

Abstract
Chronic pain is a debilitating and increasingly common medical problem with few effective treatments. In addition to the direct and indirect economic burden of pain syndromes, the concomitant increase in prescriptions for narcotics has contributed to a sharp rise in deaths associated with drug misuse – the ‘opioid crisis’. Together, these issues highlight the unmet clinical and social need for a new generation of safe, efficacious analgesics. The detection and transmission of pain stimuli is largely mediated by somatosensory afferent fibres of the dorsal root ganglia. These nociceptive cells express an array of membrane proteins that have received significant attention as attractive targets for new pain medications. Among these, a growing body of evidence supports a role for the two‐pore domain potassium (K2P) family of K+ channels. Here, we provide a concise review of the K2P channels, their role in pain biology and their potential as targets for novel analgesic agents.
Abbreviations
- CFA
complete Freund's adjuvant
- DRG
dorsal root ganglia
- SNP
single‐nucleotide polymorphisms
- TRG
trigeminal root ganglia
Introduction
More than a third of all adults in the USA require medical treatment for pain, at a cost that exceeds US $130 billion per year (Johannes et al., 2010; Volkow and McLellan, 2016). For this reason, opioids are among the most commonly prescribed drugs in the USA, with more than 245 million prescriptions dispensed during 2014 alone (Volkow and McLellan, 2016). Although opioids are effective analgesics, particularly for moderate to severe pain, their use is associated with deleterious side effects, including respiratory depression, tolerance and dependence. The prevalence and misuse of these drugs has fuelled the current ‘opioid crisis’, which is characterized by 750 000 emergency medical cases and ~19 000 deaths annually, in the USA alone (Compton et al., 2016). Despite intense and sustained biomedical research and drug‐development efforts, safe, efficacious alternatives to opioids are yet to succeed in clinical trials. This deficit reflects the heterogeneous nature of the disease processes that elicit pain, the complex physiology of pain signalling and the challenges associated with translating laboratory findings into clinically efficacious pharmaceuticals (Hughes et al., 2012; von Hehn et al., 2012). Many new candidate analgesics in development target membrane proteins, particularly GPCRs or ion channels in nociceptive sensory neurons, involved in pain signalling (Waxman and Zamponi, 2014; Yekkirala et al., 2017). Among these, the two‐pore domain potassium (K2P) channels are perhaps the least well explored. Multiple lines of evidence implicate K2P channels in the regulation of multiple types of pain, such as inflammatory, neuropathic, mechanical and thermal, migraine and cancer pain.
A brief introduction to K2P channels
The K2P channels are a diverse family of K+ selective ion channels that contribute to background or leak currents in excitable and non‐excitable tissues (Enyedi and Czirjak, 2010). Although the importance of background K+ flux to cellular physiology had been appreciated for more than a century (Bernstein, 1902), specific proteins that mediate background K+ currents were not identified until the mid‐1990s (Goldstein et al., 1996; Lesage et al., 1996).
In mammals, K2P subunits are encoded by 15 KCNK‐genes and are stratified into six subgroups based on structural and functional similarity (Table 1) (Czirjak and Enyedi, 2010; Plant 2012). Although K2P channels pass currents across the physiological voltage range, their gating is highly regulated (for detailed reviews, please see Goldstein et al., 2001; Honore, 2007; Enyedi and Czirjak, 2010). Physicochemical factors or drugs that inhibit K2P channels, including pH, oxygen‐tension, specific cellular signalling pathways and membrane lipids, increase cellular excitability. In contrast, factors that potentiate currents, such as mechanical stretch, heat, anaesthetics and phosphoinositides, dampen excitability (Chemin et al., 2007a,b). This basic operational paradigm is true for heterologous cells. However, it might not reflect the action of drugs on native cells, tissue or whole animal phenotypes, which has bearing on the development of pharmaceuticals that target K2P channels. In native cells, depolarization might decrease excitability if the magnitude and timescale is sufficient to inactivate voltage‐gated ion channels. At the systems level, overall excitability is determined by the interplay of inhibitory and excitatory circuits, which are in turn dependent on the relative expression levels of a panoply of ion channels and regulatory proteins.
Table 1.
The 15 K2P channels expressed in mammals
| Channel name | Gene name | Common name | Expression in DRG neurons |
|---|---|---|---|
| K2P1 | KCNK1 | TWIK1 | Large/medium fibres, IB4‐ C‐fibrese |
| K2P2 | KCNK2 | TREK1 | TRPV1+, C and medium fibresa , e |
| K2P3 | KCNK3 | TASK1 | TRPV1+ fibresa , e |
| K2P4 | KCNK4 | TRAAK | Highly expresseda , e |
| K2P5 | KCNK5 | TASK2 | Highly expresseda |
| K2P6 | KCNK6 | TWIK2 | Highly expresseda |
| K2P7 | KCNK7 | Kcnk8 | N/A |
| K2P9 | KCNK9 | TASK3 | TRPV1‐, IB4‐ C‐fibresa , e |
| K2P10 | KCNK10 | TREK2 | IB4+ C‐fibresa , d |
| K2P12 | KCNK12 | THIK2 | Small > medium, large fibresa , b , e |
| K2P13 | KCNK13 | THIK1 | Highly expresseda , b |
| K2P15 | KCNK15 | TASK5 | N/A |
| K2P16 | KCNK16 | TALK1 | N/A |
| K2P17 | KCNK17 | TALK2 | N/A |
| K2P18 | KCNK18 | TRESK | Highly expressed in C‐fibres of DRG and TRGa , c , e |
Mammals express 15 distinct K2P channels. Channel names are designated by IUPHAR (K2PX); however, the gene names (KCNKX) and historic names based on biophysical or pharmacological properties remain in common use. Abbreviations: TWIK, tandem of P‐domains in a weak inward rectifying K+ channel; TREK, TWIK‐related K+ channel; TASK, two‐pore domain, acid‐sensitive K+ channel; TRAAK, two‐pore domain‐related arachidonic acid‐activated K+ channel; THIK, two‐pore domain halothane‐inhibited K+ channel; TALK, two‐pore domain alkaline‐activated K+ channel; and TRESK, TWIK‐related spinal cord potassium channel. N/A is not applicable. See Figure 2. K2P6, 7, 12 and 15 channels pass little or no current when expressed in experimental cells. For more information, see the IUPHAR Guide to Pharmacology: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=79. The expression pattern of K2P channels in DRG and TRG is summarized from the following:
Marsh et al. (2012);
Haskins et al. (2017);
Lafrenière and Rouleau (2011);
Acosta et al. (2014);
Pollema‐Mays et al. (2013).
