

Komori et al. demonstrate that a multilayered gene regulation system terminates self-renewal gene activity at all levels in uncommitted stem cell progeny in the fly neural stem cell lineage.
Keywords: Notch signaling, asymmetric stem cell division, exit from stemness, neuroblasts, post-translational regulation
Abstract
Self-renewal genes maintain stem cells in an undifferentiated state by preventing the commitment to differentiate. Robust inactivation of self-renewal gene activity following asymmetric stem cell division allows uncommitted stem cell progeny to exit from an undifferentiated state and initiate the commitment to differentiate. Nonetheless, how self-renewal gene activity at mRNA and protein levels becomes synchronously terminated in uncommitted stem cell progeny is unclear. We demonstrate that a multilayered gene regulation system terminates self-renewal gene activity at all levels in uncommitted stem cell progeny in the fly neural stem cell lineage. We found that the RNA-binding protein Brain tumor (Brat) targets the transcripts of a self-renewal gene, deadpan (dpn), for decay by recruiting the deadenylation machinery to the 3′ untranslated region (UTR). Furthermore, we identified a nuclear protein, Insensible, that complements Cullin-mediated proteolysis to robustly inactivate Dpn activity by limiting the level of active Dpn through protein sequestration. The synergy between post-transcriptional and transcriptional control of self-renewal genes drives timely exit from the stem cell state in uncommitted progenitors. Our proposed multilayered gene regulation system could be broadly applicable to the control of exit from stemness in all stem cell lineages.
Exit from the stem cell state serves as a molecular switch for uncommitted stem cell progeny to initiate the commitment to differentiate. The mechanisms that drive normal stem cell progeny to exit from the stem cell state likely could also promote tumor stem cells to commit to differentiate and reduce tumor burden (Lan et al. 2017; Park et al. 2017). Thus, insights into the control of exit from the stem cell state will significantly improve our understanding of how stem cell progeny choose to remain undifferentiated or commit to differentiate in the normal as well as the tumorigenic state.
Self-renewal genes function to prevent stem cells from prematurely committing to differentiate. During asymmetric stem cell division, self-renewal gene products, including mRNAs and proteins synthesized in proliferating stem cells, segregate into uncommitted stem cell progeny and must be post-transcriptionally disposed to allow for exit from the stem cell state. Failure to dispose inherited self-renewal gene products will delay or prevent uncommitted stem cell progeny from commiting to differentiation. Thus, multiple layers of regulatory mechanisms must be in place to coordinately down-regulate self-renewal gene activity at post-transcriptional levels in uncommitted stem cell progeny. Relative to our understanding of the transcriptional control, little is known about how post-transcriptional regulatory mechanisms terminate self-renewal gene activity during the exit from the stem cell state.
The eight type II neuroblast lineages in the fly larval brain provide an excellent in vivo paradigm for investigating the termination of self-renewal gene activity due to the wealth of knowledge about the lineage hierarchy and the temporal requirement of gene functions (Fig. 1A) as well as the availability of powerful genetic tools. Asymmetric type II neuroblast division generates a neuroblast and an uncommitted intermediate neural progenitor (immature INP), which initiates the commitment to differentiate <60 min after birth (Janssens et al. 2017). In type II neuroblasts, Notch and its target genes—deadpan (dpn), Enhancer of splits mγ [E(spl)mγ], and klumpfuss (klu)—constitute the regulatory network that promotes self-renewal by maintaining the master regulator of differentiation earmuff in a poised state (San-Juán and Baonza 2011; Berger et al. 2012; Xiao et al. 2012; Zacharioudaki et al. 2012, 2016; Zhu et al. 2012; Janssens et al. 2014, 2017). In the newly born immature INP, the asymmetric inheritance of the Notch inhibitor Numb prevents continued Notch activation, terminating self-renewal gene transcription (Haenfler et al. 2012). In parallel, the conserved TRIM-NHL protein Brain tumor (Brat) also asymmetrically segregates into the newly born immature INP and down-regulates dpn and klu function (Bowman et al. 2008; Xiao et al. 2012; Janssens et al. 2014). Brat binds the Brat-responsive element (BRE) in the 3′ untranslated regions (UTRs) of target transcripts, including dpn and klu, and represses their expression (Laver et al. 2015; Loedige et al. 2015; Reichardt et al. 2018). The 3′ UTRs of thousands of transcripts in the fly genome contain multiple BREs (Arvola et al. 2017). However, the specificity by which Brat recognizes its target mRNAs remains unknown, and the mechanisms by which Brat represses self-renewal gene expression are not fully understood.
Figure 1.

Brat represses dpn expression in newly born immature INPs, likely by promoting mRNA decay. (A) Schematic showing the expression pattern of self-renewal proteins activated by Notch [Dpn, Klu, and E(spl)mγ] and the Dpn reporter in the type II neuroblast (NB) lineage. NB-Gal4 (Wor-Gal4+Ase-Gal80) overexpresses UAS transgenes in type II neuroblasts and immature INPs. (B) Reducing brat function enhances the supernumerary neuroblast phenotype in numb hypomorphic (numbhypo) brains. (numbhypo) numbNP2301/15. (C) Reducing numb function enhances the supernumerary neuroblast phenotype in brat hypomorphic (brathypo) brains. (brathypo) bratDG19310/11. (D) Reducing tis11 function enhances the supernumerary neuroblast phenotype in brat hypomorphic brains by increasing Dpn activity. (E,F) A newly born immature INP (white arrowhead) shows ectopic Dpn expression in tis11-null brains but not in wild-type brains. (White arrow) Neuroblast. (G) Reducing pop2, Not1, pan2, pan3, or me31B gene dosage enhances the supernumerary neuroblast phenotype in brat hypomorphic brains. (H,I) Reducing pop2 or pan2 function leads to ectopic Dpn expression in a newly born immature INP. (J) Overexpressing wild-type, but not enzymatically inactive Pan2, rescues increased supernumerary neuroblast formation induced by the heterozygosity of pan2 in brat hypomorphic brains. Bars, 10 µM. Bar graphs are represented as mean ± standard deviation. (**) P < 0.05; (***) P < 0.005.
Because Dpn is the fly homolog of the vertebrate Hes1 protein, post-translational control mechanisms that regulate Hes1 activity during vertebrate neurogenesis likely also contribute to the termination of Dpn activity in the newly born immature INP. In proliferating mouse neural stem cells, the Cullin 1 (Cul1) ubiquitin E3 ligase complex promotes proteasome-dependent degradation of Hes1 (Imayoshi and Kageyama 2014; Chen et al. 2017). In differentiating neuronal precursors, the Hes1 antagonist Hes6 down-regulates Hes1 activity by sequestering Hes1 monomers in inactive complexes (Bae et al. 2000; Gratton et al. 2003). The combined effects of protein sequestration and Cul-based proteolysis provide an ideal strategy for terminating Dpn activity in newly born immature INPs. Defining the mechanisms that terminate Dpn activity in the newly born immature INP will lead to a generalizable model for multimodal post-translational control of Hes family proteins in various Notch-regulated developmental transitions.
Here, we used the regulation of dpn as a paradigm to demonstrate a multilayered regulatory mechanism in which the synergy between transcriptional and post-transcriptional control synchronously terminates self-renewal gene activity in the newly born immature INP. We focused on post-transcriptional control and showed that Brat selects dpn transcripts for mRNA decay by recognizing the 3′ UTR and recruiting the RNA-binding protein Tis11 and multiple deadenylases. Furthermore, we identified a novel incomplete Hes family protein, Insensible (Insb), that limits the level of active Dpn during asymmetric neuroblast division by protein sequestration. Insb-mediated protein sequestration together with Cul1-based proteolysis rapidly terminates Dpn activity. Brat-mediated decay and the multimodal post-translational regulatory mechanisms function synergistically with transcriptional control to ensure timely termination of the stem cell program in the newly born immature INP. Our proposed multilayered gene regulation system is likely broadly applicable to the control of the commitment to differentiate in all stem cell lineages and in the regulation of numerous cell fate decisions during normal development.
