

Abstract
Purpose:
Waldenström’s macroglobulinemia is an incurable lymphoproliferative disorder driven by an L265P mutation in the Myeloid differentiation primary response gene 88 (MYD88), which activates downstream Nuclear factor kappa-B (NF-κB) signaling through the Myddosome. As this pathway depends in part on activity of Interleukin-1 receptor-associated kinases (IRAK)-1 and −4, we sought to evaluate the potential of the IRAK1/4 inhibitor R191 in pre-clinical models.
Experimental design:
Patient-derived cell lines and primary samples were used in both in vitro and in vivo experiments to model Waldenström’s macroglobulinemia and its response to IRAK1/4 inhibitors.
Results:
R191 induced a dose- and time-dependent reduction in viability of BCWM.1 and MWCL-1 Waldenström’s cell lines, and suppressed activation of IRAK1/4. This was associated with cell cycle arrest at G0/G1, reduced levels of Cyclin-dependent kinases 4 and 6, and induction of apoptosis in cell lines and primary patient samples. Further downstream, R191 exposure led to reduced activation of NF-κB, and of Protein kinase B/Akt/mammalian target of rapamycin signaling, while expression of a constitutively active Akt mutant induced R191 resistance. Gene expression profiling and gene set enrichment analysis revealed a signature consistent with inhibition of c-Myc and activation of the endoplasmic reticulum stress response. In both subcutaneous and systemic murine models of Waldenström’s, R191 showed anti-tumor activity. Finally, the activity of R191 was enhanced when it was combined with novel chemotherapeutics such as bortezomib, afuresertib, and ibrutinib.
Conclusion:
Taken together, these data support the translation of R191 as an approach to target IRAK1/4 to the clinic for Waldenström’s macroglobulinemia patients.
Introduction
Waldenström’s macroglobulinemia is diagnosed in patients who have at least 10% bone marrow involvement with lymphoplasmacytic lymphoma and a serum monoclonal IgM paraprotein (1, 2). While some patients have asymptomatic disease which can be followed without treatment, others present with, or progress to symptomatic Waldenström’s due to bulky disease, cytopenias, constitutional symptoms, or hyperviscosity, all of which require treatment. Chemotherapy for this disease has traditionally been based on the use of various combinations of drugs, including alkylating agents, anti-CD20 monoclonal antibodies, corticosteroids, nucleoside analogues, and proteasome inhibitors (1, 2). These regimens typically are effective at reducing disease burden and provide good long-term outcomes, but complete responses are rare and Waldenström’s is still considered incurable.
Understanding of the molecular pathobiology of Waldenström’s took a huge leap forward with the finding that the vast majority of patients harbor an L265P mutation in the Myeloid differentiation primary response gene 88 (MYD88)(3, 4). MYD88 is recruited to activated Toll-like receptors (TLRs) and Interleukin-1 receptor (IL-1R)(5, 6), resulting in formation of MYD88 homodimers, which then interact with Interleukin-1 receptor-associated kinase 4 (IRAK4) and IRAK2 to form the so-called Myddosome (7). Subsequent recruitment to, and phosphorylation of IRAK1 by the Myddosome is required for binding of IRAK1 to Tumor necrosis factor receptor–associated factor 6 (TRAF6), an E3 ubiquitin ligase that mediates activation of the Inhibitor of NF-κB kinase (IKK). Once IKK is activated, it phosphorylates the Inhibitor of NF-κB (IκB), prompting its ubiquitination and proteasome-mediated degradation, freeing NF-κB to translocate to the nucleus and activate its gene expression program (reviewed in (7)) to promote B cell survival and proliferation (8–15). The L265P mutation within the MYD88 Toll/interleukin-1 receptor (TIR) homology domain changes its structure and induces receptor activation-independent Myddosome formation, leading to constitutive NF-κB activation (16).
In Waldenström’s models, MYD88L265P forms complexes with and activates Bruton’s tyrosine kinase (BTK), providing a rationale for the use of tool compound BTK inhibitors, which showed activity against pre-clinical models (3).These findings set the stage for testing of the BTK inhibitor ibrutinib, whose remarkable clinical activity against relapsed and/or refractory Waldenström’s (17) prompted its regulatory approval.Since IRAK1/4 remains an interesting target for this disease, we set out to evaluate the activity of a novel set of potent and specific inhibitors of these kinases (18).As reported herein, these agents reduced viability and activated apoptosis in Waldenström’s models, inhibited NF-κB and Protein kinase B/Akt signaling, showed efficacy against primary Waldenström’s cells and in vivo, and acted synergistically with other drugs. Thus, these studies provide a good rationale for moving these IRAK1/4 inhibitors to the clinic for patients with Waldenström’s macroglobulinemia.
Materials and Methods
Reagents
The IRAK1/4 inhibitors R191, R568, and R630 were provided by Rigel Pharmaceuticals, Inc. (South San Francisco, CA)(18). R191 was profiled in a Panlabs Hit Profiling Screen (30 targets), and gave significant (>50%) inhibition of three targets at 10 μM other than IRAK1/4: Adenosine A1 (51%), Cannabinoid CB1 (97%), and Serotonin (5-Hydroxltryptamine) 5-HT2B (96%).In follow-up cellular assays at Panlabs, R191 did not show any significant effect on Cannabinoid CB1-induced GTPγS binding in CHO-K1 cells, but did antagonize Serotonin 5-HT2B-induced Inositol monophosphate (IP1) release.R191 blocked IP1 release with an EC50 of 1.93 μM, which may be due in part to cell cytotoxicity, as R191 caused cytotoxicity in CHO-K1 cells with an EC50 of 8.46 μM, but R191 did not significantly inhibit a panel of related Serotonin receptors. The effect of R191 was also evaluated on the human potassium channel using human embryonic kidney (HEK) 293 cells transfected with a human Ether-a-go-go-related gene (hERG), and the maximal inhibition by R191 of the potassium-selective IKr current was 36.6% at 10 μM. With regard to selectivity, R191 was profiled against a panel of 76 kinases representative of the different sub-families of the full kinome.In addition to IRAK1 and IRAK4 (IC50 3nM), R191 inhibited the kinase activity of eleven kinases by >80% at 250 nM and two kinases at 50 nM.The primary off-target activity of R191 was against Fms-related tyrosine kinase (FLT)-3, which was confirmed in cellular assays (Flt3 ligand-induced Extracellular signal-regulated kinase-1/2 phosphorylation and MV411 (Flt3-internal tandem duplication) proliferation). In addition, R191 inhibited the cellular activity of additional targets (TAK1, AXL receptor tyrosine kinase, TANK binding kinase 1, and Bruton’s tyrosine kinase) at concentrations of ~400 nM or above, at least 20-fold over its activity on IRAK1/4 signaling.
Afuresertib, bortezomib, and ibrutinib were purchased from Selleck Chemicals (Houston, TX). Other reagents were from Sigma-Aldrich Corporation (St. Louis, MO) unless otherwise indicated below.
Cell lines and patient samples
The Waldenström’s cell line BCWM.1 was a kind gift from Dr. Steven Treon (Dana Farber Cancer Institute; Boston, MA).These cells are CD5−/CD10−/CD19+/CD20+/CD23+/CD27−/CD38+/CD138+/CD40+/CD52+/CD70+/CD117+/cIgM+/cIgG−/cIgA−/ckappa−, and clambda+, harbor the MYD88 L265P mutation, and express low levels of wild-type CXCR4 (19). MWCL-1 Waldenström’s cells were generously provided by Dr. Stephen Ansell (The Mayo Clinic, Rochester, MN), and are CD3−/CD19+/CD20+/CD27+/CD38+/CD49D+/CD138+/cIgM+, and κ+. They also harbor the MYD88 L265P mutation and a wild-type CXCR4, but they have a TP53 missense mutation at exon 5 (V143A, GTG>GCG)(19).Cells were propagated in RPMI 1640 media (Thermo Fisher/Life Technologies, Waltham, MA) supplemented with 10% fetal calf serum (FCS; GE Healthcare/HyClone, Pittsburgh, PA), 2 mM L-glutamine (Thermo Fisher/Invitrogen, Waltham, MA), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (both from Mediatech, Manassas, VA). Cell line authentication was performed by the MD Anderson Characterized Cell Line Core Facility using short tandem repeat DNA fingerprinting with the AmpFlSTR kit (Thermo Fisher/Applied Biosystems, Waltham, MA).
