

Abstract
An increase in oxidative protein damage is a leading contributor to the age-associated decline in oocyte quality. By removing such damaged proteins, the proteasome plays an essential role in maintaining the fidelity of oocyte meiosis. In this study, we established that decreased proteasome activity in naturally aged, germinal vesicle (GV) mouse oocytes positively correlates with increased protein modification by the lipid aldehyde 4-hydroxynonenal (4-HNE). Furthermore, attenuation of proteasome activity in GV oocytes of young animals was accompanied by an increase in 4-HNE–modified proteins, including α-tubulin, thereby contributing to a reduction in tubulin polymerization, microtubule stability, and integrity of oocyte meiosis. A decrease in proteasome activity was also recapitulated in the GV oocytes of young animals following exposure to oxidative insults in the form of either hydrogen peroxide (H2O2) or 4-HNE. We also observed that upon oxidative insult, 4-HNE exhibits elevated adduction to multiple proteasomal subunits. Notably, the inclusion of the antioxidant penicillamine, to limit propagation of oxidative stress cascades, led to a complete recovery of proteasome activity and enhanced clearance of 4-HNE–adducted α-tubulin during a 6-h post-treatment recovery period. This strategy also proved effective in reducing the incidence of oxidative stress–induced aneuploidy following in vitro oocyte maturation, but was ineffective for naturally aged oocytes. Taken together, our results implicate proteasome dysfunction as an important factor in the accumulation of oxidatively induced protein damage in the female germline. This discovery holds promise for the design of therapeutic interventions to address the age-dependent decline in oocyte quality.
Keywords: oocyte, oxidative stress, aging, proteasome, lipid peroxidation, tubulin, microtubule, chromosomes, aneuploidy, fertility
Introduction
Human oocytes are established as primordial follicles during embryonic life and thereafter remain arrested in an extended prophase I until they are either recruited into the growing follicle pool for ovulation or, alternatively, undergo atresia some decades later (1). The balance of evidence indicates that, without intervention, this pool of oocytes is finite (1, 2). It is therefore concerning that maternal aging is associated with a precipitous decline in oocyte quality such that as many as 35% of the oocyte reserve in women aged 40 are the subject of aneuploidy, compared with only 2% for that of a woman in her 20s (3, 4). This loss of oocyte quality is associated with a reduction in female fertility, with the live birthrate per oocyte declining from 26% in younger women (<35 years of age) to just 1% for women at 42 years of age (5). Notwithstanding the undoubtedly complex mechanistic basis behind this age-associated phenomenon, several studies have converged on the notion that the accumulation of oxidative damage throughout the oocyte's extended life is a major contributing factor (6–9).
In support of the role of oxidative stress in the age-related decline in oocyte quality, an increase in intraovarian reactive oxygen species (ROS)3 has been convincingly correlated with advanced maternal age (10–13), decreased in vitro fertilization (IVF), and pregnancy success rates (13–18). Moreover, several studies have drawn a compelling link between oxidative stress and the decline in oocyte quality, with observed deficiencies in meiotic completion (17, 19), as well as age-related phenotypes such as spindle integrity, chromosome alignment (20–22), ploidy status (6, 23), and embryonic development (10, 15, 24–27). Despite the pervasive impact of oxidative stress on oocyte quality, the mechanisms by which this insult inflicts such damage are still being actively debated. In this context, recent studies have identified elevated production of lipid aldehydes accompanying the induction of oxidative stress in oocytes and have shown that these highly reactive entities contribute, in part, to the loss of oocyte quality (28, 29). This situation mirrors the response of somatic cells in which the induction of oxidative stress precipitates the peroxidation and breakdown of membrane lipids (including glycolipids, phospholipids, and cholesterol) (30–32), with ω-6 polyunsaturated fatty acids such as arachidonic and linoleic acids being particularly susceptible (33, 34). As this oxidative cascade proceeds, a number of by-products are generated, including lipid peroxyl radicals, hydroperoxides, and a suite of electrophilic aldehydes, with one of the most prominent and cytotoxic of these being 4-hydroxynonenal (4-HNE) (30–32). Following production, electrophilic aldehydes can covalently adduct to the nucleophilic functional groups of proteins, such as cysteine, histidine, and lysine residues (35, 36), and thereby perturb protein structure, induce protein cross-linking and aggregation, and, if left unresolved, culminate in a loss of cell viability (37–42).
The contribution of reactive aldehydes to the deterioration of the aging oocyte has been alluded to on the basis of elevated 4-HNE levels detected in the ovarian tissue of naturally aged mice (11, 43). Our own research has uncovered a similar increase in 4-HNE accumulation in pre-ovulatory germinal vesicle (GV) and post-ovulatory metaphase II (MII)-staged oocytes of aged mice, when compared with oocytes recovered from young mice (29). Additionally, we were able to establish a correlative link between elevated levels of 4-HNE and an increase in age-associated phenotypes, with in vitro 4-HNE challenge inducing pronounced spindle defects and aneuploidy in the oocytes of young mice. Perhaps most intriguing was the observation that these phenotypic changes were positively correlated with 4-HNE modification of a subset of vulnerable oocyte proteins, including those of the tubulin family. Moreover, interventions designed to limit 4-HNE bioavailability, and thus reduce tubulin adduction, were able to ameliorate the deleterious effect of oxidative stress on oocyte quality (29). Such findings are in accord with the key role of the microtubule network in supporting faithful meiotic completion, with defects in these cytoskeletal elements associated with elevated rates of oocyte aneuploidy (44, 45). They also agree with independent studies in which site-specific 4-HNE adduction to cysteine and lysine residues in the primary structure of α- and β-tubulin have been previously reported in human sperm cells, human THP-1 monocytic cells, and purified tubulin from bovine brain (46–48). More specifically, these studies reported the rapid disappearance of microtubule networks (49), tubulin cross-linking, and an inhibition of polymerization (48, 50), as well as the spontaneous generation of tubulin dimers (51) as a consequence of 4-HNE exposure.
Notwithstanding these data, the oocyte is endowed with an array of mechanisms to protect the fidelity of the female germline from the oxidative damage it may encounter throughout its extended life span. As with somatic cells, an active proteasomal system plays a fundamental homeostatic role in the oocyte, because of its ability to degrade irreversibly damaged, dysfunctional, and/or misfolded proteins (52–54), as well as a more specialized role during meiotic resumption (55, 56). Typically, proteins destined for proteasomal proteolysis are covalently tagged with multiubiquitin chains for degradation by the 26S proteasome holoenzyme. This enzyme comprises a 19S regulatory subunit that directs the entry of ubiquitin-tagged proteins into the 20S catalytic core for proteolysis. However, mounting evidence from somatic cell literature suggests that the degradation of 4-HNE–modified proteins is most often mediated by proteasomal pathways via the 20S proteasome alone, independent of the 19S subunit, ATP, and/or ubiquitin (52–54). The 20S core proteasome is composed of four stacked heptameric rings comprising seven structural subunits in the outer rings and seven β subunits making up the inner two rings and containing the active sites responsible for proteolysis. In mammals, the β1, β2, and β5 subunits are catalytic and are responsible for chymotrypsin-like, trypsin-like, and post-glutamyl peptide–hydrolyzing activities, respectively (57, 58).
An age-related decline in proteasome activity has been demonstrated in a multitude of mammalian tissues and cells (59–67). In addition, transgenic mice with decreased chymotrypsin-like activity exhibit a shortened life span associated with premature age-related phenotypes (68). Concomitantly, an increase in the levels of oxidized proteins, glycated proteins, and proteins modified by 4-HNE is also observed within mammalian somatic tissues with increasing age (62, 69, 70). In seeking to link these data, it has been shown that several components of the proteasome are themselves targeted for 4-HNE modification. Indeed, with the exception of β2, all other α and β subunits represented in the proteasome core have been reported as being vulnerable to 4-HNE adduction and have been associated with an attendant reduction in the trypsin, chymotrypsin, and peptidylglutamyl peptide hydrolase activities of the proteasome (71–78), ultimately suppressing the cell's ability to resolve alternative oxidatively-damaged proteins. In seeking to determine whether similar mechanisms contribute to declining oocyte quality, here we have investigated the efficacy of proteasomal clearance of 4-HNE–modified proteins in aged oocytes. Specifically, we have been able to show that the proteasomal pathway is compromised by oxidative challenge within oocytes and thereafter permits the accumulation of 4-HNE–modified proteins, which ultimately contribute to the functional demise of the aged oocyte. These novel discoveries hold promise for the design of therapeutic intervention strategies to combat age-dependent female infertility.
Results
Proteasome activity is compromised in oocytes of naturally aged mice
Given the significant accumulation of 4-HNE–adducted proteins in oocytes as a consequence of aging, we initially set out to determine whether aging is linked to the attenuation of proteasomal activity and an attendant reduction in the capacity of oocytes to degrade aldehyde-adducted proteins. For the purpose of this study, we elected to focus on GV oocytes because this is the stage of development in which human oocytes remain arrested for decades; GV oocytes are therefore likely to be placed at heightened vulnerability to the cumulative impact of oxidative damage. Utilizing a fluorescence-based assay, we confirmed that overall total proteasome activity was significantly reduced in GV oocytes derived from aged F1 mice (14 months) compared with young controls (4–6 weeks) (p = 0.0316; Fig. 1A). Accompanying this ∼1.5-fold reduction in proteasome activity, we also recorded a commensurate increase in the ubiquitination status of several oocyte proteins. In this context, immunocytochemical analyses with pan-ubiquitin antibodies revealed the expression of ubiquitin uniformly throughout the cytosol of both young and aged GV oocytes, with slightly exaggerated localization to the periphery of the oocytes (Fig. 1B). Notably, however, the basal levels of ubiquitin detected in oocytes were significantly elevated in oocytes recovered from aged mice compared with their younger counterparts (14 months) (p = 0.0184; Fig. 1B). The age-dependent increase in intracellular ubiquitin labeling was paralleled by equivalent accumulation of 4-HNE in the aged oocytes (p = 0.0144; Fig. 1B). This positive correlation raised the prospect that 4-HNE adduction may target vulnerable proteins for ubiquitination in preparation for degradation via a proteasomal pathway. Consistent with this hypothesis, we noted strong co-localization between 4-HNE and ubiquitin throughout the ooplasm and the oocyte periphery (Fig. 1B). Moreover, immunoblotting of oocyte lysates with anti-UB–K48 antibodies, which detect polyubiquitination linkages on lysine 48 that selectively direct proteins toward a proteasomal degradation pathway (79), revealed a significant increase in this form of post-translational modification in aged oocytes (p = 0.0340; Fig. 1C). In this context, anti-UB–K48 antibodies labeled a myriad of oocyte proteins ranging in size from ∼30 to 460 kDa; however, only those of very high molecular weight (i.e. >250 kDa) presented with increased labeling in aged versus young oocytes (Fig. 1C). On the basis of these combined data, we infer that proteasome-mediated proteolysis is compromised in aged oocytes.
Figure 1.

Aged oocytes have decreased proteasome activity that correlates with an increase in 4-HNE expression. GV oocytes were collected from nonhormonally stimulated young animals (4–6 weeks) and aged animals (14 months). A, significant decrease in proteasome activity was detected in aged oocytes using a fluorometric proteasome activity assay kit. An internal control was used during this assay to distinguish proteasome activity from other protease activity in the cell lysates. One unit of proteasome activity is defined as the amount of proteasome that generates 1.0 nmol of the fluorescently tagged AMC per min at 37 °C. This experiment was repeated across three independent biological and technical replicates using 100 oocytes per assay, pooled from a minimum of three young animals and between 12 and 15 aged animals. B, increase in ubiquitin, correlating with elevated levels of 4-HNE, was detected in aged oocytes through the use of immunocytochemistry with anti-4-HNE (red) and anti-UBI-1 (green) antibodies. Nuclei were counterstained with Hoechst (blue). Fluorescence intensity of 4-HNE and UBI-1 was quantified for aged cells against their young counterparts for reference. Scale bar = 20 μm. This experiment was repeated across three independent biological replicates, with each mouse contributing a minimum of seven oocytes and representing an independent technical replicate. C, increase in the ubiquitin in aged oocytes was also confirmed via immunoblotting with anti- UB-K48 antibodies and was quantified for aged cell lysates against their young counterparts for reference. Whole lanes were used for densitometry analysis. Immunoblots were stripped and re-probed with anti-GAPDH antibodies as a loading control. Immunoblots were performed in biological and technical triplicate using 100 oocytes per lane pooled from a minimum of three young animals and between 12 and 15 aged animals. Data are presented as mean of three replicates ± S.E. Statistical analyses were performed using Student's t test, *, p ≤ 0.05. AU, arbitrary units.
