

In this study, Yu et al. found that nuclear receptor corepressor-1 (NCoR1) is a critical component of the DRED repressor that acts as a scaffold to unite the DNA-binding and epigenetic enzyme components (e.g. DNMT1 and LSD1) that elicit DRED function. They also describe a potent new regulator of γ-globin repression: The deubiquitinase BAP1 is a component of the repressor complex whose activity maintains NCoR1 at sites in the β-globin locus, and BAP1 inhibition in erythroid cells massively induces γ-globin synthesis.
Keywords: fetal globin induction, DRED complex, TR2, TR4, NCoR1, BAP1, ubiquitin
Abstract
Human globin gene production transcriptionally “switches” from fetal to adult synthesis shortly after birth and is controlled by macromolecular complexes that enhance or suppress transcription by cis elements scattered throughout the locus. The DRED (direct repeat erythroid-definitive) repressor is recruited to the ε-globin and γ-globin promoters by the orphan nuclear receptors TR2 (NR2C1) and TR4 (NR2C2) to engender their silencing in adult erythroid cells. Here we found that nuclear receptor corepressor-1 (NCoR1) is a critical component of DRED that acts as a scaffold to unite the DNA-binding and epigenetic enzyme components (e.g., DNA methyltransferase 1 [DNMT1] and lysine-specific demethylase 1 [LSD1]) that elicit DRED function. We also describe a potent new regulator of γ-globin repression: The deubiquitinase BRCA1-associated protein-1 (BAP1) is a component of the repressor complex whose activity maintains NCoR1 at sites in the β-globin locus, and BAP1 inhibition in erythroid cells massively induces γ-globin synthesis. These data provide new mechanistic insights through the discovery of novel epigenetic enzymes that mediate γ-globin gene repression.
The fetal-to-adult developmental switch in the human β-globin gene locus involves the alteration in transcription from synthesis of the two nearly identical fetal γ-globin genes to the adult β-globin gene, a process that is elicited through the cumulative activities of transcriptional activators and repressors that bind to their cognate cis-regulatory elements in the β-globin locus (Bulger and Groudine 1999; Engel and Tanimoto 2000).
Previously, we identified a multisubunit transcriptional repressor complex that we named DRED (direct repeat erythroid-definitive). DRED binds with high affinity to direct repeat (DR1) elements located in the human embryonic ε-globin and fetal γ-globin, but not the adult β-globin, promoters and represses γ-globin transcription in definitive adult erythroid cells (Tanabe et al. 2002, 2007). Based on the results of protein affinity purification followed by mass spectrometric analysis, we proposed that the “core” DRED complex was a tetramer composed of a heterodimer of the orphan nuclear receptors TR2 (NR2C1) and TR4 (NR2C2), which bind directly to the DR1 elements in the ε-globin and γ-globin gene promoters, and the two corepressor enzymes DNA methyltransferase 1 (DNMT1) and lysine-specific demethylase 1 (LSD1 or KDM1a) (Cui et al. 2011).
We and others have shown that pharmacological inhibition of the enzymatic activities of these DRED corepressor enzymes—DNMT1 (with 5-azacytidine or decitabine) and LSD1 (with tranylcypromine or RN-1)—results in the induction of fetal γ-globin synthesis in adult definitive erythroid cells (DeSimone et al. 1982; Clegg et al. 1983; Ley et al. 1983; McCaffrey et al. 1997; Shi et al. 2013; Cui et al. 2015a,b; Rivers et al. 2015, 2016; Molokie et al. 2017). These inhibitory strategies were proposed as possible pathways that could lead to robust fetal hemoglobin (HbF) induction and therefore a potentially effective therapeutic strategy to treat the β-globinopathies (sickle cell anemia and β-thalassemia) (Suzuki et al. 2014). However, all of the corepressor enzymes identified to date are widely or ubiquitously expressed and are therefore certain to participate in other biological functions. Therefore, their potential as therapeutic agents for HbF induction may be dependent on their tissue abundance and exposure to the bloodstream.
Hydroxyurea is the current treatment standard for sickle cell disease but has significant associated problems, and no other treatment options are currently available that might achieve sufficient levels of HbF induction that would ameliorate both the symptoms and the pathophysiology caused by red blood cell (RBC) sickling (Wood et al. 1976; Noguchi et al. 1988; Ngo et al. 2012). Similarly, there is no current pharmacological treatment for patients with βmajor-thalassemia that would improve imbalanced hemoglobin chain synthesis. Thus, the generation of safer, more robust, and more specific HbF inducers is highly desirable.
We hypothesized that a more detailed analysis of the interactions within the DRED complex might reveal new potential HbF inducers through two different possible outcomes. First, detailed information of protein–protein interactions that take place in the complexes might provide information about subunit interfaces that could provide alternative strategies for pharmacologic targeting between corepressors that have already been identified, and this information could then be used to create small molecules that would disrupt specific (possibly unique) protein interaction interfaces within the DRED complex. Second, the original mass spectrometry survey of DRED complex components (Cui et al. 2011) could easily have missed weakly or transiently interacting but nonetheless important proteins that participate in the repression mechanism, and novel γ-globin regulators might be identified that would serve as either better or additional HbF targets for possible therapeutic intervention. To investigate this rationale, we used a proximity-dependent biotin identification (BioID) strategy—a method that maps transient or low-solubility protein interactions (Lambert et al. 2015) and complements our previous affinity purification strategy—to reanalyze the DRED complex in human umbilical cord blood-derived erythroid progenitor (HUDEP-2) cells, which primarily produce adult β-globin-containing RBCs in vitro.
Using the BioID strategy identified the nuclear receptor corepressor-1 (NCoR1) as a TR4-interacting protein, a cofactor that was recovered at low abundance in the original affinity purification report (Cui et al. 2011). Furthermore, by using yeast two-hybrid (Y2H) assays, we discovered that NCoR1 binds directly to both TR2 and TR4. We also found that NCoR1, but not TR2 or TR4, binds to enzymatic DRED corepressor subunits (e.g., DNMT1 and LSD1), suggesting that NCoR1 might serve as the scaffold upon which the TR2/TR4 DNA-binding proteins indirectly convey these epigenetic modifying enzymes to act at specific chromatin sites. This hypothesis was tested by site-specific CRISPR–Cas9 mutagenesis of NCoR1 in definitive adult HUDEP-2 cells that altered five amino acids predicted to disrupt the NCoR1 interaction interface with TR2 and TR4. The NCoR1 mutant protein as well as the DRED repressor component LSD1 failed to be recruited to their normal binding sites in the β-globin locus, confirming that NCoR1 is an adaptor for the DRED complex.
Finally, NCoR1 has been shown to be regulated through post-translational ubiquitination, which reduces its recruitment to specific genomic sites (Perissi et al. 2004, 2008; Catic et al. 2013; Mottis et al. 2013). Our BioID survey also identified new DRED components, including the deubiquitinase BRCA1-associated protein-1 (BAP1), as potentially novel members of the complex. shRNA knockdown of BAP1 increased NCoR1 ubiquitination and significantly reduced the recruitment of NCoR1 to sites within the globin locus that could be rescued by proteasome inhibitor treatment, indicating that this deubiquitinase plays an important role in NCoR1 activity and therefore, in turn, DRED complex regulation. Consistent with its presumptive regulatory activity, both BAP1 knockdown and BAP1+/− heterozygous mutant clones generated by CRISPR/Cas9 editing in HUDEP-2 cells significantly derepressed γ-globin transcription, leading to robust induction of HbF synthesis. These experiments not only detail the vital nature of previously identified protein–protein interactions that mediate γ-globin repression but also identify novel corepressor subunits that may serve as additional therapeutic targets for future treatment of the β-globinopathies.
Results
We reported previously that the orphan nuclear receptor heterodimer TR2:TR4 (NR2C1:NR2C2) recruited multiple epigenetic cofactors, including LSD1 (KDM1A) and DNMT1, to the promoters of the ε-globin and γ-globin genes, causing their transcriptional repression in adult definitive erythroid cells (Tanabe et al. 2002, 2007; Cui et al. 2011, 2015b). However, the precise composition as well as the number and identity of the proteins that participate in the DRED repressor complex have been only superficially defined. To address this concern, we used BioID, a proximity-dependent labeling technique (Roux et al. 2012; Kim et al. 2016) that can detect even weak and transient protein interactions, which are often difficult to determine in standard affinity purification experiments. BioID is based on the activity of a mutant form of the prokaryotic BirA biotin ligase (BirA*). To initiate the procedure, any protein of interest is first fused to BirA*, which, when expressed in cells, will promiscuously biotinylate all proteins in its immediate spatial vicinity regardless of their affinity for the protein to which the BirA* is fused. Those labeled proteins can then be collected and identified by mass spectrometry (Fig. 1A illustrates the strategy).
Figure 1.
