


Abstract
MicroRNAs (miRNAs) modulate the post-transcriptional regulation of target genes and are related to biology of complex human traits, but genetic landscape of miRNAs remains largely unknown. Given the strikingly tissue-specific miRNA expression profiles, we here expand a previous method to quantitatively evaluate enrichment of genome-wide association study (GWAS) signals on miRNA–target gene networks (MIGWAS) to further estimate tissue-specific enrichment. Our approach integrates tissue-specific expression profiles of miRNAs (∼1800 miRNAs in 179 cells) with GWAS to test whether polygenic signals enrich in miRNA–target gene networks and whether they fall within specific tissues. We applied MIGWAS to 49 GWASs (nTotal = 3 520 246), and successfully identified biologically relevant tissues. Further, MIGWAS could point miRNAs as candidate biomarkers of the trait. As an illustrative example, we performed differentially expressed miRNA analysis between rheumatoid arthritis (RA) patients and healthy controls (n = 63). We identified novel biomarker miRNAs (e.g. hsa-miR-762) by integrating differentially expressed miRNAs with MIGWAS results for RA, as well as novel associated loci with significant genetic risk (rs56656810 at MIR762 at 16q11; n = 91 482, P = 3.6 × 10−8). Our result highlighted that miRNA–target gene network contributes to human disease genetics in a cell type-specific manner, which could yield an efficient screening of miRNAs as promising biomarkers.
INTRODUCTION
Complex human traits are products of orchestration of high dimensional multi-omics layers, including genome, epigenome, transcriptome, proteome and metabolome. Genome-wide association study (GWAS) has discovered thousands of genomic loci associated with these traits (1,2). Integration of such large-scale genetic data with multi-layered omics information has successfully identified cell-type specific and context-dependent regulatory mechanism of diseases (3). While previous trans-omics approach mostly focused on integration with transcriptome (e.g. RNA-seq) (4) and epigenome data (e.g. ChiP-seq and Hi-C) (3,5), innovative construction of the analytic pipeline to integrate additional omics layers has been warranted to further elucidate complex biology of the traits.
Non-coding regions in the human genome constitute one of the unrevealed layers in biology. MicroRNAs (miRNAs), short non-coding RNA molecules of 21–25 nucleotide long, are key players in post-transcriptional gene regulation (6,7). Numerous studies have shown their critical role in the pathogenesis of various human diseases (8,9) and application of miRNAs as a biomarker or a therapeutic target is ongoing and promising (10,11). Nevertheless, it has been difficult to detect comprehensive association signals of miRNAs as compared with those of protein-coding genes and mRNAs, because genomic region that encodes miRNAs is relatively small. Biological roles of a miRNA should also be interpreted in a tissue specific context along with its target gene. On the arrival of recent high throughput sequencing technologies, comprehensive catalog of miRNA expression profile was created and revealed that the expression levels of miRNAs varied greatly according to tissues and were highly skewed (12).
Harnessing this tremendous work, here we extended our method to quantitatively evaluate enrichment of GWAS polygenic signals on miRNA–target gene networks (MIGWAS; miRNA–target gene networks enrichment on GWAS) that we have previously reported (13) to further decipher tissue-specific contribution of miRNA function in each trait. The MIGWAS enables us to study the tissue-specific landscape of post-transcriptional regulation, and to identify candidate miRNAs that are essential in pathophysiology. Our method can also conduct in silico screening of the miRNAs that can be used as novel biomarkers or therapeutic targets on the traits, which was empirically validated by the subsequent case-control analysis of differentially expressed miRNAs obtained from clinical subjects and the large-scale genetic association analysis of the lead variants.
MATERIALS AND METHODS
Calculation of gene- and miRNA- P values from GWAS summary statistics
We converted GWAS SNP association signals into a gene- or miRNA- level P value (i.e. PGene and PmiRNA, respectively). Similarly to the method Segrè et al. used in MAGENTA software (14), the best P value of a set of SNP P values mapped onto each gene or miRNA was corrected for the confounding effects of physical and genetic properties of genes or miRNAs on the P value (Figure 1A). We excluded genes and miRNAs located in the major histocompatibility complex (MHC) region to avoid the influence from its long linkage disequilibrium and complex architecture (15).
Figure 1.

Overview of MIGWAS approach. (A) GWAS summary statistics are converted to gene- and miRNA- level P values (PGene and PmiRNA). (B) MiRNA expression data from various human tissues was quantile-normalized, and a set of highly and tissue-specifically expressed miRNAs was defined in each cell type. (C) Cell-type specific enrichment of GWAS signal in miRNA–target gene network was assessed by a permutation procedure (see Materials and Methods).
Curation of miRNA expression data and miRNA–target gene network information
Tissue-specific miRNA expression data from the FANTOM5 consortium (12) excluding candidate novel ones was downloaded from a web site (see URLs). Expression count per million values of mature miRNAs derived from 1842 pre-miRNAs in 179 human cell types were quantile-normalized using preprocessCore v1.34.0 package in R software. These cell types were classified as 18 tissue type categories; bone (n = 10), brain (n = 14), cardiac (n = 3), eye (n = 3), fat (n = 15), fetal (n = 14), gastrointestinal (GI; n = 7), genitourinary (GU; n = 6), immune (n = 22), joint (n = 1), kidney (n = 7), liver (n = 4), lung (n = 10), muscle (n = 8), pancreas (n = 1), skin (n = 6), vascular (n = 15) and others (n = 33; Supplementary Table S1) based on the organs from which these cells were collected. The result of the principal component analysis describing the expression pattern of miRNAs in each cell is shown in Supplementary Figure S1. Next, we calculated a TSI as Ludwig et al. previously described (16). In brief, the TSI for jth miRNA was calculated as
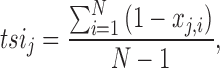 |
where N corresponds to the total number of cells measured and xj,i is the expression amount of ith cell normalized by the maximal expression of any cell for jth miRNA.