4-HNE elicits a reduction in kinetochore–microtubule stability and tubulin polymerization
The accumulation of 4-HNE–modified tubulin proteins has previously been correlatively associated with the age-related deterioration in oocyte quality (29). Furthermore, we have identified tubulin proteins as the predominant protein family targeted for 4-HNE modification in oocytes (Fig. S1). We therefore elected to investigate the impact that elevated 4-HNE exposure has on microtubule stability and tubulin polymerization. Consistent with our previous observations (29), 4-HNE co-localized with α-tubulin within the vicinity of the meiotic spindle in both MI and MII stage oocytes (Fig. 2A). Moreover, cold-shock destabilization assays revealed that 4-HNE–treated MI (p = 0.0015; Fig. 2B) and MII stage oocytes (p = 0.0023; Fig. 2B) experienced a significant 1.3- and 1.7-fold decrease in corrected total fluorescence associated with the spindle, compared with untreated control oocytes, respectively. On the basis of these data, we infer that elevated 4-HNE adduction leads to an attendant decrease in the stability of oocyte kinetochore–microtubules. Furthermore, the application of proximity ligation assay (PLA) between “anti-α-tubulin and anti-β-tubulin antibodies revealed an approximate 1.3-fold decrease in the intensity of fluorescence foci formed in MI (p = 0.0002; Fig. 2C) and MII stage oocytes (p = 0.0027; Fig. 2C) exposed to 4-HNE indicating a decreased interaction between α-tubulin and β-tubulin isoforms. In addition, the application of an in vivo microtubule assay enabled us to document a significant decrease between 2.1- and 2.3-fold in the ratio of polymerized tubulin (high-speed pellet; HSP) versus free tubulin (high-speed supernatant; HSS) in MI (p = 0.0324; Fig. 2D) and MII (p = 0.0154; Fig. 2D) oocytes exposed to 4-HNE, respectively. Taken together, these data support the hypothesis that 4-HNE modification of tubulin proteins impairs tubulin polymerization and has the potential to contribute to the age-related decline in oocyte quality.
Figure 2.

Acute exposure to 4-HNE at prophase I arrest reduces kinetochore–microtubule stability and tubulin polymerization. GV oocytes were treated with 20 μm 4-HNE for 2 h, underwent IVM to MI or MII, and were prepared for immunofluorescence analysis, tubulin polymerization assays, and PLA alongside their untreated (UT) counterparts. A, 4-HNE was localized to the spindle of MI and MII oocytes using anti-4-HNE (red) and anti-tubulin (green) antibodies. Nuclei were counterstained with Hoechst (blue). Scale bar = 20 μm. B, MI and MII oocytes were also fixed following a cold shock at 4 °C for 7 min to examine k-mt stability. Immunofluorescence analysis using anti-α-tubulin (green), anti-CREST (red), and Hoechst (blue) revealed a significant decrease in the presence of stable k-mt in the spindles of 4-HNE–exposed oocytes. Scale bar = 5 μm. C, PLA with anti-α-tubulin and anti-β-tubulin antibodies (red) revealed a decrease in tubulin polymerization in MI and MII oocytes following acute 4-HNE exposure. Nuclei were counterstained with Hoechst (blue). Scale bar = 20 μm. immunofluorescence experiments were repeated across three independent biological replicates, with a minimum of 10 oocytes, pooled from a minimum of three animals. D, decrease in the ratio of polymerized (high speed pellet; HSP) versus free tubulin (high speed supernatant; HSS) was also detected in MI and MII oocytes after 4-HNE exposure, using a microtubule/tubulin in vivo assay. In vivo microtubule/tubulin assays were performed in biological and technical triplicate using 150 oocytes pooled from a minimum of four young animals. Samples were resolved on the same gel and transferred to the same membrane for immunoblot analysis and have been cropped for presentation. Statistical analyses were performed using Student's t test, *, p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001. Data are presented as mean of three replicates ± S.E. AU, arbitrary units.
Proteasomal pathway mediates the processing of 4-HNE–modified α-tubulin
Given the essential role of the proteasome in the clearance of 4-HNE–modified proteins in somatic cells (52–54), we hypothesized that attenuation of proteasomal activity in aged oocytes would lead to an accumulation of 4-HNE–modified proteins. For the purpose of this study, we elected to focus on the clearance of 4-HNE–modified α-tubulin through immunofluorescence analyses of total 4-HNE and α-tubulin as well as immunoblotting and PLA determinations in oxidatively-stressed oocytes. A concentration of 35 μm H2O2 was selected as it has previously been titrated to induct the generation of 4-HNE without compromising oocyte vitality (29). These oocytes were pre-treated with 35 μm H2O2 and incubated for 6 h with a canonical proteasome inhibitor, MG132 (50 μm) (80). Given that there is some evidence that the inhibition of proteasomal pathways in other cell types can result in a compensatory activation of lysosomal pathways (81, 82), these experiments also featured an additional control where the lysosomal pathways were inhibited (chloroquine; 100 μm (83, 84)) independently and in conjunction with proteasomal inhibition.
Immunocytochemical analyses of H2O2-treated oocytes revealed an anticipated increase in intracellular 4-HNE labeling as observed in Fig. 1B. Notably, however, 4-HNE labeling was further elevated upon proteasomal inhibition. Thus, 4-HNE expression was significantly increased 1.22-fold above that of H2O2-treated oocytes (p = 0.007; Fig. 3A). In addition, lysosomal inhibition contributed to a similar increase in 4-HNE accumulation (1.2-fold) (p = 0.0126; Fig. 3A), with the simultaneous inhibition of both pathways leading to the most dramatic increase in total 4-HNE (1.41-fold) (p < 0.0001; Fig. 3A). Interestingly, proteasome inhibition also resulted in a significant, albeit modest, elevation of 4-HNE expression in untreated oocytes (Fig. S2). Furthermore, in all treatment groups, we observed strong co-localization between α-tubulin and 4-HNE around the periphery of the oocyte. To further explore the relationship between 4-HNE and α-tubulin under proteolytic inhibition we utilized PLA, a modified form of co-localization in which punctate fluorescent signals are only generated if the two targeted antigens reside within a maximum of 40 nm to each other (85, 86). Although total α-tubulin expression remained essentially unchanged in oocytes from all treatment groups, the increase in positive PLA fluorescence detected in H2O2-challenged oocytes confirmed that this protein was being targeted for additional 4-HNE adduction. Similarly, PLA fluorescence was further elevated upon proteasomal and lysosomal inhibition, with the simultaneous inhibition of both pathways yielding the highest proportion of 4-HNE–modified α-tubulin (p ≤ 0.0066; Fig. 3A). These trends were reflected in immunoblot analyses in which the expression of the 50-kDa 4-HNE band, previously identified as tubulin (29), was significantly elevated upon the induction of oxidative stress, and it was further elevated upon proteasomal and lysosomal inhibition (p < 0.0377; Fig. 3B). On the basis of these data, we infer that these proteolytic pathways may hold an important role in the metabolism of 4-HNE–modified α-tubulin in oxidatively-stressed oocytes.
Figure 3.

Degradation of 4-HNE–modified α-tubulin is mediated by the proteasomal and lysosomal systems under conditions of oxidative stress. GV oocytes were treated with 35 μm H2O2 (H2O2) for 1 h to induce oxidative stress and then incubated in the presence or absence of the proteasome inhibitor MG132 (50 μm; PI) and/or the lysosomal inhibitor chloroquine (100 μm; LI) for 6 h. Oocytes were then prepared for immunofluorescence analysis, PLA, and immunoblotting analysis alongside their untreated (UT) counterparts to determine the degradation pathway for 4-HNE–modified α-tubulin. A, immunofluorescence analysis using anti-α-tubulin (green) and anti-4-HNE (red) antibodies revealed consistent levels of α-tubulin expression throughout all treatments groups, which co-localized the significantly increasing expression 4-HNE, particularly at the periphery of the oocyte under conditions of oxidative stress and proteasome and lysosome inhibition (ANOVA; p ≤ 0.0190). Similarly, PLA also indicated a significant increase in the 4-HNE modification of α-tubulin at the periphery of the oocyte under conditions of oxidative stress and proteasome and lysosome inhibition (ANOVA; p ≤ 0.0066). Nuclei were counterstained with Hoechst (blue). Scale bar = 20 μm. These experiments were repeated across three independent biological replicates, with a minimum of 10 oocytes, pooled from a minimum of three animals. B, consistent expression of α-tubulin with the increase in 4-HNE modification of α-tubulin upon proteasome and lysosome inhibition was also confirmed via immunoblotting with anti-4-HNE and anti-α-tubulin antibodies and was quantified for treated cell lysates against their untreated counterparts for reference (ANOVA; p ≤ 0.0377). Immunoblots were run on the same gel and transferred onto the same membrane for immunoblot analysis and have been cropped for presentation. Only the 50-kDa α-tubulin band was used for densitometry analysis. Immunoblots were stripped and re-probed with anti-GAPDH antibodies as a loading control. Immunoblots were performed in biological and technical triplicate using 100 oocytes per lane pooled from a minimum of three animals. Data are presented as mean of three replicates ± S.E. AU, arbitrary units.
Proteasome activity is compromised in oxidatively-stressed young oocytes
In seeking to account for compromised proteasome activity in aged oocytes, we elected to exploit a model of oxidatively-stressed young oocytes that recapitulates many of the pathologies witnessed during the aging process (29). Specifically, we focused on acute oxidative insults delivered by H2O2 or 4-HNE at concentrations that have previously been titrated to reduce oocyte quality without an associated loss of viability (29). Under these conditions, we recorded a significant, dose-dependent reduction in oocyte proteasome activity in response to both H2O2 (p ≤ 0.0203; Fig. 4A) and 4-HNE (p ≤ 0.047; Fig. 4D). Furthermore, immunocytochemical staining of these oocytes revealed a reciprocal increase in total ubiquitin in response to both H2O2 (p ≤ 0.0185; Fig. 4B) and 4-HNE (p ≤ 0.00001; Fig. 4E) compared with their untreated controls. Similar to results obtained using aged oocytes (Fig. 1B), we also noted an elevation in the levels of 4-HNE detected in response to both H2O2 (p ≤ 0.0337; Fig. 4B) and 4-HNE (p ≤ 0.0010; Fig. 4E) treatments and strong co-localization between 4-HNE and ubiquitin throughout the ooplasm and the oocyte periphery (Fig. 4E). Moreover, immunoblotting of oocyte lysates confirmed a significant 2.3- and 2.8-fold increase in Lys-48 polyubiquitination in response to both 35 and 50 μm H2O2 (p ≤ 0.0025; Fig. 4C), respectively. Even more pronounced, 2.6- and 3.3-fold increases in the level of polyubiquitination harbored by oocytes was evident following direct exposure to 20 and 30 μm 4-HNE (p ≤ 0.0013; Fig. 4F), respectively. In both instances, however, anti-UB–K48 antibody labeling was primarily restricted to a subset of proteins of >250 kDa, consistent with the profile of proteins targeted for enhanced Lys-48 polyubiquitination in aged oocytes (Fig. 1C). In view of these data, 35 μm H2O2 was chosen for use in all subsequent experiments as the oxidative burden delivered under these circumstances clearly recapitulated the responses documented in aged oocytes (i.e. proportional changes in proteasome activity and overall ubiquitination).
Figure 4.

Oxidative stress inhibits proteasome activity. GV oocytes were treated with H2O2 (35 and 50 μm) for 1 h or 4-HNE (20 and 30 μm) for 2 h to induce oxidative stress and were prepared for immunofluorescence analysis or immunoblotting alongside their untreated (0 μm) counterparts. A and D, significant decrease in proteasome activity was detected in oocytes exposed to both forms of oxidative stress using a fluorometric proteasome activity assay kit (ANOVA; p ≤ 0.0203 and p ≤ 0.0470). An internal control was used during this assay to distinguish proteasome activity from other protease activity in the cell lysates. One unit of proteasome activity is defined as the amount of proteasome that generates 1.0 nmol of the fluorescently tagged AMC per min at 37 °C. B and E, dose-dependent increase in ubiquitin, correlating with elevated levels of 4-HNE, was detected in H2O2 and 4-HNE–treated oocytes through the use of immunocytochemistry with anti-4-HNE (red) and anti-UBI-1 (green) antibodies, and nuclei were counterstained with Hoechst (blue) (H2O2 ANOVA; p ≤ 0.0337 and p ≤ 0.00185) (4-HNE ANOVA; p ≤ 0.0010 and p ≤ 0.0001). Fluorescence intensity of 4-HNE and UBI-1 was quantified for treated cells against their untreated counterparts for reference. Scale bar = 20 μm. This experiment was repeated across three independent biological replicates, with a minimum of 10 oocytes, pooled from a minimum of three animals. C and F, increase in ubiquitin in oxidatively-stressed oocytes was also confirmed via immunoblotting with anti-UB-K48 antibodies and was quantified for treated cell lysates against their untreated counterparts for reference (ANOVA; p ≤ 0.0025 and p ≤ 0.0013). Immunoblots were run on the same gel and transferred onto the same membrane for immunoblot analysis and have been cropped for presentation. Whole lanes were used for densitometry analysis. Immunoblots were stripped and re-probed with anti-GAPDH antibodies as a loading control. Immunoblots were performed in biological and technical triplicate using 100 oocytes per lane pooled from a minimum of three animals. Data are presented as mean of three replicates ± S.E. AU, arbitrary units.
