

Abstract
Immune checkpoint blockade therapy has been successful in treating some types of cancers but has not shown clinical benefits for treating leukemia 1. This result suggests that leukemia exploits unique escape mechanisms. Certain immune inhibitory receptors that are expressed by normal immune cells are also present on leukemia cells. It remains unknown whether these receptors can initiate immune-related primary signaling in tumor cells. Here we show that LILRB4, an ITIM-containing receptor and a monocytic leukemia marker, supports tumor cell infiltration into tissues and suppresses T cell activity via ApoE/LILRB4/SHP-2/uPAR/Arginase-1 signaling axis in acute myeloid leukemia (AML) cells. Blocking LILRB4 signaling using knockout and antagonistic antibody approaches impeded AML development. Thus, LILRB4 orchestrates tumor invasion pathways in monocytic leukemia cells by creating an immune-suppressive microenvironment. LILRB4 represents a compelling target for treatment of monocytic AML.
To identify novel mechanisms for AML development and immune regulation, we analyzed the relationship between gene expression of known co-stimulating and co-inhibitory receptors and the overall survival of AML patients as documented in the TCGA database. The expression of the mRNA encoding leukocyte immunoglobulin-like receptor B4 (LILRB4), an immune inhibitory receptor restrictively expressed on monocytic cells 2–4 and monocytic AML cells (FAB M4 and M5 AML subtypes) 5 ranked on the top of the list for negative correlation with AML patient survival (Fig. 1a and Extended Data Fig. 1a-d and Supplementary Table 1). Importantly, LILRB4 levels were higher on monocytic AML cells than on normal monocytes (Fig. 1b).
Figure 1. LILRB4 expressed on leukemia cells suppresses T cell proliferation.
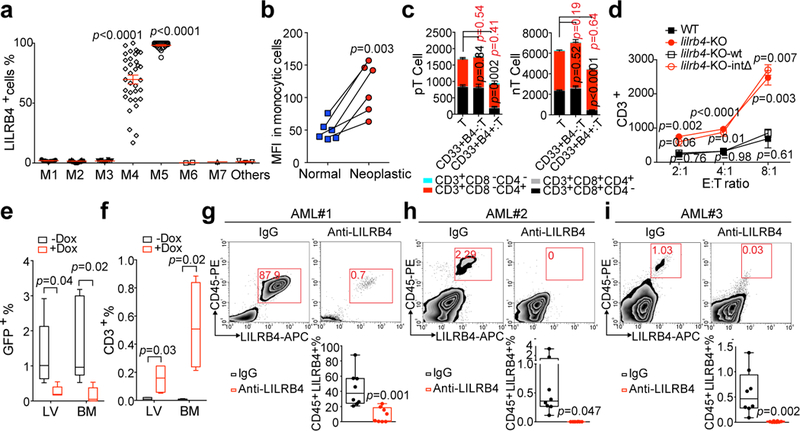
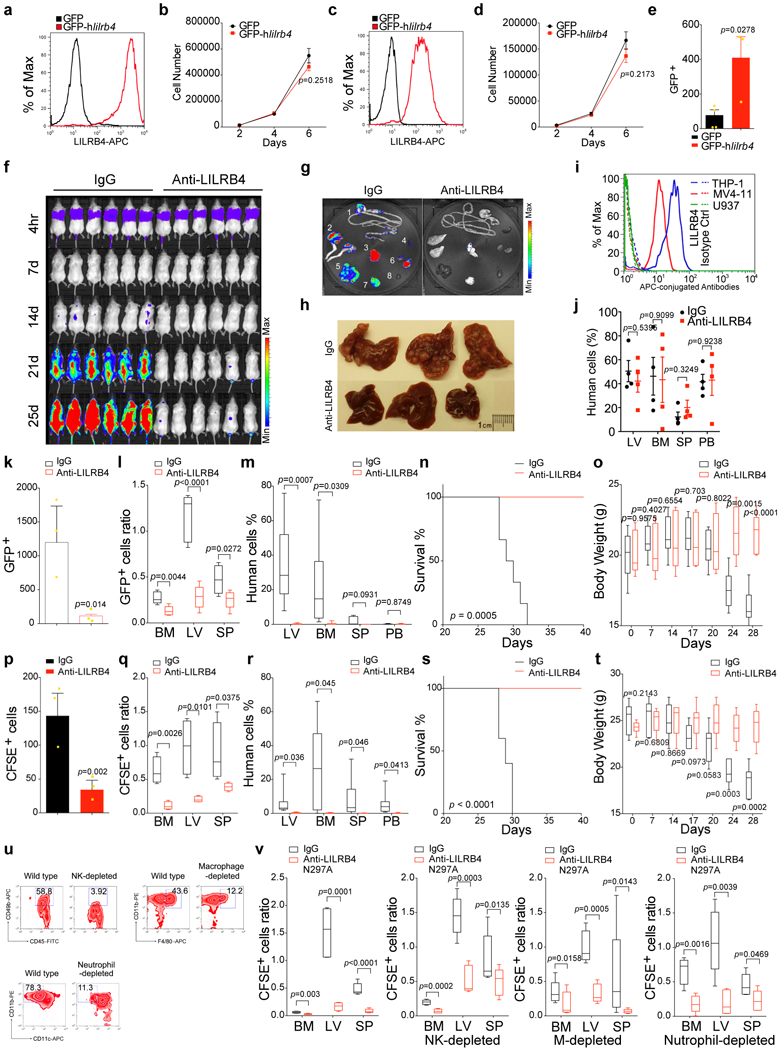
a, LILRB4 surface expression was quantified by flow cytometry analysis of samples from 105 AML patients (n=1 to 34 for each classification (see Methods) with mean and s.e.m.). b, LILRB4 surface expression was compared on normal monocytes and neoplastic monocytes from the same AML patients (n=6 independent patients). MFI: mean fluorescence intensity. c, Autologous T cells (pT, patient T cells) isolated from a monocytic AML patient (AML#19) or allogeneic T cells (nT, normal T cells) isolated from a healthy donor were incubated with irradiated lilrb4-positive or lilrb4-negative primary leukemia cells from the AML patient (AML#19) (n=3 biologically independent samples with mean and s.e.m..Also see Source Data Figure 1.). d, T cells (E: effector cells) isolated from healthy donors were incubated with indicated irradiated THP-1 cells (T: target cells) in cell-contact manner (n=3 biologically independent samples with mean and s.e.m.). e-f, Engraftment of human T cells and i.v. transplanted Doxycycline (Dox)-inducible lilrb4-knockout THP-1 cells (GFP+) in NSG mice (n=5 mice). LV, liver; BM, bone marrow. g-i, Representative flow plots and quantification of human primary monocytic AML-xenografted mouse bone marrow after anti-LILRB4 antibody or control IgG treatment (n=8 biologically independent samples). This experiment was repeated with 16 independent patient samples with similar results (also see Extended Data Fig. 4a). See Methods for definition of box plot elements in e-i. All p values were from two-tailed student t-test.
A previous study reported that the extracellular domain of LILRB4 inhibited T cell activities 6. To test whether LILRB4 expressed on AML cells has T cell-suppressive function, we co-cultured LILRB4-positive leukemia cells, LILRB4-negative leukemia cells, or normal hematopoietic cells with either autologous T cells or T cells from healthy donors. Only LILRB4-positive monocytic AML cells significantly suppressed T cell proliferation (Fig. 1c and Extended Data Fig. 1e-f). We then deleted lilrb4 from human monocytic AML THP-1 and MV4–11 cells and found that the T cell suppressive ability of AML cells was significantly reduced upon lilrb4 knockout (lilrb4-KO) and was restored by forced expression of wild-type lilrb4 (as lilrb4-KO-wt), but not by a mutant lilrb4 with deleted intracellular domain (as lilrb4-KO-intΔ) (Fig. 1d and Extended Data Fig. 2a-g). Moreover, when wild-type THP-1 cells and human T cells were cultured in separate transwells, LILRB4-mediated T cell inhibition was also observed and was able to be reversed by anti-LILRB4 blocking antibodies (Extended Data Fig. 2h-p). Blocking LILRB4 resulted in increase of cytotoxicity T cells and cytokine releasing (Extended Data Fig. 2q-u). These in vitro data suggest that, instead of the extracellular domain 6, the intracellular signaling of LILRB4 in AML cells is required for suppression of T cell activity.
Next, we used humanized mouse xenograft models and an immunocompetent mouse model to investigate LILRB4 function in immune checkpoint blockade. Subcutaneous implantation of THP-1 cells, but not the lilrb4-KO THP-1 cells, resulted in AML development in human T cell reconstituted mice, which was blocked by anti-LILRB4 treatment 7 (Extended Data Fig. 3a-i). Doxycycline-induced LILRB4 deletion in an established disseminated leukemia model in humanized mice also impaired leukemia development and restored T cells (Fig. 1e-f and Extended Data Fig. 3j-l). In addition, we subcutaneously implanted human LILRB4-expressing mouse C1498 AML cells (hlilrb4-C1498) into C57BL/6 mice to establish a syngeneic immunocompetent mouse model. To exclude the anti-tumor effects from Fc effector functions, we treated tumor-bearing mice with anti-LILRB4 with the Fc glycosylation site N297A mutation 8. LILRB4 blockade effectively lowered tumor burden and prolonged survival; depleting CD8+ T cells eliminated the anti-tumor effects of the anti-LILRB4 antibody (Extended Data Fig. 3m-r). These results suggest that the tumor-supportive effect of LILRB4 depends on host T cell inhibition. The anti-LILRB4 antibody treatment generated tumor-specific memory T cells (Extended Data Fig. 3s). Similar results were obtained in disseminated hlilrb4-C1498 syngeneic mouse model (Extended Data Fig. 3x-z). Finally, the blockade of LILRB4 significantly reduced leukemia development in primary human monocytic AML-derived xenografts (Fig. 1g-i and Extended Data Fig. 4a) and increased the numbers of engraftable autologous human T cells (Extended Data Fig. 4b). Together, our in vitro and in vivo results indicate that LILRB4 signaling in monocytic AML cells suppresses T cell-mediated anti-tumor immunity.
One of the characteristic features of monocytic AML is enhanced extramedullary infiltration of tumor cells 9. We observed that the antibody blockade of LILRB4 results in significant decrease of leukemic infiltration into internal organs, including bone marrow, liver, and brain (Extended Data Fig. 3t-v). Although anti-LILRB4 antibody treatment did not reduce the size of subcutaneous C1498 tumors in C57BL/6 mice depleted of CD8+ T cells (Extended Data Fig. 3m), treatment with anti-LILRB4 antibody did lead to decreased leukemia cell infiltration into liver (Extended Data Fig. 3w). We hypothesized that, in addition to T cell inhibition, LILRB4 promotes leukemia infiltration. To test this hypothesis, we performed trans-endothelial migration and homing assays and monitored leukemia infiltration relative to LILRB4 expression on leukemia cells. Human AML THP-1 cells depleted of LILRB4 had lower trans-endothelial migration in vitro than cells that expressed LILRB4 (Fig. 2a). Deletion of lilrb4 reduced homing and engraftment of AML cells to hematopoietic organs (Fig. 2b-c), and resulted in prolonged survival of xenografted mice (Fig. 2d) and delayed body weight loss (Fig. 2e). In contrast, forced expression of human LILRB4 in mouse AML C1498 or WEHI-3 cells had the opposite effects (Figs. 2f-j and Extended Data Fig. 5a-e). Antibody-mediated LILRB4 blockade showed the same effect as lilrb4 knockout in LILRB4-expressing AML cells (Extended Data Fig. 5f-t). This effect was depended on LILRB4 expression and its intracellular signaling in leukemia cells (Fig. 2k) but not the Fc effector functions of the antibody (Extended Data Fig. 5u-v). Furthermore, LILRB4 blockade reduced infiltration ability of primary monocytic AML cells (Fig. 2l-n and Extended Data Fig. 4c-e). Our results are concordant with previous studies showing that the frequency of circulating LILRB4+ AML blasts is significantly lower than that of the LILRB4- AML blasts 5 and that LILRB4+ chronic lymphocytic leukemia cells are associated with lymphoid tissue involvement 10. Bone marrow, liver, and brain to which LILRB4+ AML cells tend to migrate are known to have certain immune privileges 11–13. Thus, LILRB4-mediated migration, which supports enhanced extramedullary infiltration of monocytic AML cells, may also contribute to immune evasion.
Figure 2. LILRB4 promotes AML cell migration and infiltration.
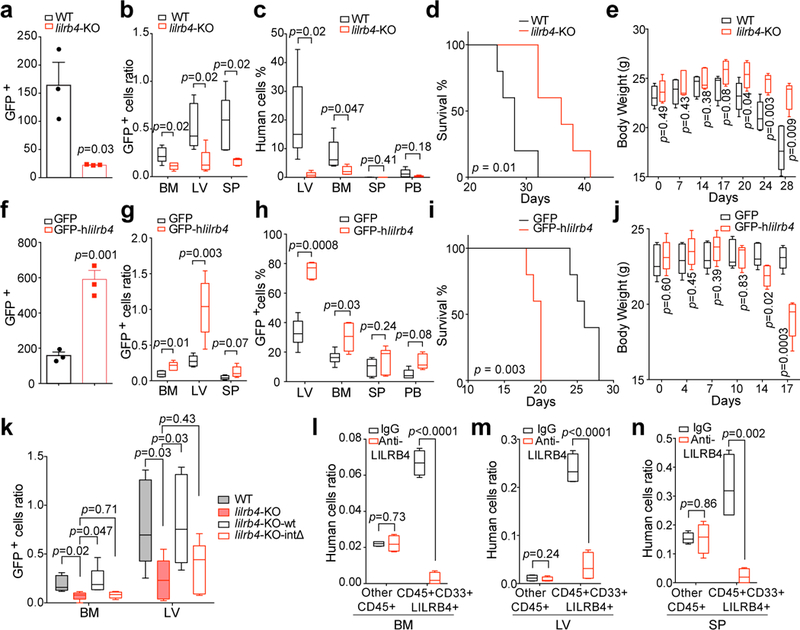
a, Comparison of the transendothelial migration abilities of WT and lilrb4-KO THP-1 cells (GFP+) in a transwell assay (n=3 biologically independent samples with mean and s.e.m.). b, Comparison of the short-term (20 hrs) infiltration of WT or lilrb4-KO THP-1 cells in NSG mice (n=5 mice). The numbers of leukemia cells (GFP+) in liver (LV), spleen (SP), and bone marrow (BM) determined by flow cytometry and normalized to number in peripheral blood (PB). c-e, Comparison of the long-term (21 days) infiltration of WT or lilrb4-KO THP-1 cells in NSG mice (n=5 mice). Shown are percentages of THP-1 cells (hCD45+) engrafted in indicated organs at day 21 post-transplant (c), and overall survival (d) and body weights (e) as a function of time. f-j, Comparison of the transendothelial migration (f, n=3 biologically independent samples with mean and s.e.m.), short-term (20 hrs, g, n=5 mice) and long-term (16 days, h-j, n=5 mice) infiltration (h) of hlilrb4-C1498 or control C1498 cells (GFP+), and overall survival (i) and body weight (j) as a function of time. k, Comparison of the short-term (20 hrs) infiltration of indicated WT or modified THP-1 cells in NSG mice (n=5 mice). l-n, Comparison of the short-term (20 hrs) infiltration of human primary monocytic AML cells (AML#21) in NSG mice (n=4 mice) after treatment with anti-LILRB4 antibody or IgG control. See Methods for definition of box plot elements in b-c, e, g-h, j-n. All p values (except of d and i from long-rank test) were from two-tailed student t-test.