Three primary samples were obtained from patients undergoing bone marrow aspiration after they had provided informed consent in compliance with the Declaration of Helsinki and according to an Institutional Review Board-approved protocol. CD20+ primary cells were purified by positive selection using magnetic-activated cell sorting with CD20+ microbeads (Miltenyi Biotec, Cologne, Germany).
Peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coats of healthy donors after standard Ficoll-Paque gradient separation, and all the experiments were performed in RPMI 1640 medium.
Cell viability assays
The cells were seeded at 20,000 cells/well on 96-well plates and treated with vehicle alone, or vehicle with increasing concentrations of R191 for 24–72 hours. Cell viability was tested using the Premix WST-1 Cell Proliferation Assay according to the manufacturer’s instructions (Clontech Laboratories, Mountain View, CA). Conversion of WST-1 to the formazan dye was measured at 650 nm using a Victor 3V plate-reader (Perkin Elmer, Waltham, MA). Data provided are representative of at least 3 independent experiments expressed as the mean ± standard deviation (SD) for samples assayed in triplicate.
Flow cytometry
Cell cycle analysis based on DNA content was performed using propidium iodide (PI) staining. Briefly, cells were collected and fixed in 70% cold ethanol in phosphate-buffered saline (PBS) for 2 hours at 4°C. After two subsequent washes with cold PBS, cells were incubated with PI staining solution (1× PBS, 100 μg/ml RNAse A, and 40 μg/ml propidium iodide) for 30 minutes. For analysis of apoptosis, cells were stained with the Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s protocol. Once harvested, cells were resuspended in a binding buffer containing Annexin V-FITC and PI or TO-PRO-3 and incubated for 15 minutes in the dark. Stained cells were analyzed by flow (FACSCalibur; BD Biosciences, San Diego, CA, USA) using FlowJo software (Tree Star, Ashland, OR).
Western blotting
Cells were harvested, washed in PBS, and lysed with buffer (Cell Signaling Technology, Danvers, MA) containing protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN). Lysates were sonicated and clarified by centrifugation, and protein concentrations were determined using the DC Assay Kit (Bio-Rad, Hercules, CA). 50 μg of protein were boiled after addition of loading buffer (Cell Signaling Technology), separated by gel electrophoresis, transferred onto nitrocellulose, and blocked with Tris-buffered saline with 0.01% Tween 20 and 5% non-fat milk prior to incubation with primary and secondary antibodies. Proteins were visualized using the Enhanced Chemiluminescence Kit (Thermo Fisher/Pierce, Waltham, MA) according to the manufacturer’s instructions and exposed on Hyperfilm-ECL (GE Healthcare Biosciences, Pittsburgh, PA). Protein band intensity was analyzed by densitometry using the ChemiDoc™ Touch Imaging System (Bio-Rad) and normalized to β-actin as a control.
Primary antibodies used included those detecting p44/42 Mitogen-activated protein kinase (MAPK; #9102) and phospho-Thr202/Tyr204 p44/42 MAPK (#4370), Akt (#9272) and phospho-Ser473 Akt (#4058), NF-κB p65 (#8242) and phospho-Ser536 NF-κB p65 (93H1, #3033), phospho-Ser32 IκBα (#2859), IRAK1 (#4359S), IRAK4 (#4363) and phospho-Thr345/Ser346 IRAK4 (#11927), Cyclin-dependent kinase (CDK)-4 (#12790) and −6 (#13331), phospho-Ser235/236 S6 kinase (#2211), and phospho-Ser21/9 Glycogen synthase kinase (GSK)-3α/β (#9327S), all from Cell Signaling Technology. Primary antibodies detecting TRAF6 (ab33915), V-myc avian myelocytomatosis viral oncogene homolog (c-Myc; ab178457), and TATA binding protein (TBP; ab63766) were from Abcam (Cambridge, MA). Primary antibodies detecting phospho-Thr209 IRAK1 (#SAB4504246), Tumor protein p53 inducible nuclear protein-1 (TP53INP1; #SAB4503641), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 (PFKFB4; #SAB1300132), and β-actin (A1978) were from Sigma-Aldrich.
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA extracted using TRIzol (Thermo Fisher/Life Technologies) underwent first strand cDNA synthesis with the High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Grand Island, NY). TaqMan Gene Expression Master Mix and probes used to perform quantitative PCR (qPCR) were from Applied Biosystems, with Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) used as internal controls. Reactions were carried out in triplicate in the ABI PRISM 7900 HT Sequence Detection System (Thermo Fisher/Life Technologies), and relative amounts of products were determined by the comparative Ct method.
Lentiviral transduction and transfection studies
Luciferase was excised from pGL3-basic (Promega; Madison, WI) and ligated into pCDH-CMV-MCS-EF1-copGFP (Systems Bioscences; Palo Alto, CA). Constitutively-active (Akt-DD, T308D/S473D)(20) and dominant-negative Akt (Akt-AAA, K179A/T308A/S473A)(21) mutants from Addgene (Cambridge, MA) were cloned into pCDH-CMV-MCS-EF1-puro (Systems Bioscences). Trono vectors (22)(Addgene) expressing shRNA targeting the human c-myc sequences TGAGACAGATCAGCAACAA (shRNA 1–1) and AACGTTAGCTTCACCAACA (shRNA 2–5) were used to knock down cellular c-myc. The Lentiviral vectors were then used for production of recombinant Lentiviruses by transient transfection of 293T cells. Briefly, subconfluent 293T cells were co-transfected with 20 μg of an expression vector (with or without inserted sequences), 15 μg of pAX2, and 5 μg of pMD2G-VSVG by calcium phosphate precipitation. Medium was changed after 16 hours, and recombinant Lentiviruses were harvested 24- and 48-hours later. Raw virus supernatants were concentrated by poly-ethyleneglycol precipitation, and target cells were transduced in growth medium containing 6 μg/mL polybrene. Transduced cells were subjected to drug selection, or selected based on Green fluorescent protein (GFP) expression by flow cytometry at 5-days post-transduction.
Two IRAK4 loss-of-function mutants (23) were a kind gift from Dr. Andrei Medvedev (University of Connecticut Health Center, Farmington, CT). The first contains a C-to-T substitution at nucleotide 877 (877–879), creating a premature stop codon at amino acid 293, while the second has an AC deletion at nucleotides 620–621. They were introduced into the WT pRK7-IRAK4 vector by site-directed mutagenesis and verified by sequencing. These constructs were cloned into Lentiviral vectors and used for production of BCWM.1-derived stable cell lines.
Gene expression and reverse phase protein analyses
Gene expression profiling (GEP) and gene set enrichment analyses (GSEA) were performed as described previously (24). For reverse phase protein array (RPPA) analysis, two replicates of BCWM.1 and MWCL-1 cells were treated with vehicle or 1.25 μM R191 for 24 hours. Cells were lysed in RIPA buffer and lysates were centrifuged at 14,000 rpm for 10 min at 4 °C. The supernatants were mixed with 4× SDS sample buffer (250 mM Tris, pH 7.4, 2% w/v SDS, 25% v/v glycerol and 10% v/v 2-mercaptoethanol) and boiled for 5 min. The RPPA analysis was performed by the MD Anderson RPPA Core Facility (http://www.mdanderson.org/education-and-research/resources-for-professionals/scientific-resources/core-facilities-and-services/functional-proteomics-rppa-core/index.html) as described previously (25).