Results
Multiple layers of control mechanisms drive exit from the neuroblast state in immature INPs
Timely exit from the neuroblast state in newly born immature INPs necessitates a mechanism that synchronously terminates self-renewal factor activity at all levels of gene expression. Therefore, we hypothesized that a mild increase in self-renewal gene transcription and translation would lead to a higher frequency of immature INPs reverting to supernumerary neuroblasts than the additive effect of these manipulations alone. Indeed, increasing self-renewal gene translation by reducing brat gene dosage enhanced the supernumerary neuroblast phenotype in numb hypomorphic brains, where aberrantly activated Notch signaling triggers ectopic self-renewal gene transcription in immature INPs (Fig. 1B; Supplemental Fig. S1A). Similarly, increasing self-renewal gene transcription by reducing numb gene dosage enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains (Fig. 1C). Thus, multiple layers of control mechanisms coordinately terminate self-renewal gene activity at transcriptional and post-transcriptional levels for the timely exit from the neuroblast program in newly born immature INPs.
Brat functions with mRNA decay machinery to repress dpn expression
mRNA translation is a key transition in the hierarchical control of gene expression from DNA to proteins. Therefore, we took a genetic approach to investigate how multilayered control mechanisms coordinately terminate self-renewal gene activity in immature INPs in brat hypomorphic brains. The supernumerary neuroblast phenotype in brat hypomorphic brain can be suppressed by reducing dpn or klu gene dosage, providing a sensitive readout for their activity (Fig. 1D; Xiao et al. 2012). We used a deficiency collection covering the X, second, and third chromosomes of the fly genome to screen for loci that, when heterozygous, alter the supernumerary neuroblast phenotype in brat hypomorphic brains (Supplemental Table 1). We hypothesized that reducing the function of genes critical for terminating dpn or klu activity should enhance the supernumerary neuroblast phenotype in brat hypomorphic brains.
Our genetic screen identified the tis11 locus as a genetic modifier of the supernumerary neuroblast phenotype in brat hypomorphic brains (Supplemental Fig. S1B). By using gene-specific alleles, we confirmed that reduced tis11 function enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains in a dosage-dependent manner (Fig. 1D). These results suggest that Tis11 plays a role in terminating dpn or klu activity in the newly born immature INP. In further support of this notion, we found (1) that reducing dpn dosage suppressed increased supernumerary neuroblast formation induced by the heterozygosity of tis11 in brat hypomorphic brains (Fig. 1D) and (2) that the newly born immature INPs in tis11-null brains displayed ectopic Dpn expression (Fig. 1E,F). Thus, Tis11 is required for repressing dpn expression in the newly born immature INP. Because tis11-null brains did not contain supernumerary neuroblasts (Fig. 1D), Tis11 likely functions together with Brat to robustly terminate dpn activity in the newly born immature INP.
Tis11 and its vertebrate homolog, Tristetraprolin, are RNA-binding proteins and repress the translation of target mRNAs by promoting RNA decay (Vindry et al. 2012; Choi et al. 2014). The first step of the RNA decay process is shortening the poly(A) tails of target mRNAs or deadenylation, which is catalyzed by the evolutionarily conserved CCR4–NOT and Pan2–Pan3 deadenylase complexes (Temme et al. 2014; Wolf and Passmore 2014; Yan 2014). From our screen, we found that reducing the dosage of genes encoding the core components of the CCR4–NOT or Pan2–Pan3 complex also enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains (Fig. 1G; Supplemental Fig. S1C). Similar to tis11, reducing the function of either complex led to ectopic Dpn expression in the newly born immature INP (Fig. 1H,I). Furthermore, reducing the level of Me31B, which indirectly promotes mRNA decay by repressing translation (Götze et al. 2017; Wang et al. 2017), enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains (Fig. 1G). Together, these data suggest that Brat functions together with the mRNA decay machinery to terminate dpn activity in the newly born immature INP.
To confirm that deadenylase activity is required for terminating dpn activity in the newly born immature INP, we overexpressed wild-type or enzymatically inactive Pan2 in brat hypomorphic brains that are pan2 heterozygous. We found that overexpressing wild-type Pan2, but not enzymatically inactive Pan2, rescued increased supernumerary neuroblast formation induced by the heterozygosity of pan2 in brat hypomorphic brains (Fig. 1J). These data strongly suggest that Brat terminates dpn activity by promoting mRNA decay in the newly born immature INP.
Brat promotes the decay of dpn transcripts by recognizing the 3′ UTR BRE–ARE (AU-rich element) motif
Brat directly binds the BREs in the 3′ UTRs of multiple self-renewal gene transcripts, including dpn and klu, and represses reporter expression controlled by these 3′ UTRs (Loedige et al. 2015). Tis11 recognizes the AUUUA pentamer sequence (ARE) in the 3′ UTRs of target transcripts (Spasic et al. 2012; Vindry et al. 2012). Thus, we hypothesize that Brat and Tis11 physically interact and that the Brat–Tis11 complex promotes the decay of self-renewal gene transcripts in the newly born immature INP by recognizing the BREs and AREs in their 3′ UTRs. We first tested whether Brat and Tis11 physically interact in Drosophila S2 cells. We overexpressed Myc-tagged Brat and Flag-tagged Tis11 in S2 cells and performed coimmunoprecipitation assays. Indeed, Brat coimmunoprecipitated with Tis11 (Fig. 2A; Supplemental Fig. S2B). Because the B-boxes are required for Brat to terminate self-renewal gene activity in the newly born immature INP (Komori et al. 2014b), we tested whether they are required for the Brat–Tis11 interaction. We found that BratΔB-boxes failed to coimmunoprecipitate with Tis11 in S2 cell lysate (Fig. 2A; Supplemental Fig. S2B). Thus, Brat and Tis11 physically interact, and the B-boxes are required for their interaction.
Figure 2.

The Brat–Tis11 complex targets dpn mRNAs for decay by recognizing the BRE–ARE motif in their 3′ UTRs. (A) An abbreviated summary of Brat functional domains. The B-boxes of Brat mediate physical interaction with Tis11 in S2 cell lysates. (B) A neuroblast-specific promoter (NB) drives the expression of a destabilized V5-tagged reporter controlled by a minimal 3′ UTR or the dpn 3′ UTR. (C–F) A minimal 3′ UTR or the dpn 3′ UTR carrying all mutant BREs cannot repress reporter activity in a newly born immature INP (white arrowhead). (White arrow) Neuroblast. (G) Quantification of reporter expression shown in C–F. Relative pixel intensity was determined by the ratio of reporter expression in immature INPs relative to neuroblasts. (H) Destabilized V5-tagged reporters controlled by a minimal 3′ UTR or a synthetic 3′ UTR carrying repeated BRE–ARE motifs. (I–L) A synthetic 3′ UTR carrying wild-type BRE–ARE motifs was sufficient to repress reporter expression in a newly born immature INP (white arrowhead). Mutating BREs derepressed reporter expression more strongly than mutating AREs. (M) Quantification of reporter expression shown in I–L. (N) Summary of functional domains in Tis11. Overexpression of RNA-binding-defective Tis11 (Tis11ΔRNA) or Not1-binding-defective Tis11 (Tis11ΔNot1) can partially rescue increased supernumerary neuroblast formation induced by the heterozygosity of tis11 in brat hypomorphic brains. (O) Schematic showing that Brat promotes the decay of inherited self-renewal gene transcripts, likely by recruiting multiple deadenylase complexes to their 3′ UTRs. Bars, 10 µM. Bar graphs are represented as mean ± standard deviation. (*) P < 0.5; (**) P < 0.05; (***) P < 0.005.
We next tested whether the BREs and AREs in the dpn 3′ UTR are sufficient for repressing gene expression. We generated transgenic fly lines carrying reporters driven by a neuroblast-specific promoter and controlled by various 3′ UTRs (Fig. 2B). All of the reporters were highly expressed in type II neuroblasts (Fig. 2C–F). Reporter expression controlled by a minimal 3′ UTR containing a poly(A) tail was not terminated in the newly born immature INP (Fig. 2C,G). In contrast, reporter expression controlled by a wild-type dpn 3′ UTR was robustly repressed (Fig. 2D,G). Importantly, mutating all BREs in the dpn 3′ UTR derepressed reporter expression in the newly born immature INP much more strongly than mutating all AREs (Fig. 2E–G). Tis11 binds tandemly repeated AREs with high affinity (Spasic et al. 2012). Because AREs in the dpn 3′ UTR are not tandemly repeated, it is unlikely that Tis11 binds the dpn 3′ UTR alone (Fig. 2B). We conclude that BREs play a major role in repressing the dpn 3′ UTR activity.
