

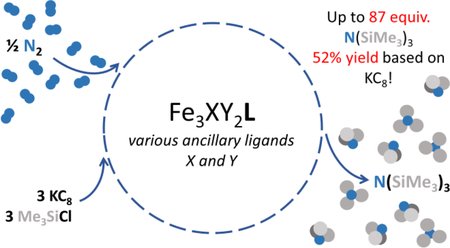
Abstract
A series of triiron complexes supported by a tris(β-diketiminate)cyclophane (L3–) catalyze the reduction of dinitrogen to tris(trimethylsilyl)amine using KC8 and Me3SiCl. Employing Fe3Br3L affords 83 ± 7 equiv. NH4+/complex after protonolysis, which is a 50% yield based on reducing equivalents. The series of triiron compounds tested evidences the subtle effects of ancillary donors, including halides, hydrides, sulfides, and carbonyl ligands, and metal oxidation state on N(SiMe3)3 yield, and highlight Fe3(μ3-N)L as a common species in product mixtures. These results suggest that ancillary ligands can be abstracted with Lewis acids under reducing conditions.
Keywords: N2 reduction, N2 silylation, iron clusters, trinuclear clusters, multinucleating ligand
Graphical Abstract
Industrialization of ammonia production was realized from Haber’s and Bosch’s discoveries over 100 years ago, and the eponymous process is hitherto largely un-changed.1–4 Despite the high efficiency and yields afforded by the Haber-Bosch process, several environmental and economic problems remain unresolved (e.g., carbon foot-print and transportation and distribution costs). Developing new approaches for N2 fixation that address these problems are, therefore, of great interest. Many current strategies are inspired by the proposed mechanism of N2 fixation by the FeMo co-factor of nitrogenase.5,6 There are several reports of molecular complexes that yield ammonia from electron and proton sources; however, these catalysts usually require cryogenic temperatures and proton reduction can be competitive with N2 reduction.7–12 To circumvent H2 production, trimethylsilyl chloride (Me3SiCl) has been employed as a proton surrogate in these reactions to afford tris(trimethylsilyl)amine instead of ammonia; N(SiMe3)3 can be readily converted to NH4+ by protonolysis.13,14 A number of complexes employing Group 5–9 metals have been utilized for the catalytic silylation of dinitrogen, with the highest turnovers reported by Masuda and coworkers (270 N(SiMe3)3 equiv. per complex).9,15,16
We have previously shown that halide-bridged Fe3 clusters are competent for the stoichiometric reduction of dinitrogen. However, we were unable to release the bound dinitrogen-derived amide or imide ligands by protonolysis without complex decomposition,17 and we postulated that Me3Si+ instead of H+ might be compatible with our complexes. Herein, we report that a family of triiron complexes housed within a tris(β-diketiminate) cyclophane effect the catalytic conversion of N2 to N(SiMe3)3 using KC8 and Me3SiCl. Notably, the series of complexes span varying oxidation states and ancillary bridging ligands (Figure 1) allowing for the first systematic evaluation of the effect of nitrogenase-relevant ancillary ligands on dinitrogen activation.17–26 Our results demonstrate that the non-cyclophane donors are labile under reducing conditions as a putative μ3-nitridotriiron(II) complex is observed as a major and common product of most reactions.
Figure 1.

Catalytic silylation of N2 (top) for the formation of N(SiMe3)3 and a series of planar triiron clusters (bottom) supported by L3– employed here.
Using previously reported protocols for catalytic silylation of N2 with KC8 and Me3SiCl,27–30 reaction of 500 equiv. KC8 and 500 equiv. Me3SiCl in the presence of 0.2 mol% Fe3Br3L generated N(SiMe3)3, corresponding to 33 ± 3 equiv. of N(SiMe3)3 per cluster (N(SiMe3)3/Fe3) and a 20% yield based on KC8. The tris(trimethylsilyl)amine was confirmed as a product by GC and quantified indirectly as NH4+ in acid digested product mixtures by 1H-NMR spec-troscopy (Supporting Information, Figure S14). Encouraged by this result, we sought to evaluate the effect of time, solvent, and catalyst loading using the number of N(SiMe3)3/Fe3 as the reporter.