In each cell type, we defined a set of highly and specifically expressed miRNAs that satisfy two conditions; (i) normalized expression value falls within the top 10 percentile of those obtained from all miRNAs and (ii) TSI > 0.7 (Figure 1B). TSI > 0.7 threshold made it possible to include miRNAs with a wide range of expression levels and a variety of miRNAs, while retaining the power (Supplementary Figure S2). This set of highly and specifically expressed miRNAs was used to partition miRNA’s heritability signal into various human cell types.
We obtained four kinds of major target gene prediction algorithms; TargetScan Human, DIANA-TarBase, PITA, and miRDB (see URLs; Supplementary Table S2). For each of the prediction algorithms, we made a prediction score matrix with a row corresponding to genes, and a column corresponding to miRNAs.
Quantifying the enrichment of miRNA–gene network to a trait in a specific cell type
In order to empirically evaluate cell type-specific enrichment of miRNA–target gene network to a trait, we extended MIGWAS, which we have previously described (Figure 1C).
Let 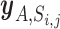 be the set of miRNA and gene pairs that satisfy the following four conditions: (i) target prediction score of the miRNA and gene is above jth threshold in ith prediction algorithm, (ii) the above mentioned PmiRNA is below a nominal signal (α = 0.01), (iii) the Pgene is also below α and (iv) the miRNA is included in a set of highly and specifically expressed miRNAs in cell A.
be the set of miRNA and gene pairs that satisfy the following four conditions: (i) target prediction score of the miRNA and gene is above jth threshold in ith prediction algorithm, (ii) the above mentioned PmiRNA is below a nominal signal (α = 0.01), (iii) the Pgene is also below α and (iv) the miRNA is included in a set of highly and specifically expressed miRNAs in cell A. 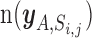 denotes the number of pairs included in the set
denotes the number of pairs included in the set 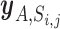 . We estimated the enrichment signal in cell A by comparing this metric
. We estimated the enrichment signal in cell A by comparing this metric 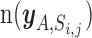 with the empirical null distribution using a permutation procedure (20 000 times). Under this null distribution, we assumed that there is no association among tissue-specific miRNA expression, miRNA–target gene network and GWAS signal. In each iteration step, we randomly shuffled P values of both all genes and all miRNAs while retaining the miRNA–target gene relationships, and calculated the metric of the kth permutation step as
with the empirical null distribution using a permutation procedure (20 000 times). Under this null distribution, we assumed that there is no association among tissue-specific miRNA expression, miRNA–target gene network and GWAS signal. In each iteration step, we randomly shuffled P values of both all genes and all miRNAs while retaining the miRNA–target gene relationships, and calculated the metric of the kth permutation step as 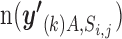 (i.e. the number of miRNA–gene pairs that met all the four condition (i)–(iv) with the shuffled P values). The significance
(i.e. the number of miRNA–gene pairs that met all the four condition (i)–(iv) with the shuffled P values). The significance 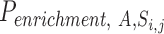 of the metric was defined as
of the metric was defined as 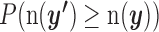 . We then sequentially slid the threshold j of the i th prediction algorithm from top one percentile to 0.1 percentile with eight partitions. When integrating the statistics, we adopted the results obtained only if the mean of
. We then sequentially slid the threshold j of the i th prediction algorithm from top one percentile to 0.1 percentile with eight partitions. When integrating the statistics, we adopted the results obtained only if the mean of 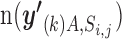 is more than five because the estimation of the null distribution can be biased when
is more than five because the estimation of the null distribution can be biased when 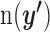 s are sparse. In order to compensate for the uncertainty existing within a single target prediction algorithm, we also integrated the results of four prediction algorithms to obtain one robust enrichment P value in cell A by meta-analyzing the enrichment significance as follows:
s are sparse. In order to compensate for the uncertainty existing within a single target prediction algorithm, we also integrated the results of four prediction algorithms to obtain one robust enrichment P value in cell A by meta-analyzing the enrichment significance as follows:
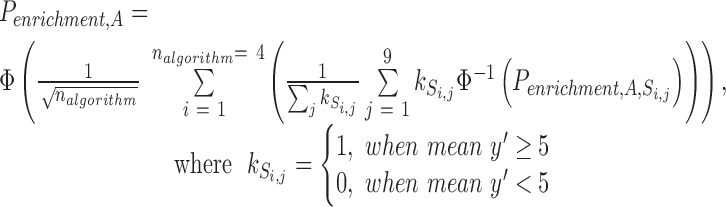 |
Φ represents the cumulative distribution function of the normal distribution. Permutation P value < 0.05 was interpreted as statistically significant in all the following analyses.
In addition to quantifying tissue specific enrichment signal, we recorded the pairs of a miRNA and a gene included in the set 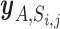 to be considered as tissue-specific trait associated pairs of a miRNA and a gene.
to be considered as tissue-specific trait associated pairs of a miRNA and a gene.
We have released a python source code together with formatted miRNA expression materials to allow users to perform MIGWAS using any GWAS summary statistics on their own computers (see URLs). A multithreading option using multiprocessing module in python is implemented to shorten the computing time (∼6.50 h when applied to summary statistics of one GWAS with ∼7 million variants for 179 cell types using four CPU cores with 2.3 GHz). Selection of the algorithms to predict miRNA–target genes, TSI thresholds, and permutation numbers can be optimized by the users.
