

Abstract
Background
Overexpression of the RAN GTP (RAN) gene has been shown to be linked to metastatic activity of MDA-MB231 human breast cancer cells by increasing Ras/MEK/ERK and PI3K/Akt/mTORC1 signalling. The aim of this study was to investigate the potential of polymeric nanoparticles to deliver two novel shRNA sequences, targeted against the RAN gene, to MDA-MB231 cells grown in culture and to assess their effects in a range of biological assays.
Methods
Biodegradable PLGA nanoparticles, loaded with shRNA-1 and shRNA-4, were fabricated using a double emulsion solvent evaporation technique and characterised for size, zeta potential and polydispersity index before testing on the MDA-MB231 cell line in a range of assays including cell viability, migration, invasion and gene knock down.
Results
shRNA-loaded nanoparticles were successfully fabricated and delivered to MDA-MB231 cells in culture, where they effectively released their payload, causing a decrease in both cell invasion and cell migration by knocking down RAN gene expression.
Conclusion
Results indicate the anti-RAN shRNA-loaded nanoparticles deliver and release biological payload to MDA-MB231 cells in culture. This works paves the way for further investigations into the possible use of anti-RAN shRNA-loaded NP formulations for the treatment of breast cancer in vivo.
Keywords: Breast cancer, Intracellular delivery, Nanotechnology, PLGA, shRNA
Introduction
Genetic instability in cellular function has a significant bearing on the transition from normal to cancerous growth patterns [1]. Of concern is the overexpression of mutated genes that enables tumour cells to metastasise to distant sites and establish secondary growth [2]. It is widely accepted that metastatic disease is associated with high rates of mortality observed in cancer patients and inhibiting metastasis is, therefore, subject to intense investigation [3]. These investigations focus on the function and mutation in specific genes [3]. Of particular interest to our group is the overexpression of the RAN gene in breast cancer cells, which has attracted much attention, with results demonstrating its clear role in the metastatic process [4]. This functional loss of cell cycle machinery causes cells to acquire aggressive and invasive characteristics, enabling invasion at distant sites.
Ran is a small GTPase involved in various cellular processes, including nucleocytoplasmic transport, apoptosis, mitotic spindle organisation and nuclear envelope formation [5,6]. Moreover, it is overexpressed in many human tumours, including stomach, lungs, head and neck, pancreas, ovarian, colorectal and kidney, but not in non-tumour tissue [4,7–14]. Importantly, RNAi-mediated knockdown of Ran in various tumour cell types cause aberrant mitotic spindle formation, mitochondrial dysfunction and apoptosis, while knockdown in non-tumour cells is well tolerated and does not lead to mitotic defects or loss of cell viability [9]. Therefore, overexpression of the RAN gene mediates invasiveness in human cancer cells, brought about by increasing Ras/MEK/ERK and PI3K/Akt/mTORC1 signalling [4,15]. Targeting the function of RAN is a feasible therapeutic target, leading to possible disruption in both cell proliferation and invasion [4]. This overexpression can be inhibited by RNA interference (RNAi), whereby the over-expressed gene is silenced using small oligonucleotide sequences, such as shRNA. However, the effective delivery of naked shRNA is problematic as it crosses the plasma membrane barrier poorly [16]. To circumvent this difficulty, cellular translocation of ribonucleic acid is assisted using nano-sized drug carriers, ranging from liposomes to polymeric nanoparticles (NP), which have been widely exploited in this field of therapeutics [17–21]. More specifically, polymeric NP, loaded with shRNA, have been shown to facilitate intracellular delivery [21]. Furthermore, the ability of polymeric NP to encapsulate and deliver a drug substance to the required target site without affecting its biological efficacy is a key advantage [22].
Recent studies show that viral-mediated shRNA or siRNA delivery inhibit cell invasion and metastasis by silencing RAN in MDA-MB231 human breast cancer cells [4,23,24]. For instance, lentiviral vectors have been used to deliver oligonucleotides that target RAN effectively [4] . Studies performed by the El-Tanani group demonstrate that this lentiviral mediated delivery of specific shRNA sequences, developed in their laboratory and known as shRNA-1 and shRNA-4, effectively silenced the RAN gene and inhibited MDA-MB231 cell invasion [4]. In addition to this, in silico analysis was carried out where shRNA-1 and shRNA-4 sequences complimented with the exons region on the targeted RAN gene, which provided further evidence of gene silencing via shRNA-1 and shRNA-4 mediated RNA interference. However, viral-mediated delivery is associated with toxicity and so an aim of this study was to use a simple polymeric vector based on poly(lactide-co-glycolide) (PLGA) [25]. This polymer has an acceptable toxicity profile and regulatory approval [26]. Results from recent studies show that NP prepared from PLGA and its PEGylated co-block variants (PLGA-PEG) are effective drug delivery vehicles for shRNA/siRNA and bring about silencing of targeted genes [27,28]. For example, the Bcl-xl gene in breast cancer cells was silenced using shRNA-PLGA-PEI NP and PLGA-siRNA NP have been effective in silencing a model gene (fire-fly luciferase) in MDA-kb2 cells [29–32]. Therefore, in this study, we report for the first time the encapsulation of two specific shRNA sequences (shRNA-1 and shRNA-4) in PLGA NP and demonstrate silencing of RAN. A modified double emulsion solvent evaporation technique was used to fabricate nucleotide-loaded NP with efficient encapsulation and these were evaluated using cell culture. The effects of both shRNA-loaded NP variants on MDA-MB231 human breast cancer cells were assessed by a range of in vitro assays, such as cell viability, migration assay, invasion assay and qRT-PCR, to determine the effect of RAN knockdown on metastatic potential.
Materials and Methods
Materials
Poly(d,l-lactide-co-glycolide) (PLGA) with a lactic:glycolic ratio of 50:50 (Resomer® RG 503H, MW 34 kDa), poly[(d,l-lactide-co-glycolide)-co-PEG] diblock (Resomer® RGP d 5055 (5% PEG) and Resomer® RGP d 50,105 (10% PEG)) were purchased from Boehringer-Ingelheim (Ingelheim, Germany). Poly(vinyl alcohol) (PVA) 87–89% hydrolysed (MW 31,000-50,000) and potassium chloride were purchased from Sigma-Aldrich (Dorset, UK).
shRNA-1 sequence clone IDs NM_006325.2-697s1c1 (sequence_CCGGGCACA GTATGAGCACGACTTACTCGAGTAAGTCGTGCTCATACTGTGCTTTTTG), shRNA-4 clone IDs NM_006325.2-697s1c1 (sequence_ CCGGGACCCTAACTTGGAATTTGTTCTC GAGAACAAATTCCAAGTTAGGGTCTTTTT) were purchased from Addgene (Cambridge, Massachusetts, USA). Quant-iT™ RiboGreen® RNA Assay Kit, RNA zap and diethyl pyrocarbonate (≥99%) (DEPC) were obtained from Thermo Fisher Scientific Ltd. (Paisley, UK). All the other chemicals and reagents were of appropriate analytical grade.
Cell culture studies were performed on the MDA-MB231 breast cancer cell line, which was obtained from Sigma-Aldrich Ltd. (Dorset, UK). Dulbecco’s Modified Eagle Medium (DMEM), foetal bovine serum (FBS), penicillin streptomycin, optimum reduced serum media, serum-free media and trypsin were obtained from Gibco, Life Technologies (Paisley, UK). Phosphate-buffered saline (PBS) was prepared from tablet form and purchased from Oxoid Ltd. (Hampshire, UK).