Plots of equivalents of fixed nitrogen for reactions conducted in toluene and using 0.2 mol% catalyst versus time are logarithmic, and do not reach completion even after 120 h (Figure S16). As a means of standardizing our optimization protocol, we elected to evaluate product yields after 24 h. We probed the relationship between N(SiMe3)3 production compared to molar equivalents of KC8 and Me3SiCl used in the reaction. In all cases, we maintained an equimolar ratio of reductant to silyl reagent, and observed a positive correlation between the N(SiMe3)3 yield and KC8/Me3SiCl equivalents (Figure 2). However, the efficiency of converting electrons from KC8 into N(SiMe3)3 decreases with increasing KC8 and Me3SiCl equivalents from 28% for 125 equiv. KC8 to 16% for 1000 equiv. KC8. This decreased efficacy remains under investigation but could arise from accelerated catalyst decomposition or an increased rate of disilane formation from transient Me3Si• radicals. To investigate the possibility of catalyst decom-position under catalytic conditions, we compared the yield from portionwise additions of KC8 and Me3SiCl to the catalyst with that for a single addition; 125 equiv. KC8 and Me3SiCl were added at 6 h intervals, and the reaction quenched after 24 h. Following this approach, the yield was 32 equiv. N(SiMe3)3 per complex after quenching. This value is comparable to that obtained from a single-addition of 500 equiv. KC8/Me3SiCl, suggesting a catalyst fidelity up to 500 equiv. of reductant. We also evaluated how the concentration of Fe3Br3L effected the yield of N(SiMe3)3 (see Figure S17). The concentration of the catalyst has little or no influence on the obtained yields; more detailed mechanistic studies will be required and are the focus of ongoing work. Taken together, these results support that catalysts decomposition is unlikely responsible for the reduced efficiency with respect to KC8.
Figure 2.

Effect of KC8/ Me3SiCl equivalents on N(SiMe3)3 production (blue squares) using Fe3Br3L catalytic system and corresponding yields based on KC8 (red circles). Solid lines are included as visual guides for the general trend. Reaction conditions: equimolar amounts of KC8 and Me3SiCl in toluene at room temperature for 24 h.
With respect to the effect of solvent on catalysis, N(SiMe3)3 yields were determined after 24 h for reactions under analogous conditions, utilizing toluene, THF, and Et2O. Given the poor solubility of Fe3Br3L in Et2O, a standard approach was applied in which an aliquot of a toluene stock solution of Fe3Br3L was diluted ten-fold in the appropriate solvent (for a final 9:1 mixture). Reactions employing Et2O afforded higher yields than those with exclusively toluene for all tested triiron complexes (vide infra) with yield enhancements of two- or three-fold in some cases whereas the lowest yields were obtained with THF (15 equiv. N(SiMe3)3 for Fe3Br3L). We, therefore, provide only data for PhMe and 9:1 Et2O:PhMe in Table 1. This improvement using Et2O can be rationalized as a combination of possible factors, such as an increased solubility of N2 in Et2O and solubility differences for triiron intermediates.31–33 Whereas the yields obtained in the 9:1 Et2O:PhMe mixture are comparable to other iron-based catalysts for this reaction under ambient conditions,11,27,29,30,34–36 the yield based on KC8 – that is, the efficiency with which reducing equivalents are converted into N(SiMe3)3 – is the highest of any reported system (Table S2). Lowering the reaction temperature to −34 °C results in only 33 ± 3 equiv. of N(SiMe3)3 per complex – a 20% yield based on KC8 – in 9:1 Et2O:PhMe after 24 h. Extending the reaction time to 96 h at −34 °C, however, affords 83 ± 6 N(SiMe3)3/Fe3 N (SiMe3)3, which correlates with a 50% yield based on reducing equivalents (Figure S18). This temperature effect on dinitrogen silylation is similar to that reported by Masuda and coworkers for a Co complex.16 To our knowledge, these triiron compounds are the most effective reported catalytic systems for this reaction, independent of metal ion type or complex nu-clearity (Table S3).9,16,28,37
Table 1.
