

Abstract
Mitochondrial genomes (mitogenomes) of five Cyrtodactylus were determined. Their compositions and structures were similar to most of the available gecko lizard mitogenomes as 13 protein-coding, two rRNA and 22 tRNA genes. The non-coding control region (CR) of almost all Cyrtodactylus mitogenome structures contained a repeated sequence named the 75-bp box family, except for C. auribalteatus which contained the 225-bp box. Sequence similarities indicated that the 225-bp box resulted from the duplication event of 75-bp boxes, followed by homogenization and fixation in C. auribalteatus. The 75-bp box family was found in most gecko lizards with high conservation (55–75% similarities) and could form secondary structures, suggesting that this repeated sequence family played an important role under selective pressure and might involve mitogenome replication and the likelihood of rearrangements in CR. The 75-bp box family was acquired in the common ancestral genome of the gecko lizard, evolving gradually through each lineage by independent nucleotide mutation. Comparison of gecko lizard mitogenomes revealed low structural diversity with at least six types of mitochondrial gene rearrangements. Cyrtodactylus mitogenome structure showed the same gene rearrangement as found in most gecko lizards. Advanced mitogenome information will enable a better understanding of structure evolution mechanisms.
Keywords: Mitogenome, Control region, Repeated sequence, Mitochondrial gene rearrangement, mtDNA
Introduction
Vertebrate mitochondrial genomes (mitogenomes) are double-stranded circular DNAs typically 16–17 kb in size that encode two rRNA genes, 22 tRNA genes, and 13 protein-coding genes. They also possess non-coding control regions (CRs) which contain signals for the initiation of replication and transcription (Boore, 1999; Fernández-Silva, Enriquez & Montoya, 2003). Mitogenomes are maternally inherited as haploid genomes with multiple copy numbers in a cell. Compared with nuclear DNA, mitogenome gene content is likely conserved with very low levels of recombination, while mitogenome sequences evolved rapidly (Elson & Lightowlers, 2006; Gissi, Iannelli & Pesole, 2008). These features are very useful tools for the resolution of taxonomic controversies (Srikulnath et al., 2012; Sun, Cheng & Zhang, 2015; Supikamolseni et al., 2015; Laopichienpong et al., 2016; Thongtam na Ayudhaya et al., 2017; Prakhongcheep et al., 2018). Simultaneously, nucleotide sequences of the CR containing repeated sequences known as variable number of tandem repeat (VNTR), show high mutation rate which is advantageous for population and phylogeographic studies (Wang, Zhou & Nie, 2011; Lapbenjakul et al., 2017). Although mitogenome organization is likely conserved in vertebrates (Pereira, 2000), mitochondrial gene rearrangements have been found in several groups (Macey et al., 1997; Mauro et al., 2006; Kurabayashi et al., 2008). The majority of these rearrangements result from gene shuffling (such as tRNA genes), translocations and/or duplications of genes, and loss of gene or genome segments (Boore, 1999; Amer & Kumazawa, 2008; Metallinou et al., 2012; Kumazawa et al., 2014). This variation of rearrangements reflects the different functional and evolutionary constraints among taxa (Boore, 1999; Gissi, Iannelli & Pesole, 2008; Pereira et al., 2008; Bernt & Middendorf, 2011).
Recently, the understanding of mitogenomes in squamate reptiles has increased, and technical advances in sequencing have led to rapid accumulation of complete mitogenome sequences (Kumazawa et al., 2014). However, only 25 species have been sequenced from the infraorder of squamate reptiles, Gekkota, which contains more than 1,200 species (Uetz & Hošek, 2018). Approximately 20% of gecko lizard mitogenomes contain various gene rearrangements including tandem duplication of the gene block and loss and reassignment of tRNA genes which were not very common in vertebrate mitogenomes (Fujita, Boore & Moritz, 2007; Kumazawa et al., 2014; Li et al., 2015). This finding leads us to question whether mitochondrial genome rearrangements show different functional roles and phenotypes and the possibility of other variations occurring in gecko lizard mitogenomes. Information is urgently required for a more comprehensive understanding of these issues. Cyrtodactylus comprises a large and diverse genus of gecko lizards in the family Gekkonidae (gekkonids), with over 200 species in Southern and Southeast Asia, Northern Australia and Oceania (Wood et al., 2012; Nazarov et al., 2014). Molecular phylogenies of Cyrtodactylus reveal a pattern of diversification and basal divergence that correlates with the India-Asia collision, and the genus is a good candidate for exploring the faunal impacts of this collision (Grismer, Grismer & Chav, 2010; Siler et al., 2010; Welton et al., 2010; Shea et al., 2011). Surprisingly, although the diversity of this lizard is critical, whole mitogenome sequences and structures of Cyrtodactylus remain uncertain. The mitogenome sequencing project is thus important to provide a ‘backbone’ for the genus by offering a major framework contribution regarding comparative analyses of biogeography, morphology, diversity, and genome evolution. Here, five complete mitogenome structures of Cyrtodactylus as Cyrtodactylus peguensis, C. tigroides, C. thirakhupti, C. auribalteatus, and C. chanhomeae were characterized. Comparisons between structural variations of the Cyrtodactylus mitogenome and other gecko lizards were also discussed.
Materials and Methods
Specimen collection and DNA extraction
Individual specimens of each of the five species (C. peguensis, C. tigroides, C. thirakhupti, C. auribalteatus, and C. chanhomeae), taken from Nakhon Ratchasima Zoo during 2012–2014, were stored in 100% ethanol and detailed information is presented in Table 1. Individual species were classified based on their morphology (Boulenger, 1893; Bauer, Sumontha & Pauwels, 2003; Pauwels et al., 2004; Sumontha, Panitvong & Deein, 2010). A piece of muscle clipped from each sample was collected to provide a source of DNA. Whole genomic DNA was extracted following the standard salting-out protocol as described previously (Supikamolseni et al., 2015). Animal care and all experimental procedures were approved by the Animal Experiment Committee, Kasetsart University, Thailand (approval no. ACKU-SCI-022), and conducted in accordance with the Regulations on Animal Experiments at Kasetsart University.
Table 1. Species used with accession numbers.
| Family | Genus | Species | GenBank accession number | Mitochondrial gene arrangement | Reference |
|---|---|---|---|---|---|
| Gekkonidae | Cyrtodactylus | Cyrtodactylus peguensis (CyPeNRZ001)a | AP018114 | Type I | In this study |
| Gekkonidae | Cyrtodactylus | Cyrtodactylus thirakhupti (CyThNRZ001)a | AP018115 | Type I | In this study |
| Gekkonidae | Cyrtodactylus | Cyrtodactylus auribaltealus (CyAuNRZ001)a | AP018116 | Type I | In this study |
| Gekkonidae | Cyrtodactylus | Cyrtodactylus chanhomeae (CyChNRZ001)a | AP018117 | Type I | In this study |
| Gekkonidae | Cyrtodactylus | Cyrtodactylus tigroides (CyTiNRZ001)a | AP018118 | Type I | In this study |
| Gekkonidae | Gekko | Gekko chinensis | NC_027191.1 | Type I | Hao, Ping & Zhang (2016) |
| Gekkonidae | Gekko | Gekko gecko | NC_007627.1 | Type I | Zhou et al. (2006) |
| Gekkonidae | Gekko | Gekko swinhonis | NC_018050.1 | Type I | Li et al. (2013) |
| Gekkonidae | Gekko | Gekko japonicus | NC_028035.1 | Type I | Kim et al. (2015) |
| Gekkonidae | Gekko | Gekko vittatus | NC_008772.1 | Type I | Kumazawa (2007) |
| Gekkonidae | Hemidactylus | Hemidactylus bowringii | NC_025938.1 | Type I | – |
| Gekkonidae | Hemidactylus | Hemidactylus frenatus | NC_012902.2 | Type I | Yan, Li & Zhou (2008) |
| Gekkonidae | Heteronotia | Heteronotia binoei | NC_010292.1 | Type VI | Fujita, Boore & Moritz (2007) |
| Gekkonidae | Cnemaspis | Cnemaspis limi | NC_020039.1 | Type I | Yan et al. (2014b) |
| Gekkonidae | Paroedura | Paroedura picta | NC_028326.1 | Type I | Starostová & Musilová (2016) |
| Gekkonidae | Tropiocolotes | Tropiocolotes tripolitanus | NC_025780.1 | Type II | Kumazawa et al. (2014) |
| Gekkonidae | Stenodactylus | Stenodactylus petrii | NC_025784.1 | Type III | Kumazawa et al. (2014) |
| Gekkonidae | Uroplatus | Uroplatus ebenaui | NC_025783.1 | Type IV | Kumazawa et al. (2014) |
| Gekkonidae | Uroplatus | Uroplatus fimbriatus | NC_025779.1 | Type V | Kumazawa et al. (2014) |
| Gekkonidae | Lepidodactylus | Lepidodactylus lugubris | NC_025782.1 | Type I | Kumazawa et al. (2014) |
| Gekkonidae | Phelsuma | Phelsuma guimbeaui | AB661664.1 | Type I | Kumazawa et al. (2014) |
| Eublepharidae | Eublepharis | Eublepharis macularius | NC_033383.1 | Type I | – |
| Eublepharidae | Hemitheconyx | Hemitheconyx caudicinctus | NC_018368.1 | Type I | Jonniaux, Hashiguchi & Kumazawa (2012) |
| Eublepharidae | Goniurosaurus | Goniurosaurus luii | NC_026105.1 | Type I | Li et al. (2015) |
| Eublepharidae | Coleonyx | Coleonyx variegatus | NC_008774.1 | Type I | Kumazawa (2007) |
| Phyllodactylidae | Phyllodactylus | Phyllodactylus unctus | NC_020038.1 | Type I | Yan et al. (2014a) |
| Phyllodactylidae | Tarentola | Tarentola mauritanica | NC_012366.1 | Type I | Albert et al. (2009) |
| Pygopodidae | Aprasia | Aprasia parapulchella | NC_024557.1 | Type I | MacDonald et al. (2005) |
| Sphaerodactylidae | Teratoscincus | Teratoscincus keyserlingii | AY753545.1 | Type I | Macey et al. (2005) |
| Sphaerodactylidae | Teratoscincus | Teratoscincus roborowskii | KP115216.1 | Type I | – |
| Iguanidae | Iguana | Iguana iguana | NC_002793.1 | – | Janke et al. (2001) |
| Scincidae | Plestiodon | Plestiodon egregius | NC_000888.1 | – | Kumazawa & Nishida (1999) |
| Varanidae | Varanus | Varanus salvator | NC_010974.1 | – | Castoe et al. (2008) |
Notes.