Beyond sequence diversity, the correlation of specific K2P channels with currents in native tissues has been hampered by the broad range of transcript processing and post‐translational modification mechanisms that diversify channel function. Modifications include phosphorylation, SUMOylation, glycosylation and alternative translation initiation (Goldstein et al., 2001; Lopes et al., 2005; Thomas et al., 2008; Plant et al., 2012). Further, a growing body of evidence shows that the functional attributes of K2P channels can be diversified by heterodimerization of specific pairs of subunits to form channels with distinct physiological and pharmacological properties (Czirjak and Enyedi, 2002; Plant et al., 2012; Blin et al., 2014, 2016). Thus, the multiscale nature of physiology can hamper the prediction of how pharmaceuticals designed to act at K2P channels might affect pathophysiological processes or cause off‐target effects.
There are currently no approved drugs that have a selective action at K2P channels. This deficit reflects several factors that have stymied the development of K2P‐selective pharmacophores. Until crystal structures of K2P1 and K2P4 were solved in 2012 (Brohawn et al., 2012; Miller and Long, 2012), the most prominent bottleneck in the development of drugs that act at K2P channels was a lack of structural data. The unique secondary structure of K2P subunits prohibited easy extrapolation from existing data available for voltage‐gated (KV), inwardly rectifying (KIR) K+ channels, or the canonical K+ channel, KcsA (Doyle et al., 1998; Long et al., 2005; Nishida et al., 2007). KcsA, KIR and KV subunits have a single pore‐forming P‐loop with two, two and six transmembrane domains, respectively, while K2P subunits have 4‐transmembrane domains (M1–M4) and two pore‐forming loops – thus, ‘two‐pore domain’ (Goldstein et al., 2001). While KcsA, KIR and KV are tetramers, K2P channels are formed by two subunits that come together to create a single, central K+ selective pore (Figure 1B) (Plant et al., 2017).
Figure 1.
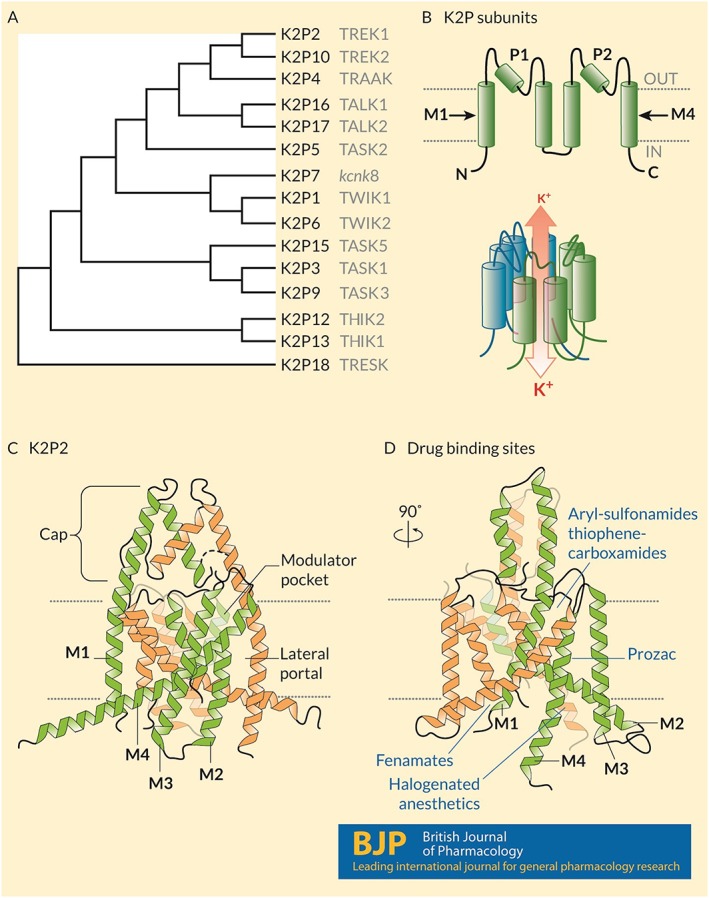
Structure and known drug interaction sites on K2P channels. (A) A phylogenetic tree calculated based on the primary sequences of the 15 K2P subunits expressed in humans. Common names appear in grey text. (B) (Upper) Humans express 15 K2P subunits with similar topologies: Intracellular N‐ and C‐termini, four‐transmembrane domains (M1–M4) and two re‐entrant pore loops (P1 and P2) that contribute to the K+ selective pore. (Lower) Two subunits come together as homodimers, and in some cases heterodimers, to form a K+ selective channel. Under physiological conditions, K+ ions move through the channel down their electrochemical gradients from the inside to the outside of the cell. (C) The crystal structure of mouse K2P2 (Lolicato et al., 2017) showing the architecture of the channel including the arrangement of the transmembrane domains, the extracellular cap above the plane of the membrane that bifurcates the K+ permeation pathway, the lateral portal and the modulator pocket. (D) A 90° rotation in the pose of K2P2 showing the location of four distinct drug binding sites: fenamates bind to the N‐terminus; aryl‐sulfonamides and thiophene‐carboxamides interact with the modulator pocket; fluoxetine (Prozac) interacts at the lateral port; and halogenated anaesthetics require the proximal C‐terminus of the channel and interact in a manner that involves Gαq proteins.
The most prominent structural motif observed in K2P channels is an extracellular cap‐domain formed by the first external loop of each subunit that extends above the outer leaflet of the plasma membrane, bifurcating the K+ permeation pathway at the outer mouth of the pore (Figure 1C, D). First described in the structures of K2P1 and K2P4 channels (Brohawn et al., 2012; Miller and Long, 2012), the cap‐domain is also apparent in the structures of K2P2 and K2P10 (Dong et al., 2015; Lolicato et al., 2017) and is thus expected to be observed throughout the K2P‐family. The position of the cap‐domain is proposed to create steric hindrance that renders K2P channels and background K+ currents in native cells largely insensitive to classical toxin peptides and pore blocking molecules like tetraethylammonium ions that act by blocking the pore of delayed‐rectifier KV channels (Niemeyer et al., 2016).
