

Abstract
Though phenotypic and target-based high-throughput screening approaches have been employed to discover new antibiotics, the identification of promising therapeutic candidates remains challenging. Each approach provides different information, and understanding their results can provide hypotheses for mechanism of action (MoA) and reveal actionable chemical matter. Here we describe a framework for identifying efficacy targets of bioactive compounds. High throughput biophysical profiling against a broad range of targets coupled with machine learning was employed to identify chemical features with predicted efficacy targets for a given phenotypic screen. We validate the approach on data from a set of 55,000 compounds in 24 historical internal antibacterial phenotypic screens and 636 bacterial targets screened in high-throughput biophysical binding assays. Models were built to reveal the relationships between phenotype, target, and chemotype, which recapitulated mechanisms for known antibacterials. We also prospectively identified novel inhibitors of dihydrofolate reductase with nanomolar antibacterial efficacy against Mycobacterium tuberculosis. Molecular modeling provided structural insight into target-ligand interactions underlying selective killing activity toward mycobacteria over human cells.
Graphical Abstract
Introduction
Finding novel, efficacious antibacterials is essential to combat growing threats of resistant infections. Conventional drug discovery approaches, namely high-throughput screens, have proven largely ineffective at expanding our current antibiotic armamentarium1,2. This has been attributed both to challenges that are unique to bacterial targets, such as permeating the bacterial cell wall and the persistent threat of resistance, as well as general screening pitfalls, such as limited molecular composition of screening libraries and gaps in validation and follow-up methodologies1,2. The traditional dichotomy in high-throughput screening, target-based versus whole-cell or phenotypic-based screening, is inherently limited - active biochemical inhibitors may fail to cross the cell membrane and engage their targets in the cellular milieu, while phenotypic screen actives provide little information about the modulated target(s). New screening paradigms to overcome these pitfalls, such as pathway-based3, synthetic lethal4, and high-content screens5 have yielded successful results, but are typically challenging to establish and difficult to scale up when pursuing multiple targets of interest.
Affinity-based methods for target deconvolution have helped elucidate mechanism of action for eukaryotic phenotypic actives6, but have had limited application to antibacterial discovery7. ALIS (Automated Ligand Identification System)8, which rapidly identifies biophysical interactions of compounds with proteins using affinity mass spectrometry in vitro, offers a unique technology to systematically assess the binding of bioactive small molecules across many targets. However, the challenge remains to uncover the modulated target(s) underlying a phenotype in the context of multiple detected interactions. We implemented machine learning to solve this problem, by identifying key chemical motifs jointly associated with both bioactivity and compound binding to specific “enriched targets”, i.e. targets whose small molecule binders are enriched in the bioactives for a given phenotypic screen. We reasoned that this strategy would address two fundamental limitations to single screening paradigms, eliminating prioritization of compounds without specific targets (such as nonspecific membrane disruptors) and target binders without bioactivity (for example, compounds unable to permeate the bacterial cell wall to engage their target in vivo). This approach was validated by examining assembled data from historical antibacterial phenotypic screens at our company and bacterial targets screened in high-throughput ALIS-based biophysical binding assays. We retrospectively identified antibiotics that modulate dihydrofolate reductase or the ribosome, then prospectively applied our methodologies to identify compounds active against Mycobacterium tuberculosis through inhibition of dihydrofolate reductase. These results illustrate the power of applying cheminformatic modeling in antimicrobial drug discovery to facilitate target and compound identification and prioritization across diverse screening datasets.
Results & Discussion
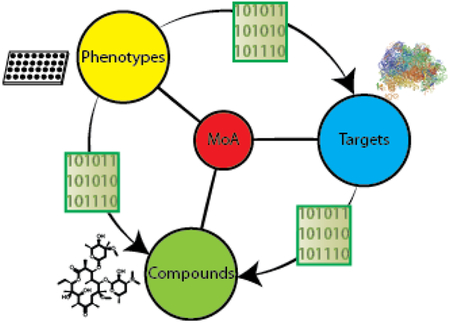
To investigate the potential to utilize target-based chemogenomic data to predict efficacy targets for antibacterial phenotypic screens, we first assembled a rich data set that enabled us to connect compounds, phenotypes, and targets (Figure 1). As a source of chemical matter, we employed an Enriched Antibacterial set comprising compounds previously active in at least one antibacterial campaign at our company9–12, as well as over 100 clinically employed antibiotics and antibacterial tools reported in the literature. We then assembled historical phenotypic assay data for these 55,000 compounds across 24 internal high-throughput screens, accumulating over 1,100,000 measurements of growth or death across 7 bacterial species. The next step was to obtain target association data for the Enriched Antibacterial set. ALIS employs mass spectrometry in high-throughput to identify small molecule binders after dissociation from their purified cognate targets. Though this format disfavors detection of covalent interactions and compounds that ionize poorly, we were able to detect the biophysical interactions of 19 chemically diverse and well-characterized antibiotics with their canonical targets (Table 1). These initial results supported our use of the ALIS platform to profile the Enriched Antibacterial set for binding across a diverse panel of 636 bacterial targets (originating from 41 different organisms and over 100 distinct metabolic and signaling pathways, see Methods), and lead to the detection of over 120,000 total interactions.