Samples from Nakhon Ratchasima Zoo, Thailand.
Complete mitogenome sequencing
Complete mitogenome sequences were obtained using a PCR (polymerase chain reaction)-based strategy to amplify overlapping mitochondrial fragments. Forty-six PCR primer pairs were designed based on five squamate reptile mitogenome sequences: Gekko chinensis (NC_027191.1), G. gecko (NC_007627.1), G. japonicas (NC_028035.1), G. swinhonis (NC_018050.1), and Hemidactylus bowringii (NC_025938.1). Nineteen primer pairs were also taken from Kumazawa & Endo (2004) (Table S1). PCR amplification was performed using 20 µl of 1×ThermoPol buffer containing 1.5 mM MgCl2, 0.2 mM dNTPs, 5.0 µM of primers, 0.5 U of Taq polymerase (Apsalagen Co. Ltd., Bangkok, Thailand), and 25 ng of genomic DNA. PCR conditions were as follows: initial denaturation at 94 °C for 3 min, followed by 35 cycles of 94 °C for 30 s, 45–60 °C for 30 s, 72 °C for 1 min 30 s, and then a final extension at 72 °C for 5 min. PCR products were detected by electrophoresis on 1% agarose gels. No multiple bands were found in any of the PCR products. PCR products were purified using FavorPrep GEL/PCR Purification Mini Kit (Favorgen Biotech Corp., Ping-Tung, Taiwan), and nucleotide sequences of the DNA fragments were determined using a 3500 Genetic Analyzer (Life Technology, California, USA). BLASTn and BLASTx programs (http://blast.ncbi.nlm.nih.gov/Blast.cgi) were used to search for nucleotide sequences in the National Center for Biotechnology Information database to confirm the identity of the DNA fragments amplified in the present study.
Nucleotide sequence annotation and analysis
Sequence assembly was performed to combine all overlapping PCR fragments into one contig strand using SeqScape software v 2.5 (Life Technology, California, USA) and re-checked carefully by visual inspection to ensure accuracy of the variable sites and avoid the problems of repeated sequences identified by the program in the CR. Gene structures of all mitogenomes were annotated using web-based MITOS (Bernt et al., 2013) with manual inspections to compare DNA or amino acid sequences with known sequences from several gecko lizards. ExPASy-translate tool (Gasteiger et al., 2003) was used to characterize the nucleotide sequences of encoded genes. To identify mitochondrial tRNA genes, the nucleotide sequences were searched for regions which could form characteristic secondary structures using tRNA Scan-SE 2.0 by parameter sequence source: vertebrate mitochondrial genome search mode (http://lowelab.ucsc.edu/tRNAscan-SE), the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) and UNFOLD (http://unafold.rna.albany.edu/) under fold algorithms as basic options is minimum free energy (MFE) and partition function (Pereira et al., 2008). Overlapping regions and intergenic spacers between genes were counted manually. The CR domains comprising the extended termination associated sequence (ETAS) domain, central conserved region (CCR) domain, and conserved sequence block (CSB) domain were characterized following Douzery & Randi (1997). Tandem repeated sequences in CR were identified using the program ‘Tandem Repeats Finder’ with default parameters (Benson, 1999). The most thermodynamically stable putative secondary structures of VNTR were determined using the UNFOLD and RNAfold web servers. All nucleotide sequences were then deposited in the DNA Data Bank of Japan (DDBJ) (Table 1). Base composition and codon usage were analyzed using the default parameters of Molecular Evolutionary Genetics Analysis 7 (MEGA7) software (Center for Evolutionary Functional Genomics, The Biodesign Institute, Tempe, AZ, USA; Kumar, Stecher & Tamura, 2016). The A +T% and G +C% values, and the AT-skew and GC-skew (Perna & Kocher, 1995) for the H-strand were also calculated for five Cyrtodactylus mitogenomes, another 25 gecko lizard mitogenomes, and Iguana iguana (NC_002793.1), Plestiodon egregious (NC_000888.1), and Varanus salvator (NC_010974.1) mitogenomes as the outgroup. Values obtained were subsequently mapped as a scatter plot.
Phylogenetic placement
Multiple sequence alignment of concatenated heavy-strand encoded protein-coding genes was performed on 33 sequences for 30 gecko lizards with the outgroup (I. iguana, P. egregious, and V. salvator) using the default parameters of MEGA 7 software. Additionally, the ND6 gene was used separately to perform alignment from phylogenetic analyses of the datasets as it was encoded by the L-strand and possessed base composition bias (Dong & Kumazawa, 2005; Wang et al., 2009). The dataset of sequence lengths of concatenated heavy-strand encoded protein-coding genes ranged from 10,766 to 10,899 bp, while ND6 sequence lengths ranged from 510 to 546 bp. All unalignable and gap-containing sites were carefully removed and trimmed from the datasets. Datasets of aligned concatenated heavy-strand encoded protein-coding genes and ND6 fragments showed 10,813 and 548 nucleotides, respectively. Level of sequence divergence between species was estimated using uncorrected pairwise distances (p-distances) as implemented in MEGA7. We also performed multiple sequence alignment comprising gap-containing sites (as insertion and deletion) using the GUIDANCE2 Server (Sela et al., 2015). Datasets of aligned concatenated heavy-strand encoded protein-coding genes and ND6 fragments showed 11,322 and 607 nucleotides, respectively. Phylogenetic analysis was performed using Bayesian inference (BI) and maximum likelihood (ML). The best-fit model of DNA substitution was determined for each gene using Kakusan4 (Tanabe, 2011). The GTR+I+G model gave the best-fit of each locus for BI and ML analyses. BI analysis was performed with MrBayes v 3.2.6 (Huelsenbeck & Ronquist, 2001). The Markov chain Monte Carlo process was used to run four chains simultaneously for one million generations. Log likelihood and parameter values were accessed through Tracer version 1.6 (Rambaut et al., 2015). After the log-likelihood value stabilized, a sampling procedure was performed every 100 generations to obtain 10,000 trees and a majority-rule consensus tree with average branch lengths was generated. All sample points prior to attaining convergence were discarded as burn-in, and the Bayesian posterior probability in the sampled tree population was calculated as a percentage. ML analyses for all datasets were performed using RAxML (Stamatakis, 2014), followed by executing 10 runs of random additional sequences generating 1,000 rapid bootstrap replicates. By combining the definite phylogenetic relationships of different species with the distribution pattern of mitogenome types in gecko lizards, evolutionary processes of the mitogenome structure were determined. Mitochondrial gene rearrangements with similar components and order were classified as identical. All mitogenome types were visualized by linearized organization and drawn by OrganellarGenomeDRAW v 1.2 (OGDraw) (Lohse et al., 2013).