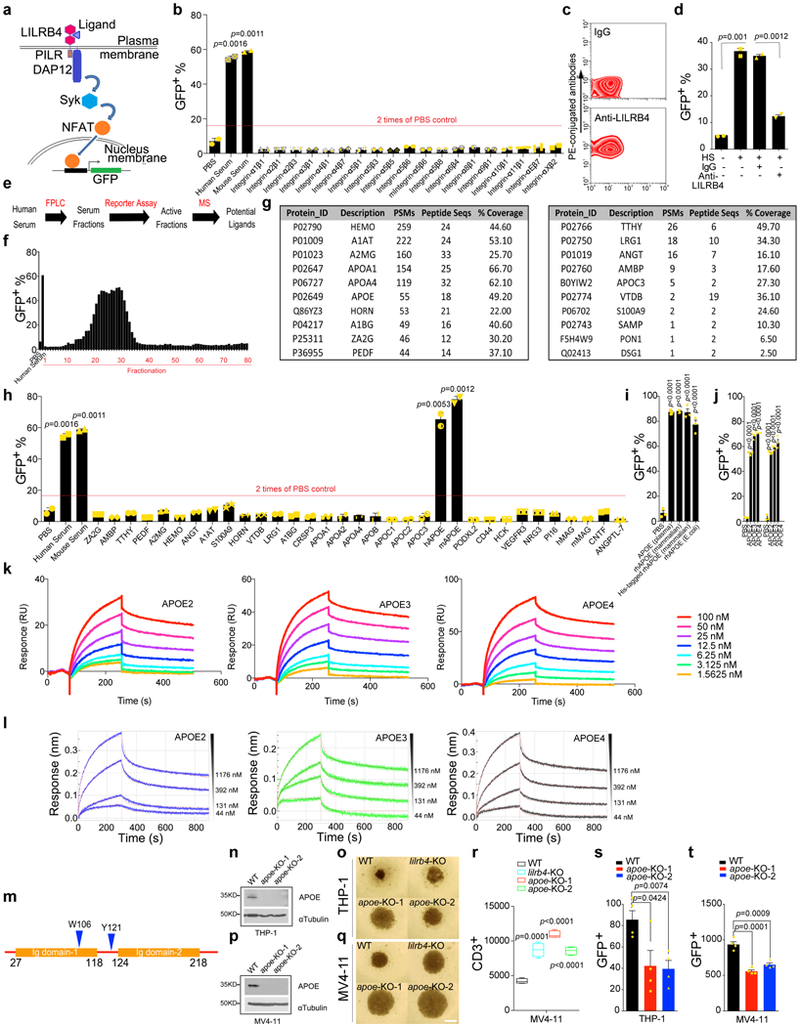
The anti-LILRB4 antibody blockade of immune inhibitory and migration functions of AML cells suggests that those functions of LILRB4 are regulated by extracellular mechanisms. Integrin-αvβ3, was previously identified as the ligand for gp49B1, the mouse LILRB4 orthologue 14. However, a variety of integrin-αβ complexes did not activate human LILRB4 reporter cells (Extended Data Fig. 6a-b). Surprisingly, human serum and mouse serum were capable of activating the LILRB4 reporter but not reporters for other LILRBs (Fig. 3a). Through protein liquid chromatography fractionation followed by reporter assays and mass spectrometry, we identified APOE that specifically activated the reporters of LILRB4 and mouse PirB (Fig. 3b and Extended Data Fig. 6c-j). The serum from wild-type but not APOE-null mice activated the LILRB4 reporter (Fig. 3c). In addition, both liposome-reconstituted APOE protein (APOE-POPC) and lipid-free APOE activated LILRB4 reporter cells (Fig. 3d). The binding of APOE to THP-1 cells was significantly decreased by lilrb4-KO (Fig. 3e). The specific binding of recombinant APOE to LILRB4 was confirmed using microscale thermophoresis (MST), surface plasmon resonance (SPR), and bio-layer interferometry (Octet). The dissociation constant was 210 nM as determined by MST (Fig. 3f and Extended Data Fig. 6k-l). Mutagenesis studies showed that the N-terminal domain of APOE, and P35 and W106 in the first Ig-domain and Y121 in the linker region between two Ig-domains of LILRB4 are critical for APOE-mediated activation of LILRB4 (Fig. 3g-h and Supplementary Table 2 and Extended Data Fig. 6m).
Figure 3. APOE is an extracellular binding protein of LILRB4.
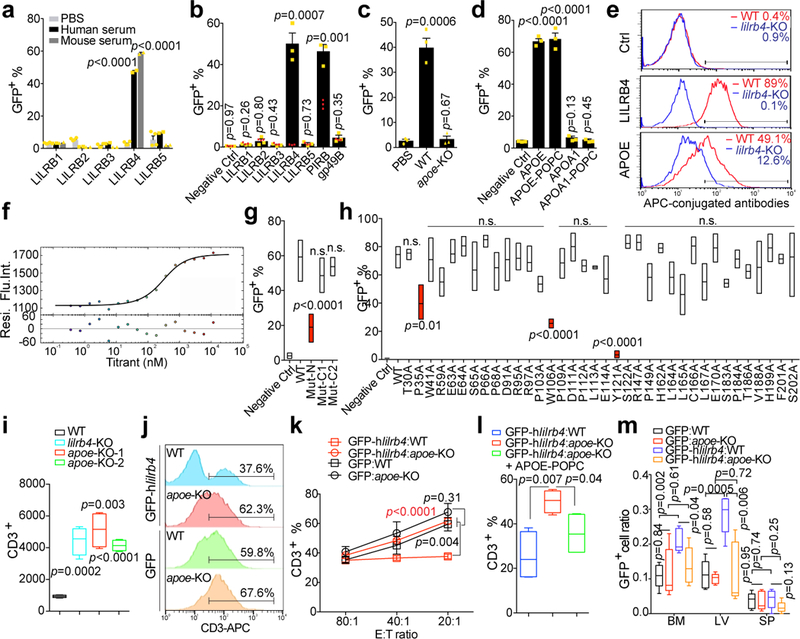
a, Percentages of indicated LILRB reporter cells activated (GFP+) in the presence of 10% human serum, 10% mouse serum, or PBS control. b, Percentages of indicated LILRB reporter cells activated by recombinant APOE (10 μg/ml). Red dots indicate the PBS treatment of each indicated reporter cell line. c, Percentages of LILRB4 reporter cells activated by 10% mouse serum collected from wild-type or apoe-knockout KO mice or PBS control. d, Percentages of LILRB4 reporter cells activated by 10 μg/ml of APOE, APOE-POPC, APOA1, or APOA1-POPC. n=3 biologically independent samples with mean and s.e.m. in (a-d). e, Binding of His-tagged APOE to WT and lilrb4-KO THP-1 cells. f, Binding kinetics of human His-tagged APOE-3 to LILRB4-ECD were measured using microscale thermophoresis (MST). Upper panel: fluorescence intensity (Flu.Int.) plot and regression of the binding; lower panel: the corresponding residuals (Resi.) versus fits plot. g, Percentages of LILRB4 reporter cells activated by WT and mutant APOE proteins. Mut-N, R142A/K143A/R145A/K146A/R147A/R150A; Mut-C1, deletion of residues 245–299; and Mut-C2, deletion of residues 279–299. h, Percentages of indicated LILRB4 mutant reporter cells activated by APOE proteins. Data on LILRB4 mutants that interfere with ApoE activation are highlighted in red in (g-h, n=4 biologically independent samples, low-to-high outline and line at mean in box plots). i, T cells isolated from healthy donors were incubated with indicated irradiated THP-1 cells with or without lilrb4- or apoe-KO. T cells were analyzed by flow cytometry after 7 days (n=4 biologically independent samples). j-l, C57bl/6 mouse spleen cells (E) were incubated with irradiated human lilrb4-expressing (GFP-hlilrb4) or control (GFP) C1498 cells (T) at indicated E:T ratios. Cells were supplemented with 5% serum collected from WT or apoe-KO mice, cultured with anti-CD3/CD28-coated beads for 60 hours, and then stained with anti-CD3 antibody. Shown are representative flow plots from samples at E:T of 20:1 (j), percentages of CD3+ T cells (k, n=4 biologically independent samples with mean and s.e.m.), and the effects of APOE-POPC rescue of apoe-KO serum (l, n=4 biologically independent samples). m, Expression of human lilrb4 in mouse leukemia C1498 cells increases leukemia cell infiltration in WT recipient mice but not in apoe-KO recipient mice (n=5 mice). (e, j) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in i, l-m. All p values were from two-tailed student t-test. n.s., not significant.
The finding that APOE activates the immune inhibitory receptor LILRB4 is in agreement with the well-documented immune-suppressive function of APOE 15,16. To determine whether T cell suppressive activity of LILRB4 depends on APOE, we examined proliferation of T cells co-cultured with control or apoe-knockout human AML cells. AML cells deficient in APOE restored proliferation of T cells and suppressed migration of leukemia cells (Fig. 3i and Extended Data Fig. 6n-t). Moreover, the percentage of T cells in co-culture was significantly lower when the LILRB4-ectopically-expressing C1498 cells were treated with wild-type mouse serum compared to those treated with apoe-knockout mouse serum (Fig. 3j-k). Addition of liposome-reconstituted APOE to co-culture of mouse spleen cells and LILRB4-expressing AML cells decreased the T cell percentage (Fig. 3l). Furthermore, expression of LILRB4 significantly increased C1498 cells infiltrating to bone marrow and liver in wild-type mice but not in APOE-null recipients (Fig. 3m). These data indicate that APOE activates LILRB4 on human monocytic AML cells to suppress T cell proliferation and support AML cell migration.
We sought to identify the signaling downstream of LILRB4 required for T cell suppression and leukemia infiltration. Phosphatases SHP-1, SHP-2, and SHIP can be recruited to the intracellular domain of LILRB 2. The level of phosphorylation of SHP-2 but not of SHP-1 or SHIP was lower in lilrb4-KO AML cells than in wild-type cells (Fig. 4a and Extended Data Fig. 7a). Loss of SHP-2, but not loss of SHP-1 or SHIP, rescued T cell suppression by THP-1 cells (Fig. 4b and Extended Data Fig. 7b-c), and decreased short-term (20 hrs) and long-term (21 days) infiltration of THP-1 cells (Fig. 4c-d). Our results suggest that SHP-2 is a mediator of LILRB4 signaling.
Figure 4. LILRB4-mediated intracellular signaling controls AML cell migration and T cell suppression.
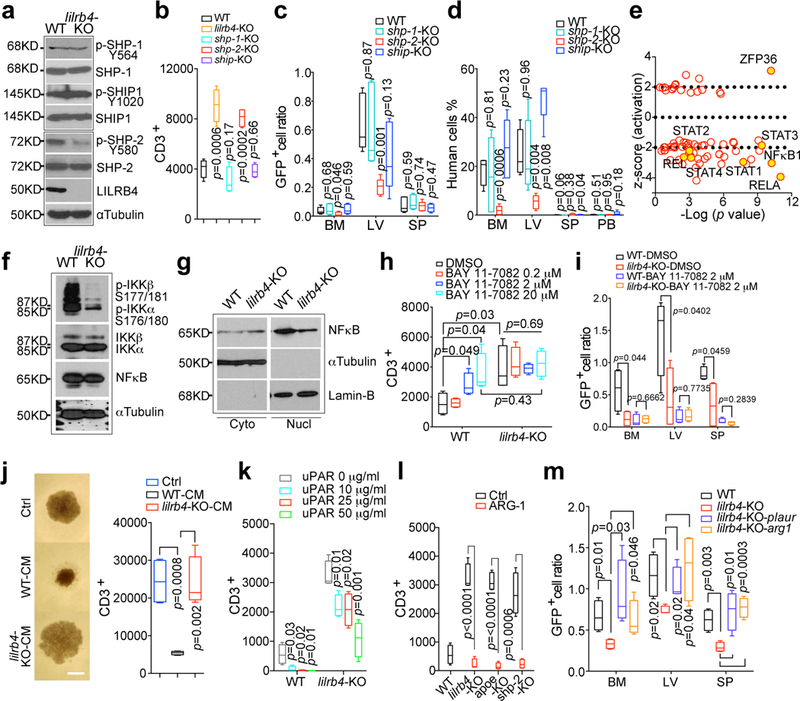
a, Expression and phosphorylation of three phosphatases in wild-type and lilrb4-KO THP-1 cells. b, Primary T cells and irradiated indicated THP-1 cells were cultured in the lower and upper chambers respectively. T cells were analyzed by flow cytometry after 7 days. n=4 biologically independent samples. c-d, Knockout of shp-2 reduces THP-1 cell short-term (20 hrs) and long-term (21 days) infiltration in NSG mice (n=5 mice). e, Upstream transcription factor analysis of RNA-seq data generated from lilrb4-KO and WT THP-1 cells (n=2 biologically independent samples). Yellow dots highlighted the transcription factors involved in JAK/STATs and NF-κB pathways. f, Decreased phosphorylation of IKKα/β in lilrb4-KO THP-1 cells. g, Decreased NFκB in the nuclear fraction in lilrb4-KO THP-1 cells. h-i, The NF-κB inhibitor reversed T cell suppression by THP-1 cells (h) and decreased infiltration of MV4–11 cells (i) in an LILRB4-dependent manner (n=4 biologically independent samples). j, T cells isolated from healthy donors were supplemented with 25% condition medium (CM) of WT or lilrb4-KO THP-1 cells. Representative cells were photographed (scale bar, 100 μm) and T cells were analyzed by flow cytometry (n=4 biologically independent samples). k-l, T cells were incubated with irradiated indicated THP-1 cells supplemented with indicated concentration of recombinant uPAR (k) or ARG-1 (l) proteins for 7 days and were analyzed by flow cytometry (n=4 biologically independent samples). m, Overexpression of uPAR (plaur) or ARG1 rescued infiltration defect of lilrb4-KO MV4–11 cells (n=5 mice). (a, f-g, j) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in (b-d, h-m). All p values were from two-tailed student t-test.
Our Ingenuity Pathway Analysis showed that the activity of key transcription factors NFkB1 and RELA in the NF-κB pathway 17, which is positively regulated by SHP-2 18, was most significantly inhibited by loss of lilrb4 (Fig. 4e and Supplementary Tables 3 and 4). Consistently, the phosphorylation of IKKα/β and levels of nuclear NF-κB were decreased in lilrb4-KO AML cells (Fig. 4f-g and Extended Data Fig. 7a). Inhibition of NF-κB signaling restored T cell suppression and reduced AML cell infiltration in a LILRB4-dependent manner (Fig. 4h-i and Extended Data Fig. 7e-f). Therefore the effects of LILRB4 activation are mediated through the NF-κB pathway, which is particularly robust in monocytic AML among AML subtypes 19.
Consistent with our result that AML cells inhibit T cell proliferation in transwells (Extended Data Fig. 2o-p), the conditioned medium from wild-type THP-1 cells suppressed T cell activity but that medium from lilrb4-KO cells did not (Fig. 4j). Among proteins that were present higher in the conditioned medium of WT THP-1 cells than the lilrb4-KO counterparts (Extended Data Fig. 7g-i), uPAR is highly expressed by monocytic AML cells 20. uPAR, an NF-κB target, is well known to promote cancer invasion, metastasis, survival, and angiogenesis 21, 22. The addition of recombinant uPAR decreased proliferation of T cells co-cultured with lilrb4-KO THP-1 cells in a dose-dependent manner (Fig. 4k and Extended Data Fig. 7j). This activity of uPAR was likely mediated by downstream effectors in AML cells because uPAR does not effectively decrease T cell proliferation directly (Extended Data Fig. 7k).
The expression of Arginase-1 (ARG1), like uPAR, was significantly lower in lilrb4-KO AML cells than in wild-type cells (Extended Data Fig. 7l-m). ARG1 is up-regulated by uPAR-mediated signaling and inhibits T cell proliferation 23,24, and can be elevated by APOE 25 and NF-κB 26 for immune-suppressive functions. We hypothesized that ARG1 is a key downstream effector of LILRB4-NF-kB-uPAR signaling. ARG1 can be secreted by AML cells to inhibit T cell activity 27. Recombinant ARG1 decreased T cell proliferation in the co-culture with lilrb4-KO, apoe-KO, and shp-2-KO AML or primary AML cells (Fig. 4l and Extended Data Fig. 7n-p). Moreover, addition and overexpression of either uPAR or ARG1 rescued the migration ability of lilrb4-KO AML cells in vitro and in vivo, respectively (Extended Data Fig. 7q and Fig. 4m). Together, our results indicate that LILRB4/SHP-2/NF-κB/uPAR/ARG1 is a signaling pathway in monocytic AML cells (Extended Data Figs. 8–9) that suppresses immune activity and supports leukemia migration.