Figure 1:

Overview of our cheminformatic approach to mechanism of action prediction for antimicrobial drug discovery. Naïve Bayes models were generated for a series of phenotypic and biophysical binding screens using a joint Enriched Antibacterial training set of ~55,000 compounds (see Methods). Models identified protein targets that were enriched in phenotypic assays (binders of that target were enriched as phenotypic actives). Models linking a phenotypic screen and enriched target of interest to chemical matter identified chemotypes enriched for both binding to the target and phenotypic screen activity (bacterial killing). Compounds possessing the selected chemotypes were hypothesized to achieve efficacy in killing bacteria by acting through the enriched efficacy target.
Table 1:
Conventional antibiotic target pairs detected via ALIS
| Antibiotic | Class | Canonical Target | Originating Organism for Nominal Target | Number Targets Bound |
|---|---|---|---|---|
| Novobiocin | Aminocoumarin | Gyrase | L. monocytogenes | 4* |
| Trimethoprim | Antifolate | Dihydrofolate reductase | B. henselae, E. coli | 4* |
| Diaveridine | Antifolate | Dihydrofolate reductase | B. henselae, E. coli | 2* |
| Pyrimetriamine | Antifolate | Dihydrofolate reductase | B. henselae, E. coli | 2* |
| Clindamycin | Lincosamide | Ribosome | M. tuberculosis | 1 |
| Rosaramicin/Rosamicin | Macrolide | Ribosome | M. tuberculosis | 1 |
| Azathramycin | Macrolide | Ribosome | M. tuberculosis | 1 |
| Ervthromycylamine | Macrolide | Ribosome | M. tuberculosis | 1 |
| Lankacidin | Macrolide | Ribosome | M. tuberculosis | 1 |
| L-701677 | Macrolide | Ribosome | M. tuberculosis | 1 |
| Lexithromycin | Macrolide | Ribosome | M. tuberculosis | 1 |
| Azithromycin | Macrolide | Ribosome | M. tuberculosis | 1 |
| Clarithromycin | Macrolide | Ribosome | M. tuberculosis | 1 |
| Linezolid | Oxazolidinone | Ribosome | M. tuberculosis | 1 |
| Sutezolid | Oxazolidinone | Ribosome | M. tuberculosis | 1 |
| VRT-752586 | Other | Gyrase | L monocytogenes | 2 |
| CHIR-090 | Other | LpxC | B. ambifaria | 58 |
| Actinonin | Peptide | Peptide deformylase | A. phagocytophilum | 1 |
| Doxycycline | Tetracycline | Ribosome | M. tuberculosis | 3 |
Includes 2 target homologs.
In order to elucidate meaningful connections between the compounds, phenotypic assays, and bacterial proteins from the assembled data, we constructed an informatics framework comprising three types of learned models that capture bioactivity and target associations and are joined together based on shared target and compound descriptors (Figure 1). We employed Naïve Bayesian modeling, as high-throughput measurements are inherently noisy13, and this form of machine learning is less sensitive to false negative rates than several alternatives14. The first models we derived linked phenotypic assays and compounds - for each phenotypic assay, we classified phenotypic activity of compounds using their chemical features as descriptors. These models facilitate accurate identification and prioritization of key chemical features correlating with favorable bioactivity (bacterial killing) for each screen.
The next connections we derived were between targets and compounds. Again, we built Naïve Bayesian models, to compensate for inherent noise in ALIS data in the form of false negatives or positives. We generated models for each target, where binding of compounds was classified using their chemical features as descriptors. Of the 636 total targets screened in the ALIS panel, 322 satisfied our minimal threshold for model building of possessing at least 20 nonpromiscuous binders (see Methods). These models empowered us to select chemical features most associated with binding to each particular target.
The final connections were between phenotypes and targets. A given set of screen actives often has many target associations, precluding the straightforward determination of efficacy target(s). Indeed, for our ALIS data, 57.4% of 11,505 compounds detected as binders had multiple target associations (Supplementary Figure 1). To prioritize targets, we built Naïve Bayesian models for each phenotypic screen - these models featured ALIS targets bound by each small molecule as its descriptors, and phenotypic activity for that molecule as its binary classification. We then used the model weights derived for each target to prioritize targets in a given phenotypic assay. If compounds that bound a target of interest tended to be active, the weight for that target was positive. Likewise, if compounds that bound the target tended to be inactive, the weight for the target was negative.
We anticipated integrating this framework (Figure 1) as follows:
Select a phenotypic assay of interest
Select one or more prioritized targets associated with the assay
Select compounds with features that are both enriched for activity in the assay and features associated with binding one of the previously selected targets
We hypothesized compounds selected in such a fashion would be active in the screen and could be associated with an efficacy target, namely, the target selected in step 2.