Results
Total mitogenome sequences of the five Cyrtodactylus species ranged between 16,795 and 17,068 bp in length. Their genome content comprised 13 protein-coding genes, two rRNA genes, 22 tRNA genes, and the CR (Table 2). There were 28 genes encoded in the majority strand (H-strand) and nine in the minority strand (L-strand), whereas the CR was surrounded by tRNA-Pro and tRNA-Phe in the genome. Average A+T% of the mitogenomes was 52.2 ± 0.61. Highest A+T% was recorded by C. chanhomeae (54.5%) with the lowest from C. peguensis (50.8%). By contrast, the average AT-skew was 0.17 ± 0.01, ranging from 0.15 (C. chanhomeae) to 0.19 (C. auribalteatus), with the average GC-skew −0.35 ± 0.01, ranging from −0.37 (C. auribalteatus) to −0.31 (C. chanhomeae) (Fig. 1).
Table 2. Summary of mitochrondrial genome composition of five Cyrtodactylus species.
| Species | GenBank accession number | Size (bp) | CRa size (bp) | %Nucleotide composition | G+C% content | GC-skew | A+T% content | AT- skew | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | T | G | C | ||||||||
| Cyrtodactylus peguensis | AP018114 | 16,988 | 1,653 | 29.4 | 21.5 | 16.1 | 33.1 | 49.2 | −0.3455 | 50.8 | 0.1552 |
| Cyrtodactylus thirakhupti | AP018115 | 16,795 | 1,454 | 30.4 | 21.5 | 15.3 | 32.8 | 48.0 | −0.3638 | 52.0 | 0.1715 |
| Cyrtodactylus auribalteatus | AP018116 | 16,795 | 1,445 | 31.1 | 21.1 | 15.0 | 32.9 | 47.9 | −0.3737 | 52.1 | 0.1916 |
| Cyrtodactylus chanhomeae | AP018117 | 17,068 | 1,728 | 31.2 | 23.3 | 15.6 | 29.9 | 45.5 | −0.3143 | 54.5 | 0.1450 |
| Cyrtodactylus tigroides | AP018118 | 16,929 | 1,557 | 30.3 | 21.5 | 15.6 | 32.6 | 48.2 | −0.3527 | 51.8 | 0.1699 |
Notes.
CR indicates control region.
Figure 1. AT-skew versus A+T% and GC-skew versus G+C% in Cyrtodactylus and gecko lizard mitochondrial genomes (mitogenomes).

Values are calculated on heavy strands for the full length of mitogenomes. The x-axis indicates the skew values, the y-axis provides the A+T% or G+C% values.
Boundaries between the protein-coding genes were determined by aligning their sequences and identifying the transcription initiation and termination codons with those of known gecko lizards. Common start codons of protein-coding genes were ATG, ATA, and GTG (Table S2). Nine protein-coding genes ended with complete stop codons: TAA, TAG, AGA, and AGG, except for ND3 in all Cyrtodactylus species and Cytb in C. auribalteatus with TA, whereas the remaining protein-coding genes (COII, COIII, and ND4) ended with the incomplete stop codon T, which appeared to be commonly created by post-transcriptional polyadenylation in vertebrate mitogenomes (Ojala, Montoya & Attardi, 1981). The longest protein-coding gene in all Cyrtodactylus species was ND5, whereas the shortest was ATPase8. Average comparison of nucleotide diversity among the five Cyrtodactylus mitogenomes was determined at 20.6 ± 0.3%. Highest level of sequence diversity (p-distance) in protein-coding genes was recorded for ATPase8 at 40.9% (between C. peguensis and C. chanhomeae), while the lowest was COIII at 14.7% (between C. tigroides and C. auribaltealus). Nucleotide diversity of ND2 was 21.0 ± 1.2%, COI 18.3 ± 0.9%, and Cytb 20.2 ± 1.1%. These three genes are often used for molecular phylogenic analysis (Castoe et al., 2008; Supikamolseni et al., 2015; Laopichienpong et al., 2016). Comparing nucleotide sequences of the five Cyrtodactylus mitogenomes with those of other gecko lizards provided a clear pattern of nucleotide diversity for whole mitogenomes, ND2, COI, and Cytb in the families Gekkonidae, Eublepharidae, Pygopodidae, Phyllodactylidae and Sphaerodactylidae, and infraorder Gekkota (Table 3).
Table 3. Comparison of sequence divergence among gecko lizards based on whole mitochondrial genome and three mitochondrial gene sequences (ND2, COI, and Cytb).
| Sample group | Average genetic distance ±standard error | |||
|---|---|---|---|---|
| Mitochondrial genome | ND2 | COI | Cytb | |
| Cyrtodactylus (5)a | 0.206 ±0.003 | 0.210 ±0.012 | 0.183 ±0.009 | 0.202 ±0.011 |
| Intrafamily Gekkonidae (16)a | 0.334 ±0.004 | 0.346 ±0.014 | 0.243 ±0.010 | 0.298 ±0.013 |
| Intrafamily Eublepharidae (4)a | 0.306 ±0.003 | 0.350 ±0.014 | 0.231 ±0.010 | 0.269 ±0.012 |
| Intrafamily Phyllodactylidae(2)a | 0.313 | 0.362 | 0.236 | 0.299 |
| Intrafamily Pygopodidae (1)a | – | – | – | – |
| Intrafamily Sphaerodactylidae (2)a | 0.200 | 0.171 | 0.215 | 0.182 |
| Infraorder Gekkota (30)a | 0.315 ±0.005 | 0.351 ±0.015 | 0.239 ±0.010 | 0.274 ±0.015 |
Notes.
Integers in parenthesis indicate the number of species analyzed.
A total of 22 tRNAs were interspersed throughout the mitogenome. Their size ranged from 54 bp (tRNA-Tyr) to 74 bp (tRNA-Phe). Most tRNAs could be folded into the canonical cloverleaf secondary structure, except for tRNA-Cys and two tRNA-Ser which appeared to lack the dihydrouridine arm. A characteristic stem and loop structure of an origin for light-strand replication (OL) was present between the tRNA-Asn and tRNA-Cys of the WANCY tRNA gene cluster. The size of 16S rRNA ranged from 1,533 bp (C. chanhomeae) to 1,545 bp (C. tigroides), with 12S rRNA ranging from 946 (C. peguensis) to 958 (C. thirakhupti). Total numbers of intergenic spacers were 13, 12, 10, 9, and 7 in the mitogenomes of C. tigroides, C. chanhomeae, C. peguensis, C. auribalteatus, and C. thirakhupti, respectively (Tables S3–S7). Interestingly, the 28 bp spacer between COI and tRNA-Ser was observed in C. tigroides but not for other Cyrtodactylus species.
The size of the CR for the five Cyrtodactylus species ranged from 1,445 to 1,728 bp (Table 4), and average A+T% was 58.9 ± 1.0. Lengths of the ETAS domain varied from 492 bp to 647 bp among the five Cyrtodactylus species. The TAS (termination-associated sequence) was identified by the conserved pentanucleotides (5′-TACAT-3′). The TAS elements were also located in repeated sequences of VNTR. These tandem repeats were identified in the ETAS domain containing six repeated sequences of a 75-bp named 75-bp box in C. peguensis, C. tigroides and C. thirakhupti, and eight 75-bp boxes in C. chanhomeae. However, two repeated sequences of a 225-bp box were found in C. auribalteatus. The tandem repeats were also able to form an “inverted repetitions” type structural conformation which was very similar to hairpins (Fig. S1). Secondary structures of single repeated sequences were retrieved based on the minimum free energy model (Zuker & Stiegler, 1981). The CCR domain ranged from 246 to 314 bp in length in all Cyrtodactylus. The consensus sequences of CSB-F were recognized as “CHCGRGAAACCAKCRACCCS”. Moreover, the size of the CSB domain ranged from 633 bp (C. auribaltealus) to 845 bp (C. peguensis). This domain contained three conserved blocks: CSB-1 (5′-KTTMATGCTCGAWRGACATAY-3′), CSB-2 (5′-AAACCCCCCTTACCCCCC-3′), and CSB-3 (5′-CGCCAAACCCCTAAAACG-3′) involving the regulation of replication and transcription (Clayton, 1991; Shadel & Clayton, 1997). Sequence similarities of CSB-2 and CSB-3 among Cyrtodactylus were higher than those of CSB-1. Microsatellite motifs: (TA)n and/or (CA)n were found between CSB-1 and CSB-2, or between CSB-3 and tRNA-Phe in Cyrtodactylus (Table S8).