Proteasomal subunits are targeted for 4-HNE adduction in oxidatively-stressed oocytes
In somatic cells, core elements of the proteasomal complex are vulnerable to direct 4-HNE adduction leading to a consequential reduction in the activity of this proteolytic pathway (71–78). Key among these are the proteasome subunits α2, α5, and α6 as well as proteasome subunits β1, β3, and β7. Thus, we sought to determine whether equivalent proteasomal subunits are similarly susceptible to 4-HNE adduction in oxidatively-stressed oocytes. For this purpose, we performed standard co-localization and PLA with validated anti-proteasomal and anti-4-HNE antibodies (Fig. S3). Subsequent immunocytochemical analysis revealed a positive fluorescent signal for each of the six proteasomal subunits within the ooplasm of GV oocytes. However, the expression profile of each subunit did not strictly overlap. Subunits α2, α6, and β7 were characterized by punctate fluorescence foci distributed uniformly throughout the cytosol, and within the nucleus, of both untreated and H2O2-treated oocytes (Fig. 5, A, E, and K). In contrast, the cytosolic labeling of subunits β1 and β3 was accompanied by intense staining around the periphery of the oocyte, presumably corresponding to the oolemma (Fig. 5, G and I). Uniquely, subunit α5 presented with uniform cytoplasmic distribution secondary to an intense perinuclear expression (Fig. 5C). Of particular interest was the demonstration of strong cytosolic co-localization between all proteasome subunits examined and 4-HNE. This co-localization also extended to the cell periphery, where it was particularly intense in oocytes exposed to H2O2 treatment. Notably, the steady-state levels of all proteasome components examined remained unchanged after acute oxidative insult (Fig. S4).
Figure 5.


4-HNE adducts to multiple subunits of the core proteasome. GV oocytes were treated with 35 μm H2O2 (H2O2) for 1 h to induce oxidative stress and were prepared for immunofluorescence analysis and proximity ligation assays alongside their untreated (UT) counterparts to examine the susceptibility of proteasome subunits to 4-HNE modification. A, C, E, G, I, and K, for immunofluorescence analysis, all proteasome subunits examined (α2, α5, and α6 and β1, β3, and β7) were detected in the oocyte and co-localized with 4-HNE, particularly at the periphery of the oocyte after H2O2 treatment using appropriate anti-proteasome (green) and anti-4-HNE (red) antibodies, and nuclei were counterstained with Hoechst (blue). B, D, F, H, J, and L, PLA between 4-HNE and several proteasome subunits (α2, α5, and α6 and β1, β3, and β7) revealed their susceptibility to 4-HNE modification with fluorescent foci (red) evident throughout the oocyte that significantly intensified after oxidative insult, with the exception of β3. Nuclei were counterstained with Hoechst (blue). Scale bar = 20 μm. These experiments were repeated across three independent biological replicates, with a minimum of 10 oocytes, pooled from a minimum of three animals. Statistical analyses were performed using Student's t test, **, p ≤ 0.01; ***, p ≤ 0.001; and **** p ≤ 0.0001. Data are presented as mean of three replicates ± S.E. AU, arbitrary units.
Analysis of GV oocytes subjected to PLA with a combination of 4-HNE and either α2, α5, α6, β1, β3, or β7 all revealed positive fluorescent foci illustrative of the susceptibility of these subunits to 4-HNE modification in both untreated and oxidatively-stressed oocytes (Fig. 5, B, D, F, H, J, and L). Notably, however, the positive PLA labeling of anti-4-HNE and each of the respective proteasomal subunits appeared to be largely restricted to the perinuclear domain of untreated oocytes. By contrast, those oocytes exposed to H2O2 generally presented with significantly more PLA labeling (Fig. 5, B, D, F, H, J, and L). Moreover, the positive red fluorescent PLA foci detected in these cells accumulated throughout the ooplasm. The one exception to this trend was that of β3 in which the H2O2-driven increase in PLA-labeling intensity did not achieve statistical significance (p = 0.1583; Fig. 5J).
In all cases, the specificity of the immunocytochemical and PLA analyses was validated through the absence of fluorescent labeling in any negative control groups consisting of either anti-mouse or rabbit IgG-negative controls or irrelevant antibody combination controls (i.e. anti-4-HNE and anti-PIWIL1 antibodies), respectively (Fig. S3B). These data support the notion that elevated levels of oxidative stress do indeed precipitate an increase in 4-HNE adduction to vulnerable proteasomal subunits in GV oocytes. Such data thereby provide a rational explanation for the loss of proteasomal activity observed in the aging oocyte as well as in young oocytes experiencing acute oxidative stress.
The nucleophile, penicillamine, allows for the processing of 4-HNE–modified tubulin
Taken together, our data raise the prospect that the decrease in oocyte quality that accompanies aging may be accentuated by reduced proteasomal activity and a consequential failure to mitigate the impact of 4-HNE–modified proteins such as α-tubulin. These findings prompted us to explore the potential for the recovery of oocyte quality post-oxidative insult following a recovery period in the presence of penicillamine, a highly efficient scavenger of 4-HNE (87). The premise for this approach centers on previous research that has shown 4-HNE is capable of eliciting a cycle of self-propagation through the dysregulation of mitochondrial enzymes (28). Thus, without intervention, 4-HNE protein adduction is unlikely to be resolved in the oocyte (Fig. S5).
In the following experiments, 4-HNE production was initiated in young oocytes via treatment with 35 μm H2O2, and thereafter, its accumulation was tracked over a 6-h period in the presence or absence of penicillamine (100 μm). Consistent with our previous data, immunofluorescence analysis revealed that total 4-HNE expression was significantly elevated (1.4-fold) directly after H2O2 treatment and remained at equivalently high levels during the ensuing 6 h of incubation (p < 0.0001; Fig. 6A). By contrast, total 4-HNE expression significantly declined 6 h post-H2O2 treatment in the presence of penicillamine (p = 0.0101; Fig. 6A), suggesting the reduced accumulation of 4-HNE–modified proteins in response to the initial insult. Accordingly, immunoblot analyses conducted with anti-4-HNE antibodies confirmed that the tubulin-specific band at ∼50 kDa (29) mirrored this trend, with an initial 1.45-fold increase in 4-HNE adduction returning to basal levels indistinguishable from those of the untreated controls by 6 h post-oxidative insult in the presence of penicillamine (p < 0.0457; Fig. 6B). Co-localization of 4-HNE and α-tubulin at the periphery of the oocyte was again observed in untreated controls and was most obvious in H2O2-treated oocytes. Interestingly, however, this peripheral co-localization was still discernible following incubation with penicillamine (Fig. 6A, 6 h treatment).
Figure 6.

Processing of 4-HNE–modified α-tubulin occurs after 6 h of recovery in the aldehyde scavenger, penicillamine. GV oocytes were treated with 35 μm H2O2 (H2O2) for 1 h to induce oxidative stress and were then immediately processed (0 h) or incubated for an additional 6 h in either the absence (6 h) or the presence of penicillamine (100 μm) (6 h + P). Oocytes were then prepared for immunofluorescence analysis, PLA, and immunoblotting analysis alongside their untreated (UT) counterparts to determine how long 4-HNE resolution takes in the oocyte under the conditions of oxidative stress. A, immunofluorescence analysis using anti-α-tubulin (green) and anti-4-HNE (red) antibodies revealed consistent levels of α-tubulin expression throughout all treatments groups, which co-localized particularly at the periphery of the oocyte with the significantly increasing expression of 4-HNE after H2O2 treatments, followed by a subsequent decrease in 4-HNE expression upon 6 h recovery in penicillamine (ANOVA; p ≤ 0.0101). Similarly, PLA also indicated a significant increase in the 4-HNE modification of α-tubulin at the periphery of the oocyte under conditions of oxidative stress, with a complete resolution observed after 6 h recovery in penicillamine (ANOVA; p ≤ 0.0001). Nuclei were counterstained with Hoechst (blue). Scale bar = 20 μm. These experiments were repeated across three independent biological replicates, with a minimum of 10 oocytes, pooled from a minimum of three animals. B, consistent expression of α-tubulin with the increase in 4-HNE modification of α-tubulin upon the induction of oxidative stress followed by a decrease upon 6 h recovery was also confirmed via immunoblotting with anti-4-HNE and anti-α-tubulin antibodies and was quantified for treated cell lysates against their untreated counterparts for reference (ANOVA; p ≤ 0.0457). Only the 50-kDa α-tubulin band was used for densitometry analysis. Immunoblots were stripped and re-probed with anti-GAPDH antibodies as a loading control. Immunoblots were performed in biological and technical triplicate using 100 oocytes per lane pooled from a minimum of three animals. Data are presented as mean of three replicates ± S.E. AU, arbitrary units.
On the basis of these data, we next utilized PLA to assess the interaction between 4-HNE and α-tubulin both prior to (i.e. 0 h) and at 6 h post-H2O2 exposure to corroborate the specific 4-HNE adduction of tubulin proteins. This strategy confirmed an initial increase in 4-HNE adduction to α-tubulin in oxidatively-stressed GV oocytes (p < 0.0001; Fig. 6A). Notably, positive PLA fluorescent foci were largely confined to the periphery of the oocyte. Consistent with our immunoblotting data, PLA analysis confirmed the initial increase in tubulin adduction 6 h post-H2O2 exposure followed by a dramatic 1.5-fold decrease in fluorescent labeling, returning to untreated levels in the presence of penicillamine (p = 0.0011; Fig. 6A).
Processing of 4-HNE–modified tubulin partially restores oocyte quality in H2O2-treated oocytes but not in naturally aged oocytes
In view of the ability of the penicillamine to block the accumulation of 4-HNE and thereby allow H2O2-treated oocytes to clear 4-HNE–modified α-tubulin, we next examined whether this intervention strategy was also capable of rescuing oocyte quality. Indeed, we observed significant resurgence in proteasome activity in the “recovered” treatment group, returning from an approximate 1.6-fold decrease in activity in H2O2-treated oocytes to levels indistinguishable from untreated controls (p ≤ 0.0462; Fig. 7A). Furthermore, immunoblotting of oocyte lysates also confirmed a significant 1.5-fold decrease in Lys-48 polyubiquitination in H2O2-treated oocyte after a 6-h recovery in penicillamine (p ≤ 0.0365; Fig. 7B), which is further indicative of recovered proteasome activity. Unfortunately, rescuing proteasome activity was not sufficient to restore oocyte quality after meiotic completion, maintaining an approximate 2-fold decrease in polar body extrusion (PBE) rates in both H2O2 and recovered treatment groups when compared with untreated controls (p ≤ 0.0046; Fig. 8A). Despite these initial data, we did observe a modest, but statistically significant, decrease in aneuploidy rates, down from 26% in H2O2-treated oocytes to 17% in recovered oocytes (p = 0.0100; Fig. 8B). Nevertheless, these levels of aneuploidy still remained significantly elevated above that of the untreated controls at 1.7% (p = 0.0005).
Figure 7.

Penicillamine restores the proteasome activity of the oxidatively-damaged oocyte. GV oocytes were treated with 35 μm H2O2 (H2O2) for 1 h to induce oxidative stress and were then immediately processed (0 h) or incubated for an additional 6 h in either the absence (6 h) or the presence of penicillamine (100 μm) (6 h + P), prior to IVM for 16 h. MII oocytes were identified by the presence of a polar body. A, decrease in proteasome activity was observed after H2O2 (ANOVA; p = 0.0087), with penicillamine treatment after H2O2 exposure able to restore the effect of H2O2 on proteasome activity (ANOVA; p = 0.0462). One unit of proteasome activity is defined as the amount of proteasome that generates 1.0 nmol of the fluorescently tagged AMC per min at 37 °C. B, increase in ubiquitin in oxidatively-stressed oocytes was detected via immunoblotting with anti-UB-K48 antibodies, with penicillamine after H2O2 exposure able to significantly decrease ubiquitination (ANOVA; p = 0.0365). Immunoblots were performed in biological and technical triplicate using 100 oocytes per lane pooled from a minimum of three animals. Whole lanes were used for densitometry analysis. Immunoblots were stripped and re-probed with anti-GAPDH antibodies as a loading control. Data are presented as mean of three replicates ± S.E. AU, arbitrary units.
Figure 8.