GWAS summary statistic data sets
We collected 49 GWAS summary statistics (Supplementary Table S3). Twenty-one of them were provided from public websites or collaborators. They include diseases consisting of four major categories (immune/allergy-related [n = 4], neuropsychiatric [n = 2], cardiovascular [n = 1] and genitourinary [n = 1]) and quantitative traits consisting of five major categories (anthropometric [n = 3], metabolic [n = 3], musculoskeletal [n = 1], cardiovascular [n = 2], kidney-related [n = 2] and hematological [n = 2]). The remaining 28 summary statistics are obtained from ongoing BioBank Japan project (17), >170 000 genome and health-related phenotype biobank of Japanese. These include diseases consisting of nine major categories (immune/allergy-related [n = 4], metabolic [n = 1], musculoskeletal [n = 2], neuropsychiatric [n = 1], eye-related [n = 2], cardiovascular [n = 5], lung-related [n = 1], liver-related [n = 2], genitourinary [n = 2], and malignancy [n = 7]), and quantitative traits consisting of 2 anthropometric traits (adult height, and body mass index [BMI]). Definition of the diseases and the process of patient registration are described elsewhere (17,18).
From these summary statistics, SNP positions based on the UCSC hg19 reference and their association P values from linear regression (quantitative trait) or logistic regression (binary trait) are collected and re-formatted.
Study populations for high throughput miRNA expression sequencing
In this case-control study, Japanese patients with early rheumatoid arthritis (RA) were enrolled together with healthy Japanese volunteers. The diagnosis of RA was based on 2010 ACR/EULAR criteria (19). Written informed consent was obtained prior to the enrollment from all the individuals. In total, 30 RA patients and 33 healthy control (HC) participants were enrolled. Detailed information on the participants is in Supplementary Table S4. This study was approved by the ethical committee of Osaka University.
MiRNA expression sequencing and differential expression analysis in RA patients
Whole blood from the subject was collected into an ethylenediaminetetraacetic acid (EDTA) tube. PBMCs were isolated using Ficoll–Paque density gradient medium. Total RNA from PBMCs was isolated using miRNeasy Micro Kit (Qiagen). Libraries for miRNA-seq were prepared using SMARTer smRNA-Seq Kit (Takara) following manufacturer instructions. RNA-seq was performed using a HiSeq 2500 (Illumina, read length of 100 bp, single-end). For QC of the sequencing data, adaptor sequences were trimmed using Cutadapt, and reads with low quality score (Phred quality score < 20 in > 80% of total bases) and reads with length >50 bp were removed. All the samples have total read counts > 1.0 × 106, and thus proceeded to further analysis. Read count information is summarized in Supplementary Table S4. Reads were aligned against the known miRNA sequences in miRBase database, using Bowtie with recommended options described in the literature (20). Differential miRNA expression analysis between RA and HC was carried out with the R package DESeq2.
Overlap enrichment analysis of differentially expressed miRNAs with the MIGWAS result
To test whether differentially expressed miRNAs between RA patients and HC significantly overlap with candidate RA-associated miRNAs obtained from the MIGWAS, we performed a permutation procedure. We randomly shuffled sample IDs of the miRNA expression table, and defined differentially expressed miRNAs (FDR-q < 0.05). The number of overlapped miRNAs included both in differentially expressed miRNAs and in the MIGWAS candidates was recorded in each permutation step. After 5000 permutation steps, empirical P value was calculated as the number of permutation steps where the number of overlapped miRNAs was equal to or exceeded that from real dataset divided by the total number of permutation steps ( = 5000 iterations).
The in silico replication study of the biomarker miRNA loci with RA risk
For each genetic locus that harbors a biomarker miRNA identified by the MIGWAS of RA and the differentially expressed miRNA analysis of the RA patients, we selected the lead SNP with the most significant association within the locus from the original trans-ethnic RA meta-analysis (19 234 RA cases and 61 565 controls) (21). By looking-up the additional two RA GWAS of Japanese (3308 RA cases and 8357 controls; Supplementary Table S5) (22), we conducted the in silico replication study of the lead SNPs with RA risk. Meta-analysis of the GWAS and replication study was conducted by an inverse-variance method assuming a fixed-effects model.
Integrative analysis with eQTL summary data and summary-level Mendelian Randomization (SMR) analysis
We hypothesized that miRNAs identified by the MIGWAS pipeline exert their effects on the trait by regulating the transcripts of their target genes, and that the target genes’ expression level should also have a dose dependent association with the trait. To test this hypothesis, we undertook two ways of analysis using publicly available eQTL summary data. First, we tested whether target genes of the identified miRNA are enriched in eGenes within whole blood when compared with those across all tissues using summary eQTL data from GTEx consortium (23). eGenes in each tissue were defined as genes that harbor cis-eQTL variants which associate with their expression level with FDR-q < 0.05. The enrichment was assessed by the binomial test, with the prior probability being the number of eGenes within whole blood divided by the number of eGenes across all tissues that were included within the scope of eQTL analysis, GWAS, and miRNA’s target prediction. Second, we performed SMR to show that the expression level of target genes that were identified through the above-mentioned analysis associates with the trait by causality or pleiotropy, and not by linkage. By integrating trans-ethnic GWAS summary statistics of RA (21) with CAGE eQTL data of peripheral blood (24), we did SMR analysis for 41 target genes of hsa-miR-762. We considered PHEIDI ≥ 0.05 as a threshold that we could not reject the null hypothesis that there was a single causal variant affecting both gene expression and trait variation (25).