Table 4. Summary of a non-coding control region composition of five Cyrtodactylus species.
| Species | GenBank accession number | CRa | ETASb | CCRc | CSBd | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRa size (bp) | A+T% content | ETASb size (bp) | A+T% content | CCRc size (bp) | A+T% content | CSB-F size (bp) | CSBd size (bp) | A+T% content | CSB-1 size (bp) | CSB-2 size (bp) | CSB-3 size (bp) | ||
| Cyrtodactylus peguensis | AP018114 | 1,653 | 57.7 | 496 | 76.6 | 312 | 52.9 | 20 | 845 | 48.4 | 21 | 18 | 18 |
| Cyrtodactylus thirakhupti | AP018115 | 1,454 | 59.4 | 495 | 76.8 | 311 | 51.5 | 20 | 648 | 49.9 | 21 | 18 | 18 |
| Cyrtodactylus auribalteatus | AP018116 | 1,445 | 57.7 | 566 | 70.9 | 246 | 48.0 | 20 | 633 | 49.8 | 21 | 18 | 18 |
| Cyrtodactylus chanhomeae | AP018117 | 1,728 | 62.5 | 647 | 74.8 | 254 | 56.7 | 20 | 827 | 54.7 | 21 | 18 | 18 |
| Cyrtodactylus tigroides | AP018118 | 1,557 | 57.2 | 492 | 73.8 | 314 | 54.2 | 20 | 751 | 47.8 | 21 | 18 | 18 |
Notes.
CR indicates control region.
ETAS indicates extended termination associated sequence domain.
CCR indicates central conserved region domain.
CSB indicates conserved sequence block domain.
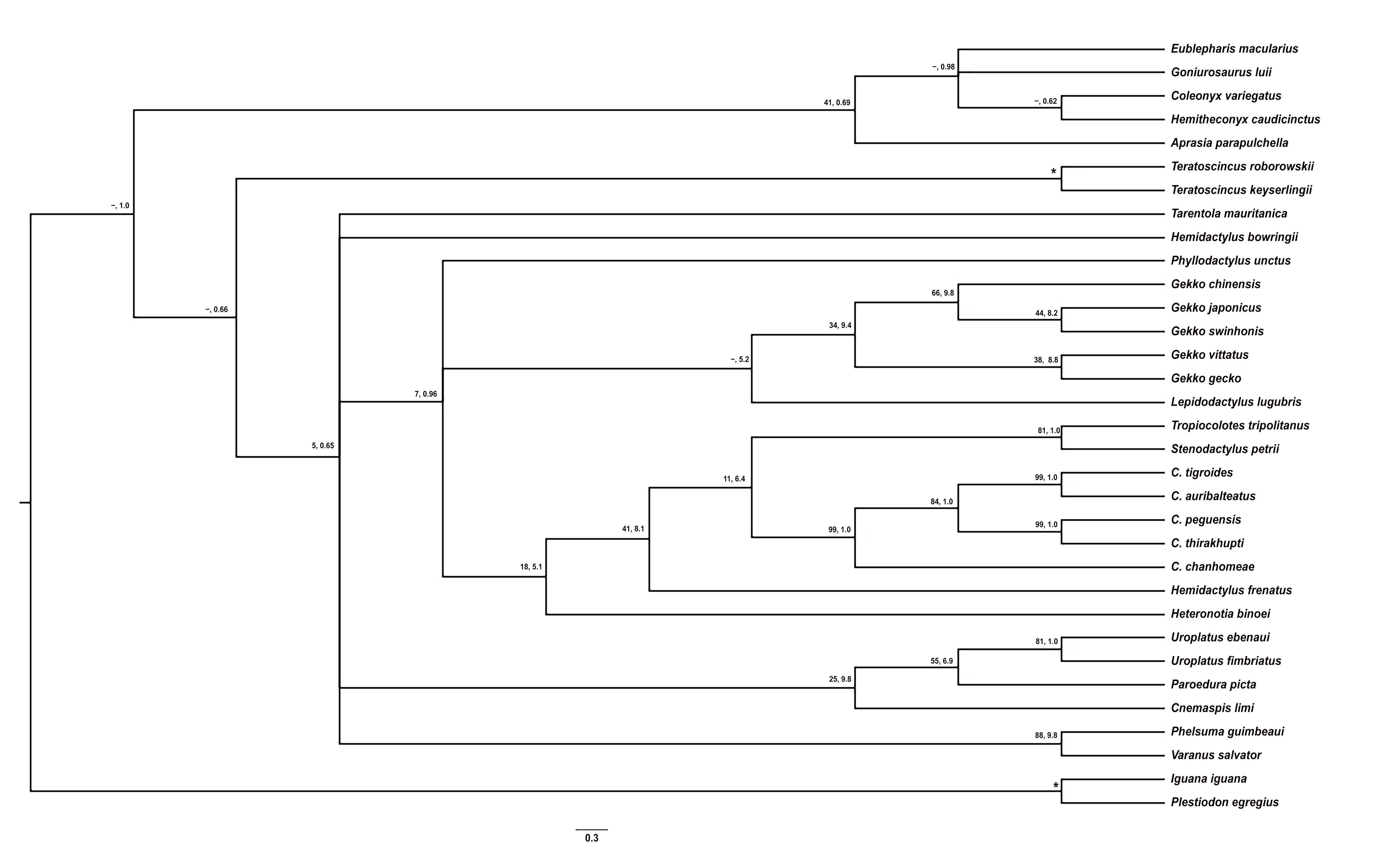
Phylogenetic positions of the five Cyrtodactylus species in gecko lizards were analyzed based on the concatenated heavy-strand encoded protein-coding genes and the ND6 gene with/without gap-containing site datasets from 30 gecko lizards. Both datasets showed highly similar tree topology (Fig. 2, Fig. S2). The pattern of phylogenies of the mitogenome sequence was monophyletic, with most species clustered in each taxonomic family. Five Cyrtodactylus species were identified within the same group. These results showed similar tree topology and strongly supported the established gecko lizard relationship (Pyron, Burbrink & Wiens, 2013; Kim et al., 2015).
Figure 2. Phylogenetic relationship among 30 gecko lizards with Iguana iguana, Plestiodon egregious and Varanus salvator as the outgroup, constructed from Bayesian inference analysis using concatenated heavy-strand encoded protein-coding gene sequences.

Support values at each node are bootstrap values from maximum likelihood (ML) (left) and Bayesian posterior probability (right). Asterisk (*) indicates full support (100%, 1.0) in both analyses, and hyphen (-) indicates no support. Detailed information of all gecko lizards is presented in Table 1. Mitochondrial gene arrangement types for gecko lizards are shown on the right hand side.
Discussion
Using the PCR method, complete mitogenomes of the five Cyrtodactylus species were determined for genome size as 16–17 kb, similar to those of other gecko lizards (Zhou et al., 2006; Kumazawa et al., 2014; Kim et al., 2015). The GC-skew values were negative in all gecko lizard mitogenomes and this phenomenon is highly conserved among vertebrates (Zhou et al., 2006; Kumazawa et al., 2014; Hao, Ping & Zhang, 2016). The GC-skew values were subjected to statistical evaluation of the asymmetric distribution of the two complementary base pair DNA strands, which suggest a higher content of C compared with G nucleotides. Comparison of A+T% for gecko lizard mitogenomes showed that the Cyrtodactylus mitogenome exhibited a low value (52.2 ± 0.6), whereas values of the AT-skew of Cyrtodactylus were higher than other gecko lizards, indicating a higher occurrence of A compared to T nucleotides. A large base composition bias arose in the Cyrtodactylus lineage after it diverged from a common ancestor around 65 million years ago in the late Paleogene (Wood et al., 2012). Although sequence diversity (p-distances) of ATPase8 was high (>40%) among Cyrtodactylus species in this study, interspecific sequence divergences of ND2, COI, and Cytb as standard DNA barcodes for squamate reptiles (Castoe et al., 2008; Supikamolseni et al., 2015; Laopichienpong et al., 2016) were about 20% (Table 3). Gene coding for the subunits of cytochrome oxidase and cytochrome b were conserved, while the most variable genes were the ND and ATPase (Meyer, 1993). Interspecific sequence divergences in Cytb were about 19–27% in gecko lizards (Lamb, Bauer & McEachran, 2001), while sequence divergences of congeneric variation were between 4.3% and 28.7% (average = 21.0 ± 4.2%) for the COI gene (Nguyen et al., 2014) and 21% for the ND2 gene in Cyrtodactylus and Geckoella (Shea et al., 2011; Agarwal et al., 2014; Agarwal & Karanth, 2015). This result agreed with sequence divergence of the three genes among Cyrtodactylus species and gecko lizards in this study, suggesting that the three genes are effective for identifying species in Cyrtodactylus and gecko lizards.