Because LILRB4 is restrictively expressed on normal monocytic cells 2 in which the LILRB4 signaling may differ from that in leukemia cells (Extended Data Fig. 9) and LILRB4 blockade did not significantly interfere with normal hematopoietic function (Extended Data Fig. 10), LILRB4 targeting may have minimal toxicity. Importantly, LILRB4 is also expressed on certain other types of cancers and myeloid-derived suppressor cells, tolerogenic dendritic cells, and tumor-associated macrophages 28,2,5,29,30. Targeting LILRB4 may thus enable combination of immunotherapy and targeted therapy in cancer treatment.
METHODS
Mice
C57 BL/6J and NOD-scid IL2Rγ null (NSG) mice were purchased from and maintained at the animal core facility of University of Texas Southwestern Medical Center (UTSW). Apoe-Knockout (apoe-KO, Apoetm1Unc) mice as previously described 31 were purchased from the Jackson Laboratory. Animal work described in this manuscript has been approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee (IACUC). For each experiment, the same sex and age-matched (4–8 weeks) mice were used and randomly allocated to each group; and For tumor size measurement and in vivo lumina imaging experiments, treatment conditions of the mice were blinded. The minimum number of mice in each group was calculated based on results from our prior relevant studies 32–36. For the subcutaneous tumor model, the tumor size was calculated by (width x width x length) cm3. The maximal tumor measurement permitted by UTSW IACUC is 2 cm in diameter of tumor. In none of the experiments were these limits exceeded (see Source Data Extended Data Figure 3). We have complied with all relevant ethical regulations with approved animal study protocols.
Cell culture
293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2 and the normal level of O2. Human umbilical vein/vascular endothelium cells (HUVECs) (ATCC, CRL-1730) were cultured in endothelial cell growth medium plus growth factor, cytokines and supplements (EGM-BulletKit, Lonza) at 37 °C in 5% CO2 and the normal level of O2. Human monocytic AML cells, THP-1 (ATCC, TIB-202), MV4–11 (ATCC, CRL-9591), and U937 (ATCC, CRL-1593.2), and mouse AML cells, WEHI-3 (ATCC, TIB-68), were cultured in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% FBS at 37 °C in 5% CO2 and the normal level of O2. Mouse AML cells, C1498 (ATCC, TIB-49) were cultured in DMEM supplemented with 10% FBS at 37 °C in 5% CO2 and the normal level of O2. All cell lines were routinely tested using a mycoplasma-contamination kit (R&D Systems).
Primary human leukemia cells
Primary human AML and B-ALL samples were obtained from the tissue banks at UTSW and University of Texas MD Anderson Cancer Center (MDACC). Informed consent was obtained under protocols reviewed and approved by the Institutional Review Board at UTSW and MDACC (IRB STU 122013–023 by UTSW and LAB10–0682 by MDACC). The UTSW cohort included 105 AML patients representative of AML subtypes by the French-American-British (FAB) classification, acute myeloblastic leukemia with minimal maturation (M1, n=9), acute myeloblastic leukemia with maturation (M2, n=34), acute promyelocytic leukemia (M3, n=10), acute myelomonocytic leukemia (M4, n=34), acute monocytic leukemia (M5, n=25), acute erythroid leukemia (M6, n=2), and acute megakaryoblastic leukemia (M7, n=1) and patients with undifferentiated leukemia (AUL; n=1) and transient myeloproliferative disorder (TAM; n=2) (Supplementary Table 1). Samples were frozen in FBS with 10% DMSO and stored in liquid nitrogen. Primary leukemia samples used in PDX, co-culture, short-term infiltration assay and western blotting were summarized in Supplementary Table 5. We have complied with all relevant ethical regulations with approved study protocols.
Human normal monocytes and macrophages
Human normal monocytes (CD14+ cells) were isolated by the AutoMACS Pro Separation System (Miltenyi Biotech, Auburn, CA) from the mononuclear cells fraction of normal peripheral blood. Briefly, buffy coat was purchased from Interstate Blood Bank (Memphis, TN) and the mononuclear cell layer was separated by Ficoll Hypaque (17144003, GE Lifesciences) density gradient separation. Mononuclear cells were treated with red blood cell lysis buffer to remove red blood cells and then incubated with CD14 microbead-conjugated antibody (130–050-201, Miltenyi Biotech, Auburn, CA) for 15 min at 4°C. CD14 positive cells were then isolated using the positive selection program according to the manufacturer’s protocol. One million CD14+ cells were plated in macrophage culture media, Iscove’s modified Dulbecco’s medium (IMDM) (12440053, Thermo fisher) supplemented with 10% human AB serum (MT35060CI, Fisher Scientific), 1% NEAA (11–140-050, Fisher), 2 μM L-alanine-L-glutamine (SH3003402, Fisher), per each well of a 6-well plate and cultured for 7 days. After incubation, most of the cells were adherent to the plastic surface and stained positive for CD14 and other markers specific for macrophages.
TCGA analyses
Data were obtained from the TCGA acute myeloid leukemia database (version: August 16, 2016). The patients were classified into AML subtypes (FAB classification) M0 (undifferentiated acute myeloblastic leukemia) (n=16), M1 (n=42), M2 (n=39), M3 (n=16), M4 (n=35), M5 (n=18), M6 (n=2), M7 (n=3); two cases were not classified by subtype. The mRNA levels of indicated genes were determined by RNA-seq (polyA+ IlluminaHiSeq). RESM-normalized counts are reported, and data were analyzed and visualized with UCSC Xena (xena.ucsc.edu). For analysis of overall survival, 160 patients with available survival data were separated into three groups based on whether they had high, moderate, or low gene expression and then analyzed by Xena Kaplan Meier plot (http://xena.ucsc.edu/survival-plots/).
Flow cytometry
Primary antibodies including anti-human CD45-PE (BD Pharmingen, HI30, 1:100), CD45-FITC (BD Pharmingen, HI30, 1:100), CD45-APC (BD Pharmingen, HI30, 1:100), anti-human CD34-FITC (BD Pharmingen, 55582, 1:100), anti-human CD19-PE (eBioscience, HIB19, 1:100), anti-human CD20-PE (BD Pharmingen, 555623, 1:100), anti-human CD11b-APC (eBioscience, ICRF44, 1:100), anti-human LILRB4-APC (eBioscience, ZM4.1, 1:100), anti-human LILRB4-PE (Biolegend, ZM4.1, 1:100), anti-human CD14-APC (eBioscience, 61D3, 1:100), anti-human CD33-APC (Biolegend, P67.6, 1:100), anti-human CD4-APC (eBioscience, RPA-T4, 1:100), anti-human CD3-FITC (BioLegend, HIT3a, 1:100), anti-human CD3-Pacific blue (BD Pharmingen, SP34–2, 1:100) anti-human CD8-PE (BD Pharmingen, 555367, 1:100), anti-human CD28-APC (eBioscience, CD28.2, 1:100), anti-human CD40L-APC (eBioscience, 24–31, 1:100), anti-human PD1-APC (Biolegend, EH12.2H7, 1:100), anti-human TIM3-APC (eBioscience, F38–2E2, 1:100), anti-human TIGIT-APC (eBioscience, MBSA43, 1:100), anti-human LAG3-APC (eBioscience, 3DS223H, 1:100), anti-human FasL-PE (eBioscience, 24–31, 1:100), anti-uPAR-APC (Biolegend, VIM5, 1:100), anti-mouse CD3-APC (BioLegend, 17A2, 1:200), anti-mouse CD8a-PE (BioLegend, 53–6.7, 1:200), anti-mouse CD45-PE (BD Pharmingen, 30-F11, 1:200), anti-mouse CD49b-APC (eBioscience, DX5, 1:200), anti-mouse CD49f-PE (eBioscience, GoH3, 1:200), anti-mouse CD11b-APC (BioLegend, M1/71, 1:200), anti-mouse CD11b-PE (BioLegend, M1/71, 1:200), anti-mouse CD11c-APC (eBioscience, N418, 1:200), anti-mouse F4/80-APC (BioLegend, BM8, 1:200), anti-His-tag-APC (R&D systems, AD1.1.10, 1:400), and IgG isotype-control-APC (eBioscience, P3.6.2.8.1, 1:400) antibodies were used. Cells were run on either Calibur for analysis or FACSAria for analysis and sorting. Flow data were analysed by Flowjo software. For analysis of human hematopoietic engraftment in NSG mice, a previously published protocol was followed 33,35,37. PI staining was used to exclude dead cells in analysis and sorting. For intracellular staining, we followed the two-step protocol for fixation/methanol from eBioscience. Briefly, human primary AML cells were stained for the surface expression of LILRB4 (anti-LILRB4-Alexa Fluor 647, Biolegend, ZM4.1, 1:100) and CD33 (anti-human CD33-FITC, Biolegend, HIM3–4, 1:100) and fixable cell viability dye eFluor 450 (Bioscience, Cat#65–0863-14, 1:100) followed by fixation (IC fixation buffer, eBioscience, Cat#00–8222) and methanol treatment. After that, cells were stained for intracellular antigens by anti-p-SHP-2 (Y580)-PE (Cell signaling, Cat#13328S, 1:100), anti-pIKKα/β (S176/180) (16A6) (Cell signaling, Cat#2697, 1:100), anti-NFκB (S529)-PE (eBioscience, B33B4WP, 1:100), anti-uPAR-PE (Biolegend, VIM5, 1:100), anti-Arginase-1 (D4E3M) (Cell signaling, Cat#93668, 1:100), rabbit IgG Isotype control-PE (Cell signaling, Cat#5742, 1:100), mouse IgG Isotype control-PE (eBioscience, m2a-15F8, 1:100) and anti-rabbit IgG-PE (Jackson Immunoresearch Lab, Cat#111–116-144, 1:400) for flow cytometry analysis.
Virus construction and infection
For retrovirus packaging, plasmid constructs XZ201-IRES-GFP and XZ201-human lilrb4 (hlilrb4)-IRES-GFP were mixed with PCL-ECO (2:1), followed by transfection into 293T cells using Lipofectamine 2000 (Invitrogen). For lentivirus packaging, CRISPER/Cas-9 based gRNA (guide RNA) constructs and other constructs for gene overexpression including – pLentiLox3.7-luciferase-IRES-GFP, ZsGreen-hlilrb4 and ZsGreen-hlilrb4-intΔ, pLVX-plaur-IRES-tdTomato, pLVX-arg1-IRES-tdTomato were mixed with psPAX2 and pMD2.G (Addgene) at a ratio of 4:3:1 and transfected into 293T cells using Lipofectamine 2000 (Invitrogen). Virus-containing supernatant was collected 48–72 hrs post-transfection and used for infection as described previously 38.
CRISPR/Cas9-based gene knockout in AML cells
Human AML cells were infected with doxycycline-inducible Cas9-expressing lentivirus (pCW-Cas9, Addgene 50661). After 1 μg/ml puromycin selection, the survived cells were infected with sgRNA-expressing lentivirus, produced by the plasmid modified from pSLQ1651 (Addgene 51024) by replacing the puro-mcherry with GFP for sorting. Scramble control sgRNA (sgRNA 5’- GAACGACTAGTTAGGCGTGTA −3’), lilrb4 targeting sgRNA (sgRNA1 5’- TGTTACTATCGCAGCCCTGT −3’; sgRNA2 5’- GTAGGTCCCCCCGTGCACTG −3’; sgRNA3 5’-CCTGTGACCTCAGTGCACGG −3’), apoe targeting sgRNA (sgRNA1 5’- CTTTTGGGATTACCTGCGC −3’; sgRNA2 5’- AACTGGCACTGGGTCGCTTT −3’), shp-1 targeting sgRNA (sgRNA1 5’- TAAGACCTACATCGCCAGCC −3’; sgRNA2 5’- GAAGAACTTGCACCAGCGTC −3’), shp-2 targeting sgRNA (sgRNA1 5’- GAGACTTCACACTTTCCGTT −3’; sgRNA2 5’- TACAGTACTACAACTCAAGC −3’), ship targeting sgRNA (sgRNA1 5’- CACGCAGAGCGCGTATGCCC −3’; sgRNA2 5’- TGGCAACATCACCCGCTCCA −3’) which were designed by an online tool (http://crispr.mit.edu), were cloned into the sgRNA plasmid, individually. After treated with 1 μg/ml doxycycline (Sigma, Cat#PHR1789) for 1 week, these cells were staining with anti-LILRB4 antibody and the LILRB4 negative cells were sorted as lilrb4-knockout cells. For apoe-, shp-1-, shp-2- and ship-knockout cells, GFP+ cells were sorted into a 96-well plate as single cell per well. After cell expanded, knockout cells were verified by western blotting. For in vivo induction of CRISPR/Cas9 to achieve gene knockout, we fed mice with doxycycline as described 39. Briefly, 7 days after Cas9/lilrb4-sgRNA-transfected THP-1 cell implantation, mice were treat with 2 mg/mouse of doxycycline via gavage daily for 5 days to achieve Cas9 expression in engrafted leukemia cells. The knockout was validated by flow cytometry.
Leukemia cell and T cell co-culture assay
In the co-culture assay, human T cells (5 × 104 per well) isolated from health donor peripheral blood (PB009–1-0, Allcells) were mixed with irradiated (28 Gy) indicated human leukemia cells in a U-bottom 96 well-plate. For non-contact co-culture of T cells with leukemia cells, leukemia cells were cultured in the upper chamber of transwell inserts (pore size, 3 μM, #09–761-80, Thermo Fisher) in U-bottom 96 well-plate. T cells isolated from healthy donors were placed in the lower chambers of a 96-well transwell plate. Irradiated indicated leukemia cells (E:T ratio = 2:1 if not indicated) were added to the upper chambers and treated with indicated antibodies, proteins and reagents. After culture with anti-CD3/CD28-coated beads (11161D, Thermo Fisher) and 50 U/ml rhIL-2 for 5~7 days, representative cells were photographed using an inverted microscope, and T cells were stained with anti-CD3 antibodies and analyzed by flow cytometry.
For primary AML or B-ALL samples, patient leukemia cells were sorted as CD33+ and CD19+ for AML and B-ALL, respectively. These leukemia cells were cultured with autologous CD3+ T cells from the same patient or allogeneic T cells from health donor (E:T ratio = 2:1). After culture with anti-CD3/CD28-coated beads (11161D, Thermo Fisher) and 50 U/ml rhIL-2 for 14 days, representative cells were photographed using an inverted microscope, and T cells were stained with anti-CD3, anti-CD4 and anti-CD8 antibodies and analyzed by flow cytometry.
For cytotoxicity assay, human CD8+ T cells (5 × 104 per well) isolated from PBMCs of a healthy donor were stimulated with anti-CD3/CD28/CD137-coated beads (11163D, Thermo Fisher) for 2 days in a 96-well plate. Then, indicated 5 × 103 leukemia cells and 50 to 500 μg/ml anti-LILRB4 antibodies or control IgG were added. Cell numbers were determined on day 7 in triplicate wells. Or indicated leukemia cells in indicated E:T ratios were cultured with T cells for 4~6 hrs in triplicate wells. Anti-CD3 and anti-CD8 were used to detect human CTL cells; indicated live THP-1 cells were positive for GFP and negative for PI. Cell supernatants from co-cultures of stimulated CTL cells and THP-1 cells treated with anti-LILRB4 or IgG were used to examine cytokine production using human cytokine arrays (AAH-CYT-6, RayBiotech).
For mouse leukemia/T cells co-culture, spleen cells from wild-type C57bl/6 were co-cultured with 2.5×104 irradiated (28 Gy) mouse leukemia C1498 cells in a U-bottom 96 well-plate for 60 hrs. Anti-CD3/CD28-coated beads (11452D, Thermo Fisher), 50 U/ml recombinant human IL-2, and 5% serum from wild-type C57bl/6 mice or that from apoe-KO mice were added to the medium. In some experiments, 50 μg/ml lipid-bound APOE proteins (APOE-POPC) were added to the medium. The lipidation of APOE recombinant protein was conducted as described 40.