Catalytic dinitrogen silylation by triiron complexes (a)
| Entry | Triiron Com- plex(b) |
N(SiMe3)3/Fe3 (yield in %) | |
|---|---|---|---|
| Solvent A | Solvent B | ||
| 1 | Fe3Br3L | 33 ± 3 (20) | 57 ± 7 (34) 83 ± 7 (50)(c) |
| 2 | Fe3Br3L(d) | 32 (19) | 63 (38) |
| 3 | Fe3F3L | 18 ± 2 (11) | 64 ± 6 (38) |
| 4 | Fe3Cl3L | 22 ± 2 (13) | 45 ± 7 (27) |
| 5 | Fe3H3L | 21 ± 3 (13) | 34 ± 5 (20) |
| 6 | Fe3H2(O2CH)L | 27 ± 5 (16) | 60 ± 7 (36) |
| 7 | (FeCO)2Fe(μ3H)L | 21 ± 3 (13) | 35 ± 3 (21) |
| 8 | Fe3Br2(μ3-N)L | 29 ± 3 (17) | 51 ± 4 (31) |
| 9 | Fe3(μ3-N)L | 31 ± 6 (19) | 43 ± 3 (26) |
| 10 | Fe3O3L | 25 ± 4 (15) | 46 ± 4 (28) |
| 11 | Fe3S3L | 31 (19) | 58 ± 1 (35) |
| 12 | 1.5 [FeClL′ ]2(e) | 17 ± 2 (10) | 46 ± 8 (28) 56 (38)(c) |
| 13 | No Catalyst | 0.7 (0) | <0.1 (0) |
| 14 | FeBr2 | 0.9 (0) | ------- |
Reaction conditions unless stated otherwise: 500 equiv. of KC8 and 500 equiv. of TMSCl after 24 h at room temperature in triplicate. Solvents A and B are PhMe and Et2 O:PhMe 9:1, respectively. N(SiMe3)3 was quantified by acidolysis followed by 1H-NMR
the synthesis of triiron complexes are reported or referenced in the supporting information;
result after 96 h at −34 °C;
portionwise method;
L′ = DIPP-nacnac.
Consistent with homogeneous catalysis, reaction filtrates retain catalytic activity whereas residues are comparatively ineffective (see Supporting Information). However, one proposed complication of utilizing Me3Si+ in lieu of H+ as an electrophile in this system is that silyl radicals – generated from one-electron reduction of Me3Si+– may react directly with N2 to generate N(SiMe3)3.13,14 Control experiments using no Fe catalyst or an equimolar amount of iron as FeBr2 evidence minimal or no activity under the conditions employed for catalytic turnover (Entry 13, Table 1). The importance of our multinucleating system is evident by the comparison of our observed turnovers to that a dimeric β-diketiminate iron(II) complex, [FeClL′ ]2 (L′ = DIPP-nacnac).38 Employing an equimolar in iron amount of [FeClL′ ]2 results in lower yields (ca. 33%) as compared to our complexes (Entry 14, Table 1), along with a likely greater contribution from heterogeneous species in catalysis (Supporting Information).
Given the family of triiron compounds at our disposal, we then explored the effect of the bridging ligand identity on yield of N(SiMe3)3 (Table 1). First, the halide-bridged complexes afford similar yields, with the order being Br ≈ F > Cl. We postulate that this ordering may reflect the relative stabilities of transient μ3-halide or di(μ-halide) complexes; access to these types of complexes to test this hypothesis remains elusive. Second, the trihydride species, Fe3H3L, affords ~50% less N(SiMe3)3 than the bromide congener whereas that of the dihydride-formate, Fe3(H)2(HCOO)L is comparable to Fe3Br3L (cf. entries 5 and 7 with 1, Table 1). This observation is noteworthy given the proposed importance of hydrides in N2 reduction for the nitrogenase cofactors and contrasts reports of di-nitrogen reduction by reported iron hydride model compounds.39–41 Third, there is no apparent correlation between metal formal oxidation states of the starting complexes and yield of amine. For example, the tri(μ-sulfido)triiron(III) and tribromotriiron(II) complexes provide similar yields, whereas the triiron(I/I/II) species is statistically lower (cf. entries 11, 1, and 7, Table 1). The ligand field differences across the three compounds tempers any general conclusions as reduction and loss of halide donors from Fe3Br3L would generate Fe(I) centers in weaker ligand field as compared to the di(μ-carbonyl)(μ3-hydride) compound.
1H-NMR spectra recorded on products from reduction reactions with varying equivalents of KC8 and Me3SiCl indicated formation of a D3h symmetric species, which we tentatively assign to a μ3-nitridotriiron(II) complex (Figures S19−S24, vide infra). The extent of accumulation of this compound is sensitive to solvent and bridging ligands in the initial triiron complex. For example, reaction of 3 equiv. KC8 and Me3SiCl with Fe3Br3L in Et2O generates predominantly the μ3-nitridotriiron(II) complex, whereas the analogous reaction with 6 equiv. KC8 and Me3SiCl affords predominantly the previously reported tri(μ-amido)triiron(II) compound, Fe3(NH2)3L (Figure S19−S20).17 There was also minimal formation of the μ3nitridotriiron(II) species upon reduction of Fe3H3L, Fe3H2(O2CH)L, or (FeCO)2Fe(μ3-H)L in the presence of Me3SiCl.
