


Abstract
Previous investigations of the impact of an imidazole-tethered thymidine in synthetic DNA duplexes, monitored using UV and NMR spectroscopy, revealed a base context dependent increase in thermal stability of these duplexes and a striking correlation with the imidazolium pKa. Unrestrained molecular dynamics (MD) simulations demonstrated the existence of a hydrogen bond between the imidazolium and the Hoogsteen side of a nearby guanosine which, together with electrostatic interactions, form the basis of the so-called pKa-motif responsible for these duplex-stabilizing and pKa-modulating properties. Here, the robustness and utility of this pKa-motif was explored by introducing multiple imidazole-tethered thymidines at different positions on the same dsDNA duplex. For all constructs, sequence based expectations as to pKa-motif formation were supported by MD simulations and experimentally validated using NOESY. Based on the analysis of the pKa values and melting temperatures, guidelines are formulated to assist in the rational design of oligonucleotides modified with imidazolium-tethered thymidines for increased thermal stability that should be generally applicable, as demonstrated through a triply modified construct. In addition, a proof-of-principle study demonstrating enhanced stability of the l-argininamide binding aptamer modified with an imidazole-tethered thymidine in the presence and absence of ligand, demonstrates its potential for the design of more stable aptamers.
INTRODUCTION
At ambient pressure and temperature, proteins often show unmatched efficiency and selectivity in the high affinity binding of specific targets (cfr. antibodies) and in catalysis (cfr. enzymes). This makes them interesting partners in the development of immunoassays and affinity purification methods on the one hand (1,2) and more energy efficient and sustainable chemical processes on the other hand (3). Besides protein based architectures the unique ability of nucleic acids to form high order single or double stranded structures has led these to enjoy considerable attention as artificial binders and enzymes, so-called aptamers and (deoxy)ribozymes. Aptamers are single stranded nucleic acids selected by an iterative in vitro selection process called ‘SELEX’ (Systematic Evolution of Ligands by EXponential enrichment) and feature a specific tertiary structure featuring double stranded regions as well as hairpin and loop regions, capable of recognising and binding a target molecule with great selectivity and affinity. They are considered as promising alternatives for antibodies and continue to find new applications as therapeutics (4,5), biosensors (6,7), drug delivery systems (8,9) or as bio-imaging (9,10) and diagnostic tools (9,11). Shortly after the development of nucleic acid aptamers, catalytically active deoxyribozymes were generated from random libraries of oligonucleotides by a combinatorial in vitro selection approach (12), similar to SELEX. The scope of deoxyribozymes has expanded from reactions with nucleic acid substrates such as cleavage (13–15) and ligation (16,17) in the early beginning, to diverse chemical transformations including ester hydrolysis (18), Diels–Alder cycloaddition (19) and Friedel–Crafts alkylation (20).
Despite the power of directed evolution approaches in the creation of functional nucleic acids, the success rates of aptamer and (deoxy)ribozyme development is substantially constrained by the limited physicochemical diversity of native nucleic acids, especially when compared to proteins. This can be addressed by introducing protein-like functionalities through modification of the phosphate backbone, sugar moiety or nucleobase. According to literature, this leads to aptamers with increased target affinity (21–29), selectivity (30,31) and pharmacokinetic parameters (32) as well as metal-ion independent deoxyribozymes that outperform their unmodified counterparts (33–36). The established way to introduce extra functionalities is the use of modified (2′-deoxy-)nucleoside triphosphates in the selection protocol (37,38). Alternatively, modifications can be embedded selectively, by using modified monomers during solid phase oligonucleotide synthesis (23,39). Chemical synthesis offers a far larger functional versatility and spatial control when it comes to the introduction of unnatural nucleotides. In addition, it allows for (semi-)rational approaches to develop new functional nucleic acids with improved properties, as an alternative to randomized selection.
The imidazole group is of particular interest in this context and is an often-recurring functionality in functional nucleic acids obtained from SELEX (24,34,40–43) or chemical synthesis (23,44,45) alike. This results from its versatile physicochemicall features, including reversible protonation and the capacity for hydrogen bond formation. In its protonated, imidazolium state, it allows to place a positive charge within the negatively charged duplex environment associated with the phosphate backbone. The resulting favourable electrostatic interaction has been invoked to attribute the stabilising effect of tethered cationic modifications, including imidazole, on DNA/DNA or DNA/RNA duplexes, albeit without further investigation, for instance regarding the influence of the neighboring base pair context (46–50).
We previously described an in-depth study of the effects of incorporating a single histamine-based imidazole-modified thymidine (TImH+, Figure 1A) into a 14 base pair duplex model with a sequence optimized to allow a diversity of thymine positions for substitution while ensuring sufficient spectral resolution for NMR investigation (Figure 1B). Histamine was tethered to the C5 position of 2′-deoxyuridine using an amide bond orienting the imidazole ring in the major groove. By combining UV spectroscopy for thermal denaturation analysis and NMR spectroscopy together with in silico molecular modelling for structural characterization, the impact of the local base pair context on the overall duplex structure and stability and the imidazolium pKa was studied (51). This revealed the possibility for a single TImH+ to increase the thermal stability (Tm) of the model duplex by 5–6°C when present in a particular three base pair sequence motif composed of the TImH+A base pair followed by a GC/CG sequence such as present in the T8ImH+ dsDNA sequence (Figure 1B). Its presence introduces a hydrogen bond between the imidazolium NHε2 and the O6 on the Hoogsteen side of the guanine at position n+2 on the opposite strand (Figure 1C). This places the imidazolium moiety in the major groove and effectively introduces a non-covalent, reversible clip between both strands (Figure 1D). This situation is referred to as an in-motif duplex hereafter. Its formation is accompanied by a significant reduction in the acidity of the imidazolium moiety by 1.6 to 1.9 pKa units, hence we conveniently refer to the three base pair sequence (Figure 1B), leading to duplex stabilization and reduced imidazolium acidity as the ‘pKa-motif’ (51). Importantly, when the TImH+A base pair in the pKa-motif is flipped to ATImH+, thus creating the T21ImH+ construct when starting from the T8ImH+ one (Figure 1B) a non-motif duplex is obtained wherein the imidazolium is solvent exposed, ΔTm is limited to ∼2°C while the acidity change ΔpKa is only about ∼0.5, thereby illustrating the specific impact of the hydrogen bond between T8ImH+ and G19 on the physicochemical properties. Throughout the text, we will refer to in-motif and non-motif duplexes as defined above, but also specify the imidazolium or TImH+ nucleotide involved as an in-motif and non-motif imidazolium or TImH+, respectively.
Figure 1.

(A) Structure of the TImH+ nucleoside building block. (B) Overview of the single TImH+ model duplex systems used as reference when comparing melting temperatures and pKa values. T* is used to represent the TImH+ nucleotide in all sequence representations. For the in-motif T8ImH+ sequence the pKa-motif is shown enclosed within the box. The arrow indicates which guanine is engaged via its Hoogsteen side in the hydrogen bond with the imidazolium moiety, thereby creating an interstrand hydrogen bond. (C) Detail of the structure aspects of the pKa-motif using in-motifT8ImH+. The hydrogen bond between the imidazolium functionality of T8ImH+ and the carbonyl oxygen (O6) of G19 on the opposite strand is highlighted. (D) The pKa-motif places the imidazolium functionality within the major groove as illustrated by the T8ImH+ system. Blue and red colours indicate positive and negative (partial) charges with variable intensity scaling with the charge size (visualization with VMD 1.9.1)
The possibility to modulate the pKa of the imidazolium functionality by introducing either in-motif or non-motif TImH+ nucleotides hints towards the possibility to develop new artificial deoxyribozymes by rational decoration of DNA duplexes with catalytic combinations of imidazole functionalities in different charge states and nucleophilicities. Compared to this long term endeavour, a more short term and versatile opportunity for application exploits the stabilizing impact of an in-motif imidazole modification to heighten the thermal stability and/or improve the binding affinity of DNA duplex sequence of specific interest or DNA aptamers. Obviously, progress towards either development requires prior and systematic exploration of the effects on structure, thermal stability and acidity caused by introducing two or more TImH+ nucleotides at various positions in a model dsDNA sequence. Indeed, compared to duplexes with a single TImH+ studied so far, combinations of multiple in-motif and/or non-motif TImH+ nucleotides are expected to produce further thermal stabilization but will also introduce the possibility for electrostatic repulsion between imidazolium units, causing destabilization and mutual pKa modulation that will depend on the relative position of the TImH+ units in the oligonucleotide sequence. To proceed towards a more general deployment of TImH+ in DNAzyme and aptamer research, a set of guidelines or design rules to guide the rational introduction of multiple TImH+ nucleotides for non-covalent stabilization or to create a particular pKa combination is required. To this end, we report our investigation of a set of five duplexes based on the same model dsDNA sequence and equipped with two or three TImH+ nucleotides. We demonstrate the rather constant and largely additive contribution of each pKa-motif to the thermal stability of a dsDNA duplex and analyze the extent to which interactions between the multiple TImH+ interfere with thermal stabilization and modulate the imidazolium pKa. Using a set of guidelines derived from our analysis we conclude with a proof-of-concept study wherein rational introduction of a single TImH+ nucleotide in the l-argininamide DNA binding aptamer affords an increase in thermal stability exceeding 10°C while maintaining its binding capacity.
MATERIALS AND METHODS
Building block synthesis
The synthesis of the 2′-deoxy-5-(N-(2-(imidazol-4-yl)ethyl)-carbamoyl)-uridine-derivative was performed according to the procedure of Holmes and Gait (52). In the first step, histamine was coupled to 5-iodo-2′-deoxyuridine using a palladium-catalyzed one-pot carboxamidation reaction. After protecting the imidazole moiety with a tert-butoxycarbonyl group, the 5′-hydroxylgroup was selectively protected with 4,4′-dimethoxytrityl chloride. After conversion to the corresponding phosphoramidite, the imidazole modified thymidine building block was conveniently incorporated into oligonucleotides without further purification (details, see supplementary data).
