacterial pathogen that is reliant on its host epithelial cell for survival, and HeLa cells are routinely used for Chlamydia-based experiments. In the context of the protocol, HeLa–Chlamydia can be substituted for any host–bacteria system of interest. The protocol includes all steps for total RNA sequence quality control and trimming, the in silico rRNA depletion and segregation of host and bacteria reads, distinct sequence alignment and sorting techniques for host and bacteria data, alignment visualization, read quantification and normalization and the separate statistical analysis of host and bacteria data (Figure 1).
Figure 1.
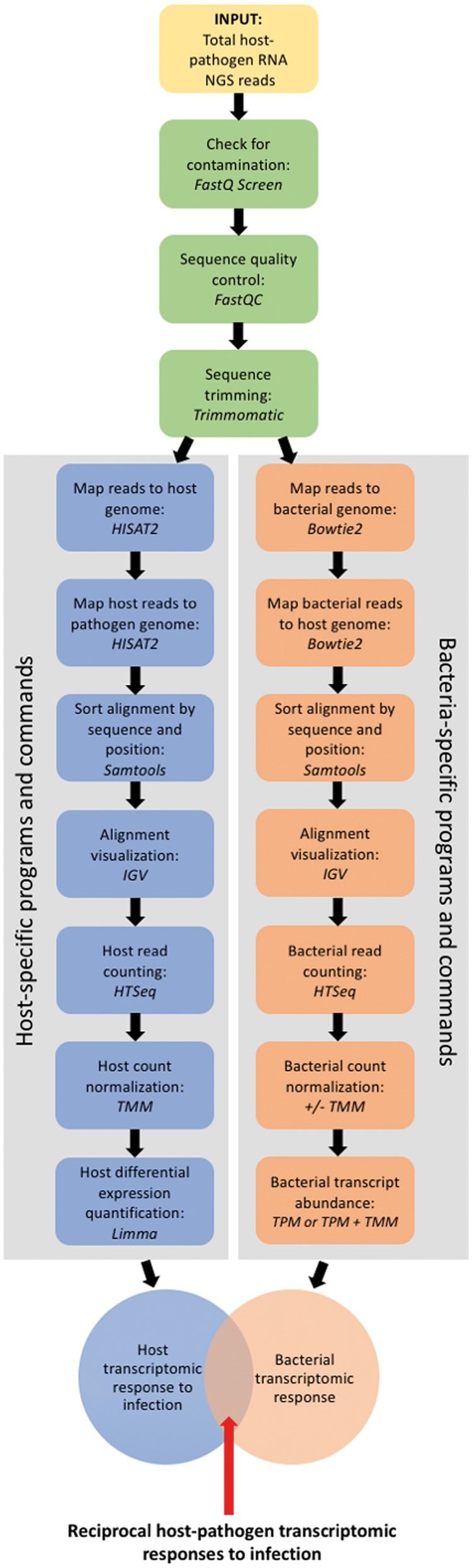
Flow chart for the bioinformatic data analysis of dRNA-Seq of host and bacteria.
The protocol
The analysis of dRNA-Seq data sets is a daunting task, especially when presented with the ever-expanding number of software packages and statistical methodologies. In response, we have a prepared a detailed, yet easy-to-follow protocol to describe each bioinformatic step for a complete dRNA-Seq analysis. The protocol is based on our experience with dRNA-Seq and has been refined over time to ensure its reproducibility and accuracy. While it has been designed to be applicable to any host–bacteria system, the possibility for alternative approaches is also discussed. The reader is encouraged to also consider these when designing a dRNA-Seq experiment, depending on their research goals, resources and experimental design.
Experimental design
The experiment should be designed to address the biological question(s) of interest, and key initial questions include the type and relevance of the host cell to be used and the RNA species to be investigated (i.e. mRNA, miRNA, small nuclear RNA, etc.). To capture sufficient RNA from both organisms, the ratio of bacteria to host genome size is a useful starting point followed by an estimation of the desired fold coverage. This can be determined by considering the number of replicates, the expected influence of housekeeping and structural RNA [rRNA and transfer RNA (tRNA)], the possibility of host:bacteria sequence overlap, the number of time points and the multiplicity of infection (MOI). We suggest at least three biological replicates for each sample rather than technical replicates taken from the same sample to minimize Type I and II errors and ensure an adequate estimation of within- and between-sample variation.
As ∼95% of the total RNA will be ribosomal, a method of rRNA depletion and/or mRNA enrichment is recommended to prevent uninformative ‘noise’ from biasing the analysis. There are several commercial kits available for hybridization-based depletion or poly(A) depletion of rRNA during the RNA extraction stage, but these differ in their efficiency and should be evaluated carefully [4]. An alternative (or supplementary) approach that we describe in the protocol is the in silico removal of rRNA, but sufficient sequencing depth is critical to ensure that the remaining ‘informative’ reads can be statistically supported. Deeper sequencing will also be necessary for the detection of low copy number transcripts, alternate isoforms or bacterial reads at early time points, and a depth of 50X coverage is usually recommended. However, increased sequencing depth can also increase the detection of transcriptional noise, spurious cDNA transcripts or genomic DNA contamination, so careful consideration is required [31, 32].
The time points of interest should be carefully considered as the initiation, and period of transcriptional response can differ between host and bacteria [33]. Ideally, multiple time points should be collected to suitably capture the dynamic host–bacteria transcriptional landscape. Finally, a suitable bacteria MOI should be selected to maximize the transcriptional signal from both host and bacteria while reducing bias toward the uninfected cells that will flourish at the later time points. Importantly, a high(er) MOI may also lead to a heightened and/or distorted host response with decreased biological relevance, depending on the system under investigation. Optionally, the addition of RNA spike-ins and unique molecular identifiers can be useful for the quantitative calibration of RNA levels [34, 35].
Data preparation
Sequence data from dRNA-Seq comprises cDNA as input from the experiment, with the majority derived from the eukaryotic host (depending on the experimental conditions and system under study). Thus, careful attention is required to accurately segregate the reads from each organism. For paired-end sequencing, host and bacteria read data are generally provided as two FASTQ format files, which are composed of a unique read identifier, the sequence read, an optional alternate identifier and the quality scores for each read position. There are a number of approaches to detect sequence contamination, including an assessment of alignment statistics with SAMtools, as described below. Another popular method is to submit a subset of FASTQ files to the BLAST database to confirm that the hits are in agreement with the expected organisms. We find that the most unambiguous approach is to use FASTQ Screen (http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/), which accurately screens the total reads against a sequence database and will identify the expected host and bacterial reads as well as any contaminating organisms, sequencing adapters, rRNA, as well as unknown hits (Figure 2).
Figure 2.

FASTQ Screen processing report of raw host and bacteria FASTQ sequencing reads. As expected, the majority of reads map to the human genome (70%), while 30% of the reads map to the Chlamydia genome.