Fabrication of shRNA-Loaded NP
NP loaded with shRNA (either shRNA-1 or shRNA-4) were fabricated using a double emulsion solvent evaporation technique, where a water-in-oil-in-water (w/o/w) emulsion was prepared. PLGA and diblock copolymers of PLGA and PEG containing 5% or 10% w/w of PEG 5 kDa were used in this work. Briefly, polymer (100 mg) was dissolved in 4.0 ml dichloromethane (DCM) and vortexed until fully dissolved. Thereafter, 1.0 ml of 2.50% (w/v) and 50 ml of 1.25% (w/v) PVA solution in DEPC-treated water (0.01% v/v) were prepared as internal aqueous phase and external aqueous phase, respectively. The payload (14 μg), of either shRNA-1 or shRNA-4, was dissolved in 1.0 ml of 2.50% PVA solution. This payload solution was added slowly and mixed with the polymer-DCM phase under constant homogenisation for 2 min to form the primary w/o emulsion. The primary emulsion was then mixed slowly with 50 ml of 1.25% of PVA solution to form a secondary w/o/w emulsion, which was kept under constant homogenisation (homogeniser-Silverson L5T FAR A17122) at 10000 rpm for 6 min at room temperature. Samples were left uncovered and stirred overnight at room temperature to allow evaporation of organic solvent. This was followed by a detailed three-part washing procedure, comprising centrifugation with (i) RNAse-free, DEPC-treated water, followed by (ii) 2% w/v sucrose solution and finally, (iii) with RNAase-free water. Repeated washing steps were carried out to remove any residual PVA and any shRNA that may have been absorbed onto the NP surface. After the final centrifugation step, the supernatant was discarded and the pellet dispersed in 5.0 ml RNAase-free ultra-pure water. Samples were freeze dried (Labconco freeze dryer, Mason Technology, Missouri, USA) before storage and use.
Characterisation of shRNA-Loaded NP
Size, zeta potential and polydispersity index (PDI) of loaded NP were obtained using dynamic light scattering (Zetamaster, Malvern Instruments, Malvern, UK) employing a 15 mW laser and an incident beam of 676 nm. NP (5 mg) were dispersed in distilled water, filtered (0.45 μm) and used to determine mean diameter and size distribution. The measurement of zeta potential was performed on samples treated in a similar fashion, with NP dispersed instead in 1.0 mM potassium chloride solution.
The morphology of NP was determined using scanning electron microscopy (FEI Quanta 400 FEG, Eindhoven). An ultra-thin layer of lyophilised NP powder was coated on a carbon tape metal grid. It was then sputter coated with gold for 15 min and samples were examined with high vacuum mode and a 2° electron ETD detector.
Determination of Drug Encapsulation Efficiency
The encapsulation efficiency (EE) of shRNA-loaded NP was determined using fluorescent nucleic acid staining (Quant-iT™ RiboGreen® RNA Assay kit, Thermo Fisher Scientific, Waltham, USA). Briefly, a sample of shRNA-loaded NP (5 mg) was dissolved in 1.0 M NaOH (0.25 ml) for 24 h under mild shaking and the pH adjusted to 7.0 using 0.5 M HCl. Dissolved samples were diluted in 1.0 ml TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) and then added to 1.0 ml of working reagent (1 part of ribogreen reagent in 20-fold of nuclease free DEPC-treated water). Samples were incubated in dark conditions for 15 min at 37°C and 200 μl from each transferred to a 96-well black plate and the fluorescence recorded (λex 480 nm, λem 520 nm) using a microplate reader (FLUOstar Omega, BMG LabTech, UK). The analytical method was calibrated using a standard plot (0–1 μg ml−1 shRNA), performed as per the manufacturer’s instructions. The standard calibration curve was used to determine the amount of shRNA loaded per unit mass of NP (μg per mg of NP). From this data, the encapsulation efficiency was determined, defined as the percentage ratio of the determined nucleotide loading to the theoretical maximum loading.
Drug Release Study
The drug release profile from loaded NP was determined by measuring the concentration of free shRNA in an aqueous receiver phase over a 96-h duration. To cover this 96-h period, four separate and identical samples were produced, each comprising shRNA-loaded NP (5 mg) suspended in PBS (1.0 ml, pH 7.4). All samples were rotated through an end-over-end motion, whilst being incubated at 37°C. At each 24 h-time point, a vial was selected, centrifuged at 10000 x g for 10 min, a sample of supernatant collected (200 μl) and the shRNA concentration measured using fluorescent nucleic acid staining, as described in section 2.4.
Determination of Cellular Uptake of shRNA-Loaded NP
Cellular uptake of shRNA-loaded NP was determined by fluorescently labelling the nano-particulate core. Coumarin 6 (0.5 mg) was encapsulated within the NP by addition to the dichloromethane-polymer phase used during the double emulsion procedure. MDA MB 231 breast cancer cells (1 × 105 cells per well in 1 ml media) were seeded in 2-well chamber slides and incubated for 24 h. Cells were then transfected by addition of 1 μg of shRNA-loaded NP dispersed in 1 ml transfection media (Opti-MEM® media) and then incubated for 24 h at 37°C. After 24 h, the media was aspirated and cells washed three times with PBS. Cells were then washed with 4% formaldehyde, stained with 100 μl of 0.5 mg ml−1 DAPI solution and visualised using fluorescence microscopy (Nicole Eclipse E400 Fluorescence Microscope, Nikon Y-FL, Japan).
Cell Viability Assay
A suspension of 4 × 104 cells in 1.0 ml medium (Opti-MEM®) was seeded per well in 24-well cell culture plates and left to adhere overnight at 37°C under a 5% CO2 atmosphere. After 24 h, the medium was aspirated and cells transfected with 500 μl of a nanoparticulate suspension in Opti-MEM® medium, at a concentration equivalent to 0.5 μg ml−1 (C1), 1.0 μg ml−1 (C2) and 2.0 μg ml−1 (C3) shRNA. Both shRNA-1 and shRNA-4 suspensions were prepared in this manner. Cell viability was evaluated after 24, 48, 72 and 96 h with the culture media being changed daily. Media were aspirated and cells washed with sterile PBS before addition of 500 μl media containing 15% (w/v) of an MTT dye solution (10 mg ml−1) in PBS solution was added to each well. All plates were incubated for 3 h at 37°C. The supernatant was then aspirated and formazan dissolved using dimethyl sulfoxide (DMSO) (500 μl per well). Absorbance was measured at 570 nm using a microplate reader (FLUOstar Omega, Ortenberg, Germany). Cell viability was calculated as a percentage of cell growth with respect to untreated cells and cells treated with blank nanoparticles.
In Vitro Cell Migration Assay
A suspension of 7.5 × 105 cells in 2 ml medium (Opti-MEM®) were seeded in 6-well plates and incubated at 37°C for 48 h. Once the cells had reached confluence, an induced artificial gap, or scratch, was made across the confluent cell monolayer using a sterile pipette tip. Medium was aspirated and cells transfected using nanoparticulate suspensions equivalent to 0.5 μg ml−1, 1.0 μg ml−1 or 2.0 μg ml−1 of shRNA dispersed in 2 ml Opti-MEM® media. Each well was viewed using light microscopy after 0, 24, 48 and 72 h to determine scratch closure, which was calculated using the formula below. Data were analysed using ImageJ.software (Cambridge, Massachusetts, USA).
