

Abstract
Polycystin-1 (PC1), encoded by the PKD1 gene that is mutated in the autosomal dominant polycystic kidney disease, regulates a number of processes including bone development. Activity of the transcription factor RunX2, which controls osteoblast differentiation, is reduced in Pkd1 mutant mice but the mechanism governing PC1 activation of RunX2 is unclear. PC1 undergoes regulated cleavage that releases its C-terminal tail (CTT), which translocates to the nucleus to modulate transcriptional pathways involved in proliferation and apoptosis. We find that the cleaved CTT of PC1 (PC1-CTT) stimulates the transcriptional coactivator TAZ (Wwtr1), an essential coactivator of RunX2. PC1-CTT physically interacts with TAZ, stimulating RunX2 transcriptional activity in pre-osteoblast cells in a TAZ-dependent manner. The PC1-CTT increases the interaction between TAZ and RunX2 and enhances the recruitment of the p300 transcriptional co-regulatory protein to the TAZ/RunX2/PC1-CTT complex. Zebrafish injected with morpholinos directed against pkd1 manifest severe bone calcification defects and a curly tail phenotype. Injection of messenger RNA (mRNA) encoding the PC1-CTT into pkd1-morphant fish restores bone mineralization and reduces the severity of the curly tail phenotype. These effects are abolished by co-injection of morpholinos directed against TAZ. Injection of mRNA encoding a dominant-active TAZ construct is sufficient to rescue both the curly tail phenotype and the skeletal defects observed in pkd1-morpholino treated fish. Thus, TAZ constitutes a key mechanistic link through which PC1 mediates its physiological functions.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in the genes that encode polycystin-1 (PC1) and polycystin-2 (PC2). PC1 is an extremely large membrane protein, with a molecular mass exceeding 460 kDa and 11 predicted transmembrane spans (1,2). PC1 has been implicated in a variety of signaling pathways (3–5), including G-protein signaling, oxygen sensing (6) and the Wnt, AP-1, NFAT and JAK-STAT cascades (7–14). The PC2 protein has a predicted molecular weight of ∼110 kDa and six putative membrane spanning regions (15,16). PC2 is a Ca2+ permeable non-selective cation channel and belongs to the transient receptor potential family of cation channels (17,18). PC2 is thought to participate in mediating the release of calcium from intracellular stores and may contribute to the transduction of mechanostimulatory sensations communicated via the primary cilium (19,20). The 200 amino acid C-terminal cytoplasmic tail of PC1 contains a predicted coiled-coil domain that mediates this protein’s interaction with PC2 (21,22). PC2 appears to be involved in several signaling pathways (23,24), including those that have been attributed to PC1 (8).
PC1 is cleaved at sites in both its N- and C-terminal domains (3). N-terminal cleavage occurs at the G protein-coupled receptor proteolytic (GPS) site, near the first transmembrane domain (25). This cis-autoproteolytic cleavage occurs as PC1 traverses the secretory pathway and is stimulated by PC2 (25–29). The cleaved N-terminus remains non-covalently attached to the membrane-bound C-terminal fragment (30). Expression of mutant PC1 that cannot undergo GPS cleavage does not rescue the cystic phenotype in PKD1-/- mice (31). In addition, a related missense mutation causes ADPKD (25,31,32). At least three other cleavages liberate portions of the cytoplasmic CTT of PC1. One of these cleavages releases a ∼35 kDa soluble portion of the tail that accumulates in the nucleus (33–36) and that influences transcriptional pathways (10,12). Another more distal cleavage releases a 17 kDa fragment that interacts with the transcriptional activators STAT3 and STAT6 and the coactivator p100 (35,37). Flow cessation increased this cleavage as well as nuclear translocation of both the PC1 tail (33) and STAT6 (35,37). A fragment with six transmembrane domains that regulates store-operated calcium entry (38) has also been identified.
TAZ (transcriptional coactivator with PDZ binding motif), also known as WWTR1 (WW domain containing transcription factor 1), acts as a transcriptional co-regulatory molecule for a variety of transcription factors. The activities of TAZ and its homolog YAP (Yes-associated protein) are negatively influenced by the Hippo signaling pathway, which is a major regulator of cell proliferation and organ size (39,40). Perturbations in Hippo signaling are associated with a variety of tumors (41), and aberrant Hippo signaling has also been detected in animal models of ADPKD (42). TAZ reduces the activity of PPARγ to inhibit adipocyte differentiation, it acts to stimulate TEAD/TEF, and it similarly enhances the activity of TTF-1 in the differentiation of respiratory epithelium (43–46). TAZ plays a critical role in cell fate-determination during mesenchymal differentiation, where it acts as a switch between adipocyte and osteoblast cell development (47). TAZ promotes osteoblast differentiation via activation of the transcription factor RunX2 and inhibition of PPARγ (48). Runx2 is the master transcription factor regulating skeletogenesis, and is essential for directing the differentiation of mesenchymal precursor cells into osteoblasts (49–51). Targeted disruption of Runx2 results in the complete loss of bone calcification (50). Overexpression of RunX2 results in accelerated endochondral ossification due to premature chondrocyte maturation (52). RunX2 expression is upregulated during chondrocyte differentiation into osteoblasts, but is later downregulated in end-stage mature osteoblasts (53).
While much remains to be learned about the functions of the polycystin proteins, it is clear that in renal epithelial cells PC1 and PC2 interact with one another and localize together to the primary cilium (54). This ciliary localization appears to be critically important in aspects of PC1 and 2 function and in preventing cystic disease (55). Osteoblasts have been shown to possess primary cilia and express both PC1 and 2 (56). Furthermore, PC1 plays an important role in skeletogenesis via regulation of the bone master transcription factor Runx2 (57). PC1 is highly expressed in adult cartilage, and Pkd1 knockout mice demonstrated severe skeletal compromise (58,59). Skeletal development has been difficult to analyze in Pkd1-/- mice due to the embryonic lethality of this genotype. Studies of a heterozygous Pkd1m1Bei mouse model, which carries one copy of an inactivating Pkd1 mutant allele and survives to adulthood without polycystic kidney disease, demonstrate the presence of osteopenia and impaired osteoblastic differentiation (57). Furthermore, conditional disruption of Pkd1 in osteoblasts results in decreased bone mineral density, as well as decreased trabecular bone volume and cortical thickness (60). Selective inactivation of Pkd1 at early stages of osteoblast development is associated with decreased bone formation and increased accumulation of fat in the marrow (61). A recent study demonstrated that Pkd1 and TAZ compound heterozygotes exhibited additive decrements in bone mineral density, which was attributed to the interaction of TAZ and the cleaved CTT of PC1, leading to increased Runx2-mediated osteogenic expression (62).
TAZ knockout in zebrafish results in complete failure of bone formation, cardiac abnormalities and early embryonic death (48). Surprisingly, TAZ knockout mice demonstrate only minor defects in skeletogenesis, while other mesenchymal-derived tissues, including the kidney and lung, are profoundly disrupted, resulting in the development of polycystic kidney disease and pulmonary emphysema (63–65). TAZ knockout mice demonstrate renal cyst formation as early as embryonic day 15.5 with prominently dilated Bowman’s capsules, multi-cystic kidneys, hydronephrosis and severe concentration defects leading to polyuria (64).
The bone and kidney phenotypes associated with perturbations of both PC1 and TAZ expression suggest the interesting possibility that these proteins may participate together in a common signaling pathway. In the present study we find that PC1 and one of its C-terminal cleavage fragments substantially increase TAZ activity, and with it the activity of the RunX2 transcriptional pathway. We find that TAZ associates with the PC1-CTT to form a functional complex. The interaction between the PC1-CTT and TAZ increases the association between TAZ and RunX2 as well as the recruitment of the p300 transcriptional co-regulatory protein to the TAZ/RunX2/PC1-CTT complex. Finally, we show that the PC1-CTT is sufficient to rescue the tail curvature and skeletal defects resulting from loss of pkd1 in zebrafish. This effect requires the presence of TAZ and can be mimicked by the expression of a constitutively active form of TAZ, suggesting that TAZ is a critical downstream component of the cellular machinery through which the PC1 protein mediates its functions.
Results
A coactivator trap screen reveals that TAZ is a positively regulated target of the PC1-CTT
The observation that Pkd1 regulates skeletogenesis via stimulation of RunX2 (57,60) led us to search for novel regulatory targets that could mediate this influence. To identify transcription factors regulated by the PC1-CTT, we employed a ‘coactivator trap’ screen, in which over 1400 transcription factors are fused to the DNA-binding domain of Gal4 (12,66). After co-transfection of each transcription factor-Gal4 construct and a Gal4-driven luciferase reporter vector into HEK293 cells, luciferase assays were performed to establish baseline activities for each transcription factor. The PC1-CTT was then co-transfected and its effect on each transcription factor’s activity was measured as a change in luciferase production as compared with each transcription factor’s respective baseline level. Transfection with the PC1-CTTΔNLS construct, which encodes the PC1-CTT lacking the 20 amino acids that encode its putative nuclear localization sequence (NLS), was also performed. PC1-CTT∆NLS does not enter the nucleus, and thus serves as a negative control for PC1-CTT-associated transcriptional regulatory activity (33). While the majority of the transcription factors that were affected by PC1-CTT expression exhibited an inhibitory effect (12), the activities of a smaller subset of transcription factors were stimulated in the presence of the PC1-CTT. Among these the most dramatically stimulated was TAZ/Wwtr1, which showed a 36-fold increase in activity in cells expressing the PC1-CTT as compared with those expressing the PC1-CTT∆NLS negative control (Supplementary Material, Table S1).