Oligonucleotide synthesis
Modified oligonucleotides were synthesized on an ABI 394 DNA synthesizer using the standard automated phosphoramidite-based solid phase synthesis protocol (53,54) for a 1 μmol synthesis scale (details, see supplementary data). The oligonucleotides were purified by solid phase extraction (SPE) using Waters C18 Sep-Pak cartridges. This affords an easy way to purify oligonucleotides from impurities such as protecting group by-products and truncated sequences. RP-HPLC analyses were carried out on an Agilent 1100 Series instrument equipped with a Phenomenex Clarity 5μM Oligo-RP column (250 × 4.6 mm, 5 μm, 110 Å) or a Waters XBridge Oligonucleotide BEH C18 column (50 × 4.6 mm, 2.5 μm, 130 Å) and used a linear increasing acetonitrile gradient (5–35% in 15 min) against a triethylammonium acetate buffer (0.1 M TEAA, pH 7) at 50°C. The identity of the synthesized sequences was confirmed using ESI or MALDI mass spectrometry. After purification by solid phase extraction further purification and exchange of remaining triethylammonium counter ions for sodium ions was achieved by precipitation of the oligonucleotides in a 95/5 mixture of isopropanol and 3 M NaOAc(aq) solutions according to Andrus and Kuimelis (55). Unmodified sequences were purchased from Integrated DNA Technologies (IDT, Belgium) and were also desalted using the aforementioned protocol. The concentration of the oligonucleotides was determined spectrophotometrically using a Trinean DropSense® 96 equipped with a DropPlate® reader. The double stranded oligonucleotides were hybridized by mixing equimolar amounts (typically ±300 nmol) of complementary single stranded oligonucleotides. Samples were heated to 95°C in 30 minutes and kept at this temperature for 2 min. After removing the heating source, the sample was allowed to slowly reach room temperature. Following this annealing step, samples were dialyzed for at least 24 h against pure water using a dialysis membrane with a molecular weight cut-off of 3.5 kDa to remove remaining single strands or other impurities. The single stranded aptamer sequences did not require the preceding annealing step and were dialyzed immediately after the isopropanol/NaOAc precipitation procedure. Oligonucleotides were at –18°C either as a pellet or starting from an aqueous solution.
UV thermal denaturation experiments
The absorbance was measured at 260 nm in a temperature range from 5°C to 90°C with a heating/cooling rate of 0.3°C/min on a Varian Cary 300 Bio UV/VIS spectrophotometer equipped with a thermostated multicell holder. The annealed oligonucleotide strands (1 μM) were dissolved in a buffer solution containing 100 mM NaCl and 10 mM phosphate (pH 7.0). The melting temperatures were obtained from the melting curves using the first derivative approach in the Cary 300 Bio software. The reported Tm values represent average values of three consecutive melting traces and are reported with one standard deviation. For the aptamer in the presence of ligand, the same conditions were used with a 10 mM l-Arm concentration, expected to generate close to 100% saturation of the aptamer binding site in the unmodified system (56).
NMR studies
Samples used for the assignment of the non-labile protons consisted of the appropriate dsDNA or L-Arm aptamer (approximately 250 nmol) dissolved in D2O (550 μL) containing 100 mM NaCl, 0.1 mM EDTA, 0.05 mM NaN3 and 0.05 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) for internal chemical shift referencing. Spectra were recorded in D2O at 25°C (298.15 K) with a pH set at 5.0 (corrected for the isotope effect) by adding small aliquots of deuterated sodium hydroxide or deuterium chloride, this to ensure full protonation of the imidazolium group. NMR spectra were measured at a 1H frequency of 700.13 MHz on a Bruker Avance II spectrometer equipped with a standard 5 mm inverse TXI-Z ATMA probe head operating under Topspin 3.1pl6. Standard pulse sequences from the Bruker library shown between brackets hereafter, were used throughout. All spectra in D2O had a spectral width of 12.0 ppm and were recorded using excitation sculpting (zgesgp) for the suppression of the residual HDO signal when necessary (57). Typically, 128 scans of 16K data points each were accumulated. Prior to Fourier transformation, the data was apodized with a squared cosine bell function and zero filled to 32K. Zero-order polynomial baseline corrections were applied. 2D measurements consisted of COSY (cosydfesgpphpp), TOCSY (clmlevesgpph) and NOESY (noesyesgpph) spectra, recorded with 512 t1 increments of 4K data points, 64 scans each. Mixing times for the TOCSY and NOESY were 75 and 200 ms, respectively. Apodization with a squared cosine bell function, followed by zero filling and Fourier transformation to a 2K × 2K data matrix followed by polynomial baseline correction yielded the final spectra. The CCPN data model (58) was used to assist the assignment of all systems, to provide chemical shift data for the non-exchangeable nucleic acid protons and to allow identification of the nOe contacts involving the imidazolium-tethered nucleotides. For all dsDNA constructs, non-exchangeable resonances were assigned with the usual methods for unlabelled oligonucleotiudes (51,59) As usual all were assigned sequence-specifically except for the the H4′ and H5′,H5′ resonances which are isolated from the remainder of the ribose spins system due to a small 3JH3H5 scalar coupling. The NMR spectra, in terms of sequential nOe contacts and intraresidue COSY intensities, are consistent with a right-handed double helix in the B-family of conformations. For the l-Arm aptamers, partial assignments were limited to those nucleotides present in the double stranded stem. The assignments for all constructs were used for the chemical shift perturbation analysis collected in the supplementary material section.
pKa titration protocol
dsDNA (∼250 nmol) was dissolved in 550 μl H2O/D2O (9/1) containing 100 mM NaCl, 0.1 mM EDTA, 0.05 mM NaN3 and 0.05 mM DSS for internal chemical shift referencing, while the water signal was suppressed using excitation sculpting. Spectra were measured over 25.0 ppm using 32K data points. The pH of the solutions was measured directly in the NMR tube using a 3 mm diameter Ag/AgCl pH electrode. The pH was adjusted by adding small aliquots of concentrated aqueous solutions of HCl or NaOH. The chemical shift of the aromatic (non-exchangeable) proton resonances of Hε1 and Hδ2 resonances were monitored while gradually increasing the pH from ∼5–10 (59,60). The identification of the Hδ2 and Hϵ1 resonances throughout the titration was facilitated by exploiting the long-range 4J(Hδ2, Hϵ1) coupling to generate a distinctive cross-peak in a 60 ms 2D TOCSY in a spectral region otherwise devoid of other cross-peaks as described previously (51). pKa values were calculated by fitting the chemical shift data derived from the Hϵ1 proton to a Henderson-Hasselbalch equation as this proton is generally observed without signal overlap. Error-values were calculated using an in-house written Monte Carlo based algorithm (61). Whenever the calculation of the pKa values was possible from Hδ2 data, they agreed within error limits with those derived from Hε1.
Molecular dynamics simulations
Initial models of the TImH+-modified duplexes were constructed in Discovery Studio (Accelrys Software Inc., San Diego USA, release 4.0) using standard canonical B-DNA parameters. These structures were neutralized with Na+ ions and solvated in a truncated octahedral box of TIP3P water molecules with the edges of the box not closer than 13 Å to any solute atom. Topology files for the TImH+ nucleotides were the ones described before (51) and were constructed using xLEAP and AmberTools 12 (62). The solvent molecules and sodium ions were first minimized using the steepest descent minimization (1000 steps) followed by the conjugate gradient minimization method (1000 steps) while applying a large harmonic restraint force constant of 500 kcal.mol−1.Å2 on all nucleic acid atoms. In a second minimization step the entire system was minimized using steepest descent minimization (1000 steps) followed by conjugate gradient minimization (1500 steps) without positional restraints. All systems were then heated to 300 K using the Langevin temperature equilibration scheme during a 20 ps NVT run with weak harmonic restraints on the oligonucleotide strands (10 kcal.mol−1.Å2). Finally, the system was equilibrated for 100 ps (NPT ensemble) before performing a 50 ns molecular dynamics simulation. Simulations were performed using the NPT ensemble and applying the SHAKE algorithm (63) to constrain all covalent bonds involving hydrogen enabling a 2.0 fs time step. All simulations were carried out using periodic boundary conditions and the PME method with a 15 Å cut-off (64). Langevin dynamics (65) were applied for temperature control using a collision frequency of 1.0 ps−1. Snapshots were collected every 2 ps, so generating 25 000 frames for each 50 ns run. Isotropic position scaling with a pressure relaxation time of 2 ps was used to maintain an average pressure of 1 atm. All MD simulations were run on a single Nvidia GTX680 GPU using the GPU implementation of PMEMD included in the AMBER v12 simulation package (66,67). After every equilibration and MD run several general system parameters (pressure, temperature, kinetic and potential energy) were extracted from the calculated trajectories and evaluated for errors or inconsistencies. VMD 1.9.2 (68) was used for the visualization of the simulated trajectories, hydrogen bond analysis was performed using the HBonanza python script (69). Hydrogen bonds were determined by the following requirements: (i) the distance between the donor and the acceptor heavy atom (D•••A) must not exceed 3.0 Å and (ii) the angle DHA between the donor heavy atom, the hydrogen atom and the acceptor heavy atom must not be inferior to 150°.