The reads are then checked for quality using FASTQC, a Java-based software that reports several quality control statistics and a judgment on each metric (pass, warn and fail) (Figure 3) [36]. Of particular importance is the per base sequence quality plot, which should indicate a lower quartile >10 (corresponding to 90% accuracy), while the per sequence quality score should have a mean base quality of ≥25. The per base sequence content plot should indicate an even proportion of each base, and the per sequence GC content plot should demonstrate a normal distribution of GC content; an abnormal distribution is likely evidence of contamination. Sequences that fall outside these parameters may benefit from trimming to remove problematic ends, but the user should bear in mind that over-trimming of low- or medium-coverage data can introduce biases and reduce the statistical significance of DEGs. Nevertheless, we find Trimmomatic to be most useful tool for processing low-quality reads and adapter removal, which calculates an average quality score and associated cutoff if reads fall below a predetermined threshold [37]. Other available QC tools available include PRINSEQ [38] and FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/).
Figure 3.

FASTQC report for per base sequence quality and adapter content. (A). Sequence quality before removal of adapters with Trimmomatic. (B). Sequence quality after removal of adapters with Trimmomatic.
The organisms of interest and experimental question will dictate which mapping software is most appropriate; we currently use HISAT2 [39], a powerful yet efficient program capable of identifying the splice junctions between exons that are characteristic of eukaryotic data, while the short-read aligner, Bowtie2 [40], is sufficient for bacterial read mapping. Other non-splice-aware aligners for bacterial reads include SEAL [41] and SOAP2 [42], and alternative splice-aware aligners for host reads include MapSplice [43], STAR [44] and Tophat2 [45]. It is important to note that read aligners are an active area of research, with new tools and updates frequently appearing [46].
The combined host and bacteria reads are mapped to the host reference genome with specific settings to preserve unmapped (bacterial) reads, which are then mapped to the bacteria reference genome. Assembly to a reference transcriptome is also possible, but this relies on the accuracy of annotated gene models, which may restrict the discovery of novel genes and isoforms. If no reference genome is available, de novo assembly to a transcriptome can be completed, but this also limits the identification of novel transcripts. Mapping to closely related genomes is not recommended as this often leads to errors, lower coverage and ultimately unreliable assemblies. If available for the organisms of interest, reference genomes and the annotation file can be obtained from either NCBI [47], UCSC [48] or Ensembl [49]. Each repository formats these files slightly differently, and so it is important to obtain both files from the same source. This protocol uses the GRCh38 release of the Homo sapiens genome and annotation file from Ensembl and the C.trachomatis serovar D genome from NCBI (NC_000117.1).
The resulting alignment files for both host and bacteria are sorted by position (i.e. chromosomal location) with SAMtools [50] to produce alignment quality statistics, including the number of mapped reads, the number of mapped first mates and second mates (for reads from paired-end sequencing), reads with multiple hits in the genome and host reads mapping to exonic, intronic and intergenic regions. Ideally, >70% of the host reads should map to exonic regions of the genome, while <5% of the reads mapping to intronic regions and <1% of the reads mapping to intergenic regions [44]. The alignment file is further converted to a BigWig format for the visualization of the number of reads aligned to every single base position in the genome using Integrated Genome Viewer (IGV) (Figure 4) [51], or other visualization tools such as UCSC Genome Browser or JBrowse [52]. Using IGV, the coverage of aligned reads across the genome for both host and bacteria can be visualized to identify genomic regions of high/low coverage that could indicate technical or biological errors, as well as host exon–intron boundaries, splice sites, exon junction read counts and read strand [53]. The alignment files are then sorted by read name to facilitate feature counting.
Figure 4.

Screen shot of IGV showing host mapped reads the associated GTF annotation file. The first bar labeled ‘chr1’ indicates which portion of the human genome (or chromosome) is displayed, with the length (8764 bp) and specific genomic region shown underneath. The graphs indicate read coverage, and the sequence alignment tracks are shown below this. The bottom row is the GTF annotation file indicating, which annotated transcripts the reads are aligning to.
Feature counting and normalization
Both host and bacteria counts are generated from their respective alignment files using the python wrapper script htseq-count from the HTSeq package [54]. This process quantitates the number of reads that align to a biologically meaningful feature such as exons, transcripts or genes [54], and is guided by the reference annotation file. At this point, in silico rRNA depletion is recommended, especially if no depletion or enrichment step was completed before sequencing. For this, the reference annotation file is edited to remove all rRNA (as well as tRNA) sequence annotations, which is a computationally inexpensive approach to prevent those features from being counted. The remaining reads are then quantified on a gene level, where a gene is considered the union of its exons. Any reads that map to several genomic locations are automatically discarded by HTSeq, and we generally take a conservative approach to also discard reads that overlap with more than one gene.
Sample read counts are collapsed into a single file containing a matrix of genes (rows) and samples (columns), with one file each for host and bacteria (Figure 5). To minimize statistical noise and enable better adjusted P-values, the matrix is prefiltered, so that greater than three counts remain in more than two of the samples [36]. Additionally, the last five lines of the matrix containing statistics for ambiguous counts from htseq-count are removed. Raw counts are normalized to minimize technical bias because of transcript length and sequencing depth; there are several normalization methods available, including Reads Per Kilobase Per Million (RPKM) [24], EDASeq [55], conditional quantile normalization [56], upper quartile [57] and transcripts per million (TPM) [58], and each have their benefits. Methods that divide the total number of mapped reads from a library, such as RPKM, should be used with caution, as they have difficulty in dealing with highly abundant transcripts (including rRNA), and can significantly bias the analysis. Instead, we prefer the trimmed mean of M-values (TMM), which corrects for differences in RNA composition and sample outliers, while providing better across-sample comparability [59]. For an in-depth assessment of normalization techniques, see Eder, et al. [60].
Figure 5.

The count matrix. Following read quantification with HTSeq, the count files are combined to form the matrix of raw counts for each sample and replicate in the data set.
Data analysis
Before differential expression analysis, both a multidimensional scaling (MDS) plot and hierarchical cluster plot are constructed to visualize the distances between samples and help identify problematic and outlying samples (Figures 6 and 7). A metadata table is generated to list the experimental variables, which in turn guides the construction of design and contrast matrices, which are mathematical representations of the experimental design and a description of the relevant treatment comparisons, respectively. This protocol describes a simple design and contrast matrix that allows differential expression comparisons to be made between infected and uninfected cells within each time point. Additional time points and other experimental factors would lend themselves to more complex design matrices and may be added by the user if required.
Figure 6.

MDS plot. This is a two-dimensional plot that visualizes the similarity between samples and replicates across conditions. It enables the identification of problematic samples that may obscure the subsequent statistical analysis. In this case, all replicates cluster together as expected.
Figure 7.

Hierarchical clustering dendrogram. An extension of the MDS plot, the hierarchical clustering dendrogram illustrates sample similarity. As expected, all replicates for each condition cluster together.