Transendothelial migration assays
To measure the ability of AML cells to migrate through endothelial cells, 3 × 105 HUVEC cells were cultured on the transwell membrane (pore size is 8 μm). After 3 days, 1 × 105 indicated leukemia cells were seeded in the upper chamber. In indicated experiments, leukemia cell were treated with antibodies or proteins in the upper chamber. After 18 h, cells in lower chamber were counted.
Short-term infiltration assay of leukemia cells and homing assay of hematopoietic stem/progenitor cells (HSPCs)
Cells (5 × 106 cells per mouse) were injected intravenously into NSG mice. Animals were treated with 10 mg/kg of anti-LILRB4 antibodies or control IgG immediately after injection of leukemia cells. Mice were sacrificed after 20 hrs. Peripheral blood, bone marrow, liver, and spleen were harvested, and single-cell suspensions were examined by flow cytometry. CFSE, GFP or indicated markers such as anti-human CD45 and anti-human CD33 was used to detect target leukemia cells in indicated experiments. Numbers of leukemia cells in recipient liver, spleen, and bone marrow are reported as a ratio relative to cell numbers in peripheral blood.
To test the infiltration ability of mouse leukemia cells, 5 × 106 C1498-GFP-hLILRB4 cells or C1498-GFP were injected intravenously into wild-type C57BL/6J or APOE-null mice. Mice were sacrificed after 20 h. GFP was used to detect leukemia cells by flow cytometry. The number of leukemia cells in recipient liver, spleen, and bone marrow were normalized to numbers in peripheral blood and are reported as a ratio.
To test HSPCs homing ability, 1 × 107 human cord blood mononuclear cells were injected intravenously into an NSG mouse. Mice were treated with 10 mg/kg of anti-LILRB4 antibodies or control IgG immediately after injection of mononuclear cells and were sacrificed after 20 hrs. Anti-human CD45 and anti-human CD34 were used to detect human HSPCs by flow cytometry. Similarly, to test the infiltration ability of normal human monocytes, 5 × 106 CD14-positive selected monocyte from health donor PBMC were labeled by CFSE and injected intravenously into an NSG mouse. Mice were treated with 10 mg/kg of anti-LILRB4 antibodies or control IgG immediately after injection of monocytes and were sacrificed after 20 hrs. CFSE-positive cells were analyzed by flow cytometry.
Innate immune cell depletion
NK cell depletion was done by i.p. injection of 50 μl anti-asialo GM1 antibodies (CL8955, Cedarlane) 3 days before leukemia cell implantation, which resulted in >90% depletion of CD45+CD49b+ NK cells in the circulation of NSG mice. Macrophages were depleted by treating NSG mice with clodronate (dichloromethylene bisphosphonate) liposomes (SKU8909, Clodrosome) (200 μl of stock solution 3 days before leukemia cell implantation), resulting in >70% depletion of CD45+CD11b+F4/80+ macrophages in the circulation of NSG mice. NSG mice were rendered neutropenic by i.p. injection of 200μg anti-Ly-6G mAb (BP0075–1, Bioxcell) on days −3, −2, −1, and 0 post leukemia cell implantation, resulting in >80% depletion of CD45.1+CD11b+CD11c- neutrophils in the circulation of NSG mice.
Human AML xenograft
Xenografts were performed essentially as described 2,3,6,7. Briefly, 6–8 week-old NSG mice were used for transplantation. 1 × 106 human leukemia cells were resuspended in 200 μl PBS for each mouse i.v. injection. Mice were immediately given 10 mg/kg of anti-LILRB4 antibodies or control IgG intravenously. Three to four weeks after transplantation, the peripheral blood, bone marrow, spleen, and liver were assessed for the engraftment. Leukemia growth was monitored over time by luminescence imaging (Max, 3×108 p/sec/cm2/sr; Min, 5×106 p/sec/cm2/sr). For survival curve experiments, the death of mice was recorded when the moribund animals were euthanized. For primary patient-derived xenograft (PDX), each NSG mouse was given 5 to 10 × 106 human primary peripheral blood or bone marrow mononuclear cells, which contain leukemia cells and other normal compartments such as normal hematopoietic stem progenitor cells and autologous T cells, via tail-vein injection. Mice were immediately given 10 mg/kg of anti-LILRB4 antibodies or control IgG intravenously and were treated twice a week until euthanization. For AML#11, mice were given 10 mg/kg of anti-LILRB4 antibodies or control IgG intravenously 7 days after leukemia cell implantation and were treated twice a week until euthanization. Leukemia growth was monitored over time by flow cytometry of human cells in peripheral blood. More than 1% of human leukemia cells in mouse tissue were considered successful engraftment of primary AML cells. One to four months after transplantation, the peripheral blood, bone marrow, spleen, and liver were assessed for the engraftment.
For the hPBMC-humanized model, 1 × 107 human PBMCs were injected intravenously into each NSG mouse. Three weeks after implantation, mice had 30 to 50% engraftment of human T cells. At 3 weeks post implantation, 1 × 106 human AML THP-1 cells, including wild-type, lilrb4-KO THP-1 cells or THP-1 cells stably express luciferase (THP-1-Luc-GFP cells), were subcutaneously implanted. Mice were immediately given 10 mg/kg of anti-LILRB4 antibodies or control IgG intravenously and were treated twice a week until euthanization. Tumor growth was monitored over time by luminescence imaging (Max, 1×108 p/sec/cm2/sr; Min, 5×106 p/sec/cm2/sr). Tumor sizes were determined by caliper measure (width x width x length). For inducible lilrb4-knockout experiment, 1 × 106 Cas9/lilrb4-sgRNA-transfected THP-1 cells were injected in each NSG mouse by i.v., immediately followed by i.v. injection of 0.5 × 106 isolated human normal T cells from health donors. 7 days after THP-1 and T cell implantation, mice were treat with 2mg/mouse of doxycycline via gavage daily for 5 days to achieve Cas9 expression in engrafted THP-1 cells. At 3 weeks post implantation, the peripheral blood, bone marrow, spleen, and liver were assessed for the engraftment.
For the human Cord blood (hCB)-xenograft model, 2 × 104 human CD34+ hCB cells were injected intravenously into each NSG mouse. Six weeks after implantation, mice had 10 to 50% engraftment of human cells. 1 × 106 THP-1 cells that stably express luciferase (THP-1-Luc-GFP cells) were intravenously implanted. Mice were immediately given 10 mg/kg of anti-LILRB4 antibodies or control IgG intravenously. Tumor growth was monitored over time by luminescence imaging (Max, 1×108 p/sec/cm2/sr; Min, 5×106 p/sec/cm2/sr). Lineages of human normal blood cells were analyzed by flow cytometry.
Mouse AML allograft
The procedure of mouse AML allograft was similar to that of human AML xenograft. Briefly, 6–8 week-old wild-type C57bl/6 mice were used for transplantation. 1 × 106 mouse leukemia cells expressing human LILRB4 were resuspended in 200 μl PBS for each mouse intravenously or subcutaneously implantation. Mice were given 10 mg/kg of anti-LILRB4-N297A antibodies or control IgG intravenously 7 days after leukemia cell implantation and were treated twice a week until euthanization. Three weeks after transplantation, the peripheral blood, bone marrow, spleen, and liver were assessed for the engraftment. For subcutaneously implant mice, tumor sizes were determined by caliper measure (width x width x length). For survival curve experiments, the death of mice was recorded when the moribund animals were euthanized. For CD8+ T depletion, 10mg/kg anti-CD8 antibodies (YTS 169.4.2, Bioxcell) were i.v. injected 3 days after leukemia cell implantation and were treated for additional two time every 3 days. To determine whether anti-LILRB4 antibody treatment generates tumor-specific memory T cells against the tumor or against LILRB4, we conducted adoptive transfer of spleen cells (5 × 106 /mouse) from anti-LILRB4 treated mice into normal recipient C57bl/6 mice. Four out of five transplanted mice rejected the control C1498-GFP mouse leukemia cells, and these mice were not susceptible to rechallenge with 3-fold higher numbers (3 × 106 /mouse) of C1498-GFP leukemia cells. While none of 5 mice with adoptive transfer of spleen cells from naïve mice reject the control C1498-GFP mouse leukemia cells.
Chimeric receptor reporter assay
We constructed a stable chimeric receptor reporter cell system as described 3,4 to test the ability of a ligand to bind to the ECD of individual LILRBs, PirB, gp49B1 and LILRB4 site mutants and to trigger the activation or inhibition of the chimerically fused intracellular domain of paired immunoglobulin-like receptor β, which signals through the adaptor DAP-12 to activate the NFAT promoter. If an agonist or antagonist binds the ECD and activates or suppresses the chimeric signaling domain, an increase or decrease, respectively, in GFP expression is observed. A competition assay was used to screen LILRB4 blocking antibodies. Briefly, APOE proteins (CI02, Novoprotein; 10 μg/ml) or human AB serum (10%, diluted in PBS) were pre-coated on 96-well plate at 37 °C for 3 hrs. After two washes with PBS, 2×104 LILRB4 reporter cells were seeded in each well; meanwhile, indicated anti-LILRB4 antibodies were added into culture media. After 16 hrs, the percentage of GFP+ reporter cells was analyzed by flow cytometry. The threshold of activation is 2 times of negative control treatment.
Fast protein liquid chromatography (FPLC) and Mass Spectrum
10% human AB serum in PBS was loaded onto a 16/60 Superdex 200 gel filtration column and eluted with PBS and 2mM EDTA. Eighty Fractions (40 ml) were collected, and each fraction (0.5 ml) was analyzed by chimeric receptor reporter assay. The active fractions (#26~30) were loaded onto PAGE-gel and processed to LC-MS/MS analysis (Orbitrap Elite) for protein identification in UTSW proteomics core. Recombinant or purified proteins used for validation were ZA2G (MBS145455, MyBioSource), AMBP (13141-H08H1, Sino Biological Inc), TTHY (12091-H08H, Sino Biological Inc), PEDF (11104-H08H, Sino Biological Inc), A2MG (MBS173010, MyBioSource), HEMO (MBS143111, MyBioSource), ANGT (MBS173525, MyBioSource), A1AT (MBS173006, MyBioSource), S100A9 (pro-814, Prospecbio), HORN (EBP08267, Biotrend USA), VTDB (CSB-EP009306HU, Biotrend USA), LRG1 (pro-141, Prospecbio), A1BG (RPE570Hu01, Cloud-Clone Corp), CRSP3 (RD172262100, BioVendor), APOA1 (16–16-120101-LEL, Athens Research & Technology), APOA2 (16–16-120102, Athens Research & Technology), APOA4 (16–16-120104, Athens Research & Technology), APOB (16–16-120200, Athens Research & Technology), APOC1 (16–16-120301, Athens Research & Technology), APOC2 (16–16-120302, Athens Research & Technology), APOC3 (16–16-120303, Athens Research & Technology), hAPOE (16–16-120500, Athens Research & Technology), mAPOE (CJ05, Novoprotein), APOE2 (350–12, Peprotech), APOE3 (350–02, Peprotech), APOE4 (350–04, Peprotech), PODXL2 (1524-EG-050, R&D systems), CD44 (12211-H08H, Sino Biological Inc), HCK (PV6128, Thermo Fisher), VEGFR3 (10806-H08H, Sino Biological Inc), NRG3 (16071-H08H, Sino Biological Inc), PI16 (H00221476-P01, Novusbio), hMAG (8940-MG-050, R&D systems), mMAG (8580-MG-100, R&D systems), CNTF (303-CR-050, R&D systems), ANGPTL-7 (914-AN-025/CF, R&D systems), integrin-α1β1 (7064-AB-025, R&D systems), integrin-α2β1 (5698-AB-050, R&D systems), integrin-α2β3 (7148-AB-025, R&D systems), integrin-α3β1 (2840-A3–050, R&D systems), integrin-α4β1 (5668-A4–050, R&D systems), integrin-α4β7 (5397-A3–050, R&D systems), integrin-α5β1 (3230-A5–050, R&D systems), integrin-α5β3 (3050-AV-050, R&D systems), integrin-α5β5 (2528-AV-050, R&D systems), integrin-α5β6 (CT039-H2508H, Sino Biological Inc), mintegrin-α5β6 (CT051-M2508H, Sino Biological Inc), integrin-α5β8 (4135-AV-050, R&D systems), integrin-α6β4 (5497-A6–050, R&D systems), integrin-α8β1 (CT016-H2508H, Sino Biological Inc), integrin-α9β1 (5438-A9–050, R&D systems), integrin-α10β1 (5895-AB-050, R&D systems), integrin-α11β1 (6357-AB-050, R&D systems), integrin-αEβ7 (5850-A3–050, R&D systems), integrin-αXβ2 (CT017-H2508H, Sino Biological Inc) and normal mouse serum (NS03L, Millipore sigma).
Bio-layer interferometry
Binding interaction analyses between LILRB4-Fc with APOE2, APOE3, and APOE4 were performed on the Octet RED96 (ForteBio, Pall Corporation). All interaction studies were performed with the protein A dip-and-read biosensors (ForteBio). All binding experiments were performed using the Octet Red and kinetics buffer at 30 °C. LILRB4-Fc coated biosensors (25 μg/ml LILRB4-Fc was loaded for 420 s) were washed in kinetics buffer before monitoring of association (300 s) and dissociation (600 s) of APOEs. Background wavelength shifts were measured from reference sensors that were loaded only with LILRB4-Fc.
Surface plasmon resonance (SPR)
Biacore 2000 and CM5 chips were used to analyze binding of recombinant APOEs to the LILRB4 extracellular domain fused to hFc, using a method as we described 2. Recombinant protein A (Pierce) was pre-immobilized in two flow cells using the amine-coupling kit from GE. LILRB4-hFc was injected into one of the flow cells to be captured by the protein A. Each binding sensorgram from the sample flow cell, containing a captured LILRB4-hFc, was corrected for the protein A coupled cell control. Following each injection of an antigen solution, which induced the binding reaction, and the dissociation period during which the running buffer was infused, the protein A surface was regenerated by the injection of the regeneration solution containing 10 mM Na3PO4 (pH 2.5) and 500 mM NaCl. All captured LILRB4-hFc, with and without APOE bound, was completely removed, and another cycle begun. All measurements were performed at 25°C with a flow rate of 30 μL/min.
Microscale thermophoresis (MST)
MST experiments were performed on a Monolith NT.115 system (NanoTemper Technologies) using 80% LED and 20% IR-laser power. Laser on and off times were set at 30 s and 5 s, respectively. Recombinant LILRB4-ECD protein (SinoBio) was labeled with 4488-NHS (NanoTemper Technologies) and applied at a final concentration of 5.9 nM. A two-fold dilution series was prepared for unlabeled His-APOE (CI06, Novoprotein) in PBS, and each dilution point was similarly transferred to LILRB4-ECD solution. The final concentrations of His-APOE ranged from 0.36 nM to 12 μM. Samples were filled into standard-treated capillaries (NanoTemper Technologies) for measurement.