Animal models
Six-week-old non-obese diabetic mice with severe combined immunodeficiency (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were from Jackson Laboratories (Bar Harbor, ME). BCWM.1 GFP+/luc+ cells (1×107 cells) with Matrigel (BD Biosciences) were injected subcutaneously (SC). On day 7 post-inoculation, mice were divided into treatment groups (N=5 each) to receive intraperitoneal (IP) PBS or 20 mg/kg of R191 on days 1–5 of three consecutive 7-day cycles. Tumor size/burden was evaluated by three methods: measured by calipers bi-weekly, with tumor volume calculated using the formula for an ellipsoid sphere ((Length × Width2)/2); by bioluminescence imaging [after IP injection of 200 μl D-luciferin (15 mg/mL; Caliper Life Sciences) in PBS] using the IVIS Imaging System (Caliper Life Sciences, Hanover, MD) and Living Image 4.4.SP2 software (Caliper Life Sciences); and by ELISA (Bethyl Laboratories, Montgomery, TX) measuring human IgM levels in samples from tail veins on treatment days 14 and 21. Surviving mice were euthanized and tumor tissue was extracted for further analysis.
To develop a model of systemic Waldenström’s, BCWM.1 GFP+/luc+ cells (1×106) were injected intravenously (IV) into the tail veins of mice. On day 3 post-inoculation, mice were divided into treatment groups (N=5 each) and treated as above. Tumor size/burden was evaluated by bioluminescence imaging, and blood samples were collected for human IgM testing. Animals were euthanized and bone marrows were harvested for further analysis at the end of the study (day 21 post-inoculation, day 14 of treatment). All in vivo modeling was performed under protocols approved by the MD Anderson Institutional Animal Care and Use Committee.
Drug synergy calculations and statistical analyses
Combination indices (CI) from drug response data were calculated using CalcuSyn software (Biosoft; Cambridge, UK). Statistical analyses were performed with unpaired t tests in GraphPad, and p-values <0.05 were considered statistically significant. The t-test, ANOVA, or their corresponding nonparametric methods (Wilcoxon rank-sum test or Kruskal–Wallis test) were used to detect differences for continuous variables between groups.
Results
IRAK1/4 inhibition decreased viability in Waldenström’s cell lines.
We evaluated the potency of a panel of novel IRAK1/4 inhibitors which had been identified originally through a cell-based screen (18). R191, R568, and R630 all produced a dose-dependent reduction in viability of BCWM.1 and MWCL-1 Waldenström’s cells after a 24-hour exposure (Supplementary Figure 1) based on data from tetrazolium dye-based assays. As the median inhibitory concentration (IC50) was lowest for R191, we selected this agent for further evaluation. Over the course of a 72-hour exposure, R191 produced a reduction in viability (Figure 1A), and the time-dependence of its action was demonstrated in studies evaluating 24-, 48-, and 72-hour time points (Figure 1B). To confirm that R191 inhibited IRAK1 and IRAK4, we analyzed the expression of the activated/phosphorylated kinases and total IRAK1/4 by Western blotting. Notably, R191 significantly decreased total and phospho-Thr209 IRAK1, and of total and phospho-Thr345/Ser346 IRAK4, in both cell models in a concentration-dependent manner (Figure 1C). Finally, R191 decreased levels of the IRAK1-interacting partner TRAF6 in a dose-dependent manner (Figure 1D).
Figure 1. Viability and IRAK1/4 signaling in R191-treated cell lines.
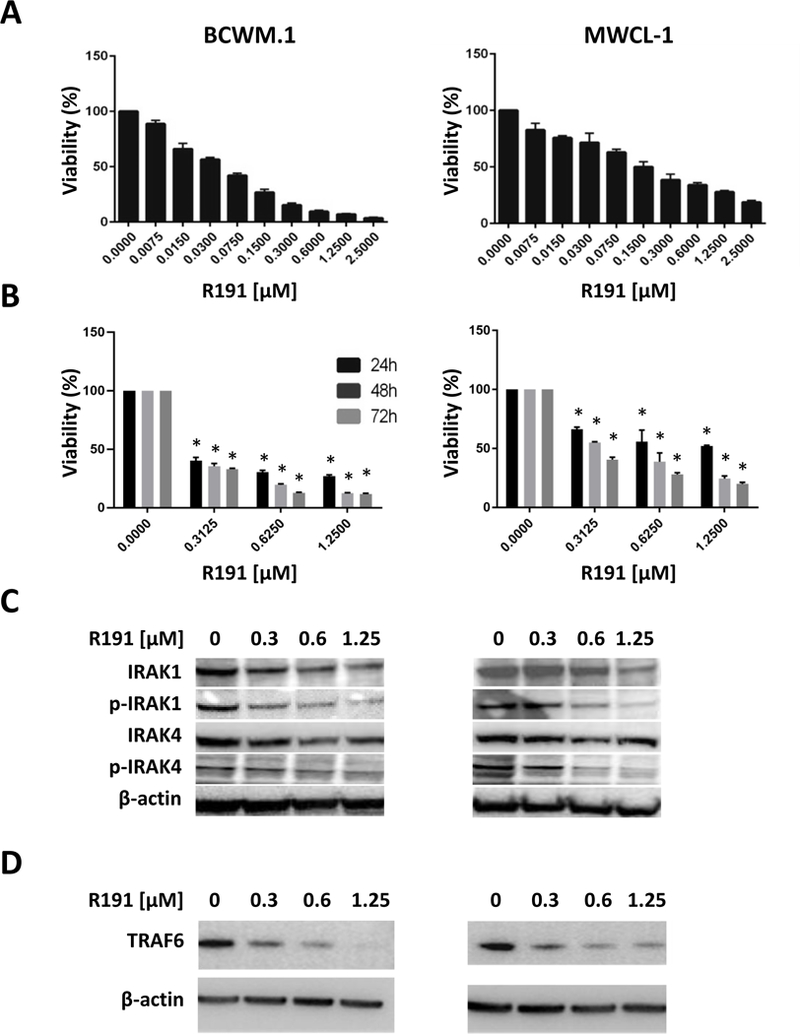
(A, B) BCWM.1 and MWCL-1 cells were treated with the indicated concentrations of R191 for 72 hours (A), or with selected concentrations of R191 for 24, 48, and 72 hours (B), and cell viability was assayed using the tetrazolium reagent WST-1. Data in A and B are expressed as the means ± the standard deviation (SD) for samples assayed in triplicates, and p-values were calculated for treated cells versus vehicle controls. p values ≤0.05 are indicated by “*” symbols.(C, D) BCWM.1 and MWCL-1 cells were treated with the indicated concentrations of R191, and the expression and phosphorylation status of IRAK1 and IRAK4 (C), or the expression of TRAF6 (D) were examined by Western blotting. β-actin was used as a loading control, and a representative blot is shown from one of two independent experiments.
R191 induced cell-cycle arrest and apoptosis.
To better understand the mechanisms by which R191 exerted its effects, we performed cell cycle analysis of Waldenström’s cells treated with R191 for 24 hours. R191 increased the fraction of cells in G0/G1 phase, and decreased the fraction in S phase in both cell lines in a concentration-dependent manner (Figure 2A; Supplementary Figure 2A). This apparent G0/G1 cell cycle arrest suggested that R191 could be modulating levels of Cyclin dependent kinase (CDK)-4 and CDK6, and Western blot analysis indeed showed decreased levels of both in BCWM.1 and MWCL-1 cells (Figure 2B). We also noticed an increased proportion of cells in the sub-G0/G1 fractions in R191-treated cells during cell cycle analysis, suggesting the activation of programmed cell death. BCWM.1 and MWCL-1 cells were therefore analyzed by flow cytometry after Annexin V-staining, and this assay confirmed that R191 induced a dose-dependent increase in apoptosis of these cells (Figure 2C, left panel; Supplementary Figure 2B), and also in a primary patient sample (Figure 2C, right panel). In order to examine the impact of R191 on normal cells, we exposed PBMCs to increasing R191 concentrations for 24 hours, and then evaluated the levels of apoptosis. Programmed cell death was activated by R191 in <20% of PBMCs even at 2.5 μM (Supplementary Figure 3). In contrast, BCWM.1 and MWCL-1 cells were more sensitive, since up to 50–60% of cells became apoptotic at this concentration (Figure 2C, left panel), and the same was true for primary cells, of which 76% were apoptotic at 1.25 μM (Figure 2C, right panel). The ability of R191 to activate apoptosis was confirmed by Western blot analysis showing an increased abundance of cleaved and activated caspases-3, −8, and −9, and of cleaved poly-(ADP-ribose) polymerase (PARP)(Figure 2D, left panel). Finally, increased levels of cleaved caspase-3 were seen in primary patient samples exposed to R191 (Figure 2D, right panel).