Owing to the nature of the host and bacteria count data, distinct statistical analyses are required for each. For bacterial transcripts, TPM is the most appropriate measure of relative transcript abundance, but this approach can suffer from biases where the calculated abundance of one transcript can affect other transcripts in the sample. Alternatively, absolute abundance may be calculated with the use of spike-in controls. Whichever method is chosen, these abundances represent a qualitative measurement of the Chlamydia transcriptome at the 1 and 24 hpi time points, which can then be interpreted in context with differential expression in the host to gain deeper insight into the host–bacteria interactome.
For the host, genes are identified that are differentially expressed between infected and time-matched noninfected samples [5]. There are multiple differential expression packages available, including BaySeq [61], Cufflinks [62], DESeq [63], edgeR [64], Salmon [65] and Kallisto [66]. The majority of these packages model RNA-Seq counts via a negative binomial distribution and apply distinct statistical methodologies to calculate reliable dispersion estimates. Alternatively, this protocol describes the use of the Linear Modeling for Microarray Data (Limma) package, which uses linear modeling to describe the expression data for each gene [67]. In contrast to these other RNA-Seq packages, Limma attempts to correctly model the mean–variance relationship between samples to achieve a more probabilistic distribution of the counts (Figure 8). This has proven to be the best method for analyzing both simple and complex experimental designs of dRNA-Seq experiments that incorporate different sample types and time points [68], but the reader is encouraged to consider the requirements of the experiment when selecting an appropriate method. For a comparison of differential expression analysis methods, see Soneson and Delorenzi [69].
Figure 8.

Limma voom plots. The mean–variance trend plot displays the gene-wise square-root residual SDs plotted against average log count, with the LOWESS fit represented by the red line. The sample-specific weights are the result of the ‘voomWithQualityWeights’ function and represents the sample-specific quality weights that can be applied to down-weight outlier samples.
The analysis of the host data set yields a list of genes that are differentially expressed compared with the uninfected control. A false discovery rate (FDR) cutoff ≤0.05 (i.e. 5% false positives), a log fold change (LFC) of at least 2-fold upregulation/downregulation, and expression levels >1 percentile in either condition (Table 1) are suitable benchmarks for identifying significant genes. These lists can then be used as input for downstream analysis of the enrichment of gene ontology and metabolic pathways using several tools, including GOSeq [70], DAVID [71] and Ingenuity Pathway Analysis Toolkit (QIAGEN Redwood City, www.qiagen.com/ingenuity).
Table 1.
Statistical output of the differential expression analysis of host reads in R
| logFC | AveExpr | t | P.Value | adj.P.Val | B | |
|---|---|---|---|---|---|---|
| ENSG00000003096 | 0.6525066 | 13.791554 | 28.40683 | 1.447731e-10 | 4.921022e-09 | 14.94091 |
| ENSG00000005483 | 0.6818259 | 14.119036 | 28.15577 | 1.574679e-10 | 4.921022e-09 | 14.85045 |
| ENSG00000003436 | 0.6141746 | 15.274943 | 28.06727 | 1.622315e-10 | 4.921022e-09 | 14.60957 |
| ENSG00000004766 | 0.5506978 | 13.234187 | 27.08358 | 2.273831e-10 | 5.172965e-09 | 14.52388 |
| ENSG00000003147 | 4.2364651 | 6.880526 | 22.53363 | 1.289647e-09 | 2.252721e-08 | 11.79033 |
| ENSG00000001630 | −1.8429486 | 11.760177 | −22.19781 | 1.485311e-09 | 2.252721e-08 | 12.61091 |
Note: The first column contains the ENSEMBL ID for the genes, logFC indicates the LFC observed, AveExpr is the expression value for each gene, t is the moderated t-statistic, P.Value is the raw P-value, adj.P.Val is the FDR-adjusted P-value and B is the log odds that the gene is differentially expressed.
Application
dRNA-Seq can be used to address a number of experimental questions. Host differential mRNA and miRNA expression, differential exon usage, alternative splicing and novel transcript and isoform discovery in response to the bacteria can be determined [5], which may be correlated with the transcriptomic response of the bacteria to determine interaction dynamics. These results can be further integrated with other sources of biological input, including genotyping data to identify genetic loci responsible for gene expression variation, epigenetic information (transcription factor binding, histone modification, methylation etc.) to highlight the influence of transcription factor binding, miRNA-Seq data to identify the regulatory mechanisms of gene expression changes via ncRNA and proteomic data to build a system-level analysis of host–bacteria regulation [72, 73].
Procedure
Materials
Hardware requirements
The analysis of dRNA-Seq experiments is a computationally intensive process that requires the manipulation of gigabytes of data. Depending on the experimental design, the size of a standard alignment file in BAM format can range from 15 to 50 GB. Access to a computer cluster, core facility or cloud service is recommended to expedite the analysis and free up resources on the local system. The time to complete the analysis will also depend on the experimental design and computing infrastructure, but the process can usually be finalized within 8 h.
Operating system
This protocol provides commands that are designed to run on a Unix-based operating system such as Linux or macOS. The protocol was specifically designed to run on the Ubuntu 16.04.1 operating system on a Linux machine. Please ensure you have administrative rights.
Command line nomenclature
This protocol assumes a basic understanding of both the Linux command line interface and the R statistical computing environment. All Linux commands are shown following a dollar sign ($), while R commands are shown following a greater-than sign (>):
$ linux command
> R command
Software requirements
FASTQ Screen: Contamination screening (http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/)
FASTQC: Sequence quality control tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
Trimmomatic: FASTQ sequence file trimming (http://www.usadellab.org/cms/?page=trimmomatic)
HISAT2: Graph-based alignment of sequences to genomes (https://ccb.jhu.edu/software/hisat2/index.shtml)
SAMtools: Manipulation of sequence alignments and mapped reads (http://www.htslib.org)
Bedtools genomecov and bedGraphToBigWig: genomic analysis tools (http://bedtools.readthedocs.io/en/latest)
IGV: Alignment and visualization tool (https://www.broadinstitute.org/igv)
HTSeq: read counting (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html)
R statistical computing environment (https://www.r-project.org)
Bioconductor packages: edgeR, limma, org.Hs.eg.db, Genomic Features and their dependencies (see below)
Bowtie2: Short-read aligner (http://bowtie-bio.sourceforge.net/index.shtml).
Always check that you are downloading and installing the latest version of each piece of software and consult the official user guide for more in-depth guidelines and options for troubleshooting any errors that may arise.
Samples and filenames
The protocol is arranged so that identical naming conventions are used for each sample and condition. For example, ‘1hpi_Host_infected_rep1’ indicates that the sample relates to the first replicate of host cells infected with Chlamydia at the 1 hpi time point, and ‘1hpi_Host_uninfected_rep1’ indicates the first replicate of host cells only (i.e. uninfected host cells) at the 1 hpi time point. Conversely, the samples relating to the bacteria are named, ‘1hpi_Bacteria_rep1’. While subsequent replicates for both host and bacteria would have names ending in ‘rep2’ and ‘rep3’, for conciseness, this protocol describes the commands using ‘1hpi_Host_infected_rep1’ as an example, and it is expected that the user will repeat the process for the remaining replicates and samples. The filenames associated with raw FASTQ sequence files will depend on the sequencing facility pipeline, and in this protocol are named ‘fastq_file_1_R1.fq’, where ‘R1’ indicates read number 1 of paired-end reads (the corresponding read file would be ‘fastq_file_1_R2.fq’). In some cases, an output directory is required, which is noted as ‘<output_directory>’ for the user to input their working directory of choice (without the ‘ < >’ symbols). Finally, reference, annotation and gene info files are prefixed with the relating organism, i.e. ‘host_reference.fa’ indicates a FASTA file containing the host reference genome.