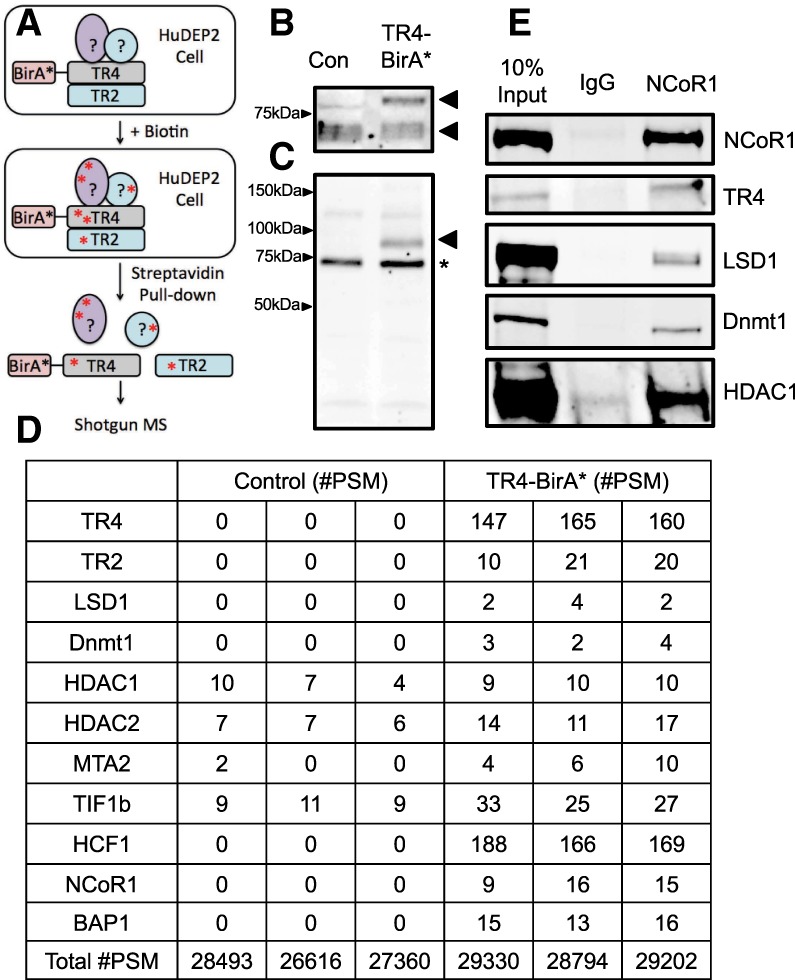
NCoR1 and BAP1 are new members of the DRED complex. (A) A schematic representation for identification of new DRED complex proteins using the BioID strategy. (B) Anti-TR4 Western blotting of whole-cell lysates (Cui et al. 2011). Successful generation of an active TR4-BirA* fusion protein was confirmed by demonstrating the presence of a new shifted band (top arrow) in comparison with endogenous TR4 (bottom arrow). (C) Streptavidin staining of whole-cell lysates prepared from TR4-BirA* transformed HUDEP-2 cells. The arrowhead indicates that the TR4-BirA* fusion protein is self-biotinylated, and the asterisk indicates the presence of a background biotinylated band detected in all cell extracts. (D) Selected proteins identified in the TR4 complex. (#PSM) Peptide spectrum matches. The total number of identified peptide sequences representing each protein is shown. (E) Coimmunoprecipitation confirmation that NCoR1 forms complexes with TR4, LSD1, DNMT1, and histone deacetylase 1 (HDAC1) in HUDEP-2 cells.
To ask whether any new proteins were identified as novel components of the DRED complex that were not copurified in our original affinity proteomics study (Cui et al. 2011), BirA* was fused to the N terminus of TR4, and then TR4-BirA* was stably transfected into HUDEP-2 cells, which, upon cytokine stimulation, produces definitive RBCs in vitro that express primarily adult β-globin mRNA and protein (Kurita et al. 2013). To circumvent potential artifactual effects that might result from massively overexpressed TR4-BirA*, the level of fusion protein was maintained near endogenous levels (Fig. 1B, bottom arrowhead [endogenous TR4] and upper arrowhead [TR4-BirA*]). Since the TR4-BirA* fusion protein was able to biotinylate itself by BirA* after staining with streptavidin (Fig. 1C, arrowhead), the data show that the fusion protein, when expressed at roughly endogenous levels, is able to identify other nearby proteins.
After incubation in the presence of biotin, the TR4-BirA* as well as parental HUDEP-2 cells were lysed, and the biotinylated proteins were isolated and identified by mass spectrometry. Among the output of total peptide spectrum matches (#PSM) from three independent experiments, all of the DRED complex proteins, including LSD1 and DNMT1, that were identified previously by affinity purification (Cui et al. 2011) were confirmed as TR4-interacting proteins (either direct or indirect). In addition to confirming the identity of proteins that were identified previously as components of the complex, the analysis also revealed new proteins that were in close spatial proximity to the TR4 BirA* tag (Fig. 1D; Supplemental Table 1). Studies described below demonstrated that two of these BioID-labeled proteins (NCoR1 and BAP1) play critical roles in γ-globin repression.
NCoR1 is a key adaptor protein in the DRED complex
NCoR1 has been shown to recruit histone deacetylases (HDACs) into large macromolecular complexes with thyroid hormone receptor, retinoic acid receptor, and other nonnuclear receptor transcription factors to mediate transcriptional repression of target genes (Perissi et al. 2008; Mottis et al. 2013). In this regard, we hypothesized that NCoR1 might serve as the adaptor between the orphan nuclear receptors TR2/TR4 and other corepressor components of the DRED complex. To test this hypothesis, protein complexes in wild-type HUDEP-2 nuclear extracts were first immunoprecipitated using an anti-NCoR1 antibody followed by Western blotting using antibodies that recognize LSD1, DNMT1, HDAC1, or TR4 (Fig. 1E). These coimmunoprecipitation (co-IP) data indicate that endogenous NCoR1 interacts with endogenous TR4 as well as each of these other epigenetic modifying enzymes in HUDEP-2 cells, consistent with the hypothesis that NCoR1 might serve as the scaffold on which the DRED repressor complex is assembled.
To address whether NCoR1 might act as the scaffold for TR2 and TR4 to recruit corepressor enzymes, we used Y2H assays to examine the interactions between these proteins using fragments of NCoR1 as bait. Successful expression of LexA bait and VP16 prey fusion proteins was confirmed by Western blots of yeast protein extracts probed with anti-LexA or anti-VP16 antibodies (Supplemental Fig. 1). Since NCoR1 is encoded in 2453 amino acids, we generated three separate overlapping fragments of NCoR1 to use as bait (Table 1). Those three fragments contained, respectively, repressive domain 1 (RD1), repressive domain 2 plus 3 (RD2/3), or the C-terminal domain (Mottis et al. 2013) of NCoR1.
Table 1.
Y2H studies of NCoR1 binding to other candidate proteins within the DRED complex
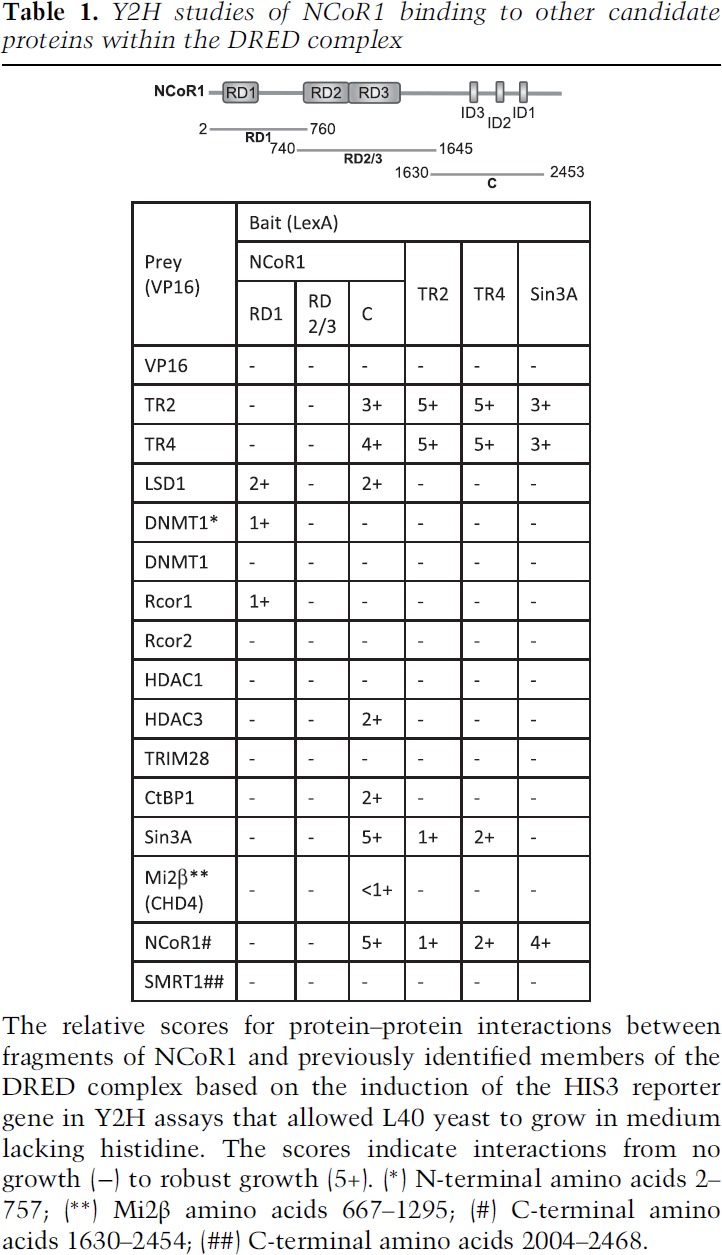
When these fragments of NCoR1 were tested in Y2H experiments, the RD1 domain of NCoR1 was found to interact with LSD1, the N terminus of DNMT1, and Rcor1 (CoREST) (Table 1). Furthermore, the C-terminal domain of NCoR1 was also found to interact with several corepressors, including LSD1, HDAC3, CtBP1, Sin3A, and mi2β (one of multiple protein constituents of the mammalian NuRD complex) as well as TR2 and TR4. None of the corepressors that we tested interacted with the NCoR1 RD2/3 domain (Table 1).
Interestingly, among all of the tested corepressor proteins, only NCoR1 (C-terminal) and Sin3A bound directly to TR2/TR4 (when TR2 or TR4 were used as Y2H bait) (Table 1), indicating that NCoR1 (and/or Sin3A) serves as an adaptor platform between TR2/TR4 and the other DRED complex subunits. Based on these data, we then addressed the possibility that Sin3A might be an additional or alternative DRED scaffold. However, when Sin3A was used as bait in additional Y2H experiments, we found that TR2, TR4, and NCoR1, but none of the epigenetic modifying corepressor enzymes, bound to Sin3A (Table 1), suggesting that either Sin3A is not an adaptor in the DRED complex or mediates interactions with additional partner proteins that have not yet been identified. Notably, the C-terminal domain of SMRT (silencing mediator of retinoic acid and thyroid hormone), which contains nuclear receptor interaction domains and also can serve in scaffold interactions between nuclear receptors and epigenetic corepressors (Mottis et al. 2013), does not directly interact with either TR2 or TR4 (Table 1), indicating that NCoR1 fulfills a unique requirement as the adaptor between TR2/TR4 and the DRED corepressor enzymes. Taken together, the data were consistent with a specific hypothesis regarding the protein constituents that might be minimally required to generate the large DRED repressor: NCoR1 serves as the central adaptor protein in which different domains recruit both the DNA-binding components (TR2 and TR4) and critical epigenetic modifying enzymes into the repressor complex.