Figure 2. Cell cycle arrest and apoptosis in R191-treated cell lines and primary cells.
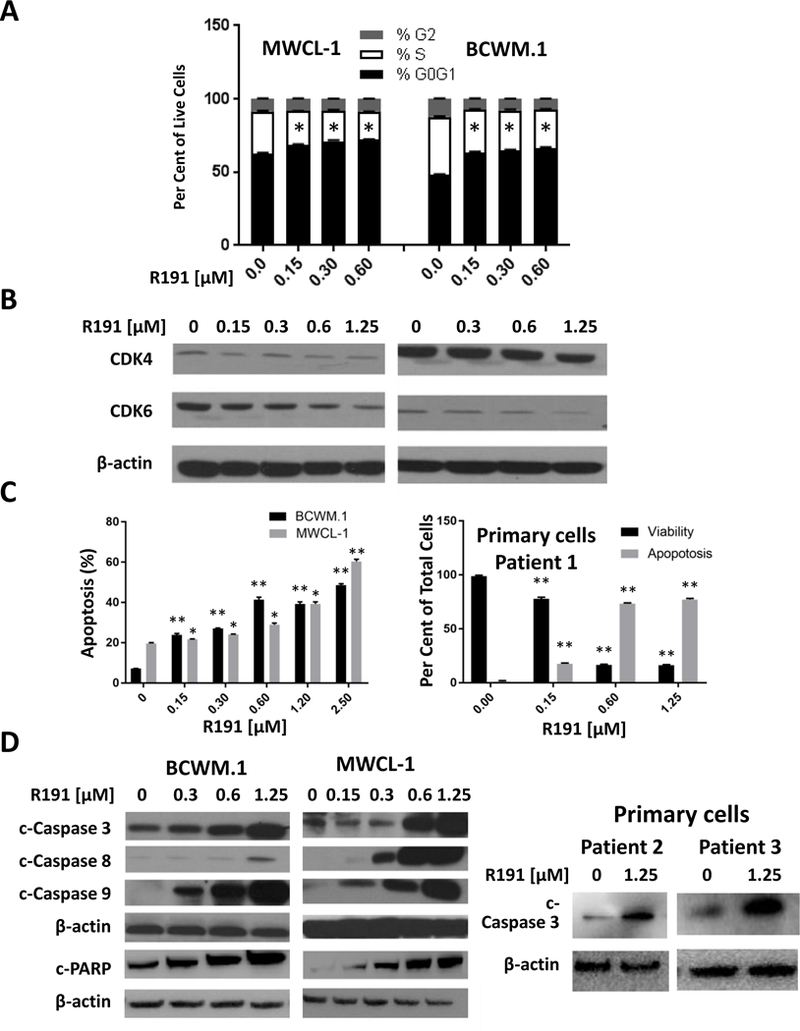
(A)BCWM.1 and MWCL-1 cells were treated with the indicated R191 concentrations for 24 hours, and cell cycle profiles were assessed by flow cytometry of PI-stained cells.An “*” indicates a significant (p-value <0.0001) reduction in the proportion of cells at G0/G1 compared to vehicle-treated controls.(B)The expression levels of CDK4 and CDK6 were examined by Western blotting using β-actin as a loading control as detailed in the legend to Figure 1. (C)Apoptosis was assessed after 24 hours of drug treatment by flow cytometry after Annexin V-staining of cell lines (left panel) or of one primary sample (right panel). Data are expressed as the means ± SD for samples assayed in triplicates, and p-values were calculated for cells treated with R191 vs. untreated controls. Significance levels achieved included “*”, indicating a p-value <0.009, and “**”, indicating a p-value <0.0005.(D) Apoptosis was assessed by Western blotting for the expression levels of cleaved and activated caspases-3, −8, and −9 (designated c-Caspase), or of PARP in cell lines (left panel), or in two new primary samples (right panel).
Inhibition of activation of NF-κB by R191.
Given the role of Myddosome and IRAK signaling in NF-κB induction, we next tested NF-κB activity by evaluating the phosphorylation status of IκBα and the NF-κB subunit p65, as well as nuclear p65 translocation. Western blotting showed decreased phosphorylation of both IκBα and p65 in BCWM.1 and MWCL-1 cells (Figure 3A). Fractionation studies showed that R191 reduced nuclear p65 levels, and enhanced its abundance in the cytoplasm (Figure 3B), consistent with inhibition of NF-κB signaling. To further confirm the inhibitory effect of R191, we used Lentiviral transduction to express the degradation-resistant IκBα-super-repressor in BCWM.1 cells and, as expected, this reduced downstream phospho-p65 levels (Figure 3C). These super-repressor expressing cells were more sensitive to R191 treatment (Figure 3D), confirming the important role of NF-κB signaling as a target for Waldenström’s therapy.
Figure 3. Inhibition of NF-κB in R191-treated cell lines.
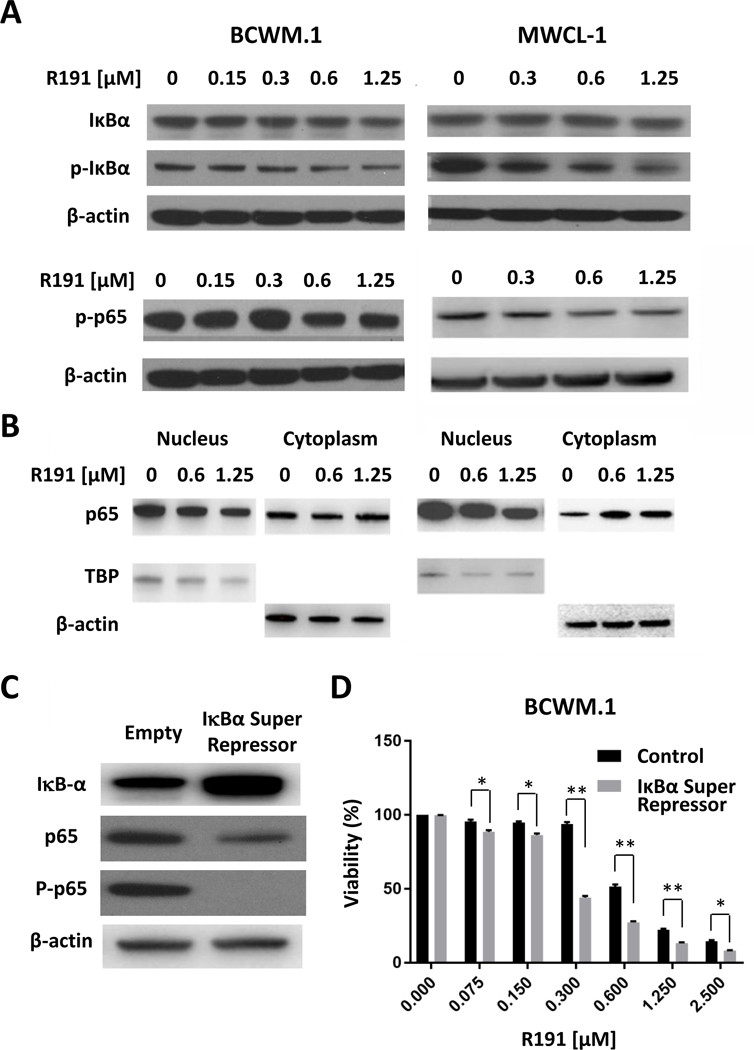
BCWM.1 and MWCL-1 cells were treated with indicated R191 concentrations for 24 hours. The NF-κB activation status was evaluated by Western blotting of whole cell lysates to determine the expression and phosphorylation status of IκBα and the p65 subunit of NF-κB (A), or of nuclear and cytoplasmic fractions for the p65 subunit (B). β-actin was used as the control for total lysates and cytoplasmic fractions, while TBP was used for the nuclear fraction. Also, BCWM.1 cells expressing an IκBα super-repressor (S32A/S36A) or vector control sequences were generated (C) in which the super-repressor reduced phospho-p65 levels. These cells were then exposed to the indicated R191 concentrations for 24 hours (D), and cell viability was determined. Data are expressed as the means ± SD for samples assayed in triplicate, and “*” indicates p-values <0.003, while “**” indicates p-values <0.0001 for the noted comparisons.