Equipment setup
Download and install the following software. Check the developer Web site to ensure you are installing the latest version and for further information about dependencies and prerequisites.
Create a directory to install program executables and add to PATH
$ mkdir $HOME/bin
$ export PATH=$HOME/bin:$PATH
$ echo “export PATH=$HOME/bin:$PATH"”≫ ∼/.bashrc
FastQ Screen installation
$ wget http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/fastq_screen_v0.11.1.tar.gz
$ tar –zxf fastq_screen_v0.11.1.tar.gz
$ cd fastq_screen_v0.11.1
$ cp fastq_screen $HOME/bin
FastQC installation
$ sudo apt-get install default-jre
$ wget http://www.bioinformatics.babraham.ac.uk/projec ts /fastqc/fastqc_v0.11.5.zip
$ unzip fastqc_v0.11.5.zip
$ cd FastQC/
$ chmod 755 fastqc
$ cp fastqc $HOME/bin
Trimmomatic installation
$ wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.36.zip
$ unzip Trimmomatic-0.36.zip
$ cd Trimmomatic-0.36
$ cp trimmomatic $HOME/bin
HISAT2 installation
$ wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads /hisat2-2.0.5-Linux_x86_64.zip
$ unzip hisat2-2.0.5-Linux_x86_64.zip
$ cd hisat2-2.0.5
$ cp hisat2 $HOME/bin
$ cp hisat2-build $HOME/bin
Samtools installation
$ sudo apt-get install samtools
Bedtools installation
$ sudo apt-get install bedtools
bedGraphToBigWig insta llation
$ mkdir bedGraphToBigWig
$ cd bedGraphToBigWig
$ wget -O bedGraphToBigWig https://github.com/ENCODE-DCC/kentUtils/blob/v302.1.0/bin/linux.x86_64/bedGraph ToBig Wig?raw = true
$ chmod 755 bedGraphToBigWig
$ cp bedGraphToBigWig $HOME/bin
IGV and IGVtools installation
$ wget http://data.broadinstitute.org/igv/projects/downloa d s /IGV_2.3.88.zip
$ unzip IGV_2.3.88.zip
$ wget http://data.broadinstitute.org/igv/projects/downloa d s /igvtools_2.3.88.zip
$ unzip igvtools_2.3.88.zip
$ cd igvtools_2.3.88
$ cp igvtools $HOME/bin
HTSeq installation
$ sudo apt-get install build-essential python2.7-dev pyth o n - numpy python-matplotlib
$ wget –no-check-certificate https://pypi.python.org/packag e s /source/H/HTSeq/HTSeq-0.6.1p1.tar.gz
$ tar –zxvf HTSeq-0.6.1p1.tar.gz
$ cd HTSeq-0.6.1p1
$ python setup.py build
$ sudo python setup.py install
$ cd scripts
$ cp htseq-count $HOME/bin
R and Bioconductor package installation
$ sudo apt-get install libcurl4-openssl-dev libxml2-dev
$ sudo apt-get update
$ echo “deb https://cran.rstudio.com/bin/linux/ubuntu xen ial/" | sudo tee -a/etc/apt/sources.list
$ gpg –keyserver hkp://keyserver.ubuntu.com:80 –recv-ke y s E084DAB9
$ gpg -a –export E084DAB9 | sudo apt-key add -
$ sudo apt-get update
$ sudo apt-get upgrade
$ sudo apt-get install r-base
Open R and install Bioconductor packages using the biocLite installation tool. All packages dependencies will automatically be installed.
$ R
> source("http://bioconductor.org/biocLite.R")
> biocLite("BiocUpgrade")
> biocLite(c("org.Hs.eg.db”, “edgeR”, “limma”, “Genomic Feat ures"))
Bowtie2 installation
$ wget https://sourceforge.net/projects/bowtie-bio/files/bo w tie2/2.2.9/bowtie2-2.2.9-linux-x86_64.zip
$ unzip bowtie2-2.2.9-linux- x86_64.zip
$ cd bowtie2-2.2.9-linux-x86_64
$ cp bowtie2 $HOME/bin
$ cp bowtie2-build $HOME/bin
File preparation
Download reference genomes and gene model annotation files
For the eukaryotic host, download the H.sapiens reference genome and Gene Transfer Format (GTF) annotation from Ensembl: http://asia.ensembl.org/info/data/ftp/index.html and rename the reference genome to ‘host_reference.fa’. For C. trachomatis, download the reference genome in FASTA format from NCBI: http://www.ncbi.nlm.nih.gov/nuccore/NC_000117 and rename the file to ‘bacteria_reference.fa’. Download the C. trachomatis GTF (000590675) file from bacteria.ensembl.org/info/website/ftp/index.html and rename file to ‘bacteria_annotation.gtf’. Save all files to your working directory. Reference genomes and annotation files should be obtained from the same repository to ensure consistent formatting and nomenclature.