In Vitro Cell Invasion Assay
A cell invasion assay was performed using a Cultrex® 96 Well BME Cell Invasion Assay kit. The assay was carried out according to the manufacturer’s instructions. Briefly, 50 μl 1x BME solution (basement membrane extract solution supplied with the kit) was added to the upper chamber of each well to produce a basement membrane-like structure and was incubated overnight at 37°C. After overnight incubation, the remaining solution was aspirated and wells were seeded at a density of 5 × 104 cells per well in 150 μl serum-free medium into the upper chamber. Cells were incubated for 24 h at 37°C and 5% CO2 to allow adhesion to the basement membrane. Cells were then transfected with 2.0 μg ml−1 shRNA-loaded PLGA NP in 200 μl serum free media. Simultaneously, 150 μl of complete medium containing 10% foetal bovine serum was added to the bottom chamber to act as a chemo-attractant. Cells were incubated for 24 and 48 h at 37°C, 5% CO2, after which time the top chamber and bottom chamber were aspirated and wells were washed with 100 μl and 200 μl PBS washing buffer, respectively. Next, a solution of calcein-AM and cell dissociation solution were prepared by adding 12 μl of calcein-AM solution to 10 ml of cell dissociation solution, supplied with the kit, to detach the cells. Calcein AM/cell dissociation solution (100 μl) was added to the bottom chamber wells and incubated for 30 min at 37°C. The cell dissociation solution was used to detach invading cells from the membrane and calcein AM was internalised by viable cells. Esterase activity inside cells converted hydrophobic calcein-acetomethyl ester into hydrophilic fluorescent calcein. Later, the fluorescence of the bottom chamber was measured at 520 nm (485 nm excitation) using a microplate reader.
qRT PCR-Gene Knockdown
A suspension of 1 × 105 cells in 2 ml medium was seeded into each well of 6-well plates and incubated overnight at 37°C to ensure adherence. Cells were transfected using 1 μg of shRNA-loaded NP (shRNA-1 and shRNA-4) in 2 ml Opti-MEM® media and then incubated at 37°C for 24 and 48 h. After each time period, the media were aspirated and cells washed several times with PBS. Cells were trypsinised using 1.0 ml of 0.5% trypsin-EDTA 10X solution in each well and incubated at 37°C for 5 min. Growth medium (1.0 ml) was added into each well, then removed and the cell suspension centrifuged at 1500 x g for 5 min. The supernatant was discarded and the pellet resuspended with sterile PBS and centrifuged again at 1500 x g for 5 min. This was repeated twice. RNA was then isolated from MDA-MB231 cells using selective binding to silica-based membranes (RNeasy® Mini Kit, Qiagen Ltd., Manchester, UK). cDNA was synthesised from 2 μg of extracted RNA using forward and reverse primer sequences based on the mRNA sequence of the human RAN gene (forward primer 5’ CCATCTTTCCAGCCTCAGTC 3′; reverse primer 5’ TACCACCATCACCAACCAAT 3′).
Statistical Analysis
All statistical analyses were performed using GraphPad PRISM software (GraphPad Prism, San Diego, USA). All experiments were performed in triplicate, unless otherwise stated. Data are presented as mean with standard deviation (SD) and significant differences were determined using a 2-tailed Student’s t test and calculated to one of three levels of significance, when appropriate.
Results
Physicochemical Characterisation of shRNA-Loaded NP
The key parameters of shRNA-loaded (shRNA-1 and shRNA-4) PEGylated and non-PEGylated NP are shown in Table I and define size, zeta potential, PDI and encapsulation efficiency. PLGA-shRNA NP were larger than the corresponding PEGylated equivalents for both the shRNA-1 and shRNA-4 formulations. Addition of PEG to the polymeric structure also affected the zeta potential and PDI of the NP formulations, with an elevation of zeta potential toward neutrality and a tightening of the distribution in mean diameter. However, the presence of PEG in the particle matrix did not significantly affect encapsulation efficiency, which was observed to exceed 66% for all formulations.
Table I.
Physical characterisation of shRNA-loaded NP, where average size, zeta potential, polydispersity index (PDI), encapsulation efficiency (EE) and encapsulated drug amount were determined*
| Copolymers |
size
(nm) |
PDI |
zeta potential
(mV) |
EE
(%) |
Drug loading
(μg per mg NP) |
|---|---|---|---|---|---|
| PLGA-shRNA-1 | 426.41 ± 26.23 | 0.414 ± 0.144 | −0.332 ± 0.66 | 74.94 ± 1.32 | 0.10 ± 0.01 |
| PLGA-PEG-5%-shRNA-1 | 251.61 ± 14.71 | 0.231 ± 0.015 | −0.224 ± 1.03 | 75.10 ± 7.67 | 0.10 ± 0.10 |
| PLGA-PEG-10%-shRNA-1 | 272.36 ± 9.82 | 0.336 ± 0.018 | −0.092 ± 0.37 | 66.31 ± 3.74 | 0.09 ± 0.05 |
| PLGA-shRNA-4 | 408.45 ± 20.18 | 0.348 ± 0.012 | −2.71 ± 0.039 | 85.18 ± 2.52 | 0.11 ± 0.03 |
| PLGA-PEG-5%-shRNA-4 | 289.93 ± 13.24 | 0.201 ± 0.020 | −1.81 ± 0.023 | 84.02 ± 5.81 | 0.11 ± 0.08 |
| PLGA-PEG-10%-shRNA-4 | 237.71 ± 23.36 | 0.265 ± 0.011 | −1.17 ± 0.044 | 80.01 ± 1.06 | 0.11 ± 0.02 |
*Data are mean ± SD with n = 6
The morphology of shRNA-loaded NP was determined using SEM and in Fig. 1 it is observed that both PLGA and PEG-PLGA NP display a smooth, spherical structure. An assessment of the mean diameters from these images is in close agreement with the dynamic light scattering data.
Fig. 1.

Surface morphology of loaded NP using scanning electron microscopy where (a) represents PLGA-shRNA-1 NP, (b) PLGA-PEG-5%-shRNA-1 NP and (c) PLGA-PEG-10%-shRNA-1 NP
Release Studies
The division of the release profile into time-related phases, as often observed in nanoparticulate formulations for cancer applications [33], was evident in Fig. 2. Although complicated profiles are known, comprising possibly triphasic patterns, the data in Fig. 2 were observed to be biphasic, which is a common finding in PLGA NP systems [26,34]. The cumulative release of shRNA NP (shRNA-1 and shRNA-4 both) comprised the initial burst, when approximately 40% of the nucleotide loading was released in the first 24 h. This was then followed by a slower and more sustained phase, which is approximately zero order until 96 h. Differences in release between the two shRNA types was not apparent in the data, with both achieving a total release of approximately 51% after 96 h. Furthermore, the additional of PEG to the NP matrix did not produce any significant increase in the drug release over the observed experimental period.
Fig. 2.
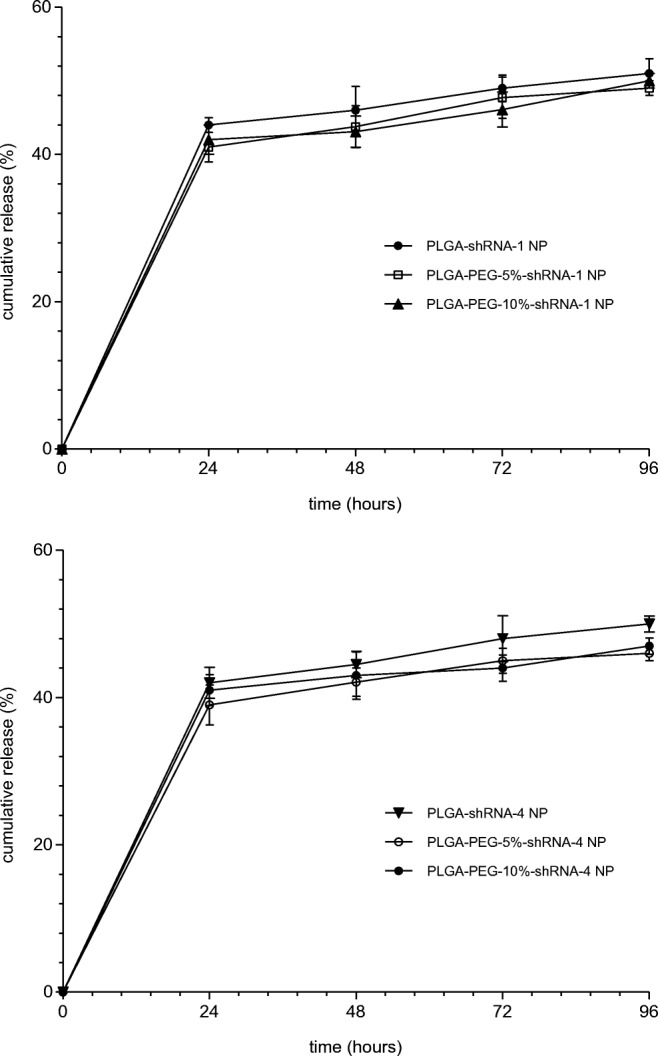
Cumulative release of shRNA-1 and shRNA-4 from loaded NP determined over a period of 96 h. (data are mean ± SD, with n = 3)
Cellular Uptake
Fluorescence imaging of coumarin-6-labelled NP following exposure to MDA-MB231 cells in shown in Fig. 3. Control experiments with free dye confirm effective nuclear staining, together with no observable cytoplasmic staining. Upon exposure to the fluorescent NP and upon inspection of the FITC channel, there is evidence of uptake into the cytoplasm. The nuclear space is dark, suggesting that nuclear uptake has not occurred. This observation would also support a lack of non-specific surface binding to the cell, which would be expected to light up the nuclear space, from either above or below the cell.