RESULTS
Overview of our MIGWAS statistical methods
The overview of our MIGWAS method is shown in Figure 1. The principal hypothesis of MIGWAS is that if the miRNA is associated with a genetic risk of a trait, the target genes of the miRNA are also associated with the trait as a miRNA functionally works by interacting with its target genes. To test this hypothesis, we first converted SNP-level association statistics in the GWAS into gene- and miRNA-level association P values (i.e. PGene and PmiRNA, respectively; Figure 1A). Then we quantitatively evaluated enrichment of pairwise association signals of miRNAs and their target genes. We note that we set the significance threshold of PGene and PmiRNA (α = 0.01) less stringent than the typical genome-wide significant level (26), to enhance sensitivity and to comprehensively incorporate the polygenic nature of complex traits (27,28).
In order to quantify tissue-specific contribution of miRNAs in each trait, we next partitioned enrichment of miRNA–target gene signals into each cell type separately. After normalization of miRNA expression data released by the FANTOM5 consortium (12), we defined a set of highly and tissue-specifically expressed miRNAs in a total of 179 human cells (Figure 1B). We performed cell type-based partitioning analysis within the defined set of miRNAs of each of the 179 cell types (Figure 1C), and thus obtained cell-type specific enrichment P value through permutation-based parallel computing. In addition our MIGWAS pipeline automatically identifies a set of trait-associated miRNA–target gene pairs for both specific tissue categories and in all tissue categories combined together.
Heterogeneous enrichment of miRNA–target gene network to human complex traits
We applied the extended MIGWAS pipeline to the 49 GWAS summary statistics to evaluate the overall contribution of miRNA–target gene networks to various complex human traits, firstly without considering tissue-specificity. The GWASs covered a wide range of traits including anthropometric (n = 4), immune/allergy (n = 8), metabolic (n = 5), musculoskeletal (n = 2), neuropsychiatric (n = 3), eye-related (n = 2), cardiovascular (n = 8), lung-related (n = 1), kidney-related (n = 2), liver-related (n = 2), genitourinary (n = 3), hematological (n = 2) and malignancy traits (n = 7). The detailed information of the GWASs is shown in Supplementary Table S3 and the Materials and Methods section. We observed nominally significant (P < 0.05) miRNA–target gene enrichment signal in three traits for five GWASs; height, RA and type 2 diabetes (Figure 2A), which was consistent and robust with our previous results despite of the update on miRNA–target gene prediction algorithms (13). We note that we observed trans-ethnically consistent results for height and type 2 diabetes (P < 0.05 in both populations), two traits for which independent summary statistics were obtained from European and Japanese populations separately (29,30), which empirically validates the robustness of the MIGWAS results.
Figure 2.

MIGWAS results of the GWASs on 49 complex human traits. An enrichment signal is shown by -log10(PMIGWAS). (A) Overall polygenic contribution of miRNA–target gene network to the traits through the tissue-naïve approach. Traits that have bars colored with pink showed nominally significant enrichment (PMIGWAS < 0.05). (B) An example of tissue-specific enrichment signals focusing on immune-related cells. A bar in each trait represents an enrichment signal of the most significant immune-related cell. (C) An enrichment signal in the strongest associated cell of each trait. The tissue category of the cell is shown by the color of the bars. Detailed descriptions on cell types and their categories are available at Supplementary Table S1. GI; gastrointestinal, GU; genitourinary.
Tissue specific MIGWAS results successfully identified the disease-relevant tissue and associated miRNAs
Motivated by the robustness of our MIGWAS pipeline supported by trans-ethnic consistency, next we performed the partitioned enrichment analysis using the tissue specific miRNA expression data. Each trait was analyzed considering 179 different human cell types in parallel, and we annotated each cell into 18 tissue categories from which it was collected from (Supplementary Table S1). We here highlighted enrichment signal to immune-related cells in each trait as an example (Figure 2B). Tissue specificity of the traits in immune-related cells showed the different spectrum of enrichment with the most significant enrichment in Graves’ disease (P = 2.6 × 10−6). Relatively high enrichment signals were also found in type 2 diabetes as well (P < 5.9 × 10−5 in both populations), where obesity-associated chronic tissue inflammation is reported to play a key contributing role (31).
Figure 2C shows the strongest associated cell in each trait. These results successfully illustrated the diverse tissue-specific nature of miRNA–target gene contribution to a trait, which was not obvious through the tissue-naïve approach. In many cases the analysis identified biologically relevant cell types in each trait (e.g. immune cells in Graves’ disease, chronic hepatitis C, systemic lupus erythematosus, Crohn's disease and asthma, adipocytes in LDL cholesterol and coronary artery disease, brain tissue in age at menarche (32)). Detailed cell names of top association in all the traits are described in Supplementary Table S6.
Considering the abundance of accumulated literature on miRNA’s associations in immune- and allergy- related traits (33), we highlighted detailed enrichment signals in immune- and allergy- related traits (Figure 3). Here again, we observed disease relevant cell types in each trait. In RA, miRNA–target gene contribution was enriched in lung, bone, and immune cells (P = 3.6 × 10−3, 9.6 × 10−3 and 1.5 × 10−2, respectively), which is consistent with biological understanding of the disease that pathogenic autoimmunity is first triggered in the lungs faced with environmental stimuli such as cigarette smoking and then causes bone erosion (34). We note that these disease-relevant tissues have not been identified by any previous methods to integrate with mRNA transcriptome and epigenome data despite their pivotal roles in RA pathophysiology (3,4,5), thereby demonstrating validity of our method to incorporate tissue-specific miRNA profiles. All the cell names with significant enrichment in immune-related traits are described in Supplementary Table S6.
Figure 3.

Tissue-specific enrichment signal of miRNA–target gene network in selected immune/allergy- related traits. Each circle represents the cell-specific enrichment signal in –log10(PMIGWAS), colored with tissue categories. Large circles pass the nominal threshold of enrichment P value < 0.05, which was indicated as dotted lines. The names of top 2 enriched cells that passed the threshold are also shown. Detailed descriptions on cell types and their categories are available at Supplementary Table S1. GI; gastrointestinal, GU; genitourinary.