IRAK1/4 inhibition suppresses Akt/mTOR signaling.
Activation of Akt signaling is important to survival and homing of Waldenström’s cells (26), and since it can be a downstream target for NF-κB, we next evaluated the impact of R191 on Protein kinase B/Akt. Judging by the level of activated, phospho-Ser473 Akt by Western blotting, R191 exposure decreased Akt activation in BCWM.1 (Figure 4A) and MWCL-1 (Figure 4B) cells. This was associated with decreased activation of downstream GSK3α/β, as judged by the levels of phospho-Ser21/9 GSK3 (Figure 4A, 4B). Similarly, other Akt signaling intermediates showed decreased activation, including PDK1, mTOR, and S6 (Figure 4A, 4B), consistent with an inhibitory effect on the entire pathway. Moreover, the use of RPPAs to provide data about the broader proteomic effects of R191 confirmed the reduction of activation of several Akt pathway intermediates (Supplementary Figure 4A, 4B). To further substantiate the effect of R191 on Akt activation, we next compared BCWM.1 cells expressing either a constitutively-active Akt mutant (Akt-DD; T308D/S473D) or a dominant-negative mutant (Akt-AAA; K179A/T308A/S473A). Expression of the Akt-DD mutant activated downstream signaling as indicated by an increase in phospho-GSK3 and phospho-S6 levels, among others, while Akt-AAA reduced these intermediates (Figure 3C). As expected, BCWM.1 cells expressing the constitutively active Akt mutant were more resistant to R191 (Figure 4D, top panel), while cells expressing dominant negative Akt showed slightly increased sensitivity. Qualitatively similar findings were noted in MWCL-1 cells expressing the Akt-DD mutant (Figure 4D, bottom panel).
Figure 4. Inhibition of Akt and mTOR pathways in R191-treated cells.
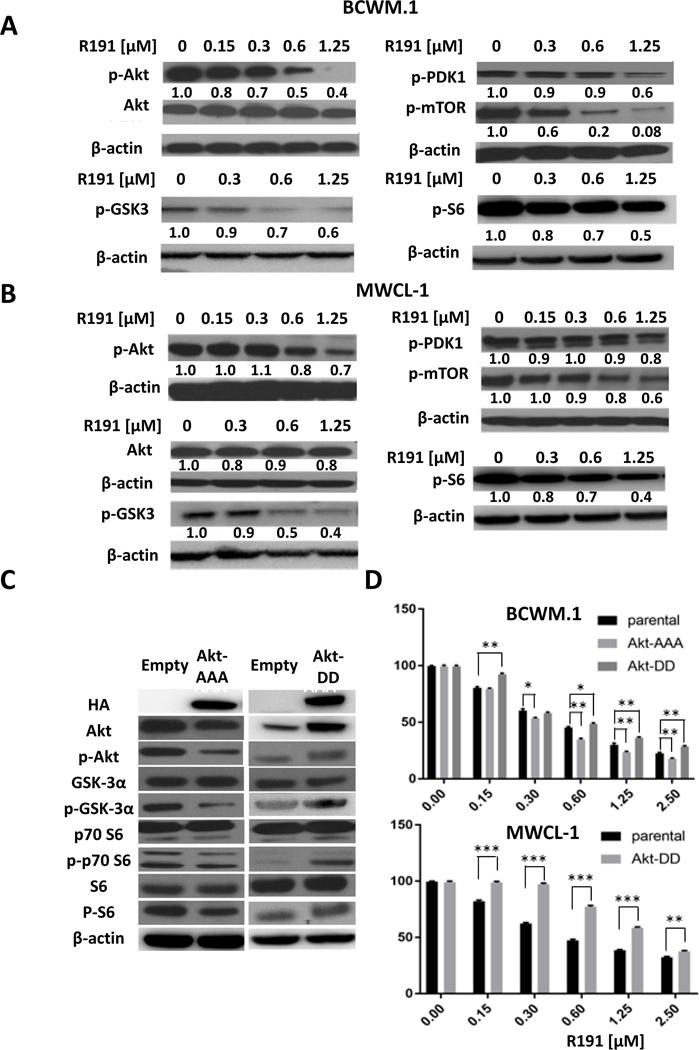
BCWM.1 (A) and MWCL-1 cells (B) were treated with R191 for 24 hours, and the expression and/or phosphorylation status of the indicated proteins was examined by Western blotting using β-actin as a loading control. Cell lines were generated that expressed either a dominant-negative Akt-AAA construct or a constitutively-active Akt-DD construct, which impacted downstream Akt signaling as would be predicted (C).BCWM.1 and MWCL-1 cells expressing these mutants were treated with R191 for 24 hours, and cell viability was evaluated (D).Data are expressed as the means ± SD for samples in triplicate, and “*” indicates p-values <0.009, while “**” indicates p-values <0.003, and “***” indicates p-values <0.0001.
Impact of R191 on c-Myc.
To understand the impact of R191 on gene expression, we next performed GEP and GSEA which confirmed inhibition of Akt/mTOR signaling (Supplementary Table 1 and Supplementary Figure 5A). Additional pathway changes of note included inhibition of the Wingless-related integration site (Wnt)/β-catenin axis, and up-regulation of CD40 signaling and the endoplasmic reticulum (ER)-stress response (not shown). Interestingly, R191 impacted the expression of a c-myc-regulated gene-set (Supplementary Figure 5B), and of genes important for cellular metabolism, including glycolysis. The effect of R191 on c-myc and PFKFB4 expression was confirmed at the mRNA level by qRT-PCR (Figure 5A and Supplementary Figure 6A, respectively), and at the protein level by Western Blotting (Figure 5B and Supplementary Figure 6B, respectively). In order to examine if c-myc suppression was important to the effects of R191, we compared the viability of control BCWM.1 cells and BCWM.1 cells in which c-myc was down-regulated by shRNAs (Figure 5C). Down-regulation of c-MYC sensitized BCWM.1 cells to the effects of R191 (Figure 5D), consistent with this possibility.
Figure 5. Expression of c-Myc in R191-treated cells.