K2P‐structures also revealed two lateral portals that expose the channel's pore to the interior of the membrane lipid, an observation that led to a proposed mechanism for the mechanosensitivity of K2P2 and K2P4 (Brohawn et al., 2014) (Figure 1C,D). In addition to visualizing channel architecture, these detailed snapshots elucidate how K2P channels interact with drugs. The structure of K2P10 was solved in complex with norfluoxetine, the active metabolite of the selective serotonin reuptake (SERT) inhibitor fluoxetine, bound within the lateral portal (Dong et al., 2015). Recent work by Lolicato et al. (2017) revealed a novel druggable site, dubbed the ‘modulator pocket’, located between the first pore loop and the M4‐helix of each subunit of K2P2 and K2P10 (Figure 1C,D).
K2P channels are expressed throughout the body, with each channel type having a distinct expression profile. The observation that several KCNK mRNA transcripts are highly expressed in somatosensory fibres (Marsh et al., 2012; Pollema‐Mays et al., 2013) led to the proposal that K2P channels regulate pain signals by determining the excitability of specific nerve fibres of dorsal and trigeminal root ganglia (DRG and TRG) (Plant, 2012).
Expression of K2P channels in dorsal root ganglia neurons
DRG comprise a heterologous population of primary afferent, somatosensory nociceptive neurons that are broadly divided into Aδ‐, Aβ‐ and C‐fibres. Aδ‐fibres are lightly myelinated, have intermediate cell body sizes and respond to acute, localized pain. Aβ‐fibres have larger cell bodies with a small nociceptive population. C‐fibres are unmyelinated, have the smallest cell bodies in DRG and respond to diffuse pain and itch (Tsunozaki and Bautista, 2009). C‐fibres are designated as peptidergic, based on the expression of CGRP or Substance P, and non‐peptidergic, based on the binding of isolectin B4 (Le Pichon and Chesler, 2014).
Numerous studies (Table 1) have identified mRNA transcripts for several members of the K2P channel family in DRG and TRG neurons with distinct expression patterns in Aδ, Aβ and C‐fibres (Figure 2) (Lafrenière and Rouleau, 2011; Marsh et al., 2012; Pollema‐Mays et al., 2013; Acosta et al., 2014; Mathie and Veale, 2015; Haskins et al., 2017).
Figure 2.
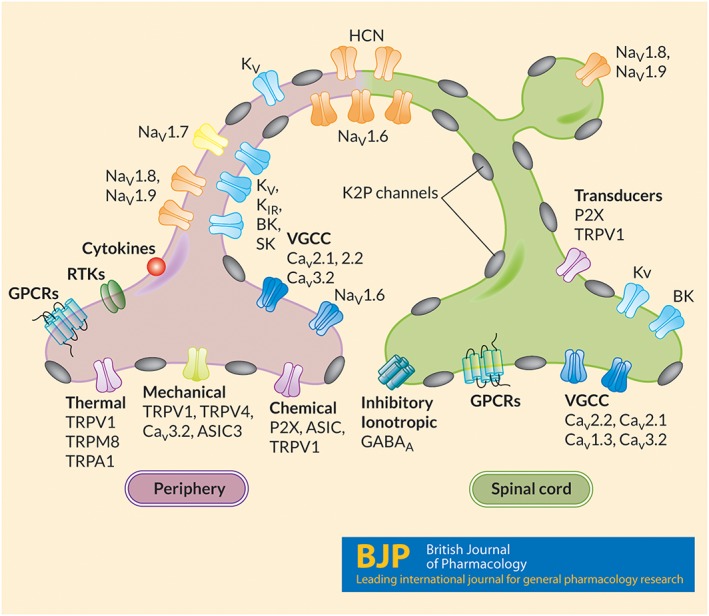
Somatosensory nociceptive fibres express several K2P channels. Somatosensory fibres in DRG and TRG are a heterogenous population of nociceptive cells that detect, integrate and transmit sensory information and pain signals. The excitability and sensory properties of peptidergic and non‐peptidergic C‐fibres as well as Aβ and Aδ‐fibre primary afferent cells are, in part, dependent on the expression patterns of numerous types of ion channels. Several K2P channels, including K2P1–6, K2P9–13 and K2P18 are highly expressed in populations of DRG and TRG somatosensory neurons, where they regulate excitability and responses to numerous pain stimuli.
Role of K2P channels in inflammatory pain
K2P channels appear to be important in the mitigation of inflammatory pain, perhaps because transcript levels of several K2P subunits are significantly altered during inflammation. In an animal model of induced inflammation using complete Freund's adjuvant (CFA), significant changes in mRNA encoding K2P3, K2P9, K2P12, K2P16 and K2P18 subunits were correlated with a decrease in spontaneous foot lifting behaviour (Marsh et al., 2012). Pain responses also correlate with changes in K2P12/13 channel expression in animal models of inflammatory injury. Thus, siRNA‐mediated knockdown of K2P12/13 channels in mouse models of CFA‐induced inflammation resulted in increased nocifensive behaviour (Haskins et al., 2017). K2P10 channels are involved in PGE2‐mediated hyperalgesia with K2P10−/− mice showing an absence of nocifensive behaviour in response to hypertonic saline injections after PGE2 sensitization (Pereira et al., 2014).
K2P18 channels in migraine
Genetic analysis of KCNK18, the gene that encodes for the K2P18 channel, identified several mutations, some of which were associated with migraine (Lafrenière et al., 2010). Rainero et al. (2014) identified several variants of KCNK18 in an Italian cohort of patients suffering from migraine with and without aura. Using in silico models, they predict that the C110R, S178T, S231P and F372L mutations in K2P18 may have deleterious effects on channel function and contribute to migraine pathogenesis (Rainero et al., 2014). Some mutations, like S231P, identified in both control samples and from unrelated migraine probands, were subsequently shown to have no overt effect on channel function (Lafrenière et al., 2010; Andres‐Enguix et al., 2012). In contrast, A34V and C110R mutations result in decreased channel currents, possibly due to their proximity to the channel pore (Andres‐Enguix et al., 2012). K2P18‐A34V channels are linked to the development of typical migraines while a frameshift mutation, F139WfsX24, that truncates K2P18 at M2, is implicated in familial migraine with aura (Lafrenière et al., 2010). Mutant/truncated K2P18 subunits have a dominant‐negative phenotype, suppressing the activity of wild‐type K2P18 channels in heterologous cell systems (Lafrenière and Rouleau, 2011; Andres‐Enguix et al., 2012) and cultured trigeminal neurons (Liu et al., 2013). In support of these reports, the overexpression of K2P18 in small TRG fibres inhibits action potential firing (Guo and Cao, 2014), presumably through K2P18‐mediated currents. A growing body of evidence suggests that many cases of migraine are polygenic in nature (Anttila et al., 2018; Gormley et al., 2018), leading to the notion that genetic variants in KCNK18 might act as risk factors, rather than as a single, penetrant ‘channelopathy’. However, the notion that activators of K2P18 channels could act as anti‐migraine medications is intriguing and worthy of further investigation.