To illustrate our ability to identify efficacy targets for bioactive screen hits using this approach, we began by recapitulating the mechanisms of action for known antibiotics. We began by investigating a phenotypic screen identifying inhibitors of E. coli growth. Out of the 636 targets screened using the ALIS platform, the most enriched target was FolA (dihydrofolate reductase, DHFR), with a normalized probability of 0.89 (Figure 2A). Of 64 total FolA binders detected in ALIS, 54 were tested in this phenotypic screen and 42 were active. Included in this subset were trimethoprim, diaveridine, and theirpara-hydroxyl derivatives, compounds with nanomolar IC50 values for FolA and known to kill E. coli by inhibiting folate metabolism15. Selecting the top-scoring chemical features from both the E.coli screen model and FolA target model generated a list of six fragments enumerating the conserved diaminopyrimidine core found in many DHFR inhibitors (green, Figure 2A). There were a total of 129 compounds in the Enriched Antibacterial set possessing at least one of these features, of which 89 were tested and 55 were active, a 2.4-fold enrichment over the bioactive rate overall for this phenotypic screen (p<0.0001, two-tailed χ2 test). In addition to successfully identifying diaveridine and trimethoprim, we also predicted bioactivity for related antibiotics iclaprim and brodimoprim that were not screened in this assay, as well as their established mechanism of action through FolA inhibition15,16, despite failure to detect their binding in ALIS. Notably, this small set of bioactive compounds bound a total 17 nonhomologous targets, illustrating the power of our approach to key in on the likely efficacy target. Thus, we retrospectively recapitulated the mechanism of action for known antibiotics targeting dihydrofolate reductase in E. coli, even for compounds that were false negatives in the ALIS binding screen.
Figure 2:

Joint identification of enriched chemotypes facilitates retrospective validation of known antibiotic mechanism of action. A) 6 enriched chemotypes for bioactivity in an E. coli live/dead phenotypic assay and for the top enriched target, FolA, yielded 4 well-known antibiotics inhibiting dihydrofolate reductase, 2 of which were active hits in the phenotypic screen. B) The M. tuberculosis ribosome was an enriched target for a S. aureus live/dead phenotypic screen, and joint identification of 19 chemotypes (8 shown) lead to identification of 14 known ribosomal antibiotics in 3 distinct chemical classes (12 active, 2 were untested in this screen).
The targets of traditional antibiotics tend to be highly conserved across bacteria17, and we wondered whether we could utilize phenotype-target connections derived from our models, even if the target originated from a different organism. One of the targets included in the ALIS screen was the fully reconstituted ribosome of M. tuberculosis, which we identified as an enriched target for phenotypic screens conducted against several organisms, including P. aeruginosa, A. baumanii, and S. aureus. For the S. aureus phenotypic screen, the ribosome was ranked as the 11th most enriched target and there were 20 chemical features highly enriched for both ribosome binding and activity against S. aureus. 425 compounds in the Enriched Antibacterial set contained at least one of these features, of which 210 were tested and 93 were active (2-fold the hit rate of the phenotypic screen, p<0.0001 two-tailed χ2 test, Figure 2B). Included in this set were 3 classes of known ribosomal inhibitors, macrolides, aminoglycosides, and a mutilin, of which 2 compounds were not screened in this assay nor were detected in ALIS, but are known to be active against S. aureus via ribosomal inhibition18,19. This demonstrated our ability to identify efficacy targets by exploiting target homology, and suggested that homology should be taken into account to reduce the total set of targets needed to thoroughly cover target space.
Encouraged by these results, we sought to assess the prediction of efficacy targets and phenotypic activities for molecules outside of the training data. Previous studies have leveraged molecular descriptors in combination with reference molecules of known mechanism of action for efficacy target inference20–22. To test the predictive ability of our models, we assembled a set of known antibiotics that were not present in the Enriched Antibacterial screening collection and scored them for both target associations and bioactivities. 48 antibiotics in this prospective set act through noncovalent inhibition of protein targets present in the ALIS screening collection, and binding was correctly predicted for 35 of these (72.9%), at an accuracy of 86.9% across all target models, with a conservative assumption of exclusive binding to the nominal target (Supplementary Table 1). Compounds with successfully predicted target associations belonged to 9 unique structural classes of antibiotics, including streptogrammins, which were not represented in the training data. For validation of predicted bioactivity, 28 antibiotics that were not part of the Enriched Antibacterial set were screened in at least one of the 24 phenotypic assays, and growth inhibitory activity was successfully predicted in 48/58 screening instances (82.8%) with an accuracy of 76.4%. These results supported the value of applying our models to predict efficacy targets and growth inhibitory activity for new molecules.
While our models successfully recovered efficacy targets for established antibiotics of distinct classes and targets, we wanted to apply our methodology to identify promising new agents. One of the screens in our collection was for compounds inhibiting the growth of Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis. In evaluating the most enriched targets for a screen assaying for compounds inhibiting the growth of Mtb, DHFR (Rv2763c) ranked 9th of 636 targets. Several compounds have been developed that potently inhibit Mtb DHFR in vitro, yet few have bioactivity23. Nevertheless, we identified 7 chemical features and 162 compounds possessing these fingerprints that were enriched for both bioactivity and target binding for Rv2763, of which 6 were active (4-fold enrichment over the phenotypic screen hit rate, p=0.0021 two-tailed χ2 test, Figure 3A). Included in this initial set of hits was 10-propargylaminopterin (10-PAP), which was previously reported to inhibit mouse dihydrofolate reductase24. We expanded this class to include structurally similar analogs in the greater screening collection at our company, and then tested this collection of 136 compounds for both Rv2763c inhibition in vitro and mycobacterial growth inhibition. For both the initial screen hits and the expansion, we observed potent inhibition of both enzymatic activity and growth with modest correlation, supporting our target hypothesis for these compounds (Figure 3B, Table 2). During our investigation, three of the tested compounds from our expansion set were reported to inhibit Rv2763c in vitro and/or inhibit growth of Mtb, and our measurements were in close agreement with reported values (Supplementary Table 2)25,26. Notably, our collection of tested compounds also included previously unreported phenyl-substituted 7H-pyrrolo[3,2-f]quinazoline-1,3-diamines (PQDs) (Figure 3, Class 4) as well as diversification of the benzyl-PQD scaffold (Classes 2,3). Thus, by searching for chemical features enriched for both Mtb growth and DHFR binding activities, then expanding to structurally related chemical matter, we were able to prospectively identify new series of antibacterials targeting Rv2763c.