Fig. 3.
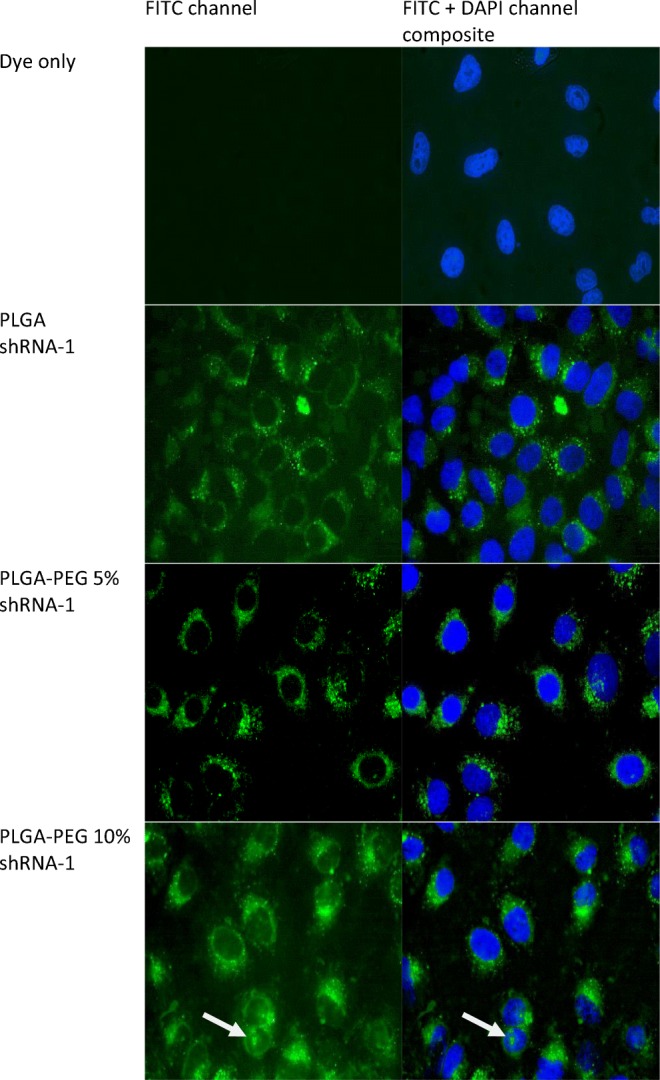
Intracellular localisation of coumarin-loaded shRNA-1 NP in MDA-MB231 cells. The DAPI channel (λex 358 nm, λem 461 nm) was used to visualise staining of the cell nucleus and the FITC channel (λex 488 nm, λem 519 nm) was used to visualise coumarin-6-loaded NP. Images taken from both channels were merged to determine the location of intracellular nanoparticulate localisation
Merged images confirm no localisation of NP in the nuclear space. There is sporadic evidence of NP occurring within the nucleus (highlighted by the white arrow), but it cannot be confirmed if these NP are either bound to the outermost cell surface or have translocated across the nuclear membrane.
Cell Viability
The results of the cell viability assay following exposure to shRNA-loaded NP are shown in Table II. Free shRNA and blank NP formulations did not cause any significant reduction (P < 0.05) in cell viability after 24 h. After longer periods, there was evidence of a modest reduction in viability, with the biggest drop observed (88%) after 96 h following exposure with PLGA-blank NP. Generally, no control formulation caused a drop in viability below 90%.
Table II.
Viability of MDA-MB231 breast cancer cells following exposure to shRNA-loaded NP (shRNA-1 and shRNA-4)
| shRNA-1* | shRNA-4* | |||||||
|---|---|---|---|---|---|---|---|---|
| 24 Hours | 48 Hours | 72 Hours | 96 Hours | 24 Hours | 48 Hours | 72 Hours | 96 Hours | |
| Cells | 100 ± 3.1 | 100 ± 10.3 | 100 ± 6.5 | 100 ± 8.0 | 100 ± 3.1 | 100 ± 0.0 | 100 ± 7.0 | 100 ± 6.0 |
| shRNA | 101.6 ± 9.1 | 105.3 ± 8.2 | 99.7 ± 7.2 | 98.2 ± 8.2 | 99.6 ± 4.0 | 101.4 ± 3.0 | 100.7 ± 7.8 | 98.2 ± 8.2 |
| PLGA-Blank NP | 96.1 ± 11.1 | 92.1 ± 6.0 | 92.7 ± 6.0 | 88.1 ± 8.1 | 96.1 ± 0.8 | 92.1 ± 3.1 | 92.7 ± 4.1 | 88.1 ± 8.1 |
| PLGA-PEG-5%-Blank NP | 97.5 ± 10.0 | 92.0 ± 5.9 | 93.3 ± 6.1 | 94.1 ± 5.1 | 95.2 ± 2.9 | 94.6 ± 2.9 | 92.0 ± 3.8 | 93.1 ± 7.1 |
| PLGA-PEG-10%-Blank NP | 96.1 ± 10.0 | 95.0 ± 4.9 | 93.6 ± 6.0 | 90.1 ± 8.0 | 96.7 ± 4.4 | 95.1 ± 3.0 | 94.7 ± 4.3 | 90.0 ± 4.2 |
| PLGA-shRNA-NP C1 | 85.6 ± 3.3 | 32.9 ± 11.7 | 63.9 ± 5.5 | 68.9 ± 7.3 | 80.6 ± 1.6 | 68.9 ± 6.4 | 74.7 ± 2.0 | 68.9 ± 7.3 |
| PLGA-shRNA-NP C2 | 45.8 ± 10.-0 | 19.23 ± 6.5 | 34.2 ± 8.7 | 37.7 ± 16.8 | 82.0 ± 3.3 | 60.2 ± 4.8 | 71.1 ± 1.7 | 37.7 ± 16.8 |
| PLGA-shRNA-NP C3 | 36.5 ± 5.5 | 16.0 ± 4.3 | 10.4 ± 4.2 | 8.0 ± 2.8 | 18.1 ± 0.5 | 41.2 ± 12.7 | 29.7 ± 3.4 | 10.0 ± 1.1 |
| PLGA-PEG-5%-shRNA-NP C1 | 43.3 ± 5.8 | 32.2 ± 4.3 | 58.6 ± 10.0 | 45.3 ± 9.3 | 92.2 ± 5.0 | 92.5 ± 1.2 | 95.1 ± 8.8 | 78.9 ± 2.7 |
| PLGA-PEG-5%-shRNA-NP C2 | 45.8 ± 7.1 | 31.5 ± 5.7 | 53.9 ± 1.2 | 42.9 ± 12.3 | 92.8 ± 4.6 | 79.2 ± 9.2 | 57.0 ± 4.4 | 47.7 ± 2.9 |
| PLGA-PEG-5%-shRNA-NP C3 | 53.5 ± 4.8 | 38.3 ± 8.4 | 35.8 ± 3.4 | 33.4 ± 3.2 | 72.0 ± 1.26 | 82.6 ± 9.4 | 71.6 ± 1.8 | 18.0 ± 5.8 |
| PLGA-PEG-10%-shRNA-NP C1 | 48.3 ± 7.1 | 32.2 ± 3.5 | 58.6 ± 9.9 | 45.3 ± 9.3 | 66.9 ± 8.6 | 80.5 ± 9.4 | 56.3 ± 10.5 | 50.7 ± 19.4 |
| PLGA-PEG-10%-shRNA-NP C2 | 45.8 ± 5.0 | 31.5 ± 8.2 | 53.9 ± 0.7 | 42.9 ± 12.3 | 34.9 ± 7.2 | 74.9 ± 7.7 | 50.7 ± 6.4 | 30.1 ± 7.2 |
| PLGA-PEG-10%-shRNA-NP C3 | 41.3 ± 6.0 | 30.9 ± 7.2 | 40.4 ± 6.9 | 41.2 ± 2.7 | 43.2 ± 3.3 | 58.4 ± 6.3 | 54.0 ± 4.9 | 25.4 ± 3.3 |
*Data are mean ± SD with n = 6
C1, C2 and C3 represents concentrations of shRNA used to transfect the cells, where C1 = 0.5 μg ml−1, C2 = 1.0 μg ml−1 and C3 = 2.0 μg ml−1 of entrapped shRNA
The encapsulation of shRNA into NP had a significant influence in cell viability. For example, cell viability reduced following transfection with increased amounts of shRNA-1 in PLGA NP. The highest concentration (C3) reduced viability to 36% after 24 h. When the addition of PEG to the NP matrix is considered, a similar reduction in cell viability was observed. This is seen in the data in Table II when PLGA-PEG-5% was used to deliver shRNA-1 to the cells. A significant decrease in cell viability was observed after incubation of the cells with PLGA-PEG-10% NP (***P < 0.001) using the lowest concentration (C1) of shRNA after 24 h, as more than 50% of the cell population was reduced. Interestingly, no further decreases in viability was observed following longer exposures. Without PEG, the effect of both longer exposure and higher concentration of shRNA-1 becomes more effective, with a cell viability of 8% using C3 and after 96 h.