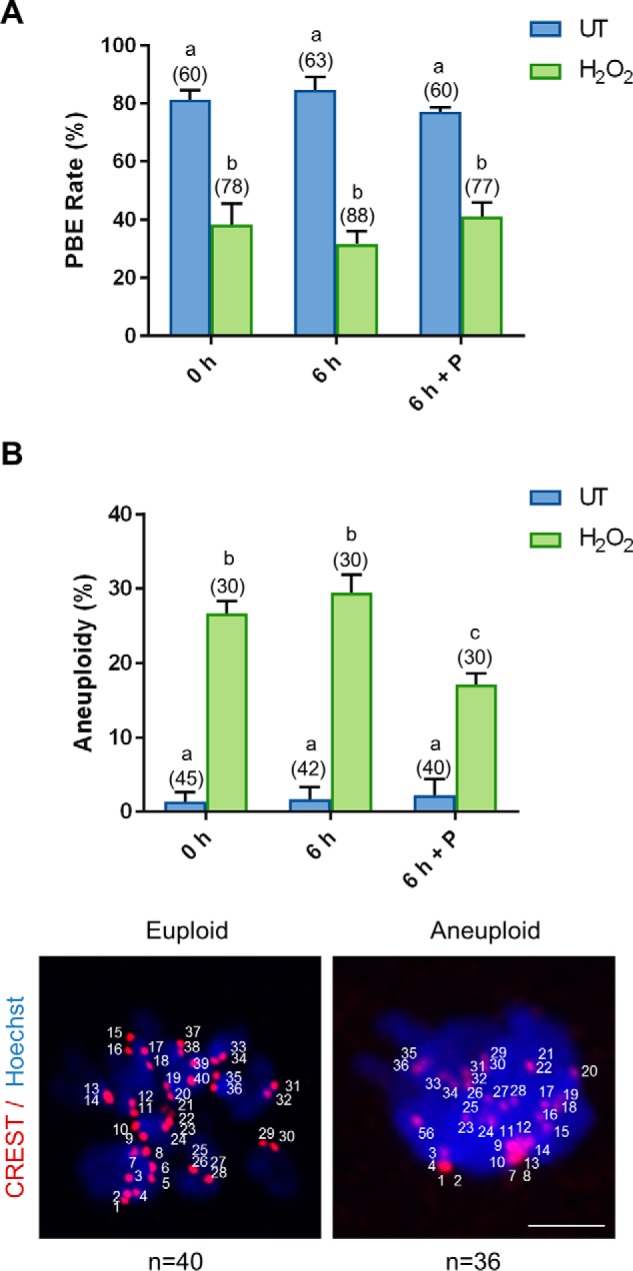
Penicillamine partially restores the quality of the oxidatively-damaged oocyte. GV oocytes were treated with 35 μm H2O2 (H2O2) for 1 h to induce oxidative stress and were then immediately processed (0 h) or incubated for an additional 6 h in either the absence (6 h) or the presence of penicillamine (100 μm) (6 h + P), prior to IVM for 16 h. MII oocytes were identified by the presence of a polar body. A, dose-dependent decrease in PBE was observed after H2O2 (ANOVA; p = 0.0033) with penicillamine treatment after H2O2 exposure unable to decrease the effect of H2O2 on meiotic completion (ANOVA; p = 0.9308). B, following IVM, spindles were collapsed and oocytes were fixed. Kinetochores were immunostained using anti-CREST antibody (red), and the chromosomes were stained with Hoechst (blue). Kinetochores of each individual oocyte were counted. An increase in aneuploid oocytes was detected at 35 μm H2O2 (ANOVA; p = 0.0001), with penicillamine after H2O2 exposure able to significantly decrease the effect of H2O2 on aneuploidy rates (ANOVA; p = 0.0100). Representative images are presented, indicating how aneuploidy was scored. Scale bar = 5 μm. IVM and aneuploidy assays were performed with three biological replicates with each replicate containing between 10 and 30 oocytes pooled from a minimum of three animals. Data are presented as mean of three replicates ± S.E.
Notably, no detectable difference in the rate of PBE was observed between young and naturally aged oocytes nor between those that experienced a 6-h recovery in penicillamine (Fig. 9A). However, as reported previously (109), we detected an approximate 1.4-fold increase in the rate of aneuploidy in aged versus young oocytes (p = 0.0035; Fig. 9B). Unlike the recovered H2O2-treated oocytes, the aneuploidy witnessed in aged oocytes proved completely refractory to a 6-h period of recovery in penicillamine, i.e. aneuploidy rates remained indistinguishable from untreated aged oocytes at levels that were 1.4-fold higher than their equivalent young counterparts (p = 0.0021; Fig. 9B).
Figure 9.
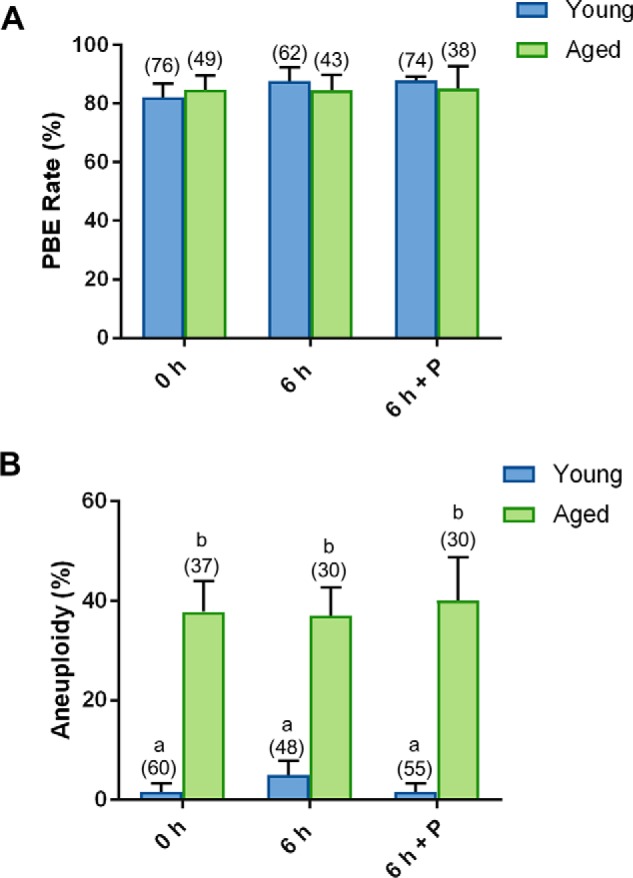
Penicillamine does not restore the quality of naturally aged oocytes. GV oocytes were collected from nonhormonally stimulated young animals (4–6 weeks) and aged animals (14 months) and incubated for an additional 6 h in either the absence (6 h) or the presence of penicillamine (100 μm) (6 h + P), prior to IVM for 16 h. MII oocytes were identified by the presence of a polar body. A, no difference in PBE was observed with penicillamine treatment. B, following IVM, spindles were collapsed, and oocytes were fixed. Kinetochores were immunolabeled with anti-CREST antibodies (red), and the chromosomes were stained with Hoechst (blue). Kinetochores of each individual oocyte were counted. An increase in aneuploidy was observed in aged oocytes (ANOVA; p = 0.0035), with penicillamine treatment proving ineffective in restoration of aneuploidy rates. IVM and aneuploidy assays were performed with three biological replicates with each replicate containing between 10 and 25 oocytes pooled from a minimum of two animals. Data are presented as mean of three replicates ± S.E.
Taken together, these data indicate that the suppression of 4-HNE–modified tubulin can partially restore oocyte quality in the context of aneuploidy. Perhaps not surprisingly, however, these data also indicate that 4-HNE adduction of tubulin is not the only mechanism contributing to the decline in oocyte quality witnessed under conditions of oxidative stress.
Discussion
Increased accumulation of the highly reactive lipid aldehyde, 4-HNE, has recently been implicated as a contributing factor in the etiology of declining oocyte quality with increasing maternal age (29). The destructive impact of 4-HNE on protein structure and function in the germline has been well documented (28, 46, 88, 89). Indeed, cytoskeletal elements such as those of the tubulin family have been identified as prominent targets for 4-HNE modification in the oocyte and have been correlatively linked with declining oocyte quality (29). Although such damage is usually mitigated by stringent surveillance and degradation pathways, this oxidative lesion can prove extremely deleterious if there is attenuation in the effectiveness of proteolysis, such as that experienced during aging (62, 69, 70). Despite this knowledge, the capacity of the proteasome to mediate clearance of 4-HNE–modified proteins in young versus aged oocytes has not yet been established. This study provides evidence that there is indeed a decrease in proteasome activity in the aged oocyte. Moreover, we have positively correlated this phenomenon with enhanced susceptibility of proteasomal subunits to 4-HNE–mediated damage under conditions of oxidative stress. We hypothesize that this situation sets in motion a cascade of events whereby reduced proteasomal activity contributes to an accumulation of damaged and/or dysfunctional 4-HNE–modified proteins (such as α-tubulin). This, in turn, contributes to deficits in the fidelity of meiosis and an attendant increase in aneuploidy rates, as is commonly observed in oocytes of women of advanced reproductive age (Fig. 10).
Figure 10.

4-HNE–mediated damage to the oocyte is exacerbated by a decrease in proteasome activity. A, under conditions of oxidative stress, ROS can elicit lipid peroxidation (B) and the generation of electrophilic aldehydes, such as 4-HNE (C). D, 4-HNE is able to covalently modify and damage a broad range of proteins, including those essential for oocyte meiosis. The proteasome is able to degrade damaged proteins, preventing them from interfering with normal cellular machinery. E, upon the induction of oxidative stress, the proteasome is also a target for 4-HNE modification, inhibiting proteasome assembly and activity (F). This ultimately results in an increase in 4-HNE–modified proteins (G) and a reduction in oocyte quality (H).
The proteasome represents a primary line of cellular defense against an array of oxidative damage. This activity is primarily attributed to its pivotal role in the degradation of irreversibly damaged, dysfunctional, and/or misfolded proteins (52–54), the abundance of which increases dramatically under conditions of oxidative stress. Indeed, when threatened with an oxidative challenge, the entire 26S holoenzyme or the 20S proteasomal core takes on the responsibility of rapid enzymatic degradation of modified proteins before they have the opportunity to accumulate and accentuate cellular damage (52–54).
Decreased proteasome activity is a common age-related phenotype reported in a multitude of mammalian cells (59–67). Our data extend these findings by not only revealing that the aged GV oocyte is vulnerable to decreased proteasome activity but also by revealing that oxidative stress is an important contributing factor toward this impaired functionality. Indeed, we demonstrate that an acute oxidative stress model could recapitulate many of the pathologies witnessed during aging (29), including a significant dose-dependent reduction in oocyte proteasome activity. This response was coupled with a marked increase in the accumulation of very high-molecular-weight polyubiquitinated proteins, mirroring those detected in aged oocytes. On the basis of these data, we infer that suppression of proteasomal activity inhibits the clearance of damaged proteins allowing the consequential formation of large ubiquitinated aggregates. This model is in agreement with the behavior of somatic cells placed under oxidative stress and thus serves to highlight the central role that the proteasome holds in maintaining normal protein homeostasis (90–94).
In cells functioning under normal physiological parameters, the activity of the proteasome is up-regulated in response to moderate levels of oxidative stress to resolve the subsequent increase in dysfunctional and misfolded proteins (95–98). In contrast, exposure to prolonged and/or intense oxidative insults results in proteasome disassembly and inhibition (53, 99). A similar scenario plays out in the presence of 4-HNE, with modest concentrations also leading to increased proteolytic flux, whereas higher concentrations exceed the protective capacity of the proteasome (100). The latter response may be attributed, at least in part, to the fact that multiple proteasome subunits are themselves vulnerable to 4-HNE adduction. Indeed, with the notable exception of β2, all other α and β subunits of the core proteasome have been shown to be readily adducted by 4-HNE. Moreover, such modifications are intimately coupled with the inhibition of the trypsin, chymotrypsin, and peptidylglutamyl peptide hydrolase activities of the proteasome (71–78). In agreement with these data, each of the subunits examined in this study proved vulnerable to 4-HNE modifications, including those responsible for maintaining the structural integrity of the proteasome (α2, α5, α6, and β7) as well as those possessing catalytic activity (β1 and β3). Taken together, these data provide a rationale for the attenuation of proteasomal activity documented in aged oocytes.
In this study, we employed immunostaining of proteasome subunits to determine the localization of the 20S and 26S proteasome as well as the immunoproteasome, with the exception of subunits β1 and β3 (which are removed from the immunoproteasome). In addition, immunolabeling also reflects the pre-assembly 20S proteasome, which accounts for the variance in the localization patterns observed between the subunits examined in our study. Indeed, there is even variation between the distribution of the core proteasome subunits due to the presence of nuclear localization signal on α subunits but not in β subunits (101, 102). Immunolocalization studies in human and rat GV oocytes have similarly documented widespread distribution of the proteasome varying from foci of enrichment within the nucleus and at the cortex of the ooplasm to perinuclear and cytoplasmic domains (56, 103).
With the goal of formulating intervention strategies that could mitigate the impact of a severely compromised proteasome in oxidatively-stressed oocytes, we observed a significant decrease in total 4-HNE–modified proteins, a complete clearance of the prominent 4-HNE target α-tubulin, and a significant recovery of proteasome activity following the provision of a 6-h (post-oxidative insult) recovery period in the presence of penicillamine. Penicillamine is a potent nucleophile and is highly efficient in reducing the bioavailability of free 4-HNE. This activity reflects the fact that the 4-HNE Michael addition kinetics for the penicillamine substrate is far greater than that of any amino acid side chain, thus enabling it to preferentially sequester free 4-HNE (28, 87) and preventing the perpetuation of further protein alkylation at this point. In addition, penicillamine acts to chelate transition metals, potentially suppressing the Fenton reactions responsible for catalyzing the lipid peroxidation cascades that culminate in reactive aldehyde production (87). In this way, penicillamine, acting as an antioxidant, can also block the initiation of cellular 4-HNE production. These two properties of penicillamine drastically limit the formation of new protein insults, thereby permitting the oocyte to eliminate already damaged proteins. However, given that these oxidatively-stressed oocytes initially harbored compromised proteasomal activity, we speculate that compensatory degradative pathways, such as those of the lysosome, may also be responsible for the resolution of 4-HNE–modified proteins observed during the recovery period. This notion is in accord with evidence that lysosomal pathways, such as autophagy, can eliminate damaged and dysfunctional proteins and thus compensate for the impairment of proteasome activity experienced under high oxidative burden (81, 82). Indeed, we have recently demonstrated that failures of placental function associated with fetal death involve 4-HNE–mediated adduction of autophagosomes (104). It is also consistent with the data presented in this study demonstrating that both proteasomal and lysosomal inhibition precipitate an accumulation of 4-HNE–modified proteins, including α-tubulin.