RESULTS AND DISCUSSION
Double introduction of TImH+ in duplexes: general analysis and selection of constructs for investigation
With the rational design of multiple imidazole-modified duplex systems in mind, understanding the mutual impact of two (or more) TImH+ modifications on duplex stability and imidazolium pKa as a function of their relative position is essential. Here, we start by a general evaluation of all configurations that may arise when two TImH+ nucleotides are introduced in an otherwise unspecified dsDNA sequence as illustrated in Figure 2. At the outset, it should be realized that a second TImH+ cannot be introduced within the 3 bp sequence corresponding to the pKa-motif (box in Figure 2B) since this would remove one of the two CG base pairs that defines the pKa-motif. Also, the specific situation that would place a second TImH+ opposite the first one would create a TT mismatch and is therefore excluded from consideration. Building on our previous work (51), we initiate our analysis based upon the expectation that introducing a first TImH+ at position n within any duplex will generate either a non-motif or in-motif imidazolium depending on the local base pair context at positions n+1 and n+2 (Figure 2). Indeed, when the base pairs at these positions do not correspond to those associated with the pKa-motif, a non-motif imidazolium will result (Figure 2A). Considering this singly modified non-motif construct, all positions n±i are in principle amenable for introduction of a second TImH+. However, when introduced at position n–2 or n–1 on the same strand or n+1 and n+2 on the complementary strand, the second TImH+ can itself only give rise to a non-motif imidazolium (Figure 2A, indicated as ‘n’ in the sequence). Indeed, when placed at either of these positions, the local base pair context of the second TImH+ will automatically include the first TImH+ and therefore prevent formation of the specific three base pair sequence of the pKa-motif. At all other positions no limitations exist and the second TimH+ can be introduced both as non-motif or in-motif (Figure 2A, indicated by ‘b’ for both), depending on the precise local base pair context. Next, we consider a singly modified in-motif construct as shown in Figure 2B. Here, the local base pair context at position n+1 and n+2 is fully defined by the sequence requirements of the pKa-motif. Similar to the non-motif case described above, placing a second TImH+ at n–1 or n–2 on the same strand again implies creation of a non-motif imidazolium, whereas either an in-motif or non-motif (indicated by ‘b’ Figure 2B) TImH+ can be introduced at any other position relative the first in-motif TImH+. When introduced on the complementary strand, all positions n–i allow for both type of motifs (indicated by ‘b’), as will positions starting at n+5. At n+4 however, the local base pair context of the first TImH+ again prevents occurrence of the pKa-motif for the second one, leading it to be non-motif. Finally, the position n+3 (Figure 2B, labelled ‘x’) represents a particular case only encountered when starting from an in-motif duplex. Indeed, it is in principle possible for a TImH+ introduced at this position to form a pKa-motif with the G at n+1 on the first strand. However, this is already part of the pKa-motif of the TImH+ at position n. Both pKa-motifs are mutually exclusive for steric reasons as they cannot simultaneously fit within the same major groove area. As we foresee this introduces an ambiguous, uncontrollable state, this position is best avoided. When two TImH+ nucleotides capable of adopting the pKa motif are introduced in the same sequence, a variety of topologies may arise, depending on the relative position of the two (T*GC·GCA) sequences and the fact whether or not the TImH+ nucleotides are on the same or opposing strands, as listed in Supplementary Figure S2.
Figure 2.

Overview of possible configurations when combining two TImH+ nucleotides within an otherwise unspecified dsDNA sequence. (A) and (B) provide a schematic evaluation of the positions amenable to introduction of a second TImH+ in the presence (indicated by T* in the sequence) of a non-motif respectively in-motif TImH+ nucleotide. The letter ‘b’ (for both) is used to indicate that a second TImH+ introduced at this position may adopt an in-motif or non-motif configuration depending on the local base-pair context. When only a non-motif configuration is possible, this is indicated with ‘n’. The pKa-motif is shown as explained in Figure 1B. The ‘x’ indicates a position where the outcome of introducing a second TImH+ creates an ambiguous situation, because two pKa-motifs overlap (see text).
Obviously, many constructs can be considered when introducing two (or more) TImH+ nucleotides in a dsDNA sequence depending on the duplex sequence used as template and their relative position. For our current investigation, we chose to start from the fully matched version of the 14mer duplex sequence of the original DNA template used in our previous study of singly modified constructs (Figure 3, model duplex) (51). This model duplex was designed with six AT base pairs, each amenable to TImH+ introduction, while affording good spectral resolution for NMR investigations. This choice also affords to use the same unmodified model sequence as a single reference construct and allows for comparison with our previous data. In total, 15 double TImH+ constructs are possible using this DNA template, too large a number for each to be subjected to our combined in silico and NMR characterization approach. Here, a total of four doubly imidazole modified constructs depicted in Figure 3 were selected for synthesis and analysis. These will be referred to as (TxTy)ImH+ throughout the text, whereby an italicized Tx indicates an in-motif TImH+ nucleotide. In our particular model duplex, non-motif TImH+ constructs arise when introducing a TImH+ at position 6 (T6ImH+) or 7 (T7ImH+), while introduction at position 8 (T8ImH+) leads to an in-motif duplex. Therefore, comparison of (T6T8)ImH+ with (T7T8)ImH+ allows to judge the impact when bringing two TImH+ nucleotides closer together within the same strand while one is in-motif. In (T6T7)ImH+ and (T7T8)ImH+ both TImH+ are immediate neighbours, but one TImH+ goes from being non-motif to in-motif respectively, allowing to judge the impact of in-motif formation while keeping relative strand position constant. A fourth construct, (T6T18)ImH+ places the in-motif TImH+ on the opposite strand. This inverts the directional positioning of the imidazolium (see arrow in Figure 3) such that it is expected to interact with G9(C20) in the major groove, i.e. spatially very near to (C10)G19, itself involved in-motif formation when a TImH+ is introduced at position 8. For this reason the (T8T18)ImH+ construct was excluded as it would have overlapping pKa-motifs (each TImH+ occupying position ‘x’ in Figure 2B with respect to the other one) creating an ambiguous state (see above). Considering all other possible doubly imidazole modified constructs based on our 14mer duplex template, the nine involving substitution of either T24 and/or T27 for a TImH+ were not considered as this would involve the first three base pairs which we judged to be less representative for the typical major groove environment well inside the sequence. Also, a TImH+ at either position would only yield a non-motif situation, leading to non/non or non/in-motif situations already covered by systems described above. This also applies to the (T7T18)ImH+ construct which was a priori judged insufficiently distinct from the already selected ones to be explicitly considered. Potential consideration of a (T8T18)ImH+ was discarded in view of the mutual exclusion of one pKa motif by the other as can be derived from Figure 2. No unambiguous double in-motif construct can be designed without introducing changes to the model sequence, a disadvantage of the model duplex which results from the fact that the pKa motif was only discovered after the model's inception (53). While we could have considered alternative sequences for in/in-motif investigations, the results obtained from analyzing non/non and non/in-motifs described above (T7T8T23)ImH+, led us to immediately move to study a triply modified system that carries two in-motif modifications, each flanking one non-motif imidazole. All these systems were compared to the singly-modified in-motif reference duplex system T8ImH+ complemented by the non-motif T21ImH+ one (Figure 1B). Using all these constructs, the impact of various spatial proximities between two or more imidazolium functionalities on pKa and duplex stability was investigated.
Figure 3.

Summary of all duplex systems featuring double and triple introduction of a TImH+ nucleotide (represented by T*). The top sequence shows the model duplex used in this and previous work (51). The box indicates pKa-motifs and the arrow indicates which guanine is engaged via its Hoogsteen side in the hydrogen bond with the imidazolium moiety. TImH+ (T*) are colour labelled as orange or blue depending on whether an in-motif respectively non-motif situation is expected to result at this position taking into account the base pair context in this particular duplex. The bottom sequence represents the triply modified system featuring two adjacent pKa-motifs separated by a single non-motif TImH+. In the labels naming the various constructs, in-motif TImH+ are italicized for easy recognition.
In silico evaluation of the doubly modified duplex constructs
Molecular Dynamics (MD) simulations of the four (TxTy)ImH+ constructs were performed to computationally validate the expectations regarding the presence or absence of the pKa-motif in the presence of multiple TImH+, as outlined above. Additionally, this also provides three-dimensional insight in the behavior of the duplex and the imidazolium moieties at the atomic level, thereby assisting the interpretation of experimental data. Given that previous pKa values are ∼8 or higher in singly modified systems while experimental data is recorded at pH 7, simulations were only performed with both modified nucleotides in their protonated states.
The pKa-motif entails proximity of the imidazolium to the Hoogsteen side of the guanine at position n+2 on the opposite strand and specifically the formation of a TImH+(NHε2)---G(O6) hydrogen bond across both strands (Figure 1C). Since calculations are initiated with the imidazolium directed towards the solvent, this distance is typically between 12 and 14 Å to start with, providing unbiased starting positions to evaluate in-motif configurations. Several descriptors were evaluated from the MD simulations and are collected in Table 1. As before, we characterized the ‘occurrence’ of this hydrogen bond in the TImH+ modified duplex constructs using the fraction (in %) of the simulation time where both the associated distance d(D---A) between donor and acceptor as well as angle θ(D–H---A) fall within the required range, specified as ≤3.0 Å and ≥150° respectively. Here, we also use the ‘persistence’ in spatial proximity between the interacting partners in the major groove as the fraction (in %) of the simulation time where only the distance criterion was satisfied. Finally, the ‘excursion’ defines the fraction of the simulation time where this distance is larger than 6 Å, indicating an excursion out of the major groove towards the solvent. Using the latter two descriptors, cases where frequent and/or longer time excursions of the imidazolium moiety well away from the guanine O6 acceptor occur during the simulation can be distinguished from those where the hydrogen bond is no longer optimal, yet the imidazolium remains very near to the O6 acceptor (Supplementary Figure S3). For instance, the singly modified in-motif T8ImH+ reference duplex, features an occurrence of 27.4% with a persistence of 57.3% and an excursion of only 10.5% (51). These numbers indicate that shortly after the start of the simulation, typically within the first 2–3 ns, the imidazolium engages with the Hoogsteen face of G19 and remains very close, forming geometrically sound hydrogen bonds for considerable time. These values are considered to reflect an in-motif situation.
Table 1.
In silico analysis of hydrogen bond properties for pKa-motifs in singly, doubly and triply modified model duplex systems
| H-bond[a] | ||||
|---|---|---|---|---|
| System | Motif | Occurrence (%) | Persistence (% ≤ 3.0 Å) | Excursion (% ≥ 6.0 Å) |
| T21ImH+ | non | n.a. | n.a | n.a |
| T8ImH+ | in | 27.4 | 57.3 | 10.5 |
| T18 ImH+ | in | 10.3 | 23.4 | 27.6 |
| (T6T8)ImH+ | non/in | 32.8 | 64.9 | 7.5 |
| (T7T8)ImH+ | non/in | 17.5 | 40.4 | 3.7 |
| (T6T18)ImH+ | non/in | 28.5 | 64.9 | 3.7 |
| (T6T7)ImH+ | non/non | n.a | n.a | n.a |
| (T7T8T23)ImH+ | non/in/in | 20.3/40.1 | 49.2/77.0 | 6.2/16.2 |
[a]Values are determined for the H-bond between the in-motif TxImH+Nϵ2 hydrogen atom and the guanine carbonyl O6 at position n+2 on the opposite strand. In-motif TImH+ are in italics. For the definition of occurrence, persistence and excursion, see text. n.a. signifies not-applicable.
Considering the various doubly modified (TxTy)ImH+ constructs, the excursions stay well below 10% in all cases, while the persistencies and occurrences show values that further support the presence of an in-motif situation for T8ImH+ or T18ImH+. Thus, all expected in-motif situations are indeed found to occur in silico.