Figure 3:

Identification of compounds that target DHFR to inhibit the growth of M. tuberculosis. A) 7 enriched chemotypes yielded 6 compounds with growth inhibitory activity and hypothesized to act through Rv2763c (Mtb DHFR). B) Correlation between in vitro inhibition of purified Mtb DHFR and inhibition of bacterial growth. Methotrexate (yellow) was used as a control for inhibiting the enzyme but having no efficacy against bacterial growth. Example class members are listed in the table, along with their IC50 values for the Mtb enzyme, MITC95 values, the correlation between these, and their toxicity observed at 99μM in HeLa cells.
To investigate the potential for progression of these series into antibiotic development, we assessed cytostatic activity and toxicity for a subset of 110 compounds against human cells. All tested phenyl-PQDs and bicyclic-PQDs (Figure 3, classes 3,4) caused a reduction in both the number of viable HeLa cells and in total incorporation of 5-ethynyl-2’-deoxyuridine (EDU) with low micromolar EC50. The majority of tested members of the folate analog (class 1) and benzyl-substituted PQD classes (class 2) that did not kill human cells also lost antibacterial activity, though many retained potent enzyme inhibition. However, 2 folic acid derivatives possessed EC50>99 μM against human cells and retained bioactivity, including 10-PAP. In order to gain structural insight into engagement of DHFR by the different inhibitor classes, we docked several compounds into both Mtb and human enzyme crystal structures (PDB 1DF7, 1OHJ)27,28. We observed that docked inhibitors adopted poses overlapping the natural substrate (dihydrofolate) and crystalized inhibitors (Figure 4, Supplementary Figure 2) for both enzymes. The shared pyrimidine cores all exhibited a common protonation state supported by previous studies29,30, and in agreement with previous docking experiments, amino groups of the planar quinazoline and pteridine cores engaged in hydrogen bonds with Rv2763c residues I5, D27, and I94 (corresponding to human DHFR I7, E30, and V115)25. The planar paraminobenzoate moieties of dihydrofolate, 10-PAP, and related class 1 inhibitors were observed to fold back toward inhibitor cores at an angle of 21–26°. While benzyl- and bicyclic substituted derivatives (classes 2,3) exhibited a ~10° distortion in this angle, phenyl-PQDs of class 4 were shifted ~120° due to their loss of rotational freedom provided by the benzyl carbon. These distortions retained π-π stacking interactions between the phenyl and benzyl PQD substituents with active site residue F31 in the human enzyme, supporting engagement of human DHFR as a possible cause for observed toxicity. In contrast, docking results exhibited steric clashes between the 10-propargyl substituent of 10-PAP and human DHFR S59 that were absent in the active site of Mtb DHFR (Figure 4), which may support bulky substitution at this position as a selectivity determinant for future analogs. Taken together, the results of our docking support targeting of DHFR by identified inhibitors and selective targeting of mycobacterial DHFR over the human enzyme by 10-PAP through exploitation of key differences in an otherwise conserved active site.
Figure 4:

A) Overlay of 10-PAP and dihydrofolate ligands in the active sites of both Mtb and human DHFR. B) Key interactions are illustrated between the diaminopyrimidine pharmacophore and conserved active site residues. Hydrogen bonds between Mtb DHFR Arg60 and the 10-PAP glutamate moiety were omitted for clarity. Other classes can be found in Supplementary Figure 2. Figure prepared using Maestro (Schrodinger version 2017–1).
Our ability to assign mechanisms of action for the cases discussed above was facilitated by the inclusion of these targets in the ALIS screening collection. To assess the performance of our approach across screens and its dependency on the protein set included in ALIS, we determined the percentage of active compounds with assigned efficacy target hypotheses for each phenotypic assay. We found significant differences across bacterial species and assays (Figure 5A, Supplementary Figure 3), noting that while greater percentages of active compounds could be assigned efficacy target hypotheses as more ALIS targets and their corresponding interactions were included in the data set, the maximum percentage ranged between 0.5–26.2%. This maximum percentage was largely independent of both the total number of actives for a screen (Supplementary Figure 4) and the number of ALIS proteins that originated from that organism (Supplementary Figure 5). Integrating high quality chemogenomics data from our internal CHEMGENIE database improved efficacy target associations, in some cases doubling the total number of active compounds with an efficacy target hypothesis (Figure 5B). This analysis highlights a key limitation of our approach, and suggests that inclusion of additional proteins in ALIS would improve our association of active compounds with efficacy target(s).
Figure 5:

Assessment of target space coverage to generate efficacy target hypotheses for phenotypic actives. A) Percentage of phenotypic assay actives (vertical axis) that could be associated with a potential efficacy target for each organism. The horizontal axis is the percentage of efficacy targets randomly selected for each screen, and each value is the average of 10 replicates. B) Adding CHEMGENIE annotations (green line) improves the percentage of bioactives for each screen with an efficacy target hypothesis, as compared to ALIS data alone (blue line). Shown are the results for M. tuberculosis, other organisms can be found in Supplementary Figure 2.