K2P channels in neuropathic pain
K2P18 channels mediate the largest component of the background K+ current in DRG neurons (Tulleuda et al., 2011; Plant, 2012), but the channel is down‐regulated in animals with spinal cord injury. This phenotype is associated with significantly decreased thresholds for withstanding mechanical pain (allodynia), activation of astrocytes and microglia and up‐regulation of connexin‐36 and connexin‐43, components of neuronal and astrocyte‐oligodendrocyte gap junctions respectively (Zhou et al., 2017). Down‐regulation of K2P18 in nerve injury occurs in combination with an increase in MAPK, ERK and p38, which are also implicated in the pathogenesis of neuropathic pain. This phenotype can be reproduced in vivo by shRNA knockdown of K2P18 channels and is rescued when K2P18 channels are overexpressed using adenoviral vectors (Zhou et al., 2017). Similarly, increased nocifensive behaviour is observed after complete axotomy of the sciatic nerve and in K2P18−/− animals (Tulleuda et al., 2011). Further support for the role of decreased K2P18 channel current activity in neuropathic pain and nerve injury comes from the observation that activation of K2P18 balances depolarizing stimuli through TRPV1 channels during nerve injury through lysophosphatidic acid signalling in DRG neurons (Kollert et al., 2015).
Changes in the activity of other K2P channels are also involved in the pathology of nerve injury. In spared sciatic nerve injury, K2P9 and K2P1 channels were down‐regulated in L4‐L5 DRG's ipsilateral to the nerve lesion while K2P3 expression remained constant. The K2P9 channels returned to homeostatic levels within weeks, although the K2P1 channels remained depleted for months after the initial injury (Pollema‐Mays et al., 2013). In a similar model, K2P13 channel knockdown increased the response to inflammation in the form of longer spontaneous foot lifting times. Of note, K2P2−/− and K2P2/4/10−/− triple knockout mice showed significantly increased sensitivity to mechanical stimuli compared to wild‐type animals but with no difference between knockout models, suggesting that K2P10 channels might play a protective role against mechanical allodynia (Pereira et al., 2014).
The TREK‐family of channels in thermal and mechanical pain
In line with the role of leak K+ channels in reducing cellular excitability, expression of TREK‐family channels (K2P2, K2P4 and K2P10) decreased the thermal sensitivity of DRG neurons. In a model of unilateral peripheral mononeuropathy, DRG from K2P2−/− mice showed increased excitability when exposed to temperatures in the 30–45°C range (Alloui et al., 2006). In a similar animal model of neuropathy, K2P2/4−/− mice showed increased cold sensitivity at 15–20°C, compared with K2P4−/− and wild‐type animals (Noël et al., 2009). C‐fibres taken from K2P2−/− and K2P2/4/10−/− animals fire action potentials at lower temperatures than wild‐type mice and display increased firing frequencies in the 30–50°C temperature range. K2P10−/− mice also showed increased sensitivity to temperatures (40–45°C) that were agreeable to wild‐type animals (Pereira et al., 2014). K2P10 channels are also implicated in the mediation of cold hypersensitivity following treatment with oxaliplatin, a drug commonly used to treat colon cancer (Pereira et al., 2014). K2P4−/− animals have a decreased heat threshold in comparison to wild‐type animals. Heat‐induced hyperalgesia in K2P2−/− and K2P2/4/10−/− mice is congruent with inflammation and neuropathy‐induced hyperalgesia in wild‐type mice (Noël et al., 2009), probably indicating a decrease in the expression of K2P2 and K2P4 channels after nerve injury. Cold sensing C‐fibres have bimodal thresholds with one population responding to temperatures above 17°C while the other group of fibres is pain‐sensing and only fires at temperatures below 17°C (Reid, 2005). In K2P2/4−/− mice, the latter population of fibres is depleted and the higher cold threshold fibres predominate (Noël et al., 2009).
K2P9 channels in cancer
The expression of several K2P channels is altered in different cancers (Williams et al., 2013). KCNK9, the gene encoding K2P9, is reportedly a proto‐oncogene, promoting tumour growth and hypoxia resistance (Dookeran and Auer, 2017). Conversely, down‐regulation of K2P9 channels through PKC activation, increased cancer metastasis by enhancing cell migration (Lee et al., 2012). K2P9 channels appear to have a dual role as drug targets in decreasing breast cancer pain as well as arresting cancer metastasis. Two single‐nucleotide polymorphisms (SNP) in the K2P9 gene (KCNK9‐rs3780039 and rs11166921) have been linked to an increase in the incidence of pre‐operative breast pain, in breast cancer patients. Individuals homozygous for KCNK9‐rs11166921 were more than twice as likely to report breast pain before surgery while individuals heterozygous and homozygous for KCNK9‐rs3780039 were twice as likely to experience breast pain (Langford et al., 2014). It is not known if this SNP affects K2P9 channel function or whether the SNP results in a dominant negative phenotype. While the mechanistic association between K2P9 and breast cancer pain is unclear, it is possible that variant K2P9 channels in somatosensory fibres are more responsive to signalling inflammatory factors in the penumbra of the tumour (Langford et al., 2014). Further, it is possible that these intronic SNPs may affect channel expression and warrant further investigation.
Pain management through K2P channels?
Drugs and small molecules
The number of drugs and small molecules recognized to interact with K2P channels is steadily increasing (Figure 1D). Halogenated volatile anaesthetics such as isoflurane and sevoflurane activate several K2P channels, including K2P2, K2P3, K2P4, K2P9 and K2P18, perhaps via the disruption of an inhibitory interaction between Gαq and the proximal C‐terminus of the channel (Chen et al., 2006). Recent data suggest that DAG produced following activation of Gq‐coupled receptors is the active inhibitory factor, rather than Gαq itself (Wilke et al., 2014). An interaction between K2P channels and DAG, however, does not preclude an interaction with Gq and remains an open question in the field. Volatile anaesthetics are proposed to promote membrane hyperpolarization via an increase in channel open probability and K+ flux (Patel et al., 1999; Plant, 2012). Thus, activation of K2P channels, in addition to GABAA receptors, is proposed to play an important role in the mediation of anaesthesia and analgesia by halothane‐like agents.