Having effectively rescued proteasome activity and thus resolved the bulk of 4-HNE–adducted α-tubulin, we anticipated that penicillamine treatment may have also been effective in ameliorating meiotic defects in oxidatively-stressed and aged oocytes. Notably, however, this was not the case, with H2O2/penicillamine–treated oocytes being characterized by decreased PBE rates indistinguishable from those of H2O2-treated oocytes without a recovery period. Similarly, the incidence of aneuploidy was only modestly improved in H2O2-penicillamine–treated oocytes. Such incomplete rescue of oocyte function likely indicates that the impact of oxidative stress, and/or 4-HNE generation, on oocyte quality is more pervasive than the overt modification of protein substrates such as α-tubulin. In this context, it is well established that oxidative stress and 4-HNE can readily damage nucleic acids (i.e. DNA and RNA) as well as key organelles such as the mitochondria (105). With this knowledge, it is possible that oxidatively stressed oocytes sustained damage to either their nuclear and/or mitochondrial genomes (105), neither of which would be expected to be improved by short-term penicillamine supplementation. Indeed, the increase in meiotic arrest observed in our studies is consistent with the pathology of oocytes harboring significant DNA damage (106–108). The utility of antioxidant therapies to alleviate oxidative burden in oocytes is therefore likely to be predicated on prophylactic in vivo application to suppress the cumulative burden of age-related oxidative stress, rather than as a late stage intervention to reduce cellular damage in the oocyte, once such stress has already manifested. This notion is further reinforced by our demonstration that the elevated aneuploidy rates documented in aged oocytes remained completely refractory to acute penicillamine treatment.
However, it is also notable that decreased PBE rates induced by acute exposure to oxidative stress in young oocytes did not recapitulate data from naturally aged oocytes. This discrepancy between the PBE rates of young H2O2-treated and aged oocytes may indicate that exposure to acute oxidative stress elicits effects beyond those associated with aging. Alternatively, it is possible that aged oocytes progress through MI, despite presenting with an elevated burden of oxidative stress, because of a defective spindle assembly checkpoint (SAC) (106, 109).
Although tubulin, as the dominant target for 4-HNE modification, has been the focus of this study, it is important to recognize that there are likely additional targets of 4-HNE within the oocyte proteome that may also contribute to the deterioration of oocyte quality under conditions of oxidative stress and aging. In this context, compelling evidence implicates compromise of bivalent cohesion (110, 111) in conjunction with reduced fidelity of the SAC (106, 109) as leading causes of age-induced aneuploidies. Thus, a more detailed characterization of the oocyte 4-HNE adductome, focusing on the potential for oxidative modification of components of the cohesion–SAC complexes, is warranted to refine our understanding of the mechanistic links between oxidative stress and age-related compromise of oocyte function.
Taken together, our data have established a synergistic relationship between decreased proteasome activity in the aged oocyte and an increased accumulation of 4-HNE–modified proteins. More specifically, our data implicate 4-HNE modification of proteasome subunits as a causal agent responsible for compromised protein homeostasis in the aged oocyte, with implications for the etiology of age-dependent aneuploidy. These data afford novel insight into the age-dependent decline in oocyte quality, with potential consequences for the development of rational therapeutic interventions to address this pathology.
Experimental procedures
Ethics approval
Research animals in this study were handled, monitored, and euthanized in accordance with New South Wales Animal Research Act 1998, New South Wales Animal Research Regulation 2010, and the Australian Code for the Care and Use of Animals for Scientific Purposes (8th Ed.) as approved by the University of Newcastle Animal Care and Ethics Committee (approval number A-2011-162). C57/BL6×CBA F1 hybrid female mice were bred and held at the Institute's Central Animal House with food and water ad libitum. Animals were housed under a 12-h light/12-h dark cycle at a constant temperature of 21–22 °C and euthanized immediately before use via cervical dislocation.
Reagents
All chemicals and reagents used were of research grade and were supplied by ThermoFisher Scientific (Waltham, MA) or Sigma unless otherwise specified. All details concerning the purchase and use of primary antibodies in immunolocalization and immunoblotting assays are reported in Table S1. Appropriate rabbit anti-mouse (catalog no. Ab6728) and goat anti-rabbit (catalog no. DC03L) HRP-conjugated secondary antibodies were obtained from Abcam (Melbourne, Victoria, Australia) and Calbiochem. Alexa Fluor 488-conjugated goat anti-mouse (catalog no. A-11001), Alexa Fluor 555-conjugated goat anti-human (catalog no. A-21433), and Alexa Fluor 633-conjugated goat anti-rabbit (catalog no. A-21070) were purchased from ThermoFisher Scientific.
Oocyte collection
Oocytes were isolated as described previously (113). Briefly, mice between 4 and 6 weeks of age were administered intraperitoneal injections of 7.5 IU equine chorionic gonadotropin (eCG) (Intervet, Sydney, New South Wales, Australia) to stimulate in vivo oocyte maturation to mature GV stage. Forty eight hours following eCG injection, animals were euthanized, and ovaries were removed. Pre-ovulatory follicles were repeatedly punctured with a 27-gauge needle to release mature GV oocytes as cumulus–oocyte complexes into pre-warmed (37 °C) M2 media supplemented with 2.5 μm milrinone to maintain GV arrest. C57Bl/6×CBA hybrid cross (F1) mice at 14 months of age were utilized for the study of aged oocytes as approximately half of these cells exhibit aneuploidy, which is equivalent to that of a woman in her 40s (3, 4, 114–116). Aged mice and young controls were not hormonally stimulated. Only oocytes with an intact layer of cumulus cells were recovered. Cumulus cells were mechanically removed via repeated aspiration with a narrow pipette at 37 °C.
Induction of oxidative stress
To examine the effects of oxidative stress on proteasome activity and 4-HNE adduct clearance, GV oocytes were treated with either H2O2 (0–50 μm) for 1 h or 4-HNE (0–30 μm) for 2 h at 37 °C in M2 media supplemented with 2.5 μm milrinone held under mineral oil. These concentrations were selected as they elicit 4-HNE generation approximate to what is observed in the aged oocyte and disrupt oocyte quality but not vitality (29). In addition, these concentrations are also comparable with those of moderate levels of physiological oxidative stress (100 μm H2O2 and 5 mm 4-HNE have been reported in biological fluids and cellular membranes, respectively) (117, 118). Following treatment, GV oocytes were either processed immediately or underwent in vitro maturation (IVM).
In vitro maturation
For IVM, oocytes were washed out of milrinone by aspiration through four 50-μl droplets of α-MEM (catalog no. 11900024, ThermoFisher Scientific) supplemented with 20% (v/v) fetal calf serum, 50 units/ml penicillin, and 50 μg/ml streptomycin. Oocytes were then placed into a single-well IVF dish (catalog no. 353653), containing 500 μl of α-MEM. Oocytes were cultured at 37 °C in an atmosphere of 5% CO2 for 7.5 h for MI or 16 h for MII stage oocytes. All media and mineral oil were equilibrated at 37 °C in an atmosphere of 5% CO2 for a minimum of 3 h before use. Following IVM, oocyte maturation was scored, with GV oocytes being identified by the presence of a nuclear envelope and nucleolus and MII oocytes identified via the presence of the first polar body (119, 120). For cold shock destabilization assays, oocytes were exposed to 4 °C for exactly 7 min in pre-chilled M2 media prior to fixation.
Immunocytochemistry
Following the induction of oxidative stress, oocytes were washed in phosphate-buffered saline (PBS) containing 3 mg/ml polyvinylpyrrolidone (PVP) before being fixed and permeabilized in 2% paraformaldehyde (w/v) diluted in PHEM, 0.5% Triton X-100 (v/v) for 30 min. Fixed oocytes were blocked in 7% goat serum (v/v) and 1% BSA (w/v) prepared in PBS, 0.1% Tween 20 (PBST) for 1 h at room temperature. Cells were then incubated with anti-4-HNE, anti-α-tubulin, anti-CREST, or anti-proteasome subunit antibodies diluted to appropriate concentrations (Table S1) in 1% BSA (w/v)/PBST overnight at 4 °C. After washing in 1% BSA (w/v)/PBST, oocytes were incubated with appropriate Alexa Fluor-conjugated secondary antibodies (diluted 1:1000 in 1% BSA (w/v)/PBST) for 1 h at room temperature. All experiments included negative control groups in which the primary antibody was substituted with the appropriate concentration of either anti-mouse or anti-rabbit IgG (catalog no. sc-2027; sc-2025) in 1% BSA (w/v)/PBST. Oocytes were counterstained with Hoechst 33258 (20 μg/ml) diluted in PBS/PVP for 15 min at room temperature. Finally, oocytes were mounted on Menzel Gläser microscope slides (ThermoFisher Scientific) in Citifluor Glycerol Solution AF2 (catalog no. AGR1321, Citifluor Ltd., London, UK). To ensure accurate fluorescence quantification, all oocytes used in a single experimental replicate were collected, treated, fixed, and permeabilized concurrently. Immunostaining protocols were also performed with all treatment groups concurrently with equivalent antibody concentrations, volumes, and incubation times. Oocytes were then mounted in equal volumes of Citifluor to minimize quenching of fluorescence signals during image capture using an Olympus FV1000 confocal microscope.
Assessment of oocyte aneuploidy status
The aneuploidy status of oocytes was assessed following treatment with either H2O2 or 4-HNE, as described previously (121, 122). Briefly, following H2O2 or 4-HNE exposure at the GV stage of development, oocytes were subjected to IVM. The resultant populations of oocytes were then incubated with 200 μm monastrol in α-MEM for 2 h at 37 °C in 5% CO2. Oocytes were fixed, and kinetochores were immunostained with anti-CREST antibodies as described above.
Proximity ligation assays
Proximity ligation assays were performed on fixed oocytes using the Duolink In Situ Red Starter kit as per the manufacturer's instructions (catalog no. DUO92101-1KT, Sigma). Briefly, oocytes were blocked with Duolink blocking solution for 30 min at 37 °C and incubated with appropriate pairs of primary antibodies (i.e. anti-4-HNE and either anti-α-tubulin or anti-proteasomal subunit antibodies) diluted in Duolink antibody buffer overnight at 4 °C. Labeled oocytes were then washed thoroughly with PBS/PVP prior to incubation with oligonucleotide-conjugated secondary antibodies (PLA probes) for 1 h at 37 °C. After additional washes, the ligation and amplification of PLA probes were conducted in accordance with the manufacturer's instructions. Finally, oocytes were counterstained with Hoechst 33258, and images were acquired using an Olympus FV1000 confocal microscope. Negative control incubations used to confirm specificity of the assay included antibody pairs targeting proteins that would not be expected to interact (i.e. anti-PIWIL1 and anti-4-HNE antibodies) as well as the omission of each primary antibody.
Confocal imaging
All cell images were captured using high-resolution confocal microscopy on an Olympus FV1000 confocal microscope. Oocytes were imaged under a ×60 oil immersion lens with a z-resolution of 0.5 μm (CREST) or 1 μm (α-tubulin). Fluorochromes were imaged sequentially to avoid bleed through. Oocytes from each treatment group were imaged with identical parameters on the same day to minimize fluorescence fading.
Quantification of fluorescence intensity
For GV oocytes, images chosen for quantification were those captured through the mid-section of the oocyte, positioned to incorporate the center of the nucleolus and thus encompass the center of the nucleus as well as the cytoplasm. The entire area within the oocyte was used for immunocytochemical and PLA quantification. For cold-shock destabilization assays, whole spindles were z-stacked for quantification. Fluorescence intensity for immunocytochemistry was measured using ImageJ (National Institutes of Health). The integrated fluorescence intensity of a mid-section (encompassing the DNA) of the whole oocyte was determined, and the background fluorescence was measured at four locations on the image and averaged. For determination of fluorescence intensity in captured images, the corrected total cell fluorescence (CTCF), or normalized fluorescence, was used as described in the following equation: CTCF = integrated fluorescence intensity − (area of selected cell × average background fluorescence). This measurement considers differences in the size of oocytes via correction of the background staining intensity for the size of the cell. Data collected from individual experimental replicates were normalized to appropriate untreated controls.
Microtubule/tubulin in vivo assay
To assess the impact of 4-HNE modification on the microtubule versus free-tubulin content in oocytes, a microtubule/tubulin in vivo assay was performed as per the manufacturer's instructions (catalog no. BK038; Cytoskeleton, Inc., Denver, CO). Briefly, 150 oocytes per sample were lysed in 50 μl of microtubule stabilization buffer supplemented with 100 μm GTP, 1 mm ATP, and a protease inhibitor mixture. Oocyte lysates were centrifuged for 5 min at 1000 × g at 37 °C. The resultant supernatant was aspirated and re-centrifuged at 100,000 × g for 60 min at 37 °C to separate microtubules (HSP) from the nonpolymerized tubulin (HSS). The HSP and HSS were resuspended separately in SDS sample buffer in preparation for separation by SDS-PAGE and immunoblotting with anti-tubulin antibodies to determine the ratio of tubulin incorporated into the microtubules versus the free-tubulin found in the oocyte cytosol.