Mitochondrial gene rearrangement of Cyrtodactylus species in gecko lizards
Generally, organization of the 37 genes and the CR of most gecko lizards tend to be conserved in squamate reptiles (Zhou et al., 2006; Kumazawa et al., 2014; Kim et al., 2015; Hao, Ping & Zhang, 2016). However, various rearrangement patterns of mitogenomes have been found in snakes (Yan, Li & Zhou, 2008), varanid lizards (Amer & Kumazawa, 2008), and acrodont lizards (Okajima & Kumazawa, 2010) as a consequence of the shuffling of tRNA gene clusters, translocations and/or duplications of genes, and gene loss (Anderson et al., 1981; Fujita, Boore & Moritz, 2007; Kumazawa et al., 2014; Li et al., 2015). Comparison of gecko lizard mitogenomes revealed six types of mitochondrial gene rearrangements as I, II, III, IV, V, and VI based on available gecko lizard mitogenome sequences. Type I is distributed in most gecko lizards (80%) including Cyrtodactylus species. Types II and III are found in Tropiocolotes tripolitanus and Stenodactylus petrii, respectively. Type II was derived from the Type I by duplication of tRNA-Gln and ND4, whereas Type III showed multiple copies of OL within the WANCY cluster. Type IV was observed in Uroplatus fimbriatus, resulting from the translocation of tRNA-Glu from 5′ proximal ND 6 to 5′ proximal tRNA-Pro (Fig. 2). Deletion of tRNA-Glu was found in Uroplatus ebenaui as Type V, while duplication of 5′ proximal ND4- to 5′ proximal tRNA-Ile was observed in Heteronotia binoei as Type VI. Our phylogenetic placement indicated that gecko lizard mitogenome Types II–VI were found at the terminus of the tree in Gekkonidae, suggesting that structural variation of mitogenomes has occurred independently in gekkonids. Rare changes of mitochondrial gene rearrangement in gecko lizard lineages may indicate the possibility of homoplasy-free datasets. However, many family-level taxa are represented by a few species; thus, more gecko lizard mitogenome sequences are required to better understand the evolutionary history in this lineage.
Dynamics of CR in Cyrtodactylus
Although the CRs of the five Cyrtodactylus species were mainly composed of high A+T%, distribution of AT was not homogenous among the three domains. The AT content of the CSB domain was lower than the ETAS and CCR domains (Table 4). The size of CRs varied from 1,445 to 1,728 bp among Cyrtodactylus species and tallied with those of gecko lizards. This feature was mainly caused by the duplication of tandem repeat at VNTR and frequently occurs in ETAS and/or CSB domains (Larizza et al., 2002). In the ETAS domain, different copy numbers of the 75-bp box tandem repeat were found in most Cyrtodactylus with at least 76% similarities, except for C. auribalteatus containing 225-bp box. These results collectively suggest that the 75-bp box tandem repeat was present in ancestral mitogenomes before divergence of Cyrtodactylus species. Here, the 75-bp box is designated as 75-bp box family. Variable copy numbers have resulted from independent duplication in each species, a factor commonly observed in other vertebrate groups (Zhou et al., 2006; Kumazawa et al., 2014; Starostová & Musilová, 2016).
Comparison of 75-bp and 225-bp boxes among Cyrtodactylus species revealed 69% similarities. The 225-bp box is likely to contain two 75-bp boxes tandemly and one 75-bp box-like (Fig. 3). This result suggests that the presence of 225-bp box resulted from duplication of 75-bp boxes with nucleotide substitution, followed by homogenization and fixation in C. auribalteatus after it diverged from Cyrtodactylus. Interestingly, both the sequence of 75-bp and 225-bp boxes could form stable secondary structures that might be responsible for increasing possible misalignment (H-strand and L-strand), involving rearrangement in CR during DNA replication of mitochondrial DNA (Broughton & Dowling, 1997; Pereira et al., 2008). Secondary structures of VNTR are believed to be termination sequences in the replication process in fish (Terencio et al., 2013). Although the function of these secondary structures remains completely unknown, they were probably retained in Cyrtodactylus under selective pressure. Structural and functional studies are required to explain this molecular mechanism. To investigate the distribution of the 75-bp box family in gecko lizards, a comparative search (BLAST) was conducted for similar sequences housed in the GenBank. A tandem repeat with 55–75% similarities was found in most gecko lizards, even for Eublepharidae and Pygopodidae as the basal group. This result suggests that the tandem repeat of 75-bp box family was acquired in the genome of the common ancestor of gecko lizards and evolved gradually through independent nucleotide mutation in each lineage.
Figure 3. Schematic representation of a non-coding control region (CR) structure of Cyrtodactylus species.

Abbreviations: TAS, Termination Associated Sequence; CSB, Conserved Sequence Block; VNTR, variable number of tandem repeats. The square color box indicates a repeated sequence. Same color boxes show grid pattern box indicating sequence similarity of more than 65%.
The CCR domain is highly conserved among gecko lizards compared to ETAS and CSB domains. The CSB-F block characterized the CCR domain in Cyrtodactylus and different levels of similarity (59.26–96.15%) were observed when compared with other gecko lizards. However, two blocks (CSB-E and/or CSB-D) observed in other vertebrates were not identified in Cyrtodactylus and other gecko lizards (Anderson et al., 1981; Broughton & Dowling, 1994; Lee et al., 1995). This result suggests plausible plasticity across vertebrates concerning the CSB-F, CSB-E, and CSB-D blocks. The CSB domain in Cyrtodactylus and gecko lizards is characterized by the three conserved blocks CSB-1, CSB-2, and CSB-3 which involve regulation of replication and transcription (Li et al., 2013; Kumazawa et al., 2014). These three blocks have also been identified in vertebrate mitogenomes (Liu, 2002; Brehm et al., 2003; Zhang et al., 2009; Xiong et al., 2010). The sequence of CSB-3 blocks is the most highly conserved in Cyrtodactylus and other gecko lizards, whereas the CSB-1 block showed the most variability. Repeated sequences as minisatellites and/or microsatellites (TA)n or (CA)n were found between CSB-1 and CSB-2 or between CSB-3 and tRNA-Phe in Cyrtodactylus and other gecko lizards (Table S8). Minisatellites were found in four gecko lizards (Hemitheconyx caudicinctus, Gekko japonicas, U. fimbriatus, and U. ebenaui) which did not contain tandem repeats in the ETAS domain. This result suggests that repeated sequences are detected in at least ETAS or CSB domains of CR. Thus, VNTRs can be used as molecular markers to provide phylogenetic information in population genetics, species identification, genetic diversity, and conservation (Zardoya & Meyer, 1998; Zhang et al., 2009). However, VNTRs differ in length, copy number, and base composition even in very closely related species, and may be shared by distantly related species but not by close ones, such as in C. auribalteatus. This result suggests that the presence/absence of repeated sequences might not be a reliable phylogenetic marker.
Conclusions
Here, complete mitogenomes of five Cyrtodactylus species were sequenced and characterized with those of gecko lizards to examine their structural patterns. Six types of mitochondrial gene rearrangements indicated low diversification in gecko lizards, whereas nucleotide sequence diversity among gecko lizards was exhibited at considerably high levels and might be useful as a tool to resolve taxonomic controversies. The CR evolutionary pattern strongly suggests high conservation level of the 75-bp repeated sequence family in gecko lizards. These repeated sequences also form secondary structures, indicating that repeated sequence families played an important role under selective pressure. This might have resulted in involvement of these structures in mitogenome replication which caused rearrangements in CR. However, since many family-level taxa are represented by a limited number of species, the detailed knowledge of the phylogeny of gecko lizard mitogenomes requires further sampling and sequencing exports.
Supplemental Information
{kind=link}
Support values at each node are bootstrap values from maximum likelihood (ML) (left) and Bayesian posterior probability (right). An asterisk (*) indicates full support (100%, 1.0) in both analyses and a hyphen (-) indicates no support. Detailed information of all gecko lizards is presented in Table 1.
{kind=link}
Acknowledgments
We would like to thank Nakhon Ratchasima Zoo and the Conservation Research and Education Division, Zoological Park Organization, Dusit, Bangkok, Thailand for sample collection.
Funding Statement
This study was financially supported by grants from the National Research Council of Thailand (NRCT; No. 2560096003012), the Fellowship of Capacity Building for Kasetsart University on Internationalization at Kasetsart University (No. 0513.10109/8384), Faculty of Medicine, Ramathibodi Hospital Scholarship (No. 678/2558), Thailand Research Fund (TRF; no. PHD60I0014), the Center for Advanced Studies in Tropical Natural Resources, National Research University-Kasetsart University (CASTNAR, NRU-KU; no. 6/2558), the Center of Excellence on Agricultural Biotechnology, Science and Technology Postgraduate Education and Research Development Office, Office of Higher Education Commission, Ministry of Education (AG-BIO/PERDO-CHE), and the Science Achievement Scholarship of Thailand (SAST; no. 5717400071) from the Office of the Higher Education Commission, Thailand. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Prapatsorn Areesirisuk conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Narongrit Muangmai conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, approved the final draft.