To confirm the results of the initial screen the TAZ-Gal4 fusion construct was co-transfected with either PC1-CTT or PC1-CTT∆NLS into HEK293 cells. Activation of TAZ was measured by Gal4-induced expression of a co-transfected luciferase reporter gene. Expression of the PC1-CTT resulted in a significant increase in TAZ-Gal4 activity, while the PC1-CTT∆NLS construct had no effect on TAZ-Gal4 luciferase activity (Fig. 1A).
Figure 1.
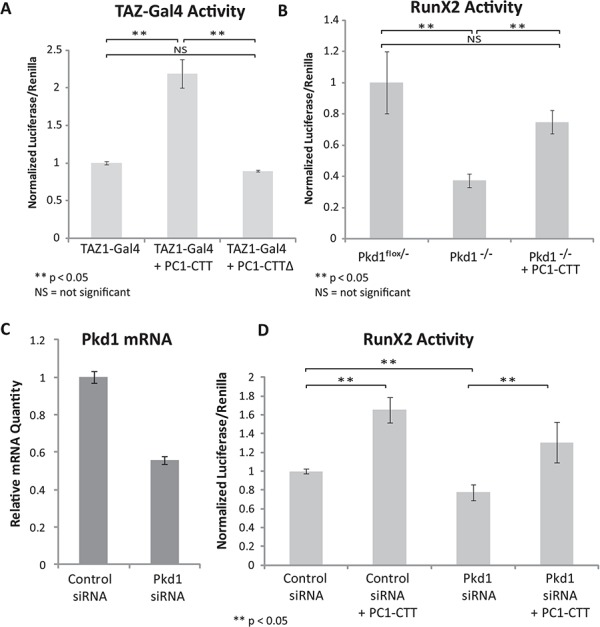
TAZ and RunX2 activities are stimulated by the PC1-CTT. (A) HEK293 cells were transfected with TAZ-Gal4, UAS-Luciferase and Renilla luciferase reporter constructs alone or in the presence of the PC1-CTT or PC1-CTT∆NLS and luciferase activity was measured 24 h later. (B) Pkd1flox/- and Pkd1-/- cells stably expressing HA-PC1-CTT in a TET-Off inducible vector were transfected with RunX2-luciferase and Renilla reporter constructs in the presence or absence of doxycyclin to induce expression of the PC1-CTT and luciferase activity was measured 24 h later. (C) C3H10T1/2 cells were reverse transfected (2× serial transfections) with either siControl (non-targeting RNA) or siRNA directed against mouse Pkd1. mRNA knockdown efficiency was assessed by qRT-PCR after 72 hrs. (D) C3H10T1/2 cells were first reverse-transfected with either siControl or siPkd1 RNAi, then super-transfected with RunX2-luciferase and Renilla-luciferase alone or in the presence of the PC1-CTT. Luciferase values were measured 24 h after RunX2-lucferase transfection (72 h total after initial siRNA treatment). Results are expressed as mean ± standard error (SE) from nine biological replicates of three independent experiments.
PC1 stimulates RunX2 activity via its CTT
To measure the effects of full length PC1 protein expression on RunX2 activation, Pkd1flox/- and Pkd1-/- cells were transfected with a RunX2 reporter construct containing six repeats of the RunX2-binding element found upstream of the osteocalcin promoter region, which drives expression of a luciferase reporter gene. The Pkd1-/- cells express no PC1 protein, whereas the Pkd1flox/- cells possess a functional Pkd1 allele and express functional PC1 protein (67). The Pkd1-/- cells exhibited significantly less RunX2 reporter activity than did the Pkd1flox/- cells. Expression of the PC1-CTT in the Pkd1-/- cells partially restored RunX2 reporter activity to levels similar to those observed in the Pkd1flox/- cells (Fig. 1B). These data suggest that the CTT of PC1 is sufficient to mediate the PC1 protein’s stimulatory effects on RunX2 activity.
We next examined the effects of PC1 and PC1-CTT expression in mesenchymal stem cells (C3H10T1/2) (68), which are capable of undergoing differentiation and commitment to osteoblast or adipocyte cell fates (69). C3H10T1/2 cells were sequentially transfected (2x) with short interfering RNA (siRNA) directed against Pkd1, achieving ∼50% knockdown of Pkd1 mRNA as determined by real-time quantitative reverse transcription PCR (qRT-PCR) (Fig. 1C). This partial knockdown of Pkd1 expression in C3H10T1/2 cells resulted in a modest but significant decrease in RunX2 activity as compared with that measured in cells treated with scrambled siRNA. Expression of the PC1-CTT stimulated RunX2 activity in siControl cells, and partially restored RunX2 activity in Pkd1 knockdown cells (Fig. 1D).
TAZ is required for PC1-CTT mediated RunX2 activation
TAZ activity has been extensively characterized in the regulation of bone growth and development, during which it is responsible for directing pluripotent mesenchymal stem cells toward the osteoblastic lineage (via stimulation of RunX2) and away from the adipocyte cell lineage (via inhibition of PPARγ). Thus, we next set out to investigate the relationship between TAZ and the PC1-CTT in the context of RunX2 activation. Overexpression of FLAG-RunX2 was used to validate the efficacy of the RunX2 reporter construct in the C3H10T1/2 cells and, as expected, it produced a significant increase in the luciferase signal (Fig. 2A, right panel). Expression of the PC1-CTT stimulates RunX2 activity, while the PC1-CTT∆NLS construct has no effect. Consistent with reports in the literature, TAZ expression increases RunX2 activity, while co-expression of TAZ with the PC1-CTT resulted in a further stimulation of in RunX2 activity (Fig. 2A, left panel).
Figure 2.
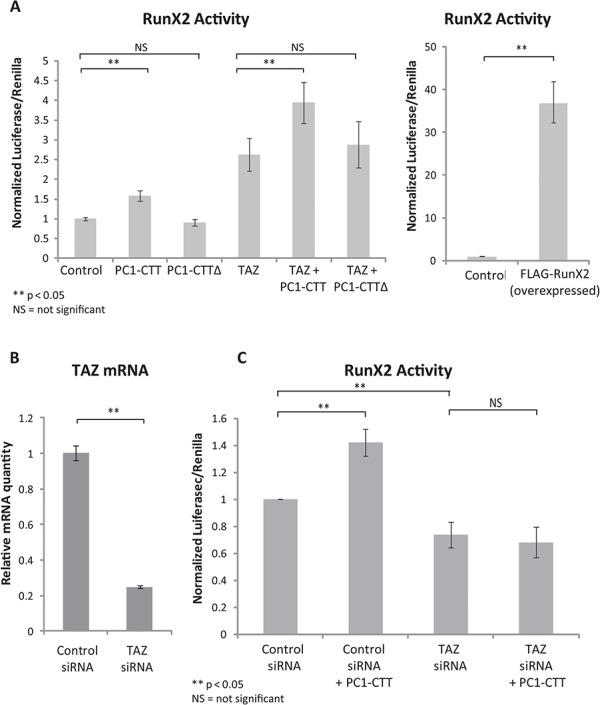
TAZ is required for PC1-CTT mediated stimulation of RunX2. (A) C3H10T1/2 cells were transfected with RunX2-luciferase, PC1-CTT, PC1-CTT∆NLS (negative control) and TAZ; luciferase activity was measured after 24 h. FLAG-RunX2 was over-expressed as a positive control for the RunX2-luciferase reporter (right panel). (B) C3H10T1/2 cells were reverse-transfected (2x serial transfections) with either siControl (non-targeting RNA) or siRNA directed against mouse TAZ. mRNA knockdown efficiency was assessed by qRT-PCR after 72 h. (C) C3H10T1/2 cells were first reverse-transfected with either siControl or siTAZ RNAi, then super-transfected with RunX2-luciferase and Renilla alone or in the presence of the PC1-CTT. Luciferase values were measured 24 hrs after RunX2-lucferase transfection (72 h total after initial siRNA treatment). Results are expressed as mean ± SE from nine biological replicates of 3 independent experiments.
To assess whether there is a requirement for TAZ in PC1-CTT mediated RunX2 activation, C3H10T1/2 cells were sequentially transfected (2x) with siRNA directed against TAZ. Approximately 80% knockdown of TAZ mRNA was obtained, as determined by qRT-PCR (Fig. 2B). RunX2 activity was decreased in C3H10T1/2 cells receiving siRNA directed against TAZ, as compared with that measured in siControl cells. While expression of the PC1-CTT stimulated RunX2 activity in siControl cells, the PC1-CTT displayed no stimulatory effect on RunX2 activity in C3H10T1/2 cells treated with siRNA against TAZ (Fig. 2C). These data implicate TAZ as the critical mechanistic link required for PC1-mediated RunX2 stimulation.