TREK‐family channels are inhibited by the SERT inhibitors, fluoxetine and norfluoxetine, via an interaction in the lateral‐portal – see Introduction (Dong et al., 2015). Clinical evidence supports the analgesic activity of numerous anti‐depressants (Obata, 2017), whereas inhibition of K2P channels is expected to augment pain signalling (Kennard et al., 2005). Hence, further studies will be required to determine the clinical pharmacology of K2P channel‐antidepressant interactions.
The fenamate class of non‐steroidal anti‐inflammatory drugs are proposed to mediate their analgesic action through the inhibition of pro‐excitatory ion channels while selectively activating K+ channels, including K2P channels (Takahira et al., 2005). This effect is proposed to involve an interaction with the N‐terminus of K2P channels (Veale et al., 2014).
Recently, Lolicato et al. identified a new druggable site, the K2P modulator pocket in K2P2, 4 and 10 channels. The aryl‐sulfonamide, ML335 and the thiophene‐carboxamide ML402 activate the channels via specific cation–π and π–π interactions with residues behind the K+ selectivity filter (Lolicato et al., 2017).
K2P2 channel activation by the μ‐opioid receptor is a component of morphine‐induced signalling and is integral to its analgesic effects but does not contribute to the adverse effects of morphine (Devilliers et al., 2013). In keeping with this finding, a series of acrylic acid compounds that selectively activate K2P2 channels has recently been reported to show significant pain mitigation in vivo (Vivier et al., 2017). Similarly, a selective activator of K2P2 and K2P10, GI‐530139, has been reported to be effective in hyperpolarizing DRG neurons by increasing channel activity (Loucif et al., 2018).
Natural compounds
In addition to low MW compounds, the activity of K2P channels is also modulated by several naturally occurring compounds. The analgesic effects of traditional medicines like Schezuan peppers and aristolochic acid have been attributed to their modulation of K2P channels. Schezuan peppers have been used as analgesics in folk medicine, due to their numbing ability and as flavouring agents to elicit a tingling‐like pungency. Hydroxy‐α‐sanshool, the active component of Schezuan peppers, mediates these effects by activating nociceptive and light‐touch sensory neurons; depolarization is mediated by inhibition of K2P3, 9 and 18 channels (Bautista et al., 2008). In vivo studies with knockout animals may yield further information as to the range and selectivity of these effects. Aristolochic acid is a natural remedy used in the Balkan region for pain relief potential via inhibition of K2P18 and activation of K2P2 and K2P10 channels (Veale and Mathie, 2016). However, the pharmacology of aristolochic acid is complex, with links to nephritis and carcinogenesis (Chen et al., 2012; Schmeiser et al., 2014).
Ion channels in pain mediation
K2P channels are a relatively recent addition to a list of ion channels that serve as potential pharmaceutical targets for novel analgesics (Figure 2). The exploration of ion channels as targets for pain management has been ongoing for some time but has resulted in limited progress in producing novel analgesics. Here, we provide a concise summary of progress, but more extensive reviews can be found elsewhere (Waxman and Zamponi, 2014; Yekkirala et al., 2017).
Transient receptor potential (TRP) channels
The peripheral termini of sensory neurons innervate target tissues and express several types of transducer ion channels including numerous members of the TRP family. Of these, TRPV1 channels are activated by acidification, temperatures greater than ~42°C, membrane depolarization, several arachidonic acid metabolites and capsaicin, the pungent chemical in hot chilli peppers (Caterina et al., 1997; Julius, 2013). TRPV1 channel antagonists have been proposed as analgesic agents. However, in clinical trials, blockade of TRPV1 channels modulated core body temperature, providing a significant problem in the development of TRPV1 channel antagonists (Skerratt and West, 2015). Interestingly, TRPV1 channel agonists such as capsaicin can produce paradoxical analgesia by causing channel desensitization, facilitating the use of capsaicin formulations in the treatment of osteoarthritis pain (Alexander et al., 2017a). Recently, a blocker of TRPM8 channels has been described in the attenuation of cold‐related pain (Andrews et al., 2015). A gain of function mutation of TRPA1 channels has been linked to familial episodic pain syndrome, and at least two candidate drugs targeting this channel are in clinical trials for the treatment of diabetic neuropathy and inflammatory pain (Skerratt and West, 2015).
Voltage‐gated calcium channels
Calcium channels are crucial to the transmission of pain signals from the primary sensory neurons to neurons in the dorsal ganglia (Yekkirala et al., 2017). Blockade of CaV2.2 channels by ziconitide (a formulation of ω‐conotoxin MVIIA) administered intrathecally provides potent analgesia, under conditions where opioids are counter‐indicated. However, ziconitide‐induced analgesia is short‐lived and fraught with several side effects, leading to restricted use of this drug (Patel et al., 2018). Gabapentin and pregabalin are both inhibitors of α2δ subunit‐containing CaV channels, and the latter is currently the drug‐of‐choice in the treatment of pain associated with diabetic neuropathy (Skerratt and West, 2015). Several new compounds targeting calcium channels have since entered clinical trials but have failed to reproduce the efficacy displayed in preclinical models. The applications and limitations of calcium channels in pain pathophysiology are dealt with in detail by Patel et al. (2018).
Voltage‐gated sodium channels
Significant effort is underway to identify novel NaV channel blockers that could dampen the excitability of somatosensory neurons. Similarly, medications that are currently available for the treatment of other NaV channel‐related maladies are being investigated for their utility as analgesics. For example, the anti‐arrhythmic agent, ranolazine, also decreases mechanical allodynia and cold hypersensitivity after nerve injury (Gould et al., 2009).
Significant interest in the role of NaV channels in pain signalling arises from the observation that specific mutations in the SCN9A gene that encodes the NaV1.7 channel result in congenital pain indifference syndromes (Cox et al., 2006) as well as increased pain sensitivity in the case of loss‐of‐function mutations (Mathie, 2010). Several compounds targeting NaV1.7 channels are currently undergoing clinical trials for their efficacy in managing neuropathic pain (Skerratt and West, 2015). NaV1.8 channels are under investigation for roles in inflammatory and neuropathic pain, particularly mechanical allodynia and heat hypersensitivity associated with inflammation (Joshi et al., 2006; Joshi and Honore, 2006; Wang et al., 2011). A ligand targeting NaV1.8 channels has, however, failed to show efficacy in a post‐surgical dental pain clinical trial, raising questions about the role of these channels in pain pathogenesis (Wang et al., 2011). NaV1.9 channels are also expressed in peripheral neurons and has been proposed as a druggable target for the treatment of hyperalgesia following inflammation. However, low channel expression in heterologous systems is proving to be a significant hindrance to research efforts in this direction (Yekkirala et al., 2017). Of note, a recent report by Osteen et al. (2016) revealed a role for NaV1.1 channels in mechanical pain, including in a mouse model of irritable bowel syndrome.