Western blotting and co-immunoprecipitation
Whole cells were lysed in Laemmli sample buffer (Sigma-Aldrich) supplemented with protease inhibitor cocktail (Roche Diagnostics). Samples were separated on SDS–PAGE gels (Bio-Rad) and transferred on nitrocellulose membranes (Bio-Rad) for protein detection. Primary antibodies including Anti-SHP-1 (Cell signaling, 3759, 1:1000), anti-phospho-SHP-1 Tyr564 (Cell signaling, 8849, 1:500), anti-phospho-SHP-1 Tyr564 (Invitrogen, PA537708, 1:500), anti-SHP-2 (Cell signaling, 3397, 1:1000), anti-phospho-SHP-2 Tyr580 (Cell signaling, 3703, 1:500), anti-SHIP1 (Cell signaling, 2727, 1:1000), anti-phospho-SHIP1 Tyr1020 (Cell signaling, 3941, 1:500), anti-NFκB p65 (Cell signaling, 8242, 1:1000), anti-IKKα (Cell signaling, 11930, 1:1000), anti-IKKβ (Cell signaling, 8943, 1:1000), anti-phospho-IKKα/β Ser176/180 (Cell signaling, 2697, 1:500), anti-IκBα (Cell signaling, 4814, 1:1000), anti-phospho-IκBα Ser32 (Cell signaling, 2859, 1:500), anti-Lamin-B2 (Cell signaling, 12255, 1:1000) and anti-Arginase-1 (Cell signaling, 9819, 1:1000), anti-uPAR (Invitrogen, MON R-4–02, 1:500), anti-LILRB4 (Santa cruz, sc-366213, 1:200), anti-APOE (Creative diagnostics, DCABH-2367, 1:250), anti-β-actin (Sigma-Aldrich, A2066, 1:1000) and anti-α-tubulin (Sigma-Aldrich, MABT205, 1:1000), as well as horseradish peroxidase (HRP) conjugated secondary antibodies (Cell signaling, 7074, 1:1,000, and 7076, 1:1,000) and chemi-luminescent substrate (Invitrogen), were used. Specific cellular compartment fractionations were carried out using the NE-PER nuclear/cytoplasmic extraction kit (Thermo fisher, 78833) or the plasma membrane protein extraction kit (Abcam, ab65400). Proteins from plasma membrane fraction were further incubated with anti-LILRB4 antibodies and dynabeads protein A (Thermo fisher, 10001D) for further immunoprecipitation and western blotting.
Immunohistochemistry
Hematoxylin staining and immunostaining were performed on paraffin sections of tumors. Antibodies used were against LILRB4 (lab produced, 1:100), CD3 (Abcam, ab16669, 1:100), PD-1 (Thermo Fisher, J116, 14–9989-82, 1:100) and Arginase-1 (Cell signaling, 9819S, 1:100). The images were visualized using the Hamamatsu NanoZoomer 2.0-HT (Meyer instruments Inc., Houston, TX) and viewed in NPDview2 software (Hamamatsu, Japan).
Cytokine antibody array and arginase activity assay
To examine the secreted protein from leukemia cells, condition media were applied to a human cytokine antibody array (AAH-CYT-1000, RayBio) for the semi-quantitative detection of 120 human proteins. Image J (NIH) was used for quantification. Arginase activity was determined in condition media of indicated leukemia cells by a QuantiChrom Arginase assay kit (DARG-100, BioAssay system).
RNA-seq analysis
RNA was purified from sorted cells with Qiagen RNeasy Mini Kit and then reverse-transcribed with SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. RNA-seq was performed at the UTSW Genomics and Microarray Core Facility. The cDNA was sonicated using a Covaris S2 ultrasonicator, and libraries were prepared with the KAPA High Throughput Library Preparation Kit. Samples were end-repaired, and the 3′ ends were adenylated and barcoded with multiplex adapters. PCR-amplified libraries were purified with AmpureXP beads and validated on the Agilent 2100 Bioanalyzer. Before being normalized and pooled, samples were quantified by Qubit (Invitrogen) and then run on an Illumina Hiseq 2500 instrument using PE100 SBS v3 reagents to generate 51-bp single-end reads. Before mapping, reads were trimmed to remove low-quality regions in the ends. Trimmed reads were mapped to the human genome (HM19) using TopHat v2.0.1227 with the UCSC iGenomes GTF file from Illumina.
Methods for data normalization and analysis are based on the use of “internal standards” that characterize some aspects of the system’s behavior, such as technical variability, as presented elsewhere. Genes with log2 (fold change) > 2, P < 0.01 and RPKM > 0.1 were deemed to be significantly differentially expressed between the two conditions and were used for pathway analysis and upstream transcription factor analysis. Pathway analysis was conducted using the DAVID (https://david.ncifcrf.gov/tools.jsp). Upstream transcription-factor analysis was conducted using QIAGEN’s Ingenuity tool (http://www.ingenuity.com/).
Molecular docking of LILRB4 with APOE
Docking of LILRB4 with APOE was performed on ZDOCKpro module of the Insight II package. The general protocol for running ZDOCK includes two consecutive steps of calculation described as geometry search and energy search, running in program ZDOCK and RDOCK, respectively. LILRB4 crystal structure (3P2T) and APOE3 structure (2L7B) were obtained from PDB database. Top 50 ZDOCK poses were submitted to RDOCK refinement. Poses with high score both in ZDOCK and RDOCK were selected as candidate complex for LILRB4/APOE interaction analysis (Supplementary Table 2).
Statistical analyses
Representative data from four independent experiments or indicated independent samples are presented as dot plots (means ± s.e.m.) or as box-and-whisker plots (median values (line), 25th–75th percentiles (box outline) and minimum and maximum values (whiskers)). Statistical significance for two samples-comparison was calculated by two-tailed Student’s t-test. Statistical significance for survival was calculated by the log–rank test. The multivariate analysis of TCGA data was analyzed by Cox regression. The difference was considered statistically significant if p < 0.05. n.s., not significant; p values are represented as precise values. The Pearson’s correlation analyses were performed with the RStudio software (the R Foundation).
Code availability
The custom code for Pearson’s correlation analysis in RStudio is showed below.
setwd(“~/file paths”)
draw.graph = function(sam) {
file = paste(sam, “.tsv”, sep =““)
df1 = read.table(“file name A.tsv”, sep = “\t”, header = T)
df2 = read.table(file, sep = “\t”, header =T)
df = merge(df1, df2, by.x = “sample”, by.y = “sample”)
df = df[!is.na(df[,2]),]
r = cor.test(df[,2], df[,3])
p = cor.test(df[,2], df[,3])$p.value
jpeg(filename = paste(sam,”.jpeg”, sep = ““),
width = 280, height = 280, units = “px”, pointsize = 12,
quality = 75)
#bg = “white”, res = 300)
plot(df[[2]], df[[3]], xlab = “Title”, ylab = sam,
pch = 16, cex = 1, col = “red”,
main = paste(“r = “,round(r$estimate,2), “\n”,”p = “,round(p,8), sep = ““))
reg = lm(df[[2]]~df[[3]])
abline(reg)
dev.off()
}
sample = c(“file name B”)
for (s in sample) {
draw.graph(s)
}
Extended Data
Extended Data Figure 1. LIRB4 expression in human AML patients and negatively correlated with patient overall survival and T cell proliferation.
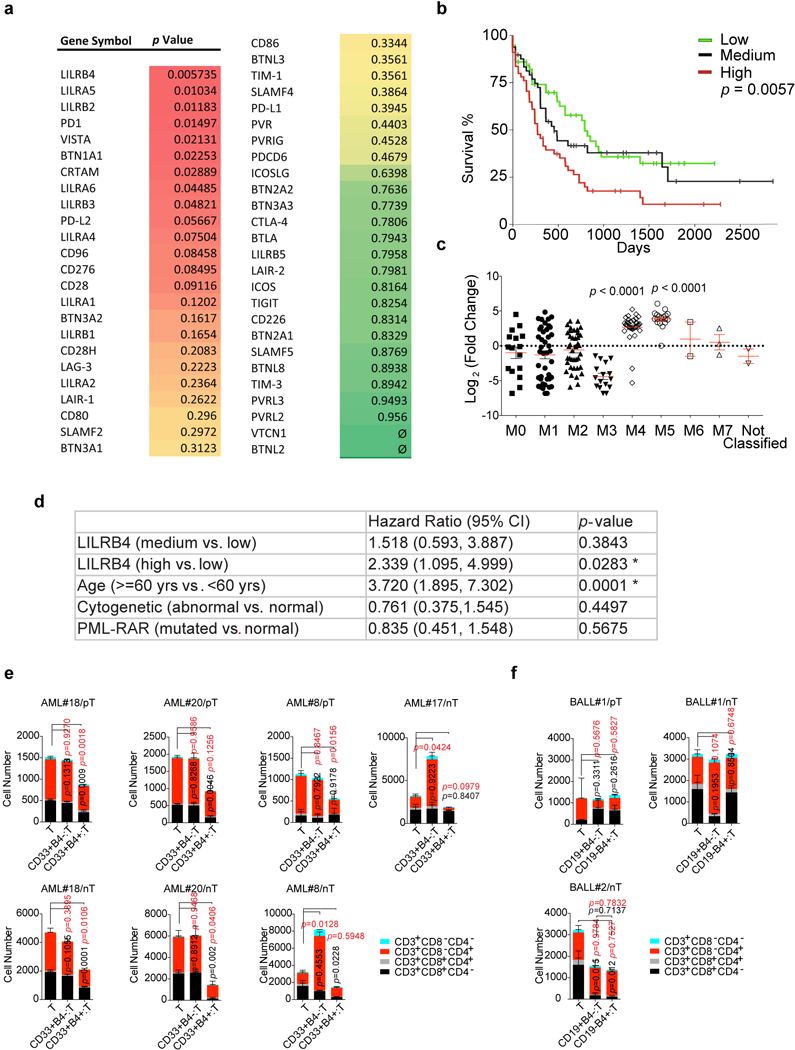
a, Analysis of correlation between mRNA levels of immune modulating molecules and the overall survival of AML patients (n=160, divided into three groups based on gene expression) in TCGA database (https://xena.ucsc.edu) by Kaplan-Meier long-rank test. b, Kaplan-Meier analysis of correlations between lilrb4 mRNA level and the overall survival of AML patients (n=160) from the TCGA database was performed in Xena browser (https://xena.ucsc.edu). Low, n=57; Medium, n=48; High, n=55. The p value was from Kaplan-Meier long-rank test. c, mRNA expression data from the TCGA database was analyzed as a function of patient AML subtype. M0, n=16; M1, n=42; M2, n=39; M3, n=16; M4, n=35; M5, n=18; M6, n=2; M7, n=3; and two not-classified AML samples. Pairwise comparisons between M4 and each one of the other subtypes (all of the p-values are <0.0001), as well as between M5 and each one of the other subtypes (all of the p-values are <0.0001), using two-sample t-test. Mean and s.e.m. values were shown. d, A multivariable Cox regression analysis to assess the association, with adjustment for confounders that include age, cytogenetics, and PML-RAR mutation in TCGA database. The total sample size was 79. *, p<0.05 is considered as significant. e-f, Autologous T cells isolated from individual monocytic AML or B-ALL patients were incubated with irradiated lilrb4-positive or lilrb4-negative primary leukemia cells from the same patients. pT, patient T cells. Allogeneic T cells isolated from healthy donors were incubated with irradiated lilrb4-positive or lilrb4-negative primary leukemia cells from indicated AML or B-ALL patients at an E:T of 10:1. nT, normal T cells. After culture with anti-CD3/CD28/CD137-coated beads and rhIL-2 for 14 days, T cells were stained with anti-CD3, anti-CD4, and anti-CD8 antibodies and analyzed by flow cytometry. (e and f) p values were from two-tailed student t-test. p values in black indicate significance of CD3+CD8+ cells; p values in red indicate significance of CD3+CD4+ cells. e-f, n=2 or 3 biologically independent samples with mean and s.e.m. See raw data of e and f in Source Data Extended Data Figure 1.
Extended Data Figure 2. LILRB4 suppresses T cell proliferation in vitro.
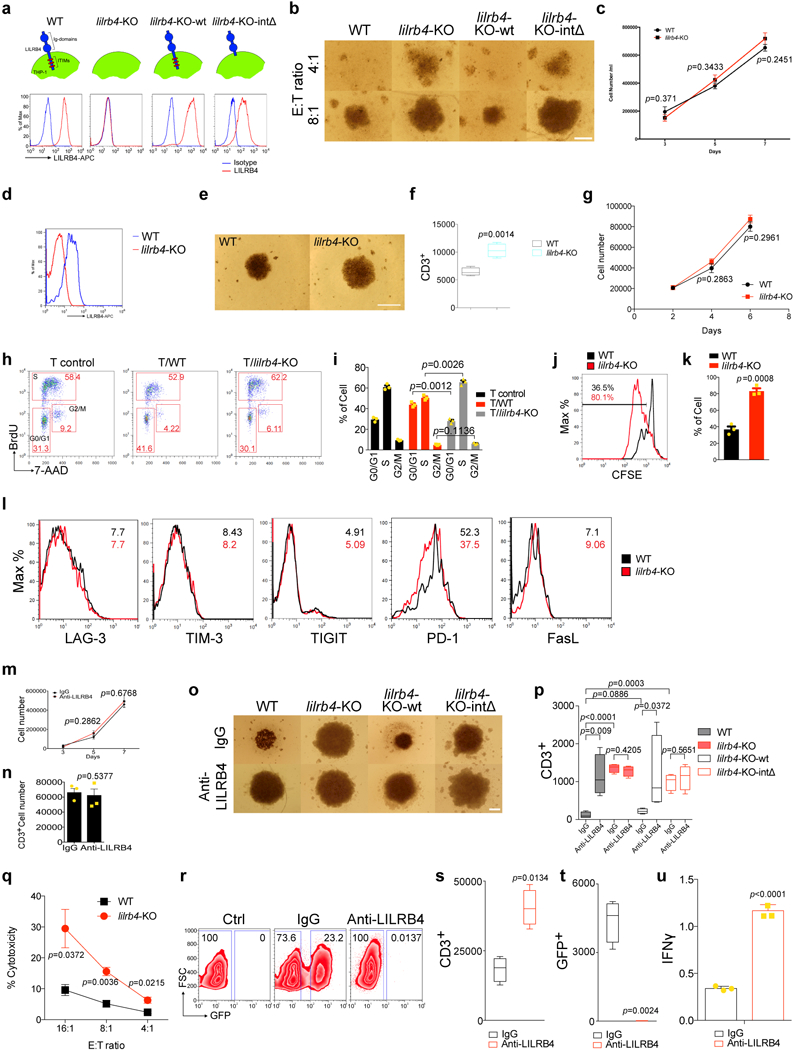
a, Schematic of preparation of lilrb4-modulated THP-1 cells and examination of LILRB4 expression on the cell surfaces by flow cytometry. WT, THP-1 cells treated with scrambled control; lilrb4-KO, lilrb4-knockout THP-1 cells; lilrb4-KO-wt, forced expression of wild-type lilrb4 on lilrb4-KO THP-1 cells; lilrb4-KO-intΔ, forced expression of the intracellular domain-deleted mutant lilrb4 on lilrb4-KO THP-1 cells. b, Loss of lilrb4 on THP-1 cells reduces T cell suppression. Representative photograph of Fig. 1c (scale bar, 100 μm). c, Loss of lilrb4 on THP-1 cells does not affect cell proliferation (n=3 biologically independent samples with mean and s.e.m.). d, Examination of LILRB4 expression on cell surface of lilrb4-KO MV4–11 cells by flow cytometry. e-f, Loss of lilrb4 on MV4–11 cells reduces T cell suppression. T cells isolated from healthy donors incubated in the lower chambers of a 96-well transwell plate with irradiated MV4–11 cells (E:T of 2:1) in the upper chamber separated by a membrane with 3 μm pores. After culture with anti-CD3/CD28-coated beads and rhIL-2 for 7 days. Representative cells were photographed using an inverted microscope (scale bar, 100 μm) (e) and T cells were stained with anti-CD3 and analyzed by flow cytometry (f). n=4 biologically independent samples. g, Loss of lilrb4 on MV4–11 cells does not affect cell proliferation (n=3 biologically independent samples with mean and s.e.m.). h-i, T cells (E: effector cells) isolated from healthy donors were incubated with indicated irradiated THP-1 cells (T: target cells) without direct contact in transwells for 2 days. E:T=2:1. T cells were treated with BrdU for 30 mins followed by BrdU and 7-AAD staining for flow cytometry analysis. Representative flow cytometry plots are shown in h and the cell cycle status is summarized in i. T control, T cells were cultured without THP-1 cells. n=3 biologically independent samples with mean and s.e.m. j-k, T cells (E: effector cells) isolated from healthy donors were stained with CFSE and incubated with indicated irradiated THP-1 cells (T: target cells) without direct contact in transwells for 2 days. A representative flow cytometry plot is shown in j and the percentages of proliferating T cells indicated by CFSE-low staining is shown in k. n=3 biologically independent samples with mean and s.e.m. l, LILRB4 increases PD-1 expression on T cells in coculture of leukemia cells and T cells. T cells (E: effector cells) isolated from healthy donors were incubated with indicated irradiated THP-1 cells (T: target cells) in a non-contact manner for 5 days. E:T=2:1. T cells were stained with anti-LAG-3, anti-TIM-3, anti-TIGIT, anti-PD-1 and anti-FasL antibodies for flow cytometry analysis. Shown are representative flow cytometry plots and the mean of fluorescence intensities at the right-upper corner (black, WT; red, KO). Experiments were performed three times with similar results. m-n, Anti-LILRB4 antibody had no effect on proliferation of THP-1 cells (m) or T cells (n). m, The growth of THP-1 cells during 7 days treatment with IgG or anti-LILRB4 antibody (n=3 biologically independent samples with mean and s.e.m.). n, The activation status of human primary T cells after 5 days treatment of IgG or anti-LILRB4 antibody in vitro (n=3 biologically independent samples with mean and s.e.m.). o-p, Primary T cells and irradiated THP-1 cells (E:T ratio, 2:1) were placed to the lower chambers and upper chamber respectively and treated with 10 μg/ml control IgG or anti-LILRB4 antibodies. Shown in o are representative T cells photopgraphed (scale bar, 100 μm) and T cells were stained with anti-CD3 and analyzed by flow cytometry (p). n=4 biologically independent samples. q, Primary T cells stimulated with anti-CD3/CD28/CD137-coated beads were co-cultured with WT or lilrb4-KO-THP-1 cells with indicated E:T ratios for 4 hrs (n=3 biologically independent samples with mean and s.e.m.). Cytotoxity of leukemia cells was determined by PI staining in flow cytometry analysis. r-u, CD8+ T cells (5×104 cells) stimulated with anti-CD3/CD28/CD137-coated beads were co-cultured with 5×103 THP-1 cells that stably express GFP and treated with 100 μg/ml anti-LILRB4 antibodies or control IgG for 5 days. s-t, n=4 biologically independent samples; u, n=3 biologically independent samples with mean and s.e.m. Shown are representative flow plots (r) of the percentages of T cells (GFP-) and surviving leukemia cells (GFP+), and quantification of T cells (s), GFP+ leukemia cells (t), and secretion of IFNγ (u). (b, d-e, h, j, o and r) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in f, p, and s-t. All p values were from two-tailed student t-test.