The PC1-CTT physically interacts with TAZ
To determine if the PC1-CTT could exert its regulatory effects on TAZ through a physical interaction, we performed co-immunoprecipitation experiments between FLAG-TAZ or FLAG-YAP constructs and hemagglutinin (HA)-tagged PC1-CTT (HA-PC1CTT) expressed in HEK293 cells. FLAG immunoprecipitation (FLAG-IP) demonstrated a clear association between the PC1-CTT and TAZ (Fig. 3A), but not with YAP, which shares significant sequence homology with TAZ. Using HA-conjugated beads to immunoprecipitate the PC1-CTT, we were able to confirm the TAZ-PC1-CTT interaction (Fig. 3B). To further refine the interaction site between TAZ and the PC1-CTT we generated a construct containing only the final 91 amino acids of PC1, which includes the coiled-coil domain (p91). Glutathione S-transferases-p91 (GST-p91) was produced in BL21 bacteria, purified on glutathione 4B beads and exposed to lysate from HEK293 cells expressing FLAG-TAZ. Elution of the washed beads revealed GST-p91 in a complex with FLAG-TAZ, suggesting that the final 91 amino acids of PC1 are sufficient to mediate its interaction with TAZ (Fig. 3C).
Figure 3.
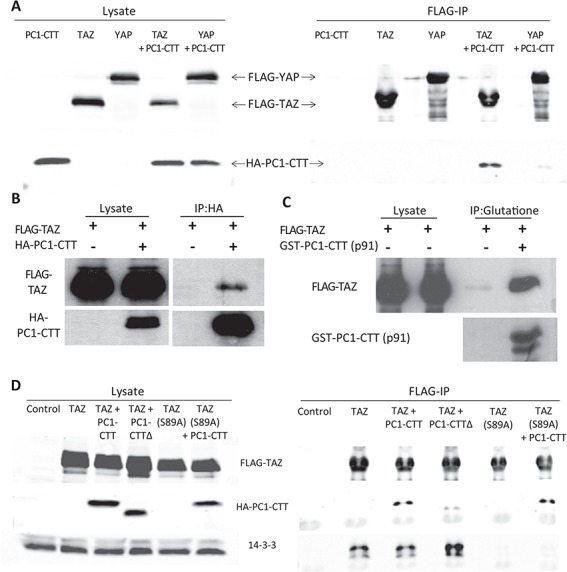
PC1-CTT binds to TAZ without disrupting the TAZ-14-3-3 interaction. (A) HEK293 cells were co-transfected with FLAG-TAZ or FLAG-YAP and HA-PC1-CTT. Cell lysates were subjected to immunoprecipitation using anti-FLAG sepharose, then blotted with the indicated antibodies. (B) HEK293 cells were co-transfected with FLAG-TAZ and HA-PC1-CTT. Cell lysates were subjected to immunoprecipitation using anti-HA sepharose, then blotted with the indicated antibodies. (C) A GST-tagged construct containing the C-terminal 91 amino acids of the PC1-CTT (p91) was produced in BL21 bacteria and purified on glutathione-sepharose 4B beads. The GST-p91 coated glutathione beads were then exposed to lysates from HEK293 cells expressing FLAG-TAZ and the resulting complexes were blotted with the indicated antibodies. (D) HEK293 cells were co-transfected with FLAG-TAZ(WT) or FLAG-TAZ(S89A) and HA-PC1-CTT. Cell lysates were subjected to immunoprecipitation using anti-FLAG sepharose, then blotted with the indicated antibodies.
The PC1-CTT does not change exogenous TAZ protein expression levels, 14-3-3 binding or nuclear localization
TAZ activity is regulated by phosphorylation at a number of sites that influence its stability and subcellular localization (44). Phosphorylation by Casein Kinase 1 at Ser314 targets TAZ for interaction with the SCF E3 ubiquitin ligase and degradation. TAZ phosphorylated at Ser89 interacts with 14-3-3, which prevents it from being translocated into the nucleus and results in its accumulation in the cytosol. We wondered whether association with the PC1-CTT might enhance the activity of TAZ by blocking its interaction with 14-3-3, by preventing its degradation or by directly inducing the nuclear translocation of TAZ through the activity of the PC1-CTT protein’s intrinsic NLS. To address these possibilities, HEK293 cells were transfected with FLAG-TAZ alone or together with HA-PC1-CTT. Expressed alone, TAZ demonstrates a robust interaction with 14-3-3 (Fig. 3D) and exhibits a primarily cytoplasmic localization (Fig. 4), in agreement with published reports (44). Co-expression of TAZ with the PC1-CTT did not increase levels of exogenous TAZ protein expression (Fig. 3D). Furthermore, PC1 co-expression did not result in a significant change in TAZ binding to 14-3-3, nor did it cause TAZ to redistribute into the nucleus (Figs 3D and 4). In addition, endogenous RunX2 levels were not altered by the expression of the HA-PC1-CTT, of TAZ or of both of these proteins together (Supplementary Material, Fig.1). Mutation of TAZ serine 89 to alanine results in the production of a dominant-active TAZ(S89A) construct, which is predominantly located in the nucleus (44). TAZ(S89A) shows no interaction with 14-3-3 but binds to the PC1-CTT at similar levels as does the WT TAZ (Fig. 3D). TAZ(S89A) expression simulates a 1.5-fold increase in RunX2-Luc activity, consistent with its nuclear localization. Co-expression of TAZ(S89A) with the PC1-CTT results in a further increase of 2.3-fold stimulation of RunX2 activity (Supplementary Material, Fig.2). TAZ(S89A) also interacts with the PC1-CTTΔNLS protein, although apparently at a somewhat lower level than it does with PC1-CTT (Fig. 3D). Interestingly, expression of TAZ(S89A) together with the construct encoding the PC1-CTT∆NLS protein resulted in the accumulation within the nucleus of the of the usually non-nuclear PC1-CTT∆NLS protein (Supplementary Material, Fig.3), providing additional evidence for the existence and the functional significance of the interaction between TAZ and the PC1 CTT. Taken together, these data suggest that the PC1-CTT regulates nuclear TAZ activity by a mechanism that does not involve enhancing its expression or inducing the redistribution of TAZ to the nucleus.
Figure 4.

Expression of the PC1-CTT does not affect TAZ nuclear localization. HEK293 cells were transfected with FLAG-TAZ(WT) and FLAG-TAZ(S89A) alone or co-transfected with HA-PC1-CTT. The cells were fixed and stained with anti-FLAG (green) and anti-HA (red) antibodies and with DAPI (blue) to reveal the locations of nuclei.
The transcriptional co-regulatory molecule p300 is required for the stimulatory effects of the PC1-CTT on RunX2 activity
The PC1-CTT does not appear to stimulate TAZ activity by enhancing its nuclear entry. The ubiquitously expressed transcriptional co-regulatory protein p300 has been reported to interact with both TAZ and RunX2, and is essential for RunX2 transcriptional activation (70–72). Thus, p300 interaction with the TAZ-RunX2 complex may represent a potential point of regulation of TAZ by the PC1-CTT that could alter TAZ activity rather than induce nuclear redistribution of TAZ. To test this hypothesis we knocked down p300 using shRNA with an efficiency of approximately 50% as determined by qRT-PCR analysis (Fig. 5A). RunX2-luciferase activity was significantly reduced in cells receiving shRNA against p300, when compared with cells receiving control shRNA. Expression of the PC1-CTT stimulated RunX2 activity in shControl cells. This stimulatory effect, however, was completely abolished in cells with reduced levels of p300 (Fig. 5B).
Figure 5.
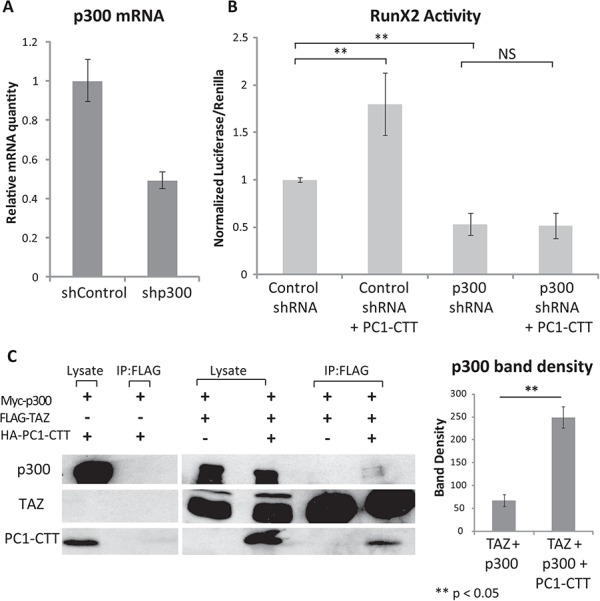
PC1-CTT activation of RunX2 requires p300, and PC1-CTT enhances the TAZ-p300 association (A) HEK293 cells were transfected with either control shRNA (shControl) or shRNA directed against human p300 (shp300). mRNA knockdown efficiency was assessed by qRT-PCR. (B) HEK293 cells were transfected with shControl or shp300. After 48h, the cells were super-transfected with RunX2-luciferase and Renilla-luciferase alone, or in the presence of the PC1-CTT. Luciferase values were measured 24 h after RunX2-lucferase transfection (72 h total after initial shRNA treatment). (C) HEK293 cells were transfected with Myc-p300, FLAG-TAZ, and HA-PC1-CTT where indicated. Cell lysates were incubated with anti-FLAG beads, complexes were eluted in SDS-PAGE loading buffer and run on a 10% SDS- polyacrylamide gel and blotted with the indicated antibodies. Densitometry was performed on the immunoprecipitated p300 band using image analysis software (right panel). Results are expressed as mean ± SE from four independent experiments.