Differential expression of miRNAs among rheumatoid arthritis patients enriches in MIGWAS result and pinpoints novel causal mechanisms
Through the MIGWAS pipeline, we also obtained a list of candidate trait-associated miRNAs and target genes as candidate biomarkers, which was defined systematically to satisfy the criteria used in the enrichment analysis (i.e. both miRNA and its predicted target gene are associated with the trait with a nominal significance [P value < 0.01]) with all tissue categories combined together. Given the previous studies showing the association of miRNAs in the biology of RA (13,35,36), we decided to focus on RA as a target of in vivo validation. To validate candidate novel miRNAs of which association with RA was identified by MIGWAS, we profiled miRNA expression in peripheral blood mononuclear cells (PBMCs) from 30 patients with early RA and 33 control subjects using high throughput miRNA sequencing. PBMCs were selected for the miRNA expression profiling because they have been shown to harbor much of heritability in the pathology of RA (3,32), and also because they can be collected in the clinical setting. We found 94 differentially expressed miRNAs with a false discovery rate (FDR) of 0.05 (Figure 4A). Of these, four miRNAs (hsa-miR-93-5p, hsa-miR-106b-5p, hsa-miR-301b-3p and hsa-miR-762) overlapped those identified by the MIGWAS pipeline applied for the RA GWAS. Permutation procedures in which sample IDs of the miRNA expression data were randomly shuffled 5000 times revealed that the observed overlap of as many as four miRNAs was much larger than would be expected by chance (98.0-fold enrichment [four miRNAs divided by the mean simulated overlap, 0.0408], permutation P value = 0.0010 [five simulation steps with equal or more overlap than the observed overlap divided by total simulation steps], Materials and Methods for permutation procedure; Table 1 and Supplementary Figure S4). One of the overlapped miRNAs, hsa-miR-762, was significantly highly expressed in RA patients (log2 fold change = 1.15 and FDR-q = 0.043) and has a prominent expression specific to immune-related cells (tissue specificity index [TSI] = 0.982, Figure 4B and C). This would implicate the utility of hsa-miR-762 as a future target of clinical validation for a biomarker of RA. On the other hand, the other three miRNAs showed repressed expressions in the RA patients with ubiquitous expression profiles among the cell types (TSI < 0.40).
Figure 4.

Results of differentially expressed miRNA analysis between RA patients and healthy controls. (A) Volcano plot displaying differentially expressed miRNAs between RA and healthy controls. The x-axis corresponds to the log2 fold change value and the y-axis corresponds to –log10(FDR-q). The blue dots represent the differentially expressed miRNAs (FDR-q < 0.05). The pink dots represent differentially expressed miRNAs that overlapped with those identified in MIGWAS analysis. (B) The distribution of tissue specificity index (TSI) among all the miRNAs in expression data by FANTOM5. Bars colored with pink show the bins which the overlapped candidate miRNAs belong to. (C) Quantile-normalized expression values of the overlapped miRNAs in 179 cell types (x-axis), scaled as fold changes of the expression levels from the mean levels among all the cell types (y-axis). Bar colors represent the tissue into which each cell is categorized. GI; gastrointestinal, GU; genitourinary. (D) CIRCOS plot that shows a Manhattan plot of the RA GWAS marked by MIR762 and its target genes in red. (E) Regional plots of the RA GWAS illustrating variants neighboring MIR762 (left) and SYNGR1 (right top). Diamonds represent the leading variants, and also shown are their identifiers, linkage disequilibrium structure and association statistics. Two diamonds in the left panel represent the P value of rs2069235 from the original RA GWAS summary statistics (PGWAS) (21) and one from the new meta-analysis described in this paper (PMETA_GWAS). Right bottom, eQTL P values from CAGE dataset for a probe tagging SYNGR1.
Table 1.
Candidate biomarker miRNAs identified both in the differentially expressed miRNA study and in the MIGWAS analysis (all cell types combined)
| RA cases versus controls | ||||
|---|---|---|---|---|
| Candidate biomarker miRNA | log2 fold change in expression | FDR-q | TSI | Target genes |
| hsa-miR-93–5p | -0.522 | 0.00022 | 0.254 | ANKH, ANKRD52,ARCN1,BCL2L15,C7orf43,CASP8, CDKN1A, DENND1B, FAM126B, FAM133B, FYCO1, IKZF4, KIAA1109,KLF2,LDLR, MAGI3, PEX13,PHTF1,PRDX5, RAB5B, RAG1, RSBN1,SAR1B, SEC24A, SLC12A5, SNN, STK38, TAGAP, TRAK2, ZBTB10 |
| hsa-miR-106b-5p | -0.811 | 0.00022 | 0.269 | ANKH, ANKRD52,BCL2L15,C7orf43,CASP8, CDK6, CDKN1A,CEP76, FAM126B,FAM167A, FAM65B, FYCO1, ICOS,KLF2,LDLR, MAGI3, PA2G4, PAPOLG,PDGFB, RAB5B, RSBN1,RTKN2,SAR1B, STK38, TMEM151B, TNFAIP3, VPS37C,ZFP36L1 |
| hsa-miR-301b-3p | -0.954 | 0.0090 | 0.397 | C5orf30, CDK6, DDX6,ITSN1,LDLR, PAN3,SAR1B, SERBP1, SNRPE, SRSF3,TMEM50B, ZBTB10 |
| hsa-miR-762 | 1.154 | 0.043 | 0.982 | ARHGAP20, C11orf20, C11orf9, C1orf93, C7orf59, CPNE5,DAP,FADS2,GATS,GNAI2, HDAC5,INPP5B,IQGAP1,IRF5,ITSN1,PADI2,PFKL,PGAP3, PHF15, PHKG2, PHLDB1, PKNOX2,PPIL2, RAF1, RAVER1, SLC25A23,SLC44A2, STAC2,SYNGR1, TAB1, TMEM151B,TMPRSS3, TNFRSF14, TRAF1,TSPAN33,UBASH3A, UPK2,UTP11L, VPS37C,YDJC,ZNF594 |
Each miRNA’s target genes were defined to have top one percentile of target prediction score in at least two prediction algorithms. Genes in bold face are those overlapped with eGenes of GTEx eQTL data of whole blood. TSI; tissue specificity index, RA; rheumatoid arthritis.