Extended Data Figure 3. Inhibition of LILRB4 reduces leukemia development in humanized immunocompromised mice and syngeneic mice.
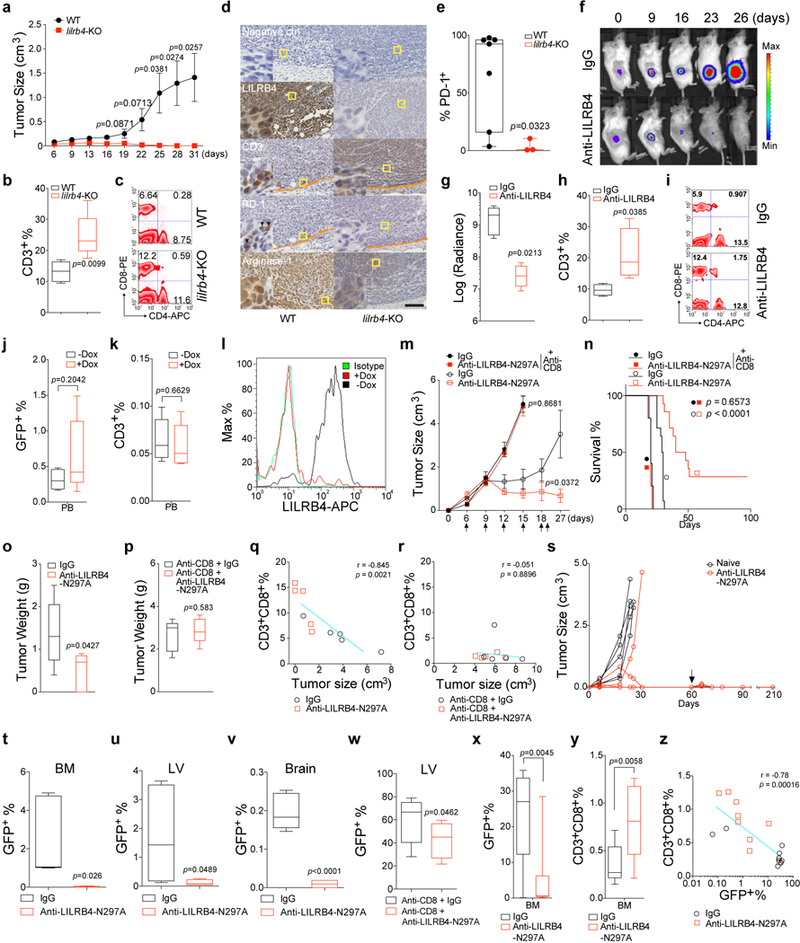
a-c, WT or lilrb4-KO THP-1 cells (3×106 cells/mouse) were subcutaneously implanted into hPBMC-repopulated NSG mice (WT, n=14 mice with mean and s.e.m.; lilrb4-KO, n=10 mice with mean and s.e.m. Also see Source Data Extended Data Figure 3.). Shown are tumor size (a), quantitation of CD3+ at day 31 in peripheral blood of recipient mice (b) and representative flow plots showing CD4+ and CD8+ T cells (c). d-e, LILRB4 increases PD-1 expression on tumor-infiltrated T cells. WT or lilrb4-KO THP-1 cells were subcutaneously implanted into hPBMC-repopulated NSG mice. Three weeks after implantation, 7 out of 10 WT-group mice had large tumors and 3 out of 10 KO-group mice had tiny tumors. These tumors were dissected for immunohistochemistry and flow cytometry staining with anti-LILRB4, anti-CD3, anti-PD-1 or anti-Arginase-1 antibodies. Left corner images were magnified from the yellow highlighted regions. In CD3 and PD-1 staining images, orange dash lines indicate the tumor boundary. Black arrowheads indicate PD-1 positive cells. Scale bar, 100 μm. Shown in e, tumors were dissected and cells in tumor region were stained with anti-CD3 and anti-PD-1 antibodies for flow cytometry analysis. The percentages of PD-1+ T cells (Ratio of PD-1+CD3+cells/CD3+ cells) were calculated. f-i, THP-1 cells were transplanted into hPBMC-repopulated NSG mice, and mice were treated with control IgG or anti-LILRB4 antibody after 6 days (10 mg/kg; n=5). Leukemia development was monitored by luminescence imaging (f); luminescence flux (radiance) at day 26 (g; n=5) and T cell numbers at day 26 in representative mice (h-i) were also shown. j-k, Engraftment of human T cells and i.v. transplanted Doxycycline (Dox)-inducible lilrb4-knockout THP-1 cells (GFP+) in NSG mice at day 7 before Dox administration (n=5). l, Representative flow plot shows LILRB4 was successfully deleted in engrafted leukemia cells in bone marrow of Dox-fed mouse at the endpoint. n.s., not significant. m-w, Mouse AML C1498 cells (3×106 cells/mouse) that stably express LILRB4-IRES-GFP were s.c. implanted into C57bl/6 mice. Anti-LILRB4-N297A antibodies or control IgG were i.v. injected at 6, 9, 12, 15, 18, and 21 days post implantation of tumor cells. Two groups of mice were treated with anti-CD8 antibodies at 3, 6, 9, and 12 days post implantation of tumor cells to achieve CD8+ T cell depletion. m, Tumor growth of subcutaneously implanted human LILRB4-expressing mouse AML C1498 cells (hlilrb4-C1498) in C57BL/6 mice with anti-LILRB4-N297A antibody or control antibody treatment (n=5 mice). Also see Source Data Extended Data Figure 3. n, Survival curve of subcutaneous hlilrb4-C1498-tumor-bearing mice (n=12 mice). Same as tumor size, anti-LILRB4 antibodies decreased the tumor weight (o, n=5 mice) but did not in the absence of CD8+ T cells (p, n=5 mice). The percentage of CD8+ T cells in spleen is significantly negatively correlated with tumor size (q, n=5 mice) but not in the absence of CD8+ T cells (r, n=5 mice). s, Adoptive transplantation of spleen cells from control mice or tumor-bearing mice that were cured by anti-LILRB4-N297A treatment (n=5 mice). Tumor size was monitored as a function of time. Arrow indicates day of rechallenge in mice that had eliminated leukemia with 3-times number of AML cells (n=4 mice). Also see Source Data Extended Data Figure 3. Anti-LILRB4 antibodies reduced the leukemia cells infiltrating into host tissues (t-v, n=5 mice) and even CD8+ cells were depleted (w, n=5 mice). x-z, C57bl/6 mice were i.v. implanted with human LILRb4-expressing mouse AML C1498 cells (3×106 cells/mouse) that express GFP. Anti-LILRB4-N297A antibodies (n=9 mice) or control IgG (n=9 mice) were i.v. injected at 6, 9, 12, 15 and 18 days post implantation of tumor cells. Anti-LILRB4 antibodies decreased the percentage of leukemia cells in bone marrow (x). Anti-LILRB4 antibodies increased CD8+ T cells (y). The percentage of CD8+ T cells in bone marrow is significantly negatively correlated with the percentage of leukemia cells (z). (c, i and l) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in b, e, g-h, j-k, o-p and t-y. All p values (except of n from long-rank test; and except of q-r and z from Pearson’s correlation) were from two-tailed student t-test.
Extended Data Figure 4. Anti-LILRB4 antibodies reduce leukemia development by restore of autologous T cells in PDX mice and inhibition of primary AML cell infiltration.
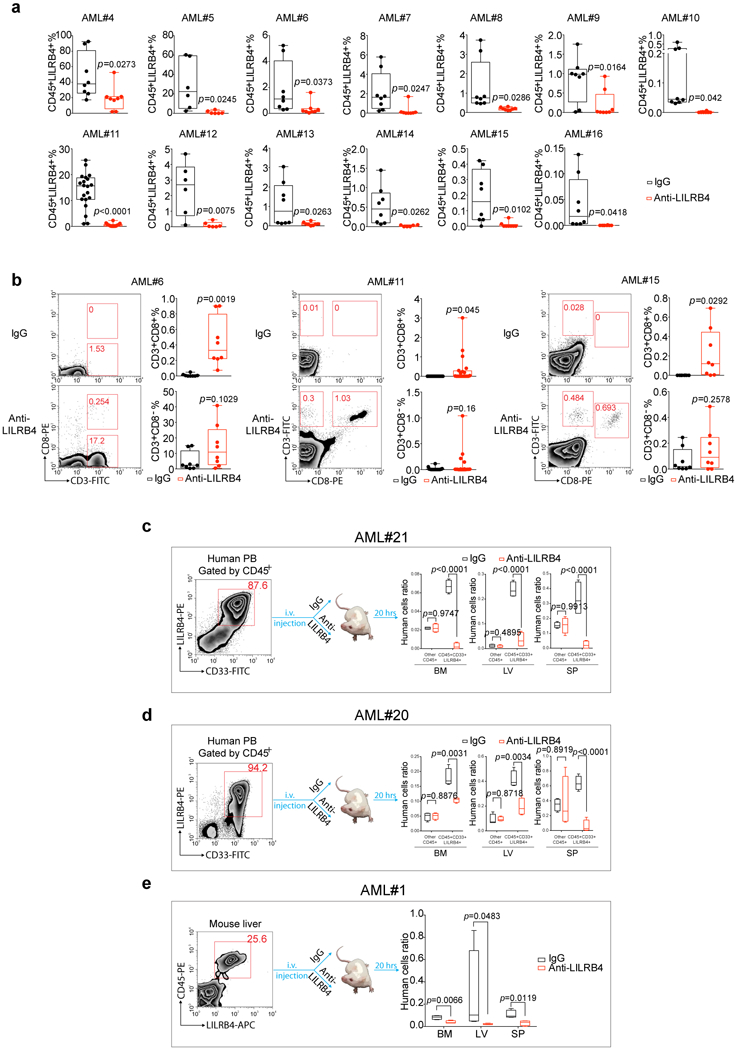
a, Primary peripheral blood or bone marrow mononuclear AML cells (5×106 −1×107 cells/mouse) from each of sixteen human patients (three shown in Fig.1g-i, also see Supplementary Table 5) were injected into NSG mice followed by treatment with IgG or anti-LILRB4 antibody (10mg/kg. twice a week by i.v. injection). Shown are percentages of human CD45+LILRB4+ AML cells harvested from hematopoietic tissues including bone marrow, spleen, liver and peripheral blood at 2~4 months after transplantation as determined by flow cytometry. b, Shown are percentages of autologous human T cells harvested from hematopoietic tissues including bone marrow, spleen, liver and peripheral blood at 2~4 months after transplantation as determined by flow cytometry; and representative flow plots of CD3+CD8+ T cells in bone marrow of mice in three PDXs. n=8 biologically independent samples for all PDXs except AML#11 (n=20 biologically independent samples) in a-b. c-e, Comparison of infiltration of human primary monocytic AML cells in NSG mice (n=5 mice) after treatment with anti-LILRB4 antibody or IgG control. c-d, Primary human peripheral blood mononuclear cells from monocytic AML patients were injected. The quantifications in c are also shown in Fig. 2l-n. e, Mouse liver cells with xenografted primary human monocytic AML cells (human CD45+LILRB4+ cells) were injected. See Methods for definition of box plot elements in a-e. All p values were from two-tailed student t-test.
Extended Data Figure 5. LILRB4 promotes infiltration of AML cells.
a and c, Examination of LILRB4 expression on mouse AML cells, C1498 (a) or WEHI-3 (c) that stably express lilrb4. b and d, Forced expression of LILRB4 did not affect cell proliferation of mouse AML cells, C1498 (b, n=3 biologically independent samples with mean and s.e.m.) or WEHI-3 (d, n=3 biologically independent samples with mean and s.e.m.). e, Forced expression of human LILRB4 promoted transendothelial migration of mouse AML WEHI-3 cells (n=3 biologically independent samples with mean and s.e.m.). f, NSG mice (n=6 mice) were injected with 1×106 THP-1 cells followed immediately by IgG or anti-LILRB4 antibody treatment and were monitored by bioluminescence imaging. g-h, Anti-LILRB4 antibodies decreased AML cells infiltration into internal organs. Mice were sacrificed at 21 days for ex vivo bioluminescence imaging of internal organs after transplantation of 1×106 luciferase-expressed THP-1 cells. Images of luminescence flux (radiance) from representative mice are shown (g). 1: GI tract; 2: legs; 3: lung; 4: spleen; 5: liver; 6: kidneys; 7: brain; 8: heart. Infiltrated leukemia cells formed tumor nodules in liver (h). i-j, Anti-LILRB4 antibodies did not have effect on LILRB4-negative cancer cells. LILRB4 is expressed on THP-1 and MV4–11 human AML cells but not on U937 cells as analyzed by flow cytometry (i). Isotype IgG was used as control. NSG mice were injected with U937 human AML cells, which do not express LILRB4, and then treated with anti-LILRB4 antibodies (j). IgG served as control antibodies. Mice were sacrificed at day 25 post-transplant for analysis of LV, BM, SP, and PB by flow cytometry. The presence of human AML cells was detected by anti-human CD45 antibody staining (n=4 mice with mean and s.e.m.). k-t, Anti-LILRB4 antibodies decreased infiltration of THP-1 (k-o) or MV4–11 (p-t) human AML cells. Comparison of transendothelial migration abilities of GFP-expressing THP-1 (k) or CFSE-labeled MV4–11 (p) cells after treatment with anti-LILRB4 antibody or IgG control in a transwell assay (n=3 biologically independent samples with mean and s.e.m.). Comparison of the homing abilities of CFSE-labeled MV4–11 cells (5×106 per mouse) that were injected into NSG mice followed immediately by IgG or anti-LILRB4 antibody treatment at 20 hr post-injection (n=5 mice). Numbers of leukemia cells (GFP+ in l or CFSE+ in q) in LV, SP, and BM normalized to that in PB as determined by flow cytometry are shown. NSG mice were injected with 1×106 THP-1 or MV4–11 cells followed immediately by IgG or anti-LILRB4 antibody treatment (n=6 mice for THP-1 or 5 mice for MV4–11 xenografts). Shown are percentages of MV4–11 cells (stained with anti-human CD45) as determined by flow cytometry in indicated organs at day 21 post-transplant (m and r), overall survival (n and s), and body weights as a function of time (o and t). u, Targeted immune cell populations were depleted in NSG mice. Representative flow cytometry plots demonstrating successful reduction of NK cell (CD45+CD49b+), macrophage (CD11b+F4/80+), and neutrophil (CD11b+CD11c-) frequency in NSG mice depleted of the respective immune cell subtype by treatment with anti-asialo GM1 antibodies, clodronate liposomes, and anti-Ly6G antibodies, respectively, compared to non-depleted (wild-type) NSG mice. v, CFSE-labeled MV4–11 cells (5×106 per mouse) were injected into NSG mice in that respective innate immune cells were depleted, followed immediately by IgG or anti-LILRB4-N297A antibody treatment (n=5 mice). Numbers of leukemia cells (CFSE positive) in LV, SP, and BM normalized to that in PB at 20 hr post-injection are shown. (a, c, i and u) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in l-m, o, q-r, t and v. All p values (except of n and s from long-rank test) were from two-tailed student t-test.