SDS-PAGE and immunoblotting
Electrophoretic resolution of oocyte proteins was conducted by SDS-PAGE using conventional procedures with minor modifications (123). Briefly, protein was extracted from isolated oocytes via direct incubation in SDS extraction buffer comprising 2% SDS (w/v), 10% sucrose (w/v) in 0.1875 m Tris, pH 6.8, and supplemented with ProteCEASE protease inhibitors (catalog no. 786-326, G-Biosciences) and boiling (100 °C for 5 min). Entire protein lysates recovered from 100 oocytes were diluted into SDS-PAGE loading buffer containing 2% β-mercaptoethanol and bromphenol blue before being resolved on NuPAGE 4–12% BisTris gels (catalog no. NP0321BOX, ThermoFisher Scientific) and transferred using an XCell Blot Module (catalog no. EI9051, ThermoFisher Scientific) onto nitrocellulose membranes (catalog no. 10600002, GE Healthcare, Buckinghamshire, UK). Membranes were blocked by incubation in 3% BSA (w/v)/Tris-buffered saline (TBS; pH 7.4) and 0.1% Tween 20 (TBST) for 2 h at room temperature before being incubated with anti-4-HNE, anti-α-tubulin, UB-K48, or anti-GAPDH antibodies each diluted in 1% BSA (w/v)/TBST overnight at 4 °C. Membranes were washed three times with TBST and incubated with horseradish peroxidase-conjugated secondary antibody diluted into 1% BSA (w/v)/TBST for 1 h. Following three washes in TBST, labeled proteins were detected using an enhanced chemiluminescence kit (catalog no. RPN2106, GE Healthcare) and visualized using ImageQuant LAS 4000 (Fujifilm, Tempe, AZ). Band density was quantified in three replicate blots using ImageJ software (version 1.48 v; National Institutes of Health, Bethesda, MD) and expressed relative to GAPDH labeling intensity.
Proteasome activity assay and inhibitor studies
Proteasome activity assays were performed as previously reported with minor modifications (112). To assess oocyte proteasome activity in response to aging, H2O2, and 4-HNE treatment, 100 oocytes per group were collected and washed three times in PBS/PVP. All cell suspensions were lysed in 20 μl of protein extraction buffer composed of 150 mm sodium chloride, 50 mm Tris, and 0.5% Triton X-100 for 30 min under constant rotation at 4 °C. After centrifugation at 16,300 × g for 15 min, supernatants were transferred to a clean tube and then assessed for proteasome activity using a commercial proteasome assay kit, which utilizes a 7-amino-4-methylcoumarin–tagged peptide substrate that releases highly fluorescent AMC upon proteolytic cleavage (Abcam, ab107921). Briefly, 10 μl of each sample was loaded into a 96-well plate in duplicate, alongside a Jurkat cell lysate-positive control (supplied) and AMC protein standards. A total of 50 μm proteasome inhibitor MG132 was added to one well of each sample to differentiate proteasome activity from other protease activity that may be present in the samples.
Plates were incubated for 60 min and then analyzed on a BMG Fluostar Optima plate reader (BMG Labtech, Mornington, Victoria, Australia) at an excitation/emission of 350/440 nm. Following a further 30-min incubation at 37 °C, plates were analyzed a second time to allow the change in relative fluorescence units to be calculated for each sample. Data were analyzed following the manufacturers' instructions, and proteasome activity was calculated such that one unit of proteasome activity is equivalent to the amount of proteasome activity that generates 1.0 nmol of AMC per min at 37 °C.
To further investigate the role of the proteasome, oocytes were treated with either a proteasome inhibitor MG132 (50 μm) and/or a lysosome inhibitor (chloroquine: 100 μm) for up to 6 h post-induction of oxidative stress with 35 μm H2O2 for 1 h at 37 °C in M2 media. Oocytes were then prepared for immunocytochemical and immunoblotting experiments with anti-α-tubulin and anti-4-HNE antibodies (as above).
Penicillamine treatment
In an effort to examine 4-HNE metabolism and to explore the possibility that oocyte quality could be restored after the induction of oxidative stress, oocytes were allowed to recover after the initial oxidative insult. For this purpose, GV oocytes were exposed to 35 μm H2O2 for 1 h in M2 media and allowed to recover for 6 h in α-MEM at 37 °C in 5% CO2 (as above) supplemented with 100 μm of the nucleophile, d-penicillamine (catalog no. P4875-1G, Sigma), to limit the bioavailability of 4-HNE and prevent additional 4-HNE modifications from occurring (28, 88, 89). After recovery, the extent of 4-HNE adduction to tubulin, polar body extrusion, and aneuploid status of the oocytes after IVM was assessed, relative to untreated controls.
Statistical analysis
Statistical analysis was performed using two-tailed unpaired Student's t tests and one-way analyses of variance (ANOVA) with Tukey's post-hoc multiple comparison using Graphpad Prism 7 software (San Diego). A p value of <0.05 was considered significant. Experiments were performed in biological triplicate unless otherwise stated. Statistical analyses were performed using the mean of each biological replicate ± S.E.
Author contributions
B. P. M. data curation; B. P. M. formal analysis; B. P. M. investigation; B. P. M., E. G. B., J. M. S., G. N. D. I., E. A. M., R. J. A., and B. N. methodology; B. P. M. writing-original draft; B. P. M., E. G. B., J. M. S., G. N. D. I., E. A. M., R. J. A., and B. N. writing-review and editing; E. A. M. and B. N. conceptualization; E. A. M. and B. N. supervision; E. A. M. and B. N. project administration; B. N. funding acquisition.
Supplementary Material
Acknowledgments
We gratefully acknowledge Nicole J. Camlin, Jacinta H. Martin, and Shaun D. Roman for their technical advice and critical feedback.
This work was supported in part by the University of Newcastle's Priority Research Centre for Reproductive Science and the Hunter Medical Research Institute's (HMRI) Pregnancy and Reproduction Program. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Table S1.
- ROS
- reactive oxygen species
- 4-HNE
- 4-hydroxynonenal
- GV
- germinal vesicle
- MI
- metaphase I
- MII
- metaphase II
- IVF
- in vitro fertilization
- IVM
- in vitro maturation
- PBE
- polar body extrusion
- k-mt
- kinetochore–microtubule
- H2O2
- hydrogen peroxide
- IB
- immunoblotting
- PLA
- proximity ligation assays
- ANOVA
- analysis of variance
- eCG
- equine chorionic gonadotropin
- ICC
- immunocytochemistry
- IB
- immunoblot
- PLA
- proximity ligation assay
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- α-MEM
- α-minimal essential medium
- 4-HNE
- 4-hydroxynonenal
- HSP
- high-speed pellet
- HSS
- high-speed supernatant
- SAC
- spindle assembly checkpoint
- PVP
- polyvinylpyrrolidone
- AMC
- 7-amino-4-methylcoumarin
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. McLaughlin E. A., and McIver S. C. (2009) Awakening the oocyte: controlling primordial follicle development. Reproduction 137, 1–11 10.1530/REP-08-0118 [DOI] [PubMed] [Google Scholar]
- 2. Kerr J. B., Myers M., and Anderson R. A. (2013) The dynamics of the primordial follicle reserve. Reproduction 146, R205–R215 10.1530/REP-13-0181 [DOI] [PubMed] [Google Scholar]
- 3. Pan H., Ma P., Zhu W., and Schultz R. M. (2008) Age-associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev. Biol. 316, 397–407 10.1016/j.ydbio.2008.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiang T., Schultz R. M., and Lampson M. A. (2012) Meiotic origins of maternal age-related aneuploidy. Biol. Reprod. 86, 1–7 10.1095/biolreprod.111.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silber S. J., Kato K., Aoyama N., Yabuuchi A., Skaletsky H., Fan Y., Shinohara K., Yatabe N., and Kobayashi T. (2017) Intrinsic fertility of human oocytes. Fertil. Steril. 107, 1232–1237 10.1016/j.fertnstert.2017.03.014 [DOI] [PubMed] [Google Scholar]
- 6. Tarín J. J. (1995) Aetiology of age-associated aneuploidy: a mechanism based on the ‘free radical theory of ageing’. Hum. Reprod. 10, 1563–1565 10.1093/HUMREP/10.6.1563 [DOI] [PubMed] [Google Scholar]
- 7. Tarín J. J. (1996) Potential effects of age-associated oxidative stress on mammalian oocytes/embryos. Mol. Hum. Reprod. 2, 717–724 10.1093/molehr/2.10.717 [DOI] [PubMed] [Google Scholar]
- 8. Harman D. (1956) Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 10.1093/geronj/11.3.298 [DOI] [PubMed] [Google Scholar]
- 9. Harman D. (1972) The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 20, 145–147 10.1111/j.1532-5415.1972.tb00787.x [DOI] [PubMed] [Google Scholar]
- 10. Elizur S. E., Lebovitz O., Orvieto R., Dor J., and Zan-Bar T. (2014) Reactive oxygen species in follicular fluid may serve as biochemical markers to determine ovarian aging and follicular metabolic age. Gynecol. Endocrinol. 30, 705–707 10.3109/09513590.2014.924100 [DOI] [PubMed] [Google Scholar]
- 11. Lim J., and Luderer U. (2011) Oxidative damage increases and antioxidant gene expression decreases with aging in the mouse ovary. Biol. Reprod. 84, 775–782 10.1095/biolreprod.110.088583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeo S., Kimura K., Shirasuna K., Kuwayama T., and Iwata H. (2017) Age-associated deterioration in follicular fluid induces a decline in bovine oocyte quality. Reprod. Fertil. Dev. 29, 759–767 10.1071/RD15228 [DOI] [PubMed] [Google Scholar]
- 13. Wiener-Megnazi Z., Vardi L., Lissak A., Shnizer S., Reznick A. Z., Ishai D., Lahav-Baratz S., Shiloh H., Koifman M., and Dirnfeld M. (2004) Oxidative stress indices in follicular fluid as measured by the thermochemiluminescence assay correlate with outcome parameters in in vitro fertilization. Fertil. Steril. 82, 1171–1176 10.1016/j.fertnstert.2004.06.013 [DOI] [PubMed] [Google Scholar]
- 14. Babuška V., Cedíková M., Rajdl D., Racek J., Zech N. H., Trefil L., Mocková A., Ulčová-Gallova Z., Novotný Z., and Králičková M. (2012) Comparison of selective oxidative stress parameters in the follicular fluid of infertile women and healthy fertile oocyte donors. Ceska Gynekol 77, 543–548 [PubMed] [Google Scholar]
- 15. Das S., Chattopadhyay R., Ghosh S., Ghosh S., Goswami S. K., Chakravarty B. N., and Chaudhury K. (2006) Reactive oxygen species level in follicular fluid—embryo quality marker in IVF? Hum. Reprod. 21, 2403–2407 10.1093/humrep/del156 [DOI] [PubMed] [Google Scholar]
- 16. Nuñez-Calonge R., Cortés S., Gutierrez Gonzalez L. M., Kireev R., Vara E., Ortega L., Caballero P., Rancan L., and Tresguerres J. (2016) Oxidative stress in follicular fluid of young women with low response compared with fertile oocyte donors. Reprod. Biomed. Online 32, 446–456 10.1016/j.rbmo.2015.12.010 [DOI] [PubMed] [Google Scholar]
- 17. Tamura H., Takasaki A., Miwa I., Taniguchi K., Maekawa R., Asada H., Taketani T., Matsuoka A., Yamagata Y., Shimamura K., Morioka H., Ishikawa H., Reiter R. J., and Sugino N. (2008) Oxidative stress impairs oocyte quality and melatonin protects oocytes from free radical damage and improves fertilization rate. J. Pineal Res. 44, 280–287 10.1111/j.1600-079X.2007.00524.x [DOI] [PubMed] [Google Scholar]
- 18. Agarwal A., Aponte-Mellado A., Premkumar B. J., Shaman A., and Gupta S. (2012) The effects of oxidative stress on female reproduction: a review. Reprod. Biol. Endocrinol. 10, 49 10.1186/1477-7827-10-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chaube S. K., Prasad P. V., Thakur S. C., and Shrivastav T. G. (2005) Hydrogen peroxide modulates meiotic cell cycle and induces morphological features characteristic of apoptosis in rat oocytes cultured in vitro. Apoptosis 10, 863–874 10.1007/s10495-005-0367-8 [DOI] [PubMed] [Google Scholar]
- 20. Choi W. J., Banerjee J., Falcone T., Bena J., Agarwal A., and Sharma R. K. (2007) Oxidative stress and tumor necrosis factor-α-induced alterations in metaphase II mouse oocyte spindle structure. Fertil. Steril. 88, 1220–1231 10.1016/j.fertnstert.2007.02.067 [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., Wu X. Q., Lu S., Guo Y. L., and Ma X. (2006) Deficit of mitochondria-derived ATP during oxidative stress impairs mouse MII oocyte spindles. Cell Res. 16, 841–850 10.1038/sj.cr.7310095 [DOI] [PubMed] [Google Scholar]
- 22. Liu L., and Keefe D. L. (2002) Ageing-associated aberration in meiosis of oocytes from senescence-accelerated mice. Hum. Reprod. 17, 2678–2685 10.1093/humrep/17.10.2678 [DOI] [PubMed] [Google Scholar]