While genetic variants located nearby the identified biomarker miRNAs confer suggestive associations in the original RA case-control GWAS (19 234 RA cases and 61 565 controls, PGWAS≥ 3.2 × 10−7) (21), overlap with the RA case-control miRNA-seq analysis strongly prioritizes true-positive associations of the lead variants in such loci. Thus, we conducted an in silico replication study using additional 3308 RA cases and 8357 controls (Supplementary Table S5) (22). Of these, two loci that harbor the three miRNAs satisfied the genome-wide significance threshold when GWAS and the replication study was meta-analyzed (22 119 cases and 69 363 controls; PMETA_GWAS = 3.3 × 10−8 for rs34130487 at MIR95-MIR106B at 7q22 and P = 3.6 × 10−8 for rs56656810 at MIR762 at 16q11; Figure 4D, E, Table 2, and Supplementary Table S5), of which RA associations were novel findings.
Table 2.
RA case-control associations of the SNPs located at the biomarker miRNA loci
| No. subjects | Allele 1 frequency | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| rsID | Chr | Position | Allele 1/2 | Gene | RA cases | Controls | RA cases | Controls | OR (95%CI) | P |
| rs34130487 | 7 | 99 759 205 | C/T | MIR95-MIR106B | 16 633 | 54 807 | 0.638 | 0.623 | 1.08 (1.05–1.11) | 3.3×10–8 |
| rs56656810 | 16 | 30 788 759 | A/C | MIR762 | 22 119 | 69 363 | 0.210 | 0.196 | 1.09 (1.05–1.12) | 3.6×10–8 |
| rs11089637 | 22 | 21 979 096 | C/T | MIR301B | 22 119 | 69 363 | 0.280 | 0.273 | 1.07 (1.05–1.10) | 3.7×10–7 |
RA, rheumatoid arthritis; OR, odds ratio; 95%CI, 95% confidence interval.
Detailed results are available at Supplementary Table S5.
We further listed target genes of these miRNAs (Table 1). As for hsa-miR-762, 41 genes were identified as potential target genes that work synergistically to cause RA (Figure 4D). To investigate whether those target genes have an eQTL effect in immune-related cells where hsa-miR-762 is supposed to be functioning, we referred to the summary eQTL statistics in human tissues released by GTEx consortium (23). We observed that target genes of hsa-miR-762 harbor cis-eQTL variants within whole blood as a proxy of immune-related cells more frequently than expected when compared to those in all available human tissues (1.46-fold enrichment with binomial P value = 0.014). This consistently suggests that miRNAs should modulate the association with the trait through interfering the transcriptome, and that the targeted gene's association with the trait should be driven by regulation on expression within a specific trait-associated tissue.
In order to clarify the connection between eQTL signals of the variants which hsa-miR-762’s target genes harbor and their neighboring association signals in RA GWAS, we performed the summary data-based Mendelian randomization (SMR) (25). This analysis can test whether the expression level of the target genes associates with RA by causality or pleiotropy, and not by linkage. In many of the target genes of hsa-miR-762, we could finemap the potentially causal or pleiotropic eQTL variants that not only existed nearby GWAS genes but also both mediated gene-expression levels and were associated with RA risk (Supplementary Table S7 for the summary statistics). As an illustrative example, genetic variants at SYNGR1 locus, which was one of the target genes of hsa-miR-762 and showed the strongest association statistics in the SMR analysis, are associated with RA risk (upper right panel at Figure 4E). In expression data from Consortium for the Architecture of Gene Expression (CAGE) in PBMCs (24), SYNGR1 locus harbors cis-eQTL variants (lower right panel at Figure 4E). The SMR analysis reveals that GWAS signal of the SNP (rs2069235, PGWAS = 7.6 × 10−13, PeQTL = 1.3 × 10−38) in SYNGR1 is strongly and significantly mediated by eQTL effect (PSMR = 3.5 × 10−10), which was not driven by linkage (PHEIDI = 0.27). The MIGWAS pipeline and miRNA expression profiles from clinical subjects successfully pinpointed the causal mechanism of MIR762’s association with RA, where increased expression of hsa-miR-762 in immune-related cells interferes with transcripts such as SYNGR1, whose expression levels are associated with causing RA.
DISCUSSION
By integrating the large scale GWAS data and miRNA–target gene prediction algorithms together with comprehensive tissue-specific miRNA expression profiles, here we successfully extended a method MIGWAS to evaluate enrichment of GWAS signals on miRNA–target gene networks and to partition them into a tissue specific context. The application of the MIGWAS pipeline to a wide range of genetics of complex human traits depicted cell type-specificity of the trait biology, as well as identification of biomarker miRNAs and novel genetic risk loci such as hsa-miR-762 encoded in the MIR762 locus at 16q11 for RA. The implementation of genetic information on miRNAs will certainly serve to help investigators elucidate pathophysiology and forward clinical application, as well as further development of novel therapeutic targets.