TR4 and NCoR1 genomic binding site occupancy overlaps in K562 cells
The data reported here indicate that NCoR1 might serve as the scaffold between TR2/TR4 and other DRED complex proteins. If this hypothesis is correct, then NCoR1 and TR2/TR4 ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) peaks would be predicted to significantly overlap in the erythroid genome. To address this hypothesis, we examined the ChIP-seq signatures of NCoR1 (NCoR1-1 and NCoR1-2), EGFP-TR2, and EGFP-TR4 in K562 cells (a human embryonic myeloerythroid cell line) from the ENCODE database (http://genome.ucsc.edu/encode/downloads.html). The common peaks shared between two different data sets (NCoR1-1 and NCoR1-2) were accumulated as NCoR1 peaks to calculate NCoR1 and TR2/TR4 peak overlap. Among the 17,417 NCoR1 peaks, 6195 (35.5%) and 6932 (39.8%) peaks colocalized with TR2 and TR4, respectively, and 4538 (26%) peaks overlapped both TR2 and TR4 (Fig. 2A). Additionally, among the 22,243 TR2 and 26,557 TR4 peaks, 6195 (27.9%) and 6932 (26.1%) peaks were found to be co-occupied by NCoR1 (Fig. 2A).
Figure 2.
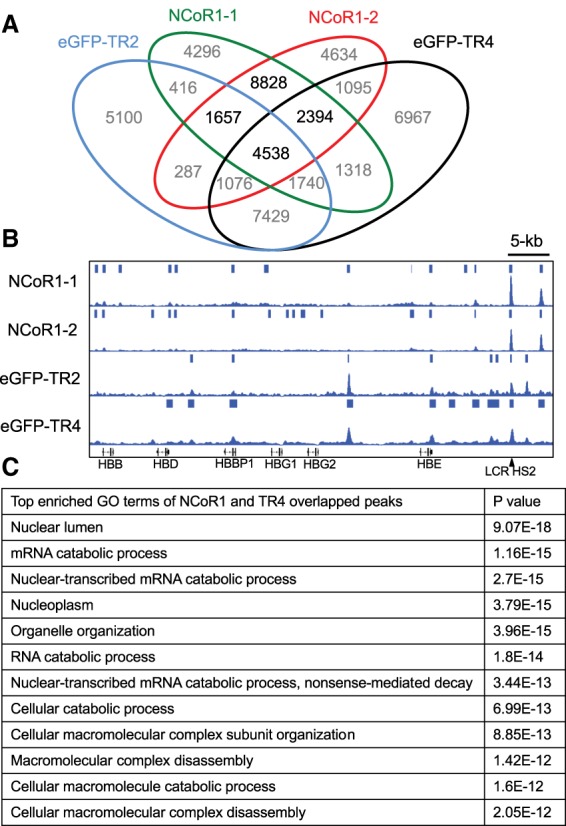
TR4 and NCoR1 significantly overlap in genome-wide binding site distribution. (A) Venn diagram of TR4, TR2, and NCoR1 ChIP-seq peaks in human erythroleukemia K562 cells. (B) Representative NCoR1, TR2, and TR4 overlapping peaks in the β-globin locus. (C) The top enriched gene ontology terms extracted from the TR4, NCoR1-1, and NCoR1-2 overlapping peak-associated genes.
The β-globin locus and the genes that regulate RNA catabolism and cell cycle were chosen as representative loci for association with all of these proteins, and there are clearly overlapping as well as unique binding site signatures (Fig. 2B; Supplemental Fig. 2). The strongest overlap among all three chromatin-immunoprecipitated proteins in the β-globin locus in K562 cells was at a DR2 element (GCTGACCACCTGACTAAA) in LCR HS2. Taken together, the ChIP-seq data indicate that NCoR1 and TR2/TR4 extensively overlap in the erythroid genome, consistent with the concept that these proteins often function together in a complex for a significant fraction of time (e.g., at LCR HS2) (Fig. 2B). Among the top gene ontology (GO) enrichment pathway terms among the peaks shared by NCoR1 and TR4 (Fig. 2C), there are multiple terms describing enrichment of proteins involved in macromolecular catabolism and cell organelle organization, suggesting that the DRED complex likely plays additional unexplored roles in these pathways.
NCoR1 interaction domain 3 (ID3) binds to the ligand-binding domains (LBDs) of TR2 and TR4
To begin to detail potential mechanisms of transcriptional repression elicited by the DRED complex, we characterized specific protein–protein interaction interfaces between TR2, TR4, and NCoR1. A series of TR2 or TR4 truncations was generated and tested by Y2H in order to map the domains that mediate their interactions with the C-terminal domain of NCoR1 (NCoR1-C) (Table 1). The expression of LexA-bait and VP16-prey fusion proteins was confirmed by Western blots using anti-LexA and anti-VP16 antibodies (Supplemental Fig. 1). These results show that the LBDs (EF domains) (Table 2) of both TR2 and TR4 were responsible for the interaction with NCoR1-C in a manner similar to other nuclear receptor/NCoR1 associations (Jepsen and Rosenfeld 2002) and that this interaction does not require the N-terminal domain (regions A/B), DNA-binding domain (region C), or hinge region (region D) of either orphan receptor (Table 2). Additionally, the N-terminal domain (A/B) of TR4, but not of TR2, is capable of independently interacting with NCoR1-C; thus, two different domains of TR4 can interact with NCoR1-C.
Table 2.
Identification of the binding interface between NCoR1 and TR2 or TR4
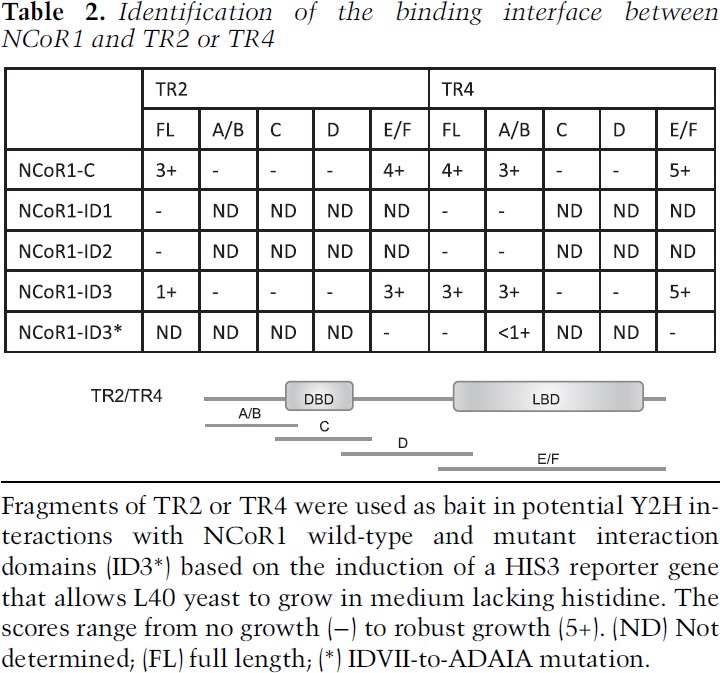
The C terminus of NCoR1 has three IDs (ID1–ID3) (Table 1) that were reportedly responsible for its interaction with other nuclear receptors (Cohen et al. 2001). Each of the IDs contains a corepressor–nuclear receptor (CoRNR) box bearing the five-amino-acid consensus sequence (I/L)XX(V/I)I (where X is any amino acid) that preferentially interacts with nuclear receptors (Hu and Lazar 1999; Perissi et al. 1999). To assess which IDs of NCoR1 interact with the LBDs of TR2 and TR4, three truncated fragments containing each of the NCoR1-IDs were tested in Y2H assays. As shown in Table 2, the LBDs of TR2 and TR4 specifically interact with NCoR1-ID3, demonstrating an ID3 preference for TR2/TR4:NCoR1 interactions. Furthermore, mutation of three amino acids in the ID3-CoRNR box (IDVII to ADAIA) fully abolished interactions with both TR2 and TR4 (Table 2), demonstrating that the ID3-CoRNR box of NCoR1 is both necessary and sufficient for interactions with the DRED orphan nuclear receptors. Taken together, these results clearly demonstrate that NCoR1 uses a CoRNR box in its ID3 domain to specifically bind to sequences in the LBDs of both TR2 and TR4.
NCoR1 is the adaptor for the DRED complex in HUDEP-2 cells
By identifying the amino acids in the NCoR1-ID3 that are critical for TR2/TR4 interaction, we next used CRISPR–Cas9 genome editing to generate site-directed mutants of NCoR1-ID3 in HUDEP-2 cells that were predicted to block interactions with wild-type TR2/TR4. Two homozygous mutant (IDVII to A5) NCoR1 clones were generated, and the site-specific mutations in both clones were verified by Sanger sequencing (Fig. 3A). Consistent with the Y2H data shown in Table 2, mutation of the five amino acids that comprise the CoRNR box completely disrupted NCoR1 interaction with TR4 in HUDEP-2 cells, whereas the interaction between the mutant NCoR1 protein and LSD1 is unperturbed (Fig. 3B).
Figure 3.