Conclusion
We have demonstrated the power of machine learning to determine mechanism of action for molecules with antibacterial activities. Our approach in building and integrating models for high-throughput biophysical and phenotypic screening data enables us to pinpoint chemical fingerprints enriched for bioactivity and for binding to enriched efficacy targets. Chemical matter possessing these molecular features can then be assessed for bioactivity, target engagement, and toxicity against human cells in follow-up assays. Applying our methodology to historical screening data at our company recapitulated the mechanisms of action for known antibiotics across several structural classes and acting on diverse targets. Prospective application in a screen for inhibition of M. tuberculosis growth identified Rv2763c (Mtb DHFR) as a potential efficacy target. Follow-up experiments yielded 4 classes of Rv2763c inhibitors, several of which exhibited both nanomolar enzymatic inhibition and sub-micromolar potency in inhibiting Mtb growth. We observed that the majority of PQD-containing compounds exhibited toxicity toward mammalian cells. PQDs have also been reported as micromolar thrombin receptor antagonists30, suggesting that future development of these series as therapeutics may require mitigation of off-target activities. Nevertheless, we identified Class 1derivatives as effective Mtb DHFR inhibitors with potent antimycobacterial activity and little or no measurable toxicity.
Though we were only able to predict mechanism of action for a subset of antibiotics, and were able to assign an efficacy target hypothesis to a limited number of screen actives, it is anticipated that increasing the size of the compound and target sets will improve model accuracy and utility. Indeed our analysis suggests that the ALIS platform might benefit from including additional proteins from the body of emerging and resistant targets31. It is important to note that models can readily be regenerated to incorporate newly acquired data, and that incorporating models from other forms of high-throughput biophysical data could assist in addressing the target scope limitations of ALIS (e.g. membrane proteins, proteins with covalent inhibitors such as beta-lactamases, and non-protein targets for antibiotics, such as lipid II). It is anticipated that our approach can be used to identify and prioritize both new chemical matter and novel targets to aid in the development of antimicrobials across a broad range of bacterial pathogens.
Methods
Compound Collection Assembly
In order to supply our screens with rich chemical matter and positive controls for bioactivity and binding, we employed an “Enriched Antibacterials” set of approximately 55,000 small molecules from known antibiotics as well as compounds with bioactivity in at least one of a set of historical high-throughput, cell-based bacterial screens at our company, from 1996–20119-12.
Historical Antibacterial Screens
A set of 24 primary and confirmatory high-throughput antibacterial screens was included in our analysis, including several that have been previously described9–11, with the prerequisite that at least 1,000 compounds from the Enriched Antibacterials set were assayed. For each of these screens, we selected the corresponding data for the subset of compounds present in the Enriched Antibacterials set. Compounds that were not screened were excluded from Naïve Bayes (NB) models built for that screen.
Biophysical Screen of 636 Bacterial Targets
The full Enriched Antibacterials set was screened in the ALIS platform against 636 bacterial targets with modification of previously reported conditions8. Purified proteins were obtained from the Seattle Structural Genomics Center for Infectious Disease (SSGCID) and the Center for Structural Genomics of Infectious Diseases (CSGID). Targets were curated into pathways using UniProt, or manually when annotation was unavailable. To enable the screening of tens of thousands of compounds against hundreds of targets, an arrayed screening format was developed, which entails screening mixtures of compounds against mixtures of proteins and deconvoluting detected binding interactions to single compound-protein pairs across pools.
CHEMGENIE Database
To supplement bacterial ALIS data with pharmacological evidence for Enriched Antibacterial compound activities, we integrated experimental evidence from our company’s Chemical Genetic Interaction enterprise (CHEMGENIE) Database32, which contains data curated from internal bacterial and eukaryotic biochemical and biophysical assays, other ALIS target screens, high-throughput screening campaigns, as well as external results acquired from Metabase, PDB, and CHEMBL. Biochemical data were filtered to only include IC50 or equivalent inhibitory activities at ≤1μM.
Construction of Naïve Bayes Models for Bioactivity and Binding
In order to determine chemical features enriched for bioactivity or target-binding among the Enriched Antibacterial set, we built NB models based on extended connectivity fingerprints (ECFP4)33 in Pipeline Pilot v17.0 for each phenotypic assay and each target binding assay. Target models were generated for 325 proteins that satisfied the criterion of binding at least 20 compounds, after removal of data for 705 promiscuous compounds (defined as binding >10% of all targets screened). Compounds were annotated as active in a phenotypic screen if they induced ≥80% inhibition of bacterial growth. Models built to identify efficacy targets for a phenotypic assay were learned using targets as features, with ALIS target-compound associations as inputs and phenotypic activity (as defined above) as a test for good.
Each model output a normalized probability for every learning feature present in the observed data, normalized by the total occurrences of that feature across the training data set. Mathematically expressed,
| 34 | (Eqn. 1) |
where Hi and Ti are the number of active and total molecules possessing a feature, respectively, H is the total number of features present for active molecules, and T is the total number of features present in the screening set for the assay.
Models were assessed for performance by calculating a leave-one-out cross-validated ROC score and enrichment factor for the top 1% of actives (EF1%). For the 24 models linking phenotypic activity to chemical matter, these values averaged 0.81 and 45.8%, respectively. ALIS binding models had an average leave-one-out cross-validated ROC AUC score of 0.83 and an average EF1% of 51.9%. Models connecting assays with efficacy targets had an average cross-validated ROC AUC score of 0.68 and EF1% = 11.1%.