Expression of the PC1-CTT increases the extent of the TAZ-RunX2 and the TAZ-p300 interactions
Co-expression of the Abl tyrosine kinase enhances the formation of a stable and functional RunX2-TAZ complex and consequently stimulates osteoblast differentiation (73). We hypothesized that PC1-CTT might produce its stimulation of RunX2 activity through a similar mechanism, resulting in the formation of a larger quantity of the transcriptionally active RunX2/TAZ/p300 complex. To test this possibility, we performed co-immunoprecipitation experiments from cells transfected to express Myc-tagged p300, FLAG- tagged TAZ and HA-tagged PC1-CTT to assess whether PC1-CTT expression enhanced the association of TAZ with p300. We first performed a control experiment which demonstrated that, in the absence of FLAG-TAZ expression, neither the p300 protein nor the PC1-CTT was recovered in an anti-FLAG immunoprecipitation (Fig. 5C, left blot). When FLAG-TAZ was expressed in the absence of the PC1-CTT, little p300 was detected in complex with TAZ. In contrast, expression of the PC1-CTT resulted in the appearance of a faint but detectable band corresponding to p300 in complex with TAZ (Fig. 5C, right blot). Quantification of these results demonstrated a significant increase in the association of p300 with TAZ in the presence of the PC1-CTT (Fig. 5C, right panel). We next assessed the level of complex formation between TAZ and RunX2 in cells that did or did not express the PC1-CTT. Lysates from HEK293 cells co-transfected with RunX2, TAZ and HA-PC1-CTT were subjected to immunoprecipitation with antibodies directed against RunX2 or against the TAZ protein, and the recovered proteins were analyzed by western blot. The quantity of RunX2 that co-precipitated with TAZ (Fig. 6A) and the quantity of TAZ that co-precipitated with RunX2 (Fig. 6B) were both substantially increased in cells that co-expressed the PC-CTT. Overexpression of the PC1-CTT-HA results in an increase in endogenous TAZ expression, which can be observed in the lysate and IP lanes of Figure 6B as a band running slightly below the overexpressed TAZ band. The increased expression of endogenous TAZ in the presence of PC1-CTT likely facilitates immunoprecipitation of RunX2 without the addition of exogenous TAZ (Fig. 6A).
Figure 6.
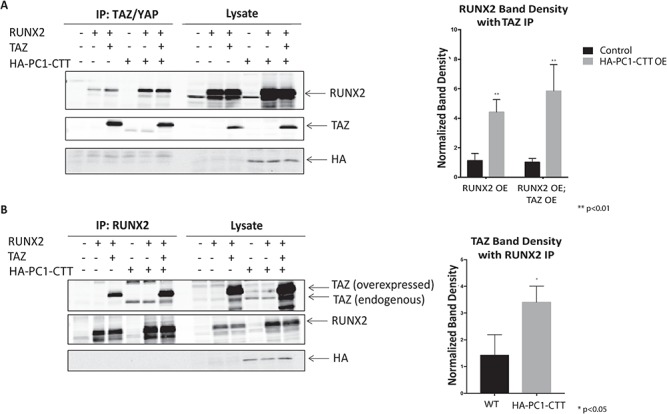
PC1-CTT enhances the TAZ-RunX2 association. (A) HEK293 cells were transfected with RunX2, TAZ and HA-PC1-CTT where indicated. Cell lysates were incubated with anti- TAZ antibody, complexes were eluted in SDS-PAGE loading buffer and run on a 10% SDS- polyacrylamide gel and blotted with the indicated antibodies. Densitometry was performed on the immunoprecipitated RunX2 band using image analysis software (right panel) (OE = Over Expressed). Results are expressed as mean ± SE from four independent experiments. (B) HEK293 cells were transfected with RunX2, TAZ and HA-PC1-CTT where indicated. Cell lysates were incubated with anti-RunX2 antibody, complexes were eluted in SDS-PAGE loading buffer and run on a 10% SDS- polyacrylamide gel and blotted with the indicated antibodies. Densitometry was performed on the immunoprecipitated TAZ band using image analysis software (right panel). Results are expressed as mean ± SE from four independent experiments.
Morpholino-mediated disruption of Pkd1a/b gene expression in zebrafish results in decreased jaw bone mineralization and curly tails, which can be rescued by expression of the PC1-CTT or of constitutively active TAZ
The phenotype of zebrafish injected with morpholinos directed against pkd1a and pkd1b includes a dramatic upward curling of the tail as well as significant skeletal abnormalities, the most prominent of which is a disruption of jaw bone growth and mineralization (74). To determine the capacity of the PC1-CTT to rescue the phenotypes associated with impaired pkd1 gene expression in vivo, zebrafish embryos were injected with pkd1a/b morpholinos alone, or in association with mRNA encoding the PC1-CTT or mRNA encoding dominant active TAZ(S89A). Knockdown of pkd1a/b results in a significant decrease in jaw bone mineralization, as assayed by calcein staining at 7 days post fertilization (Fig. 7A and B). As expected, knockdown of pkd1a/b also produces significant tail curvature (74,75), scored as previously described (12) (Fig. 7C and D). Concurrent injection of the PC1-CTT restored jaw bone mineralization and suppressed tail curvature in the pkd1a/b knockdown fish. Injection of PC1-CTT mRNA was unable to rescue jaw bone mineralization or tail curvature in pkd1a/b-knockdown zebrafish lacking expression of TAZ. Expression of dominant active TAZ(S89A) was sufficient to rescue both bone density and tail curvature in pkd1a/b-knockdown fish (Fig. 7A–D).
Figure 7.
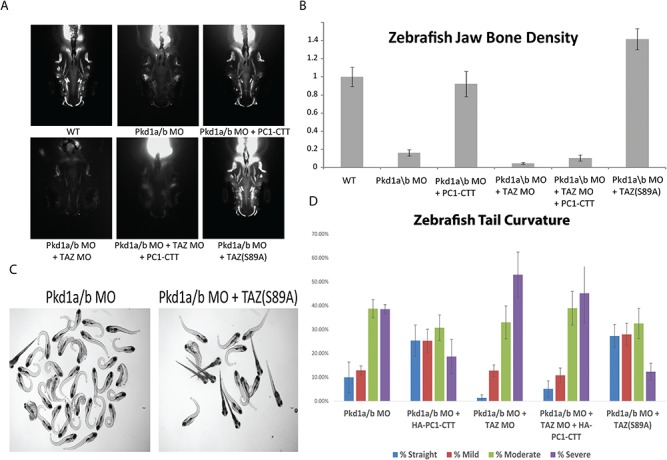
Loss of Pkd1 expression in zebrafish results in decreased jaw bone mineralization and tail curvature, which can be rescued with PC1-CTT or TAZ(S89A) overexpression. (A) Morpholinos corresponding to the zebrafish Pkd1a and Pkd1b PC1 or TAZ genes were injected into zebrafish embryos at the 1-2 cell stage to impair expression of the two Pkd1 genes and/or of TAZ. The embryos were subsequently injected with 300nM of mRNA encoding HA-PC1-CTT or TAZ(S89A), where indicated. At 7dpf the embryos were stained for calcified bone using the fluorescent calcein dye dissolved in deionized water, mounted in agarose and imaged. (B) Bone mineralization was quantified on Image-J using thresholded images. The data represent averages of three independent experiments (n = 40–100 embryos per condition); error bars represent standard error of the mean (SEM). (C and D) Morpholinos corresponding to the zebrafish Pkd1a and Pkd1b PC1 or TAZ genes were injected into zebrafish embryos at the 1–2 cell stage to impair expression of the two Pkd1 genes and/or of TAZ. The embryos were subsequently injected with 300nM of mRNA encoding HA-PC1-CTT or TAZ(S89A), where indicated. Images of embryos injected with Pkd1a/b morpholinos alone or followed by injected of mRNA encoding TAZ(S89A) are presented in Panel C. Tail curvature was scored and the results were binned into straight, mild, moderate and severe categories, as previously described (12). Knockdown of Pkd1a and Pkd1b produce a curly tail phenotype whose severity is partially ameliorated by expression of the PC1-CTT. Loss of TAZ expression prevents the PC1-CTT mediated rescue, but expression of TAZ(S89A) is sufficient to rescue the curly tail phenotype even in the absence of PC1-CTT expression.