Our MIGWAS pipeline highlights four innovative features: (i) quantitatively assessing the miRNA–target gene contribution to genetics of a trait, (ii) deciphering tissue specificity to identify causal tissues on a trait, (iii) efficiently prioritizing genetic loci for a follow-up study that harbor true-positive associations with disease risk and (iv) providing candidate biomarker miRNA–target gene lists that might contribute to the understanding of disease biology. Our work firmly supports the idea that the miRNA’s contribution to genetics of complex human traits occurs in a cell type-specific manner in the same way as gene expression (4) and epigenetic regulation (3,32). A list of candidate miRNA–target gene pairs provides an ideal resource for experimental validation aiming at the discovery of biomarkers or potential therapeutics. The MIGWAS enables us to pinpoint a causal miRNA–target gene network to a trait, which was not achieved by GWAS alone or previous approaches to integrate GWAS and epigenome or transcriptome data.
We acknowledge that our study has several limitations. First, tissue specific MIGWAS analysis was performed based on an expression data from healthy individuals, while miRNA expression profile might be different depending on the disease status of individuals. We consider that a compromising approach to use summary expression data from healthy individuals as ours has been shown to successfully capture enrichment of the trait heritability to biologically-relevant tissue (37). However, to have further insights into the disease biology, future application of expression profiles from pathologically altered tissues or inclusion of specifically depleted miRNAs should be warranted. Second, the in silico prediction of a miRNA and a target gene confers ambiguity dependent on the algorithms. In order to obtain unbiased prediction scores and to assure the robustness of the analysis, we integrated multiple different algorithms. When compared with a recently updated database miRTarBase (38), which provides the largest amount of miRNA–target gene interactions with experimental validations to date, our approach of multiple thresholding and multiple algorithms indeed contributed to the better functional prediction in vitro (Supplementary Figure S3). Further integration of such experiment-based target gene information into the MIGWAS pipeline would be warranted. Third, since our method is based on the GWAS results, enrichment signal could be inflated due to the inflated GWAS summary statistics. To address this point, we took a permutation approach to estimate the null distribution, which made the MIGWAS results robust against the inflation of the GWAS itself (13). Forth, we did not incorporate tissue-specific expression profile of genes in our method. The interaction of both miRNAs’ and their target genes’ expression levels is beyond this study because it suffers from exponential combinations and computational intensiveness. Nevertheless, future integration would contribute to deciphering fine nature of tissue specificity.
In conclusion, MIGWAS demonstrated that miRNA–target gene networks contribute to human disease genetics in the context of cell type-specific expressions, and successfully identified miRNAs as promising biomarkers.
URLs
The URLs for data presented herein are as follows:
MIGWAS source code, https://github.com/saorisakaue/MIGWAS
FANTOM5 consortium, http://fantom.gsc.riken.jp/5/
The BioBank Japan Project (BBJ), https://biobankjp.org/english/index.html
miRBase, http://www.mirbase.org/
miRDB, http://www.mirdb.org/
TargetScan Human, http://www.targetscan.org/vert_72/
DIANA-TarBase, http://diana.imis.athena-innovation.gr/DianaTools/index.php
PITA, https://genie.weizmann.ac.il/pubs/mir07/mir07_data.html
GTEx consortium, https://www.gtexportal.org/home/
CAGE eQTL data, http://cnsgenomics.com/shiny/CAGE/
R DESeq2 package, https://bioconductor.org/packages/release/bioc/html/DESeq2.html
R preprocessCore package, https://bioconductor.org/packages/release/bioc/html/preprocessCore.html
SMR software, cnsgenomics.com/software/smr/
CIRCOS, http://circos.ca/
Cutadapt, cutadapt.readthedocs.io/en/stable/
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Kiyoshi Takeda and Dr Miho Kaneda for their kind supports on the study. We would like to thank all members of the FANTOM5 consortium for contributing to generation of samples and analysis of the data-set and thank GeNAS for data production.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science (JSPS) KAKENHI [15H05670, 15H05911, 15K14429, 16KT0196]; Japan Agency for Medical Research and Development [17ek0410047h0001, 18gm6010001h0003, 18ek0410041h0002]; Takeda Science Foundation; Nakajima Foundation; SECOM Science and Technology Foundation, Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives, Osaka University, Bioinformatics Initiative of Osaka University Graduate School of Medicine, and Inoue Foundation for Science. FANTOM5 was made possible by a Research Grant for RIKEN Omics Science Center from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to Y.H.); MEXT for the RIKEN Preventive Medicine and Diagnosis Innovation Program (to Y.H.); Grant of the Innovative Cell Biology by Innovative Technology (Cell Innovation Program) from the MEXT, Japan (to Y.H.); Grant from MEXT to the RIKEN Center for Life Science Technologies and Grant from MEXT to RIKEN Center for Integrative Medical Sciences; J.H. is an employee of TEIJIN PHARMA LIMITED; N.S. is an employee of Ono Pharmaceutical Co., Ltd. Funding for open access charge: Japan Agency for Medical Research and Development [18ek0410041h0002].
Conflict of interest statement. None declared.