In order to predict performance for compounds outside training data, a model score was first calculated for each compound as the sum of the model’s normalized probabilities for each chemical feature present in that compound. A score cut-off was then calculated for each target or assay model by picking a split that minimized the sum of the percent misclassified for category members (binders or actives) and for category nonmembers (non-binders or inactive compounds), using the cross-validated score for each sample. For assay performance prediction, compounds scoring above the cut-off for a given assay model were predicted to be active. For target prediction, compounds scoring above the cut-off for a given target model were predicted to bind. Only one model was evaluated in cases of targets with multiple homologs present in the protein screening collection. Multitarget antibiotics (ralitrexed and pemetrexed) were evaluated independently for each nominal target, and for performance calculations, all remaining compounds were assumed to exclusively bind their nominal targets.
To determine the percentage of phenotypic screen bioactives with an enriched efficacy target hypothesis, fixed percentages of total targets were then selected randomly from pharmacogenomic (CHEMGENIE) and/or biophysical data, and the corresponding target-compound associations used to identify a fraction of the total bioactives for the phenotypic screen that were associated with an enriched efficacy target (in this case defined as scoring in the top 10% of targets for a phenotypic screen model). These target models were built in the same manner as the ALIS models connecting phenotypic assays and targets, with the exception that additional target-compound associations were included using filtered pharmacogenomic data (vide supra). The combined CHEMGENIE+ALIS models had an average cross-validated ROC AUC score of 0.69 and EF1% = 21.57%.
M. tuberculosis Growth Inhibition Assay
In order to test the growth inhibitory effect of predicted DHFR inhibitors on mycobacteria, M. tuberculosis ATCC 27294 was grown to an OD600 of 0.2–0.3 then diluted 1:1000 in a 96 well plate containing 7H9 media supplemented with 0.5% BSA Fraction V, .081% NaCl, 0.5% (v/v) Tyloxapol, 0.4% glucose and inhibitors in dilution series. Plates were incubated in a sealed bag for 2 weeks at 37°C. 10% (v/v) Alamar blue was then added and wells were scored for growth after 24 hrs incubation. Isoniazid was used as a positive control for growth inhibition.
DHFR Inhibition Assay
In order to test the growth inhibitory effect of predicted DHFR inhibitors on mycobacteria, M. tuberculosis PMSP12 expressing green fluorescence protein (GFP) was grown to OD600 0.2, then diluted 1:1500 in a black, flat bottom 96 well plate containing 7H9 media supplemented with 0.5% BSA Fraction V, 0.081% NaCl, 0.05% (v/v) Tyloxapol, 0.4% glucose or 0.01% cholesterol and inhibitors in dilution series. Plates were incubated in a sealed bag for 1 weeks at 37°C. GFP fluorescence was measured by PerkinElmer EnVision™ multilabel Plate Reader (λex485 nm and λem 520 nm). Isoniazid was used as a positive control for growth inhibition.
HeLa Cytostatic and Toxicity Assays
The effects of test compounds on Human cervical adenocarcinoma cell (HeLa, ATCC, Manassas, VA) DNA synthesis and growth were assessed using the Click-iT EdU Alexa Fluor 488 HCS Assay (Invitrogen, Grand Island, NY). DNA synthesis as well as total cell counts were assessed following incubation with test compounds and 5 μM 5-ethynyl-2’ deoxyuridine (EdU) at 37°C for 24 hours. Cells were fixed and EdU was click-labeled with Alexa Fluor 488 azide. Microplates were analyzed on an Acumen eX3 laser scanning plate cytometer (TTP Labtech, Inc.; Melbourne, UK). Data were analyzed using a 4-parameter curve fitting algorithm.
Docking
The binding pockets of PDB structure 1DF7 (M. tuberculosis) and 1OHJ (H. sapiens) were first aligned and preprocessed to add hydrogens, remove waters and glycerol, and minimize residue energies using the Protein Preparation Wizard functionality in Schrodinger software suite version 2017–1. Ligands were prepared using LigPrep (Schrodinger Inc., v 2017–1) allowing for multiple protonation and tautomerization states. GLIDE (Schrodinger Inc., version 2017–1) with extra precision and default parameters was used to predict poses of ligands within a bounding box defined by the methotrexate ligand of 1DF7. The final pose for each ligand was selected from among the top 5 scoring poses using visual inspection ensuring correct overlap between diaminopyrimidine cores with the crystallized ligand.
Supplementary Material
Acknowledgments
We would like to thank the laboratory of J. Sacchetini for providing purified proteins of the Mtb ribosome for ALIS screening. Other protein targets used in this work were generously provided by the Chicago Center for Structural Genomics of Infectious Disease (www.CSGCID.org, funded by the National Institute of Allergy and Infectious Diseases of NIH, Contracts No. HHSN272200700058C and HHSN272201200026C), and the Seattle Structural Genomics Center for Infectious Disease (www.SSGCID.org, supported by Federal Contract No. HHSN272201200025C from the National Institute of Allergy and Infectious Diseases, NIH). This work was funded in part by the Intramural Research Program of NIAID.