Cell viability observed for shRNA-4 was generally in agreement to that determined for shRNA-1. However, there is greater influence of concentration and time of exposure for all formulations, with the presence of PEG having a greater effect. As with the shRNA-1 formulation, the PLGA NP with no PEG was found to have the biggest effect on viability (10%) after 96 h and at 2.0 μg ml−1 of entrapped shRNA-4.
Cell Migration
The results from the cell migration assay confirm that scratch closure was significantly inhibited by both shRNA-1 and shRNA-4-loaded NP and the rate of migration was affected by the concentration of shRNA inside the NP. Free, non-encapsulated shRNA and blank PLGA NP did not have any effect on migration. Both Figs. 4(B) and 4 (B) confirm a concentration dependence on the % scratch closure, so that after 24 h, for example, 79% of the scratch width was observed in PLGA-shRNA-1 C1 treated cells, whereas with PLGA-shRNA-1 C3 treatment, the same response was found to be 34%. This result showed that cell migration was slower when more shRNA was available in the NP formulation. The data in Fig. 4 show that the patterns of cell retardation over time were similar for both shRNA types, with the shRNA-1 demonstrating the larger reduction in scratch closure after 72 h.
Fig. 4.

Scratch assay performed on confluent MDA-MB231 cells showing (A) light microscopy examination of the effect of PLGA-shRNA-1-NP and PLGA-shRNA-4-NP at 2.0 μg ml−1 on cell migration and (B) graphical representation of the dimensional change over 72 h using PLGA-shRNA-1-NP at three different concentrations and (C) graphical representation of the dimensional change over 72 h using PLGA-shRNA-4-NP at three different concentrations). Data are mean ± SD with n = 6. *P < 0.05, **P < 0.01, ***P < 0.001, C1, C2 and C3 are 0.5 μg ml−1, 1.0 μg ml−1 and 2.0 μg ml−1 of entrapped shRNA-4
Cell Invasion Assay
Invasiveness is a property of MDA-MB231 cells that contributions to their aggressive growth pattern. Attenuation of this characteristic is of particular interest in devising novel therapeutic interventions [35]. Therefore, an invasion assay was carried out to study the anti-invasive properties of shRNA-loaded NP and the results are shown in Fig. 5 . It was observed that PLGA-shRNA-1 NP (2.0 μg ml−1) inhibited significantly the invasion of the cells up to 57% after 48 h (***P < 0.001). In contrast, treatment with blank PLGA NP and non-encapsulated shRNA-1 showed no significant effect on invasion. Similarly, shRNA-4-loaded NP slowed the invasion of cells with a reduction in invasion of 20% after 24 h and 38% after 48 h, when compared to controls. A greatest significant effect is observed after 48 h and using shRNA-1 (P < 0.001). Although a reduction in invasiveness is significant after 48 h using encapsulated shRNA-4, the data in Fig. 5 (A) shows that is reduction is not as great as the result obtained using shRNA-1. Nevertheless, both shRNA types, when encapsulated in PLGA NP, reduced cell invasion.
Fig. 5.

Effect of 2.0 μg ml−1 shRNA delivered using (A) PLGA-shRNA-1 NP and (B) PLGA-shRNA-4 on the invasion of MDA-MB231 cells. Data are mean ± SD with n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, where significance is determined by comparison with invasion following treatment with blank NP
qRT-PCR-Gene Knockdown
In this work, real time quantitative PCR was used to determine the effect of shRNA-1 and shRNA-4 on RAN inside the cell. Knockdown (95%) of RAN was observed when cells were transfected with PLGA-shRNA-1 NP and knockdown of 85% was observed following transfection with PLGA-shRNA-4 NP after 48 h, as shown in Fig. 6 . In order to demonstrate a maximal effect, the concentration of shRNA (shRNA-1 and shRNA-4) used for transecting cells was 2.0 μg ml−1. The expression of RAN was reduced significantly in both cases, but encapsulated shRNA-1 was found to be more effective than shRNA-4 as evident from the percentage of reduction of RAN expression. These results demonstrate RNA interference [36] and verifies the specificity of the shRNA variants used in this work towards RAN.
Fig. 6.