Experimental validation of the pKa-motif in doubly modified constructs
Based on our previous work on singly modified TImH+ duplex constructs, four experimental criteria were defined that accompany the effective presence of the pKa-motif. These are: (i) an increase in UV measured Tm value of 6 ± 1°C compared to the unmodified duplex; (ii) an increase in imidazolium pKa to a value around 8.5–9.0, i.e. an increase with approximately one unit compared to the non-motif situation where values are in the 7.5–8.0 range, itself above the value of the individual nucleotide at 7.2; (iii) a significant downfield shift of the imidazolium Hϵ1 proton in the 1H NMR spectrum to 9.0 ppm compared to the 8.7 ppm of the Hϵ1 resonance when the group is solvent-exposed and (iv) a clear set of nOe contacts between the Hϵ1 proton of the imidazolium group at position n to the nucleotide at position n+3 on the complementary strand. The latter criterion is very well illustrated by the detail of the NOESY spectrum of the T8ImH+ in-motif reference system showing multiple specific short range nOe contacts (Figure 4A). In contrast, the corresponding flipped T21ImH+ system used as non-motif reference system shows a dearth of such nOe's (not shown, see (51)).
Figure 4.

Overview of the specific nOe contacts of the Hε1 protons to adjacent base pairs in TImH+ -modified duplex systems. (A) The trace of specific nOe contacts associated with the pKa-motif in a single TImH+ modified duplex as illustrated by the T8ImH+ system. nOe's are assigned as labelled and mostly involve the ribose and base hydrogens of T18, i.e. at position n+3 on the opposite strand relative to the TImH+ nucleotide considered at position n. (B–F) demonstrate the presence of in-motif and non-motif TImH+ nucleotides within doubly (B–E) and triply (F) modified duplex constructs. In most cases, the amide NH (indicated by ◊) of the imidazole linker is visible, and displays clear intra-residue nOe's (dashed boxes) with the ethylene part of the chain or the neighboring thymine methyl group (in B). All 2D NOESY were recorded at 700 MHz and 25°C using a 200 ms nOe mixing time in 90%H2O/D2O.
Unfortunately, the first three criteria can no longer be used as proxy for the presence of the pKa-motif when considering constructs with two or more TImH+ residues, because they are not exclusively sensitive to the individual formation of each pKa-motif. Indeed, the introduction of a second positively charged TImH+ will also contribute to the Tm value, change the environment and therefore the Hϵ1 chemical shift, while electrostatic interactions between both imidazolium moieties are bound to shift the acid-base equilibrium and therefore the pKa of either imidazolium, all in ways that cannot be predicted. Since nOe contacts require a close proximity of the interacting 1H’s, these do provide an independent measure of motif formation for each individual TImH+, independent from the presence of another TImH+ nucleotide. Therefore, 1H resonance assignment was performed for all constructs, followed by assigning the nOe contacts involving the imidazolium moieties, as shown in Figure 4. In most cases, the NH resonance of the amide in the linker of both TImH+ constructs is also visible. For constructs where in-motif TImH+ are predicted and in silico validated (vide supra), i.e. (T6T8)ImH+, (T7T8)ImH+ and (T6T18)ImH+, clear nOe's connecting Hϵ1 with sugar and base 1H resonances of the nucleotide at position n+3 on the opposing strand, a thymine in all cases, are visible (Figure 4B–D). In addition, intra-nucleotide nOe's involving the amide NH of the linker are also clearly present and mostly involve the ethylene protons of the linker located between 2 and 3 ppm, or a neighbouring thymine methyl group (only Figure 4B). For imidazolium groups proposed to be non-motif these intranucleotide nOe's are the only ones present, supporting their orientation towards the solvent. In addition, analysis of the chemical shift perturbations caused by the introduction of TImH+ compared to the unmodified sequences (Figures S4-S10) indicates a characteristic and remarkably consistent shift by 0.45 ± 0.03 ppm to lower value for the 6-CH3 group of the thymine located at the position complementary to n+3 (i.e. the position marked by x in Figure 2), showing nOe's with the TImH+ residue. This characteristic feature was already observed in singly modified systems and can be attributed to an aromaticity induced shift resulting from the immediate spatial vicinity of the interacting imidazolium moieties to the thymine methyl group. It constitutes therefore an independent marker for the occurrence of an in-motif system. We conclude therefore that all predicted in-motif and non-motif TImH+ nucleotides in the singly and doubly modified constructs are experimentally observed, again demonstrating the general validity of the pKa-motif. Using these results, we can now interpret the impact on duplex stability (Tm) and imidazolium pKa while being certain about the motif-state of the particular imidazolium of interest, thus providing guidance for further analysis.
Initial analysis of duplex stability and imidazolium pKa in doubly modified systems
The Tm values and pKa for the imidazolium functions, the latter obtained from monitoring their aromatic resonances as a function of pH, are collected in Table 2. As reported previously (51), the singly modified non-motif T21ImH+ reference system shows a ΔTm of 2.1°C and a pKa of 7.62, while the in-motif T8ImH+ gives characteristically higher values of both 5.2°C and 8.72 respectively. For completeness, and to allow comparison with the (T6T18)ImH+ construct, we also determined these properties for the in-motif T18ImH+ construct as this had not been investigated before. Gratifyingly, it features, within error, an identical ΔTm and similarly increased pKa value as for the in-motif T8ImH+ construct (Table 2).
Table 2.
Trends in thermal stability using experimental and predicted melting temperatures and overview of experimental pKa values in singly, doubly and triply modified model duplex systems
| Duplex | Motif | T m a | ΔTmb | pKa | pKa | 0 ≤ fi ≤ 1c | ΔmaxTmp d | ΔTmp e | (ΔTmp – ΔTm) |
|---|---|---|---|---|---|---|---|---|---|
| (°C) | (°C) | Ti | Tj>i | (°C) | (°C) | (°C) | |||
| Model | n.a. | 58.9 ± 0.2 | ref | ||||||
| T21-model | n.a. | 57.2 ± 0.2 | ref* | ||||||
| T8 ImH+ | in | 64.1 ± 0.6 | 5.2 ± 0.6 | 8.72 ± 0.02 | n.a. | ∼1 | 5.2 | ref | ref |
| T18 ImH+ | in | 64.2 ± 0.6 | 5.3 ± 0.6 | 8.57 ± 0.03 | n.a. | ∼1 | 5.2 | 5.2 | -0.1 |
| T21ImH+ | non | 59.3 ± 0.5 | 2.1 ± 0.5* | 7.62 ± 0.03 | n.a. | 0.81 | 2.6 | ref | n.a. |
| (T6T8)ImH+ | non/in | 65.0 ± 0.2 | 6.1 ± 0.3 | 7.82 ± 0.05 | 8.90 ± 0.05 | 0.87 | 7.8 | 7.6 | 1.5 |
| (T7T8)ImH+ | non/in | 63.9 ± 0.4 | 5.0 ± 0.4 | 6.96 ± 0.08 | 8.70 ± 0.07 | 0.48 | 7.8 | 6.5 | 1.5 |
| (T6T18)ImH+ | non/in | 64.7 ± 0.3 | 5.8 ± 0.4 | 8.01 ± 0.04 | 8.53 ± 0.03 | 0.91 | 7.8 | 7.7 | 1.9 |
| (T6T7)ImH+ | non/non | 59.6 ± 0.1 | 0.7 ± 0.2 | 7.50 ± 0.05 | 7.64 ± 0.04 | 0.76/0.82 | 5.2 | 4.1 | 3.4 |
| T7T8T23-model | n.a. | 62.5 ± 1.5 | 3.6 ± 1.7 | ||||||
| (T7T8T23)ImH+ | non/in/in | 71.8 ± 0.9 | 9.3 ± 1.8∧ | 7.40 ± 0.08 | 8.59 ± 0.08 | 0.72 | 13.0 | 12.3 | 3.0 |
| 9.03 ± 0.02 |
a1 μM strand concentration, 100 mM NaCl, 10 mM sodium phosphate, pH 7.0 The reported errors are one standard deviation as described in the materials section.
bThe reference system used for calculations is always the model except when T21-model is used as indicated by an * or when the triply modified system is used indicated by ∧.
cProtonation degree calculated using the experimental pKa; underlined pKa values are those for in-motif imidazolium moieties.
dMaximum predicted increase in Tm and [e] predicted increase taking protonation in c into account.
Before evaluating the ΔTm values observed in the doubly modified systems (Figure 3), the pKa values need to be addressed as they allow to assess the protonation state of the imidazolium moieties in the duplex constructs at conditions used for Tm determination, i.e. at pH 7. For all in-motif TImH+, the pKa values in doubly modified constructs remain within the expected 8.5–9.0 interval, very close or identical to the values for the respective singly modified reference systems (Table 2). Thus, it appears that irrespective of the presence of a second, non-motif TImH+, the in-motif pKa values are well maintained and in all cases, the in-motif imidazolium approaches 100% protonation. We conclude therefore that the acid-base equilibrium for an in-motif TImH+ is mostly insensitive to the presence of a non-motif TImH+ even when they are immediately adjacent on the same strand. In contrast, non-motif TImH+ pKa values show a distinct behavior depending on the nature and position of the second TImH+ construct. When a second non-motif is introduced immediately adjacent to one on the same strand, like in (T6T7)ImH+, the pKa values appear little affected compared to that of the non-motif T21ImH+ reference system (Table 2). However, when an in-motif is introduced immediately adjacent a non-motif TImH+ on the same strand, as in (T7T8)ImH+ the pKa is significantly lowered. At 6.96, the non-motif pKa value of T7ImH+ is even lower than that of the isolated TImH+ nucleoside, and the lowest value reported so far for any TImH+ modified duplex. On the other hand, separating both TImH+ by one nucleotide while maintaining the non/in-motif combination, as in (T6T8)ImH+, causes a moderate raise in pKa of the non-motif T6ImH+ compared to this reference value. The fact that moving the non-motif TImH+ from position 6 to 7 with respect to in-motif T8ImH+ lowers the pKa by almost one order of magnitude while only having a marginal effect (within error) on the T8ImH+ pKa is quite striking. While singly modified T6ImH+ and T7ImH+ analogues were not investigated, there is no reason to believe this variation is linked to a pKa variation caused by the local base pair context when shifting the non-motif TImH+ along the strand. Indeed, the previously reported difference in pKa values between singly modified but TT mismatched sequences mT6ImH+ and mT7ImH+ is less than 0.20 pKa units (51). Finally, comparing (T6T8)ImH+ with (T6T18)ImH+, where both in-motifs involve a guanine in the same region of the major groove (G19 opposite C10, versus G9) but on opposing strands, the pKa for the non-motif T6ImH+ is close to identical within error, indicating that the orientation of the pKa-motif does not play a role in determining the pKa of the non-motif TImH+. We conclude therefore that a pKa difference of two units can be achieved within a duplex environment, providing opportunities for catalysis design.