Remove rRNA annotations from the GTF file
To prevent any rRNA reads from being counted, remove all lines in the GTF annotation file annotated as rRNA:
$ grep -wv rRNA Homo_sapiens.GRCh38.87 > host_ annot ation.gtf
Method: Host
1. Examine a subset of FASTQ sequence files for contamination using FASTQ Screena:
$ fastq_screen –aligner bowtie2 fastq_file_1_R1.fq
2. Check the quality of FASTQ sequences using FASTQCb:
$ fastqc –noextract –o <output _direc tory> fastq_file _1_R1. f q
3. Remove sequencing adapters and low- quality reads using Trimmomaticc:
$ java -jar trimmomatic-0.36.jar PE -threads 6 -phred33 fastq_file_1_R1.fq fastq_file_1_R2.fq fastq_file_1_ R1_pai re d_ trimmed.fq fastq_file _1_R1_unpaired _trimmed.fq fastq _ file_1_ R2_paired _trimmed.fq fastq_file_1_ R2_ unpaired_trimmed.fq ILL U M INACLIP: adapters.fa:2: 30: 10 LEADING:3 TRAILING:3 SLIDIN GW INDOW:4:15 MINLEN:36
4. Build host transcriptome index and align host sequence reads to reference using HISAT2d:
$ hisat2-build host_reference.fa host_reference.index
$ hisat2 –x host_reference.index –un-conc pair1_unmapped.fastq -1 fastq_file_1_.R1.trim.fq -2 fastq_file_1_. R2. trim.fq | samtools view –bS - > accepted_hits.bam
5. Sort BAM files generated by HISAT2 by both name and position using SAMtoolse:
$ samtools sort accepted_hits.bam –o 1hpi_Host_ infected_ rep1.sorted_position
$ samtools sort –n accepted_hits.bam –o 1hpi_Host_ in f e c t e d _ rep1.sorted_name
6. Convert ‘sorted by position’ BAM file to BigWig formatf:
$ samtools faidx host_reference.fa
$ cut –f1,2 host_reference.fa.fai > host_reference.genome
$ bedtools genomecov –split –bg –ibam 1hpi_Host_ infected_rep1.sorted_position.bam –g host_reference.genome > 1hpi_Host_infected_rep 1.sorted_position.bedGraph
$ bedGraphToBigWig 1hpi_Host_infected_rep1.so rted_po sition.bedGraph host_reference.genome 1hpi_Hos t_in fe cted_rep1.sorted_position.bigWig
7. Index the ‘sorted by position’ BAM file for visualization in IGVg:
$ samtools index 1hpi_Host_infected_ rep1.sorted_ position. bam
8. Index the GTF file for visualization in IGVh:
$ igvtools sort host_annotation.gtf host_annotation_ sorted .gtf
$ igvtools index host_annotation_sorted.gtf
9. Visualize alignments with IGVi:
$ java –jar igv.jar
10. Create count matrix with HTSeqj:
$ htseq-count –s no –a 10 –r name –f bam 1hpi_ Host_infected_rep1.sorted_name.bam Host_annotation.gtf > 1hpi_Host_infected_rep1.sorted_name.count
11. Set working directory in Rk:
> R
> data = setwd(…)
12. Create data frame in R containing experiment metadatal:
> group = factor(c(rep(“1hpi_mock”, 3), rep(“1hpi_infected”, 3), rep(“24hpi_mock”, 3),rep(“24hpi_infected”, 3)))
13. Combine count files into a DGEList in Rm:
> library(edgeR)
> counts.host = readDGE(list.files(pattern = “.count“), data, col u mns = c(1,2))
14. Remove the last five rows from the count matrixn:
> counts.host$counts = counts.host$counts[1:(nrow(counts.host$counts)-5),]
15. Filter counts to exclude low- expressing genes°:
> counts.host$counts = rowSums(cpm(counts.host$ counts ) > 2) > = 2
16. Inspect the count matrixp:
> head(counts.host$counts, 20)
> dim(counts.host$counts)
17. Apply TMM normalization to the raw countsq:
> counts.host = calcNormFactors(counts.host)
18. Create the design matrix and define the contrasts of interestr:
> design = model.matrix(∼0 + group)
> rownames(design) = colnames(counts.host$counts)
> contrasts = makeContrasts (“Host_1hpi“ = group1hpi_i nfe ct e d–group1hpi_mock, “Host_24hpi“ = group24hpi_ infected–group24hpi_mock, levels = design)
19. Apply voom transformation to normalized countss:
> library(limma)
> png(“host_voom.png“)
> y = voomWithQualityWeights(counts = counts.host, design = design, plot = TRUE)
> dev.off()
20. Construct an MDS plot to identify any outlier samplest:
> plot.colors = c(rep(“blue”, 3), rep(“red”, 3), rep(“orange”, 3), rep(“ black”, 3))
> png(“host_MDS.png“)
> plotMDS(counts.host, main = “MDS Plot for Count Data”, labels =colnames(counts.host$counts), col = plot. colors, cex = 0.9, xlim = c(-2,5))
> dev.off()
21. Construct a hierarchical clustering plot to visualize sample groupingsu:
> counts.host.mod = t(cpm(counts.host))
> dist = dist(counts.host.mod)
> png(“host_HC.png”)
> plot(hclust(dist), main=“Hierarchical Clustering Dendr ogram”)
> dev.off()
22. Fit the modelv:
> fit = lmFit(y, design)
> fit = contrasts.fit(fit, contrasts)
> fit = eBayes(fit)
23. Print the differentially expressed transcripts for both the 1 and 24 hpi time pointsw:
> top_1hpi = topTable(fit, coef = “Host_1hpi”, adjust = “fdr”, number = “Inf”, p.value = 0.05, sort.by = “P”)
> top_24hpi = topTable(fit, coef = “Host_24hpi”, adjust = “fdr”, number = “Inf”, p.value = 0.05, sort. by = “P”)
24. Annotate the differentially expressed transcript tables with gene symbol, description and type informationx:
> library(org.Hs.eg.db)
> gene.info = select(org.Hs.eg.db, key = rownames(top_ 1h pi), keytype = “ENSEMBL”, columns = c(“ENSEMBL”, “SYM BOL”, “GENE NAME”))
> gene.info = gene.info[!duplicated(gene.info$ENSEMBL),]
> rownames(gene.info) = gene.info$ENSEMBL
> identical(rownames(top_1hpi), rownames(gene.info))
> gene.info = gene.info[, −1]
> host_DEG_table = cbind(top_1hpi, gene.info)
25. Write the annotated differentially expressed transcript table to the local hard drivey:
> write.table(host_DEG_table, file = “Host_DEG_annotated. csv”, sep = “,”, col.names = NA
Method: Bacteria
26. Build bacteria reference index file and map the unmapped reads (bacterial reads) from HISAT2 to the bacteria reference genome with Bowtie2z:
$ bowtie2-build -f bacteria_reference.fa bacteria_ reference_ index
$ bowtie2 -q pair1_unmapped.fastq bacteria_reference_index
27. Repeat Steps 6 – 10 from the host-specific protocol above.
Sort the BAM files by both name and position, convert the ‘sorted by position’ BAM files to BigWig format and visualize with IGV. Create count matrix with HTseq.
28. Repeat Steps 13 – 16 from the host-specific protocol above.
Combine the count files into a DGEList, remove the last five rows from the counts, filter counts to remove low expression genes, and inspect the counts for errors.
29. Apply TMM normalization to countsaa
> dge.bacteria gecalcNormFactors(dge_bacteria)
> bacteria.cpm = cpm(dge.bacteria, normalized.lib. sizes = TRUE)
30. Calculate gene lengthsbb
> library(GenomicFeatures)
> txdb = makeTxDbFromGFF(“bacteria_annotation.gtf”, format = “gtf”)
> exons = exonsBy(txdb, by = “gene”)
> gene.length = sum(width(reduce(exons)))
> gene.length = as.data.frame(gene.length)
31. Define a function to calculate TPMcc
> TPM = function(counts, lengths){rate = counts/lengthsrate/sum(rate) * 1e6 }
> final.tpm = apply(bacteria.cpm, 2, function(x) TPM(x, gene. length))
> final.tpm = as.data.frame(final.tpm)
> colnames(final.tpm) = colnames(bacteria.cpm)
32. Write TPM values to file:
> write.table(final.tpm, file = “Ct_relativeabundance.csv”, sep = “,”, col.names = NA
Command reference
aThis command will run FASTQ Screen on the chosen FASTQ file, checking against locally prebuilt databases for possible sources of contamination. ‘fastq_screen’ runs the software, –aligner Bowtie2 specifies the aligner used to create the databases and ‘fastq_file_1_R1.fq’ is the input file. This step should be repeated for a random number of samples. To generate a database, the genomes of each species which to test against should be downloaded. Using the host and bacterial genomes already downloaded from the earlier steps, Bowtie2 (or other aligners) is used to build an index (see Step 4). Once built, the location and index should be added to the FASTQ Screen configuration file (Figure 2).