Kirati Kunya contributed reagents/materials/analysis tools, approved the final draft.
Worapong Singchat and Siwapech Sillapaprayoon analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Sorravis Lapbenjakul, Watcharaporn Thapana and Attachai Kantachumpoo performed the experiments, approved the final draft.
Sudarath Baicharoen contributed reagents/materials/analysis tools, approved the final draft.
Budsaba Rerkamnuaychoke and Surin Peyachoknagul conceived and designed the experiments, approved the final draft.
Kyudong Han conceived and designed the experiments, contributed reagents/materials/analysis tools, approved the final draft.
Kornsorn Srikulnath conceived and designed the experiments, performed the experiments, analyzed the data, contributed reagents/materials/analysis tools, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Animal Ethics
The following information was supplied relating to ethical approvals (i.e., approving body and any reference numbers):
Animal care and all experimental procedures were approved by the Animal Experiment Committee, Kasetsart University, Thailand (approval no. ACKU-SCI-022), and conducted in accordance with the Regulations on Animal Experiments at Kasetsart University.
DNA Deposition
Data Availability
The following information was supplied regarding data availability:
The raw data are provided in the Supplemental Files and shows mitochondrial genome characterization of all species.
References
- Agarwal et al. (2014).Agarwal I, Bauer AM, Jackman TR, Karanth KP. Insights into Himalayan biogeography from geckos: a molecular phylogeny of Cyrtodactylus (Squamata: Gekkonidae) Molecular Phylogenetics and Evolution. 2014;80:145–155. doi: 10.1016/j.ympev.2014.07.018. [DOI] [PubMed] [Google Scholar]
- Agarwal & Karanth (2015).Agarwal I, Karanth KP. A phylogeny of the only ground-dwelling radiation of Cyrtodactylus (Squamata, Gekkonidae): diversification of Geckoella across peninsular India and Sri Lanka. Molecular Phylogenetics and Evolution. 2015;82:193–199. doi: 10.1016/j.ympev.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Albert et al. (2009).Albert EM, Mauro DS, García-París M, Rüber L, Zardoya R. Effect of taxon sampling on recovering the phylogeny of squamate reptiles based on complete mitochondrial genome and nuclear gene sequence data. Gene. 2009;441:12–21. doi: 10.1016/j.gene.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Amer & Kumazawa (2008).Amer SAM, Kumazawa Y. The mitochondrial genome of the lizard Calotes versicolor and a novel gene inversion in South Asian draconine agamids. Molecular Biology and Evolution. 2008;24:1330–1339. doi: 10.1093/molbev/msm054. [DOI] [PubMed] [Google Scholar]
- Anderson et al. (1981).Anderson S, Bankier AT, Barrell BG, De Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Bauer, Sumontha & Pauwels (2003).Bauer AM, Sumontha M, Pauwels OS. Two new species of Cyrtodactylus (Reptilia: Squamata: Gekkonidae) from Thailand. Zootaxa. 2003;376:1–18. doi: 10.11646/zootaxa.376.1.1. [DOI] [Google Scholar]
- Benson (1999).Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt et al. (2013).Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- Bernt & Middendorf (2011).Bernt M, Middendorf M. A method for computing an inventory of metazoan mitochondrial gene order rearrangements. BMC Bioinformatics. 2011;12:S6. doi: 10.1186/1471-2105-12-S9-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore (1999).Boore JL. Animal mitochondrial genomes. Nucleic Acids Research. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenger (1893).Boulenger GA. Concluding report on the reptiles and batrachians obtained in Burma by Signor L. Fea, dealing with a collection made in Pegu and Karin Hills in 1887–1888. Doriana. 1893;13:304–347. [Google Scholar]
- Brehm et al. (2003).Brehm A, Jesus J, Spínola H, Alves C, Vicente L, Harris DJ. Phylogeography of the Madeiran endemic lizard Lacerta dugesii inferred from mtDNA sequences. Molecular Phylogenetics and Evolution. 2003;26:222–230. doi: 10.1016/S1055-7903(02)00310-X. [DOI] [PubMed] [Google Scholar]
- Broughton & Dowling (1994).Broughton RE, Dowling TE. Length variation in mitochondrial DNA of the minnow Cyprinella spiloptera. Genetics. 1994;138:179–190. doi: 10.1093/genetics/138.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton & Dowling (1997).Broughton RE, Dowling TE. Evolutionary dynamics of tandem repeats in the mitochondrial DNA control region of the minnow Cyprinella spiloptera. Molecular Biology and Evolution. 1997;14:1187–1196. doi: 10.1093/oxfordjournals.molbev.a025728. [DOI] [PubMed] [Google Scholar]
- Castoe et al. (2008).Castoe TA, Jiang ZJ, Gu W, Wang ZO, Pollock DD. Adaptive evolution and functional redesign of core metabolic proteins in snakes. PLOS ONE. 2008;3:e2201. doi: 10.1371/journal.pone.0002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton (1991).Clayton DA. Nuclear gadgets in mitochondrial DNA replication and transcription. Trends in Biochemical Sciences. 1991;16:107–111. doi: 10.1016/0968-0004(91)90043-U. [DOI] [PubMed] [Google Scholar]
- Dong & Kumazawa (2005).Dong S, Kumazawa Y. Complete mitochondrial DNA sequences of six snakes: phylogenetic relationships and molecular evolution of genomic features. Journal of Molecular Evolution. 2005;61:12–22. doi: 10.1007/s00239-004-0190-9. [DOI] [PubMed] [Google Scholar]
- Douzery & Randi (1997).Douzery E, Randi E. The mitochondrial control region of Cervidae: evolutionary patterns and phylogenetic content. Molecular Biology and Evolution. 1997;14:1154–1166. doi: 10.1093/oxfordjournals.molbev.a025725. [DOI] [PubMed] [Google Scholar]
- Elson & Lightowlers (2006).Elson JL, Lightowlers RN. Mitochondrial DNA clonality in the dock: can surveillance swing the case. Trends in Genetics. 2006;22:603–607. doi: 10.1016/j.tig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Fernández-Silva, Enriquez & Montoya (2003).Fernández-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Experimental Physiology. 2003;88:41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- Fujita, Boore & Moritz (2007).Fujita MK, Boore JL, Moritz C. Multiple origins and rapid evolution of duplicated mitochondrial genes in parthenogenetic geckos (Heteronotia binoei; Squamata, Gekkonidae) Molecular Biology and Evolution. 2007;24:2775–2786. doi: 10.1093/molbev/msm212. [DOI] [PubMed] [Google Scholar]
- Gasteiger et al. (2003).Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Research. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissi, Iannelli & Pesole (2008).Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- Grismer, Grismer & Chav (2010).Grismer JL, Grismer LL, Chav T. New species of Cnemaspis strauch 1887 (Squamata: Gekkonidae) from Southwestern Cambodia. Journal of Herpetology. 2010;44:28–36. doi: 10.1670/08-211.1. [DOI] [Google Scholar]
- Hao, Ping & Zhang (2016).Hao S, Ping J, Zhang Y. Complete mitochondrial genome of Gekko chinensis (Squamata, Gekkonidae) Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2016;27:4226–4227. doi: 10.3109/19401736.2015.1022751. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck & Ronquist (2001).Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Janke et al. (2001).Janke A, Erpenbeck D, Nilsson M, Arnason U. The mitochondrial genomes of the iguana (Iguana iguana) and the caiman (Caiman crocodylus): implications for amniote phylogeny. Proceedings of the Royal Society Biological Sciences Series B. 2001;268:623–631. doi: 10.1098/rspb.2000.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonniaux, Hashiguchi & Kumazawa (2012).Jonniaux P, Hashiguchi Y, Kumazawa Y. Mitochondrial genomes of two African geckos of genus Hemitheconyx (Squamata: Eublepharidae) Mitochondrial DNA. 2012;23:278–279. doi: 10.3109/19401736.2012.668898. [DOI] [PubMed] [Google Scholar]
- Kim et al. (2015).Kim IH, Park J, Cheon K-S, Lee HJ, Kim JK, Park D. Complete mitochondrial genome of Schlegel’s Japanese gecko Gekko japonicus (Squamata: Gekkonidae) Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2015;27:3684–3686. doi: 10.3109/19401736.2015.1079855. [DOI] [PubMed] [Google Scholar]