Relative contributions of in-motif versus non-motif TImH+ on duplex stability
Our experimental data shows that like in all previously reported singly modified constructs, modification does not destabilize the duplex compared to the unmodified construct. In all cases a higher Tm value with ΔTm ranging from 0.7°C up to 6.1°C is observed. To explore the contribution of stabilizing and destabilizing interactions in doubly modified constructs, we start with the assumption that the change in Tm values of +5.2 and +2.1°C observed in the singly modified reference systems may be considered as characteristic ‘reference’ values for an in-motif and non-motif TImH+ at pH 7. If we first assume the lack of any kind of mutual interference between TImH+ nucleotides these contributions may be used in an additive fashion to predict ΔmaxTmp, the maximum predicted value for doubly (and higher) modified constructs:
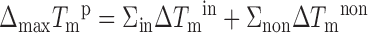 |
(1) |
where ΔTmin and ΔTmnon represent the increase observed in the singly modified reference systems. However, care must be taken to account for the impact of the degree of protonation fi of the imidazolium groups in the various constructs. Here, these are accessible from the experimental pKa values (Table 2). For in-motif imidazolium groups in singly (51) and doubly modified constructs, pKa values are above 8.5 and the degree of protonation is of no concern as it approaches fi = 1. For a single non-motif group with pKa around 7.5 however, the protonation degree drops to approximately 0.75. Thus, in/non motif combinations will lead to two distinct duplex populations at pH 7 depending on whether the non-motif TImH+ is protonated or not. This situation is further complicated for double non-motif combinations like (T6T7)ImH+, where four species corresponding to doubly non-protonated, singly protonated or doubly protonated forms co-exist in solution thereby further convoluting the measurement and interpretation of the data. This notwithstanding, a simplified approach can be devised to correct ΔTmp values for the actual protonation degree. We propose that in most cases, ΔTmp, i.e. the predicted increase in Tm of a doubly imidazole-modified duplex compared to the corresponding unmodified sequence taking into account the protonation degree can be approximated as follows:
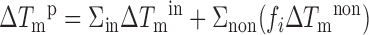 |
(2) |
Herein, the assumption is made that any in-motif TImH+ is fully protonated (fi = 1) while partial protonation of the non-motif TImH+ is handled by scaling ΔTmnon with the protonation degree of the associated imidazolium. Based on the ΔTm values observed for the T8ImH+ and T21ImH+ reference systems, we propose to set ΔTmin to 5.2°C, while for ΔTmnon a value of 2.6°C is proposed for a fully protonated non-motif system based on the 2.1°C increase occurring at 80% protonation of the singly modified T21ImH+ construct. Note that this approach implicitly assumes that non-protonated states do not contribute to change the Tm value. The values of ΔmaxTmp and ΔTmp for each specific construct is listed in Table 2. Because ΔTmnon is only 2.6°C, inclusion of the protonation degree when calculating ΔTmp causes a small i.e. −0.1 to −0.2°C to moderate reduction i.e. −0.9 to −1.3°C, the latter due to the low pKa (fi = 0.48) of T7ImH+ in (T7T8)ImH+. These protonation degree corrected ΔTmp values can now be compared with the experimental ΔTm values, by calculating (ΔTmp – ΔTm) as collected in Table 3.
Table 3.
Experimental trends in thermal stability of the aptamer constructs in presence and absence of the L-Arm ligand
| System | Motif | T m (°C)a | ΔTm (°C) | Change (reference)c |
|---|---|---|---|---|
| apt | n.a. | 55.2 ± 1.6* | ref* | – |
| apt(ImH+) | Non | 56.4 ± 1.3 | 1.2 ± 2.0 | + T3ImH+ (*) |
| f2-apt | n.a. | 53.5 ± 0.7∧ | f2-ref∧ | – |
| −1.7 ± 2.0 | + double flip (*) | |||
| + L-Armb | 62.2 ± 1.7 | 8.7 ± 1.9 | + L-Arm (∧) | |
| f2-apt (ImH+) | in | 64.9 ± 1.1 | 11.4 ± 1.3# | + T3ImH+ (∧) |
| + L-Arm[b] | 69.4 ± 0.8 | 4.5 ± 1.4 | + L-Arm (#) | |
| 15.9 ± 1.3 | +(T3ImH+ & L-Arm) (*) |
a1 μM strand concentration, 100 mM NaCl, 10 mM sodium phosphate, pH 7.0 The reported errors are one standard deviation as described in the materials section.
bThe L-Arm concentration used was 10 mM representing a 10 000:1 L-Arm/aptamer ratio.
c *, ∧ and # refer to the same labels introduced in column Tm in order to identify the temperature used as a reference to gauge the impact of the change mentioned.
Relatively good agreement is found when an in/non-motif construct is considered, as the difference between experiment and prediction is quite close, on average −1.6 ± 0.20°C. The similar difference suggests that some mutual interference occurs between the in- and non-motif imidazolium in these systems causing a destabilising ‘cost’ in melting temperature. The electrostatic repulsion between the positively charged imidazolium groups is the most obvious choice for this destabilising effect. Therefore, we evaluated whether the distance between both imidazolium moieties in the simulation trajectory can be correlated with the observed trends in Tm values. The average distance between the centroid of both imidazolium rings is 7.3 Å for (T6T8)ImH+ and 7.4 Å for (T7T8)ImH+ whereas for (T6T18)ImH+ this is about 10.7 Å. For the double non-motif case, ΔTmp is simplistically calculated from the protonation weighted contribution of each individual TImH+ to be 4.1°C. Here, the discrepancy with the experimental ΔTm value is −3.4°C, more than double the value seen for the other systems. This not only reflects the simplicity of the approximation in this case, but likely in also indicates that a considerable electrostatic repulsion occurs between both solvent-exposed charged rings, which are one average 7.3 Å from each other in the simulation. However, assuming the MD trajectories are representative for the actual solution behavior we can only conclude that a simple distance dependent electrostatic term only involving two fully charged imidazolium moieties does not suffice. An in-depth in silico investigation involving a full scale electrostatic computation which includes solvent screening effects is thus warranted but falls beyond the scope of current investigation.
First conclusions and guidelines
From all the above, a number of conclusions can be made concerning the mutual impact of two TImH+ modifications on duplex stability and imidazolium pKa as a function of their relative position in doubly modified (TxTy)ImH+ systems. With respect to the impact on thermal stability, a first general guideline is that simple additivity of the individual stabilizing contributions ΔTmin and ΔTmnon observed in the non-motif and in-motif singly modified reference systems represents an excellent first estimate for the maximum gain in stability that can be expected experimentally. Indeed, even when both TImH+ nucleotides are in close sequence proximity, i.e. like in (T6T8)ImH+ or in (T7T8)ImH+, predictions overestimate the experimental ΔTm values by 1.5 to 2°C only depending on whether protonation degrees are taken into account or not. While not explicitly investigated, we suggest that as the distance is increased between the TImH+ nucleotides in a particular duplex, the experimental ΔTm may even further tend towards the predicted ones. The only important exception that must be considered occurs when two non-motif TImH+ nucleotides are immediately adjacent. Here more severe deviations can be expected as strikingly illustrated by (T6T7)ImH+.
With respect to mutual effects on the acidity of the imidazolium moiety, the observation that an in-motif imidazolium is only weakly sensitive (on the order of |ΔpKa| < 0.3) to the presence of other nearby TImH+ irrespective whether that occurs in a pKa-motif or not is the basis for the second guideline: the pKa for in-motif TImH+ is expected to be >8.5 and little affected by nearby TImH+ residues. The case for non-motif imidazolium moieties on the other hand, is quite different. Indeed, here one should recognize that the only guideline that can be formulated is that it is not possible to predict what to expect.
Investigation of (T7T8T23)ImH+—the first triply modified duplex
To build upon these guidelines and demonstrate their applicability we chose to investigate a triply modified system using the same approach outlined for the doubly modified ones. Obviously, many other systems could be considered, but we chose to start from the current doubly modified ones, to allow for optimal comparison. We settled for the (T7T8T23)ImH+ construct since it allows to investigate the outcome of adding a second pKa-motif by introducing a TImH+ close to the non-motif/in-motif combination already investigated in the (T7T8)ImH+ construct (Figure 3). In addition, it creates the possibility to address the response of the non-motif T7ImH+ acidity to another nearby in-motif imidazolium possibly decreasing it even further below the pKa value of 6.96 in (T7T8)ImH+. To this end the T6A23 and C4G25 base pairs were flipped while the intermediate A5T24 base pair was replaced by a CG one compared to the model duplex (Figure 3). A priori in silico analysis is consistent with the formation of two pKa-motifs (Figure 5), involving hydrogen bonds between T8ImH+ and G19, respectively T23ImH+ and G4 (Figure 3) as evident from Table 1. In the triply modified construct, values for T23ImH+ reach the highest values for occurrence and persistence observed so far, the latter reaching 77%. At 16%, the excursion degree is in fact completely located within the first 8 ns of the simulation, representing the time it took for the imidazolium to lodge itself into the major groove (Supplementary Figure S3). The presence of both pKa-motifs is evident from the intense and specific nOe contacts connecting the imidazole Hϵ1 of T8ImH+ and T23ImH+ with sugar and base 1H resonances of T18 respectively C3 (Figure 4F), the bases located on the opposite strand and complementary to position n+3 from the respective TImH+ (Figure 3). As before, T7ImH+ does not show such contacts. Gratifyingly, the 6-CH3 of T18 and the H5 of C3 both show the expected perturbation in chemical shift by 0.43 and 0.40 ppm respectively (Supplementary Figure S10), indicating this feature is quite general and not dependent on the nature of the pyrimidine base as the n+3 position.