Discussion
The PC1 protein influences a wide variety of signaling pathways, but the downstream effectors that connect PC1 expression to these pathways are largely unknown (3,5). A number of studies have suggested that PC1 plays a role in modulating bone formation by regulating the activity of the RunX2 transcription factor, (57,60), that disruption of TAZ expression results in the development of renal cystic disease (63,64) and that the cleaved PC1-CTT interacts with TAZ to promote osteogenesis (62); however, the mechanistic link between PC1 and TAZ had not been fully elucidated. The data presented here demonstrate that the PC1-CTT binds to and activates the transcriptional coactivator TAZ to facilitate PC1-mediated RunX2 activation in the process of bone growth and development. Using a ‘coactivator trap’ screen, we identified the transcriptional co-regulatory protein TAZ as a target of the PC1-CTT. The majority of the transcriptional pathways influenced by the PC1-CTT were inhibited by PC1-CTT expression (12). Among the short list of transcription factors that were activated by the PC1-CTT, TAZ was the most dramatically stimulated. These data indicate that the PC1-CTT is not simply a general transcriptional repressor, but rather selectively represses some targets while activating others. The results of this initial TF-Gal4 screen were confirmed in HEK293 cells, where the PC1-CTT showed a significant stimulatory effect on TAZ-Gal4. Importantly, TAZ-Gal4 activity was unaffected by expression of the PC1-CTT∆NLS, which does not translocate into the nucleus. These data indicate that nuclear localization of the PC1-CTT, or at least the presence of the putative NLS, is essential for its regulatory effects on TAZ activity. Taken together with the results of the coactivator trap screen presented here, these observations suggest the interesting possibility that PC1 and TAZ participate in a common physiological pathway.
To assess the activity of the PC1-CTT on RunX2 in the context of bone development, we utilized a mouse mesenchymal stem cell line (C3H10T1/2). These cells have the potential to differentiate into the bone lineage (chondrocytes and osteoblasts) or the fat lineage (adipocytes) (68). This developmental fate choice is controlled by TAZ, which directs the cells away from the adipocyte lineage (via inhibition of PPARγ) and toward the chondrocyte/osteoblast lineage (via co-stimulation of RunX2 target genes). Thus, these cells represent a physiologically relevant model in which to assess the regulatory effects of the PC1-CTT on TAZ and RunX2 activity. Furthermore, we employed a RunX2-luciferase reporter construct that facilitated measurement of the activity of endogenous RunX2. Using this reporter construct we observed a decrease in RunX2 activity upon siRNA-mediated knockdown of Pkd1 in the C3H10T1/2 mesenchymal cell line, which is further substantiated by the recent report demonstrating that endogenously cleaved PC1-CTT, released from full-length membrane-associated PC1, regulates TAZ/RunX2 activity to stimulate osteogenesis and inhibit adipogenesis in C3H10T1/2 cells (62). This supports the hypothesis that PC1 plays a crucial role in the process through which RunX2 influences mesenchymal stem cell fate determination, driving them to pursue the chondrocyte and osteoblast lineages.
Using the RunX2-luciferase reporter system in C3H10T1/2 cells, we found that expression of either TAZ or the PC1-CTT alone each induces an increase in endogenous RunX2 activity, and that their co-expression results in a further increase in RunX2 activity. These data are consistent with the possibility that the PC1-CTT may act upstream of TAZ or alternatively that the two proteins could work cooperatively to activate RunX2. To discriminate among these possibilities we performed siRNA-mediated knockdown of TAZ, which abrogated the stimulatory effects of the PC1-CTT on RunX2. This observation indicates that the PC1-CTT requires TAZ for RunX2 activation, and thus that the two proteins are operating sequentially in the same signaling pathway.
The PC1-CTT may regulate TAZ via direct binding, as evidenced by our demonstration of a physical complex that involves TAZ and the PC1-CTT. We further narrowed the interacting domain to the last 91 amino acids of PC1, which contains its coiled-coil domain. While the last 91 amino acid residues of PC1 are sufficient to interact with TAZ, they lack the NLS domain that we found to be required in order for the PC1-CTT to exert its influence on TAZ activity. This observation is consistent with the interesting possibility that the PC1-CTT must be present in the nucleus in order for it to modulate TAZ function. Alternatively, the NLS may affect intra-molecular interactions within the structure of the PC1-CTT such that, in its absence, the interaction motif that is present in p91 is unavailable for assembly with TAZ. It is interesting to note that we do not detect any evidence that the PC1-CTT protein expressed in HEK293 cells is further processed to produce the 17 kDa fragment of the PC1 protein (35,37). It remains to be determined whether there is any sort of precursor–product relationship between the CTT fragment of PC1 and the smaller 17 kDa fragment of PC1. It is quite possible 17 kDa fragment is not the product of further cleavage of the CTT protein, but rather that both the CTT and the 17 kDa fragment are produced by cleavage directly from the full length PC1 protein. Since 17 kDa fragment should contain the 91 amino acid coiled-coil domain sequence, it will be interesting to assess in future studies whether the 17 kDa protein is capable of interacting with TAZ and, if so, whether this interaction is dependent upon its localization to the nucleus.
The PC1-CTT-HA clearly associates with FLAG-TAZ following immunoprecipitation using anti-FLAG; however, this interaction was not detected with antibodies directed against endogenous TAZ. This may be due to relatively high antibody availabity of the N-terminal FLAG tag versus a possible physical obstruction of the endogenous epitope through the formation of a TAZ-containing multi-protein complex. We have presented evidence that the PC1-CTT significantly increases the association between TAZ and RunX2; however, it is entirely possible that this final protein assembly does not include the PC1-CTT. Rather it may be that the influence of the PC1-CTT on RunX2-TAZ complex formation may be due to its effects on the expression levels and conformation of TAZ and RunX2.
The PC1-CTT robustly associates with TAZ but not with its close homolog YAP, highlighting the specificity of the PC1-CTT-TAZ interaction, and suggesting that the protein domains unique to TAZ may be involved in PC1-CTT binding. In this context it is worth noting that, while YAP and TAZ share overlapping transcriptional targets and regulatory mechanisms, their physiological and pathophysiological properties are clearly not identical. This fact is well illustrated by the renal phenotypes observed in TAZ and YAP knockout mice. Whereas TAZ knockout results in polycystic kidneys (63,64), YAP knockout interferes with nephron formation and morphogenesis (76). Furthermore, YAP activity, as revealed by immunocytochemical detection of nuclear YAP protein, appears to be elevated in a mouse model of ADPKD. These observations suggest the hypothesis that TAZ and YAP may play opposing roles in the processes that lead to renal cyst formation and that the PC1-CTT may serve as a critical factor in determining the relative magnitudes of their respective influences.
In the Hippo signaling pathway the activity of TAZ is regulated by a phosphorylation event that determines this protein’s subcellular distribution. Phosphorylation of serine 89 on TAZ promotes its association with 14-3-3, leading to its cytoplasmic retention (44). The stimulatory effect of the PC1-CTT on RunX2 activity might thus be attributable to promotion of TAZ nuclear import, bringing it into contact with RunX2. To address this hypothesis we examined the localization of TAZ in the presence and absence of the PC1-CTT and found that expression of the PC1-CTT did not cause a dramatic redistribution of TAZ into the nucleus. Furthermore, the PC1-CTT was capable of further stimulating RunX2 activity in the presence of TAZ(S89A). These data suggest that the PC1-CTT stimulates TAZ activity via interactions in the nucleus, rather than by promoting its nuclear redistribution.
The transcriptional activity of RunX2 on its target genes is dependent upon its formation of a complex both with TAZ and with the transcriptional coactivator p300 (48,70). Both TAZ and RunX2 have been shown to bind directly to p300 (71,72). The data presented here show that PC1-CTT-mediated stimulation of RunX2 is abolished upon knockdown of p300. Furthermore, we found that expression of the PC1-CTT enhances the interaction of TAZ both with p300 and with RunX2, suggesting that the PC1-CTT activates RunX2 by enhancing its ability interact with TAZ and, in turn, to recruit p300.
Studies in zebrafish (74) reveal that PC1 expression plays an important role in bone development and mineralization in this animal model. We took advantage of this system to test whether the PC1-CTT is capable of playing an important role in bone growth and mineralization in vivo. Expression of the PC1-CTT is sufficient to rescue both the jaw bone mineralization defect and the curly tail phenotype produced by morpholino-mediated of knockdown of pkd1a/b expression in zebrafish (74,75). Furthermore, both of these rescue effects of PC1-CTT expression are dependent upon the endogenous expression of TAZ and both effects can be recapitulated in the absence of the PC1-CTT by expression of the S89A constitutively active form of TAZ. These data indicate that the PC1-CTT is capable of recapitulating significant components of the activity of full-length PC1 in an in vivo system, highlighting the physiological significance of the PC1-CTT in a living organism.
Previous studies have established the importance of PC1 in RunX2 signaling, but have not illustrated a direct link between the two proteins. Here, we have shown that the PC1-CTT associates with and enhances the activity of TAZ, which in turn stimulates RunX2, possibly via enhanced recruitment of p300. Thus, the evidence presented in this study constitutes a compelling mechanism that accounts for activation of RunX2 by PC1 in the process of bone growth and development. Furthermore, these findings are consistent with the conclusion that TAZ serves as an obligate downstream component of the PC1 protein’s effector machinery.