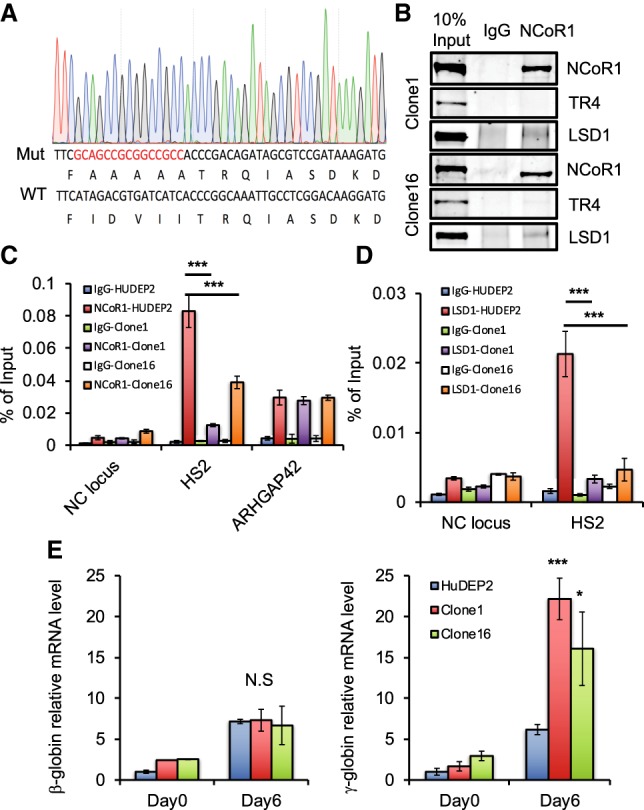
NCoR1 is the direct adaptor between TR2/TR4 and other DRED complex corepressors. (A) Design and generation of an NCoR1 mutant with deficient TR4 interaction. Red letters represent the mutant nucleic acid sequence generated by CRISPR targeting that destroys the ID3-CoRNR box (Hu and Lazar 1999; Perissi et al. 1999). (B) Confirmation that the TR4:NCoR1 interaction is abolished, whereas the LSD1:NCoR1 interaction is maintained in two individual NCoR1 CoRNR mutant clones by co-IP in HUDEP-2 cells. (C) CRISPR/Cas9-generated NCoR1 mutants in HUDEP-2 cells (clones 1 and 16) fail to be recruited to an endogenous TR2/4-binding site in the β-globin locus. (D) LSD1 occupancy is significantly reduced at the HS2 TR2/4-binding site in the β-globin locus in the absence of a robust NCoR1:TR4 interaction in undifferentiated HUDEP-2 cells. (Blue bar) Wild-type HUDEP-2 with IgG control; (red bar) wild-type HUDEP-2 cells with anti-NCoR1 or anti-LSD1; (green bar) NCoR1 mutant HUDEP-2 clone 1 with IgG control; (purple bar) NCoR1 mutant HUDEP-2 clone 1 with anti-NCoR1 or anti-LSD1; (white bar) NCoR1 mutant HUDEP-2 clone 16 with IgG control; (orange bar) NCoR1 mutant HUDEP-2 clone 16 with anti-NCoR1 or anti-LSD1. (E) Disruption of the TR2/TR4:NCoR1 interaction within the DRED complex derepresses γ-globin transcription in HUDEP-2 cells. β-Globin (left) and γ-globin (right) relative mRNA abundances in undifferentiated or differentiated (for 6 d) wild-type or NCoR1 mutant HUDEP-2 cells. Data are shown as the mean ± SD from three independent experiments. (*) P < 0.05; (***) P < 0.001, unpaired Student's t-test.
In the β-globin locus, wild-type NCoR1 binds most prominently to LCR HS2 (Figs. 2B, 3C). However, recruitment of the AAAAA mutated NCoR1 to its most prominent β-globin locus-binding site in HS2 was significantly reduced in both CRISPR mutant clones (Fig. 3C), indicating that TR2/4 plays a central role in NCoR1 recruitment to globin locus chromatin. Recruitment of NCoR1 to an unrelated regulatory element (in the ARHGAP42 gene, where it is tethered by transcription factor AP-1) was hardly affected (Fig. 3C). Consistent with the concept that NCoR1 is the adaptor between TR2/4 and multiple corepressor enzymes, LSD1 recruitment at HS2 of the β-globin locus was also significantly reduced in NCoR1 mutant HUDEP-2 cells (Fig. 3D). Taken together, these data confirm that NCoR1 serves as a primary adaptor to aggregate TR2/4 with DRED corepressor enzymes.
Disruption of the TR2/4:NCoR1 interaction derepresses γ-globin transcription
The DRED complex is critical for γ-globin repression in adult RBCs (Suzuki et al. 2014), and NCoR1 knockdown by shRNA in CD34+ cell erythroid differentiation cultures has been shown to induce γ-globin mRNA synthesis (Xu et al. 2013). To test for possible functional deficiencies that are due to NCoR1 loss of function in γ-globin repression, we determined globin mRNA levels in undifferentiated (day 0) or differentiated (day 6) HUDEP-2 cells bearing NCoR1 mutations that disrupt its interaction with TR2/4 (Fig. 3E). Upon examining either independent NCoR1 mutant HUDEP-2 clone, both β-globin expression and γ-globin expression were very similar in wild-type and mutant cells in the absence of differentiation induction. However, after 6 d of erythroid differentiation induction, while β-globin expression was essentially unchanged from its level in wild-type cells, γ-globin mRNA in the mutant clones increased by twofold to threefold, demonstrating that disruption of the TR2/TR4:NCoR1 interface specifically derepresses γ-globin expression in differentiated erythroid progenitor cells.
BAP1 regulates β-globin locus NCoR1 recruitment
In addition to the previously defined DRED complex corepressors and NCoR1 (identified here using BioID), we detected strong HCF1 association with TR4 (Fig. 1D), which was in agreement with the interactome of TR2/TR4 that was demonstrated previously in mouse erythroleukemia (MEL) cells (Cui et al. 2011). Interestingly, the well-characterized HCF1-interacting tumor suppressor BAP1 was also identified as another new component in the DRED complex (Fig. 1D). BAP1 is a nuclear-localized ubiquitin C-terminal hydrolase (Dey et al. 2012; Lee et al. 2014; Zarrizi et al. 2014; Qin et al. 2015), which has been shown to stabilize nuclear proteins through its deubiquitinase activity. Among the multiple corepressors already identified in the DRED complex, NCoR1 has been explicitly shown to be regulated by ubiquitination (Perissi et al. 2004, 2008; Catic et al. 2013; Mottis et al. 2013), and the ubiquitination of NCoR1 has been proposed to modulate its stability and genome recruitment through proteasome degradation (Catic et al. 2013). Based on previous studies as well as the earlier demonstration that both NCoR1 and BAP1 interact with TR4 (Fig. 1D), we investigated the possibility that BAP1 might regulate NCoR1 activity through its deubiquitinase activity.
To test this hypothesis, we immunoprecipitated HCF1 from wild-type HUDEP-2 nuclear extracts and probed those immune complexes by Western blotting with TR4, NCoR1, and BAP1 antibodies. These co-IP experiments indicated that all four of these proteins can be found in complex (Fig. 4A), consistent with the proteomics data. Next, the HCF1 co-IP was performed using the AAAAA NCoR1 mutant HUDEP-2 cell nuclear extracts in which the TR4 interaction with NCoR1 was disrupted (Table 2; Fig. 3B). In these cells, mutant NCoR1 is still able to bind to HCF1 (Supplemental Fig. 3), indicating that NCoR1:HCF1 binding is not dependent on TR4:NCoR1 interaction.
Figure 4.
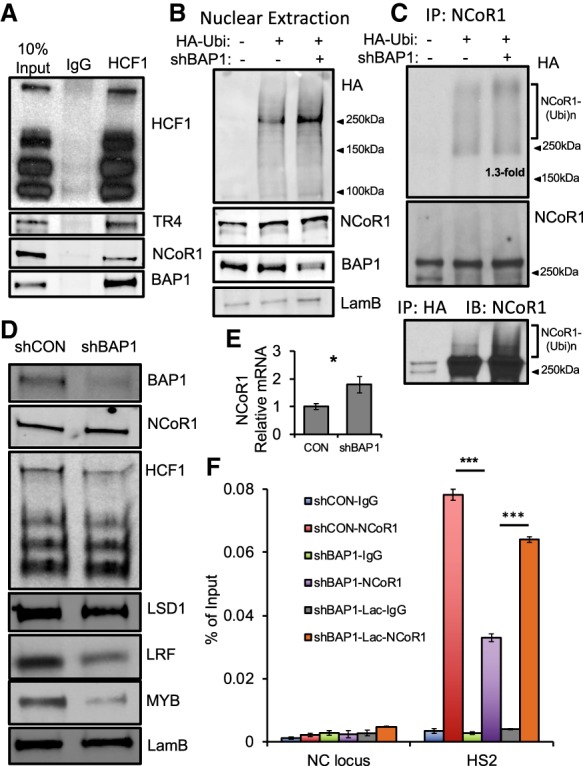
HCF1 and BAP1 are new components of the DRED complex and regulate NCoR1 activity in the β-globin locus. (A) Immunoprecipitated HCF1 was interrogated on Western blots using antibodies recognizing HCF-1, TR4, NCoR1, or BAP-1 in wild-type HUDEP-2 cells. (B) BAP1 knockdown enhances nuclear protein ubiquitination with virtually no change in NCoR1 abundance in 293T cells. (C) shRNA knockdown of BAP1 modestly increased NCoR1 ubiquitination in 293T cells. 293T nuclear extraction from wild type (lane 1), HA-ubiquitin expression plasmid (lane 2), or the combination of HA-ubiquitin and shBAP1 plasmids (lane 3) was immunoprecipitated with anti-NCoR1 or anti-HA followed by immune blotting with anti-HA or anti-NCoR1 antibodies. (D) BAP1 shRNA knockdown in HUDEP-2 cells affects NCoR1 and HCF1 protein abundance and slightly reduces LSD1 and LRF protein levels but significantly reduces MYB protein abundance. (E) NCoR1 mRNA level increased by only 1.7-fold after BAP1 knockdown. (F) NCoR1 occupancy at the TR2/4-binding site in HS2 is significantly reduced after BAP1 knockdown in undifferentiated HUDEP-2 cells, and the reduced NCoR1 occupancy could be partially rescued by proteasome inhibitor lactacystin treatment. (Blue bar) Control scrambled shRNA-infected HUDEP-2 with IgG control; (red bar) control HUDEP-2 cells with anti-NCoR1; (green bar) BAP1 knockdown HUDEP-2 with IgG control; (purple bar) BAP1 knockdown HUDEP-2 cells with anti-NCoR1; (gray bar) BAP1 knockdown HUDEP-2 after 3 h of treatment with 25 µM lactacystin (IgG control); (orange bar) NCoR1 detection in BAP1 knockdown HUDEP-2 cells after 3 h of treatment with 25 µM lactacystin. Data are shown as the means ± SD from three independent experiments (*) P < 0.05; (***) P < 0.001, unpaired Student's t-test.
To examine whether NCoR1 is directly ubiquitinated, we next used an in vivo ubiquitination assay in 293T cells. 293T cells were transfected with either an HA-tagged ubiquitin expression vector or HA-ubiquitin plus anti-BAP1 shRNA plasmids. BAP1 knockdown enhanced overall nuclear protein ubiquitination but did not significantly alter total NCoR1 abundance (Fig. 4B). Wild-type as well as transfected 293T nuclear extracts were next immunoprecipitated with anti-NCoR1. The immune complex was then examined by Western blot by using an anti-HA antibody (Fig. 4C, top and middle panels). The data indicate that HA-ubiquitin was coimmunoprecipitated at a higher-molecular-weight position than unmodified NCoR1, which slightly increased by 1.3-fold after BAP1 knockdown. Next, we immunoprecipitated HA followed by NCoR1 Western blotting (Fig. 4C, bottom panel). A significant amount of unmodified NCoR1 was detected, suggesting either that NCoR1 forms complexes with other ubiquitinated proteins or that ubiquitination might be a normal steady-state housekeeping modification that is reproduced in HUDEP-2 cells (Supplemental Fig. 4). Notably, there is also a small amount of NCoR1 that appears in diffuse higher-molecular-weight forms, and BAP1 knockdown increases those species (Fig. 4C, bottom panel). Taken together, these assays indicate that NCoR1 is directly regulated through ubiquitination and that BAP1 plays an important role in NCoR1 deubiquitination.