- Kumar, Stecher & Tamura (2016).Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumazawa (2007).Kumazawa Y. Mitochondrial genomes from major lizard families suggest their phylogenetic relationships and ancient radiations. Gene. 2007;388:19–26. doi: 10.1016/j.gene.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Kumazawa & Endo (2004).Kumazawa Y, Endo H. Mitochondrial genome of the Komodo dragon: efficient sequencing method with reptile-oriented primers and novel gene rearrangements. DNA Research. 2004;11:115–125. doi: 10.1093/dnares/11.2.115. [DOI] [PubMed] [Google Scholar]
- Kumazawa et al. (2014).Kumazawa Y, Miura S, Yamada C, Hashiguch Y. Gene rearrangements in gekkonid mitochondrial genomes with shuffling, loss, and reassignment of tRNA genes. BMC Genomics. 2014;15:930. doi: 10.1186/1471-2164-15-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumazawa & Nishida (1999).Kumazawa Y, Nishida M. Complete mitochondrial DNA sequences of the green turtle and blue-tailed mole skink: statistical evidence for archosaurian affinity of turtles. Molecular Biology and Evolution. 1999;16:784–792. doi: 10.1093/oxfordjournals.molbev.a026163. [DOI] [PubMed] [Google Scholar]
- Kurabayashi et al. (2008).Kurabayashi A, Sumida M, Yonekawa H, Glaw F, Vences M, Hasegawa M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Molecular Biology and Evolution. 2008;25:874–891. doi: 10.1093/molbev/msn031. [DOI] [PubMed] [Google Scholar]
- Lamb, Bauer & McEachran (2001).Lamb T, Bauer AM, McEachran JD. Mitochondrial phylogeny of namib day geckos (Rhoptropus) based on Cytochrome b and 16S rRNA sequences. Copeia. 2001;2001:775–780. doi: 10.1643/0045-8511(2001)001[0775:MPONDG]2.0.CO;2. [DOI] [Google Scholar]
- Laopichienpong et al. (2016).Laopichienpong N, Muangmai N, Supikamolseni A, Twilprawat P, Chanhome L, Suntrarachun S, Peyachoknagul S, Srikulnath K. Assessment of snake DNA barcodes based on mitochondrial COI and Cytb genes revealed multiple putative cryptic species in Thailand. Gene. 2016;594:238–247. doi: 10.1016/j.gene.2016.09.017. [DOI] [PubMed] [Google Scholar]
- Lapbenjakul et al. (2017).Lapbenjakul S, Thapana W, Twilprawat P, Muangmai N, Kanchanaketu T, Temsiripong Y, Unajak S, Peyachoknagul S, Srikulnat K. High genetic diversity and demographic history of captive Siamese and Saltwater crocodiles suggest the first step toward the establishment of a breeding and reintroduction program in Thailand. PLOS ONE. 2017;12:e0184526. doi: 10.1371/journal.pone.0184526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larizza et al. (2002).Larizza A, Pesole G, Reyes A, Sbisà E, Saccone C. Lineage specificity of the evolutionary dynamics of the mtDNA D-Loop region in rodents. Journal of Molecular Evolution. 2002;54:145–155. doi: 10.1007/s00239-001-0063-4. [DOI] [PubMed] [Google Scholar]
- Lee et al. (1995).Lee WJ, Conroy J, Howell WH, Kocher TD. Structure and evolution of teleost mitochondrial control regions. Journal of Molecular Evolution. 1995;41:54–66. doi: 10.1007/BF00174041. [DOI] [PubMed] [Google Scholar]
- Li et al. (2013).Li HM, Zeng DL, Guan QX, Qin PS, Qin XM. Complete mitochondrial genome of Gekko swinhonis (Squamata, Gekkonidae) Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2013;24:86–88. doi: 10.3109/19401736.2012.717938. [DOI] [PubMed] [Google Scholar]
- Li et al. (2015).Li W, Zhang XC, Zhao J, Shi Y, Zhu XP. Complete mitochondrial genome of Cuora trifasciata (Chinese three-striped box turtle), and a comparative analysis with other box turtles. Gene. 2015;555:169–177. doi: 10.1016/j.gene.2014.10.060. [DOI] [PubMed] [Google Scholar]
- Liu (2002).Liu HZ. The structure and evolution of the mtDNA control region in fish: taking example for acheilognathinae. Progress in Natural Science. 2002;12:266–270. [Google Scholar]
- Lohse et al. (2013).Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Research. 2013;41:W575–W581. doi: 10.1093/nar/gkt289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald et al. (2005).MacDonald AJ, Knopp T, Pepper M, Keogh JS, Sarre SD. The first complete mitochondrial genome of Pygopodidae (Aprasia parapulchella Kluge) Australian Journal of Zoology. 2005;63:111–114. doi: 10.1071/ZO14092. [DOI] [Google Scholar]
- Macey et al. (2005).Macey JR, Fong JJ, Kuehl JV, Shafiei S, Ananjeva NB, Papenfuss TJ, Boore JL. The complete mitochondrial genome of a gecko and the phylogenetic position of the Middle Eastern Teratoscincus keyserlingii. Molecular Phylogenetics and Evolution. 2005;36:188–193. doi: 10.1016/j.ympev.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Macey et al. (1997).Macey JR, Larson A, Ananjeva NB, Fang Z, Papenfuss TJ. Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Molecular Biology and Evolution. 1997;14:91–104. doi: 10.1093/oxfordjournals.molbev.a025706. [DOI] [PubMed] [Google Scholar]
- Mauro et al. (2006).Mauro DS, Gower DJ, Zardoya R, Wilkinson M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Molecular Biology and Evolution. 2006;23:227–234. doi: 10.1093/molbev/msj025. [DOI] [PubMed] [Google Scholar]
- Metallinou et al. (2012).Metallinou M, Arnold EN, Crochet P-A, Geniez P, Brito JC, Lymberakis P, Baha El Din S, Sindaco R, Robinson M, Carranza S. Conquering the Sahara and Arabian deserts: systematics and biogeography of Stenodactylus geckos (Reptilia: Gekkonidae) BMC Evolutionary Biology. 2012;12:258. doi: 10.1186/1471-2148-12-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer (1993).Meyer A. Evolution of mitochondrial DNA in fishes. In: Mochachka PW, Mommsen TP, editors. Biochemistry and molecular biology of fishes. Elsevier Press Amsterdam; New York: 1993. pp. 1–38. [Google Scholar]
- Nazarov et al. (2014).Nazarov RA, Poyarkov NA, Orlov NL, Nguyen SN, Milto KD, Martynov AA, Konstantinov EL, Chulisov AS. A review of genus Cyrtodactylus (Reptilia: Sauria: Gekkonidae) in fauna of Laos with description of four new species. Proceedings ZIN. 2014;318:391–423. [Google Scholar]
- Nguyen et al. (2014).Nguyen SN, Yang JX, Le TNT, Nguyen LT, Orlov NL, Hoang CV, Nguyen TQ, Jin JQ, Rao DQ, Hoang TN, Che J, Murphy RW, Zhang Y-P. DNA barcoding of Vietnamese bent-toed geckos (Squamata: Gekkonidae: Cyrtodactylus) and the description of a new species. Zootaxa. 2014;3784:48–66. doi: 10.11646/zootaxa.3784.1.2. [DOI] [PubMed] [Google Scholar]
- Ojala, Montoya & Attardi (1981).Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- Okajima & Kumazawa (2010).Okajima Y, Kumazawa Y. Mitochondrial genomes of acrodont lizards: timing of gene rearrangements and phylogenetic and biogeographic implications. BMC Evolutionary Biology. 2010;10:141. doi: 10.1186/1471-2148-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels et al. (2004).Pauwels OS, Bauer AM, Sumontha M, Chanhome L. Cyrtodactylus thirakhupti (Squamata: Gekkonidae), a new cave-dwelling gecko from southern Thailand. Zootaxa. 2004;772:1–11. doi: 10.11646/zootaxa.772.1.1. [DOI] [PubMed] [Google Scholar]
- Pereira (2000).Pereira SL. Mitochondrial genome organization and vertebrate phylogenetics. Genetics and Molecular Biology. 2000;23:745–752. doi: 10.1590/S1415-47572000000400008. [DOI] [Google Scholar]
- Pereira et al. (2008).Pereira F, Soares P, Carneiro J, Pereira L, Richards MB, Samuels DC, Amorim A. Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Molecular Biology and Evolution. 2008;25:2759–2770. doi: 10.1093/molbev/msn225. [DOI] [PubMed] [Google Scholar]
- Perna & Kocher (1995).Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Journal of Molecular Evolution. 1995;41:353–358. doi: 10.1007/BF01215182. [DOI] [PubMed] [Google Scholar]