This μ3-nitridotriiron(II) complex could be independently synthesized by reduction of the reported Fe3Br2(μ3-N)L using two equivalents of KC8 or KHBR3 (R = Et or sec-But).22,42 Attempts to obtain single crystals of sufficient quality for structural characterization have been as yet unsuccessful; crystals obtained hitherto are typically twinned, which we were unable to resolve in the data analysis.43 The formulation, Fe3NL, is consistent with ESI-MS data collected on THF solutions of this compound, although the data evidence adventitious oxidation of complexes during analysis. 1H-NMR spectra of Fe3(μ3-N)L are consistent with D3h symmetry and the resonances agree with those of the common species observed in the catalytic silylation product mixtures mentioned above. Taken together, our data support a structure in which a nitride ligand is present in a μ3-bridging mode and a structure analogous to our previously reported chalco-genide-bridged tricopper complexes.21
Our data indicates, surprisingly, that bridging ligand identity has minimal influence on fixed-nitrogen yield. This result implies that, under reducing conditions, the initial bridging ligands may undergo silylation for formate, sulfide, nitride, or oxide and dissociation leading to compounds with single atom μ3-donors (e.g., sulfide) or one μ3- and one μ-donor. These species are likely reactive towards dinitrogen to install nitride donors; however, silylation of the remaining donors (e.g., sulfide) instead of the dinitrogen-derived nitride provides a pathway towards Fe3(μ3-N)L (Figure 3). Subjecting Fe3(μ3-N)L to the same reaction conditions as described above results in comparable albeit lower N(SiMe3)3 yields (Table 1). In addition, treatment of Fe3(μ3-N)L with up to 20 equiv. of KC8 and Me3SiCl results in recovery of the nitride complex, suggesting stability of this complex under the reaction conditions (Figure S25−S27). We postulate, then, that the starting bridging ligands can be diluted from a catalytically-active species as the reaction proceeds, and ultimately funnel towards the μ3-nitride species. Indeed, we can correlate the greater accumulation of the Fe3(μ3-N)L in those systems with less covalent metal-ligand interactions (e.g., Fe3Br3L) as compared to those with greater covalency (e.g., Fe3S3L). Therefore, we do not have definitive evidence for any specific benefit for supporting ligands on reaction yield. Our data do not suggest substitution of our ancillary ligands by chloride as Fe3Cl3L or C2v-symmetric species containing chlorides were not identified in our studies; we cannot rigorously exclude an additive effect by halide ions, although precipitation of potassium halides is expected to be highly favored. Another possible explanation relies on the fact that these ligands may influence recyclability and the reaction kinetics, but the predicted diverse speciation of the metal complexes during early turnover likely complicate this analysis (i.e., speciation changes as bridging ligands are exchanged for nitride). To validate the specific consequence of a particular bridging ligand, single turnover reactivity studies of the dinitrogen-reactive triiron complexes will be required rather than the precatalysts reported here. Candidate compounds include Fe3(μ3-X)L and Fe3(μ-Y)(μ3-X)L in which X and Y are varied systematically. Our results agree with previous reports by Nishibayashi and collaborators that showed minimal dependence of carbonyl or cyclo-pentadienyl ligands on the catalytic activity of iron(0) compounds attributed to ligand release under cycling conditions.34 In addition, our results are surprisingly complementary to recent work on the nitrogenase cofactors that evidence ligand dissociation – specifically, sulfide – upon reductive activation of the cluster.44 Release of bridging ligands to generate open coordination sites in weak-field ligated multiiron species is likely a common theme, and suggests new opportunities in the design of such catalysts.
Figure 3.

Proposed reaction pathways during the catalytic cycle of a generic Fe3XY2L complex.
In summary, a series of triiron clusters are competent for the catalytic fixation of dinitrogen to produce N(SiMe3)3 from Me3SiCl and KC8 under a dinitrogen atmosphere. Turnovers ranged from 20 to 83 ± 7 N(SiMe3)3/catalyst, with higher yields obtained in a mixture of Et2O/PhMe 9:1. To date, members of this triiron series exhibit the highest yields of N(SiMe3)3 based on KC8 for this reaction.