Purinoreceptors
ATP‐activated P2X receptors are cation channels that contribute to pain signalling by enhancing neuronal excitation (Mathie, 2010). The P2X4 subtype is involved in the mediation of neuropathic pain via the activation of microglia and the subsequent release of BDNF and pro‐inflammatory cytokines which are known to exacerbate pain after nerve injuries (Ulmann et al., 2008; Mathie, 2010).
Acid‐sensing ion channels
Acid‐sensing ion channels (ASICs) are cation‐permeable channels that are activated by extracellular acidic pH. The anti‐hypertensive and diuretic drug, amiloride, can inhibit various ASIC subunits and alleviated cutaneous and migraine pain. A corpus of literature links ASIC channel blockade to analgesia and is succinctly summarized in Wemmie et al. (2013).
Challenges and future perspectives on the pharmacology of K2P channels
With the renewed effort to develop non‐opioid pain medications, K2P channels are evolving into exciting potential targets for novel analgesics. Although K2P channels have a paucity of specific blockers and activators to guide drug development, two decades of study have provided powerful tools for understanding the pathophysiological relevance of these proteins. More recently, this nascent field has been invigorated by the opening of an exciting new frontier in K2P channel research – combining drug development with atomic resolution structures and models of specific K2P channels. We predict that this approach will lead to fruitful advances in the understanding of structure–activity relationships for K2P channels and the next generation of K2P‐pharmacophores with clinical application as analgesic agents.
Despite these advances, significant questions remain unanswered. Perhaps chief among these is the safety of drugs that target K2P channels. Although the expression of K2P18 channels appears to be restricted to DRG and TRG fibres, other pain‐related K2P channels are expressed in multiple tissues. Broad tissue expression could lead to off‐target effects of drugs that target K2P channels. For example, K2P2 channels are highly expressed in GABAergic interneurons, the prefrontal cortex, the hippocampus and dorsal raphe nuclei neurons, as well as in the cardiovascular system and gastrointestinal tract (Fink et al., 1996; Talley et al., 2001; Honore, 2007; Thomas et al., 2008; Plant et al., 2012). Several studies using knockout animal studies have been undertaken to provide specific information about the expression and function of individual K2P channels. However, the knockdown of one channel may cause up‐regulation of other K2P channels and necessitates double or triple knockout models or perhaps conditional knockout models, as well as rigorous controls to validate study results. The distribution and regulation of the different K2P channels is catalogued extensively elsewhere (Enyedi and Czirjak, 2010; Plant et al., 2017).
More rigorous studies in heterologous and primary cell systems are needed to define the composition of K2P subunits in specific cell types during development and in the context of specific pathophysiological states. Concurrently, evidence is mounting to support the observation first reported by Czirjak and Enyedi (2002) that at least some K2P channels are heterodimers. Because of the reported differences in function of heterodimers versus homodimeric K2P channels, it is possible that the pain phenotype will be altered or that drug discovery efforts will require modification to accommodate the expanded role of homodimeric and heterodimeric K2P channels in the mediation of different types of pain.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d,e).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors are grateful to Diomedes Logothetis and the Logothetis lab for vibrant discussion and feedback. K.G. is supported by Northeastern University, Bouvé College of Health Sciences.
Gada, K. , and Plant, L. D. (2019) Two‐pore domain potassium channels: emerging targets for novel analgesic drugs: IUPHAR Review 26. British Journal of Pharmacology, 176: 256–266. 10.1111/bph.14518.
This article, contributed by members of the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC‐IUPHAR) subcommittee for the Two‐pore domain potassium (K2P) channels, confirms the existing nomenclature for these channels, and reviews our current understanding of their structure, pharmacology and functions, and likely physiological roles in health and disease. More information on these channels can be found in the Concise Guide to PHARMACOLOGY (http://onlinelibrary.wiley.com/doi/10.1111/bph.13884/full), and in the corresponding open access IUPHAR/BPS Guide to PHARMACOLOGY database (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=79).
References
- Acosta C, Djouhri L, Watkins R, Berry C, Bromage K, Lawson SN (2014). TREK2 expressed selectively in IB4‐binding C‐fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci 34: 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters J, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J et al (2006). TREK‐1, a K+ channel involved in polymodal pain perception. EMBO J 25: 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres‐Enguix I, Shang L, Stansfeld PJ, Morahan JM, Sansom MSP, Lafrenière RG et al (2012). Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci Rep 2: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews MD, Af Forselles K, Beaumont K, Galan SR, Glossop PA, Grenie M et al (2015). Discovery of a selective TRPM8 antagonist with clinical efficacy in cold‐related pain. ACS Med Chem Lett 6: 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Bulik‐Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L et al (2018). Analysis of shared heritability in common disorders of the brain. Science 360: eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Sigal YM, Milstein AD, Garrison JL, Zorn JA, Tsuruda PR et al (2008). Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two‐pore potassium channels. Nat Neurosci 11: 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein J (1902). Untersuchungen zur Thermodynamik der bioelektrischen Ströme. Archiv für die gesamte Physiologie des Menschen und der Tiere 92: 521–562. [Google Scholar]
- Blin S, Ben Soussia I, Kim EJ, Brau F, Kang D, Lesage F et al (2016). Mixing and matching TREK/TRAAK subunits generate heterodimeric K2P channels with unique properties. Proc Natl Acad Sci U S A 113: 4200–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin S, Chatelain FC, Feliciangeli S, Kang D, Lesage F, Bichet D (2014). Tandem pore domain halothane‐inhibited K+ channel subunits THIK1 and THIK2 assemble and form active channels. J Biol Chem 289: 28202–28212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, del Marmol J, MacKinnon R (2012). Crystal structure of the human K2P TRAAK, a lipid‐ and mechano‐sensitive K+ ion channel. Science 335: 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, Su Z, MacKinnon R (2014). Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci U S A 111: 3614–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Delmas P, Sachs F, Lazdunski M, Honore E (2007a). Regulation of the mechano‐gated K2P channel TREK‐1 by membrane phospholipids. Curr Top Membr 59: 155–170. [DOI] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Duprat F, Sachs F, Lazdunski M, Honore E (2007b). Up‐ and down‐regulation of the mechano‐gated K(2P) channel TREK‐1 by PIP (2) and other membrane phospholipids. Pflugers Arch 455: 97–103. [DOI] [PubMed] [Google Scholar]