bIn this command, ‘-noextract’ tells FASTQC to not uncompress the output file, while ‘-o’ defines the output directory. ‘fastq_file_1_R1.fq’ is the FASTQ sequencing file. These commands produce a quality report with results saved to the directory defined by ‘<output_directory> ’. The results are reported in both illustrated form (the ‘fastqc_report.html’ file) and text form (the ‘summary.txt’ file). Repeat for all FASTQ files. As the FASTQ files are derived from total RNA sequencing, this step includes both host and bacterial sequences (Figure 3).cRun this command from the Trimmomatic installation directory. The command specifies PE as paired-end data, six threads and the FASTQ files are encoded with Phred + 33 quality scores. ‘fastq_file_ 1_R1.fastq.gz’ and ‘fastq_file_1_R2.fastq.gz’ specify the input FASTQ files to use. As paired-end data are inputted, four output files are needed to store the reads. Two ‘paired’ files from which both reads survived after processing, and two ‘unpaired’ files from which a single read survived, but the corresponding mate did not. ‘ILLUMINACLIP:adapters.fa’ uses the ‘adapters.fa’ file containing sequences and names of commonly used adapters to remove. ‘2:30:10’ are three parameters used in the ‘palindrome’ mode of Trimmomatic to identify the supplied adapters, regardless of their location within a read. For a detailed description of the best use of these three parameters, consult the Trimmomatic manual. ‘LEADING:3’ and ‘TRAILING:3’ remove a base from either the start or end position if the quality is below ‘3’. ‘SLIDINGWINDOW:4:15’ performs trimming based on a sliding window method, ‘4’ is the window size and ‘15’ is the required average quality. By examining multiple bases, if a single low-quality base is encountered, it will not cause high-quality data later in the read to be removed. Finally, ‘MINLEN:36’ removes any remaining reads that are <36 bases long. Repeat for all FASTQ files. As above, this step includes both host and bacterial sequences.
dThis ‘-x’ specifies the path to the index previously built by Bowtie2: ‘host_reference.index’. The ‘—
un-conc’ argument tells HISAT2 to write a FASTQ file containing all unmapped reads (‘pair1_unmapped.fastq’), and ‘-1’ and ‘-2’ specify the paired-end FASTQ file mates. The ‘samtools view –bS -’ argument converts to the output file from SAM to BAM format. Ensure that the HISAT2 output files, ‘accepted_hits.bam’ and ‘pair1_unmapped.fastq’, are preserved in the working directory, as these are required for bacterial read mapping.
eThe first command takes the ‘accepted_hits.bam’ file and sorts it by position, with the output file called ‘1hpi_ Host _infected_rep1.sorted_position’. In the second command, ‘-n’ tells SAMtools to sort the ‘accepted_hits.bam’ file by name, with the output file called ‘1hpi_Host_infected _rep1. sorted_name’. Repeat for all BAM files.
fThe first command indexes the reference genome by creating a ‘host_reference.fa.fai’ output file. The second command extracts the first two fields (sequence ID and sequence length) to generate the ‘host_reference.genome’ file. The third command generates a histogram illustrating alignment coverage according to the reference genome. The ‘-split’ argument tells genomecov to take into account spliced BAM alignments (as we used the splice-aware aligner HISAT2 for the host reads), while the ‘-bg’ argument tells genomecov to report genome-wide coverage in bedGraph format. ‘ibam 1hpi_Host_ infected _rep1.sorted_ position.bam’ is the input file in BAM format, ‘-g host_reference.genome’ is the reference genome in FASTA format and ‘1hpi_Host_infected _rep1.sorted _position. bedGraph’ is the output file in bedGraph format. The fourth command converts this bedGraph file to BigWig format for use with IGV (below).
gThis command takes the ‘1hpi_Host_infected_ rep1. sorted_ position’ BAM file created above and creates an indexed ‘1hpi_Host_infected_ rep1. sorted_ position.bai’ file for use with IGV.
hThe first command sorts the GTF file, specifying an input file and output file. The second command creates an index of the sorted GTF file.
iRun this command from the IGV installation directory. Within the software, load the ‘host_reference.fa’ reference genome by clicking on ‘Genomes’ and then ‘Load genome from file’. Load the sample BAM files from Step 9, the indexed GTF annotation file (Step 10) and the bigwig file (Step 8) by clicking on File, then Open. Inspect the mapped reads and visualize their alignment to the reference genome to ensure whole-genome coverage and that they align with exons as defined by the GTF file. The sample BAM files and index files must be in the working directory (Figure 4).
jThis command calls the htseq-count python wrapper script, which performs the gene-level counts. ‘-s no’ indicates that the reads are unstranded and ‘-a 10’ sets the minimum mapping quality for a read to be counted as 10. ‘-r name’ indicates that the input file is sorted by name, and ‘-f bam’ indicates that this input file is in BAM format and named ‘1hpi_Host_ infected_rep1.sorted_name.bam’. ‘host_annotation.gtf’ is the GTF annotation file from Ensembl, and ‘1hpi_Host_ infected_ rep1.sorted_name.count’ is the output file produced. Repeat command for each BAM file, which will produce a series of text files counting the gene-level reads for each sample. The last five lines of each file contain a list of reads that were not counting because of alignment ambiguities, multimapping or low alignment quality.
kThe first command opens R, and the second command sets the working directory to the folder location containing all the relevant files from Step 12. Replace ‘…’ with the complete path to this location, for example: setwd(‘/home/username/dRNA-Seq/HTSeq_counts’).
lThis creates the metadata table containing all the experimental variables, including sample name, treatment and time point.
mA DGEList is an R object from the edgeR package that efficiently compiles the count data set and experimental variables that is fed into subsequent downstream analyses. The first command loads edgeR into the current R workspace. The second command creates a variable called counts_host, which is a DGEList containing all data from Columns 1 and 2 from all files in the current working directory ending in ‘.count’ (Figure 5).
nThis command removes the last five rows of the count matrix, which contain a summary of the ambiguous and noncounted reads from htseq-count.