Materials and Methods
Antibodies, plasmids and cell lines
The following antibodies and labeling reagents were used: anti-HA antibody, anti-FLAG and anti-cMyc (Sigma, St. Louis, MO), anti-GST (Amersham Biosciences, Piscataway, NJ), anti-YAP/TAZ (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-14-3-3, anti-TAZ and anti-RunX2 (Cell Signaling, Danvers, MA). For laser-scanning confocal fluorescence microscopy, dye-coupled Alexa antibodies (Alexa-488, 594; Molecular Probes, Eugene OR) were used as secondary reagents.
The sequence encoding the final 200 amino acids of human PC1 (4102–4302), containing a 2x HA tag at the N-terminus, was cloned into the pcDNA3.1 zeo vector. The sequence for human PC1-CTT (residues 4102-4302 of Pkd1) was modified by deleting residues 4134–4154, corresponding to the putative NLS to generate the PC1-CTTΔNLS (33). Stable cell lines were generated by transfection using Lipofectamine 2000 (Invitrogen, Waltham, MA) and selection with 350 μg ml−1 zeocin (Invitrogen, Waltham, MA). Constructs encoding 3xFLAG-TAZ(WT), 3xFLAG-TAZ(S89A) and 3xFLAG-YAP were purchased from addgene (ID# 24809, #24815, #19045). RunX2-Luciferase (6OSE2-Luc) was a generous gift from Dr Roland Baron and Dr Eric Hesse, Harvard School of Dental Medicine. pRL-TK, a vector constitutively expressing Renilla luciferase, was included as an internal control to normalize for transfection differences. The sequence encoding human p300 (FL or AA 1-664) was cloned from pCMVβ (Upstate Biotechnology, Lake Placid, NY) into the pCMV-Tag 3B vector (Stratagene, La Jolla, California) to generate Myc-p300. HEK293 cells (33), LLC-PK1 cells and Pkd1flox/- and Pkd1-/- TSLargeT renal proximal tubule cells (67,77) were maintained as described. C3H10T1/2 mesenchymal stem cells were a generous gift from Dr Roland Baron and Dr Eric Hesse, and were maintained in α-MEM.
Coactivator trap screen
The coactivator trap screen was performed as previously described (66). A pcDNA3.1 construct expressing HA-PC1-CTT was co-transfected with each of 837 transcription factor-Gal4 fusion proteins and the GAL4 luciferase and Renilla reporter plasmids into HEK293 cells in a 384-well plate. Cells were cultured for 24 h in a humidified incubator at 37°C in 5% CO2. BrightGlo (Promega, Madison, WI) reagent (35 μl) was added to each well, and luciferase luminescence was measured with an Acquest plate reader (LJL Biosystems, Sunnyvale, CA).
Transient transfection and luciferase assay
C3H10T1/2 cells, HEK293 cells, Pkd1flox/- and Pkd1-/- cells were reverse transfected using Lipofectamine 2000 (Invitrogen, Waltham, MA) and added to a 24-well plate to achieve 80–100% confluency after 24 h. 6OSE2-Luc vector (RunX2-Luciferase) and pRL-TK (Renilla) were mixed with 2 uL Lipofectamine and transfection mixtures were added drop wise to cell culture media and mixed, after which the appropriate number of cells were added to each well and incubated at 37°C for 24 h. The amount of DNA in each well was equalized through the addition of a control plasmid, pcDNA3.1, which was also used for mock transfection. Transfected cells were harvested with PBS and lysed with 100 μL of passive lysis buffer (Promega, Madison, WI). Luciferase levels were assayed using the Dual Luciferase Assay Reagent kit (Promega, Madison, WI). Luciferase signals were determined in a GloMaxTM 20/20 luminometer (Promega, Madison, WI).
siRNA and shRNA treatment
C3H10T1/2 cells or HEK293 cells were transfected in 24-well plates with 100 nM target-specific siRNA, with control siRNA or with 0.8ug shRNA using Lipofectamine 2000. Control siRNA: Silencer Negative Control siRNA#1 (#AM4611, Ambion, Invitrogen, Waltham, MA), siWwtr1 (#4390771 ID#s206974, Ambion, Invitrogen, Waltham, MA), siPkd1 (#4390771 ID#s717171, Ambion, Invitrogen, Waltham, MA). Oligonucleotides corresponding to p300 were cloned into the pSUPER vector as previously described (78).
Immunoprecipitation, immunoblot and GST pulldown
Cells were lysed by probe sonication for 3× 10 s bursts at 40% intensity in 25 mm Tris pH 7.4, 150 mm NaCl, 1% NP-40, 1 mm Ethylenediaminetetraacetic acid (EDTA) with protease inhibitors (Roche, Branchburg, NJ). Precleared lysates (18 000g, 30 min) were incubated at 4°C overnight with either monoclonal-anti-FLAG-M2 agarose (Sigma, St. Louis, MO) or glutathione-sepharose 4B beads (Amersham Biosciences, Piscataway, NJ) pre-bound with the indicated GST fusion protein constructs harvested from BL21 bacteria by standard procedures (Stratagene, La Jolla, California). Beads were collected by centrifugation and the pellets were washed in lysis buffer 4 × for 10 min with rotation at 4°C. Immunoprecipitates were eluted in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (25 mm TrisHCL pH 6.7, 10% glycerol, 1% SDS, 50 mm DTT, bromophenol blue). For the RunX2/TAZ co-immunoprecipitation experiments, cells were lysed by probe sonication for 3× 4 seconds bursts at 40% intensity in 50 mm Tris pH 7.5, 100 mm NaCl, 1% Triton X-100, 1 mm EDTA with protease inhibitors (Roche). Lysates were incubated at 4°C overnight with either monoclonal anti-RUNX2 (Cell Signaling, Danvers, MA) and protein A agarose beads (Thermo Scientific, Waltham, MA), or monoclonal anti-YAP/TAZ (Santa Cruz Biotechnology, Santa Cruz, CA) and protein G agarose beads (Thermo Scientific, Waltham, MA). Beads were collected by centrifugation and the pellets were washed in lysis buffer 5× for 5 min with rotation at 4°C. Immunoprecipitates were eluted in SDS-PAGE loading buffer (25 mm Tris–HCL pH 6.7, 10% glycerol, 1% SDS, 50 mm DTT, bromophenol blue).
Proteins were separated on a 10–12% SDS-polyacrylamide gel and then electrophoretically transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA), incubated in blocking buffer (150 mm NaCl, 20 mm Tris, 5% (w/v) powdered milk, 0.1% Tween) for 60 min, and then incubated with one of the following primary antibodies at 4°C overnight: monoclonal anti-HA (Rat) antibody (1:5000, Roche), polyclonal anti-FLAG (1:5000, Sigma, St. Louis, MO), polyclonal anti-cMyc (1:5000, Sigma, St. Louis, MO), polyclonal anti-GST (Goat) (1:10 000 Amersham Biosciences, Piscataway, NJ) or other antibodies as specified in the text. Subsequently, primary antibody binding was detected using infrared (IR)-conjugated (1:10 000−1:15 000; Li-Cor Biosciences, Lincoln, Nebraska) or horseradish peroxidase-conjugated secondary antibodies (1:5 000–10:000; Jackson Labs, Bar Harbor, ME). Membranes blotted with IR-conjugated secondaries were visualized on an Odyssey Infrared Imager (Li-Cor Biosciences, Lincoln, Nebraska) and quantitated using the associated software package; horseradish peroxidase (HRP)-blotted membranes were visualized with an enhanced chemiluminescence detection kit (ECL, Amersham Biosciences, Piscataway, NJ) and quantitated using ImageJ software.
Zebrafish morpholino antisense oligonucleotide injection, mRNA preparation/injections and calcein staining
Morpholino-induced knockdown of pkd1a, pkd1b and TAZ expression was performed as previously reported (48,74). Wild-type embryos at the 1- to 2-cell stage were microinjected with 4.6 nl of a 0.15 mm antisense morpholino oligonucleotide dissolved in 1x Daneau buffer (58 mm NaCl, 0.7 mm KCl, 0.4 mm MgSO4, 0.6 mm Ca(NO3)2, 5 mm HEPES pH 7.6) (Gene Tools LLC, Philomath, OR) with 0.1% Phenol Red using a nanoject2000 microinjector (World Precision Instruments, Sarasota, FL). The sequences of the morpholinos targeting pkd1a and pkd1b were identical to those that have been previously described (74); briefly, the splice donor-blocking oligonucleotide sequences were pkd1a MO ex8: 5′-GATCTGAGGACTCACTGTGTGATTT-3′; pkd1b MO ex45: 5′-ACATGATATTTGTACCTCTTTGGTT-3′. Gene Tools standard negative control morpholino was used as an injection control and demonstrated no effect on development. mRNA encoding 2xHA-PC1-CTT, and TAZ(S89A) were transcribed, using the mMESSAGE mMACHINE Kit (Invitrogen, Waltham, MA), from the corresponding genes inserted into the pCS2 vector.