The dpn 3′ UTR contains one predicted BRE (with a mean score of 6.243) that is ranked in the top 10% of 100 7-mers likely bound by Brat based on the RNAcompete matrix, strongly suggesting that this BRE is a high-affinity Brat-binding site (Laver et al. 2015). This putative BRE is located in tandem with an ARE (Supplemental Fig. S2A). A similar “BRE–ARE motif,” in which a high-affinity Brat-binding site (mean score of 6.096) is located in tandem with an ARE, is also present in the klu 3′ UTR (Supplemental Fig. S2A). Thus, we hypothesized that the BRE–ARE motif provides a primary sequence that prioritizes gene transcripts for Brat–Tis11-mediated decay. We tested this hypothesis by generating transgenic fly lines carrying reporters driven by a neuroblast-specific promoter and controlled by synthetic 3′ UTRs carrying repeated wild-type or mutant BRE–ARE motifs (Fig. 2H). We confirmed that all reporters were highly expressed in type II neuroblasts (Fig. 2I–M). Reporter expression controlled by the minimal 3′ UTR containing a poly(A) tail was not repressed in the newly born immature INP, unlike reporter expression controlled by the 3′ UTR carrying wild-type BRE–ARE motifs, which was robustly repressed (Fig. 2I,J,M). Importantly, mutating only the BREs in the BRE–ARE motif derepressed reporter expression in the newly born immature INP much more strongly than mutating only the AREs (Fig. 2K–M). These data support our hypothesis that Brat recognizes and targets dpn transcripts for decay by recruiting Tis11 to their 3′ UTRs.
brat-null brains contain thousands of supernumerary type II neuroblasts, but tis11-null brains do not (Fig. 1D). Because Tis11 can directly recruit the CCR4–NOT deadenylase complex to target mRNAs (Choi et al. 2014), we hypothesized that the Brat–Tis11 complex concurrently recruits multiple deadenylases to the dpn 3′ UTR. To test this hypothesis, we generated transgenes for overexpressing wild-type or mutant Tis11 (Supplemental Fig. S2C). Overexpression of RNA-binding-defective Tis11 (Tis11ΔRNA), which carries a H198Q mutation disrupting Tis11–RNA contact (Choi et al. 2014), partially rescued increased supernumerary neuroblast formation induced by the heterozygosity of tis11 in brat hypomorphic brains (Fig. 2N). This result is consistent with analyses of reporter expression controlled by the dpn 3′ UTR and the synthetic 3′ UTR containing mutant AREs and strongly suggests that Tis11 contributes to the termination of self-renewal gene activity partly through RNA binding (Fig. 2F,G,L,M). Overexpressing Not1-binding-defective Tis11 (Tis11ΔNot), which lacks amino acids 418–422, partially rescued the increased supernumerary neuroblast formation induced by the heterozygosity of tis11 in brat hypomorphic brains (Fig. 2N). This result suggests that Tis11 contributes to Brat-mediated termination of self-renewal gene activity by recruiting multiple deadenylases to the 3′ UTR of self-renewal gene transcripts. A previous study reported that Brat coimmunoprecipitates with Not1 in fly embryonic lysate (Temme et al. 2010). Thus, the Brat–Tis11 complex mechanistically links mRNA decay to the termination of self-renewal gene expression in the newly born immature INP by concurrently recruiting multiple deadenylases to their 3′ UTRs (Fig. 2O).
Cul1-mediated proteolysis and Brat-mediated mRNA decay function synergistically to terminate dpn activity in immature INPs
The termination of Dpn activity in the newly born immature INP likely requires a multitude of post-translational control mechanisms similar to the regulation of Hes1 activity during vertebrate neurogenesis (Kobayashi and Kageyama 2014). The proteasome system directed by the Cul1-based ubiquitin E3 ligase complex promotes Hes1 degradation in mice and regulates asymmetric fly neuroblast division (Li et al. 2014; Chen et al. 2017). To test whether Cul1 plays a role in terminating Dpn activity in the newly born immature INP, we first compared the efficiency of knocking down cul1 function by expressing an inducible RNAi transgene with a previously described cul1 hypomorphic allele in type II neuroblasts. We found that knocking down cul1 function by RNAi led to a stronger supernumerary neuroblast phenotype than the cul1 hypomorphic allele (Fig. 3A). Thus, expressing an UAS-cul1-RNAi transgene can efficiently knock down cul1 function. We next tested whether knocking down cul1 function during asymmetric type II neuroblast division leads to ectopic Dpn expression in the newly born immature INP. Indeed, knocking down cul1 function reproducibly led to ectopic Dpn expression in newly born immature INPs in all type II neuroblast lineages examined (Fig. 3B,C). Furthermore, the heterozygosity of dpn completely suppressed the supernumerary neuroblast phenotype induced by cul1 knockdown (Fig. 3A). Thus, Cul1 is required for terminating Dpn activity in the newly born immature INP.
Figure 3.
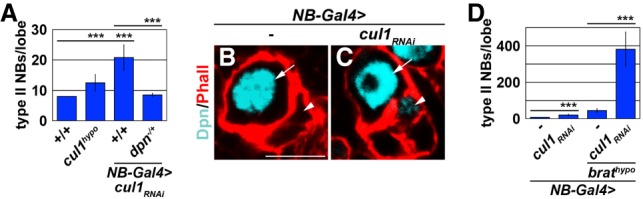
Cul1 promotes termination of Dpn activity in the newly born immature INP. (A) Overexpressing a UAS-cul1RNAi transgene led to a stronger supernumerary type II neuroblast phenotype than a cul1 hypomorphic allelic combination (cul1hypo). The heterozygosity of dpn suppressed supernumerary neuroblast formation induced by cul1 knockdown. (Cul1hypo) cul1EY11668/Ex. (B,C) Knocking down cul1 function led to mild ectopic Dpn expression in a newly born immature INP (white arrowheads). Bar, 10 µM. (White arrows) Type II neuroblast. (D) Knocking down cul1 function drastically enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains. Bar graphs are represented as mean ± standard deviation. (***) P < 0.005.
We next tested whether Cul1-mediated proteolysis of Dpn and Brat-mediated decay of dpn transcripts function synergistically to drive the timely exit from the stem cell state in the newly born immature INP. We knocked down cul1 function by RNAi in brat hypomorphic brains and quantified the formation of supernumerary neuroblasts. Knockdown of cul1 or the brat hypomorphic genetic background led to a mild supernumerary neuroblast phenotype (Fig. 3D). Under identical conditions, the severity of the supernumerary neuroblast phenotype induced by cul1 knockdown in brat hypomorphic brains far exceeded the supernumerary neuroblast phenotype in either single mutant brains alone (Fig. 3D). Thus, Cul1-mediated proteolysis functions synergistically with Brat-mediated mRNA decay to terminate dpn activity in the newly born immature INP.
Insb intrinsically regulates Dpn activity during asymmetric neuroblast division
Our results strongly suggest that multiple modes of post-translational control are required for robust termination of Dpn activity in the newly born immature INP. In our genetic screen, we identified several deficiency stocks removing the insb locus that, when heterozygous, enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains. By using insb-specific alleles, we confirmed that reduced insb function indeed enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains in a dosage-dependent manner (Fig. 4A). Furthermore, a bacterial artificial chromosome transgene containing the insb locus where the gfp coding sequence is fused in-frame with the insb-coding sequence (insb::gfp(g)) rescued increased supernumerary neuroblast formation induced by the heterozygosity of insb in brat hypomorphic brains (Fig. 4A). These results suggest that reduced insb function increases self-renewal gene activity. Analyses of insb::gfp(g) expression indicated that Insb is expressed in the nuclei of type II neuroblasts and newly born immature INPs (Fig. 4B). Thus, Insb likely antagonizes self-renewal gene activity during asymmetric neuroblast division. Consistent with this hypothesis, reducing dpn gene dosage suppressed the increased supernumerary neuroblast formation induced by the heterozygosity of insb in brat hypomorphic brains (Fig. 4A). Thus, Insb likely down-regulates dpn activity during asymmetric neuroblast division.