- 23. Tarín J. J., Vendrell F. J., Ten J., Blanes R., van Blerkom J., and Cano A. (1996) The oxidizing agent tertiary butyl hydroperoxide induces disturbances in spindle organization, c-meiosis, and aneuploidy in mouse oocytes. Mol. Hum. Reprod. 2, 895–901 10.1093/molehr/2.12.895 [DOI] [PubMed] [Google Scholar]
- 24. Bhattacharya S., Maheshwari A., and Mollison J. (2013) Factors associated with failed treatment: an analysis of 121,744 women embarking on their first IVF cycles. PLoS ONE 8, e82249 10.1371/journal.pone.0082249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tarín J. J., Gómez-Piquer V., Pertusa J. F., Hermenegildo C., and Cano A. (2004) Association of female aging with decreased parthenogenetic activation, raised MPF, and MAPKs activities and reduced levels of glutathione S-transferases activity and thiols in mouse oocytes. Mol. Reprod. Dev. 69, 402–410 10.1002/mrd.20180 [DOI] [PubMed] [Google Scholar]
- 26. Schwartz D., and Mayaux M. J. (1982) Female fecundity as a function of age: results of artificial insemination in 2193 nulliparous women with azoospermic husbands. Federation CECOS. N. Engl. J. Med. 306, 404–406 10.1056/NEJM198202183060706 [DOI] [PubMed] [Google Scholar]
- 27. Devroey P., Godoy H., Smitz J., Camus M., Tournaye H., Derde M. P., and Van Steirteghem A. (1996) Female age predicts embryonic implantation after ICSI: a case-controlled study. Hum. Reprod. 11, 1324–1327 10.1093/oxfordjournals.humrep.a019380 [DOI] [PubMed] [Google Scholar]
- 28. Lord T., Martin J. H., and Aitken R. J. (2015) Accumulation of electrophilic aldehydes during postovulatory aging of mouse oocytes causes reduced fertility, oxidative stress, and apoptosis. Biol. Reprod. 92, 33 [DOI] [PubMed] [Google Scholar]
- 29. Mihalas B. P., De Iuliis G. N., Redgrove K. A., McLaughlin E. A., and Nixon B. (2017) The lipid peroxidation product 4-hydroxynonenal contributes to oxidative stress-mediated deterioration of the ageing oocyte. Sci. Rep. 7, 6247 10.1038/s41598-017-06372-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yin H., Xu L., and Porter N. A. (2011) Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 111, 5944–5972 10.1021/cr200084z [DOI] [PubMed] [Google Scholar]
- 31. Girotti A. W. (1998) Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 39, 1529–1542 [PubMed] [Google Scholar]
- 32. Kanner J., German J. B., and Kinsella J. E. (1987) Initiation of lipid peroxidation in biological systems. Crit. Rev. Food Sci. Nutr. 25, 317–364 10.1080/10408398709527457 [DOI] [PubMed] [Google Scholar]
- 33. Ayala A., Muñoz M. F., and Argüelles S. (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 360438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Esterbauer H. (1993) Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 57, S779–S786 10.1093/ajcn/57.5.779S [DOI] [PubMed] [Google Scholar]
- 35. Monroy C. A., Doorn J. A., and Roman D. L. (2013) Modification and functional inhibition of regulator of G-protein signaling 4 (RGS4) by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 26, 1832–1839 10.1021/tx400212q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uchida K., and Stadtman E. R. (1992) Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. U.S.A. 89, 4544–4548 10.1073/pnas.89.10.4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kinnunen P. K., Kaarniranta K., and Mahalka A. K. (2012) Protein-oxidized phospholipid interactions in cellular signaling for cell death: from biophysics to clinical correlations. Biochim. Biophys. Acta 1818, 2446–2455 10.1016/j.bbamem.2012.04.008 [DOI] [PubMed] [Google Scholar]
- 38. Pizzimenti S., Ciamporcero E., Daga M., Pettazzoni P., Arcaro A., Cetrangolo G., Minelli R., Dianzani C., Lepore A., Gentile F., and Barrera G. (2013) Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 4, 242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dalleau S., Baradat M., Guéraud F., and Huc L. (2013) Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 20, 1615–1630 10.1038/cdd.2013.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doorn J. A., and Petersen D. R. (2002) Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol. 15, 1445–1450 10.1021/tx025590o [DOI] [PubMed] [Google Scholar]
- 41. Sayre L. M., Lin D., Yuan Q., Zhu X., and Tang X. (2006) Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab. Rev. 38, 651–675 10.1080/03602530600959508 [DOI] [PubMed] [Google Scholar]
- 42. Esterbauer H., Schaur R. J., and Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 [DOI] [PubMed] [Google Scholar]
- 43. Aiken C. E., Tarry-Adkins J. L., and Ozanne S. E. (2015) Transgenerational developmental programming of ovarian reserve. Sci. Rep. 5, 16175 10.1038/srep16175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Compton D. A. (2011) Mechanisms of aneuploidy. Curr. Opin. Cell Biol. 23, 109–113 10.1016/j.ceb.2010.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Q., and Sun Q. Y. (2007) Evaluation of oocyte quality: morphological, cellular and molecular predictors. Reprod. Fertil. Dev. 19, 1–12 10.1071/RD06103 [DOI] [PubMed] [Google Scholar]
- 46. Baker M. A., Weinberg A., Hetherington L., Villaverde A. I., Velkov T., Baell J., and Gordon C. P. (2015) Defining the mechanisms by which the reactive oxygen species by-product, 4-hydroxynonenal, affects human sperm cell function. Biol. Reprod. 92, 108 [DOI] [PubMed] [Google Scholar]
- 47. Chavez J., Chung W.-G., Miranda C. L., Singhal M., Stevens J. F., and Maier C. S. (2010) Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: protein carbonylation is diminished by ascorbic acid. Chem. Res. Toxicol. 23, 37–47 10.1021/tx9002462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stewart B. J., Doorn J. A., and Petersen D. R. (2007) Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 20, 1111–1119 10.1021/tx700106v [DOI] [PubMed] [Google Scholar]
- 49. Kokubo J., Nagatani N., Hiroki K., Kuroiwa K., Watanabe N., and Arai T. (2008) Mechanism of destruction of microtubule structures by 4-hydroxy-2-nonenal. Cell Struct. Funct. 33, 51–59 10.1247/csf.07038 [DOI] [PubMed] [Google Scholar]
- 50. Neely M. D., Sidell K. R., Graham D. G., and Montine T. J. (1999) The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J. Neurochem. 72, 2323–2333 [DOI] [PubMed] [Google Scholar]
- 51. Wang X., Yang Y., and Huycke M. M. (2015) Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 64, 459–468 10.1136/gutjnl-2014-307213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shang F., Nowell T. R. Jr, and Taylor A. (2001) Removal of oxidatively damaged proteins from lens cells by the ubiquitin-proteasome pathway. Exp. Eye Res. 73, 229–238 10.1006/exer.2001.1029 [DOI] [PubMed] [Google Scholar]
- 53. Davies K. J. (2001) Degradation of oxidized proteins by the 20S proteasome. Biochimie 83, 301–310 10.1016/S0300-9084(01)01250-0 [DOI] [PubMed] [Google Scholar]
- 54. Shringarpure R., Grune T., Mehlhase J., and Davies K. J. (2003) Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 278, 311–318 10.1074/jbc.M206279200 [DOI] [PubMed] [Google Scholar]
- 55. Huo L. J., Fan H. Y., Zhong Z. S., Chen D. Y., Schatten H., and Sun Q. Y. (2004) Ubiquitin-proteasome pathway modulates mouse oocyte meiotic maturation and fertilization via regulation of MAPK cascade and cyclin B1 degradation. Mech. Dev. 121, 1275–1287 10.1016/j.mod.2004.05.007 [DOI] [PubMed] [Google Scholar]
- 56. Josefsberg L. B., Galiani D., Dantes A., Amsterdam A., and Dekel N. (2000) The proteasome is involved in the first metaphase-to-anaphase transition of meiosis in rat oocytes. Biol. Reprod. 62, 1270–1277 10.1095/biolreprod62.5.1270 [DOI] [PubMed] [Google Scholar]
- 57. Elofsson M., Splittgerber U., Myung J., Mohan R., and Crews C. M. (1999) Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide α′,β′-epoxyketones. Chem. Biol. 6, 811–822 10.1016/S1074-5521(99)80128-8 [DOI] [PubMed] [Google Scholar]
- 58. Berkers C. R., Verdoes M., Lichtman E., Fiebiger E., Kessler B. M., Anderson K. C., Ploegh H. L., Ovaa H., and Galardy P. J. (2005) Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat. Methods 2, 357–362 10.1038/nmeth759 [DOI] [PubMed] [Google Scholar]
- 59. Bulteau A. L., Petropoulos I., and Friguet B. (2000) Age-related alterations of proteasome structure and function in aging epidermis. Exp. Gerontol. 35, 767–777 10.1016/S0531-5565(00)00136-4 [DOI] [PubMed] [Google Scholar]
- 60. Carrard G., Dieu M., Raes M., Toussaint O., and Friguet B. (2003) Impact of ageing on proteasome structure and function in human lymphocytes. Int. J. Biochem. Cell Biol. 35, 728–739 10.1016/S1357-2725(02)00356-4 [DOI] [PubMed] [Google Scholar]
- 61. Chondrogianni N., Stratford F. L., Trougakos I. P., Friguet B., Rivett A. J., and Gonos E. S. (2003) Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 278, 28026–28037 10.1074/jbc.M301048200 [DOI] [PubMed] [Google Scholar]
- 62. Petropoulos I., Conconi M., Wang X., Hoenel B., Brégégère F., Milner Y., and Friguet B. (2000) Increase of oxidatively modified protein is associated with a decrease of proteasome activity and content in aging epidermal cells. J. Gerontol. A. Biol. Sci. Med. Sci. 55, B220–B227 10.1093/gerona/55.5.B220 [DOI] [PubMed] [Google Scholar]
- 63. Wagner B. J., and Margolis J. W. (1995) Age-dependent association of isolated bovine lens multicatalytic proteinase complex (proteasome) with heat-shock protein 90, an endogenous inhibitor. Arch. Biochem. Biophys. 323, 455–462 10.1006/abbi.1995.0067 [DOI] [PubMed] [Google Scholar]
- 64. Bardag-Gorce F., Farout L., Veyrat-Durebex C., Briand Y., and Briand M. (1999) Changes in 20S proteasome activity during ageing of the LOU rat. Mol. Biol. Rep. 26, 89–93 10.1023/A:1006968208077 [DOI] [PubMed] [Google Scholar]
- 65. Bulteau A. L., Szweda L. I., and Friguet B. (2002) Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 397, 298–304 10.1006/abbi.2001.2663 [DOI] [PubMed] [Google Scholar]
- 66. Conconi M., Szweda L. I., Levine R. L., Stadtman E. R., and Friguet B. (1996) Age-related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat-shock protein 90. Arch. Biochem. Biophys. 331, 232–240 10.1006/abbi.1996.0303 [DOI] [PubMed] [Google Scholar]
- 67. Shibatani T., Nazir M., and Ward W. F. (1996) Alteration of rat liver 20S proteasome activities by age and food restriction. J. Gerontol. A. Biol. Sci. Med. Sci. 51, B316–B322 [DOI] [PubMed] [Google Scholar]
- 68. Tomaru U., Takahashi S., Ishizu A., Miyatake Y., Gohda A., Suzuki S., Ono A., Ohara J., Baba T., Murata S., Tanaka K., and Kasahara M. (2012) Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 180, 963–972 10.1016/j.ajpath.2011.11.012 [DOI] [PubMed] [Google Scholar]
- 69. Reeg S., and Grune T. (2015) Protein oxidation in aging: does it play a role in aging progression? Antioxid. Redox Signal. 23, 239–255 10.1089/ars.2014.6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grune T., Shringarpure R., Sitte N., and Davies K. (2001) Age-related changes in protein oxidation and proteolysis in mammalian cells. J. Gerontol. A. Biol. Sci. Med. Sci. 56, B459–B467 10.1093/gerona/56.11.B459 [DOI] [PubMed] [Google Scholar]
- 71. Ferrington D. A., and Kapphahn R. J. (2004) Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 578, 217–223 10.1016/j.febslet.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 72. Hyun D. H., Lee M. H., Halliwell B., and Jenner P. (2002) Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: a mechanism contributing to neurodegeneration? J. Neurochem. 83, 360–370 10.1046/j.1471-4159.2002.01125.x [DOI] [PubMed] [Google Scholar]
- 73. Okada K., Wangpoengtrakul C., Osawa T., Toyokuni S., Tanaka K., and Uchida K. (1999) 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 274, 23787–23793 10.1074/jbc.274.34.23787 [DOI] [PubMed] [Google Scholar]
- 74. Shringarpure R., Grune T., Sitte N., and Davies K. J. (2000) 4-Hydroxynonenal-modified amyloid-β peptide inhibits the proteasome: possible importance in Alzheimer's disease. Cell. Mol. Life Sci. 57, 1802–1809 10.1007/PL00000660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Farout L., Mary J., Vinh J., Szweda L. I., and Friguet B. (2006) Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and depandant on 20S proteasome subtypes. Arch. Biochem. Biophys. 453, 135–142 10.1016/j.abb.2006.02.003 [DOI] [PubMed] [Google Scholar]