References
- 1.Payne DJ, Gwynn MN, Holmes DJ, & Pompliano DL (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Disc 6, 29–40. [DOI] [PubMed] [Google Scholar]
- 2.Tomassi R, Brown DG, Walkup GK, Manchester JI, & Miller AA (2015) ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Disc 14, 529–542 [DOI] [PubMed] [Google Scholar]
- 3.Matano L, Morris HG, Wood BM, Meredith TC, & Walker S (2016) Accelerating the Discovery of Antibacterial Compounds Using Pathway-Directed Whole Cell Screening. BioorgMed Chem 24(24), 6307–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasquina L, Santa Maria JP Jr., Wood BM, Moussa SH, Matano LM, Santiago M, Martin SES, Lee W, Meredith TC, & Walker S (2016) A Synthetic Lethal Approach for Compound and Target Identification in Staphylococcus aureus. Nat Chem Biol 12(1), 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nonejuie P, Burkart M, Pogliano K, & Pogliano J (2013) Bacterial Cytological Profiling Rapidly Identifies the Cellular Pathways Targeted by Antibacterial Molecules. Proc Natl Acad Sci USA 110(40), 16169–16174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schirle M & Jenkins JL (2016) Identifying Compound Efficacy Targets in Phenotypic Drug Discovery. Drug Disc Today 21(1), 82–89. [DOI] [PubMed] [Google Scholar]
- 7.Ruzin A, Singh G, Severin A, Yang Y, Dushin RG, Sutherland AG, Minnick A, Greenstein M, May MK, Shlaes DM, & Bradford PA (2004) Mechanism of Action of the Mannopeptimycins, a Novel Class of Glycopeptide Antibiotics Active Against Vancomycin-Resistant Gram-Positive Bacteria. Antimicrob Age Chemo 48(3), 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kutilek VD, Andrews CL, Richards MP, Xu Z, Sun T, Chen Y, Hashke A Smotrov N, Fernandez R, Nickbarg EB, Chamberlin C, Sauvagnat B, Curran PJ, Boinay R, Saradjian P, Allen SJ, Byrne N, Elsen NL, Ford RE, Hall DL, Kornienko M, Rickert KW, Sharma S Shipman JM, Lumb KJ, Coleman K, Dandliker PJ, Kariv I, & Beutel B (2016) Integration of Affinity Selection-Mass Spectrometry and Functional Cell-Based Assays to Rapidly Triage Druggable Target Space within the NF-κ B Pathway. JBiomol Screening 21(6), 608–619. [DOI] [PubMed] [Google Scholar]
- 9.Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, Malinvemi JC, Mayhood T, Villafania A, Nahvi A, Murgolo N, Barbieri CM, Mann PA, Carr D, Xia E, Zuck P, Riley D, Painter RE, Walker SS, Sherborne B, de Jesus R, Pan W, Plotkin MA, Wu J, Rindgen D, Cummings J, Garlisi CG, Zhang R, Sheth PR, Gill CJ, Tang H, & Roemer T (2015) Selective small-molecule inhibition of an RNA structural element. Nature 526(7575), 672–677. [DOI] [PubMed] [Google Scholar]
- 10.Lee SH, Wang H, Labroli M, Koseoglu S, Zuck P, et al. (2016) TarO-specific Inhibitors of Wall Teichoic Acid Biosynthesis Restore β-lactam Efficacy Against Methicillin-Resistant Staphylococci. Sci TranslMed. 8(329), 329ra32. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo M, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Halley J, Mcguinness D, Tan CM, Flattery A, Walker S, Black T, & Roemer T (2013) Discovery of Novel Wall Teichoic Acid Inhibitors as Effective anti-MRSA β-lactam Combination Agents. Chem Biol 20(2), 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker SS, Degen S Nickbarg E, Carr D, Soriano A, Mandal MB, Painter RE, Sheth PR, Xiao L, Sher X, Murgolo N, Su J, Olsen DB, Ebright RH, & Young K (2017) Affinity Selection-Mass Spectrometry Identifies a Novel Antibacterial RNA Polymerase Inhibitor. ACS Chem Biol, 12(5), 1346–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diller DJ, & Hobbs DW (2004) Deriving Knowledge through Data Mining High-Throughput Screening Data. J Med Chem 47, 6373–6383. [DOI] [PubMed] [Google Scholar]
- 14.Glick M, Jenkins JL, Nettles JH, Hitchings H, & Davies JW (2006). Enrichment of High-Throughput Screening Data with Increasing Levels of Noise Using Support Vector Machines, Recursive Partitioning, and Laplacian-Modified Naïve Bayesian Classifiers. J Chem Inf Model 46, 193–200. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Hilgers M, Cunningham M, Chen Z, Trzoss M, Zhang J, Kohnen L, Lam T, Creighton C, GC K, Nelson K, Kwan B, Stidham M, Brown-Driver V, Shaw KJ, & Finn J (2001) Structure-based design of new DHFR-based antibacterial agents: 7-aryl-2,4-diaminoquinazolines. BioorgMed Chem Lett 21, 5171–5176. [DOI] [PubMed] [Google Scholar]
- 16.Selassie CD, Li R-L, Poe M & Hansch C (1991) On the Optimization of Hydrophobic and Hydrophilic Substituent Interactions of 2,4-Diamino-5-(substituted-benzyl)pyrimidines with Dihydrofolate Reductase. J Med Chem 34, 46–54. [DOI] [PubMed] [Google Scholar]