To test whether BAP1 and ubiquitination alter NCoR1 recruitment to chromatin (as reported in a different cellular context) (Catic et al. 2013), we reduced the abundance of BAP1 mRNA using an shRNA-containing lentivirus followed by NCoR1 ChIP assays in the β-globin locus in HUDEP-2 cells. The efficiency of BAP1 knockdown was confirmed by Western blotting (Fig. 4D). We found that reduced BAP1 abundance does not significantly alter the level of total NCoR1 protein (Fig. 4D), whereas the NCoR1 mRNA level slightly increased (Fig. 4E). Additionally, other DRED components (HCF1 and LSD1) were slightly diminished after BAP1 degradation, while, interestingly, other globin regulatory proteins (LRF and c-Myb) appeared to be more significantly depleted (Fig. 4D). When compared with infection with a control scrambled shRNA, BAP1 mRNA reduction significantly depleted the amount of NCoR1 at its most prominent binding site in the β-globin locus: LCR HS2 (Fig. 4F, purple vs. red bars).
Ubiquitination of NCoR1 destabilized its occupancy on chromatin, presumably by acting through proteasome-mediated protein degradation. Treatment with the proteasome inhibitor lactacystin (Lac) has been reported to significantly enhance NCoR1 recruitment at specific chromosomal binding sites by preventing its degradation (Catic et al. 2013). To test whether BAP1 knockdown would reduce NCoR1 recruitment to HS2 through a proteasome-mediated process, we first treated uninfected HUDEP-2 cells with Lac for 3 h, which significantly enhanced accumulation of ubiquitinated protein in a dose-dependent manner, while total cellular NCoR1 abundance was essentially unchanged (Supplemental Fig. 5; Catic et al. 2013). When anti-BAP1 shRNA lentivirus-infected HUDEP-2 cells were treated with Lac for 3 h, NCoR1 occupancy at HS2 was largely rescued (Fig. 4F, orange vs. purple bars), indicating that proteasome-mediated NCoR1 degradation plays a key role in NCoR1 chromatin occupancy. Taken together, the data suggest that both BAP1 and NCoR1 are involved in DRED complex activity and that BAP1 is a central regulator of NCoR1 recruitment to specific chromatin sites in the β-globin locus.
Reduced BAP1 expression robustly induces γ-globin transcription
Since BAP1 was identified here as a novel regulator of DRED complex activity that appears to be important for NCoR1 recruitment to β-globin locus HS2 (Fig. 4E), we next asked whether BAP1 knockdown would functionally affect globin gene expression. To do so, mRNA abundances were assessed in undifferentiated (day 0) or differentiated (day 6) (same as in Fig. 3E) BAP1 shRNA-infected and control (scrambled sequence) shRNA-infected HUDEP-2 cells.
BAP1 gene expression increased during erythroid differentiation (day 6 vs. day 0) in control (scrambled) shRNA-infected HUDEP-2 cells, whereas BAP1 mRNA was reduced to ∼30% of controls in shBAP1-infected cells (Fig. 5A, left panel). At day 0, BAP1 knockdown induced β-globin mRNA levels by fourfold and γ-globin by 43-fold (Fig. 5A, middle and right panels). After 6 d of erythroid differentiation, BAP1 knockdown further induced β-globin expression less than twofold, whereas γ-globin transcription was massively induced (∼90-fold) (Fig. 5A, right panel), which enhanced the γ/(γ + β) ratio by 15% (Fig. 5B). The significant derepression of γ-globin synthesis was confirmed by high-performance liquid chromatography (HPLC) monitoring of hemoglobin production in the BAP1 knockdown cells (Fig. 5C); the percentage of HbF increased from 0.1% in scrambled sequence-infected cells (shCON) to 13.5% in BAP1 shRNA-infected cells (shBAP1) after 10-d of erythroid differentiation induction.
Figure 5.
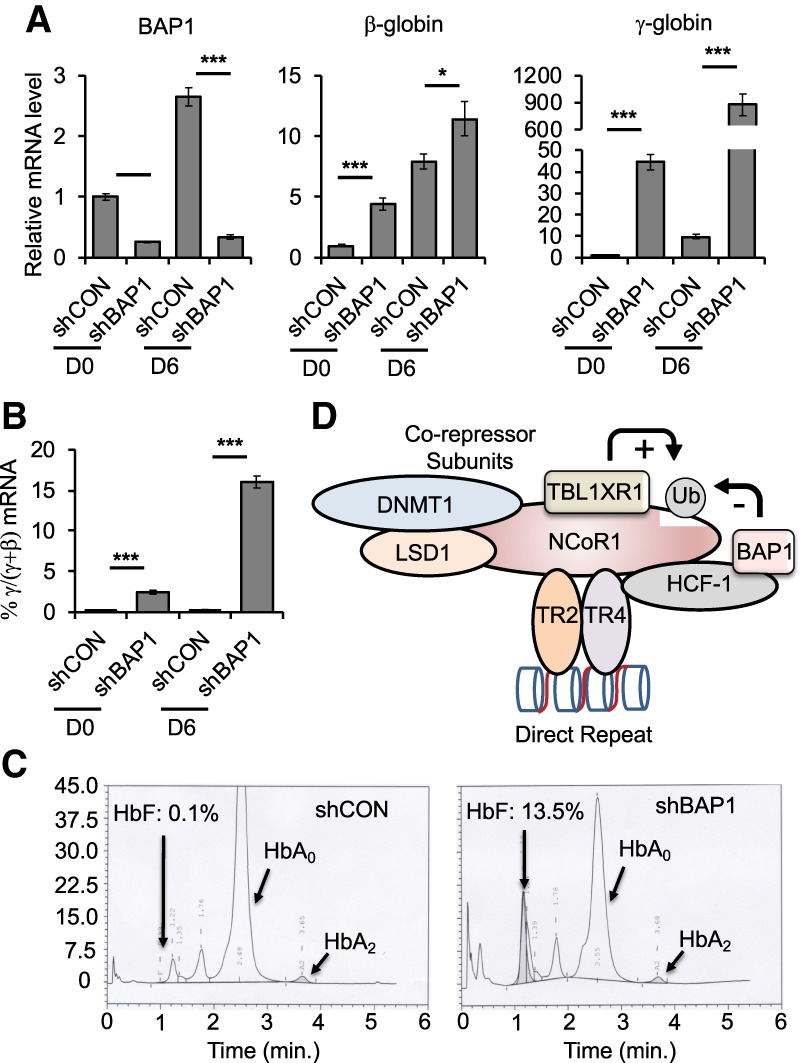
Reduced BAP1 expression robustly induces γ-globin transcription. (A) The relative mRNA abundance of BAP1, β-globin, and γ-globin in undifferentiated (D0) or differentiated (D6) HUDEP-2 cells. (B) γ/(γ + β) ratio in undifferentiated (day 0 [D0]) or differentiated (day 6 [D6]) HUDEP-2 cells. (C) Reduced BAP1 expression significantly induces HbF after 10 d of HUDEP-2 cell differentiation induction. (D) Schematic summarizing the observations that NCoR1 is the scaffold on which TR2/TR4 and the corepressor epigenetic enzymatic subunits of the DRED complex are assembled. We hypothesize that the ubiquitination of NCoR1 by TBL1XR1 or its deubiquitination by BAP1 regulates dynamic homeostasis of NCoR1 recruitment or retention in the DRED complex and subsequent globin gene repression and/or derepression (*) P < 0.05; (***) P < 0.001, unpaired Student's t-test.
To avoid possible off-target effects of shRNA treatment on γ-globin derepression, we next used CRISPR–Cas9 genome editing to generate BAP1 mutant HUDEP-2 cells. Two clones bearing 13-base-pair deletions in BAP1 exon 5 were examined (Supplemental Fig. 6A); no homozygous BAP1 knockouts were recovered. Consistent with the BAP1 shRNA knockdown data, BAP1 heterozygous mutation modestly affected adult β-globin mRNA abundance but significantly induced γ-globin expression (Supplemental Fig. 6B). Taken together, the data indicate that this newly discovered DRED complex regulator, BAP1, plays a vital role in fetal globin gene repression and that inhibition of BAP1 deubiquitinase activity may serve in the future as a novel therapeutic target for γ-globin induction.
Discussion
In this study, we found that NCoR1 serves as a central scaffold upon which the active DRED repressor is assembled. We also showed that a novel DRED repressor subunit, deubiquitinase BAP1, regulates site-specific NCoR1 recruitment within the β-globin locus and that BAP1 reduction leads to enormously increased γ-globin mRNA and protein induction (Fig. 5D).