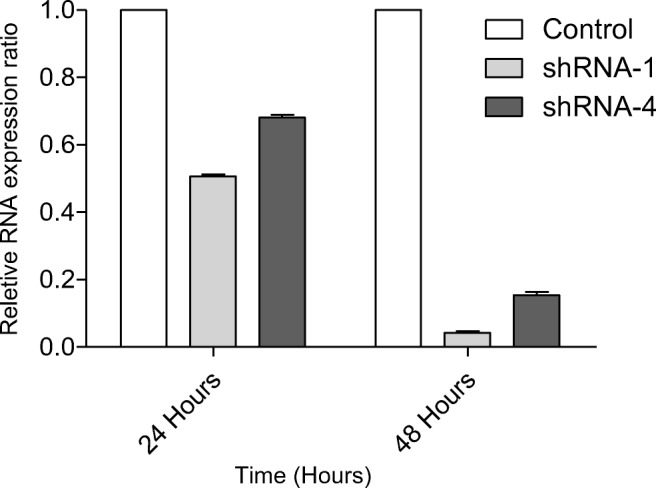
qRT-PCR for RAN gene expression after 24 and 48 h transfection of PLGA-shRNA-1 NP and PLGA-shRNA-4, where cells were transfected with 2.0 μg ml−1 shRNA-1 and 2.0 μg ml−1 shRNA-4. Control was blank PLGA-shRNA NP. Relative gene expression was measured using β-actin as a house keeping gene
Discussion
Silencing of the RAN has been shown by our group to have significant potential in reducing both invasion and proliferation in an aggressive tumour cell line [4]. Effective delivery of shRNA, which is needed to bring about silencing, is a challenging task. However, polymeric nanoparticles have been shown to assist intracellular delivery of biological payloads, but key parameters, such as the polymer type, loading and release rates, must be investigated beforehand. This was especially pertinent in this work given that it is the first report of polymeric encapsulation of shRNA-1 and shRNA-4. Therefore, as part of the initial formulation investigations, PLGA and PLGA-PEG co-block polymers were chosen as the NP matrix. The use of PLGA is an understandable choice as it is widely used for drug delivery applications, being both biodegradable and non-toxic. However, in order to optimise drug loading and subsequent release, alternations in the polymer packing and hydrophobicity are required. A common approach to do this is to use co-block addition of PEG residues, which was the approach adopted in this work. This provides a means to adjust particle size, charge and loading and is often part of the formulation studies used to optimise encapsulation efficiency and cellular uptake.
The key parameters investigated in this work, such as size, charge and drug loading are shown in Table I. Mean diameter plays a significant role in cellular uptake, with NP in the range of 50–500 nm taken up more efficiently than larger NP via endocytotic pathways [37,38]. However, given the difficulties in the delivery of naked shRNA to cells, the key focus of this work was the loading of NP with shRNA and the effective cellular delivery of the payload, not the size of the NP themselves. The double emulsion procedure used in the work was able to produce NP with a mean size within this range. Differences in the sizes of shRNA-loaded PLGA and PLGA-PEG NP were due to the difference in the chain length of PLGA polymeric moiety in the polymer [39]. On adding PEG to the PLGA polymer, the hydrophobic chain size of PLGA was condensed, which reduced the hydrophobic aspects of the NP core and the size the NP. This effect has been reported for other smaller molecular weight drug substances, such as flurbiprofen-loaded PLGA NP (190 nm), which were bigger than flurbiprofen-loaded PLGA-PEG NP (170 nm) [39].
The surface charge on a NP plays an important role in its interaction with the plasma membrane of cells. Since the plasma membrane is anionic, it will favour the absorption of cationic NP due to electrostatic interaction [40,41]. PLGA-shRNA, PLGA-PEG-5%-shRNA and PLGA-PEG-10%-shRNA NP (both shRNA-1 and shRNA-4) all possessed a negative surface charge, PLGA-shRNA exhibited the maximum negative charge followed by PLGA-PEG-5%- shRNA and PLGA-Peg-10%-shRNA. Addition of PEG to PLGA masked the negative charge on PLGA-PEG-shRNA nanoparticles and hence makes them less negative. This masking effect increased on increasing the PEG concentration in the PLGA-PEG polymer moiety. The results in Table I show that inclusion of PEG to the PLGA NP did reduce the negative zeta potential but this caused an increase in cell viability rather than a decrease as was expected (Table II). This finding has been observed in other work [42]. The data in Table I also shows that the double emulsion procedure is able to achieve impressive encapsulation efficiencies in nanoparticles that are spherical and smooth (Fig. 1). Similar studies have reported encapsulation efficiencies of siRNA in PLGA NP of approximately 51%, comparing favourably with the findings of this current study, which demonstrated efficiencies of around 80% for the PLGA NP [43,44]. However, it was observed that the encapsulation of the payload increased when PEG increased, suggesting that enhanced hydrophilicity did not improve encapsulation. Clearly, the presence of PEG in the NP core does not lead to enhanced incorporation.
The release of encapsulated drugs from NP can be sustained, or adopt a burst release, or both, depending on the NP formulation type [45]. The profiles in Fig. 2 show similarity between all formulations tested. There are indications that PLGA-shRNA-NP give higher release than PLGA-PEG-shRNA NP, but this finding is not significant. Other work has shown better release from PLGA NP when compared to PEG-PLGA NP and it has been proposed that this effect arises due to hydrophilic interaction between the drug and PEG moieties, leading to a delay in the diffusion of the drug [46]. However, Fig. 2 shows a distinctive burst release phase during the first 24 h, which is attributable to release of surface resident and adsorbed shRNA. The slower phase, occurring after 24 h and for several hours afterwards, is attributable to NP degradation and diffusion through the NP core [46].
Intracellular uptake of drug-loaded NP is an appealing property, especially if subsequent release occurs directly into the cytoplasm. Our group has described the development of two novel shRNA sequences, as used in this work, which if delivered intracellularly, can exert anti-proliferative and anti-invasive effects in MDA-MB231 breast cancer cells. Non-encapsulated forms of our shRNA sequences (shRNA-1 and shRNA-4) without assistance from delivery vectors are ineffective and did not affect cell viability. Similar results were obtained elsewhere when non-encapsulated shRNA was shown to be ineffective at transfecting LNCaP prostate cancer cells, whereas retroviral-mediated shRNA delivery caused inhibition in cell populations [47]. In this current work, blank NP were found to be non-toxic, whereas shRNA-loaded PLGA NP, shRNA-loaded PLGA-PEG-5% NP and shRNA-loaded PLGA-PEG-10% NP significantly reduced cell viability, as shown in Table II. Cell viability decreased with an increase in concentration of NP (C1 < C2 < C3). Cell viability increased at the 72 h point for PLGA-shRNA-NP C1 and some other of the formulations, which may be due to the temporal release profile of the nanoparticles, as suggested by Basu et al. [48]. They reported a similar increase in cell viability when MAPK inhibitor-loaded PLGA nanoparticles were delivered to MDA-MB231 cells. Another study by Wang et al. [49] also reported an increase in cell viability after the fifth day of a cell viability study after introduction of nanoparticles and they suggested this increase was due to the high encapsulation efficiency of the nanoparticles.
The results from fluorescence imaging, as shown in Fig. 3, suggests that these NP have translocated to the cytoplasm. PLGA was shown to be superior to its PEG-containing variants, with shRNA-1 and shRNA-4 encapsulated in PLGA NP significantly reduced cell viability to 8% and 10% after 96 h, respectively (***P < 0.001) when the highest drug loading was used (C3 = 2.0 μg ml−1). This reduction in viability was concentration dependent, particularly in the PLGA formulations. This form of concentration dependency is a common finding. For example, bicalcutamide-loaded PLGA NP in various concentrations (0.5 μg ml−1 – 50 μg ml−1) to C4–2 prostate cancer cells caused a proportional reduction in cell viability in relation to the payload concentration [50,51].
An important aim of this work was to demonstrate that shRNA-1 and shRNA-4 inhibited the migration of MDA-MB231 breast cancer cells following nanoparticulate delivery. In order to rationalise the experimental design, the formulations providing the most effective reduction in cell viability (PLGA-shRNA-1 and PLGA-shRNA-4) were taken forward for evaluation of migration and invasion. The scratch assay was used to quantify migration using a simple measure of determining gap closure, as shown in Figs. 4 and 5. Results in these figures show that both PLGA-shRNA-1 and PLGA-shRNA-4 brought about a concentration-dependent reduction in the speed at which MDA-MB231 breast cancer cells migrate and close the induced gap. shRNA-1-loaded NP slowed migration significantly when compared to blank NP, non-encapsulated shRNA-1 and untreated cells (controls). Indeed, 72 h was sufficient time to allow the scratch to close completely when no treatment was given. However, the gap was still apparent following treatment with loaded NP. Results for cell invasion followed a similar pattern to migration. Invasion of MDA-MB231 cells was significantly reduced to 60% (P < 0.001) and 68% (P < 0.01) after 48 h following treatment with PLGA-shRNA-1 NP and shRNA-4 NP, respectively, with shRNA-1 marginally superior to shRNA-4.