°This command returns the count matrix, so that there are greater than three reads in at least two replicates across all of the samples. Any nonconforming samples are removed.
pIt is often helpful to visualize the count matrix at this point to confirm that it is formatted correctly and that there are no errors. The second command provides the matrix dimensions, which is useful for determining the number of genes remaining following independent filtering.
qTMM normalization factors are calculated and incorporated into the DGEList object.
rThese commands define the design matrix and the contrasts of interest to enable differential expression to be calculated. In this case, the contrasts we are interested are the host genes differentially expressed when infected versus the host genes differentially expressed when uninfected at the 1 and at the 24 hpi time points. More complex experimental designs that include multiple samples, time points, batch effects and treatments are possible and are explained in detail in the Limma User’s Guide [74].
sThis command applies a voom transformation to the counts, by converting them to log counts per million (CPM) with associated precision weights [68]. We generally extend this by using the voomWithQualityWeights function, which applies sample-specific weights to down-weight any outlier samples. This can be especially useful if outliers were identified in the MDS plot constructed in Step 21 (below). This function takes as input the normalized count matrix (‘counts_host’) and design matrix (‘design’) and outputs two quality control plots: an estimation of the mean–variance relationship and the sample-specific weights that were applied. The output figure ‘host_voom.png’ is generated containing the voom transformation plots (Figure 8).
tThese commands generate an MDS plot by taking the host DGEList as input (‘counts_host’). The MDS plot allows the visual inspection of sample proximities to highlight possible batch effects and sample outliers that may need to be addressed. The MDS plot is saved to the working directory as ‘host_MDS.png’ (Figure 6).
uLine 1 converts the counts into CPM and then transposes the resulting matrix. The second command generates the distance matrix between each of the 12 samples, and the third command generates a hierarchical clustering dendrogram from the normalized counts in ‘counts_host’ where the most similar samples occupy closer positions in the tree. The plot is saved to the working directory as ‘host_HC.png’ (Figure 7).
vThe first two commands estimate expression fold changes and standard errors by fitting a linear model to each gene, using the comparisons defined by the contrast matrix (‘contrasts’). The third command applies empirical Bayes smoothing to the standard errors to further weaken any outliers.
wThis command prints all the DEGs with a P-value of ≤0.05 after correcting for multiple testing using the FDR (Benjamini and Hochberg) method. Additionally, a LFC threshold may be included by adding an ‘lfc = 2’ argument, which would return all DEG with a LFC in expression greater than two (Table 1).
xOften for downstream applications, it is necessary to have the gene name or identifier for each DEG. These commands are derived from the Limma documentation [74] and extract gene annotation information stored in the org.Hs.eg.db R package to annotate the DEG list with gene symbols descriptions. Repeat for the ‘Host_24hpi’ DEG list.
yThis command writes the DEG list to a comma-separated file. Repeat for the ‘Ct_24hpi’ DEG list.
zThe first command indexes the bacteria reference genome ‘-f bacteria_reference.fa’ and generates the ‘bacteria_ reference_index’ output file. The second command performs the read mapping using the unmapped reads from the host mapping step (‘pair1_unmapped.fastq’).
aaThe first command generates TMM normalization factors, and the second command converts the raw counts to normalized counts.
bbThese commands use the GenomicFeatures R package to extract the gene lengths from the ‘bacteria_annotation.gtf’ file, which are required for the calculation of TPM values (below).
ccThis command creates a function (TPM), which is used to convert the normalized counts in ‘bacteria.cpm’ to TPM.
Key Points
dRNA-Seq is an emerging technique for the simultaneous capture of both host and bacterial transcriptomes.
This is a high-throughput method that enables a deeper understanding of host–pathogen interactions.
The technique is technically challenging and requires careful consideration of the experimental goals, available resources and organisms of interest.
Acknowledgements
The authors wish to thank Alicia Oshlack and Belinda Phipson for their advice.
Funding
Sequence data generation in support of this work was funded through NIAID HHSN272200900009C, Genome Sequencing Center for Infectious Diseases (Claire Fraser, PI) at the Institute of Genome Sciences, University of Maryland School of Medicine (to G.M.) and by start-up funds from the University of Technology Sydney (to G.M.).
Biographies
James Marsh is a Postdoctoral Research Fellow in the Myers Lab at The ithree institute, University of Technology Sydney and studies the host response to chlamydial infection.
Regan Hayward is a PhD student in the Myers Lab at The ithree institute, University of Technology Sydney.
Amol Shetty is a Lead Bioinformatics Software Engineer at the Institute for Genome Sciences at the University of Maryland, Baltimore and works on the analysis of next-generation sequencing data sets including genomic, transcriptomic and epigenomic analyses.
Anup Mahurkar is the Executive Director of Software Engineering and Information Technology at the Institute for Genome Sciences at University of Maryland, Baltimore.
Michael Humphrys is the Research Laboratory Manager for the Ravel Laboratory at the Institute for Genome Sciences at University of Maryland, Baltimore.
Garry Myers is an Associate Professor and Group Leader at The ithree institute, University of Technology Sydney and studies chlamydial disease and host responses to infection by genome-scale analyses.
References
- 1. Humphrys MS, Creasy T, Sun Y, et al.Simultaneous transcriptional profiling of bacteria and their host cells. PLoS One 2013;8:e80597.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oosthuizen JL, Gomez P, Ruan J, et al.Dual organism transcriptomics of airway epithelial cells interacting with conidia of Aspergillus fumigatus. PLoS One 2011;6:e20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lister R, O'Malley RC, Tonti-Filippini J, et al.Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008;133:523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Westermann AJ, Gorski SA, Vogel J.. Dual RNA-seq of pathogen and host. Nature 2012;10:618–30. [DOI] [PubMed] [Google Scholar]
- 5. Anders S, McCarthy DJ, Chen Y, et al.Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 2013;8:1765–86. [DOI] [PubMed] [Google Scholar]
- 6. Nagalakshmi U, Wang Z, Waern K, et al.The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008;320:1344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Westermann AJ, Förstner KU, Amman F, et al.Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 2016;529:496–501. [DOI] [PubMed] [Google Scholar]
- 8. Camilios-Neto D, Bonato P, Wassem R, et al.Dual RNA-seq transcriptional analysis of wheat roots colonized by Azospirillum brasilense reveals up-regulation of nutrient acquisition and cell cycle genes. BMC Genomics 2014;15:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rienksma RA, Suarez-Diez M, Mollenkopf HJ.. Comprehensive insights into transcriptional adaptation of intracellular Mycobacteria by microbe-enriched dual RNA sequencing. BMC Genomics 2015;16:34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baddal B, Muzzi A, Censini S, et al.Dual RNA-seq of nontypeable Haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. mBio 2015;6:e01765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Avican K, Fahlgren A, Huss M, et al.Reprogramming of Yersinia from virulent to persistent mode revealed by complex in vivo RNA-seq analysis. PLoS Pathog 2015;11:e1004600-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nuss AM, Beckstette M, Pimenova M, et al.Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc Natl Acad Sci USA 2017;114:E791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aprianto R, Slager J, Holsappel S, et al.Time-resolved dual RNA-seq reveals extensive rewiring of lung epithelial and pneumococcal transcriptomes during early infection. Genome Biol 2016;17:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brogaard L, Klitgaard K, Heegaard PM, et al.Concurrent host-pathogen gene expression in the lungs of pigs challenged with Actinobacillus pleuropneumoniae. BMC Genomics 2015;16:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Avraham R, Haseley N, Fan A, et al.A highly multiplexed and sensitive RNA-seq protocol for simultaneous analysis of host and pathogen transcriptomes. Nat Protoc 2016;11:1477–91. [DOI] [PubMed] [Google Scholar]