Several lines of evidence demonstrate that BAP1 regulates NCoR1 recruitment through its deubiquitinase activity. First, NCoR1 is ubiquitinated and BAP1 is critical for NCoR1 deubiquitination in 293T cells (Fig. 4C), and the ubiquitination of NCoR1 followed by proteasome degradation was reported previously to be vital for the regulation of NCoR1 transcriptional activity (Catic et al. 2013). Second, BAP1 is found in a complex with NCoR1 (as well as with TR4 and HCF1) (Fig. 4A), and knockdown of BAP1 mRNA reduced NCoR1 recruitment at β-globin locus-associated sites and could be rescued by proteasome inhibitor administration (Fig. 4F). Third, although BAP1 erythroid ChIP-seq information is not yet available, HCF1 (to which the majority of cellular BAP1 appears to be bound [Fig. 4A] and which is found in complex with a majority (85%) of BAP1 in bone marrow-derived macrophages) (Dey et al. 2012) colocalized genome-wide with NCoR1 as well as in the globin locus in K562 erythroleukemia cells (from ENCODE ChIP-seq studies). Additionally, the F-box-like/WD repeat-containing protein TBL1XR1 that is responsible for NCoR1 ubiquitination (Perissi et al. 2004, 2008; Mottis et al. 2013) also colocalized with NCoR1 and HCF1 as well as with TR2/TR4 at sites in the β-globin locus in K562 cells (ChIP-seq in ENCODE). Taken together, the available data suggest that in vivo homeostasis of NCoR1 is achieved by an opposing ubiquitination/deubiquitination balance and that this likely plays a key regulatory role in NCoR1 activity in erythroid cells (Fig. 5D).
mRNA stability is an important mechanism that can regulate protein abundance. Globin mRNA stability is a critical determinant of protein abundance in normal erythropoiesis, since the long half-life of these mRNAs is fundamental for the continuous translation of globin proteins, which is important during later stages of erythropoiesis, when transcription is arrested and nuclei are shed (Waggoner and Liebhaber 2003). In contrast, destabilization of globin mRNA by naturally occurring mutations such as anti-termination signals (TAA to CAA in the α2-globin gene leading to accelerated mRNA decay) (Morales et al. 1997) results in α-thalassemia. Under physiological conditions, it is possible that nonsense transcripts could be generated and that the cell is able to destabilize these aberrant transcripts through various pathways, including nonsense-mediated decay (Palacios 2013), indicating that mRNA catabolism could play a role in erythropoiesis. In this regard, it is interesting to note that the GO terms shown in Figure 2 (and represented in Supplemental Fig. 2) suggest that the genomic sites of overlap between TR4 and NCoR1 are enriched in multiple pathways associated with RNA catabolism and nonsense-mediated mRNA decay, suggesting that TR4 and NCoR1 might affect erythropoiesis through the regulation of pathways that control mRNA stability.
In a previous study, we found that TR4 promotes erythroid cell proliferation in mouse bone marrow-derived erythroid progenitors (Lee et al. 2017). In this regard, it is interesting to note that both of the HUDEP-2 clones harboring an NCoR1 mutation that leads to failure to interact with TR2/TR4 grow significantly slower than parental cells (Supplemental Fig. 7), suggesting that NCoR1 might mediate cell cycle regulatory activity of TR4 as the DRED complex adaptor. By analyzing the TR2/TR4 and NCoR1 ChIP-seq database in K562 cells, we found co-occupancy of all three proteins near the promoters of E2F family transcription factors, critical regulators of G1-to-S-phase transition during the cell cycle (Supplemental Fig. 2). This suggests that TR4 might regulate cell proliferation through direct recruitment of NCoR1 and subsequent transcriptional control of E2F family members.
The switch from corepressor to coactivator function is an important gene regulatory mechanism for nuclear receptors (Perissi et al. 2010). Based on this fact (for which many examples exist) and the model proposed in this study, there are intriguing questions that remain. First, what proteins (if any) constitute the coactivator form of TR2/TR4 after depleting cells of NCoR1? A previous study suggested that PGC-1α/β might be such coactivator candidates (Cui et al. 2014). PGC-1α serves as the coactivator for several other nuclear receptors, such as PPARγ (Lin 2009), and PGC-1α/β are in fact recruited in the mouse β-globin locus, where TR2/TR4 bind and the interaction between TR2/TR4 and PGC-1α/β was confirmed by co-IP in erythroid cells (Cui et al. 2014). Furthermore, compound mutants in the PGC-1α/β genes impaired murine β-type globin gene expression, which is induced by disrupting TR2/TR4 and NCoR1 interaction (Fig. 3E), suggesting that NCoR1 and PGC-1α/β share a mutually antagonistic relationship. Taken together, these data suggest that PGC-1α/β might serve as candidate coactivators that replace ubiquitin-mediated turnover of NCoR1 to generate a TR2/4 activator from the DRED repressor form.
An additional question addresses the mechanism that could allow or promote the exchange of NCoR1 for coactivators. A study showing that ubiquitination of NCoR1 by TBL1XR1 favors the switch from corepressors to coactivators has been reported, which may be the underlying mechanism responsible for a cofactor switch (Mottis et al. 2013). NCoR1 has been shown to bind to unliganded nuclear receptors, and this interaction is lost upon ligand binding (Perissi et al. 2010). In this regard, it is interesting to note that nonphysiological concentrations of vitamin A (or, more likely, a related molecule) have been promoted as a potential TR4 ligand from in vitro analysis (Zhou et al. 2011). If there is a high-affinity ligand for either or both TR2 and TR4, such a fact could have significant clinical potential, since ligand antagonists might be identified that could induce γ-globin transcription toward the aim of benefiting patients with β-globinopathies.
In this study, we discovered that a loss of NCoR1 recruitment to β-globin locus regulatory sites induced fetal globin gene expression by twofold to threefold, whereas a 70% reduction in BAP1 enzyme abundance induces γ-globin synthesis 90-fold, indicating that there are other likely γ-globin-inductive mechanisms that are regulated by BAP1. Of note, among the critical γ-globin repressors examined, we found that LRF abundance was moderately reduced, whereas MYB abundance was significantly diminished after BAP1 knockdown (Fig. 4D), suggesting that BAP1 might play an important role in stabilizing these key γ-globin regulatory proteins. To delve more deeply into the underlying mechanism by which BAP1 mediates repression of γ-globin synthesis, it would be of great significance to determine the identity of additional BAP1 target proteins in erythroid cells in future studies. Detailed investigation of this issue is of potential clinical significance, since increased γ-globin synthesis is known to mitigate the symptoms of the β-globinopathies (Noguchi et al. 1988; Weatherall 2001; Ngo et al. 2012). Considering that multiple enzymatic activities could be simultaneously targeted for therapeutic purposes, the deubiquitinase BAP1 as well as additional epigenetic enzymatic activities that are regulated by BAP1 might provide novel targets for therapeutic intervention in inducing fetal globin synthesis for treatment of the β-globinopathies.
Materials and methods
HUDEP-2 cell culture
HUDEP-2 cells were the kind gift of Dr. Yukio Nakamura (Kurita et al. 2013). HUDEP-2 cells were expanded in StemSpan SFEM medium (Stem Cell Technologies) supplemented with 50 ng/mL human stem cell factor (SCF) (Peprotech), 3 IU/mL erythropoietin (Amgen), 1 µg/mL doxycycline (Sigma), 1 µM dexamethasone (Cayman), and 100 U/mL penicillin/streptomycin. To induce erythroid differentiation, HUDEP-2 cells were cultured in Iscove's modified Dulbecco's medium (IMDM) (Life Technologies) supplemented with 5% human AB serum (Sigma), 50 ng/mL SCF, 3 IU/mL erythropoietin (Amgen), 1 µg/mL doxycycline (Sigma), 330 µg/mL holo-transferrin (Sigma), 10 µg/mL human insulin (Sigma), 2 IU/mL heparin (Sigma), and 100 U/mL penicillin/streptomycin.
BioID
TR4-BirA*-expressing or parental wild-type HUDEP-2 cells were incubated for 24 h in StemSpan SFEM complete medium supplemented with 50 µM biotin. After washing the cells with cold PBS, cell pellets (108 cells) were resuspended in 4.5 mL of ice-cold RIPA buffer (50 mM Tris–HCl at pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 0.5% sodium deoxcycholate with 1 mM PMSF, 1 mM DTT, 1× Roche protease inhibitor cocktail). The cell lysates were sonicated twice on ice in a 15-mL conical tube for a total of 30 sec at 3 sec on/7 sec off, total pulse on 30 sec with 40% amplitude (Sonic Dismembrator model 500, Fisher Scientific). Three-hundred units of benzonase (Sigma, E1014) was added and then rotated for 60 min at 4°C. Cell lysates were cleared by 16,500g centrifugation for 10 min at 4°C. Supernatants were incubated with 250 µL of Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific, 65001; the beads were prewashed with RIPA buffer) three times overnight in a 4°C room. After incubation, the beads were washed with RIPA buffer four times followed by three washes with 50 mM fresh NH4HCO3. The beads were frozen at −80°C until mass spectrometry identification.
Protein identification by mass spectrometry and data analysis
The beads were resuspended in 50 µL of 0.1 M ammonium bicarbonate buffer (pH 8). Cysteines were reduced by adding 50 µL of 10 mM DTT and incubated for 30 min at 45°C. Samples were cooled to room temperature, and alkylation of cysteines was achieved by incubating with 65 mM 2-chloroacetamide in the dark for 30 min at room temperature. An overnight digestion with 1 µg of sequencing-grade modified trypsin was carried out at 37°C with constant shearing in a Thermomixer. Digestion was stopped by acidification, and peptides were desalted using SepPak C18 cartridges following the manufacturer's protocols (Waters). Samples were completely dried using a vacufuge; the resulting peptides were dissolved in 8 µL of 0.1% formic acid/2% acetonitrile solution, and 2 µL of the peptide solution was resolved on a nanocapillary reverse-phase column (Acclaim PepMap C18, 2 µm, 50 cm, Thermo Scientific) using a 0.1% formic acid/2% acetonitrile (buffer A) and 0.1% formic acid/95% acetonitrile (buffer B) gradient at 300 nL/min over a period of 180 min (2%–22% buffer B for 110 min, 22%–40% for 25 min, and 40%–90% for 5 min followed by holding in 90% buffer B for 5 min and requilibration with buffer A for 35 min). Eluent was directly introduced into an Orbitrap Fusion tribrid mass spectrometer (Thermo Scientific) using an EasySpray source. MS1 scans were acquired at 120,000 resolution (AGC target = 1 × 106; maximum IT = 50 msec). Data-dependent collision-induced dissociation tandem mass spectrometry (MS/MS) spectra were acquired using top speed method (3 sec) following each MS1 scan (NCE = ∼32%; AGC target = 1 × 105; maximum IT = 45 msec).
Proteins were identified by searching the MS/MS data against Homo sapiens (Swissprot, version 2016-11-30) using Proteome Discoverer (version 2.1, Thermo Scientific). Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.2 Da, two missed cleavages were allowed, carbamidimethylation of cysteine was considered a fixed modification, and oxidation of methionine and deamidation of asparagine and glutamine were considered as potential modifications. False discovery rate (FDR) was determined using Percolator, and proteins/peptides with a FDR of ≤1% were retained for final analysis.