The data in Fig. 6 confirm that RAN knockdown was observed when cells were transfected with PLGA-shRNA-1 NP and PLGA-shRNA-4 NP with relative expression rations of 96% and 85%, respectively. Effective knockdown of RAN gene demonstrates that shRNA is undergoing endosomal escape after internalisation of the NP which is crucial it these delivery vehicles are to have a therapeutic potential. These figures compare favourably to reported values of 90% knockdown of RAN using viral vector in pancreatic and melanoma cell lines [52,53]. Importantly, the concentration of shRNA used in these studies was 6 μg ml−1, which was 3 times higher than the concentrations used in this study. This comparison demonstrates the effective delivery using the PLGA carrier and is comparable to the levels achieved with more established viral vectors.
Conclusion
PLGA NP loaded with two novel shRNA sequences were of a size range that has been shown to be effective in cell uptake. The encapsulation efficiency was found to be high for all formulations with minimal loss of the payload during the nanoparticle fabrication. Variation in the PEG content allowed for adjustment in particulate properties, such surface charge and size distribution. However, the inclusion of PEG did not improve in vitro characterisations, such as migration and invasion, with pure PLGA NP loaded with both shRNA-1 and shRNA-4 producing significant reductions in migration and invasion of MDA-MB231 breast cancer cells. However, differences in shRNA-1 and shRNA-4 in terms of drug release, cell viability and RAN knockdown were small, with the former being judged as marginally superior. The results of this study demonstrate that shRNA-1 and shRNA-4 can be delivered effectively using PLGA NP and that biological activity is preserved once drug release has occurred. This delivery system is a potential therapeutic means to cell invasion and migration in cells of metastatic potential.
Compliance with Ethical Standards
Conflict of Interest
The authors declare no potential conflicts of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gupta A, Ahmad A, Dar AI, Khan R. Synthetic lethality: from research to precision Cancer Nanomedicine. Curr Cancer Drug Targets. 2018;18(4):337–346. doi: 10.2174/1568009617666170630141931. [DOI] [PubMed] [Google Scholar]
- 2.Landesman-Milo D, Ramishetti S, Peer D. Nanomedicine as an emerging platform for metastatic lung cancer therapy. Cancer Metastasis Rev. 2015;34(2):291–301. doi: 10.1007/s10555-015-9554-4. [DOI] [PubMed] [Google Scholar]
- 3.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6(6):449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 4.Yuen HF, Chan KK, Grills C, Murray JT, Platt-Higgins A, Eldin OS, O'Byrne K, Janne P, Fennell DA, Johnston PG, Rudland PS, el-Tanani M. Ran is a potential therapeutic target for cancer cells with molecular changes associated with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways. Clinical Cancer Res. 2012;18(2):380–391. doi: 10.1158/1078-0432.CCR-11-2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matchett KB, McFarlane S, Hamilton SE, Eltuhamy YS, Davidson MA, Murray JT, et al. Ran GTPase in nuclear envelope formation and cancer metastasis. Adv Exp Med Biol. 2014;773:323–351. doi: 10.1007/978-1-4899-8032-8_15. [DOI] [PubMed] [Google Scholar]
- 6.Clarke PR, Zhang C. Spatial and temporal coordination of mitosis by ran GTPase. Nat Rev Mol Cell Biol. 2008;9(6):464–477. doi: 10.1038/nrm2410. [DOI] [PubMed] [Google Scholar]
- 7.Azuma K, Sasada T, Takedatsu H, Shomura H, Koga M, Maeda Y, Yao A, Hirai T, Takabayashi A, Shichijo S, Itoh K. Ran, a small GTPase gene, encodes cytotoxic T lymphocyte (CTL) epitopes capable of inducing HLA-A33-restricted and tumor-reactive CTLs in cancer patients. Clin Cancer Res. 2004;10(19):6695–6702. doi: 10.1158/1078-0432.CCR-04-0818. [DOI] [PubMed] [Google Scholar]
- 8.Xu WJ, Zhou JJ, Xie Y, Wang WL, Zhao Y, Chen X, et al. Association between the expression and methylation of energy-related genes with helicobacter pylori infection in gastric cancer. Zhonghua Yi Xue Za Zhi. 2012;92(6):366–370. [PubMed] [Google Scholar]
- 9.Yuen HF, Gunasekharan VK, Chan KK, Zhang SD, Platt-Higgins A, Gately K, O’Byrne K, Fennell DA, Johnston PG, Rudland PS, el-Tanani M. RanGTPase: a candidate for Myc-mediated cancer progression. J Natl Cancer Inst. 2013;105(7):475–488. doi: 10.1093/jnci/djt028. [DOI] [PubMed] [Google Scholar]
- 10.Martens-de Kemp SR, Nagel R, Stigter-van Walsum M, van der Meulen IH, van Beusechem VW, Braakhuis BJ, et al. Functional genetic screens identify genes essential for tumor cell survival in head and neck and lung cancer. Clin Cancer Res. 2013;19(8):1994–2003. doi: 10.1158/1078-0432.CCR-12-2539. [DOI] [PubMed] [Google Scholar]
- 11.Xia F, Dohi T, Martin NM, Raskett CM, Liu Q, Altieri DC. Essential role of the small GTPase ran in postnatal pancreatic islet development. PLoS One. 2011;6(11):e27879. doi: 10.1371/journal.pone.0027879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barres V, Ouellet V, Lafontaine J, Tonin PN, Provencher DM, Mes-Masson AM. An essential role for ran GTPase in epithelial ovarian cancer cell survival. Mol Cancer. 2010;9:272. doi: 10.1186/1476-4598-9-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan H, Lu Y, Qin H, Zhou Y, Gu Y, Zhou J, Wang X, Fan D. High ran level is correlated with poor prognosis in patients with colorectal cancer. Int J Clin Oncol. 2013;18(5):856–863. doi: 10.1007/s10147-012-0465-x. [DOI] [PubMed] [Google Scholar]
- 14.Abe H, Kamai T, Shirataki H, Oyama T, Arai K, Yoshida K. High expression of ran GTPase is associated with local invasion and metastasis of human clear cell renal cell carcinoma. Int J Cancer. 2008;122(10):2391–2397. doi: 10.1002/ijc.23400. [DOI] [PubMed] [Google Scholar]
- 15.Yuen H-F, Chan K-K, Platt-Higgins A, Dakir E-H, Matchett KB, Haggag YA, et al. Ran GTPase promotes cancer progression via met receptor-mediated downstream signaling. Oncotarget. 2016;7(46):75854–75864. doi: 10.18632/oncotarget.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leng Q, Woodle MC, Lu PY, Mixson AJ. Advances in Systemic siRNA Delivery. Drugs of the future. 2009;34(9):721. doi: 10.1358/dof.2009.034.09.1413267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opinion Solid State Mater Sci. 2002;6(4):319–327. doi: 10.1016/S1359-0286(02)00117-1. [DOI] [Google Scholar]
- 18.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, et al. Formulation of functionalized PLGA–PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. doi: 10.1016/j.jconrel.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 20.De Jong WH, Borm PJA. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. doi: 10.2147/IJN.S596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117(12):3623–3632. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behnke T, Mathejczyk JE, Brehm R, Würth C, Ramos Gomes F, Dullin C, et al. Target-specific nanoparticles containing a broad band emissive NIR dye for the sensitive detection and characterization of tumor development. Biomaterials. 2013 2013/01/01/;34(1):160–70. [DOI] [PubMed]