Extended Data Figure 6. APOE induces LILRB4 activation to suppress T cell and support AML cell migration in vitro.
a, Schematic of the LILRB4 reporter system. b, Human and mouse integrin heterodimer proteins cannot activate LILRB4 reporter (n=3 biologically independent samples with mean and s.e.m.). Human and mouse serum were used as positive controls. The threshold of activation is 2 times of negative control treatment. c, Flow cytometry demonstrating that anti-LILRB4 antibody binds to human LILRB4 reporter cells. d, The LILRB4 activation as indicated by percentage of GFP+ cells in the presence and absence of 10% human serum (HS) with or without anti-LILRB4 antibody or control IgG (n=3 biologically independent samples with mean and s.e.m.). e, Flowchart of ligand identification of potential ligands of LILRB4 in human serum. f, Fractionation of LILRB4 stimulating activities from human serum by FPLC. The positive control was 10% human serum. g, A list of proteins identified from the LILRB4 stimulating fractions by mass spectrometry. PSMs: peptide spectrum matches. h, Both Human and mouse APOE proteins can activate LILRB4 reporter (n=3 biologically independent samples with mean and s.e.m.). Human and mouse serum were used as positive controls. The threshold of activation is 2 times of negative control treatment. i, APOE proteins from different sources all activate LILRB4. APOE (20 μg/ml) purified from human plasma, His-tagged or tag-free recombinant human APOE (rhAPOE) (20 μg/ml) expressed by 293T mammalian cells, or rhAPOE (20 μg/ml) expressed by bacteria all activate the LILRB4 reporter. These APOE all represent human APOE3 (n=3 biologically independent samples with mean and s.e.m.). j, APOE2, APOE3 and APOE4 all activate the LILRB4 reporter (n=3 biologically independent samples with mean and s.e.m.). 40 μg/ml APOEs were coated on plates or directly added in cell culture media (soluble). k-l, Three APOE isoforms binds to human LILRB4. k, Binding kinetics of APOE 2, 3, and 4 to LILRB4-Fc were measured using surface plasmon resonance (SPR). LILRB4-Fc was immobilized on Protein A biosensor tips and incubated with APOE concentrations ranging from 1.5625 nM to 100 nM. l, Binding kinetics of APOE 2, 3, and 4 to LILRB4-Fc were measured using Bio-layer Interferometry (Octet). LILRB4-Fc was immobilized on Protein A biosensor tips and incubated with APOE concentrations ranging from 44 nM to 1176 nM. m, As shown in Fig. 3h, mutation of two residues, W106 and Y121 significantly reduced activation of LILRB4 by APOE, located in the first Ig domain and in the linker between two Ig domains, respectively. n and p, Examination of APOE expression in apoe-knockout THP-1 and MV4–11 cells by immunoblots. Primary T cells and irradiated THP-1 or MV4–11 cells (E:T=2:1) were incubated in the lower and upper chambers respectively. T cells were photographed (o and q, scale bar, 100 μm) and quantified by flow cytometry (Fig. 3i and, here r, n=4 biologically independent samples) after 7 days. s-t, Loss of APOE suppresses transendothelial migration of human AML THP-1 and MV4–11 cells (n=4 biologically independent samples with mean and s.e.m.). (c, k-l and n-q) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in r. All p values were from two-tailed student t-test.
Extended Data Figure 7. LILRB4 upregulates phosphorylation of SHP-2, NF-kB signaling, and expression of uPAR and Arginase-1 to suppress T cell activity and support leukemia migration.
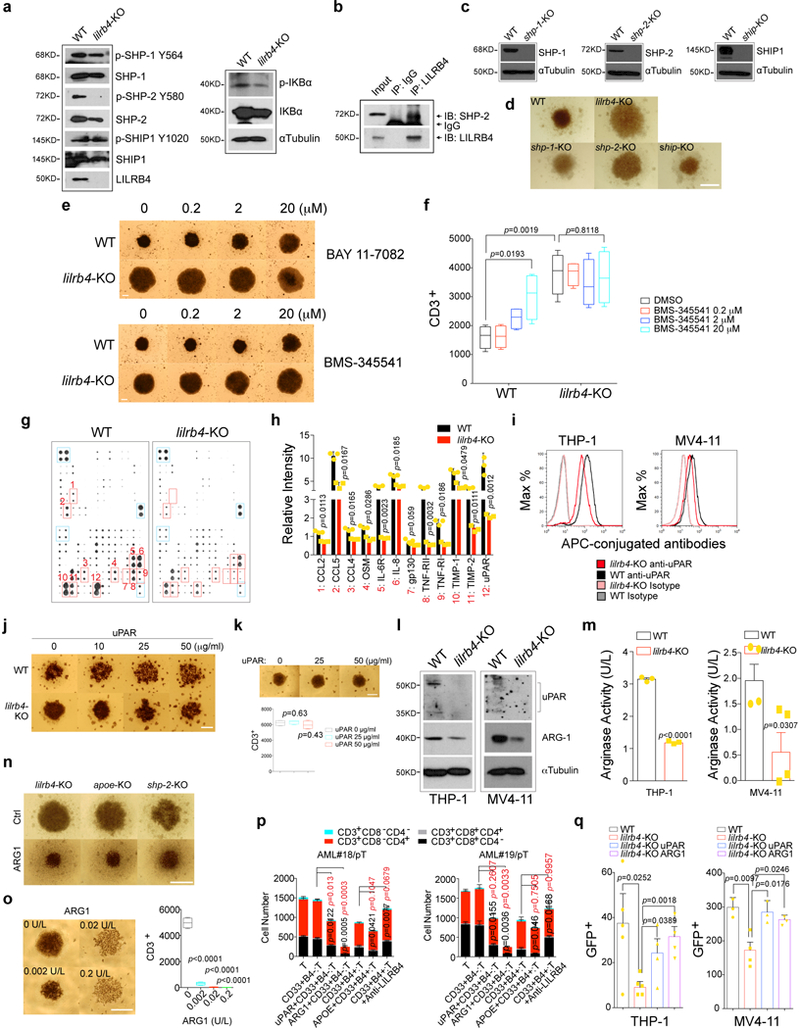
a, Phosphorylated SHP-2, phosphorylated IKB, uPAR, and ARG1 were down-regulated upon lilrb4-knockout (KO) in MV4–11 cells. b, Co-immunoprecipitation demonstrated LILRB4 interacts with SHP-2 in THP-1 cells. c, shp-1, shp-2, and ship were individually knockout by CRISP/Cas9 in THP-1 cells as detected by Western blotting. d, Primary T cells and irradiated THP-1 cells (E:T=2:1) were cultured in the lower and upper chambers respectively. T cells were photographed (scale bar, 100 μm) after 7 days. e-f, Two different NF-κB inhibitors restored T cell proliferation from the suppression by THP-1 cells in an LILRB4-dependent manner (n=4 biologically independent samples). THP-1 cells were pretreated with various doses of NF-κB inhibitors for 1 hr. Primary T cells and irradiated pretreated THP-1 cells (E:T=2:1) were cultured in the lower and upper chambers respectively. T cells were photographed (e, scale bar, 100 μm) and analyzed by flow cytometry (f) after 7 days. g-h, Loss of lilrb4 decreased secreted protein production in THP-1 cells as determined by a human cytokine antibody array (g) and the blot intensities were quantified by ImageJ software (h, n=3 biologically independent samples with mean and s.e.m.). Red boxes indicate proteins that were changed upon lilrb4-knockout; blue boxes indicate positive controls. i, Surface uPAR was downregulated in lilrb4-knockout THP-1 and MV4–11 AML cells. j, T cells were incubated with irradiated indicated THP-1 cells supplemented with indicated concentration of recombinant uPAR proteins for 7 days. T cells were photographed. k, T cells isolated from healthy donors were cultured with anti-CD3/CD28-coated beads and rhIL-2 and supplemented with indicated concentrations of uPAR proteins for 3 days (n=4 biologically independent samples). Representative cells were photographed using an inverted microscope and T cells were analyzed by flow cytometry. l, Expression of uPAR and Arginase-1 (ARG1) is downregulated in in lilrb4-knockout THP-1 and MV4–11 AML cells. m, Arginase activity as determined by a colorimetric method (DARG-100, BioAssay system) was decreased in condition medium of lilrb4-KO THP-1 and MV4–11 cells (n=3 biologically independent samples with mean and s.e.m.). n, Primary T cells and irradiated indicated THP-1 cells (E:T=2:1) were incubated in the lower and upper chambers respectively and were supplemented with 0.002 U/L recombinant ARG1 proteins for 7 days. T cells were photographed. o, T cells isolated from healthy donors were cultured with anti-CD3/CD28-coated beads and rhIL-2 and supplemented with indicated concentrations of ARG1 proteins for 3 days (n=4 biologically independent samples). Representative cells were photographed using an inverted microscope and T cells were analyzed by flow cytometry. p, Autologous T cells isolated from individual monocytic AML patients were incubated with irradiated lilrb4-positive or lilrb4-negative primary leukemia cells from the same patients at an E:T of 10:1, supplemented with recombinant anti-LILRB4 antibodies, APOE-VLDL, uPAR or ARG1. pT, patient T cells. After culture with anti-CD3/CD28/CD137-coated beads and rhIL-2 for 14 days, T cells were stained with anti-CD3, anti-CD4, and anti-CD8 antibodies and analyzed by flow cytometry. n=3 biologically independent samples with mean and s.e.m. q, Supplementation of recombinant uPAR or ARG1 to the medium rescued the decrease of transmigration ability of lilrb4-KO THP-1 or lilrb4-KO MV4–11 cells across endothelium (n=3 biologically independent samples with mean and s.e.m.). Scale bar, 100 μm. (a-e, g, i-j, l and n) These experiments were repeated independently three times with similar results. See Methods for definition of box plot elements in f, k and o. All p values were from two-tailed student t-test. See raw data of p in Source Data Extended Data Figure 7.
Extended Data Figure 8. Detection of SHP-2/NF-κB signaling and uPAR and Arginase-1 expression in primary human monocytic AML cells.
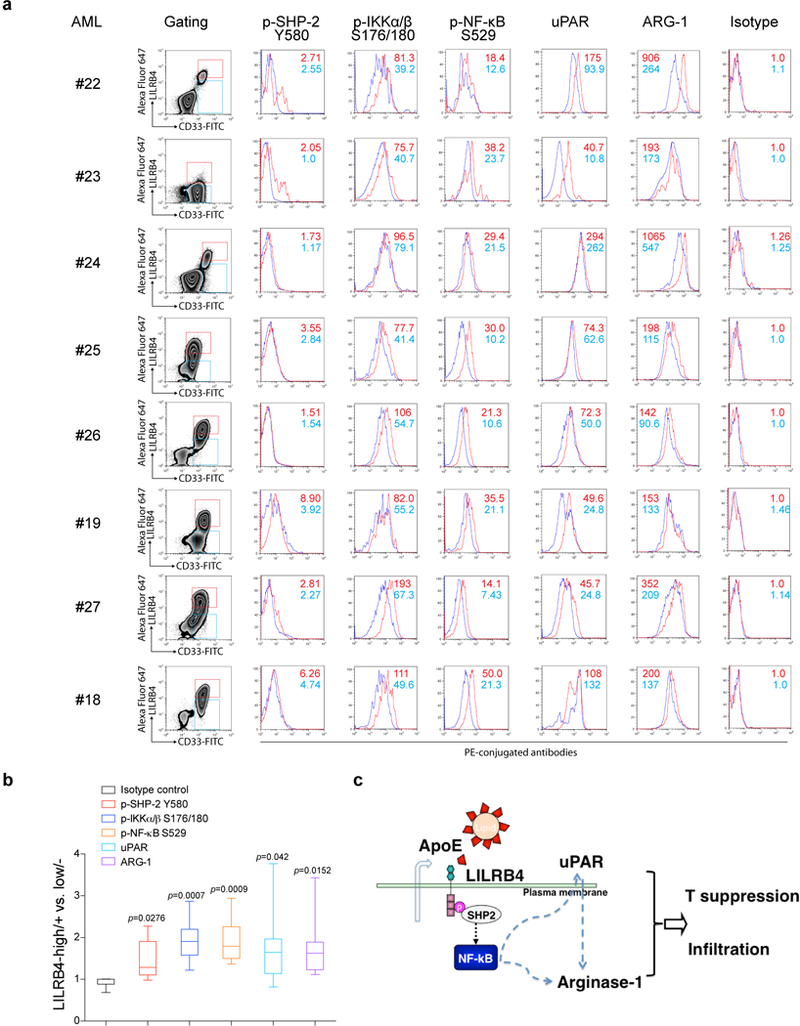
a, LILRB4-positive or -high CD33+ AML cells (red box) and LILRB4-negative or –low CD33+ AML cells (blue box) were gated for further intracellular staining of phosphorylated-SHP-2 at Y580, phosphorylated-IKKα/β at S176/S180, phosphorylated-NF-κB at S529, uPAR, and Arginase-1 (ARG1). Isotype IgG was used as negative controls. Red numbers indicate MFIs (mean fluorescence intensity) of LILRB4-positive or -high CD33+ AML cells; blue numbers indicate MFIs of LILRB4-negative or –low CD33+ AML cells. This experiment was repeated with 8 individual patient samples with similar results. b, Quantification of individual staining in LILRB4-positive or -high CD33+ AML cells versus in LILRB4-negative or low CD33+ AML cells. n=8 independent patients and see Methods for definition of box plot elements. p values were from two-tailed student t-test. c, Schematic for the mechanisms by which LILRB4 suppresses T cells and promotes leukemia infiltration.
Extended Data Figure 9. Comparison of LILRB4 mediated intracellular signaling in leukemia cells and in normal hematopoietic cells.