Calcein staining was performed according to Du et al. (79). Briefly, embryos at 7dpf were stained by immersion in a 0.2% solution of calcein in deionized water for 10 min, followed by 6× serial washes in water. Embryos were then mounted in acrylamide and imaged using an Olympus BX51 epifluorescent microscope equipped with a 10× objective. Images of the calcein stained bones were background subtracted, thresholded, and the integrated pixel density of the jaw bones was calculated using ImageJ software.
Statistical analysis
Results are expressed as means ± SE. Differences between means were evaluated using Student’s t test or analysis of variance as appropriate. Values of P < 0.05 were considered to be significant.
Supplementary Material
Acknowledgements
We would like to thank Dr Roland Baron for the generous gift of the 6OSE2-Luc construct and C3H10T1/2 cells, Lu Zhou for expertise and assistance with zebrafish embryo microinjection, Stefan Somlo for the Pkd1flox/- and Pkd1-/- mouse kidney epithelial cell lines and members of the Caplan lab for discussion and advice.
Conflict of Interest statement. None declared.
Funding
National Institute of Diabetes and Digestive and Kidney Diseases (F30DK083227-02 to D.M.M., DK57328, DK090744 to M.J.C., Z.S., S.S.); Department of Defense Peer Reviewed Medical Research Program (W81XWH-10-1-0504 to M.J.C.).
References
- 1. Harris P.C., Germino G., Klinger K., Landes G. and Adelsberg J. (1995) The PKD1 gene product. Nat. Med., 1, 493. [DOI] [PubMed] [Google Scholar]
- 2. Nims N., Vassmer D. and Maser R.L. (2003) Transmembrane domain analysis of polycystin-1, the product of the polycystic kidney disease-1 (PKD1) gene: evidence for 11 membrane-spanning domains. Biochemistry, 42, 13035–13048. [DOI] [PubMed] [Google Scholar]
- 3. Chapin H.C. and Caplan M.J. (2010) The cell biology of polycystic kidney disease. J. Cell Biol., 191, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takiar V. and Caplan M.J. (2011) Polycystic kidney disease: Pathogenesis and potential therapies. Biochim. Biophys. Acta, 1812, 1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harris P.C. and Torres V.E. (2014) Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Invest., 124, 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Padovano V., Kuo I.Y., Stavola L.K., Aerni H.R., Flaherty B.J., Chapin H.C., Ma M., Somlo S., Boletta A., Ehrlich B.E. et al. (2017) The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol. Biol. Cell, 28, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arnould T., Kim E., Tsiokas L., Jochimsen F., Gruning W., Chang J.D. and Walz G. (1998) The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J. Biol. Chem., 273, 6013–6018. [DOI] [PubMed] [Google Scholar]
- 8. Bhunia A.K., Piontek K., Boletta A., Liu L., Qian F., Xu P.N., Germino F.J. and Germino G.G. (2002) PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell, 109, 157–168. [DOI] [PubMed] [Google Scholar]
- 9. Kim E., Arnould T., Sellin L.K., Benzing T., Fan M.J., Gruning W., Sokol S.Y., Drummond I. and Walz G. (1999) The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem., 274, 4947–4953. [DOI] [PubMed] [Google Scholar]
- 10. Lal M., Song X., Pluznick J., Di Giovanni V., Merrick D., Rosenblum N., Chauvet V., Gottardi C., Pei Y. and Caplan M. (2008) Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum. Mol. Genet., 17, 3105–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le N.H., Bent P., Huls G., Wetering M., Loghman-Adham M., Ong A.C., Calvet J.P., Clevers H., Breuning M.H., Dam H. et al. (2004) Aberrant polycystin-1 expression results in modification of activator protein-1 activity, whereas Wnt signaling remains unaffected. J. Biol. Chem., 279, 27472–27481. [DOI] [PubMed] [Google Scholar]
- 12. Merrick D., Chapin H., Baggs J.E., Yu Z., Somlo S., Sun Z., Hogenesch J.B. and Caplan M.J. (2012) The γ-secretase cleavage product of polycystin-1 regulates TCF and CHOP-mediated transcriptional activation through a p300-dependent mechanism. Dev. Cell, 22, 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parnell S.C., Magenheimer B.S., Maser R.L., Zien C.A., Frischauf A.M. and Calvet J.P. (2002) Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J. Biol. Chem., 277, 19566–19572. [DOI] [PubMed] [Google Scholar]
- 14. Puri S., Magenheimer B.S., Maser R.L., Ryan E.M., Zien C.A., Walker D.D., Wallace D.P., Hempson S.J. and Calvet J.P. (2004) Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J. Biol. Chem., 279, 55455–55464. [DOI] [PubMed] [Google Scholar]
- 15. Mochizuki T., Wu G., Hayashi T., Xenophontos S.L., Veldhuisen B., Saris J.J., Reynolds D.M., Cai Y., Gabow P.A., Pierides A. et al. (1996) PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science, 272, 1339–1342. [DOI] [PubMed] [Google Scholar]
- 16. Cai Y., Maeda Y., Cedzich A., Torres V.E., Wu G., Hayashi T., Mochizuki T., Park J.H., Witzgall R. and Somlo S. (1999) Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem., 274, 28557–28565. [DOI] [PubMed] [Google Scholar]
- 17. Gonzalez-Perrett S., Kim K., Ibarra C., Damiano A.E., Zotta E., Batelli M., Harris P.C., Reisin I.L., Arnaout M.A. and Cantiello H.F. (2001) Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc. Natl. Acad. Sci. U.S.A., 98, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsiokas L., Arnould T., Zhu C., Kim E., Walz G. and Sukhatme V.P. (1999) Specific association of the gene product of PKD2 with the TRPC1 channel. Proc. Natl. Acad. Sci. U.S.A., 96, 3934–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nauli S.M., Alenghat F.J., Luo Y., Williams E., Vassilev P., Li X., Elia A.E., Lu W., Brown E.M., Quinn S.J. et al. (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet., 33, 129–137. [DOI] [PubMed] [Google Scholar]
- 20. Koulen P., Cai Y., Geng L., Maeda Y., Nishimura S., Witzgall R., Ehrlich B.E. and Somlo S. (2002) Polycystin-2 is an intracellular calcium release channel. Nat. Cell. Biol., 4, 191–197. [DOI] [PubMed] [Google Scholar]
- 21. Qian F., Germino F.J., Cai Y., Zhang X., Somlo S. and Germino G.G. (1997) PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet., 16, 179–183. [DOI] [PubMed] [Google Scholar]
- 22. Tsiokas L., Kim E., Arnould T., Sukhatme V.P. and Walz G. (1997) Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. U.S.A., 94, 6965–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Delmas P., Nomura H., Li X., Lakkis M., Luo Y., Segal Y., Fernandez-Fernandez J.M., Harris P., Frischauf A.M., Brown D.A. et al. (2002) Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J. Biol. Chem., 277, 11276–11283. [DOI] [PubMed] [Google Scholar]
- 24. Arnould T., Sellin L., Benzing T., Tsiokas L., Cohen H.T., Kim E. and Walz G. (1999) Cellular activation triggered by the autosomal dominant polycystic kidney disease gene product PKD2. Mol. Cell. Biol., 19, 3423–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qian F., Boletta A., Bhunia A.K., Xu H., Liu L., Ahrabi A.K., Watnick T.J., Zhou F. and Germino G.G. (2002) Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. U.S.A., 99, 16981–16986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chapin H.C., Rajendran V. and Caplan M.J. (2010) Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site. Mol. Biol. Cell., 21, 4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurbegovic A., Kim H., Xu H., Yu S., Cruanes J., Maser R.L., Boletta A., Trudel M. and Qian F. (2014) Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease. Mol. Cell. Biol., 34, 3341–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim H., Xu H., Yao Q., Li W., Huang Q., Outeda P., Cebotaru V., Chiaravalli M., Boletta A., Piontek K. et al. (2014) Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat. Commun, 5, 5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gainullin V.G., Hopp K., Ward C.J., Hommerding C.J. and Harris P.C. (2015) Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Invest., 125, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei W., Hackmann K., Xu H., Germino G. and Qian F. (2007) Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystic kidney disease 1 gene. J. Biol. Chem., 282, 21729–21737. [DOI] [PubMed] [Google Scholar]
- 31. Yu S., Hackmann K., Gao J., He X., Piontek K., Garcia-Gonzalez M.A., Menezes L.F., Xu H., Germino G.G., Zuo J. et al. (2007) Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. U.S.A., 104, 18688–18693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu C., Rossetti S., Jiang L., Harris P.C., Brown-Glaberman U., Wandinger-Ness A., Bacallao R. and Alper S.L. (2007) Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am. J. Physiol. Renal Physiol., 292, F930–F945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chauvet V., Tian X., Husson H., Grimm D.H., Wang T., Hiesberger T., Igarashi P., Bennett A.M., Ibraghimov-Beskrovnaya O., Somlo S. et al. (2004) Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest., 114, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bertuccio C.A., Chapin H.C., Cai Y., Mistry K., Chauvet V., Somlo S. and Caplan M.J. (2009) Polycystin-1 C-terminal cleavage is modulated by polycystin-2 expression. J. Biol. Chem., 284, 21011–21026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Talbot J.J., Shillingford J.M., Vasanth S., Doerr N., Mukherjee S., Kinter M.T., Watnick T. and Weimbs T. (2011) Polycystin-1 regulates STAT activity by a dual mechanism. Proc. Natl. Acad. Sci. U.S.A., 108, 7985–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Talbot J.J., Song X., Wang X., Rinschen M.M., Doerr N., LaRiviere W.B., Schermer B., Pei Y.P., Torres V.E. and Weimbs T. (2014) The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J. Am. Soc. Nephrol., 25, 1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Low S.H., Vasanth S., Larson C.H., Mukherjee S., Sharma N., Kinter M.T., Kane M.E., Obara T. and Weimbs T. (2006) Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell, 10, 57–69. [DOI] [PubMed] [Google Scholar]