Figure 5.

Specific hydrogen bond pattern of the T8ImH+ and T23ImH+ imidazolium functionalities with G19 respectively G4, in triply modified (T7T8T23)ImH+. The hydrogen bonds are highlighted in blue dotted lines. Visualized using VMD 1.9.1.
Using our guidelines we predict that the in-motif imidazolium moieties should have a pKa above 8.5, which is indeed the case, T23ImH+ reaching 9.03 (Table 2). The impact on the pKa of T8ImH+ is marginal as expected, being in fact identical within error to the value in the (T7T8)ImH+ construct. Remarkably, the pKa of T7ImH+ is now 7.40 ± 0.08, i.e. 0.44 pKa unit up from the value in this construct. Clearly, the expectation that the acidity of the non-motif T7ImH+ may further increase by introducing a second positively charged pKa motif in the vicinity is not borne out. It further underlines the need for a more quantitative, structure driven rather than sequence based approach to predict the pKa of the imidazolium groups in a DNA duplex. Recently, constant pH molecular dynamics have gained popularity to compute pKa values of titratable residues and study pH-dependent conformational dynamics of biomolecules (70,71), thus opening possibilities for future in silico investigation of the effects impacting the pKa of TImH+ modified oligonucleotides.
When it comes to predicting the change in melting temperature, the introduction of a third TImH+ raises the complication as to which and how and which additional mutual interactions should be taken into account. In the absence of mutual interference, a ΔmaxTmp value of (2*5.2 + 2.6) = 13.0°C can be predicted assuming full protonation of T7ImH+. Using the experimental pKa of T7ImH+, this is corrected to 12.3°C. At +9.3°C compared to the T7T8T23 reference duplex the experimental value is distinctly higher than those of singly and doubly modified TImH+ constructs, and approximately 3.0°C lower than the predicted maximum value. At first sight, one could rationalize this by noting that the imidazolium moieties of T8 and T23, being separated in space by 5 base pairs as a result of their hydrogen bonding to G4 respectively G19 i.e. in opposite directions (Figure 3) are too distant to one another to be significantly contributing to destabilizing interactions. Indeed, their mutual distance is 17.4 Å on average. In this case, the remaining interactions between T7ImH+ and T8ImH+, respectively T7ImH+ and T23ImH+, which appear equally distant along the duplex sequence, could be assumed to each contribute a destabilising term of −1.5°C or −3°C in total. While this appears to agree with the experimental finding, this is most likely coincidental. Indeed, the relative disposition of each pair is clearly different as evident from Figure 5 and features average inter-imidazolium distances of 8.9 Å respectively 16.9 Å, thus quite distinct values. Nevertheless we conclude that when introducing multiple motifs in each other's vicinity as done here, the gain in stabilization is very much evident and outweighs the loss due to possible destabilizing interactions involving the imidazolium functionalities. Thus, complementing the results from the previous section we conclude that adding multiple pKa-motifs in short to medium sized oligonucleotides duplexes should lead to sizable and mostly additive stabilization against thermal denaturation.
Broadening the scope: application of the pKa-motif for non-covalent stabilization of aptamers
The possibility to stabilize short duplex segments using the pKa-motif provides so far unchartered possibilities for DNA-based aptamer development. In literature reporting aptamer research, a ligand binding event involving an aptamer is generally presented as occurring between an unfolded state and a fully folded state. However, a three step model is generally more appropriate (56,72,73), wherein unfolded (or partially folded) states exist in an equilibrium with one or a discrete collection of pre-folded binding competent states, typically featuring one or more double stranded stems. Binding, either through induced-fit or conformational selection processes then lead to the final bound aptamer state. As demonstrated by various authors (56,72,73), this coupling of a ‘switch’ or selection (KS) and ‘sense’ (KD) equilibrium allows for tuning of dynamic range and sensitivity of the aptamer based detection. Based upon this three-state model, the introduction of a covalent or non-covalent clip or staple, a term reminiscent of the concept of stapled peptides that is often used for conformational stabilization of short peptide sequences, at the appropriate position in the aptamer that stabilizes the binding-competent state can be considered a valuable asset. Indeed, any modification that renders population of the binding competent state more robust against changes in temperature, pH, salt conditions, etc. will increase the application potential. However, the introduction of a TImH+ based non-covalent staple should not overly interfere with ligand binding, for instance through modification of the binding pocket, steric effects or repulsive interactions involving charged moieties in the ligand and the tether.
As a proof-of-principle investigation that a TImH+ nucleotide could act as such a stabilising non-covalent staple in aptamers, the well-characterized L-argininamide (L-Arm) aptamer was chosen. It consists of a 24 deoxynucleotide single strand containing a 7 bp helical stem and a 10-nucleotide loop region (Figure 6), binding L-Arm with a binding constant KA = 5998 (± 920) M−1 (74,75). Mutagenesis experiments identified nucleotides essential for binding within the loop or base pairs just below the loop and their role could be rationalized from the solution structure of the complex as determined by Patel et al. (PDB-ID 1OLD) (76). It was found that the L-argininamide is enveloped within a cavity formed by the 10-nucleotide loop and interacts through intermolecular hydrogen bonds and stacking interactions. Given the double positive charge of L-Arm, and to change the sequence of the aptamer only to a minimal extent, only T3 and T19 in the double stranded stem were considered of interest for introduction of a TImH+ residue. Using the solution structure (76) T19 was judged too close to the stem-loop interface and so the binding site, therefore we decided to introduce a TImH+ at position 3. To this effect, the C4G21 and G5C20 base pairs were flipped so as to introduce the pKa-motif in the aptamer sequence (Figure 6). Thus, a total of four aptamer constructs were assessed experimentally: the unmodified aptamer referred to as apt and the double flipped or f2-apt aptamer together with the corresponding T3ImH+ modified versions aptImH+ and f2-aptImH+, expected to represent a non-motif and in-motif aptamer respectively.
Figure 6.

(A) The sequence and secondary structure of the L-argininamide (L-arm) binding DNA aptamer (apt) and of the L-Arm ligand (at pH 7). (B) Sequences and putative secondary structures of the two imidazolium-modified apt variants aptImH+ and f2-aptImH+, featuring a non-motif and an in-motif TImH+ respectively, and the f2-apt system serving as a non-modified reference for the f2-aptImH+ construct. T* is used to indicate the TImH+ nucleotide. The pKa-motif is shown as explained in Figure 1B.
The 2D NOESY spectra show the expected pattern for the modified aptamers: no relevant NOE’s for non-motif aptImH+ while a string of nOe's connecting the Hϵ1 from T3ImH+ with T19 is clearly visible in f2-aptImH+. This includes a nOe to T19-Me6 (Figure 7)), itself displaying a notable chemical shift perturbation of 0.29 ppm (Supplementary Figures S11 and S12), both in excellent agreement with the observations for pKa-motif formation in doubly and triply modified cases described above. The more reduced value of 0.29 compared to 0.45 may reflect the impact of the shorter 7mer stem compared to the 14mer duplex studied before. With respect to thermal stability, the impact from the introduction of the double flip in f2-apt is marginal, the Tm being identical within error (Table 3). This is also the case for aptImH+ where the introduction of T3ImH+ leads to a non-motif construct. In f2-aptImH+ however, the change in melting temperature caused by introducing an in-motif T3ImH+ amounts to 11.4 ± 1.3°C when compared to the unmodified reference f2-apt reference strand, an unprecedented high value for singly modified oligonucleotides. We also note from the 1H NMR spectra that in f2-aptImH+, additional base pair resonances become visible in the stem that can be assigned to the C5A6A7/G20T19T18 base pairs, whereas these remain absent in the aptImH+ construct (see Supplementary Figures S11 versus S12). Since these are positioned at the end of the dsDNA stem forming the hairpin, the strikingly higher Tm most likely reflects a zippering up of that end of the stem, thus contrasting with the TImH+ dsDNA constructs described before, where modifications were well away from the duplex ends. While all this clearly indicates that the introduction of a single pKa-motif induces major stabilization of the aptamer through tethering of its duplex stem, this does not guarantee that ligand binding is maintained. This was checked by repeating the Tm measurements on f2-apt and f2-aptImH+ in the presence of a 100-fold excess of L-Arm under conditions where complete saturation of the unmodified aptamer is normally obtained (Table 3). Upon complexation with L-Arm, the thermal stability of f2-apt is increased by 8.7°C, a value that compares well with those for apt in the literature (75). Gratifyingly, the thermal stability of f2-aptImH+ increases further, achieving 15.9°C more than the uncomplexed and unmodified f2-apt. While the additional increase upon ligand addition to f2-aptImH+ is only half that seen for f2-apt, i.e. 4.5°C as opposed to 8.8°C, its size supports the attribution of this effect to an additional stabilization associated with ligand binding. It is interesting to note that in their original NMR investigation, Lin and Patel remarked that the DNA aptamer undergoes an adaptive conformational transition upon complex formation with L-Arm (76). They based this observation on the appearance of a structured loop but also of the A7T18 and A6T19 base pair resonances in the stem preceding the hairpin. We propose that the apparition of the same signals (vide supra) prior to L-Arm binding in the f2-aptImH+ and the unusually high ΔTm value of 11.4°C accompanying the introduction of the in-motif T3ImH+ in the f2-aptamer provide initial support for an interpretation whereby the stabilization of the duplex stem by the pKa-motif assists in pre-organizing the aptamer into a more stable, binding competent state. Both this pre-organization and the mutual electrostatic interaction between TImH+ and L-Arm may modulate the binding affinity, possibly in opposing ways. To respond to these questions, further in depth investigations should be performed.
Figure 7.

Overview of the specific nOe contacts involving the Hε1 protons of the (A) f2-aptImH+ and (B) aptImH+ constructs. A specific trace of nOe's characteristic for an in-motif configuration is evident (compare with Figure 4A) for f2-aptImH+, while these are totally absent for aptImH+ indicating a non-motif configuration. Partial resonance assignment only allows for certain nOe contacts to be assigned. 2D NOESY were recorded at 700 MHz and 25°C using a 200 ms nOe mixing time in 90%H2O/D2O.