Detailed mechanistic studies employing the nitridotriiron(II) complex as well as the targeted synthesis of possible intermediates are ongoing.
Supplementary Material
ACKNOWLEDGMENT
L.J.M.: University of Florida departmental instrumentation award NSF CHE-1048604, NSF CHE-1464876 and CHE1650652, NIH R01-GM123241. R.B.F.: University of Florida, College of Liberal Arts and Sciences Graduate Research Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- DIPP
2,6-diisopropylphenyl.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Experimental details. Characterization of new compounds. Additional results for the catalytic silylation of dinitrogen, 1H-NMR spectra of reaction mixtures. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Smil V Detonator of the Population Explosion. Nature 1999, 400, 415–415. [Google Scholar]
- 2).Mittasch A; Frankenburg W Early Studies of Multicomponent Catalysts. Adv. Catal. 1950, 2, 81–104. [Google Scholar]
- 3).Prieto G; Schüth F The Yin and Yang in the Development of Catalytic Processes: Catalysis Research and Reaction Engineering. Angew. Chem. Int. Ed 2015, 54, 3222–3239. [DOI] [PubMed] [Google Scholar]
- 4).Inoue Y; Kitano M; Kishida K; Abe H; Niwa Y; Sasase M; Fujita Y; Ishikawa H; Yokoyama T; Hara M; Ho-sono H Efficient and Stable Ammonia Synthesis by Self-Organized Flat Ru Nanoparticles on Calcium Amide. ACS Catal. 2016, 6, 7577–7584. [Google Scholar]
- 5).Smith BE Nitrogenase Reveals Its Inner Secrets. Science 2002, 297, 1654–1655. [DOI] [PubMed] [Google Scholar]
- 6).Seefeldt LC; Hoffman BM; Dean DR Mechanism of Mo-Dependent Nitrogenase. Annu. Rev. Biochem 2009, 78, 701–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Schrock RR Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Acc. Chem. Res. 2005, 38, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Arashiba K; Miyake Y; Nishibayashi Y A Molybdenum Complex Bearing PNP-Type Pincer Ligands Leads to the Catalytic Reduction of Dinitrogen into Ammonia. Nat. Chem. 2011, 3, 120–125. [DOI] [PubMed] [Google Scholar]
- 9).Nishibayashi Y Recent Progress in Transition-Metal-Catalyzed Reduction of Molecular Dinitrogen under Ambient Reaction Conditions. Inorg. Chem. 2015, 54, 9234–9247. [DOI] [PubMed] [Google Scholar]
- 10).Lindley BM; Appel AM; Krogh-Jespersen K; Mayer JM; Miller AJ M. Evaluating the Thermodynamics of Electrocatalytic N 2 Reduction in Acetonitrile. ACS Energy Lett. 2016, 1, 698–704. [Google Scholar]
- 11).Kuriyama S; Arashiba K; Nakajima K; Matsuo Y; Tanaka H; Ishii K; Yoshizawa K; Nishibayashi Y Catalytic Transformation of Dinitrogen into Ammonia and Hydrazine by Iron-Dinitrogen Complexes Bearing Pincer Ligand. Nat. Commun. 2016, 7, 12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Del Castillo TJ; Thompson NB; Peters JC A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc. 2016, 138, 5341–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Shiina K Reductive Silylation of Molecular Nitrogen via Fixation to Tris(Trialkylsilyl)Amine. J. Am. Chem. Soc. 1972, 94, 9266–9267. [Google Scholar]
- 14).Komori K; Oshita H; Mizobe Y; Hidai M Preparation and Properties of Molybdenum and Tungsten Dinitrogen Complexes. 25. Catalytic Conversion of Molecular Nitrogen into Silylamines Using Molybdenum and Tungsten Dinitrogen Complexes. J. Am. Chem. Soc. 1989, 111, 1939–1940. [Google Scholar]