- Chen CH, Dickman KG, Moriya M, Zavadil J, Sidorenko VS, Edwards KL et al (2012). Aristolochic acid‐associated urothelial cancer in Taiwan. Proc Natl Acad Sci U S A 109: 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B et al (2006). Inhibition of a background potassium channel by Gq protein alpha‐subunits. Proc Natl Acad Sci U S A 103: 3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton WM, Jones CM, Baldwin GT (2016). Relationship between nonmedical prescription‐opioid use and heroin use. N Engl J Med 374: 154–163. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K et al (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444: 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirjak G, Enyedi P (2002). Formation of functional heterodimers between the TASK‐1 and TASK‐3 two‐pore domain potassium channel subunits. J Biol Chem 277: 5426–5432. [DOI] [PubMed] [Google Scholar]
- Czirjak G, Enyedi P (2010). TRESK background K(+) channel is inhibited by phosphorylation via two distinct pathways. J Biol Chem 285: 14549–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilliers M, Busserolles J, Lolignier S, Deval E, Pereira V, Alloui A et al (2013). Activation of TREK‐1 by morphine results in analgesia without adverse side effects. Nat Commun 4: 2941. [DOI] [PubMed] [Google Scholar]
- Dookeran KA, Auer P (2017). The Emerging Role of Two‐Pore Domain Potassium Channels in Breast Cancer. J Glob Epidemiol Environ Health 2017: 27–36. [Google Scholar]
- Dong YY, Pike ACW, Mackenzie A, McClenaghan C, Aryal P, Dong L et al (2015). K2P channel gating mechanisms revealed by structures of TREK‐2 and a complex with Prozac. Science 347: 1256–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL et al (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280: 69–77. [DOI] [PubMed] [Google Scholar]
- Enyedi P, Czirjak G (2010). Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev 90: 559–605. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C et al (1996). Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15: 6854–6862. [PMC free article] [PubMed] [Google Scholar]
- Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N (2001). Potassium leak channels and the KCNK family of two‐P‐domain subunits. Nat Rev Neurosci 2: 175–184. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Price LA, Rosenthal DN, Pausch MH (1996). ORK1, a potassium‐selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 93: 13256–13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley P, Kurki MI, Hiekkala ME, Veerapen K, Happola P, Mitchell AA et al (2018). Common variant burden contributes to the familial aggregation of migraine in 1,589 families. Neuron 98 (743–753): e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould HJ 3rd, Garrett C, Donahue RR, Paul D, Diamond I, Taylor BK (2009). Ranolazine attenuates behavioral signs of neuropathic pain. Behav Pharmacol 20: 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Cao Y‐Q (2014). Over‐expression of TRESK K+ channels reduces the excitability of trigeminal ganglion nociceptors. PLOS ONE 9: e87029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskins W, Benitez S, Mercado JM, Acosta CG (2017). Cutaneous inflammation regulates THIK1 expression in small C‐like nociceptor dorsal root ganglion neurons. Mol Cell Neurosci 83: 13–26. [DOI] [PubMed] [Google Scholar]
- Honore E (2007). The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci 8: 251–261. [DOI] [PubMed] [Google Scholar]
- Hughes JP, Chessell I, Malamut R, Perkins M, Backonja M, Baron R et al (2012). Understanding chronic inflammatory and neuropathic pain. Ann N Y Acad Sci 1255: 30–44. [DOI] [PubMed] [Google Scholar]
- Johannes CB, Le TK, Zhou X, Johnston JA, Dworkin RH (2010). The prevalence of chronic pain in United States adults: results of an Internet‐based survey. J Pain 11: 1230–1239. [DOI] [PubMed] [Google Scholar]
- Joshi SK, Honore P (2006). Animal models of pain for drug discovery. Expert Opin Drug Discov 1: 323–334. [DOI] [PubMed] [Google Scholar]
- Joshi SK, Mikusa JP, Hernandez G, Baker S, Shieh CC, Neelands T et al (2006). Involvement of the TTX‐resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post‐operative, pain states. Pain 123: 75–82. [DOI] [PubMed] [Google Scholar]
- Julius D (2013). TRP channels and pain. Annu Rev Cell Dev Biol 29: 355–384. [DOI] [PubMed] [Google Scholar]
- Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A (2005). Inhibition of the human two‐pore domain potassium channel, TREK‐1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol 144: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollert S, Dombert B, Döring F, Wischmeyer E (2015). Activation of TRESK channels by the inflammatory mediator lysophosphatidic acid balances nociceptive signalling. Sci Rep 5: 12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrenière RG, Cader MZ, Poulin J‐F, Andres‐Enguix I, Simoneau M, Gupta N et al (2010). A dominant‐negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16: 1157–1160. [DOI] [PubMed] [Google Scholar]
- Lafrenière RG, Rouleau GA (2011). Migraine: role of the TRESK two‐pore potassium channel. Int J Biochem Cell Biol 43: 1533–1536. [DOI] [PubMed] [Google Scholar]
- Langford DJ, West C, Elboim C, Cooper BA, Abrams G, Paul SM et al (2014). Variations in potassium channel genes are associated with breast pain in women prior to breast cancer surgery. J Neurogenet 28: 122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pichon CE, Chesler AT (2014). The functional and anatomical dissection of somatosensory subpopulations using mouse genetics. Front Neuroanat 8: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GW, Park HS, Kim EJ, Cho YW, Kim GT, Mun YJ et al (2012). Reduction of breast cancer cell migration via up‐regulation of TASK‐3 two‐pore domain K+ channel. Acta Physiol (Oxf) 204: 513–524. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G et al (1996). TWIK‐1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15: 1004–1011. [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xiao Z, Ren F, Guo Z, Chen Z, Zhao H et al (2013). Functional analysis of a migraine‐associated TRESK K+ channel mutation. J Neurosci 33: 12810–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA et al (2017). K2P2.1 (TREK‐1)‐activator complexes reveal a cryptic selectivity filter binding site. Nature 547: 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R (2005). Crystal structure of a mammalian voltage‐dependent Shaker family K+ channel. Science 309: 897–903. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Rohacs T, Czirjak G, Balla T, Enyedi P, Logothetis DE (2005). PIP2 hydrolysis underlies agonist‐induced inhibition and regulates voltage gating of two‐pore domain K+ channels. J Physiol 564: 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loucif AJC, Saintot PP, Liu J, Antonio Brett M, Zellmer Shannon G, Yoger K et al (2018). GI‐530159, a novel, selective, mechanosensitive two‐pore‐domain potassium (K2P) channel opener, reduces rat dorsal root ganglion neuron excitability. Br J Pharmacol 175: 2272–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh B, Acosta C, Djouhri L, Lawson SN (2012). Leak K(+) channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol Cell Neurosci 49: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A (2010). Ion channels as novel therapeutic targets in the treatment of pain. J Pharm Pharmacol 62: 1089–1095. [DOI] [PubMed] [Google Scholar]