CONCLUSIONS
In previous work, the thermal stabilizing effects occurring upon introduction of a single imidazole-tethered thymidine TImH+ were reported (51). The discovery of the pKa-motif that generates a non-covalent staple mediated by hydrogen bond formation between the in-motif imidazolium moiety and the Hoogsteen side of a guanine on the opposite strand was described and shown to provide remarkable stabilizing properties, while increasing its pKa value; effects not observed for non-motif imidazolium constructs. In this study, the possibility of and effects associated with the introduction of multiple imidazole-tethered thymidine TImH+ moieties into dsDNA sequences and a model aptamer have been investigated. The possible configurations for combining in-motif and/or non-motif TImH+ have been outlined and subsequently applied to design four doubly modified sequences and one triply modified one. We demonstrated that all in- and non-motif combinations expected from the sequence design are reproduced in silico using MD simulations, and can be experimentally validated using specific nOe contacts in NOESY spectra. Irrespective of the presence of a second or third TImH+, the individual in-motif pKa values are well maintained in a pKa range of 8.5–9, approaching 100% protonation at physiological pH. In contrast, non-motif TImH+ pKa values show a distinct and currently unpredictable behavior depending on the nature and position of the other TImH+ nucleotides in the construct. Nevertheless, the fact that up to two units difference in pKa can be achieved within a duplex environment, provides interesting further opportunities for catalysis design and should motivate further computational research into the factors affecting the pKa values.
Of more immediate application potential is the thermal stabilization effect associated with the pKa-motif. In all dsDNA constructs, we observed that introduction of multiple TImH+ never decreases the thermal stability and that with a single exception, the increase in Tm can to a good approximation be predicted from suitable addition of the ΔTm values associated with singly modified in-motif (+5.2°C) and non-motif (+2.6°C at 100% protonation) constructs. It is therefore clear that introduction of multiple pKa-motifs in constructs containing short to medium sized oligonucleotide duplex sequences can be exploited to generate a predictable degree of thermal stabilization, each pKa-motif raising the Tm value stepwise as is quite strikingly illustrated in Figure 8. Indeed, modified homologous duplexes containing the same number of in-motif TImH+ nucleotides cluster together around the same Tm value, with clear stepwise changes occurring as the number of in-motif TImH+ is increased.
Figure 8.

Graph of the melting temperatures of the singly, doubly and triply modified dsDNA constructs shown along the abscis. The data is shown clustered as a function the number of in-motif TImH+ present increasing from 0 to 2.
Furthermore, we demonstrated the usefulness of the TImH+ based pKa motif for the design of a modified aptamer with enhanced stability prior to ligand binding. Next to expanding the chemical diversity, modification of aptamers with side chains reminiscent of those of amino acids has been previously shown to enhance stability against nucleases and improve target affinity (77). Here, we show that incorporation of a imidazole-tethered thymidine nucleotide at a well-considered position in the L-Arm aptamer greatly increased the thermal stability by stabilizing the dsDNA stem at the basis of the hairpin binding site. It stabilizes a conformation resembling the one competent for ligand binding, while maintaining L-Arm binding capacity. More detailed investigations into the nature of all factors contributing to this effect and the potential for stabilization of alternative aptamer–ligand complexes are ongoing and will be reported upon in due course. Given a recent report on the sequence independent increased affinity of imidazole-tethered aptamers for the negatively charged ligand ATP, we foresee interesting applications of our TImH+ motif in such systems, where additional attractive electrostatic interactions with the ligand can further enhance the aptamer performance (23).
Supplementary Material
ACKNOWLEDGEMENTS
The 700 MHz is part of the NMR Expertise Centre at UGent, and funded by the Flemish Government via the FFEU–ZWAP initiative. A.M. and J.C.M. acknowledge the University Research Council for a BOF project that funds A.d.V. The authors wish to thank Prof. K. De Waele for stimulating discussions regarding use of TImH+ in aptamer stabilization.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
University of Ghent [1.5.186.03 to V.G., B.V.G., J.V.D.B.]; Research Foundation – Flanders (FWO) [G.0422.13 to J.C.M. and A.M. for a position filled by D.B.]. Funding for open access charge: FWO [G.0422.13].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hofstetter H., Hofstetter O.. Antibodies as tailor-made chiral selectors for detection and separation of stereoisomers. TrAC - Trends Anal. Chem. 2005; 24:869–879. [Google Scholar]
- 2. Borrebaeck C.A.K. Antibodies in diagnostics - From immunoassays to protein chips. Immunol. Today. 2000; 21:379–382. [DOI] [PubMed] [Google Scholar]
- 3. Wenda S., Illner S., Mell A., Kragl U.. Industrial biotechnology—the future of green chemistry. Green Chem. 2011; 13:3007–3047. [Google Scholar]
- 4. Keefe A.D., Pai S., Ellington A.. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010; 9:537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nimjee S.M., White R.R., Becker R.C., Sullenger B.A.. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017; 57:61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu J.W., Cao Z.H., Lu Y.. Functional nucleic acid sensors. Chem. Rev. 2009; 109:1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pfeiffer F., Mayer G.. Selection and biosensor application of aptamers for small molecules. Front. Chem. 2016; 4:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Min K., Jo H., Song K., Cho M., Chun Y.S., Jon S., Kim W.J., Ban C.. Dual-aptamer-based delivery vehicle of doxorubicin to both PSMA (+) and PSMA (-) prostate cancers. Biomaterials. 2011; 32:2124–2132. [DOI] [PubMed] [Google Scholar]
- 9. Chandola C., Kalme S., Casteleijn M.G., Urtti A., Neerathilingam M.. Application of aptamers in diagnostics, drug-delivery and imaging. J. Biosci. 2016; 41:535–561. [DOI] [PubMed] [Google Scholar]
- 10. Song Y., Zhu Z., An Y., Zhang W., Zhang H., Liu D., Yu C., Duan W., Yang C.J.. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal. Chem. 2013; 85:4141–4149. [DOI] [PubMed] [Google Scholar]
- 11. Kunii T., Ogura S., Mie M., Kobatake E.. Selection of DNA aptamers recognizing small cell lung cancer using living cell-SELEX. Analyst. 2011; 136:1310. [DOI] [PubMed] [Google Scholar]
- 12. Breaker R.R., Joyce G.F.. A DNA enzyme that cleaves RNA. Chem. Biol. 1994; 1:223–229. [DOI] [PubMed] [Google Scholar]
- 13. Carmi N., Shultz L.A., Breaker R.R.. In vitro selection of self-cleaving DNAs. Chem. Biol. 1996; 3:1039–1046. [DOI] [PubMed] [Google Scholar]
- 14. Carmi N., Balkhi S.R., Breaker R.R.. Cleaving DNA with DNA. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:2233–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Silverman S.K. In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res. 2005; 33:6151–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cuenoud B., Szostak J.W.. A DNA metalloenzyme with DNA ligase activity. Nature. 1995; 375:611–614. [DOI] [PubMed] [Google Scholar]
- 17. Purtha W.E., Coppins R.L., Smalley M.K., Silverman S.K.. General deoxyribozyme-catalyzed synthesis of native 3′-5′ RNA linkages. J. Am. Chem. Soc. 2005; 127:13124–13125. [DOI] [PubMed] [Google Scholar]
- 18. Brandsen B.M., Hesser A.R., Castner M.A., Chandra M., Silverman S.K.. DNA-catalyzed hydrolysis of esters and aromatic amides. J. Am. Chem. Soc. 2013; 135:16014–16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chandra M., Silverman S.K.. DNA and RNA can be equally efficient catalysts for carbon-carbon bond formation. J. Am. Chem. Soc. 2008; 130:2936–2937. [DOI] [PubMed] [Google Scholar]
- 20. Mohan U., Burai R., McNaughton B.R.. In vitro evolution of a Friedel-Crafts deoxyribozyme. Org. Biomol. Chem. 2013; 11:2241–2244. [DOI] [PubMed] [Google Scholar]
- 21. Kimoto M., Yamashige R., Matsunaga K., Yokoyama S., Hirao I.. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 2013; 31:453–457. [DOI] [PubMed] [Google Scholar]
- 22. Imaizumi Y., Kasahara Y., Fujita H., Kitadume S., Ozaki H., Endoh T., Kuwahara M., Sugimoto N.. Efficacy of base-modification on target binding of small molecule DNA aptamers. J. Am. Chem. Soc. 2013; 135:9412–9419. [DOI] [PubMed] [Google Scholar]
- 23. Zhao J., Katsube S., Yamamoto J., Yamasaki K., Miyagishi M., Iwai S.. Analysis of ATP and AMP binding to a DNA aptamer and its imidazole-tethered derivatives by surface plasmon resonance. Analyst. 2015; 140:5881–5884. [DOI] [PubMed] [Google Scholar]
- 24. Vaught J.D., Bock C., Carter J., Fitzwater T., Otis M., Schneider D., Rolando J., Waugh S., Wilcox S.K., Eaton B.E.. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 2010; 132:4141–4151. [DOI] [PubMed] [Google Scholar]
- 25. Gold L., Ayers D., Bertino J., Bock C., Bock A., Brody E.N., Carter J., Dalby A.B., Eaton B.E., Fitzwater T. et al. . Aptamer-Based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010; 5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tolle F., Brändle G.M., Matzner D., Mayer G.. A versatile approach towards Nucleobase-Modified aptamers. Angew. Chem. Int. Ed. 2015; 54:10971–10974. [DOI] [PubMed] [Google Scholar]
- 27. Kong D., Yeung W., Hili R.. In vitro selection of diversely functionalized aptamers. J. Am. Chem. Soc. 2017; 39:13977–13980. [DOI] [PubMed] [Google Scholar]
- 28. Gawande B.N., Rohloff J.C., Carter J.D., von Carlowitz I., Zhang C., Schneider D.J., Janjic N.. Selection of DNA aptamers with two modified bases. Proc. Natl. Acad. Sci. 2017; 114:2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Renders M., Miller E., Lam C.H., Perrin D.M.. Whole cell-SELEX of aptamers with a tyrosine-like side chain against live bacteria. Org. Biomol. Chem. 2017; 15:1980–1989. [DOI] [PubMed] [Google Scholar]