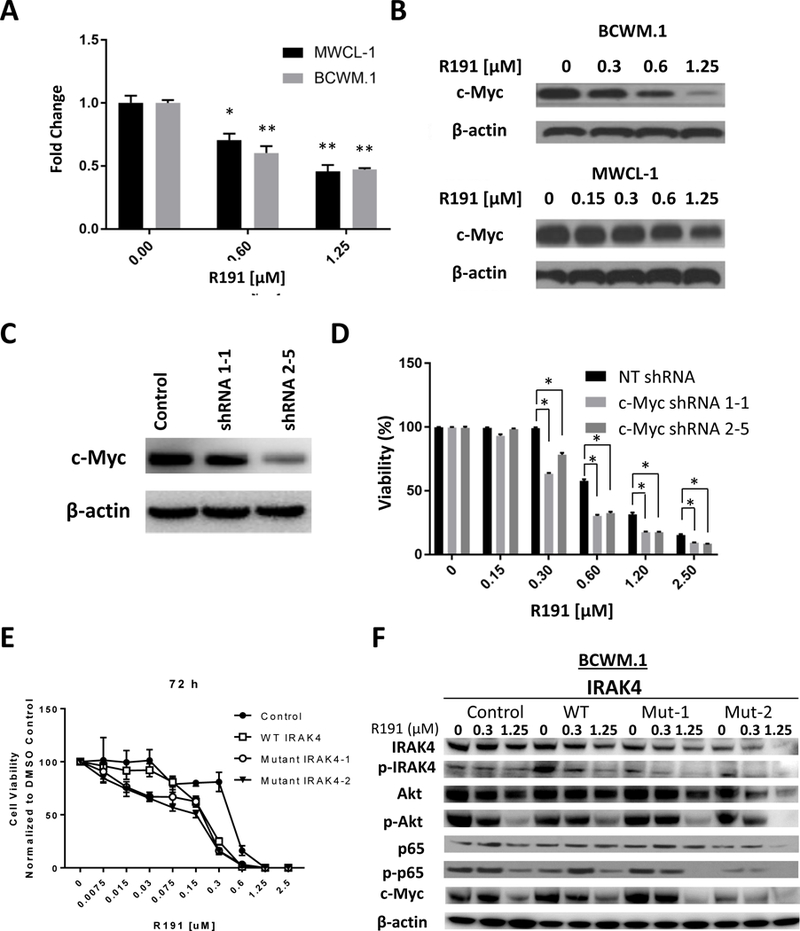
The impact of R191 treatment for 24 hours on BCWM.1 and MWCL-1 cells was studied at the mRNA level using quantitative real-time PCR (A), and at the protein level by Western blot analysis (B). Data in A are expressed as the means ± SD for samples assayed in triplicates, and “*” indicates p-values <0.003, while “**” indicates p-values ≤0.0003.Expression of c-Myc was suppressed in BCWM.1 cells using two different shRNAs (designated 1–1 and 2–5 in (C)), and these were then treated with R191 and viability was studies with the WST-1 assay (D) and compared with a non-targeting (NT) control. Data are expressed as the means ± SD for samples assayed in triplicates, and “*” indicates p-values ≤0.0001.Two loss-of-function IRAK4 mutants were expressed in BCWM.1 cells, exposed to the indicated concentrations of R191 for 72 hours, and the viability was then determined and expressed as detailed above, and compared to control cells and cells with wild-type (WT) IRAK4 (E). Western blotting was then performed to evaluate the impact of these mutants on downstream targets of interest (F).
As R191 may have off-target effects, we suppressed expression of first IRAK1 and then IRAK4 individually in BCWM.1 cells. Decreasing IRAK1 or IRAK4 sensitized these cells to R191 (Supplementary Figure 7A, 7B), as would be expected if IRAK inhibition is a major mechanism of R191’s action since lower target levels would require less drug to give a similar impact. Also, we expressed two previously described IRAK4 loss-of-function mutants (23), and studied their effect. Expression of both mutants sensitized cells to R191 compared to control cells (Figure 5E) and, at low R191 concentrations, wild-type IRAK4 over-expression protected cells. Also, the loss of function mutants contributed to inhibition of downstream signaling (Figure 5F). For example, NF-κB activation as measured by phospho-p65 levels was reduced to a greater extent by R191 in cells with these mutants than in control or wild-type cells. The same was true for phospho-Akt and c-Myc expression, which were decreased to a greater extent by R191 with the dominant-negative constructs than with either approach alone, consistent with the concept that R191 and the mutants had a similar target.
Induction of an ER stress response by R191.
GSEA analysis described above had suggested that R191 induced an up-regulation of the ER stress response, and we therefore sought to confirm if this was the case. By using qRT-PCR, R191 was indeed found to induce increased expression of the 94 kDa Glucose-regulated protein (GRP94), Activating transcription factor 4 (ATF4), and the spliced variant of X-box binding protein 1 (XBP1; Supplementary Figure 8A, upper panels). Moreover, message for CCAAT-enhancer-binding protein homologous protein (CHOP) and ER degradation enhancing alpha-mannosidase like protein 1 (EDEM) were enhanced as well (Supplementary Figure 8A, lower panels). Because eukaryotic cells often induce autophagy to overcome ER stress (27), we also evaluated the impact of R191 on the autophagic machinery. Increased levels of Microtubule-associated protein 1A/1B-light chain 3 form II were seen in BCWM.1, and to some extent in MWCL-1 cells after exposure to R191 (Supplementary Figure 8B). In addition, levels of p62/Sequestosome 1, which can be degraded upon autophagy processing (28), were decreased by R191 (Supplementary Figure 8B).
Anti-tumor activity of R191.
To test the activity of R191 against Waldenström’s in vivo, immunodeficient mice were inoculated with BCWM.1 cells subcutaneously to provide a local model, or intravenously to provide a systemic model. Mice bearing subcutaneous xenografts received three R191 cycles starting on day 3 post-inoculation, while systemic xenograft-bearing mice received two cycles starting on day 7 post-inoculation. R191 significantly delayed growth of subcutaneous Waldenström’s cells based on caliper measures of tumor size (Figure 6A, left panel), and prolonged survival of these mice (Figure 6A, right panel). Using whole-animal in vivo imaging to measure disease burden, R191 significantly delayed tumor growth in both the subcutaneous (Figure 6B, left panel; Supplementary Figure 9A) and systemic models (Figure 6B, right panel; Supplementary Figure 9B). This reduction was paralleled by a reduced level of circulating human IgM in the serum of mice with subcutaneous (Figure 6C, left panel) and systemic disease (Figure 6C, right panel). Notably, there were no significant changes in the weights of R191-treated mice throughout the in vivo experiments (Supplementary Figure 8C). As had been the case in cell line studies, R191 reduced expression of CDK6, and enhanced the levels of cleaved caspase 3 in vivo (Supplementary Figure 10). Finally, analysis of bone marrows from mice with systemic disease showed decreased levels of GFP+/CD20+ tumor cells in the R191-treated mice compared to vehicle-treated mice (Figure 6D).
Figure 6. Impact of R191 on BCWM.1 xenografts in vivo.