Quantitative RT–PCR (qRT–PCR) analysis
Total RNA recovered from HUDEP-2 cell cultures was isolated using Trizol (Thermo Fisher Scientific) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized with SuperScript III reverse transcriptase (Thermo Fisher Scientific). Total RNA from the murine ex vivo cultures was isolated using Trizol followed by the synthesis of cDNA using iScript cDNA synthesis kits (Bio-Rad). qRT–PCR was performed using FastSYBR Green master mix on an ABI Step One Plus. The abundance of human OAZ1 (Cui et al. 2015a, b) and/or mouse GAPDH mRNA was used as the normalization control. Sequences of all primers used for qRT–PCR are listed in Supplemental Table 2.
ChIP assays
HUDEP-2 cells growing in the exponential phase were cross-linked by treatment with 1% formaldehyde for 10 min at room temperature with gentle shaking. Cross-linking was terminated by adding 0.125 M glycine for 5 min with gentle shaking at room temperature. Cross-linked cells were washed twice with cold PBS, and 107 cells were aliquoted for each ChIP assay. Cells (107) were resuspended in 1 mL of lysis buffer-1 (50 mM HEPES-KOH at pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1 mM PMSF, 1× protease inhibitors [Sigma, P8340]). The cell suspension was rotated for 10 min at 4°C followed by 35 strokes in a type-B dounce homogenizer on ice. The pellet was resuspended in 1 mL of lysis buffer-2 (10 mM Tris-Cl at pH 8, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1 mM PMSF, 1× protease inhibitors) and rotated for 10 min at room temperature. The nuclei were then resuspended in 0.3 mL of lysis buffer-3 (10 mM Tris-Cl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, 0.5% N-lauroylsarcosine, 1 mM PMSF, 1× protease inhibitors) in a 1.5-mL tube and sonicated. Sonication was performed at 40% amplitude with 10-sec pulse on and 20-sec pulse off for eight cycles (Sonic Dismembrator model 500, Fisher Scientific). After sonication, Triton X-100 was added to 1%, and cell debris was removed by centrifugation at 20,817g for 10 min at 4°C. Ten percent of the supernatant containing the chromatin was saved as the input control, and the remaining 90% was brought to 500 µL by the addition of immunoprecipitation dilution buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholic acid, 1 mM EDTA, 1 mM PMSF, 1× protease inhibitors [Sigma, P8340]).
Ten microliters of protein A/G magnetic beads (Thermo Fisher Scientific, 88802) was washed twice with cold blocking solution (0.5% BSA in PBS) and resuspended in 500 µL of blocking solution followed by rotation for 2 h in a 4°C room. Two micrograms of antibody or control rabbit IgG was added to the bead solution and then rotated overnight at 4°C. The beads were then washed once with blocking solution and incubated with 107 cell equivalents of chromatin for 12–16 h at 4°C. The beads were washed gently with cold immunoprecipitation wash buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholic acid, 1 mM EDTA) three times and 4°C PBS once. After washing, 300 µL of elution buffer (100 mM NaHCO3, 1% SDS) was used to remove the chromatin from the beads by incubation with 900 rpm shaking for 2 h at 65°C. Thirty-six microliters of 5 M NaCl was then added to reverse-cross-link the chromatin, and the mixture was incubated overnight at 65°C. After reverse cross-linking, DNase-free RNaseA digestion was performed for 30 min at 37°C followed by proteinase K treatment. The DNA sample was prepared by phenol extraction followed by column purification (Thermo Scientific) and subsequently analyzed by qPCR. qPCR was performed using a FastSYBR Green master mix (20-L total reaction volume) on an ABI Step One Plus PCR machine with fast protocol settings (once for 20 sec at 95°C, 40 times for 3 sec at 95°C and 30 sec at 60°C, once for 15 sec at 95°C, and once for 60 sec at 60°C with 0.3°C/sec increases up to 95°C for dissociation curve analysis). Antibodies used for these experiments are listed in Supplemental Table 3.
Y2H assays
Analysis of protein–protein interactions in yeast was performed as described previously (Vojtek and Hollenberg 1995) using Saccharomyces cerevisiae L40 harboring HIS3 and lacZ as reporter genes. All corepressor DNA sequences were amplified by PCR using MEL cDNA samples as templates. PCR fragments were subcloned into bait (pBTM116) or prey (pVP16) plasmids to generate LexA DNA-binding domain and VP16 activation domain fusion constructs, respectively. Point mutations were introduced into the constructs by site-directed mutagenesis. All constructs were verified by DNA sequencing. Interactions were tested in Y2H based on the induction of the HIS3 reporter gene that allowed yeast to grow in medium lacking histidine. Autoactivation of the HIS3 reporter gene by bait plasmids was suppressed by addition of 3-aminotriazole into the selective medium. The expression of fusion proteins in yeast was confirmed by Western blot using antibodies against LexA DNA-binding domain or VP16 activation domain. An antibody recognizing phosphoglycerate kinase 1 was used as the loading control. Antibody information is shown in Supplemental Table 3.
Lentiviral shRNAs
The human TR4 cDNA was cloned into BioID plasmid (Addgene, 74223), and the BirA*-TR4 fusion protein was then cut out and inserted into CD550A-1 lentivirus plasmid (System Biosciences). The pLKO-puro lentiviral plasmids carrying shRNAs were obtained from Sigma-Aldrich. Human BAP1 shRNA clone TRCN0000007374 was used to knock down BAP1 expression. TRC2 pLKO.5-puro nonmammalian shRNA were used as a control. Lentiviruses were prepared as described previously (Moffat et al. 2006). To knock down BAP1, HUDEP-2 cells were transduced with lentiviruses carrying BAP1 or control (scrambled sequence) shRNAs by RetroNectin according to the product manufacturer (TaKaR, T100A/B). Transduced cells were selected with 0.25 µg/mL puromycin 48 h after transduction.
Co-IP
Nuclear extracts were prepared as described (Folco et al. 2012). For each co-IP reaction, 200 µg of nuclear extracts and 5 µg of antibody were mixed and brought to a final volume of 500 µL by the addition of sample buffer (10 mM HEPES-KOH at pH 9.0, 1.5 mM MgCl2, 0.25 mM EDTA, 20% glycerol, 0.3% NP-40, 0.5 mM DTT, 1 mM PMSF, 1× protease inhibitors [Sigma P8340]), and the mixture was rotated overnight at 4°C to promote antigen:antibody interaction. Twenty-five microliters of protein A/G magnetic beads (Thermo Fisher Scientific, 88802) was washed three times with PBS. After the final wash, the beads were blocked by treatment with blocking buffer (10 mM HEPES-KOH at pH 9.0, 1.5 mM MgCl2, 0.25 mM EDTA, 20% glycerol, 200 µg/mL chicken egg albumin) and rotated overnight at 4°C. After blocking, the antigen:antibody mixture was incubated with the protein A/G beads for 2 h at room temperature. The beads were then washed five times with wash buffer (10 mM HEPES-KOH at pH 9.0, 1.5 mM MgCl2, 0.25 mM EDTA, 20% glycerol, 0.06% NP-40, 300 mM KCl, 1 mM PMSF, 1× protease inhibitors). Proteins were eluted from the beads by the addition of 1× SDS-PAGE loading buffer (Bio-Rad, 161-0737) for 10 min at 25°C, electrophoresed, transferred to nylon membranes, and then subjected to Western blot detection. The antibodies used for co-IP are listed in Supplemental Table 3.
293T cell ubiquitination assays
The HA-ubiquitin expression plasmid was from Addgene (18712). 293T cells were transfected with HA-ubiquitin or a combination of HA-ubiquitin and BAP1 shRNA plasmids. Forty-eight hours after transfection, nuclear extracts were prepared for HA or NCoR1 immunoprecipitation.
Western blotting
Western blotting was performed as described previously (Cui et al. 2011). The antibodies used for detection are listed in Supplemental Table 3.
CRISPR mutagenesis
CRISPR–Cas9 targeted mutagenesis of NCoR1 was performed as described (Ran et al. 2013). The guide RNA (CCGGCAAATTGCCTCGGACA) was used to generate the NCoR1 mutant depicted in Figure 3A. The targeted (repairing) single-strand oligonucleotide template sequence used was 5′-ATAAAGGGCCTCCTCCAAAATCCAGATATGAGGAAGAGCTAAGGACCAGAGGGAAGACTACCATTACTGCAGCTAACTTCGCAGCCGCGGCCGCCACCCGACAGATAGCGTCCGATAAAGATGCGAGGGAACGTGGCTCTCAAAGTTCAGACTCTTCTAGTAGCTGTATGTATCTCAATCCGAGTTTCACAATGTGATGT-3′. The guide RNA (ACCCACCCTGAGTCGCATGA) was used to generate the BAP1 mutants depicted in Supplemental Figure 6A. The guide RNA was cloned in the px458 vector (Addgene, 48138), with exchange of CMV to the EF1α promoter driving Cas9 expression. The vector and repairing template were transduced into HUDEP-2 cells by electroporation. EGFP-positive cells were then sorted using a FACS Aria II, and single-cell cloning was performed to generate the mutant cells.
ChIP-seq analysis
The ChIP-seq data in K562 cells were obtained from ENCODE: NCoR1-1 (ENCSR910JAI), NCoR1-2 (ENCSR798ILC), eGFP-TR2 (ENCSR178DEG), and eGFP-TR4 (ENCSR750LYM). The extent of peak overlap was calculated using BEDtools (Quinlan and Hall 2010). GO enrichment was analyzed by ChIP-Enrich (Welch et al. 2014).
Supplementary Material
Acknowledgments
We gratefully acknowledge the support, insights, and comments of our colleagues Yogen Saunthararajah (Cleveland Clinic), Joe DeSimone, and Don Lavelle (University of Illinois at Chicago) from a National Heart, Lung, and Blood Institute (NHLBI) Excellence in Hemoglobinopathies Research Award (U01 HL117658). We also appreciate numerous insightful comments from D. Lucas (Cincinnati Children's Hospital), and the expert technical assistance of V. Basrur in the Department of Pathology Proteomics Resource Facility. We gratefully acknowledge partial support of shared facilities (sequencing and flow cytometry) from a National Cancer Institute grant to the University of Michigan Comprehensive Cancer Center (P30 CA046592). L.Y. is supported by a fellowship from the Cooley's Anemia Foundation, N.J. was supported by a Translational Scholarship from the NHLBI (U01 HL117658), and M.P.L. was supported by a post-doctoral fellowship from the National Institute of Diabetes and Digestive and Kidney Diseases (F32 DK108493).