Figure 4.

Insb likely limits the level of active Dpn during asymmetric type II neuroblast division. (A) Reducing insb function enhances the supernumerary neuroblast phenotype in brat hypomorphic brains by increasing Dpn activity. (B) Insb is expressed in type II neuroblasts (white arrow), Ase− immature INPs (white arrowhead), Ase+ immature INPs (yellow arrow), INPs (yellow arrowhead), and immature neurons. (C) Overexpressing wild-type Insb (Insb::Myc) induces premature differentiation in type II neuroblasts, which is prevented by deletion of the C-terminal 54 amino acids of Insb. (D–G) Overexpressing wild-type Insb (Insb::Myc) did not affect Notch reporter [Gbe+Su(H)-lacZ] expression. In D, relative pixel intensity was determined by the ratio of reporter expression in neuroblasts. (H,I) Overexpressed wild-type Insb in type II neuroblasts led to an abnormally high level of nuclear Dpn but derepressed Dpn reporter expression in type II neuroblasts (white arrow). (J) Loss of insb function enhanced supernumerary neuroblast formation induced by cul1 knockdown. (K) insb heterozygously enhanced the supernumerary neuroblast phenotype in numb hypomorphic brains. Bar graphs are represented as mean ± standard deviation. (***) P < 0.005. Bars, 10 µM.
Insb appears to be a rapidly evolving nuclear protein among insect species, and amino acid alignment of Insb from 12 distinct Drosophila species revealed three highly conserved regions (Supplemental Fig. S3; Coumailleau and Schweisguth 2014). To gain insights into how Insb antagonizes dpn activity, we overexpressed a series of UAS transgenes encoding wild-type Insb or truncated Insb lacking one of the three conserved motifs (Coumailleau and Schweisguth 2014). Type II neuroblasts overexpressing wild-type Insb prematurely differentiated, as did type II neuroblasts overexpressing N-terminally or centrally truncated Insb transgenic protein (Fig. 4C). In contrast, type II neuroblasts overexpressing C-terminally truncated Insb transgenic protein did not prematurely differentiate (Fig. 4C). These data indicate that Insb down-regulates dpn activity through its C terminus. A previous study suggested that Insb inhibits Notch-dependent activation of gene transcription in the fly peripheral nervous system (Coumailleau and Schweisguth 2014). To test whether Insb indirectly antagonizes dpn activity by inhibiting Notch-mediated gene activation, we overexpressed Insb in type II neuroblasts carrying a Notch reporter. Knocking down Notch function strongly reduced Notch reporter activity as compared with the control (Fig. 4D–F). In contrast, type II neuroblasts overexpressing Insb displayed a level of Notch reporter activity similar to that of the control (Fig. 4D,E,G). Thus, Insb likely directly antagonizes dpn activity during asymmetric neuroblast division.
Type II neuroblasts overexpressing wild-type Dpn give rise to supernumerary neurobalsts instead of immature INPs (Janssens et al. 2014, 2017). Despite accumulating an abnormally high level of nuclear Dpn, type II neuroblasts overexpressing Insb prematurely differentiate (Fig. 4C,I). Thus, we hypothesized that overexpressed Insb inactivates nuclear Dpn and results in the accumulation of inactive Dpn in type II neuroblast nuclei. We tested this hypothesis by overexpressing Insb in larval brains carrying a Dpn reporter (Janssens et al. 2017). In wild-type brains, Dpn reporter activity is inhibited by endogenous Dpn in type II neuroblasts and only becomes activated in immature INPs following the termination of Dpn activity (Fig. 4H). Insb overexpression robustly derepressed Dpn reporter activity in the type II neuroblast (Fig. 4I). This result supports that type II neuroblasts overexpressing insb aberrantly accumulate inactive Dpn in the nuclei and strongly suggests that Insb functions as a post-translational antagonist of Dpn activity. Consistently, removal of insb function strongly enhanced the supernumerary neuroblast phenotype in cul1 hypomorphic brains (Fig. 4J). We conclude that Insb and Cul1 are part of the multimodal post-translational control of Dpn activity during asymmetric neuroblast division.
Our data demonstrate that the synergy between Insb-mediated inactivation of Dpn activity and Brat-mediated decay of dpn mRNAs promotes the exit from the stem cell state in the newly born immature INP (Fig. 4A). We extended our analyses to test whether Insb-mediated post-translational control functions synergistically with transcriptional control of dpn expression in the newly born immature INP. Indeed, reducing insb gene dosage enhanced the supernumerary neuroblast phenotype in numb hypomorphic brains (Fig. 4K). Thus, Insb-mediated post-translational control of Dpn activity is part of a multilayered gene regulation system that terminates dpn function in the newly born immature INP.
Insb antagonizes Dpn activity through protein sequestration
Our data suggest that Insb antagonizes Dpn activity via direct protein–protein interaction. Therefore, we tested whether Insb and Dpn physically interact. We overexpressed wild-type Insb in brat-null brains, which contain thousands of supernumerary type II neuroblasts and provide an enriched source of neuroblast-specific proteins, including Dpn (Komori et al. 2014a). We found that Insb and Dpn coimmunoprecipitated from brain neuroblast lysate (Fig. 5A). Because the C-terminal 54 amino acids of Insb are required for inducing premature differentiation in type II neuroblasts (Fig. 4C), we tested whether the C terminus of Insb is required for binding to Dpn. Indeed, the C-terminally truncated Insb failed to coimmunoprecipitate with Dpn (Fig. 5A). In addition, the 54-amino-acid C-terminal of Insb alone was sufficient for binding Dpn in S2 cell lysates (Supplemental Figs. S3, S4). Together, these data indicate that Insb binds Dpn through its C terminus.
Figure 5.

Insb antagonizes Dpn activity by forming inactive dimers through the Orange motifs. (A) Overexpressed wild-type but not C-terminally truncated Insb coimmunoprecipitates with Dpn in larval brain neuroblast lysate. (B) The Orange motif of Dpn mediates the Dpn–Insb interaction in S2 cells. (C) Overexpressed wild-type but not C-terminally truncated Insb coimmunoprecipitates with Hes1 in HEK293 cell lysate. (D) Wild-type Insb overexpression alleviates Hes1-mediated repression of the Hes reporter in mouse neural stem cells. (E) The SWISS-MODEL Web tool predicts that the C terminus of Insb folds into an Orange motif based on the crystal structure of the Hey1 Orange dimer (Protein Data Bank file 2DB7). (F) Alignment of the C terminus of Insb and Hes family proteins. The residues that likely mediate Orange dimer formation are highlighted. (G) An illustration of key functional motifs in Hes proteins and Insb. “H” indicates an α helix similar to the second α helix in the basic helix–loop–helix motif of Dpn. (H–I) Overexpressed wild-type Insb prevents Dpn–Dpn homodimerization and Dpn–E(spl)mγ heterodimerization. The asterisk indicates IgG.
We next defined the motif in Dpn that mediates binding to Insb. We overexpressed wild-type Insb with wild-type or truncated Dpn in S2 cells and performed coimmunoprecipitation. We found that the Orange motif of Dpn specifically mediates binding to Insb (Fig. 5B). This result suggests that the C terminus of Insb interacts with the Orange motif of Dpn. Because all Hes family proteins have the Orange motif, we extended our analyses to test whether Insb physically and functionally interacts with Hes1, the vertebrate homolog of Dpn. Indeed, overexpressed wild-type, but not C-terminally truncated, Insb coimmunoprecipitated with overexpressed Hes1 in HEK293 cell lysates (Fig. 5C). Furthermore, overexpressed wild-type Insb antagonized Hes1-mediated repression of reporter activity much more potently than C-terminally truncated Insb in mouse neural stem cells (Fig. 5D). In this assay, Insb inhibited Hes1 function more efficiently than dnHes1 and Hes6 (Fig. 5D). Together, these data indicate that the mechanism by which the C terminus of Insb antagonizes Dpn activity can also robustly attenuate Hes1 activity.