- 15).Liao Q; Cavaillé A; Saffon-Merceron N; Mézailles N Direct Synthesis of Silylamine from N2 and a Silane: Mediated by a Tridentate Phosphine Molybdenum Fragment. Angew. Chem. Int. Ed. 2016, 55, 11212–11216. [DOI] [PubMed] [Google Scholar]
- 16).Suzuki T; Fujimoto K; Takemoto Y; Wasada-Tsutsui Y; Ozawa T; Inomata T; Fryzuk MD; Masuda H Efficient Catalytic Conversion of Dinitrogen to N(SiMe3)3 Using a Homogeneous Mononuclear Cobalt Complex. ACS Catal. 2018, 8, 3011–3015. [Google Scholar]
- 17).Lee Y; Sloane FT; Blondin G; Abboud KA; García-Serres R; Murray LJ Dinitrogen Activation Upon Reduction of a Triiron(II) Complex. Angew. Chem. Int. Ed 2015, 54, 1499–1503. [DOI] [PubMed] [Google Scholar]
- 18).Guillet GL; Sloane FT; Dumont MF; Abboud KA; Murray LJ Synthesis and Characterization of a Tris(2-Hydroxyphenyl)Methane-Based Cryptand and Its Triiron(III) Complex. Dalton Trans 2012, 41, 7866–7869. [DOI] [PubMed] [Google Scholar]
- 19).Guillet GL; Sloane FT; Ermert DM; Calkins MW; Peprah MK; Knowles ES; Čižmár E; Abboud KA; Meisel MW; Murray LJ Preorganized Assembly of Three Iron(II) or Manganese(II) β-Diketiminate Complexes Using a Cyclophane Ligand. Chem. Commun 2013, 49, 6635–6637. [DOI] [PubMed] [Google Scholar]
- 20).Lee Y; Anderton KJ; Sloane FT; Ermert DM; Abboud KA; García-Serres R; Murray LJ Reactivity of Hydride Bridges in High-Spin [3M–3(μ-H)] Clusters (M = FeII, CoII). J. Am. Chem. Soc 2015, 3, 10610–10617. [DOI] [PubMed] [Google Scholar]
- 21).(a) Di Francesco GN; Gaillard A; Ghiviriga I; Abboud KA; Murray LJ. Modeling Biological Copper Clusters: Synthesis of a Tricopper Complex, and Its Chloride- and Sulfide-Bridged Congeners. Inorg. Chem. 2014, 53, 4647– 4654. [DOI] [PubMed] [Google Scholar]; (b) Cook BJ; Di Francesco GN; Abboud KA; Murray LJ Countercations and Solvent Influence CO2 Reduction to Oxalate by Chalcogen-Bridged Tricopper Cyclophanates. J. Am. Chem. Soc 2018, 140, 5696–5700. [DOI] [PubMed] [Google Scholar]
- 22).Ermert DM; Gordon JB; Abboud KA; Murray LJ Nitride-Bridged Triiron Complex and Its Relevance to Dinitrogen Activation. Inorg. Chem. 2015, 54, 9282–9289. [DOI] [PubMed] [Google Scholar]
- 23).Lee Y; Abboud KA; García-Serres R; Murray LJ A Three-Coordinate Fe(II) Center within a [3Fe–(μ3-S)] Cluster That Provides an Accessible Coordination Site. Chem. Commun. 2016, 52, 9295–9298. [DOI] [PubMed] [Google Scholar]
- 24).Lee Y; Jeon I-R; Abboud KA; García-Serres R; Shearer J; Murray LJA [3Fe–3S]3+ Cluster with Exclusively μSulfide Donors. Chem. Commun. 2016, 52, 1174–1177. [DOI] [PubMed] [Google Scholar]
- 25).Anderton KJ; Ermert DM; Quintero PA; Turvey MW; Fataftah MS; Abboud KA; Meisel MW; Čižmár E; Murray LJ Correlating Bridging Ligand with Properties of Ligand-Templated [MnII3X3]3+ Clusters (X = Br–, Cl–, H–, MeO–). Inorg. Chem. 2017, 56, 12012–12022. [DOI] [PubMed] [Google Scholar]
- 26).Anderton KJ; Knight BJ; Rheingold AL; Abboud KA; García-Serres R; Murray LJ Reactivity of Hydride Bridges in a High-Spin [Fe3(μ-H)3]3+ Cluster: Reversible H2/CO Exchange and Fe–H/B–F Bond Metathesis. Chem. Sci 2017, 8, 4123–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Ung G; Peters JC Low-Temperature N2 Binding to Two-Coordinate L2Fe0 Enables Reductive Trapping of L2FeN2– and NH3 Generation. Angew. Chem. Int. Ed 2015, 54, 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Siedschlag RB; Bernales V; Vogiatzis KD; Planas N; Clouston LJ; Bill E; Gagliardi L; Lu CC Catalytic Silylation of Dinitrogen with a Dicobalt Complex. J. Am. Chem. Soc 2015, 137, 4638–4641. [DOI] [PubMed] [Google Scholar]