- 38. Woodward O.M., Li Y., Yu S., Greenwell P., Wodarczyk C., Boletta A., Guggino W.B. and Qian F. (2010) Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1. PLoS One, 5, e12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao B., Lei Q.Y. and Guan K.L. (2008) The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr. Opin. Cell. Biol., 20, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang H., Liu C.Y., Zha Z.Y., Zhao B., Yao J., Zhao S., Xiong Y., Lei Q.Y. and Guan K.L. (2009) TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem., 284, 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hong W. and Guan K.L. (2012) The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin. Cell. Dev. Biol., 23, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Happe H., Wal A.M., Leonhard W.N., Kunnen S.J., Breuning M.H., Heer E. and Peters D.J. (2011) Altered Hippo signalling in polycystic kidney disease. J. Pathol., 224, 133–142. [DOI] [PubMed] [Google Scholar]
- 43. Cui C.B., Cooper L.F., Yang X., Karsenty G. and Aukhil I. (2003) Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol. Cell. Biol., 23, 1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanai F., Marignani P.A., Sarbassova D., Yagi R., Hall R.A., Donowitz M., Hisaminato A., Fujiwara T., Ito Y., Cantley L.C. et al. (2000) TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J., 19, 6778–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mahoney W.J., Hong J., Yaffe M. and Farrance I. (2005) The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J., 388, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vassilev A., Kaneko K., Shu H., Zhao Y. and DePamphilis M. (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev., 15, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hong J. and Yaffe M. (2006) TAZ: a beta-catenin-like molecule that regulates mesenchymal stem cell differentiation. Cell Cycle, 5, 176–179. [DOI] [PubMed] [Google Scholar]
- 48. Hong J., Hwang E., McManus M., Amsterdam A., Tian Y., Kalmukova R., Mueller E., Benjamin T., Spiegelman B., Sharp P. et al. (2005) TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science, 309, 1074–1078. [DOI] [PubMed] [Google Scholar]
- 49. Ducy P., Zhang R., Geoffroy V., Ridall A. and Karsenty G. (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell, 89, 747–754. [DOI] [PubMed] [Google Scholar]
- 50. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R., Gao Y., Inada M. et al. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell, 89, 755–764. [DOI] [PubMed] [Google Scholar]
- 51. Otto F., Thornell A., Crompton T., Denzel A., Gilmour K., Rosewell I., Stamp G., Beddington R., Mundlos S., Olsen B. et al. (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell, 89, 765–771. [DOI] [PubMed] [Google Scholar]
- 52. Ueta C., Iwamoto M., Kanatani N., Yoshida C., Liu Y., Enomoto-Iwamoto M., Ohmori T., Enomoto H., Nakata K., Takada K. et al. (2001) Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell. Biol., 153, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Komori T. (2010) Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res., 339, 189–195. [DOI] [PubMed] [Google Scholar]
- 54. Yoder B.K., Hou X. and Guay-Woodford L.M. (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol., 13, 2508–2516. [DOI] [PubMed] [Google Scholar]
- 55. Ma M., Tian X., Igarashi P., Pazour G.J. and Somlo S. (2013) Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Gene, 45, 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xiao Z., Zhang S., Mahlios J., Zhou G., Magenheimer B., Guo D., Dallas S., Maser R., Calvet J., Bonewald L. et al. (2006) Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J. Biol. Chem., 281, 30884–30895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xiao Z., Zhang S., Magenheimer B., Luo J. and Quarles L. (2008) Polycystin-1 regulates skeletogenesis through stimulation of the osteoblast-specific transcription factor RUNX2-II. J. Biol. Chem., 283, 12624–12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boulter C., Mulroy S., Webb S., Fleming S., Brindle K. and Sandford R. (2001) Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc. Natl. Acad. Sci. U.S.A., 98, 12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu W., Shen X., Pavlova A., Lakkis M., Ward C., Pritchard L., Harris P., Genest D., Perez-Atayde A. and Zhou J. (2001) Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet., 10, 2385–2396. [DOI] [PubMed] [Google Scholar]
- 60. Xiao Z., Zhang S., Cao L., Qiu N., David V. and Quarles L. (2010) Conditional disruption of Pkd1 in osteoblasts results in osteopenia due to direct impairment of bone formation. J. Biol. Chem., 285, 1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qiu N., Xiao Z., Cao L., David V. and Quarles L.D. (2012) Conditional mesenchymal disruption of pkd1 results in osteopenia and polycystic kidney disease. PLoS One, 7, e46038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xiao Z., Baudry J., Cao L., Huang J., Chen H., Yates C.R., Li W., Dong B., Waters C.M., Smith J.C. et al. (2018) Polycystin-1 interacts with TAZ to stimulate osteoblastogenesis and inhibit adipogenesis. J. Clin. Invest., 128, 157–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hossain Z., Ali S., Ko H., Xu J., Ng C., Guo K., Qi Z., Ponniah S., Hong W. and Hunziker W. (2007) Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc. Natl. Acad. Sci. U.S.A., 104, 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Makita R., Uchijima Y., Nishiyama K., Amano T., Chen Q., Takeuchi T., Mitani A., Nagase T., Yatomi Y., Aburatani H. et al. (2008) Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am. J. Physiol. Renal Physiol., 294, F542–F553. [DOI] [PubMed] [Google Scholar]
- 65. Tian Y., Kolb R., Hong J., Carroll J., Li D., You J., Bronson R., Yaffe M., Zhou J. and Benjamin T. (2007) TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol. Cell. Biol., 27, 6383–6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Amelio A., Miraglia L., Conkright J., Mercer B., Batalov S., Cavett V., Orth A., Busby J., Hogenesch J. and Conkright M. (2007) A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl. Acad. Sci. U.S.A., 104, 20314–20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Joly D., Ishibe S., Nickel C., Yu Z., Somlo S. and Cantley L. (2006) The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells. J. Biol. Chem., 281, 26329–26339. [DOI] [PubMed] [Google Scholar]
- 68. Tang Q.Q., Otto T.C. and Lane M.D. (2004) Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. U.S.A., 101, 9607–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhao L., Li G., Chan K.M., Wang Y. and Tang P.F. (2009) Comparison of multipotent differentiation potentials of murine primary bone marrow stromal cells and mesenchymal stem cell line C3H10T1/2. Calcif. Tissue Int., 84, 56–64. [DOI] [PubMed] [Google Scholar]
- 70. Sierra J., Villagra A., Paredes R., Cruzat F., Gutierrez S., Javed A., Arriagada G., Olate J., Imschenetzky M., Van Wijnen A. et al. (2003) Regulation of the bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. Mol. Cell. Biol., 23, 3339–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kitabayashi I., Yokoyama A., Shimizu K. and Ohki M. (1998) Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. EMBO J., 17, 2994–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Murakami M., Nakagawa M., Olson E. and Nakagawa O. (2005) A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. U.S.A., 102, 18034–18039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matsumoto Y., La Rose J., Kent O.A., Wagner M.J., Narimatsu M., Levy A.D., Omar M.H., Tong J., Krieger J.R., Riggs E. et al. (2016) Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J. Clin. Invest., 126, 4482–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mangos S., Lam P.Y., Zhao A., Liu Y., Mudumana S., Vasilyev A., Liu A. and Drummond I.A. (2010) The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model. Mech., 3, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Coxam B., Sabine A., Bower N.I., Smith K.A., Pichol-Thievend C., Skoczylas R., Astin J.W., Frampton E., Jaquet M., Crosier P.S. et al. (2014) Pkd1 regulates lymphatic vascular morphogenesis during development. Cell Reports, 7, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Reginensi A., Scott R.P., Gregorieff A., Bagherie-Lachidan M., Chung C., Lim D.S., Pawson T., Wrana J. and McNeill H. (2013) Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet, 9, e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shibazaki S., Yu Z., Nishio S., Tian X., Thomson R., Mitobe M., Louvi A., Velazquez H., Ishibe S., Cantley L. et al. (2008) Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet., 17, 1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. James M.A., Lee J.H. and Klingelhutz A.J. (2006) HPV16-E6 associated hTERT promoter acetylation is E6AP dependent, increased in later passage cells and enhanced by loss of p300. Int. J. Cancer, 119, 1878–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Du S.J., Frenkel V., Kindschi G. and Zohar Y. (2001) Visualizing normal and defective bone development in zebrafish embryos using the fluorescent chromophore calcein. Dev. Biol., 238, 239–246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.