To gain mechanistic insight into Insb-mediated inhibition of Hes protein activity, we used the SWISS-MODEL Web tool to model the structure of the C terminus of Insb (Waterhouse et al. 2018). The C terminus of Insb is predicted to adopt a tertiary structure mimicking the Orange motif of Hey1 (Fig. 5E). The crystal structure of the Hey1 Orange motif suggested that Orange motif monomers can dimerize (Protein Data Bank [PDB] file 2DB7) (Eastwood et al. 2011). Alignment of the C terminus of Insb to the Orange motifs of multiple Hes family proteins revealed significant homology among the residues located at the interaction interface of Orange monomers (Fig. 5F, highlighted in green). Furthermore, the central conserved region of Insb is predicted to adopt an α-helix conformation resembling the second α helix of the basic helix–loop–helix motif in Hes proteins (Fig. 5G). The combination of protein–protein interaction and structural modeling led us to propose that Insb is a novel incomplete member of the Hes protein family.
Hes family transcription factors, including Dpn and E(spl), repress target gene transcription by forming homodimers or heterodimers with other Hes proteins (Ross et al. 2006; Kageyama et al. 2007). Thus, we hypothesized that Insb antagonizes Dpn activity by sequestering Dpn monomers and preventing Dpn dimer formation. We tested this hypothesis by overexpressing Dpn::V5, Dpn::Flag, and Insb::Myc in S2 cells and performing coimmunoprecipitation. We easily detected Dpn::Flag-Dpn::V5 dimers in S2 cell lysates in the absence of Insb::Myc overexpression (Fig. 5H). In the presence of overexpressed Insb::Myc, Dpn::Flag-Dpn::V5 dimer formation was drastically reduced, and Dpn::Flag-Insb::Myc dimers were apparent (Fig. 5H). Under identical conditions, overexpressed Insb::Myc abolished Dpn::Flag-E(spl)m::V5 dimer formation and instead formed Insb::Myc-Dpn::Flag dimers (Fig. 5I). These results demonstrate that Insb binding perturbs Dpn homodimerization and heterodimerization. Thus, we conclude that Insb limits the level of active Dpn in asymmetrically dividing neuroblasts by sequestering Dpn monomers in an inactive Insb–Dpn complex through Orange dimer formation.
Discussion
Self-renewal gene activity needs to be terminated for uncommitted progenitors to exit from the stem cell program in all stem cell lineages. Our study demonstrates that transcriptional, translational, and post-translational controls synergize to rapidly terminate the activity of the self-renewal gene dpn and drive the timely exit from the stem cell state in the newly born immature INP (Fig. 6). Many developmental signaling mechanisms must rapidly and robustly transition from an “on” to an “off” state to allow for proper patterning, proliferation, and cell identity specification (Isomura and Kageyama 2014). We believe that our proposed multilayered gene regulation system will be broadly applicable to the regulation of numerous developmental transitions.
Figure 6.
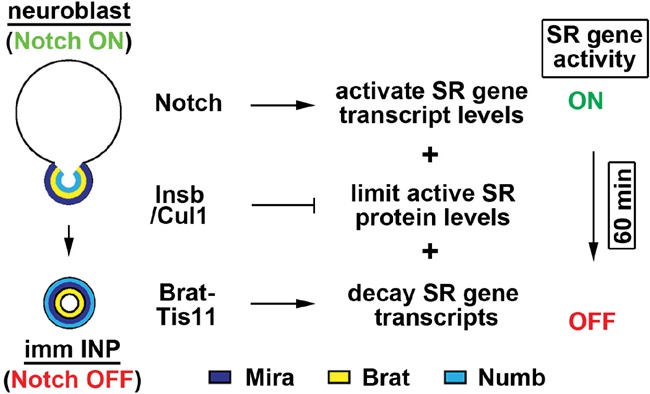
Model for multilayered regulation of self-renewal gene activity during asymmetric type II neuroblast division.
mRNA decay terminates self-renewal gene activity in uncommitted progenitors
Thousands of genes, including housekeeping and self-renewal genes, are transcribed during asymmetric type II neuroblast division (Berger et al. 2012; Carney et al. 2012). Thus, the RNA-binding proteins that recognize self-renewal gene transcripts and target them for decay play a central role in driving exit from the stem cell state in the newly born immature INP. Our data support a model in which Brat specifies the selection of self-renewal gene transcripts by recognizing the BRE–ARE motif in their 3′ UTRs and assembles RNA decay machinery by forming a complex with Tis11 (Fig. 2O).
Previous studies identified multiple putative BREs in the 3′ UTRs of self-renewal gene transcripts dpn and klu, but the physiological significance of these BREs in Brat-mediated repression is unknown (Loedige et al. 2015; Reichardt et al. 2018). By using the RNAcompete matrix (Laver et al. 2015), we found a single BRE in the dpn 3′ UTR as well as in the klu 3′ UTR that is consistently ranked in the top 10% of putative Brat-binding 7-mers and likely has high affinity for Brat binding (Fig. 2B; Supplemental Fig. S2A). Importantly, these high-affinity BREs are located in tandem with AREs and hence constitute BRE–ARE motifs (Supplemental Fig. S2A). Mutating the BRE in the synthetic 3′ UTR carrying repeated BRE–ARE motifs derepressed reporter activity much more strongly than mutating the ARE (Fig. 2I,M). In addition, overexpressed Tis11ΔRNA can substitute for wild-type Tis11 and function together with Brat to repress self-renewal gene translation in the newly born immature INP (Fig. 2N). Thus, Brat binding to the high-affinity BRE within the BRE–ARE motif, rather than Brat and Tis11 co-occupying the BRE–ARE motif, likely dictates the selection of self-renewal gene transcripts for decay.
Following the recognition of self-renewal gene transcripts, Brat must assemble the RNA decay machinery that confers robust deadenylase activity on their 3′ UTRs in the newly born immature INP. Tis11 physically interacts with Not1 and can directly recruit the CCR4–NOT deadenylase complex to target mRNAs (Choi et al. 2014). Furthermore, overexpressed Tis11ΔNot can substitute wild-type Tis11 and function together with Brat to promote the decay of self-renewal gene transcripts in the newly born immature INP (Fig. 2N). Hence, Tis11 can interact with multiple deadenylases. A previous study showed that Brat and Not1 coimmunoprecipitate in fly embryonic lysate (Temme et al. 2010). We propose that binding of the Brat–Tis11 complex to the 3′ UTRs of self-renewal gene transcripts serves as a platform to concurrently recruit multiple deadenylases, and Tis11 functions to confer robust deadenylase activity of the Brat–Tis11 complex. As such, loss of tis11 function alone will not hinder Brat-mediated decay of self-renewal gene transcripts in the newly born immature INP or lead to supernumerary neuroblast formation (Fig. 1D). In contrast, loss of tis11 function in brat hypomorphic brains simultaneously perturbs Brat- and Tis11-mediated recruitment of deadenylases to the 3′ UTRs of self-renewal gene transcripts, leading to an enhanced supernumerary neuroblast phenotype as compared with brat hypomorphic brains alone (Fig. 1D).
Multimodal post-translational control terminates self-renewal gene activity in uncommitted progenitors
Self-renewal proteins maintain type II neuroblasts in a undifferentiated state by poising the activation of the master regulator of differentiation earmuff (Janssens et al. 2017). The exit from the neuroblast state transitions earmuff from a poised state to an active state by steadily excluding self-renewal proteins Dpn, E(spl)mγ, and Klu from its cis-regulatory element. Thus, the post-translational control of the exit from the stem cell state must include mechanisms that degrade as well as sequester self-renewal proteins. The proteasome system directed by Cul-based ubiquitin E3 ligase complexes provides a conserved mechanism to degrade self-renewal proteins (Chen et al. 2017; Dubiel et al. 2018). The protein sequestration mechanisms likely play a multifaceted role by limiting active self-renewal protein levels as well as decommissioning free self-renewal proteins. We demonstrated that Insb, an incomplete Hes protein containing the Orange motif but not the basic helix–loop–helix motif (Fig. 5F), antagonizes Dpn activity by forming inactive dimers through the Orange motifs (Fig. 5G,I). As such, Insb overexpression attenuated the negative feedback autoregulation of Dpn, leading to continual dpn transcription and aberrant nuclear accumulation of inactive Dpn (Fig. 4H,I). While Insb is coexpressed with Dpn in the type II neuroblast, it remains expressed in the newly born immature INP where Dpn activity becomes terminated (Fig. 4B). We propose that Insb limits the level of Dpn dimer activity and decommissions Dpn monomers into inactive Orange dimers in the type II neuroblast and the newly born immature INP. Because proteins previously shown to antagonize Hes activity, including Id/Emc family proteins and Hes6, form inactive dimers through the basic helix–loop–helix, Orange dimer formation provides a new and novel strategy for attenuating Hes activity. Excess active Dpn dimers in insb single mutants can be cleared by robust Cul-mediated proteolysis (Fig. 4A). However, reduced insb function in brat hypomorphic brains where dpn transcripts become ectopically translated would likely overwhelm Cul-mediated proteolysis. As such, loss of insb function alone did not lead to a supernumerary neuroblast phenotype but drastically enhanced the supernumerary neuroblast phenotype in brat hypomorphic brains (Fig. 4A).