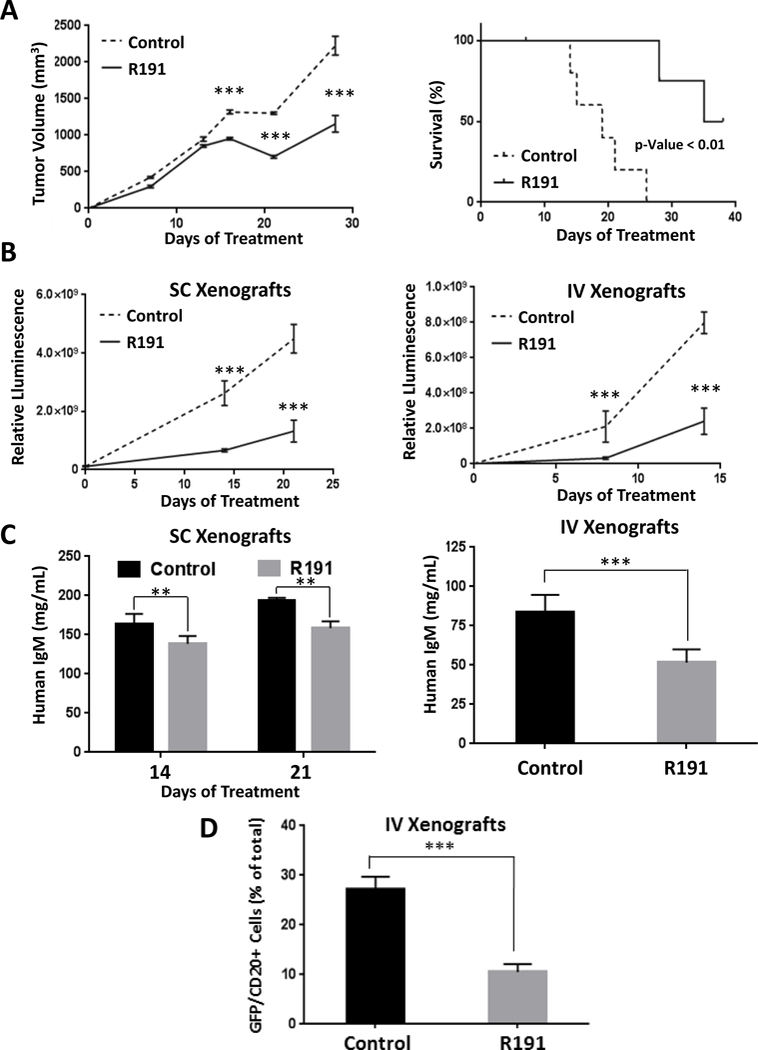
Mice were inoculated subcutaneously (SC) with 1×107 GFP+/luc+ BCWM.1 cells, or intravenously (IV) with 1×106 BCWM.1 GFP+/luc+ cells. They were then randomized and treated with intraperitoneal injections of R191 or vehicle (Control) with five mice per cohort. Growth of subcutaneous xenografts was measured by calipers and the mean values ± SD are shown (A), with “***” indicating p-values <0.0001 (left panel). The Kaplan-Meier method was also used to show survival of the xenograft-bearing mice (right panel). Survival was defined as the time until the tumors exceeded 15 mm in the largest dimension, at which point the animals were euthanized to avoid undue distress. In an independent set of experiments, tumor burden was measured by whole animal in vivo imaging in either a subcutaneous (SC; left panel) or systemic (IV; right panel) mouse model at the indicated time-points (B). The mean values ± SD are shown, and “***” indicates p-values <0.0001. As an added measure of disease burden, the serum levels of human IgM were determined by ELISA (C) in SC xenograft-bearing mice at treatment days 14 and 21 (left panel), and in IV xenograft-bearing mice at the end of the experiment (right panel). The proportion of GFP+/CD20+ cells in bone marrows of the systemic (IV) xenograft-bearing mice was evaluated by flow cytometry at the end of the experiment (3 weeks post-inoculation)(D). The mean values per group ± SD are shown, and “***” indicates p-values <0.0001.
Synergy of R191 with other chemotherapeutics.
Earlier mechanistic insights into the activity of R191 suggested that combining it with other targeted therapies could in the future provide attractive options for the clinic. In that the expression of a dominant-negative Akt sensitized cells to R191, we evaluated the Akt inhibitor afuresertib (29). R191 and afuresertib together enhanced the reduction of viability in both Waldenström’s cell lines (Supplementary Table 2), with combination indexes well below 1.0, indicating strong synergy. Since BTK is another target in the Myddosome signaling pathway, we tested the ability of ibrutinib to add to the efficacy of R191, and strongly synergistic effects were seen between the two in BCMW.1 cells (Supplementary Table 2). Because ER stress was induced by R191, we next combined it with bortezomib, another agent that works in part through inducing a stress response (30), and synergy was again seen in BCMW.1 cells (Supplementary Table 2). Finally, the alkylating agent bendamustine is one of the standards of care for initial therapy as part of a combination with rituximab for Waldenström’s patients. We therefore combined bendamustine with R191, and found synergistic effects at lower bendamustine concentrations, though at higher ones there was an additive effect, with combination indices of approximately 1.0.
Discussion
Genomic studies of Waldenström’s macroglobulinemia have identified a number of commonly recurring mutations in this disease, which clinically shares some features of a low-grade B-cell lymphoma and a plasma cell dyscrasia. Among these are mutations of CXCR4, which are found in 30–40% of patients, of AT-rich interaction domain 1A (ARID1A), which occur in 17%, and of CD79B, which can be detected in 8% to 15% of patients (31). However, the most common, and essentially pathognomonic lesion is in MYD88, which is seen in 95–97% of patients. Moreover, the downstream effects of this mutation include formation of MYD88 homodimers and, in combination with IRAKs, the so-called Myddosome. This produces constitutive NF-κB activation, and provides downstream proliferative and survival signaling to these cells. Validation of the importance of this pathway’s role in the pathobiology of this disease was provided by the strong clinical activity of the BTK inhibitor ibrutinib (17), which remains the only agent that has achieved regulatory approval for Waldenström’s. Indeed, the overall response rate of 90.5% was so impressive that this drug was approved for both newly-diagnosed and more advanced patients even though the study enrolled only those with relapsed or refractory disease. However, complete responses are rare, and treatment-emergent toxicities are not infrequent, including neutropenia in >20%, thrombocytopenia in almost 15%, and epistaxis or atrial fibrillation in up to 5%. These findings suggest that other therapeutic targets remain of relevance in this disease, and since MYD88L265P activates NF-κB through both BTK and IRAK-1/4 (32), the latter could clearly be considered in this category.
Based on the rationale above, we evaluated the therapeutic potential of R191, a newly developed IRAK1/4 inhibitor, in pre-clinical Waldenström’s models. R191 suppressed the viability of, and induced apoptosis in patient-derived cell lines and patient-derived primary samples while reducing activation of IRAKs 1 and 4 (Figures 1, 2). Moreover, R191 decreased levels of TRAF6, a crucial component of MYD88L265P–induced IRAK-mediated signaling (33), and subsequent NF-κB activation (34) in cell lines (Figures 2, 3). Beyond that, Akt signaling is critical to the survival and homing of Waldenström’s cells (26), and R191 suppressed activation of this pathway (Figure 4), providing further support for its rational use against this disease.
Beyond showing activity against cells grown in culture, R191 delayed the growth of, and induced apoptosis in murine Waldenström’s models in which tumor cells were grown either as a local, subcutaneous mass, or after systemic, intravenous injection (Figure 6). Moreover, as would be hoped for from a drug with potential for clinical application, R191 reduced human IgM levels in the sera of tumor-bearing mice, and bone marrow involvement with Waldenström’s cells (Figure 6). In addition to its ability to induce apoptosis and reduce anti-apoptotic Akt signaling, this activity may have been in part due to R191-mediated reduction of c-MYC expression (Figure 5), which correlated with cell cycle arrest and decreased CDK4 and CDK6 levels (Figure 1). Finally, R191 showed promising activity in combination with agents currently in use against Waldenström’s (Supplementary Table 2), including afuresertib, bendamustine, and bortezomib, the latter possibly due to an impact on stress pathways (Supplementary Figure 8). Calcium dysregulation, oxidative damage, and perturbations in post-translational protein modifications can lead to accumulation of misfolded/unfolded proteins and ER stress. An unfolded protein response (UPR) is triggered to expand the processing capacity of the overwhelmed ER (35). Production of large quantities of IgM by Waldenström’s cells puts a large burden on the protein processing machinery, and makes them susceptible to ER stressors. Indeed, proteasome inhibitors were shown previously to inhibit proliferation and to induce apoptosis in Waldenström’s models (36), and are therefore used against this disease in patients. Interestingly, there was also strong synergy, as measured by combination indices of 0.251–0.496, with ibrutinib itself, supporting the possible use of dual BTK and IRAK1/4 inhibitor regimens. Members of the latter category, such as PF-06650833 (37), are currently in phase I and II trials for patients with inflammatory conditions such as rheumatoid arthritis (NCT02996500), suggesting they could be applied to Waldenström’s as well.