REFERENCES
- 1. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L. et al. . The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Visscher P.M., Brown M.A., McCarthy M.I., Yang J.. Five years of GWAS discovery. Am. J. Hum. Genet. 2012; 90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S., Raychaudhuri S.. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013; 45:124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ongen H., Brown A.A., Delaneau O., Panousis N.I., Nica A.C., Dermitzakis E.T.. Estimating the causal tissues for complex traits and diseases. Nat. Genet. 2017; 49:1676–1683. [DOI] [PubMed] [Google Scholar]
- 5. Martin P., McGovern A., Orozco G., Duffus K., Yarwood A., Schoenfelder S., Cooper N.J., Barton A., Wallace C., Fraser P. et al. . Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat. Commun. 2015; 6:10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee R., Feinbaum R., Ambros V.. A short history of a short RNA. Cell. 2004; 116:S89–S92. [DOI] [PubMed] [Google Scholar]
- 7. Lee R.C., Ambros V.. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001; 294:862–864. [DOI] [PubMed] [Google Scholar]
- 8. Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A. et al. . MicroRNA expression profiles classify human cancers. Nature. 2005; 435:834–838. [DOI] [PubMed] [Google Scholar]
- 9. Brest P., Lapaquette P., Souidi M., Lebrigand K., Cesaro A., Vouret-Craviari V., Mari B., Barbry P., Mosnier J.-F., Hébuterne X. et al. . A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat. Genet. 2011; 43:242–245. [DOI] [PubMed] [Google Scholar]
- 10. Krützfeldt J., Rajewsky N., Braich R., Rajeev K.G., Tuschl T., Manoharan M., Stoffel M.. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005; 438:685–689. [DOI] [PubMed] [Google Scholar]
- 11. Titze-de-Almeida R., David C., Titze-de-Almeida S.S.. The race of 10 synthetic RNAi-based drugs to the pharmaceutical market. Pharm. Res. 2017; 34:1339–1363. [DOI] [PubMed] [Google Scholar]
- 12. De Rie D., Abugessaisa I., Alam T., Arner E., Arner P., Ashoor H., Åström G., Babina M., Bertin N., Burroughs A.M. et al. . An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017; 35:872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okada Y., Muramatsu T., Suita N., Kanai M., Kawakami E., Iotchkova V., Soranzo N., Inazawa J.. Significant impact of miRNA – target gene networks on genetics of human complex traits. Sci. Rep. 2016; 6:22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ayellet V.S., Groop L., Mootha V.K., Daly M.J., Altshuler D.. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010; 6:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okada Y., Momozawa Y., Ashikawa K., Kanai M., Matsuda K., Kamatani Y., Takahashi A., Kubo M.. Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nat. Genet. 2015; 47:798–802. [DOI] [PubMed] [Google Scholar]
- 16. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stähler C., Meese E. et al. . Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T. et al. . Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 2017; 27:S2–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirata M., Kamatani Y., Nagai A., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Kubo M., Muto K., Mushiroda T. et al. . Cross-sectional analysis of BioBank Japan clinical data: A large cohort of 200,000 patients with 47 common diseases. J. Epidemiol. 2017; 27:S9–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aletaha D., Neogi T., Silman A.J., Funovits J., Felson D.T., Bingham C.O., Birnbaum N.S., Burmester G.R., Bykerk V.P., Cohen M.D. et al. . 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010; 62:2569–2581. [DOI] [PubMed] [Google Scholar]
- 20. Ziemann M., Kaspi A., El-Osta A.. Evaluation of microRNA alignment techniques. RNA. 2016; 22:1120–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okada Y., Wu D., Trynka G., Raj T., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Yoshida S. et al. . Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014; 506:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okada Y., Suzuki A., Ikari K., Terao C., Kochi Y., Ohmura K., Higasa K., Akiyama M., Ashikawa K., Kanai M. et al. . Contribution of a Non-classical HLA Gene, HLA-DOA, to the Risk of Rheumatoid Arthritis. Am. J. Hum. Genet. 2016; 99:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. GTEx Consortium Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lloyd-Jones L.R., Holloway A., McRae A., Yang J., Small K., Zhao J., Zeng B., Bakshi A., Metspalu A., Dermitzakis M. et al. . The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet. 2017; 100:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M. et al. . Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016; 48:481–487. [DOI] [PubMed] [Google Scholar]
- 26. Kanai M., Tanaka T., Okada Y.. Empirical estimation of genome-wide significance thresholds based on the 1000 Genomes Project data set. J. Hum. Genet. 2016; 61:861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee S.H., Wray N.R., Goddard M.E., Visscher P.M.. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011; 88:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bulik-Sullivan B., Loh P.R., Finucane H.K., Ripke S., Yang J., Patterson N., Daly M.J., Price A.L., Neale B.M., Corvin A. et al. . LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015; 47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wood A.R., Esko T., Yang J., Vedantam S., Pers T.H., Gustafsson S., Chu A.Y., Estrada K., Luan J., Kutalik Z. et al. . Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014; 46:1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium Mahajan A., Go M.J., Zhang W., Below J.E., Gaulton K.J. et al. . Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet. 2014; 46:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osborn O., Olefsky J.M.. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012; 18:363–374. [DOI] [PubMed] [Google Scholar]
- 32. Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K. et al. . Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015; 47:1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J.-Q., Papp G., Szodoray P., Zeher M.. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun. Rev. 2016; 15:1171–1180. [DOI] [PubMed] [Google Scholar]
- 34. Malmström V., Catrina A.I., Klareskog L.. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat. Rev. Immunol. 2017; 17:60–75. [DOI] [PubMed] [Google Scholar]
- 35. Chen J.-Q., Papp G., Szodoray P., Zeher M.. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun. Rev. 2016; 15:1171–1180. [DOI] [PubMed] [Google Scholar]
- 36. Murata K., Yoshitomi H., Furu M., Ishikawa M., Shibuya H., Ito H., Matsuda S.. MicroRNA-451 Down-Regulates neutrophil Chemotaxis via p38 MAPK. Arthritis Rheumatol. 2014; 66:549–559. [DOI] [PubMed] [Google Scholar]
- 37. Finucane H.K., Reshef Y.A., Anttila V., Slowikowski K., Gusev A., Byrnes A., Gazal S., Loh P.R., Lareau C., Shoresh N. et al. . Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 2018; 50:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chou C.-H., Shrestha S., Yang C.-D., Chang N.-W., Lin Y.-L., Liao K.-W., Huang W.-C., Sun T.-H., Tu S.-J., Lee W.-H. et al. . miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018; 46:D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.