The role of the Orange motif in eliciting Hes protein functions in a physiological context remains poorly defined because all previous analyses were performed using truncated Hes protein lacking the entire Orange motif (Dawson et al. 1995; Leimeister et al. 2000; Nakatani et al. 2004; Taelman et al. 2004; Belanger-Jasmin et al. 2007). The amino acid sequence of the Orange motif in Hes family proteins varies greatly, with the exception of the residues located at the dimerization interface (Fig. 5E,F). The Orange motif is required for Insb binding to Hes1 in HEK293 cells, and ectopically expressed Insb potently inhibited Hes1 activity in mouse neural stem cells via an Orange motif-dependent mechanism (Fig. 5C,D). Functional analyses and structural modeling of Insb suggested that several conserved residues likely mediate Orange dimer formation (Fig. 5F). Insights into Orange dimer formation will allow us to define the role of this motif in eliciting the function of Hes family proteins and likely reveal novel conserved strategies to target Hes family proteins in translationally relevant applications.
Multilayered regulation of gene expression during developmental transitions
Although transcriptional control undoubtedly plays a key role in regulating gene expression, translational and post-translational control are also important for this process. As such, a multilayered control of gene expression provides an ideal strategy for regulating a wide variety of critical transitions, including the exit from stemness in uncommitted stem cell progeny, the maternal-to-zygotic transition, and somitogenesis. Thus, insights into the multilayered control of self-renewal gene activity during the exit from the neuroblast state in the newly born immature INP likely will be broadly applicable to many developmental transitions.
Materials and methods
Fly genetics and transgenes
Bloomington Df kit was used for the genetic screening. The whole deficiency list is available on the home page of Bloomington Drosophila Stock Center (https://bdsc.indiana.edu). The following stocks were published previously or are available in publich stock centers: Ase-Gal80, brat11, bratDG19310, cul1Ex, dpn1, Gbe+Su(H)-lacZ, numb15, Wor-Gal4, cul1EY11668, Df(1)IE35/Dp(1;Y)BSC5 (tis11Df), Df(2L)Exel8040 (bratDf), Me31Bk06607, pan2f00130, pop2MB11505, tis11G1183, TRiP.HM05197 (cul1RNAi), UAS-lacZ.NZ, P{GSV7}GS22604, numbNP2301, and PBac {SAstopDsRed}LL08100 (not1−). The following transgenic lines were generated in this study: UAS-insb-myc, UAS-insbΔ1-14-myc, UAS-insbΔ66-100-myc, UAS-insbΔ123-176-myc, UAS-pan2FL-V5, UAS-pan2D1039N,E1041Q-V5, UAS-tis11-Flag, UAS-tis11Δ418-424 -Flag, UAS-tis11H198Q-Flag, and insb::gfp(g). The DNA fragments were cloned into pattB, p{UAST}attB, or VanGlow-GL gateway destination vectors. insb::gfp(g) was generated by inserting GFP sequence in-frame with the insb-coding sequence in the BAC clone (CH322-168B11). The transgenic fly lines were generated via ϕC31 integrase-mediated transgenesis. InsbexA45 was generated by imprecise excision of P{GSV7}GS22604 that was inserted at a P element juxtaposed to the transcription start site of the insb gene.
Generation of BRE–ARE reporters
We took an identical strategy outlined below to generate E(spl)mγ-V5-dpn3′UTRBREwt,AREwt, E(spl)mγ-V5-dpn3′UTRBREwt,AREmut, E(spl)mγ-V5-dpn3′UTRBREmut,AREwt, E(spl)mγ-V5-dpn3′ UTRBREmut,AREmut, E(spl)mγ-V5-6xBREwt-4xAREwt, E(spl)mγ-V5-6xBREwt-4xAREmut, E(spl)mγ-V5-6xBREmut-4xAREwt, and E(spl)mγ-V5-6xBREmut-4xAREmut. We showed previously that a 250-base-pair (bp) enhancer [9D112–5, mut Klu/Dpn/E(spl)mγ] of the earmuff gene is constitutively activated in type II neuroblasts by the transcriptional activator PointedP1 (Janssens et al. 2017). We coupled this enhancer to a Drosophila synthetic core promoter to drive the expression of the E(spl)mγ::V5 reporter transgene controlled by the dpn 3′ UTR or a synthetic 3′ UTR carrying repeated BRE–ARE motifs. The E(spl)mγ::V5 fusion protein was created by fusing the N-terminal E(spl)mγ that carries the destruction sequence, the basic helix–loop–helix, and the Orange motif in-frame with a V5 epitope (Almeida and Bray 2005).
dpn reporter
The Dpn reporter (9D112-5-GFP::Luc (nls)) was described previously (Janssens et al. 2017).
Immunofluorescent staining and antibodies
Larva brains were dissected in PBS and fixed in 100 mM PIPES (pH 6.9), 1 mM EGTA, 0.3% Triton X-100, and 1 mM MgSO4 containing 4% formaldehyde for 23 min. Fixed brain samples were washed with PBST containing PBS and 0.3% Triton X-100. After removing the fix solution, samples were incubated with primary antibodies for 3 h at room temperature. Three hours later, samples were washed with PBST and then incubated with secondary antibodies overnight at 4°C. On the next day, samples were washed with PBST and then equilibrated in ProLong Gold anti-fade mountant (Thermo Fisher Scientific). Antibodies used in this study included chicken anti-GFP (1:2000; Aves Laboratories), rabbit anti-Ase (1:400), rabbit anti-β-gal (1:1000; MP Biomedicals), mouse anti-cMyc (1:200; Sigma), mouse anti-V5 (1:500; Thermo Fisher Scientific), and rat anti-Dpn (1:2). Secondary antibodies were from Jackson ImmunoResearch, Inc.. We used rhodamine phalloidin (Thermo Fisher Scientific) to visualize cortical actin. The confocal images were acquired on a Leica SP5 scanning confocal microscope (Leica Microsystems, Inc). More than 10 brains per genotype were used to obtain data in each experiment.
Cell lines
Drosophila S2 cell line was cultured in Schneider's Drosophila medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin at 25°C. The HEK293 cell line was cultured in Dulbecco's modified Eagle medium (DMEM) (Thermo Fisher Scientific) containing 10% FCS, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C with 5% CO2. The mouse neural stem cell line was established in a previous study (Imayoshi et al. 2013). Neural stem cells were cultured in DMEM/F12 (Thermo Fisher Scientific) supplemented with N2-Max (R&D Systems) and 20 ng/mL EGF and FGF (Wako). Culture dishes were coated to let neural stem cells adhere to the bottom of dish by treating with 2 µg/mL Laminin (Wako).
Plasmid constructions for cell culture experiments
cDNAs of dpn and E(spl)mγ were inserted into pUAST-attB vector with Flag or V5 tag fragment for S2 cell experiments. cDNA of insb was inserted into pcDNA3 or pCI vector for immunoprecipitation of HEK293 cells and luciferase assays in mouse neural stem cells.
Transfection and luciferase assay
Plasmid DNAs were transfected into cells by lipofection. Transfected cells were cultured for 48 h before luciferase assays and immunoprecipitation. Luciferase activities were assayed using the dual-luciferase assay system (Promega). All assays were performed three times in duplicate, and values are shown as mean ± standard deviation.