- Prakhongcheep et al. (2018).Prakhongcheep O, Muangmai N, Peyachoknagul S, Srikulnath K. Complete mitochondrial genome of mouthbrooding fighting fish (Betta pi) compared with bubble nesting fighting fish (B. splendens) Mitochondrial DNA Part B: Resources. 2018;3:6–8. doi: 10.1080/23802359.2017.1413294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyron, Burbrink & Wiens (2013).Pyron RA, Burbrink FT, Wiens JJ. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evolutionary Biology. 2013;13:93. doi: 10.1186/1471-2148-13-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut et al. (2015).Rambaut A, Suchard M, Xie D, Drummond A. Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer 2015
- Sela et al. (2015).Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Research. 2015;43:W7–W14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel & Clayton (1997).Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annual Review of Biochemistry. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- Shea et al. (2011).Shea G, Couper P, Worthington, Wilmer J, Amey A. Revision of the genus Cyrtodactylus Gray, 1827 (Squamata: Gekkonidae) in Australia. Zootaxa. 2011;3146:1–63. [Google Scholar]
- Siler et al. (2010).Siler CD, Oaks JR, Esselstyn JA, Diesmos AC, Brown RM. Phylogeny and biogeography of Philippine bent-toed geckos (Gekkonidae: Cyrtodactylus) contradict a prevailing model of Pleistocene diversification. Molecular Phylogenetics and Evolution. 2010;55:699–710. doi: 10.1016/j.ympev.2010.01.027. [DOI] [PubMed] [Google Scholar]
- Srikulnath et al. (2012).Srikulnath K, Thongpan A, Suputtitada S, Apisitwanich S. New haplotype of the complete mitochondrial genome of Crocodylus siamensis and its species-specific DNA markers: distinguishing C. siamensis from C. porosus in Thailand. Molecular Biology Reports. 2012;39:4709–4717. doi: 10.1007/s11033-011-1263-7. [DOI] [PubMed] [Google Scholar]
- Stamatakis (2014).Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starostová & Musilová (2016).Starostová Z, Musilová Z. The complete mitochondrial genome of the Madagascar ground gecko Paroedura picta (Squamata: Gekkonidae) Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2016;27:4397–4398. doi: 10.3109/19401736.2015.1089540. [DOI] [PubMed] [Google Scholar]
- Sumontha, Panitvong & Deein (2010).Sumontha M, Panitvong N, Deein G. Cyrtodactylus auribalteatus (Squamata: Gekkonidae), a new cave-dwelling gecko from Phitsanulok province, Thailand. Zootaxa. 2010;2370:53–64. doi: 10.11646/zootaxa.2370.1.3. [DOI] [Google Scholar]
- Sun, Cheng & Zhang (2015).Sun Z, Cheng Y, Zhang J. MITOSCISSOR: a useful tool for auto-assembly of mitogenomic datasets in the evolutionary analysis of fishes. Evolutionary Bioinformatics Online. 2015;11:115–120. doi: 10.4137/EBO.S22340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supikamolseni et al. (2015).Supikamolseni A, Ngaoburanawit N, Sumontha M, Chanhome L, Suntrarachun S, Peyachoknagul S, Srikulnath K. Molecular barcoding of venomous snakes and species-specific multiplex PCR assay to identify snake groups for which antivenom is available in Thailand. Genetics and Molecular Research. 2015;14:13981–13997. doi: 10.4238/2015.October.29.18. [DOI] [PubMed] [Google Scholar]
- Tanabe (2011).Tanabe AS. KAKUSAN: a computer program to automate the selection of a nucleotide substitution model and the configuration of a mixed model on multilocus data. Molecular Ecology Notes. 2011;7:962–964. doi: 10.1111/j.1471-8286.2007.01807.x. [DOI] [Google Scholar]
- Terencio et al. (2013).Terencio ML, Schneider CH, Gross MC, Feldberg E, Porto JIR. Structure and organization of the mitochondrial DNA control region with tandemly repeated sequence in the Amazon ornamental fish. Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2013;24:74–82. doi: 10.3109/19401736.2012.717934. [DOI] [PubMed] [Google Scholar]
- Thongtam na Ayudhaya et al. (2017).Thongtam na Ayudhaya P, Muangmai N, Banjongsat N, Singchat W, Janekitkarn S, Peyachoknagul S, Srikulnath K. Unveiling cryptic diversity of the anemonefish genera Amphiprion and Premnas (Perciformes: Pomacentridae) in Thailand with mitochondrial DNA barcodes. Agriculture and Natural Resources. 2017;51:198–205. doi: 10.1016/j.anres.2017.07.001. [DOI] [Google Scholar]
- Uetz & Hošek (2018).Uetz P, Hošek J. 2018. [15 October 2017]. The reptile database. http://www.reptile-database.org
- Wang et al. (2009).Wang H, Moore MJ, Soltis PS, Bell CD, Brockington SF, Alexandre R, Davis CC, Latvis M, Manchester SR, Soltis DE. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3853–3858. doi: 10.1073/pnas.0813376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Zhou & Nie (2011).Wang L, Zhou X, Nie L. Organization and variation of mitochondrial DNA control region in Pleurodiran turtles. Zoologia. 2011;28:495–504. doi: 10.1590/S1984-46702011000400011. [DOI] [Google Scholar]
- Welton et al. (2010).Welton LJ, Siler CD, Linkem CW, Diesmos AC, Brown RM. Philippine bent-toed geckos of the Cyrtodactylus agusanensis complex: ultilocus phylogeny, morphological diversity, and descriptions of three new species. Herpetological Monographs. 2010;24:55–85. doi: 10.1655/HERPMONOGRAPHS-D-10-00005.1. [DOI] [Google Scholar]
- Wood et al. (2012).Wood PL, Heinicke MP, Jackman TR, Bauer AM. Phylogeny of bent-toed geckos (Cyrtodactylus) reveals a west to east pattern of diversification. Molecular Phylogenetics and Evolution. 2012;65:992–1003. doi: 10.1016/j.ympev.2012.08.025. [DOI] [PubMed] [Google Scholar]
- Xiong et al. (2010).Xiong L, Nie L, Li X, Liu X. Comparison research and phylogenetic implications of mitochondrial control regions in four soft-shelled turtles of Trionychia (Reptilia, Testudinata) Genes and Genomics. 2010;32:291–298. doi: 10.1007/s13258-010-0015-8. [DOI] [Google Scholar]
- Yan, Li & Zhou (2008).Yan J, Li H, Zhou K. Evolution of the mitochondrial genome in snakes: gene rearrangements and phylogenetic relationships. BMC Genomics. 2008;9:569. doi: 10.1186/1471-2164-9-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan et al. (2014a).Yan J, Tian C, Lv L, Bauer AM, Zhou K. Complete mitochondrial genome of the San Lucan gecko, Phyllodactylus unctus (Sauria, Gekkota, Phyllodactylidae), in comparison with Tarentola mauritanica. Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2014a;25:202–203. doi: 10.3109/19401736.2013.796464. [DOI] [PubMed] [Google Scholar]
- Yan et al. (2014b).Yan J, Tian C, Zhou J, Bauer AM, Lee GL, Zhou K. Complete mitochondrial genome of the Tioman Island rock gecko, Cnemaspis limi (Sauria, Gekkota, Gekkonidae) Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis. 2014b;25:181–182. doi: 10.3109/19401736.2013.792066. [DOI] [PubMed] [Google Scholar]
- Zardoya & Meyer (1998).Zardoya R, Meyer A. Complete mitochondrial genome suggests diapsid affinities of turtles. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14226–14231. doi: 10.1073/pnas.95.24.14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al. (2009).Zhang Y, Nie L, Huang Y, Pu Y, Zhang L. The mitochondrial DNA control region comparison studies of four hinged turtles and its phylogentic significance of the genus Cuora sensu lato (Testudinata: Geoemydidae) Genes and Genomics. 2009;31:349–359. doi: 10.1007/BF03191253. [DOI] [Google Scholar]
- Zhou et al. (2006).Zhou K, Li H, Han D, Bauer AM, Feng J. The complete mitochondrial genome of Gekko gecko (Reptilia: Gekkonidae) and support for the monophyly of Sauria including Amphisbaenia. Molecular Phylogenetics and Evolution. 2006;40:887–892. doi: 10.1016/j.ympev.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Zuker & Stiegler (1981).Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Uetz P, Hošek J. 2018. [15 October 2017]. The reptile database. http://www.reptile-database.org
Supplementary Materials
Support values at each node are bootstrap values from maximum likelihood (ML) (left) and Bayesian posterior probability (right). An asterisk (*) indicates full support (100%, 1.0) in both analyses and a hyphen (-) indicates no support. Detailed information of all gecko lizards is presented in Table 1.
Data Availability Statement
The following information was supplied regarding data availability:
The raw data are provided in the Supplemental Files and shows mitochondrial genome characterization of all species.