- 30. Shoji A., Kuwahara M., Ozaki H., Sawai H.. Modified DNA aptamer that binds the (R)-isomer of a thalidomide derivative with high enantioselectivity. J. Am. Chem. Soc. 2007; 129:1456–1464. [DOI] [PubMed] [Google Scholar]
- 31. Ohsawa K., Kasamatsu T., Nagashima J.-I., Hanawa K., Kuwahara M., Ozaki H., Sawai H.. Arginine-modified DNA aptamers that show enantioselective recognition of the dicarboxylic acid moiety of glutamic acid. Anal. Sci. 2008; 24:167–172. [DOI] [PubMed] [Google Scholar]
- 32. Healy J.M., Lewis S.D., Kurz M., Boomer R.M., Thompson K.M., Wilson C., McCauley T.G.. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm. Res. 2004; 21:2234–2246. [DOI] [PubMed] [Google Scholar]
- 33. Sidorov A. V., Grasby J.A., Williams D.M.. Sequence-specific cleavage of RNA in the absence of divalent metal ions by a DNAzyme incorporating imidazolyl and amino functionalities. Nucleic Acids Res. 2004; 32:1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hollenstein M., Hipolito C.J., Lam C.H., Perrin D.M.. Toward the combinatorial selection of chemically modified DNAzyme RNase a mimics active against all-RNA substrates. ACS Comb. Sci. 2013; 15:174–182. [DOI] [PubMed] [Google Scholar]
- 35. Hollenstein M., Hipolito C.J., Lam C.H., Perrin D.M.. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+). Nucleic Acids Res. 2009; 37:1638–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou C., Avins J.L., Klauser P.C., Brandsen B.M., Lee Y., Silverman S.K.. DNA-Catalyzed amide hydrolysis. J. Am. Chem. Soc. 2016; 138:2106–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diafa S., Hollenstein M.. Generation of aptamers with an expanded chemical repertoire. Molecules. 2015; 20:16643–16671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hollenstein M. Nucleoside triphosphates - building blocks for the modification of nucleic acids. Molecules. 2012; 17:13569–13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gao S., Zheng X., Jiao B., Wang L.. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016; 408:4567–4573. [DOI] [PubMed] [Google Scholar]
- 40. Röthlisberger P., Levi-Acobas F., Sarac I., Marlière P., Herdewijn P., Hollenstein M., Tanaka Y., Kondo Y., Sawa R., Fujimoto T. et al. . On the enzymatic incorporation of an imidazole nucleotide into DNA. Org. Biomol. Chem. 2017; 15:4449–4455. [DOI] [PubMed] [Google Scholar]
- 41. Hollenstein M., Hipolito C.J., Lam C.H., Perrin D.M.. A DNAzyme with three protein-like functional groups: Enhancing catalytic efficiency of M2+-independent RNA cleavage. ChemBioChem. 2009; 10:1988–1992. [DOI] [PubMed] [Google Scholar]
- 42. Santoro S.W., Joyce G.F., Sakthivel K., Gramatikova S., Barbas C.F.. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 2000; 122:2433–2439. [DOI] [PubMed] [Google Scholar]
- 43. Lermer L., Roupioz Y., Ting R., Perrin D.M.. Toward an RNaseA mimic: a DNAzyme with imidazoles and cationic amines. J. Am. Chem. Soc. 2002; 124:9960–9961. [DOI] [PubMed] [Google Scholar]
- 44. Ma Z., Taylor J.-S.. Nucleic acid-triggered catalytic drug release. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:11159–11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Flanagan M.L., Arguello A.E., Colman D.E., Kim J., Krejci J.N., Liu S., Yao Y., Zhang Y., Gorin D.J.. A DNA-conjugated small molecule catalyst enzyme mimic for site-selective ester hydrolysis. Chem. Sci. 2018; 9:2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramasamy K.S., Zounes M., Gonzalez C., Freier S.M., Lesnik E.A., Cummins L.L., Griffey R.H., Monia B.P., Dan Cook P.. Remarkable enhancement of binding affinity of Heterocycle-modified DNA to DNA and RNA. Synthesis, characterization and biophysical evaluation of N2-imidazolylpropylguanine and N2-imidazolylpropyl-2-aminoadenine modified oligonucleotides. Tetrahedron Lett. 1994; 35:215–218. [Google Scholar]
- 47. Eritja R., Díaz A.R., Saison-Behmoaras E.. Duplex-stabilization properties of oligodeoxynucleotides containing N2- substituted guanine derivatives. Helv. Chim. Acta. 2000; 83:1417–1423. [Google Scholar]
- 48. Heystek L.E., Zhou H., Dande P., Gold B.. Control over the localization of positive charge in DNA: the effect on duplex DNA and RNA stability. J. Am. Chem. Soc. 1998; 120:12165–12166. [Google Scholar]
- 49. Soto A.M., Kankia B.I., Dande P., Gold B., Marky L.A.. Thermodynamic and hydration effects for the incorporation of a cationic 3-aminopropyl chain into DNA. Nucleic Acids Res. 2002; 30:3171–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li Z., Huang L., Dande P., Gold B., Stone M.P.. Structure of a tethered cationic 3-Aminopropyl chain incorporated into an oligodeoxynucleotide: Evidence for 3′-Orientation in the major groove accompanied by DNA bending. J. Am. Chem. Soc. 2002; 124:8553–8560. [DOI] [PubMed] [Google Scholar]
- 51. Buyst D., Gheerardijn V., Feher K., Van Gasse B., Van Den Begin J., Martins J.C., Madder A.. Identification of a pKa-regulating motif stabilizing imidazole-modified double-stranded DNA. Nucleic Acids Res. 2015; 43:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holmes S.C., Gait M.J.. Syntheses and oligonucleotide incorporation of nucleoside analogues containing pendant imidazolyl or amino Functionalities - The search for Sequence-Specific artificial ribonucleases. Eur. J. Org. Chem. 2005; 2005:5171–5183. [Google Scholar]
- 53. Caruthers M.H., Barone A.D., Beaucage S.L., Dodds D.R., Fisher E.F., McBride L.J., Matteucci M., Stabinsky Z., Tang J.Y.. Chemical synthesis of deoxyologonucleotides by the phosphoramidate method. Methods Enzymol. 1987; 154:287–313. [DOI] [PubMed] [Google Scholar]
- 54. Beaucage S.L., Caruthers M.H.. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981; 22:1859–1862. [Google Scholar]
- 55. Andrus A., Kuimelis R.G.. Overview of purification and analysis of synthetic nucleic acids. Curr. Protoc. Nucleic Acid Chem. 2000; 10:1–6. [DOI] [PubMed] [Google Scholar]
- 56. Vallée-Bélisle A., Ricci F., Plaxco K.W.. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:13802–13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stott K., Stonehouse J., Keeler J., Hwang T.-L., Shaka A.J.. Excitation sculpting in High-Resolution nuclear magnetic resonance spectroscopy: Application to selective NOE experiments. J. Am. Chem. Soc. 1995; 117:4199–4200. [Google Scholar]
- 58. Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D.. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins Struct. Funct. Genet. 2005; 59:687–696. [DOI] [PubMed] [Google Scholar]
- 59. Roberts G.C.K. NMR of Macromolecules: A Practical Approach. 1993; Oxford: Oxford University Press. [Google Scholar]
- 60. Markley J.L., Porubcan M.A.. The charge-relay system of serine proteinases: Proton magnetic resonance titration studies of the four histidines of porcine trypsin. J. Mol. Biol. 1976; 102:487–509. [DOI] [PubMed] [Google Scholar]
- 61. Alper J.S., Gelb R.I.. Standard errors and confidence intervals in nonlinear regression: comparison of Monte Carlo and parametric statistics. J. Phys. Chem. 1990; 94:4747–4751. [Google Scholar]
- 62. Case D.A., Darden T.A., Cheatham T.E., Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M. et al. . AMBER 12. 2012; San Francisco: University of California. [Google Scholar]
- 63. Ryckaert J.-P., Ciccotti G., Berendsen H.J.. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 64. Darden T., York D., Pedersen L.. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993; 98:10089–10092. [Google Scholar]
- 65. Uberuaga B.P., Anghel M., Voter A.F.. Synchronization of trajectories in canonical molecular-dynamics simulations: Observation, explanation, and exploitation. J. Chem. Phys. 2004; 120:6363–6374. [DOI] [PubMed] [Google Scholar]
- 66. Götz A.W., Williamson M.J., Xu D., Poole D., Le Grand S., Walker R.C.. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012; 8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Götz A.W., Williamson M.J., Xu D., Poole D., Le Grand S., Walker R.C.. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J. Chem. Theory Comput. 2012; 8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Humphrey W., Dalke A., Schulten K.. VMD: Visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 69. Durrant J.D., McCammon J.A.. HBonanza: A computer algorithm for molecular-dynamics-trajectory hydrogen-bond analysis. J. Mol. Graph. Model. 2011; 31:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Radak B.K., Chipot C., Suh D., Jo S., Jiang W., Phillips J.C., Schulten K., Roux B.. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017; 13:5933–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vila-Viçosa D., Silva T.F.D., Slaybaugh G., Reshetnyak Y.K., Andreev O.A., Machuqueiro M.. The membrane-induced p Kashifts in wt-pHLIP and its L16H variant. J. Chem. Theory Comput. 2018; 14:3289–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vallee-Belisle A., Ricci F., Plaxco K.W.. Engineering biosensors with extended, narrowed, or arbitrarily edited dynamic range. J. Am. Chem. Soc. 2012; 134:2876–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baker B.R., Lai R.Y., Wood M.S., Doctor E.H., Heeger A.J., Plaxco K.W.. An electronic, aptamer-based small-molecule sensor for the rapid, label-free detection of cocaine in adulterated samples and biological fluids. J. Am. Chem. Soc. 2006; 128:3138–3139. [DOI] [PubMed] [Google Scholar]
- 74. Harada K., Frankel A.D.. Identification of two novel arginine binding DNAs. EMBO J. 1995; 14:5798–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bishop G.R., Ren J., Polander B.C., Jeanfreau B.D., Trent J.O., Chaires J.B.. Energetic basis of molecular recognition in a DNA aptamer. Biophys. Chem. 2007; 126:165–175. [DOI] [PubMed] [Google Scholar]
- 76. Lin C.H., Patel D.J.. Encapsulating an amino acid in a DNA fold. Nat. Struct. Biol. 1996; 3:1046–1050. [DOI] [PubMed] [Google Scholar]
- 77. Rohloff J. C., Gelinas A. D., Jarvis T. C., Ochsner U. A., Schneider D. J., Gold L., Janjic N.. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol. Ther. Nucleic Acids. 2014; 3:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.