Immunoprecipitation and immunoblotting
Expression vectors were transfected into culture cells. Protein was extracted using lysis buffer containing 25 mM TrisHCl (pH 8.0), 0.5 mM EDTA, 1% NP40, and 150 mM Nacl with proteinase inhibitor cocktail (Roche). For immunoprecipitation, 1 µg of antibodies was incubated with cell lysates for 3 h at 4°C. Samples were incubated with 15 µL of 50% slurry of Protein G Sepharose 4 Fast Flow (GE Healthcare) for an additional 1 h at 4°C. Immunoprecipitates were washed with lysis buffer five times and denatured for 5 min at 95°C in 1× SDS loading buffer containing 62.5 mM TrisHCl (pH 7.4), 2% SDS, 10% glycerol, and 0.002% BPB. Proteins were separated by SDS-PAGE, blotted onto a PVDF membrane, and then incubated with antibodies specific for individual proteins. Blots were incubated with HRP-conjugated secondary antibodies (Thermo Fisher Scientific), and proteins were detected by Pierce ECL Western blotting substrate (Thermo Fisher Scientific) according to the manufacturer's protocol. The following antibodies were used: mouse anti-Flag, mouse anti-Myc, mouse anti-V5, rabbit anti-Hes1 (clone D6P2U, Cell Signaling Technology), and rabbit anti-Tis11 (Choi et al. 2014).
RNA extraction and RT–PCR
Total RNA was extracted from pan2f00130 homozygous mutant adults using TRIzol (Thermo Fisher Scientific), and mRNA was purified by using RNeasy microkit (Qiagen) according to the manufacturer's protocol. First strand cDNA was synthesized using a first strand cDNA synthesis kit for RT–PCR [AMV] (Roche) according to the manufacturer's protocol. cDNA was amplified by using gene-specific primers. The PCR products were resolved by electrophoresis on 2% agarose gels and visualized by ethidium bromide staining. The following individual specific primer sets were used for quantitative PCR: pan2 (5′-CCTCTTCAACATGCTGGATA-3′ and 5′-TCTTTGATGTGGTTGGGATAC-3′) and rp49 (5′-aTCGGTTACGGATCGAACAA-3′ and 5′-GACAATCTCCTTGCGCTTCT-3′).
Quantification and statistical analysis
ImageJ software was used to quantify the expression of the E(spl)mγ-V5 reporter proteins. Dpn single-channel confocal images were used to assign the area of the type II neuroblast or INP nucleus, and the pixel intensities of GFP were assessed in the same optical section.
The number of biological replicates is indicated by n = 10 in each figure legend, and standard deviation among samples is indicated by error bars. All statistical analysis was performed using a two-tailed Student's t-test, and P-values of <0.05 (*), <0.005 (**), and <0.0005 (***) are indicated in the figures.
Supplementary Material
Acknowledgments
We thank Dr. P. Blackshear, Dr. J. Treisman, Dr. K. Inoki, and Dr. T. Ikeda for providing fly stocks, cell lines, reagents, and technical advice. We thank the Bloomington Drosophila Stock Center and Kyoto Stock Center for fly stocks. We thank BestGene, Inc., for generating transgenic fly lines, and the Science Editors Network for editing the manuscript. We thank former members of the Lee laboratory for their technical and intellectual input during the course of this study. This work was supported by a National Institutes of Health grant (R01NS077914) to C.-Y.L., and Grant-in-Aid for Scientific Research on Innovative Areas from Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (16H06480) to R.K.
Author contributions: H.K., T.K., and K.L.G. conducted the experiments. H.K., T.K., R.K., and C.-Y.L. designed the experiments. H.K. and C.-Y.L. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.320333.118.
References
- Almeida MS, Bray SJ. 2005. Regulation of post-embryonic neuroblasts by Drosophila Grainyhead. Mech Dev 122: 1282–1293. 10.1016/j.mod.2005.08.004 [DOI] [PubMed] [Google Scholar]
- Arvola RM, Weidmann CA, Tanaka Hall TM, Goldstrohm AC. 2017. Combinatorial control of messenger RNAs by Pumilio, Nanos and Brain Tumor Proteins. RNA Biol 14: 1445–1456. 10.1080/15476286.2017.1306168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Bessho Y, Hojo M, Kageyama R. 2000. The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development 127: 2933–2943. [DOI] [PubMed] [Google Scholar]
- Belanger-Jasmin S, Llamosas E, Tang Y, Joachim K, Osiceanu AM, Jhas S, Stifani S. 2007. Inhibition of cortical astrocyte differentiation by Hes6 requires amino- and carboxy-terminal motifs important for dimerization and phosphorylation. J Neurochem 103: 2022–2034. 10.1111/j.1471-4159.2007.04902.x [DOI] [PubMed] [Google Scholar]
- Berger C, Harzer H, Burkard TR, Steinmann J, van der Horst S, Laurenson AS, Novatchkova M, Reichert H, Knoblich JA. 2012. FACS purification and transcriptome analysis of Drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep 2: 407–418. 10.1016/j.celrep.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman SK, Rolland V, Betschinger J, Kinsey KA, Emery G, Knoblich JA. 2008. The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev Cell 14: 535–546. 10.1016/j.devcel.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TD, Miller MR, Robinson KJ, Bayraktar OA, Osterhout JA, Doe CQ. 2012. Functional genomics identifies neural stem cell sub-type expression profiles and genes regulating neuroblast homeostasis. Dev Biol 361: 137–146. 10.1016/j.ydbio.2011.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang C, Wu H, Ma Y, Luo X, Gong X, Jiang F, Gui Y, Zhang H, Lu F. 2017. The E3 ubiquitin ligase SCFFBXL14 complex stimulates neuronal differentiation by targeting the Notch signaling factor HES1 for proteolysis. J Biol Chem 292: 20100–20112. 10.1074/jbc.M117.815001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Lai WS, Fedic R, Stumpo DJ, Huang W, Li L, Perera L, Brewer BY, Wilson GM, Mason JM, et al. 2014. The Drosophila Tis11 protein and its effects on mRNA expression in flies. J Biol Chem 289: 35042–35060. 10.1074/jbc.M114.593491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumailleau F, Schweisguth F. 2014. Insensible is a novel nuclear inhibitor of Notch activity in Drosophila. PLoS One 9: e98213 10.1371/journal.pone.0098213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson SR, Turner DL, Weintraub H, Parkhurst SM. 1995. Specificity for the hairy/enhancer of split basic helix–loop–helix (bHLH) proteins maps outside the bHLH domain and suggests two separable modes of transcriptional repression. Mol Cell Biol 15: 6923–6931. 10.1128/MCB.15.12.6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel W, Dubiel D, Wolf DA, Naumann M. 2018. Cullin 3-based ubiquitin ligases as master regulators of mammalian cell differentiation. Trends Biochem Sci 43: 95–107. 10.1016/j.tibs.2017.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood K, Yin C, Bandyopadhyay M, Bidwai A. 2011. New insights into the Orange domain of E(spl)-M8, and the roles of the C-terminal domain in autoinhibition and Groucho recruitment. Mol Cell Biochem 356: 217–225. 10.1007/s11010-011-0996-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götze M, Dufourt J, Ihling C, Rammelt C, Pierson S, Sambrani N, Temme C, Sinz A, Simonelig M, Wahle E. 2017. Translational repression of the Drosophila nanos mRNA involves the RNA helicase Belle and RNA coating by Me31B and Trailer hitch. RNA 23: 1552–1568. 10.1261/rna.062208.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton MO, Torban E, Jasmin SB, Theriault FM, German MS, Stifani S. 2003. Hes6 promotes cortical neurogenesis and inhibits Hes1 transcription repression activity by multiple mechanisms. Mol Cell Biol 23: 6922–6935. 10.1128/MCB.23.19.6922-6935.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenfler JM, Kuang C, Lee CY. 2012. Cortical aPKC kinase activity distinguishes neural stem cells from progenitor cells by ensuring asymmetric segregation of Numb. Dev Biol 365: 219–228. 10.1016/j.ydbio.2012.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, Kageyama R. 2014. Oscillatory control of bHLH factors in neural progenitors. Trends Neurosci 37: 531–538. 10.1016/j.tins.2014.07.006 [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Isomura A, Harima Y, Kawaguchi K, Kori H, Miyachi H, Fujiwara T, Ishidate F, Kageyama R. 2013. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342: 1203–1208. 10.1126/science.1242366 [DOI] [PubMed] [Google Scholar]