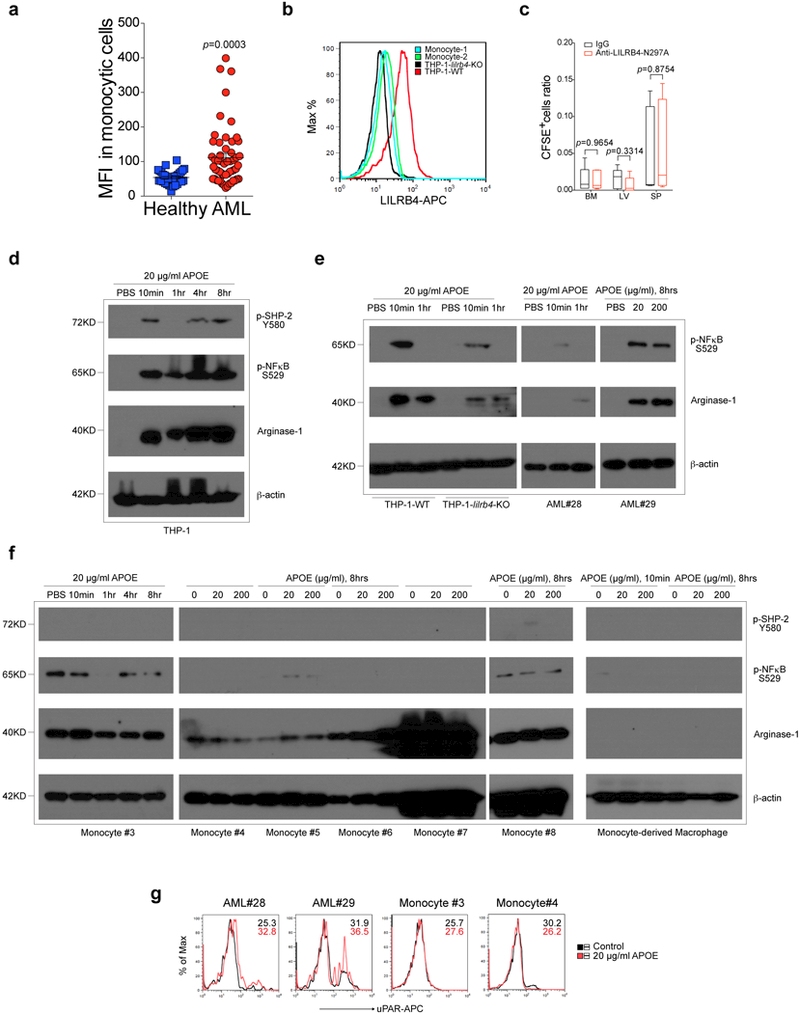
a, Comparison of LILRB4 surface expression on normal monocytes from healthy donors (n=25 individual donors with mean and s.e.m.) and neoplastic monocytic cells from AML patients (n=53 individual patients with mean and s.e.m.). MFI: mean fluorescence intensity. b, Comparison of LILRB4 surface expression on normal monocytes from two healthy donors and on WT and lilrb4-KO THP-1 cells. This experiment was repeated independently three times with similar results. c, Anti-LILRB4 antibody did not affect homing ability of normal monocytes. Human normal monocytes (as shown in b) through CD14-positive selection. These isolated monocytes were pooled and stained by CFSE. After staining, monocytes (5×106 for each mouse) were injected into NSG mice followed immediately by antibody treatment, and then the mice (n=4 mice, see Methods for definition of box plot elements) were sacrificed at 20 hrs after transplant. The number of CFSE+ cells in liver, spleen, and bone marrow were normalized to that in peripheral blood as determined by flow cytometry. d-e, APOE activates LILRB4 intracellular signaling in leukemia cells. Indicated THP-1 cells and primary AML (M5) cells were serum starved overnight and then treated with the indicated concentration of human recombinant APOE protein for indicated time. Phospho-SHP-2, phosphor-NFκB, and Arginase-1 were examined by western blotting. f, The effect of APOE on normal monocytes or in vitro differentiated macrophages. Normal monocytes were isolated from health donors and macrophages were derived from these monocytes after one-week differentiation in vitro. Cells were serum starved overnight and then treated with indicated concentrations of human recombinant APOE protein for indicated time. Phospho-SHP-2, phosphor-NFκB, and Arginase-1 were examined by western blotting. g, APOE induces uPAR upregulation on AML cells. Normal monocytes were isolated from health donors. Indicated primary AML cells and normal monocytes were serum starved overnight and then treated with 20 μg/ml human recombinant APOE protein for eight hours. Surface uPAR were examined by flow cytometry. Representative flow plots are shown and the mean fluorescence intensities were shown in right-up corner (black, PBS control; red, APOE treatment). Experiments were performed three times with similar results. p values were from two-tailed student t-test.
Extended Data Figure 10. Anti-LILRB4 does not affect engraftment of normal hematopoietic cells.
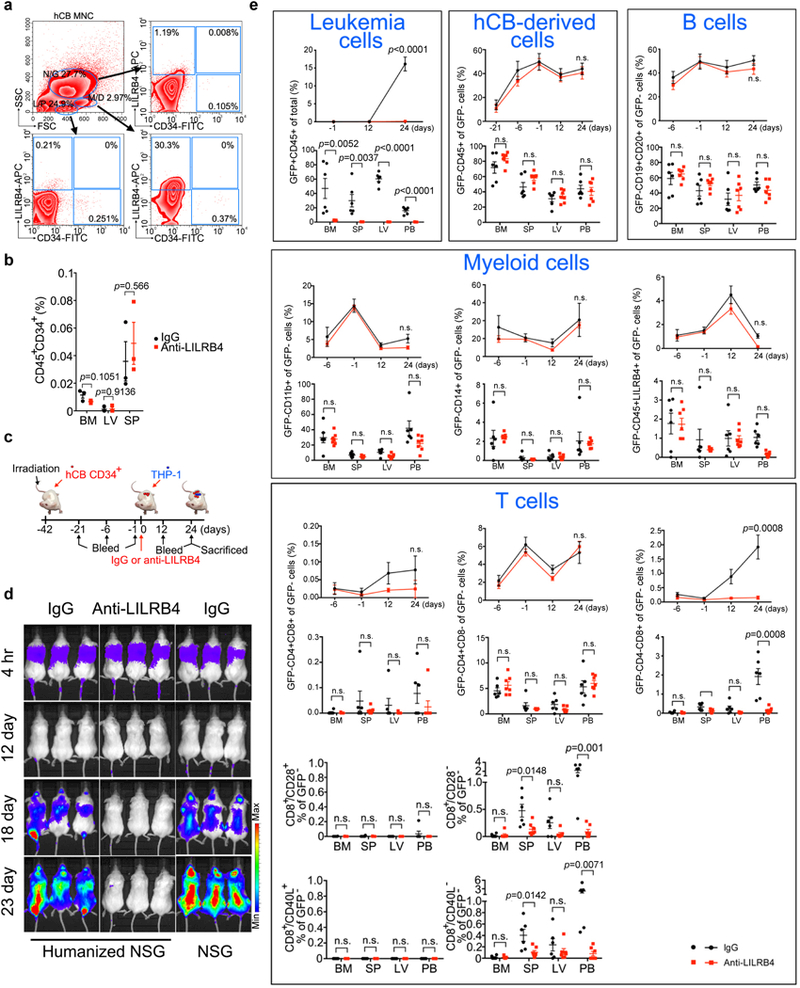
a, Shown are LILRB4 and CD34 co-staining patterns for representative samples of human cord blood mononuclear cells (hCB MNCs). N/G, neutrophils and granulocytes; M/D, monocytes, macrophages and dendritic cells; L/P, lymphocytes, hematopoietic stem and progenitor cells. This experiment was repeated independently three times with similar results. b, Anti-LILRB4 antibody did not affect homing ability of normal hematopoietic progenitor cells. Human cord blood mononuclear cells (1×107) were injected into NSG mice followed immediately by antibody treatment, and then the mice (n=3 mice with mean and s.e.m.) were sacrificed at 20 hrs after transplant. The number of CD45+CD34+ HSCs in liver, spleen, and bone marrow were normalized to that in peripheral blood as determined by flow cytometry. c-e, Anti-LILRB4 antibodies inhibited leukemia development in hCB-humanized NSG mice. c, Schematic of the experiment to test whether anti-LILRB4 antibody inhibits leukemia development in hCB-humanized NSG mice. d, Leukemia development was monitored over time by luminescence imaging. This experiment was repeated independently two times with similar results. e, Frequency of engrafted leukemia, normal human cells, including human B cells, human myeloid cells and human T cells in peripheral blood over time and hematopoietic tissues of hCB-humanized mice at the 24 days after leukemia transplantation. n=3 mice with mean and s.e.m. BM: bone marrow; LV: liver; SP: spleen; PB: peripheral blood. All p values were from two-tailed student t-test.
Supplementary Material
Acknowledgments:
We thank the National Cancer Institute (1R01CA172268 and 5P30CA142543), the Leukemia & Lymphoma Society (1024–14 and TRP-6024–14), the March of Dimes Foundation (1-FY14–201), the Cancer Prevention and Research Institute of Texas (RP140402, DP150056, RP180435, and PR150551), the Robert A. Welch Foundation (I-1834 and AU-0042–20030616), the National Natural Science Foundation of China (81570093, 81422001, and 81721004), the National Basic Research Program of China (2014CB965000), and the China Scholarship Council (201608330307) for generous support. We also thank Dr. Georgina Salazar for editing of the manuscript.
Footnotes
Data Availability Statement
The TCGA datasets analyzed are available in UCSC Xena Browser (https://xena.ucsc.edu). The RNA-seq datasets generated in the current study have been deposited in NCBI SRA database with the SRA accession number SRP155049 (https://www.ncbi.nlm.nih.gov/sra/SRP155049).
The Board of Regents of the University of Texas System has filed patent applications with PCT Application Nos. PCT/US2016/020838, which covers anti-LILRB antibodies and their uses in detecting and treating cancer, and PCT/US2017/044171, which covers the methods for identifying LILRB-blocking antibodies. Authors C.C.Z., M.D., Z.A., N.Z., X.G., and J.Z. are listed as inventors of PCT/US2016/020838. Authors C.C.Z., Z.A., N.Z., M.D., J.K. and X.G. are listed as inventors of PCT/US2017/044171. Both patent applications have been exclusively licensed to Immune-Onc Therapeutics, Inc. by the Board of Regents of the University of Texas System. Authors T.H. and X.C.L. are employees and hold equities of Immune-Onc Therapeutics, Inc.
References
- 1.Curran EK, Godfrey J & Kline J Mechanisms of Immune Tolerance in Leukemia and Lymphoma. Trends Immunol, doi:10.1016/j.it.2017.04.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang X et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 15, 25–40, doi:10.1080/15384101.2015.1121324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirayasu K & Arase H Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. Journal of human genetics, doi:10.1038/jhg.2015.64 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Trowsdale J, Jones DC, Barrow AD & Traherne JA Surveillance of cell and tissue perturbation by receptors in the LRC. Immunol Rev 267, 117–136, doi:10.1111/imr.12314 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Dobrowolska H et al. Expression of immune inhibitory receptor ILT3 in acute myeloid leukemia with monocytic differentiation. Cytometry B Clin Cytom 84, 21–29, doi:10.1002/cyto.b.21050 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Vlad G et al. Membrane and soluble ILT3 are critical to the generation of T suppressor cells and induction of immunological tolerance. Int Rev Immunol 29, 119–132, doi:10.3109/08830180903281185 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Mosier DE, Gulizia RJ, Baird SM & Wilson DB Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 335, 256–259, doi:10.1038/335256a0 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Ha S et al. Isolation and characterization of IgG1 with asymmetrical Fc glycosylation. Glycobiology 21, 1087–1096, doi:10.1093/glycob/cwr047 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Straus DJ et al. The acute monocytic leukemias: multidisciplinary studies in 45 patients. Medicine (Baltimore) 59, 409–425 (1980). [DOI] [PubMed] [Google Scholar]
- 10.Colovai AI et al. Expression of inhibitory receptor ILT3 on neoplastic B cells is associated with lymphoid tissue involvement in chronic lymphocytic leukemia. Cytometry B Clin Cytom 72, 354–362, doi:10.1002/cyto.b.20164 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Crispe IN et al. Cellular and molecular mechanisms of liver tolerance. Immunol Rev 213, 101–118, doi:10.1111/j.1600-065X.2006.00435.x (2006). [DOI] [PubMed] [Google Scholar]
- 12.Carson MJ, Doose JM, Melchior B, Schmid CD & Ploix CC CNS immune privilege: hiding in plain sight. Immunol Rev 213, 48–65, doi:10.1111/j.1600-065X.2006.00441.x (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujisaki J et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474, 216–219, doi:10.1038/nature10160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castells MC et al. gp49B1-alpha(v)beta3 interaction inhibits antigen-induced mast cell activation. Nat Immunol 2, 436–442, doi:10.1038/87749 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Grainger DJ, Reckless J & McKilligin E Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J Immunol 173, 6366–6375 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Ali K, Middleton M, Pure E & Rader DJ Apolipoprotein E suppresses the type I inflammatory response in vivo. Circ Res 97, 922–927, doi:10.1161/01.res.0000187467.67684.43 (2005). [DOI] [PubMed] [Google Scholar]
- 17.DiDonato JA, Mercurio F & Karin M NF-kappaB and the link between inflammation and cancer. Immunol Rev 246, 379–400, doi:10.1111/j.1600-065X.2012.01099.x (2012). [DOI] [PubMed] [Google Scholar]
- 18.You M, Flick LM, Yu D & Feng GS Modulation of the nuclear factor kappa B pathway by Shp-2 tyrosine phosphatase in mediating the induction of interleukin (IL)-6 by IL-1 or tumor necrosis factor. J Exp Med 193, 101–110 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumgartner B et al. Increased IkappaB kinase activity is associated with activated NF-kappaB in acute myeloid blasts. Leukemia 16, 2062–2071, doi:10.1038/sj.leu.2402641 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Bene MC et al. CD87 (urokinase-type plasminogen activator receptor), function and pathology in hematological disorders: a review. Leukemia 18, 394–400, doi:10.1038/sj.leu.2403250 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Su SC, Lin CW, Yang WE, Fan WL & Yang SF The urokinase-type plasminogen activator (uPA) system as a biomarker and therapeutic target in human malignancies. Expert Opin Ther Targets 20, 551–566, doi:10.1517/14728222.2016.1113260 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Wang Y et al. Identification of a novel nuclear factor-kappaB sequence involved in expression of urokinase-type plasminogen activator receptor. Eur J Biochem 267, 3248–3254 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Hu J et al. uPAR induces expression of transforming growth factor beta and interleukin-4 in cancer cells to promote tumor-permissive conditioning of macrophages. Am J Pathol 184, 3384–3393, doi:10.1016/j.ajpath.2014.08.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilkovitch D & Lopez DM Urokinase-mediated recruitment of myeloid-derived suppressor cells and their suppressive mechanisms are blocked by MUC1/sec. Blood 113, 4729–4739, doi:10.1182/blood-2008-08-176438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baitsch D et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol 31, 1160–1168, doi:10.1161/atvbaha.111.222745 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagemann T et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 205, 1261–1268, doi:10.1084/jem.20080108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mussai F et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 122, 749–758, doi:10.1182/blood-2013-01-480129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Goeje PL et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology 4, e1014242, doi:10.1080/2162402x.2015.1014242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang CC et al. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol 3, 237–243, doi:10.1038/ni760 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Suciu-Foca N et al. Soluble Ig-like transcript 3 inhibits tumor allograft rejection in humanized SCID mice and T cell responses in cancer patients. J Immunol 178, 7432–7441 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM & Maeda N Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci U S A 89, 4471–4475 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng J et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 485, 656–660, doi:10.1038/nature11095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang X et al. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat Cell Biol 17, 665–677, doi:10.1038/ncb3158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng M et al. A motif in LILRB2 critical for Angptl2 binding and activation. Blood 124, 924–935, doi:10.1182/blood-2014-01-549162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J et al. Ex vivo expanded hematopoietic stem cells overcome the MHC barrier in allogeneic transplantation. Cell Stem Cell 9, 119–130, doi:10.1016/j.stem.2011.06.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Z et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med 23, 79–90, doi:10.1038/nm.4252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang CC, Kaba M, Iizuka S, Huynh H & Lodish HF Angiopoietin-like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood 111, 3415–3423, doi:blood-2007-11-122119 [pii] 10.1182/blood-2007-11-122119 [doi] (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng J, Huynh H, Umikawa M, Silvany R & Zhang CC Angiopoietin-like protein 3 supports the activity of hematopoietic stem cells in the bone marrow niche. Blood 117, 470–479, doi:10.1182/blood-2010-06-291716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cawthorne C, Swindell R, Stratford IJ, Dive C & Welman A Comparison of doxycycline delivery methods for Tet-inducible gene expression in a subcutaneous xenograft model. J Biomol Tech 18, 120–123 (2007). [PMC free article] [PubMed] [Google Scholar]
- 40.Denisov IG, Grinkova YV, Lazarides AA & Sligar SG Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc 126, 3477–3487, doi:10.1021/ja0393574 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.