Author contributions: L.Y., N.J., K.-C.L., and J.D.E. conceived the study. R.K. and Y.N. obtained the resources for the study. T.H. and G.M. performed the formal analysis. L.Y., N.J., M.P.L., and Q.W. performed the investigations. L.Y., N.J., and J.D.E. wrote the original draft of the manuscript. A.B.V. and J.-F.R. supervised the study. J.D.E. acquired the funding.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.318436.118.
References
- Bulger M, Groudine M. 1999. Looping versus linking: toward a model for long-distance gene activation. Genes Dev 13: 2465–2477. 10.1101/gad.13.19.2465 [DOI] [PubMed] [Google Scholar]
- Catic A, Suh CY, Hill CT, Daheron L, Henkel T, Orford KW, Dombkowski DM, Liu T, Liu XS, Scadden DT. 2013. Genome-wide map of nuclear protein degradation shows NCoR1 turnover as a key to mitochondrial gene regulation. Cell 155: 1380–1395. 10.1016/j.cell.2013.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg JB, Weatherall DJ, Bodmer WF. 1983. 5-azacytidine for β-thalassemia? Lancet 321: 536 10.1016/S0140-6736(83)92227-4 [DOI] [PubMed] [Google Scholar]
- Cohen RN, Brzostek S, Kim B, Chorev M, Wondisford FE, Hollenberg AN. 2001. The specificity of interactions between nuclear hormone receptors and corepressors is mediated by distinct amino acid sequences within the interacting domains. Mol Endocrinol 15: 1049–1061. 10.1210/mend.15.7.0669 [DOI] [PubMed] [Google Scholar]
- Cui S, Kolodziej KE, Obara N, Amaral-Psarris A, Demmers J, Shi L, Engel JD, Grosveld F, Strouboulis J, Tanabe O. 2011. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic β-type globin promoters in differentiated adult erythroid cells. Mol Cell Biol 31: 3298–3311. 10.1128/MCB.05310-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Tanabe O, Lim KC, Xu HE, Zhou XE, Lin JD, Shi L, Schmidt L, Campbell A, Shimizu R, et al. 2014. PGC-1 coactivator activity is required for murine erythropoiesis. Mol Cell Biol 34: 1956–1965. 10.1128/MCB.00247-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Lim KC, Shi L, Lee M, Jearawiriyapaisarn N, Myers G, Campbell A, Harro D, Iwase S, Trievel RC, et al. 2015a. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood 126: 386–396. 10.1182/blood-2015-02-626259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Tanabe O, Sierant M, Shi L, Campbell A, Lim KC, Engel JD. 2015b. Compound loss of function of nuclear receptors Tr2 and Tr4 leads to induction of murine embryonic β-type globin genes. Blood 125: 1477–1487. 10.1182/blood-2014-10-605022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSimone J, Heller P, Hall L, Zwiers D. 1982. 5-azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Natl Acad Sci 79: 4428–4431. 10.1073/pnas.79.14.4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, et al. 2012. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 337: 1541–1546. 10.1126/science.1221711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JD, Tanimoto K. 2000. Looping, linking, and chromatin activity: new insights into β-globin locus regulation. Cell 100: 499–502. 10.1016/S0092-8674(00)80686-8 [DOI] [PubMed] [Google Scholar]
- Folco EG, Lei H, Hsu JL, Reed R. 2012. Small-scale nuclear extracts for functional assays of gene-expression machineries. J Vis Exp 64: pii: 4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Lazar MA. 1999. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402: 93–96. 10.1038/47069 [DOI] [PubMed] [Google Scholar]
- Jepsen K, Rosenfeld MG. 2002. Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci 115: 689–698. [DOI] [PubMed] [Google Scholar]
- Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ. 2016. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27: 1188–1196. 10.1091/mbc.e15-12-0844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita R, Suda N, Sudo K, Miharada K, Hiroyama T, Miyoshi H, Tani K, Nakamura Y. 2013. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8: e59890 10.1371/journal.pone.0059890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC. 2015. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics 118: 81–94. 10.1016/j.jprot.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Lee SA, Hur SK, Seo JW, Kwon J. 2014. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat Commun 5: 5128 10.1038/ncomms6128 [DOI] [PubMed] [Google Scholar]
- Lee MP, Tanabe O, Shi L, Jearawiriyapaisarn N, Lucas D, Engel JD. 2017. The orphan nuclear receptor TR4 regulates erythroid cell proliferation and maturation. Blood 130: 2537–2547. 10.1182/blood-2017-05-783159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, DeSimone J, Noguchi CT, Turner PH, Schechter AN, Heller P, Nienhuis AW. 1983. 5-azacytidine increases γ-globin synthesis and reduces the proportion of dense cells in patients with sickle cell anemia. Blood 62: 370–380. [PubMed] [Google Scholar]
- Lin JD. 2009. Minireview: the PGC-1 coactivator networks: chromatin-remodeling and mitochondrial energy metabolism. Mol Endocrinol 23: 2–10. 10.1210/me.2008-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey PG, Newsome DA, Fibach E, Yoshida M, Su MS. 1997. Induction of γ-globin by histone deacetylase inhibitors. Blood 90: 2075–2083. [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124: 1283–1298. 10.1016/j.cell.2006.01.040 [DOI] [PubMed] [Google Scholar]
- Molokie R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, Ibanez V, Ruiz MA, Ng KP, Woost P, et al. 2017. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med 14: e1002382 10.1371/journal.pmed.1002382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J, Russell JE, Liebhaber SA. 1997. Destabilization of human α-globin mRNA by translation anti-termination is controlled during erythroid differentiation and is paralleled by phased shortening of the poly(A) tail. J Biol Chem 272: 6607–6613. 10.1074/jbc.272.10.6607 [DOI] [PubMed] [Google Scholar]
- Mottis A, Mouchiroud L, Auwerx J. 2013. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 27: 819–835. 10.1101/gad.214023.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo DA, Aygun B, Akinsheye I, Hankins JS, Bhan I, Luo HY, Steinberg MH, Chui DH. 2012. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol 156: 259–264. 10.1111/j.1365-2141.2011.08916.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. 1988. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 318: 96–99. 10.1056/NEJM198801143180207 [DOI] [PubMed] [Google Scholar]
- Palacios IM. 2013. Nonsense-mediated mRNA decay: from mechanistic insights to impacts on human health. Brief Funct Genomics 12: 25–36. 10.1093/bfgp/els051 [DOI] [PubMed] [Google Scholar]
- Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG. 1999. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev 13: 3198–3208. 10.1101/gad.13.24.3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. 2004. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116: 511–526. 10.1016/S0092-8674(04)00133-3 [DOI] [PubMed] [Google Scholar]
- Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG. 2008. TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell 29: 755–766. 10.1016/j.molcel.2008.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. 2010. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet 11: 109–123. 10.1038/nrg2736 [DOI] [PubMed] [Google Scholar]
- Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge G, Shao M, You D, Fan Z, Xia H, et al. 2015. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat Commun 6: 8471 10.1038/ncomms9471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8: 2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers A, Vaitkus K, Ruiz MA, Ibanez V, Jagadeeswaran R, Kouznetsova T, DeSimone J, Lavelle D. 2015. RN-1, a potent and selective lysine-specific demethylase 1 inhibitor, increases γ-globin expression, F reticulocytes, and F cells in a sickle cell disease mouse model. Exp Hematol 43: 546–553.e1–3. 10.1016/j.exphem.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers A, Vaitkus K, Ibanez V, Ruiz MA, Jagadeeswaran R, Saunthararajah Y, Cui S, Engel JD, DeSimone J, Lavelle D. 2016. The LSD1 inhibitor RN-1 recapitulates the fetal pattern of hemoglobin synthesis in baboons (P. anubis). Haematologica 101: 688–697. 10.3324/haematol.2015.140749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810. 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Cui S, Engel JD, Tanabe O. 2013. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med 19: 291–294. 10.1038/nm.3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Yamamoto M, Engel JD. 2014. Fetal globin gene repressors as drug targets for molecular therapies to treat the β-globinopathies.Mol Cell Biol 34: 3560–3569. 10.1128/MCB.00714-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe O, Katsuoka F, Campbell AD, Song W, Yamamoto M, Tanimoto K, Engel JD. 2002. An embryonic/fetal β-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J 21: 3434–3442. 10.1093/emboj/cdf340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe O, McPhee D, Kobayashi S, Shen Y, Brandt W, Jiang X, Campbell AD, Chen YT, Chang C, Yamamoto M, et al. 2007. Embryonic and fetal β-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J 26: 2295–2306. 10.1038/sj.emboj.7601676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM. 1995. Ras-Raf interaction: two-hybrid analysis. Methods Enzymol 255: 331–342. 10.1016/S0076-6879(95)55036-4 [DOI] [PubMed] [Google Scholar]
- Waggoner SA, Liebhaber SA. 2003. Regulation of α-globin mRNA stability. Exp Biol Med 228: 387–395. 10.1177/153537020322800409 [DOI] [PubMed] [Google Scholar]
- Weatherall DJ. 2001. The thalassemias In The molecular basis of blood diseases (ed. Stamatoyannopoulos G, et al. ), pp. 183–226. W.B. Saunders, Philadelphia. [Google Scholar]
- Welch RP, Lee C, Imbriano PM, Patil S, Weymouth TE, Smith RA, Scott LJ, Sartor MA. 2014. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res 42: e105 10.1093/nar/gku463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood WG, Weatherall DJ, Clegg JB. 1976. Interaction of heterocellular hereditary persistence of foetal haemoglobin with β thalassaemia and sickle cell anaemia. Nature 264: 247–249. 10.1038/264247a0 [DOI] [PubMed] [Google Scholar]
- Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, Yao H, Trowbridge JJ, Mandel G, Orkin SH. 2013. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci 110: 6518–6523. 10.1073/pnas.1303976110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrizi R, Menard JA, Belting M, Massoumi R. 2014. Deubiquitination of γ-tubulin by BAP1 prevents chromosome instability in breast cancer cells. Cancer Res 74: 6499–6508. 10.1158/0008-5472.CAN-14-0221 [DOI] [PubMed] [Google Scholar]
- Zhou XE, Suino-Powell KM, Xu Y, Chan CW, Tanabe O, Kruse SW, Reynolds R, Engel JD, Xu HE. 2011. The orphan nuclear receptor TR4 is a vitamin A-activated nuclear receptor. J Biol Chem 286: 2877–2885. 10.1074/jbc.M110.168740 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.