- 29).Prokopchuk DE; Wiedner ES; Walter ED; Popescu CV; Piro NA; Kassel WS; Bullock RM; Mock MT Catalytic N2 Reduction to Silylamines and Thermodynamics of N2 Binding at Square Planar Fe. J. Am. Chem. Soc 2017, 139, 9291–9301. [DOI] [PubMed] [Google Scholar]
- 30).Imayoshi R; Nakajima K; Takaya J; Iwasawa N; Nishibayashi Y Synthesis and Reactivity of Iron- and Cobalt-Dinitrogen Complexes Bearing PSiP-Type Pincer Ligands toward Nitrogen Fixation. Eur. J. Inorg. Chem 2017, 2017, 3769–3778. [Google Scholar]
- 31).Battino R; Rettich TR; Tominaga T The Solubility of Nitrogen and Air in Liquids. J. Phys. Chem. Ref. Data 1984, 13, 563–600. [Google Scholar]
- 32).Saouma CT; Lu CC; Day MW; Peters JC CO2 Reduction by Fe(I): Solvent Control of C–O Cleavage versus C–C Coupling. Chem. Sci 2013, 4, 4042. [Google Scholar]
- 33).Hill PJ; Doyle LR; Crawford AD; Myers WK; Ashley AE Selective Catalytic Reduction of N2 to N2H4 by a Simple Fe Complex. J. Am. Chem. Soc 2016, 138, 13521–13524. [DOI] [PubMed] [Google Scholar]
- 34).Yuki M; Tanaka H; Sasaki K; Miyake Y; Yoshizawa K; Nishibayashi Y Iron-Catalysed Transformation of Molecular Dinitrogen into Silylamine under Ambient Conditions. Nat. Commun 2012, 3, 1254. [DOI] [PubMed] [Google Scholar]
- 35).Araake R; Sakadani K; Tada M; Sakai Y; Ohki Y [Fe4] and [Fe6] Hydride Clusters Supported by Phosphines: Synthesis, Characterization, and Application in N2 Reduction. J. Am. Chem. Soc. 2017, 139, 5596–5606. [DOI] [PubMed] [Google Scholar]
- 36).Ohki Y; Araki Y; Tada M; Sakai Y Synthesis and Characterization of Bioinspired [Mo2Fe2]-Hydride Cluster Complexes and Their Application in the Catalytic Silylation of N2. Chem. Eur. J 2017, 23, 13240–13248. [DOI] [PubMed] [Google Scholar]
- 37).Tanaka H; Sasada A; Kouno T; Yuki M; Miyake Y; Nakanishi H; Nishibayashi Y; Yoshizawa K Molybdenum-Catalyzed Transformation of Molecular Dinitrogen into Silylamine: Experimental and DFT Study on the Remarkable Role of Ferrocenyldiphosphine Ligands. J. Am. Chem. Soc 2011, 133, 3498–3506. [DOI] [PubMed] [Google Scholar]
- 38).Eckert NA; Smith JM; Lachicotte RJ; Holland PL Low-Coordinate Iron(II) Amido Complexes of β-Diketiminates: Synthesis, Structure, and Reactivity. Inorg. Chem 2004, 43, 3306–3321. [DOI] [PubMed] [Google Scholar]
- 39).Hoffman BM; Lukoyanov D; Dean DR; Seefeldt LC Nitrogenase: A Draft Mechanism. Acc. Chem. Res 2013, 46, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Hoffman BM; Lukoyanov D; Yang Z-Y; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Lukoyanov D; Khadka N; Yang Z-Y; Dean DR; Seefeldt LC; Hoffman BM Reversible Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride State, the E4(4H) Janus Intermediate. J. Am. Chem. Soc 2016, 138, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).A preliminary result reported in reference 22 incorrectly concluded that the nitridotriiron(II) could not be accessed by chemical reduction of the di(bromo)nitridotriiron(II/III/III).
- 43).Data reported in prior publications suggest the cyclophane ligand typically dominates crystal packing, which leads to co-crystallization of multiple compounds in a single crystal as well as a likelihood of twinning and poor crystal quality.
- 44).Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A Bound Reaction Intermediate Sheds Light on the Mechanism of Nitrogenase. Science 2018, 359, 1484–1489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.