Outside of Waldenström’s macroglobulinemia, IRAK1/4 inhibitors could have other applications. For example, a Phe196Ser mutation of IRAK1 is a common, essential driver for Kaposi sarcoma herpesvirus lymphoma (38), and the MYD88L265P mutation occurs in almost 30% of activated B-cell like diffuse large B-cell lymphomas (39). Also, IRAK1 activation and overexpression have been reported in myelodysplastic syndrome and acute myeloid leukemia (40), and may play a role in melanoma as well (41). Overall, as our data suggest that R191 effectively inhibited the BTK-IRAK1/4-NF-κB signaling axis in Waldenström’s cells in vitro and in vivo, further studies may be warranted of this agent in these other malignancies as well.
Supplementary Material
Statement of Translational Relevance.
Waldenström’s macroglobulinemia patients are currently treated with a variety of chemotherapeutics used alone or in combination, that have been adapted for use based on their activity in related disorders, including multiple myeloma and indolent non-Hodgkin lymphomas. The first, and so far only agent to receive regulatory approval in this disease is the Bruton’s tyrosine kinase ibrutinib, but only a minority of patients achieve a complete remission with this therapy, indicating a need for other rationally targeted therapies. This study provides evidence that the novel Interleukin-1 receptor-associated kinase (IRAK)-1/4 inhibitor R191 has potent activity against pre-clinical and physiologically relevant models of this disease. Since IRAK1/4 is a key signaling intermediate downstream of the pathobiologic lesion responsible for Waldenström’s, this supports the possibility that R191 could be an attractive option for translation to the clinic.
Acknowledgments
RZO, the Florence Maude Thomas Cancer Research Professor, would like to acknowledge support from the National Cancer Institute (R01 CA194264, R01 CA184464, P50 CA142509, U10 CA032102), and the Leukemia & Lymphoma Society (SCOR-12206–17). Core facilities that were key to these studies, including the Characterized Cell Line Core and the Flow Cytometry and Cellular Imaging Core, were supported by the MD Anderson Cancer Center Support Grant (P30 CA016672).
Footnotes
Authorship
Contribution: HN designed and performed most of the initial research, while FS performed studies needed for the revised manuscript; HN, VB, and HL performed the statistical data analyses; IK, HW, and RJJ performed additional laboratory studies; ZB wrote manuscript drafts and provided helpful suggestions; YH, SMA, and SPT provided cells, reagents, and valuable suggestions; SKT, and HCL consented patients for primary sample collection and provided helpful suggestions; ZW and RED performed gene expression profiling and provided helpful suggestions; and RZO supervised all the research completed herein and finalized the manuscript.
References
- 1.Gertz MA. Waldenstrom macroglobulinemia: 2017 update on diagnosis, risk stratification, and management. American journal of hematology. 2017;92:209–17. [DOI] [PubMed] [Google Scholar]
- 2.Leblond V, Kastritis E, Advani R, Ansell SM, Buske C, Castillo JJ, et al. Treatment recommendations from the Eighth International Workshop on Waldenstrom’s Macroglobulinemia. Blood. 2016;128:1321–8. [DOI] [PubMed] [Google Scholar]
- 3.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. The New England journal of medicine. 2012;367:826–33. [DOI] [PubMed] [Google Scholar]
- 4.Poulain S, Roumier C, Decambron A, Renneville A, Herbaux C, Bertrand E, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121:4504–11. [DOI] [PubMed] [Google Scholar]
- 5.Loiarro M, Gallo G, Fanto N, De Santis R, Carminati P, Ruggiero V, et al. Identification of critical residues of the MyD88 death domain involved in the recruitment of downstream kinases. J Biol Chem. 2009;284:28093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watters TM, Kenny EF, O’Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol. 2007;85:411–9. [DOI] [PubMed] [Google Scholar]
- 7.Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. British journal of cancer. 2015;112:232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, et al. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–91. [DOI] [PubMed] [Google Scholar]
- 10.Cheng H, Addona T, Keshishian H, Dahlstrand E, Lu C, Dorsch M, et al. Regulation of IRAK-4 kinase activity via autophosphorylation within its activation loop. Biochem Biophys Res Commun. 2007;352:609–16. [DOI] [PubMed] [Google Scholar]
- 11.Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li S, et al. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem. 2004;279:5227–36. [DOI] [PubMed] [Google Scholar]
- 12.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. [DOI] [PubMed] [Google Scholar]
- 13.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–58. [DOI] [PubMed] [Google Scholar]
- 14.Mu Y, Sundar R, Thakur N, Ekman M, Gudey SK, Yakymovych M, et al. TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun. 2011;2:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avbelj M, Wolz OO, Fekonja O, Bencina M, Repic M, Mavri J, et al. Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood. 2014;124:3896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. The New England journal of medicine. 2015;372:1430–40. [DOI] [PubMed] [Google Scholar]
- 18.Markovtsov VV, Lamagna C, Chan M, Yi S, Young C, Frances R, et al. Abstract 346: Potential role for R191, potent and selective IRAK4 kinase inhibitor, in treatment of hematologic malignancies. Cancer research. 2016;76:346-. [Google Scholar]
- 19.Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia. 2015;29:169–76. [DOI] [PubMed] [Google Scholar]
- 20.Watton SJ, Downward J. Akt/PKB localisation and 3’ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr Biol. 1999;9:433–6. [DOI] [PubMed] [Google Scholar]
- 21.Stoll V, Calleja V, Vassaux G, Downward J, Lemoine NR. Dominant negative inhibitors of signalling through the phosphoinositol 3-kinase pathway for gene therapy of pancreatic cancer. Gut. 2005;54:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. The Journal of experimental medicine. 2003;198:521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bjorklund CC, Ma W, Wang ZQ, Davis RE, Kuhn DJ, Kornblau SM, et al. Evidence of a role for activation of Wnt/beta-catenin signaling in the resistance of plasma cells to lenalidomide. The Journal of biological chemistry. 2011;286:11009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics. 2006;5:2512–21. [DOI] [PubMed] [Google Scholar]
- 26.Leleu X, Jia X, Runnels J, Ngo HT, Moreau AS, Farag M, et al. The Akt pathway regulates survival and homing in Waldenstrom macroglobulinemia. Blood. 2007;110:4417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith DA, Brigandi RA, et al. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014;124:2190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nature reviews Clinical oncology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter ZR, Yang G, Xu L, Liu X, Castillo JJ, Treon SP. Genomics, Signaling, and Treatment of Waldenstrom Macroglobulinemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017:JCO2016710814. [DOI] [PubMed] [Google Scholar]
- 32.Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122:1222–32. [DOI] [PubMed] [Google Scholar]
- 33.Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays. 2003;25:1096–105. [DOI] [PubMed] [Google Scholar]
- 34.Hartupee J, Li X, Hamilton T. Interleukin 1alpha-induced NFkappaB activation and chemokine mRNA stabilization diverge at IRAK1. The Journal of biological chemistry. 2008;283:15689–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. [DOI] [PubMed] [Google Scholar]
- 36.Roccaro AM, Leleu X, Sacco A, Jia X, Melhem M, Moreau AS, et al. Dual targeting of the proteasome regulates survival and homing in Waldenstrom macroglobulinemia. Blood. 2008;111:4752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee KL, Ambler CM, Anderson DR, Boscoe BP, Bree AG, Brodfuehrer JI, et al. Discovery of Clinical Candidate 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}−7-methoxyisoquinoli ne-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin-1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. Journal of medicinal chemistry. 2017;60:5521–42. [DOI] [PubMed] [Google Scholar]
- 38.Yang D, Chen W, Xiong J, Sherrod CJ, Henry DH, Dittmer DP. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for Kaposi sarcoma herpesvirus lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4762–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer cell. 2013;24:90–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Srivastava R, Geng D, Liu Y, Zheng L, Li Z, Joseph MA, et al. Augmentation of therapeutic responses in melanoma by inhibition of IRAK-1,−4. Cancer research. 2012;72:6209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.