- 23.Sheng C, Qiu J, Wang Y, He Z, Wang H, Wang Q, Huang Y, Zhu L, Shi F, Chen Y, Xiong S, Xu Z, Ni Q. Knockdown of ran GTPase expression inhibits the proliferation and migration of breast cancer cells. Mol Med Rep. 2018;18(1):157–168. doi: 10.3892/mmr.2018.8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurisetty VV, Johnston PG, Johnston N, Erwin P, Crowe P, Fernig DG, Campbell FC, Anderson IP, Rudland PS, el-Tanani MK. RAN GTPase is an effector of the invasive/metastatic phenotype induced by osteopontin. Oncogene. 2008;27(57):7139–7149. doi: 10.1038/onc.2008.325. [DOI] [PubMed] [Google Scholar]
- 25.Lee CS, Bishop ES, Zhang R, Yu X, Farina EM, Yan S, et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes & Diseases. 2017 2017/06/01/;4(2):43–63. [DOI] [PMC free article] [PubMed]
- 26.Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Risnayanti C, Jang Y-S, Lee J, Ahn HJ. PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer. Scientific Reports. 2018 2018/05/14;8(1):7498. [DOI] [PMC free article] [PubMed]
- 28.Wang L, Griffel B, Xu X. Synthesis of PLGA-Lipid Hybrid Nanoparticles for siRNA Delivery Using the Emulsion Method PLGA-PEG-Lipid Nanoparticles for siRNA Delivery. Methods in molecular biology (Clifton, NJ). 2017:7138-1_15. [DOI] [PubMed]
- 29.Ebrahimian M, Taghavi S, Mokhtarzadeh A, Ramezani M, Hashemi M. Co-delivery of doxorubicin encapsulated PLGA nanoparticles and Bcl-xL shRNA using alkyl-modified PEI into breast Cancer cells. Appl Biochem Biotechnol. 2017;183(1):126–136. doi: 10.1007/s12010-017-2434-3. [DOI] [PubMed] [Google Scholar]
- 30.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in Cancer therapy. Angew Chem Int Ed. 2014;53(46):12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- 31.Sezlev Bilecen D, Rodriguez-Cabello JC, Uludag H, Hasirci V. Construction of a PLGA based, targeted siRNA delivery system for treatment of osteoporosis. Journal of Biomaterials Science, Polymer Edition. 2017 2017/11/02;28(16):1859–73. [DOI] [PubMed]
- 32.Patil Y, Panyam J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int J Pharm. 2009;367(1–2):195–203. doi: 10.1016/j.ijpharm.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das S, Khuda-Bukhsh AR. PLGA-loaded nanomedicines in melanoma treatment: future prospect for efficient drug delivery. Indian J Med Res. 2016;144(2):181–193. doi: 10.4103/0971-5916.195024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine. 2011;6:877–895. doi: 10.2147/IJN.S18905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Y, Chen K, Gu W, Dong X, Lei R, Chang Y, et al. Small size fullerenol nanoparticles suppress lung metastasis of breast cancer cell by disrupting actin dynamics. Journal of Nanobiotechnology. [journal article]. 2018 June 23;16(1):54. [DOI] [PMC free article] [PubMed]
- 36.Babu A, Muralidharan R, Amreddy N, Mehta M, Munshi A, Ramesh R. Nanoparticles for siRNA-based gene silencing in tumor therapy. IEEE Trans Nanobioscience. 2016;15(8):849–863. doi: 10.1109/TNB.2016.2621730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dreaden EC, Austin LA, Mackey MA, El-Sayed MA. Size matters: gold nanoparticles in targeted cancer drug delivery. Ther Deliv. 2012;3(4):457–478. doi: 10.4155/tde.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotech. [Research]. 2015;33(9):941–51. [DOI] [PMC free article] [PubMed]
- 39.Vasconcelos A, Vega E, Pérez Y, Gómara MJ, García ML, Haro I. Conjugation of cell-penetrating peptides with poly(lactic-co-glycolic acid)-polyethylene glycol nanoparticles improves ocular drug delivery. Int J Nanomedicine. 2015;10:609–631. doi: 10.2147/IJN.S71198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldenberg NM, Steinberg BE. Surface charge: a key determinant of protein localization and function. Cancer Res. 2010;70(4):1277–1280. doi: 10.1158/0008-5472.CAN-09-2905. [DOI] [PubMed] [Google Scholar]
- 41.Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guedj A-S, Kell AJ, Barnes M, Stals S, Gonçalves D, Girard D, Lavigne C. Preparation, characterization, and safety evaluation of poly(lactide-co-glycolide) nanoparticles for protein delivery into macrophages. Int J Nanomedicine. 2015;10:5965–5979. doi: 10.2147/IJN.S82205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cun D, Jensen DK, Maltesen MJ, Bunker M, Whiteside P, Scurr D, Foged C, Nielsen HM. High loading efficiency and sustained release of siRNA encapsulated in PLGA nanoparticles: quality by design optimization and characterization. Eur J Pharm Biopharm. 2011;77(1):26–35. doi: 10.1016/j.ejpb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Yeo Y, Park K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Archives of Pharmacal Research. [journal article]. 2004;27(1):1–12. [DOI] [PubMed]
- 45.Xu S, Wang W, Li X, Liu J, Dong A, Deng L. Sustained release of PTX-incorporated nanoparticles synergized by burst release of DOX·HCl from thermosensitive modified PEG/PCL hydrogel to improve anti-tumor efficiency. Eur J Pharm Sci. 2014;62:267–273. doi: 10.1016/j.ejps.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 46.Dutta D, Salifu M, Sirianni RW, Stabenfeldt SE. Tailoring sub-micron PLGA particle release profiles via centrifugal fractioning. J Biomed Mater Res A. 2016;104(3):688–696. doi: 10.1002/jbm.a.35608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin S, Oh S, An S, Janknecht R. ETS variant 1 regulates matrix metalloproteinase-7 transcription in LNCaP prostate cancer cells. Oncol Rep. 2013;29(1):306–314. doi: 10.3892/or.2012.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu S, Harfouche R, Soni S, Chimote G, Mashelkar RA, Sengupta S. Nanoparticle-mediated targeting of MAPK signaling predisposes tumor to chemotherapy. Proc Natl Acad Sci. 2009;106(19):7957–7961. doi: 10.1073/pnas.0902857106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang B, Tan L, Deng D, Lu T, Zhou C, Li Z, Tang Z, Wu Z, Tang H. Novel stable cytokine delivery system in physiological pH solution: chitosan oligosaccharide/heparin nanoparticles. Int J Nanomedicine. 2015;10:3417–3427. doi: 10.2217/nnm.15.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo JUN, Wu S-H, Ren W-G, Wang X-L, Yang A-Q. Anticancer activity of bicalutamide-loaded PLGA nanoparticles in prostate cancers. Exp Ther Med. 2015;10(6):2305–2310. doi: 10.3892/etm.2015.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cosco D, Cilurzo F, Maiuolo J, Federico C, Di Martino MT, Cristiano MC, et al. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Scientific Reports. [Article]. 2015;5:17579. [DOI] [PMC free article] [PubMed]
- 52.Deng L, Shang Y, Guo S, Liu C, Zhou L, Sun Y, Nie Y, Fan D, Lu Y, Guo X. Ran GTPase protein promotes metastasis and invasion in pancreatic cancer by deregulating the expression of AR and CXCR4. Cancer Biol Ther. 2014;15(8):1087–1093. doi: 10.4161/cbt.29217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caputo E, Wang E, Valentino A, Crispi S, De Giorgi V, Fico A, et al. Ran signaling in melanoma: implications for the development of alternative therapeutic strategies. Cancer Lett. 2015;357(1):286–296. doi: 10.1016/j.canlet.2014.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]