- Mathie A, Veale EL (2015). Two‐pore domain potassium channels: potential therapeutic targets for the treatment of pain. Pflugers Arch 467: 931–943. [DOI] [PubMed] [Google Scholar]
- Miller AN, Long SB (2012). Crystal structure of the human two‐pore domain potassium channel K2P1. Science 335: 432–436. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Cid LP, Gonzalez W, Sepulveda FV (2016). Gating, regulation, and structure in K2P K+ channels: in varietate concordia? Mol Pharmacol 90: 309–317. [DOI] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R (2007). Crystal structure of a Kir3.1‐prokaryotic Kir channel chimera. EMBO J 26: 4005–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël J, Zimmermann K, Busserolles J, Deval E, Alloui A, Diochot S et al (2009). The mechano‐activated K+ channels TRAAK and TREK‐1 control both warm and cold perception. EMBO J 28: 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata H (2017). Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci 18: 2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X et al (2016). Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534: 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M (1999). Inhalational anesthetics activate two‐pore‐domain background K + channels. Nat Neurosci 2: 422–426. [DOI] [PubMed] [Google Scholar]
- Patel R, Montagut‐Bordas C, Dickenson AH (2018). Calcium channel modulation as a target in chronic pain control. Br J Pharmacol 175: 2173–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V, Busserolles J, Christin M, Devilliers M, Poupon L, Legha W et al (2014). Role of the TREK2 potassium channel in cold and warm thermosensation and in pain perception. Pain 155: 2534–2544. [DOI] [PubMed] [Google Scholar]
- Plant L (2012). A role for K2P channels in the operation of somatosensory nociceptors. Front Mol Neurosci 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Bayliss DA, Minor DL, Jr. , Czirják G, Enyedi P, Lesage F et al (2017). Two P domain potassium channels, introduction. Available from http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=79 (accessed 11/17/2018).
- Plant LD, Zuniga L, Araki D, Marks JD, Goldstein SA (2012). SUMOylation silences heterodimeric TASK potassium channels containing K2P1 subunits in cerebellar granule neurons. Sci Signal 5: ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollema‐Mays SL, Centeno MV, Ashford CJ, Apkarian AV, Martina M (2013). Expression of background potassium channels in rat DRG is cell‐specific and down‐regulated in a neuropathic pain model. Mol Cell Neurosci 57: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainero I, Rubino E, Gallone S, Zavarise P, Carli D, Boschi S et al (2014). KCNK18 (TRESK) genetic variants in Italian patients with migraine. Headache 54: 1515–1522. [DOI] [PubMed] [Google Scholar]
- Reid G (2005). ThermoTRP channels and cold sensing: what are they really up to? Pflugers Arch 451: 250–263. [DOI] [PubMed] [Google Scholar]
- Schmeiser HH, Nortier JL, Singh R, Gamboa da Costa G, Sennesael J, Cassuto‐Viguier E et al (2014). Exceptionally long‐term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int J Cancer 135: 502–507. [DOI] [PubMed] [Google Scholar]
- Skerratt SE, West CW (2015). Ion channel therapeutics for pain. Channels (Austin) 9: 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahira M, Sakurai M, Sakurada N, Sugiyama K (2005). Fenamates and diltiazem modulate lipid‐sensitive mechano‐gated 2P domain K+ channels. Pflugers Arch 451: 474–478. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Lei QB, Kim D, Bayliss DA (2001). CNS distribution of members of the two‐pore‐domain (KCNK) potassium channel family. J Neurosci 21: 7491–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Plant LD, Wilkens CM, McCrossan ZA, Goldstein SA (2008). Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 58: 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunozaki M, Bautista DM (2009). Mammalian somatosensory mechanotransduction. Curr Opin Neurobiol 19: 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X (2011). TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain 7: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F et al (2008). Up‐regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci 28: 11263–11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veale EL, Al‐Moubarak E, Bajaria N, Omoto K, Cao L, Tucker SJ et al (2014). Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol Pharmacol 85: 671–681. [DOI] [PubMed] [Google Scholar]
- Veale EL, Mathie A (2016). Aristolochic acid, a plant extract used in the treatment of pain and linked to Balkan endemic nephropathy, is a regulator of K2P channels. Br J Pharmacol 173: 1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier D, Soussia IB, Rodrigues N, Lolignier S, Devilliers M, Chatelain FC et al (2017). Development of the first two‐pore domain potassium channel TWIK‐related K+ channel 1‐selective agonist possessing in vivo antinociceptive activity. J Med Chem 60: 1076–1088. [DOI] [PubMed] [Google Scholar]
- Volkow ND, McLellan AT (2016). Opioid abuse in chronic pain – misconceptions and mitigation strategies. N Engl J Med 374: 1253–1263. [DOI] [PubMed] [Google Scholar]
- von Hehn CA, Baron R, Woolf CJ (2012). Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73: 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gu J, Li YQ, Tao YX (2011). Are voltage‐gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol Pain 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Zamponi GW (2014). Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci 17: 153–163. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Taugher RJ, Kreple CJ (2013). Acid‐sensing ion channels in pain and disease. Nat Rev Neurosci 14: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke BU, Lindner M, Greifenberg L, Albus A, Kronimus Y, Bunemann M et al (2014). Diacylglycerol mediates regulation of TASK potassium channels by Gq‐coupled receptors. Nat Commun 5: 5540. [DOI] [PubMed] [Google Scholar]
- Williams S, Bateman A, O'Kelly I (2013). Altered expression of two‐pore domain potassium (K2P) channels in cancer. PLOS ONE 8: e74589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Roberson DP, Bean BP, Woolf CJ (2017). Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 16: 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chen H, Yang C, Zhong J, He W, Xiong Q (2017). Reversal of TRESK downregulation alleviates neuropathic pain by inhibiting activation of gliocytes in the spinal cord. Neurochem Res 42: 1288–1298. [DOI] [PubMed] [Google Scholar]