- 17.Gladki A, Kaczanowski S, Szczesny P, & Zielenkiewicz P (2013) The evolutionary rate of antibacterial drug targets. BMC Bioinformatics 14, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt E (2000) Pleuromutilin antibiotics. Drugs of the Future. 25(11), 1163–1168. [Google Scholar]
- 19.Kavanagh F, Hervey A, & Robbins WJ (1951) Antibiotic Substances from Basidomycetes. VIII. Pleurotus multilus (Fr.) Sacc. And Pleurotuspasseckerianus Pilat. Proc. Nat. Sci Acad. 37(9): 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reker D, Perna AM, Rodrigues T, Schneider P, Reutlinger M, Mönch B, Koeberle A, Lamers C, Gabler M, Steinmetz H, Müller R, Schubert-Zsilavecz M, Werz Ol, & Schneider G (2014) Revealing the macromolecular targets of complex natural products. Nat. Chem 6: 1072–1078. [DOI] [PubMed] [Google Scholar]
- 21.Alversson J, Eklund M, Engkvist O, Spjuth O, Carlsson L, Wikberg JES, & Noeske T (2014) Ligand-based target prediction with signature fingerprints. J. Chem. Inf. Model 54, 2647–2653. [DOI] [PubMed] [Google Scholar]
- 22.Lounkine E Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Côte S, Shoichet BK, & Urban L (2012) Large-scale prediction and testing of drug activity on side-effect targets. Nature. 486(7403), 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nixon MR, Saionz KW, Koo M-S, Szymonifka MJ, Jung H, Roberts JP, Nandakumar M, Kumar A, Liao R, Rustad T, Sacchettini JC, Rhee KY, Freundlich JS, & Sherman DR (2014) Folate Pathway Disruption Leads to Critical Disruption of Methionine Derivatives in Mycobacterium tuberculosis. Chem. Biol 21, 819–830. [DOI] [PubMed] [Google Scholar]
- 24.Jones TR, Calvert AH, Jackman AL, Brown SJ, Jones M, and Harrap KR (1981) A Potent Antitumour Quinazoline Inhibitor of Thymidylate Synthetase: Synthesis, Biological Properties, and Therapeutic Results in Mice. Eur J Cancer 17, 11–19. [DOI] [PubMed] [Google Scholar]
- 25.Hong W, Wang Y, Chang Z, Yang Y, Pu J, Sun T, Kaur S, Sacchettini JC, Jung H, Wong WL, Yap LF, Ngeow YF, Paterson IC, & Wang H (2015) The identification of novel Mycobacterium tuberculosis DHFR inhibitors and the investigation of their binding preferences by using molecular modeling. Sci Rep 5, 15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Guardia A, Colmenarejo G, Pérez E, Ruben R, Gonzalez PT, Calvo D, Gómez RM, Ortega F, Jiménez E, Gabarro RC, Rullás J, Ballell L, & Sherman DR (2015) A focused screen identifies antifolates with activity on Mycobacterium tuberculosis. ACS Infect Dis 1, 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cody V, Galitsky N, Luft JR, Pangborn W, Rosowsky A, & Blakley RL (1997). Comparison of two independent crystal structures of human dihydrofolate reductase ternary complexes reduced with nicotinamide adenine dinucleotide phosphate and the very tight-binding inhibitor PT523. Biochemistry 36, 13897–13903. [DOI] [PubMed] [Google Scholar]
- 28.Li R, Sirawaraporn R, Chitnumsub P, Sirawaraporn W, Wooden J, Athappilly F, Turley S, & Hol WG (2000) Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J Mol Biol 295, 307–323. [DOI] [PubMed] [Google Scholar]
- 29.Matthews DA, Alden RA, Bolin JT, & Freer ST (1977) Dihydrofolate reductase: x-ray structure of the binary complex with methotrexate. Science 197, 452–455. [DOI] [PubMed] [Google Scholar]
- 30.Cocco L, Roth B, Temple C Jr., Montgomery JA, London RE, & Blakley RL (1983) Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch Biochem Biophys 226(2), 567–577. [DOI] [PubMed] [Google Scholar]
- 31.Ahn H-S, Arik L, Boykow G, Burnett DA., Caplen MA, Czarniecki M, Domalski MS, Foster C, Manna M, Stamford AW, and Wu Y (1999) Structure-activity relationships of pyrroloquinazolines as thrombin receptor antagonists. BioorgMed Chem Lett 9(14), 2073–2078. [DOI] [PubMed] [Google Scholar]
- 32.Sutterlin HA, Malinverni JC, Lee SH, Balibar CJ, & Roemer T (2017) Antibacterial New Target Discovery: Sentinel Examples, Strategies, and Surveying Success. Top Med Chem doi: 10.1007/7355_2016_31 [DOI] [Google Scholar]
- 33.Kutchukian PS, So S-S, Fischer C, & Waller CL (2015) Fragment Library Design: Using Cheminformatics and Expert Chemists to Fill Gaps in Existing Fragment Libraries Meth Mol Biol 1290, 43–53. [DOI] [PubMed] [Google Scholar]
- 34.Rogers D, & Hahn M (2010) Extended Connectivity Fingerprints. J Chem Inf Model 50, 742–754. [DOI] [PubMed] [Google Scholar]
- 35.Clark AM, Dole K, Coulon-Spektor A, McNutt A, Grass G, Freundlich JS, Reynolds RC, & Ekins S (2015) Open Source Bayesian Models. 1. Application to ADME/Tox and Drug Discovery Datasets. J Chem Inf Model 55, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.