- 76. Just J., Jung T., Friis N. A., Lykkemark S., Drasbek K., Siboska G., Grune T., and Kristensen P. (2015) Identification of an unstable 4-hydroxynonal modification on the 20S proteasome subunit α7 by recombinant antibody technology. Free Radic. Biol. Med. 89, 786–792 10.1016/j.freeradbiomed.2015.10.405 [DOI] [PubMed] [Google Scholar]
- 77. Bulteau A. L., Lundberg K. C., Humphries K. M., Sadek H. A., Szweda P. A., Friguet B., and Szweda L. I. (2001) Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J. Biol. Chem. 276, 30057–30063 10.1074/jbc.M100142200 [DOI] [PubMed] [Google Scholar]
- 78. Höhn T. J., and Grune T. (2014) The proteasome and the degradation of oxidized proteins: part III—Redox regulation of the proteasomal system. Redox Biol. 2, 388–394 10.1016/j.redox.2013.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Saez I., and Vilchez D. (2014) The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 15, 38–51 10.2174/138920291501140306113344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Han S. J., Martins J. P. S., Yang Y., Kang M. K., Daldello E. M., and Conti M. (2017) The translation of cyclin B1 and B2 is differentially regulated during mouse oocyte reentry into the meiotic cell cycle. Sci. Rep. 7, 14077 10.1038/s41598-017-13688-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ding W.-X., Ni H.-M., Gao W., Yoshimori T., Stolz D. B., Ron D., and Yin X.-M. (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 171, 513–524 10.2353/ajpath.2007.070188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- 83. Koh Y. H., von Arnim C. A., Hyman B. T., Tanzi R. E., and Tesco G. (2005) BACE is degraded via the lysosomal pathway. J. Biol. Chem. 280, 32499–32504 10.1074/jbc.M506199200 [DOI] [PubMed] [Google Scholar]
- 84. Dunmore B. J., Drake K. M., Upton P. D., Toshner M. R., Aldred M. A., and Morrell N. W. (2013) The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum. Mol. Genet. 22, 3667–3679 10.1093/hmg/ddt216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 10.1038/nmeth947 [DOI] [PubMed] [Google Scholar]
- 86. Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras K., Gústafsdottir S. M., Ostman A., and Landegren U. (2002) Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20, 473–477 10.1038/nbt0502-473 [DOI] [PubMed] [Google Scholar]
- 87. Aitken R. J., Gibb Z., Mitchell L. A., Lambourne S. R., Connaughton H. S., and De Iuliis G. N. (2012) Sperm motility is lost in vitro as a consequence of mitochondrial free radical production and the generation of electrophilic aldehydes but can be significantly rescued by the presence of nucleophilic thiols. Biol. Reprod. 87, 110 10.1093/biolreprod/87.s1.110 [DOI] [PubMed] [Google Scholar]
- 88. Bromfield E. G., Aitken R. J., Anderson A. L., McLaughlin E. A., and Nixon B. (2015) The impact of oxidative stress on chaperone-mediated human sperm-egg interaction. Hum. Reprod. 30, 2597–2613 10.1093/humrep/dev214 [DOI] [PubMed] [Google Scholar]
- 89. Aitken R. J., Whiting S., De Iuliis G. N., McClymont S., Mitchell L. A., and Baker M. A. (2012) Electrophilic aldehydes generated by sperm metabolism activate mitochondrial reactive oxygen species generation and apoptosis by targeting succinate dehydrogenase. J. Biol. Chem. 287, 33048–33060 10.1074/jbc.M112.366690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ross C. A., and Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- 91. Hochrainer K., Jackman K., Anrather J., and Iadecola C. (2012) Reperfusion rather than ischemia drives the formation of ubiquitin aggregates after middle cerebral artery occlusion. Stroke 43, 2229–2235 10.1161/STROKEAHA.112.650416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Canu N., Barbato C., Ciotti M. T., Serafino A., Dus L., and Calissano P. (2000) Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J. Neurosci. 20, 589–599 10.1523/JNEUROSCI.20-02-00589.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Haglund C., Mohanty C., Fryknäs M., D'Arcy P., Larsson R., Linder S., and Rickardson L. (2014) Identification of an inhibitor of the ubiquitin–proteasome system that induces accumulation of polyubiquitinated proteins in the absence of blocking of proteasome function. Medchemcomm. 5, 376–385 10.1039/C3MD00386H [DOI] [Google Scholar]
- 94. Casarejos M. J., Solano R. M., Gómez A., Perucho J., de Yébenes J. G., and Mena M. A. (2011) The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem. Int. 58, 512–520 10.1016/j.neuint.2011.01.008 [DOI] [PubMed] [Google Scholar]
- 95. Pickering A. M., Koop A. L., Teoh C. Y., Ermak G., Grune T., and Davies K. J. (2010) The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 432, 585–594 10.1042/BJ20100878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ding Q., Reinacker K., Dimayuga E., Nukala V., Drake J., Butterfield D. A., Dunn J. C., Martin S., Bruce-Keller A. J., and Keller J. N. (2003) Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Lett. 546, 228–232 10.1016/S0014-5793(03)00582-9 [DOI] [PubMed] [Google Scholar]
- 97. Hussong S. A., Kapphahn R. J., Phillips S. L., Maldonado M., and Ferrington D. A. (2010) Immunoproteasome deficiency alters retinal proteasome's response to stress. J. Neurochem. 113, 1481–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ding Q., Martin S., Dimayuga E., Bruce-Keller A. J., and Keller J. N. (2006) LMP2 knock-out mice have reduced proteasome activities and increased levels of oxidatively damaged proteins. Antioxid. Redox Signal. 8, 130–135 10.1089/ars.2006.8.130 [DOI] [PubMed] [Google Scholar]
- 99. Aiken C. T., Kaake R. M., Wang X., and Huang L. (2011) Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteomics 10, R110.006924 10.1074/mcp.M110.006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Grune T., and Davies K. J. (2003) The proteasomal system and HNE-modified proteins. Mol. Aspects Med. 24, 195–204 10.1016/S0098-2997(03)00014-1 [DOI] [PubMed] [Google Scholar]
- 101. Tanaka K., Yoshimura T., Tamura T., Fujiwara T., Kumatori A., and Ichihara A. (1990) Possible mechanism of nuclear translocation of proteasomes. FEBS Lett. 271, 41–46 10.1016/0014-5793(90)80367-R [DOI] [PubMed] [Google Scholar]
- 102. Nederlof P. M., Wang H. R., and Baumeister W. (1995) Nuclear localization signals of human and Thermoplasma proteasomal α subunits are functional in vitro. Proc. Natl. Acad. Sci. U.S.A. 92, 12060–12064 10.1073/pnas.92.26.12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wójcik C., Benchaib M., Lornage J., Czyba J. C., and Guerin J. F. (2000) Localization of proteasomes in human oocytes and preimplantation embryos. Mol. Hum. Reprod. 6, 331–336 10.1093/molehr/6.4.331 [DOI] [PubMed] [Google Scholar]
- 104. Maiti K., Sultana Z., Aitken R. J., Morris J., Park F., Andrew B., Riley S. C., and Smith R. (2017) Evidence that fetal death is associated with placental aging. Am. J. Obstet. Gynecol. 217, 441.e441–441.e14 10.1016/j.ajog.2017.06.015 [DOI] [PubMed] [Google Scholar]
- 105. Lord T., and Aitken R. J. (2015) Fertilization stimulates 8-hydroxy-2′-deoxyguanosine repair and antioxidant activity to prevent mutagenesis in the embryo. Dev. Biol. 406, 1–13 10.1016/j.ydbio.2015.07.024 [DOI] [PubMed] [Google Scholar]
- 106. Marangos P., Stevense M., Niaka K., Lagoudaki M., Nabti I., Jessberger R., and Carroll J. (2015) DNA damage-induced metaphase I arrest is mediated by the spindle assembly checkpoint and maternal age. Nat. Commun. 6, 8706 10.1038/ncomms9706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Collins J. K., Lane S. I., Merriman J. A., and Jones K. T. (2015) DNA damage induces a meiotic arrest in mouse oocytes mediated by the spindle assembly checkpoint. Nat. Commun. 6, 8553 10.1038/ncomms9553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ma J. Y., Ou Yang Y. C., Wang Z. W., Wang Z. B., Jiang Z. Z., Luo S. M., Hou Y., Liu Z. H., Schatten H., and Sun Q. Y. (2013) The effects of DNA double-strand breaks on mouse oocyte meiotic maturation. Cell Cycle 12, 1233–1241 10.4161/cc.24311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yun Y., Holt J. E., Lane S. I., McLaughlin E. A., Merriman J. A., and Jones K. T. (2014) Reduced ability to recover from spindle disruption and loss of kinetochore spindle assembly checkpoint proteins in oocytes from aged mice. Cell Cycle 13, 1938–1947 10.4161/cc.28897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chiang T., Duncan F. E., Schindler K., Schultz R. M., and Lampson M. A. (2010) Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr. Biol. 20, 1522–1528 10.1016/j.cub.2010.06.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lister L. M., Kouznetsova A., Hyslop L. A., Kalleas D., Pace S. L., Barel J. C., Nathan A., Floros V., Adelfalk C., Watanabe Y., Jessberger R., Kirkwood T. B., Höög C., and Herbert M. (2010) Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr. Biol. 20, 1511–1521 10.1016/j.cub.2010.08.023 [DOI] [PubMed] [Google Scholar]
- 112. Bromfield E. G., Aitken R. J., McLaughlin E. A., and Nixon B. (2017) Proteolytic degradation of heat shock protein A2 occurs in response to oxidative stress in male germ cells of the mouse. Mol. Hum. Reprod. 23, 91–105 [DOI] [PubMed] [Google Scholar]
- 113. Mihalas B. P., Western P. S., Loveland K. L., McLaughlin E. A., and Holt J. E. (2015) Changing expression and subcellular distribution of karyopherins during murine oogenesis. Reproduction 150, 485–496 10.1530/REP-14-0585 [DOI] [PubMed] [Google Scholar]
- 114. Selesniemi K., Lee H.-J., Muhlhauser A., and Tilly J. L. (2011) Prevention of maternal aging-associated oocyte aneuploidy and meiotic spindle defects in mice by dietary and genetic strategies. Proc. Natl. Acad. Sci. U.S.A. 108, 12319–12324 10.1073/pnas.1018793108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Miao Y. L., Kikuchi K., Sun Q. Y., and Schatten H. (2009) Oocyte aging: cellular and molecular changes, developmental potential and reversal possibility. Hum. Reprod. Update 15, 573–585 10.1093/humupd/dmp014 [DOI] [PubMed] [Google Scholar]
- 116. Camlin N. J., McLaughlin E. A., and Holt J. E. (2017) The use of C57Bl/6xCBA F1 hybrid cross as a model for human age-related oocyte aneuploidy. Mol. Reprod. Dev. 84, 6–7 10.1002/mrd.22766 [DOI] [PubMed] [Google Scholar]
- 117. Halliwell B., Clement M. V., Ramalingam J., and Long L. H. (2000) Hydrogen peroxide. Ubiquitous in cell culture and in vivo? IUBMB Life 50, 251–257 10.1080/15216540051080930 [DOI] [PubMed] [Google Scholar]
- 118. Uchida K. (2003) 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 42, 318–343 10.1016/S0163-7827(03)00014-6 [DOI] [PubMed] [Google Scholar]
- 119. Jennings P. C., Merriman J. A., Beckett E. L., Hansbro P. M., and Jones K. T. (2011) Increased zona pellucida thickness and meiotic spindle disruption in oocytes from cigarette smoking mice. Hum. Reprod. 26, 878–884 10.1093/humrep/deq393 [DOI] [PubMed] [Google Scholar]
- 120. Taiyeb A. M., Dees W. L., Ridha-Albarzanchi M. T., Sayes C. M., and Kraemer D. C. (2014) In vitro effects of cilostazol, a phosphodiesterase 3A inhibitor, on mouse oocyte maturation and morphology. Clin. Exp. Pharmacol. Physiol. 41, 147–153 10.1111/1440-1681.12193 [DOI] [PubMed] [Google Scholar]
- 121. Holt J. E., Lane S. I., Jennings P., García-Higuera I., Moreno S., and Jones K. T. (2012) APC(FZR1) prevents nondisjunction in mouse oocytes by controlling meiotic spindle assembly timing. Mol. Biol. Cell 23, 3970–3981 10.1091/mbc.e12-05-0352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Camlin N. J., Sobinoff A. P., Sutherland J. M., Beckett E. L., Jarnicki A. G., Vanders R. L., Hansbro P. M., McLaughlin E. A., and Holt J. E. (2016) Maternal smoke exposure impairs the long term fertility of female offspring in a murine model. Biol. Reprod. 94, 39 [DOI] [PubMed] [Google Scholar]
- 123. Nixon B., Stanger S. J., Mihalas B. P., Reilly J. N., Anderson A. L., Dun M. D., Tyagi S., Holt J. E., and McLaughlin E. A. (2015) Next generation sequencing analysis reveals segmental patterns of microRNA expression in mouse epididymal epithelial cells. PLoS ONE 10, e0135605 10.1371/journal.pone.0135605 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.