- 16. Ravasi T, Mavromatis CH, Bokil NJ, et al.Co-transcriptomic analysis by RNA sequencing to simultaneously measure regulated gene expression in host and bacterial pathogen. Methods Mol. Biol 2016;1390:145–58. [DOI] [PubMed] [Google Scholar]
- 17. Richter A, Schwager C, Hentze S, et al.Comparison of fluorescent tag DNA labeling methods used for expression analysis by DNA microarrays. Biotechniques 2002;33:620–30. [DOI] [PubMed] [Google Scholar]
- 18. Wang Z, Gerstein M, Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shendure J. The beginning of the end for microarrays? Nat. Methods 2008;5:585–7. [DOI] [PubMed] [Google Scholar]
- 20. Bertone P, Stolc V, Royce TE, et al.Global identification of human transcribed sequences with genome tiling arrays. Science 2004;306:2242–6. [DOI] [PubMed] [Google Scholar]
- 21. Kawahara Y, Oono Y, Kanamori H, et al.Simultaneous RNA-seq analysis of a mixed transcriptome of rice and BLAST fungus interaction. PLoS One 2012;7:e49423.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fu X, Fu N, Guo S, et al.Estimating accuracy of RNA-seq and microarrays with proteomics. BMC Genomics 2009;10:161.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marioni JC, Mason CE, Mane SM, et al.RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008;18:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mortazavi A, Williams BA, McCue K, et al.Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621–8. [DOI] [PubMed] [Google Scholar]
- 25. Tsuchihara K, Suzuki Y, Wakaguri H, et al.Massive transcriptional start site analysis of human genes in hypoxia cells. Nucleic Acids Res 2009;37:2249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Choi Y-J, Aliota MT, Mayhew GF, et al.Dual RNA-seq of parasite and host reveals gene expression dynamics during filarial worm-mosquito interactions. PLoS Negl Trop Dis 2014;8:e2905.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giannoukos G, Ciulla DM, Huang K, et al.Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 2012;13:R23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vogel C, Abreu R, de S, Ko D, et al.Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol 2010;6:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolf J. Principles of transcriptome analysis and gene expression quantification: an RNA‐seq tutorial. Mol Ecol Resour 2013;13:559–72. [DOI] [PubMed] [Google Scholar]
- 30. Oshlack A, Wakefield MJ.. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct 2009;4:14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haas BJ, Chin M, Nusbaum C, et al.How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes?. BMC Genomics 2012;13:734.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tarazona S, García-Alcalde F, Dopazo J, et al.Differential expression in RNA-seq: A matter of depth. Genome Res 2011;21:2213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schulze S, Henkel SG, Driesch D, et al.Computational prediction of molecular pathogen-host interactions based on dual transcriptome data. Front Microbiol 2015;6:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang L, Schlesinger F, Davis CA, et al.Synthetic spike-in standards for RNA-seq experiments. Genome Res 2011;21:1543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parekh S, Ziegenhain C, Vieth B, et al.The impact of amplification on differential expression analyses by RNA-seq. Sci Rep 2016;6:25533.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korpelainen E, Tuimala J, Somervuo P, et al.RNA-seq Data Analysis. Abingdon, Oxfordshire, UK: CRC Press, 2014. [Google Scholar]
- 37. Bolger AM, Lohse M, Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmieder R, Edwards R.. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011;27:863–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim D, Langmead B, Salzberg SL.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2015;12:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langmead B, Trapnell C, Pop M, et al.Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pireddu L, Leo S, Zanetti G.. SEAL: a distributed short read mapping and duplicate removal tool. Bioinformatics 2011;27:2159–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li R, Yu C, Li Y, et al.SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009;25:1966–7. [DOI] [PubMed] [Google Scholar]
- 43. Wang K, Singh D, Zeng Z, et al.MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010;38:e178.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dobin A, Davis CA, Schlesinger F, et al.STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trapnell C, Pachter L, Salzberg SL.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baruzzo G, Hayer KE, Kim EJ, et al.Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Methods 2017;14:135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pruitt KD, Brown GR, Hiatt SM, et al.RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014;42:D756–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kent WJ, Sugnet CW, Furey TS, et al.The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Flicek P, Amode MR, Barrell D, et al.Ensembl 2014. Nucleic Acids Res 2014;42:D749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H, Handsaker B, Wysoker A, et al.The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robinson JT, Thorvaldsdóttir H, Winckler W, et al.Integrative genomics viewer. Nat Biotechnol 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skinner ME, Uzilov AV, Stein LD, et al.JBrowse: a next-generation genome browser. Genome Res 2009;19:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Griffith M, Walker JR, Spies NC, et al.Informatics for RNA sequencing: a web resource for analysis on the cloud. PLoS Comput Biol 2015;11:e1004393.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anders S, Pyl PT, Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Risso D, Schwartz K, Sherlock G, et al.GC-content normalization for RNA-Seq data. BMC Bioinformatics 2011;12:480.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hansen KD, Irizarry RA, Wu Z.. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 2012;13:204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bullard JH, Purdom E, Hansen KD, et al.Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 2010;11:94.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wagner GP, Kin K, Lynch VJ.. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci 2012;131:281–285. [DOI] [PubMed] [Google Scholar]
- 59. Robinson MD, Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010;11:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eder T, Grebien F, Rattei T.. NVT: a fast and simple tool for the assessment of RNA-seq normalization strategies. Bioinformatics 2016; 32:3682–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hardcastle TJ, Kelly KA.. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 2010;11:422.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trapnell C, Hendrickson DG, Sauvageau M, et al.Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2012;31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Anders S, Huber W.. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Robinson MD, McCarthy DJ, Smyth GK.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patro R, Duggal G, Love M, et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;4:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bray NL, Pimentel H, Melsted P, et al.Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34:525–527. [DOI] [PubMed] [Google Scholar]
- 67. Smyth GK. Limma: linear models for microarray data In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York, NY, USA: Springer, 2005, 397–420 [Google Scholar]
- 68. Ritchie ME, Phipson B, Wu D, et al.Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43: e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Soneson C, Delorenzi M.. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 2013;14:91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Young MD, Wakefield MJ, Smyth GK, et al.Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 2010;11:R14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Huang DW, Sherman BT, Lempicki RA.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 72. Oshlack A, Robinson MD, Young MD.. From RNA-seq reads to differential expression results. Genome Biol 2010;11:220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thingholm LB, Andersen L, Makalic E, et al.Strategies for integrated analysis of genetic, epigenetic, and gene expression variation in cancer: addressing the challenges. Front Genet 2016;7:2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smyth GK, Ritchie M, Thorne N, et al.Limma: Linear Models for Microarray Data User's Guide. 2016. http://www.bioconductor.org/packages/release/bioc/html/limma.html. [Google Scholar]