- Isomura A, Kageyama R. 2014. Ultradian oscillations and pulses: coordinating cellular responses and cell fate decisions. Development 141: 3627–3636. 10.1242/dev.104497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens DH, Komori H, Grbac D, Chen K, Koe CT, Wang H, Lee CY. 2014. Earmuff restricts progenitor cell potential by attenuating the competence to respond to self-renewal factors. Development 141: 1036–1046. 10.1242/dev.106534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens DH, Hamm DC, Anhezini L, Xiao Q, Siller KH, Siegrist SE, Harrison MM, Lee C-Y. 2017. An Hdac1/Rpd3-poised circuit balances continual self-renewal and rapid restriction of developmental potential during asymmetric stem cell division. Dev Cell 40: 367–380.e7. 10.1016/j.devcel.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Kobayashi T. 2007. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development 134: 1243–1251. 10.1242/dev.000786 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Kageyama R. 2014. Expression dynamics and functions of Hes factors in development and diseases. Curr Top Dev Biol 110: 263–283. 10.1016/B978-0-12-405943-6.00007-5 [DOI] [PubMed] [Google Scholar]
- Komori H, Xiao Q, Janssens DH, Dou Y, Lee CY. 2014a. Trithorax maintains the functional heterogeneity of neural stem cells through the transcription factor Buttonhead. Elife 3: e03502 10.7554/eLife.03502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori H, Xiao Q, McCartney BM, Lee CY. 2014b. Brain tumor specifies intermediate progenitor cell identity by attenuating β-catenin/Armadillo activity. Development 141: 51–62. 10.1242/dev.099382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X, Jörg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, Guilhamon P, Lee L, Kushida MM, Pellacani D, et al. 2017. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 549: 227–232. 10.1038/nature23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver JD, Li X, Ray D, Cook KB, Hahn NA, Nabeel-Shah S, Kekis M, Luo H, Marsolais AJ, Fung KY, et al. 2015. Brain tumor is a sequence-specific RNA-binding protein that directs maternal mRNA clearance during the Drosophila maternal-to-zygotic transition. Genome Biol 16: 94 10.1186/s13059-015-0659-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimeister C, Dale K, Fischer A, Klamt B, Hrabe de Angelis M, Radtke F, McGrew MJ, Pourquié O, Gessler M. 2000. Oscillating expression of c-Hey2 in the presomitic mesoderm suggests that the segmentation clock may use combinatorial signaling through multiple interacting bHLH factors. Dev Biol 227: 91–103. 10.1006/dbio.2000.9884 [DOI] [PubMed] [Google Scholar]
- Li S, Wang C, Sandanaraj E, Aw SS, Koe CT, Wong JJ, Yu F, Ang BT, Tang C, Wang H. 2014. The SCFSlimb E3 ligase complex regulates asymmetric division to inhibit neuroblast overgrowth. EMBO Rep 15: 165–174. 10.1002/embr.201337966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loedige I, Jakob L, Treiber T, Ray D, Stotz M, Treiber N, Hennig J, Cook KB, Morris Q, Hughes TR, et al. 2015. The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of Drosophila brain-tumor-mediated gene regulation. Cell Rep 13: 1206–1220. 10.1016/j.celrep.2015.09.068 [DOI] [PubMed] [Google Scholar]
- Nakatani T, Mizuhara E, Minaki Y, Sakamoto Y, Ono Y. 2004. Helt, a novel basic-helix–loop–helix transcriptional repressor expressed in the developing central nervous system. J Biol Chem 279: 16356–16367. 10.1074/jbc.M311740200 [DOI] [PubMed] [Google Scholar]
- Park NI, Guilhamon P, Desai K, McAdam RF, Langille E, O'Connor M, Lan X, Whetstone H, Coutinho FJ, Vanner RJ, et al. 2017. ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells. Cell Stem Cell 21: 209–224.e7. 10.1016/j.stem.2017.06.004 [DOI] [PubMed] [Google Scholar]
- Reichardt I, Bonnay F, Steinmann V, Loedige I, Burkard TR, Meister G, Knoblich JA. 2018. The tumor suppressor Brat controls neuronal stem cell lineages by inhibiting Deadpan and Zelda. EMBO Rep 19: 102–117. 10.15252/embr.201744188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DA, Hannenhalli S, Tobias JW, Cooch N, Shiekhattar R, Kadesch T. 2006. Functional analysis of Hes-1 in preadipocytes. Mol Endocrinol 20: 698–705. 10.1210/me.2005-0325 [DOI] [PubMed] [Google Scholar]
- San-Juán BP, Baonza A. 2011. The bHLH factor deadpan is a direct target of Notch signaling and regulates neuroblast self-renewal in Drosophila. Dev Biol 352: 70–82. 10.1016/j.ydbio.2011.01.019 [DOI] [PubMed] [Google Scholar]
- Spasic M, Friedel CC, Schott J, Kreth J, Leppek K, Hofmann S, Ozgur S, Stoecklin G. 2012. Genome-wide assessment of AU-rich elements by the AREScore algorithm. PLoS Genet 8: e1002433 10.1371/journal.pgen.1002433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman V, Van Wayenbergh R, Sölter M, Pichon B, Pieler T, Christophe D, Bellefroid EJ. 2004. Sequences downstream of the bHLH domain of the Xenopus hairy-related transcription factor-1 act as an extended dimerization domain that contributes to the selection of the partners. Dev Biol 276: 47–63. 10.1016/j.ydbio.2004.08.019 [DOI] [PubMed] [Google Scholar]
- Temme C, Zhang L, Kremmer E, Ihling C, Chartier A, Sinz A, Simonelig M, Wahle E. 2010. Subunits of the Drosophila CCR4–NOT complex and their roles in mRNA deadenylation. RNA 16: 1356–1370. 10.1261/rna.2145110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme C, Simonelig M, Wahle E. 2014. Deadenylation of mRNA by the CCR4–NOT complex in Drosophila: molecular and developmental aspects. Front Genet 5: 143 10.3389/fgene.2014.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindry C, Lauwers A, Hutin D, Soin R, Wauquier C, Kruys V, Gueydan C. 2012. dTIS11 Protein-dependent polysomal deadenylation is the key step in AU-rich element-mediated mRNA decay in Drosophila cells. J Biol Chem 287: 35527–35538. 10.1074/jbc.M112.356188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ly M, Lugowski A, Laver JD, Lipshitz HD, Smibert CA, Rissland OS. 2017. ME31B globally represses maternal mRNAs by two distinct mechanisms during the Drosophila maternal-to-zygotic transition. Elife 6: e27891 10.7554/eLife.27891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303. 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J, Passmore LA. 2014. mRNA deadenylation by Pan2-Pan3. Biochem Soc Trans 42: 184–187. 10.1042/BST20130211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Komori H, Lee CY. 2012. klumpfuss distinguishes stem cells from progenitor cells during asymmetric neuroblast division. Development 139: 2670–2680. 10.1242/dev.081687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan YB. 2014. Deadenylation: enzymes, regulation, and functional implications. Wiley Interdiscip Rev RNA 5: 421–443. 10.1002/wrna.1221 [DOI] [PubMed] [Google Scholar]
- Zacharioudaki E, Magadi SS, Delidakis C. 2012. bHLH-O proteins are crucial for Drosophila neuroblast self-renewal and mediate Notch-induced overproliferation. Development 139: 1258–1269. 10.1242/dev.071779 [DOI] [PubMed] [Google Scholar]
- Zacharioudaki E, Housden BE, Garinis G, Stojnic R, Delidakis C, Bray S. 2016. Genes implicated in stem cell identity and temporal programme are directly targeted by Notch in neuroblast tumours. Development 143: 219–231. 10.1242/dev.126326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Wildonger J, Barshow S, Younger S, Huang Y, Lee T. 2012. The bHLH repressor Deadpan regulates the self-renewal and specification of Drosophila larval neural stem cells independently of Notch. PLoS One 7: e46724 10.1371/journal.pone.0046724 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.