

Abstract
The dioxin-like PCB126 elicits toxicity in various target organs. In rat liver an alteration in the transcript levels of several genes involved in glucose and fatty acid metabolism provides insights into the origin of its hepatotoxicity. To explore these, male Sprague-Dawley rats, fed AIN-93G diet were injected with PCB126 (1 or 5 μmol/kg) or corn oil and euthanized after 2 weeks. PCB126 significantly decreased serum glucose levels and the transcript levels of genes of many gluconeogenic and glycogenolytic enzymes under the transcriptional control of a nuclear transcription factor, cAMP response element-binding protein (CREB). As a novel finding, we show that PCB126 significantly decreases CREB phosphorylation, which is important for regulating both gluconeogenesis and fatty acid oxidation in the liver and explains CREB’s integrative effects on both carbohydrate and lipid metabolism in PCB126 toxicity.
Keywords: PCB126, dioxin, CREB, gluconeogenesis, glycogenolysis, metabolic disruptor, hepatotoxicity, fatty liver, glucose
Graphical Abstract
Introduction:
Exposure and serum levels of a PCB (Polychlorinated biphenyl) congener, PCB126 and other dioxin-like compounds in human populations are positively correlated with altered blood glucose levels, risk of diabetes and non-alcoholic fatty liver disease (NAFLD).1–3 PCB126 is resistant to biotransformation, accumulates in the liver and causes dioxin-like toxicity through activation of nuclear transcription factor aryl hydrocarbon receptor (AhR).4 PCB126 binds to AhR and trans-activates the expression of genes and proteins that possess xenobiotic recognition elements (XREs) in their promoters.5 The rapid transcription of enzymes involved in xenobiotic metabolism and the associated oxidative stress, does not completely explain the observed metabolic disruption and fatty liver in PCB126 exposure.6, 7 We and several others have reported alterations of genes involved in gluconeogenesis and fatty acid oxidation.8, 9 Despite the indication of effects of dioxin-like chemicals on intermediary metabolism and gluconeogenesis in early reports, very little is known about the mechanisms.10–12 Hence, to study the genes that are involved in hepatic glucose production and consequent hypoglycemia, we have analyzed dose-dependent changes in the livers of rats acutely exposed to PCB126.
Materials and methods:
Animal studies:
All experiments were conducted with the approval from Institutional Animal Care and Use Committee of the University of Iowa. Male Sprague-Dawley (SD) rats weighing 75–100 g at an age of 4–5 weeks were fed a defined AIN-93G diet purchased from Harlan Laboratories (Indianapolis, Indiana) and acclimatized for three weeks. The animals were housed individually in wire hanging ages with free access to feed and water and in a controlled environment of 22°C in a 12 h light-dark cycle. Animals were randomly divided in 3 groups (6–7 rats per group) that received a single i.p. injection of vehicle (corn oil; 5 ml/kg body weight) or PCB126 at a dose 1 μmol/kg (326 μg/ kg body weight) or 5 μmol/kg (1.63 mg/ kg body weight) body weight. The PCB126 used in the injections was prepared by an improved Suzuki-coupling method as previously described.13 The tocopherol stripped corn oil used to prepare PCB126 and vehicle injections was purchased from Acros Chemical Company (Pittsburgh, Pennsylvania). Two weeks after injection, all the animals were euthanized at the same time without fasting, using carbon-dioxide asphyxiation followed by cervical dislocation. Serum fractions separated from the whole blood obtained from the heart and the livers were harvested and flash frozen in liquid nitrogen and stored at – 80°C until further analysis.
Serum preparation and glucose measurements.
The collected blood was allowed to clot in the non-anticoagulant coated tubes and the serum fractions were separated by centrifugation at 1500 × g for 10 min. The glucose levels in the serum were measured by using Glucose hexokinase (HK) assay reagent kit (G 3293) purchased from Sigma-Aldrich; glucose measurements were performed as described by the manufacturer. Briefly, the frozen serum was thawed on ice and diluted (1:10) with 1X PBS. 100 μl of each sample was added to clear bottom microtiter plate along with HK reagent and was incubated for 15 min on shaker. Absorbance of the samples was measure at 340 nm using a plate reader.
Gene expression analysis.
Total RNA of each rat liver sample or cell fraction was extracted using the RNeasy extraction kit from Qiagen Inc (Valencia, California). Briefly, 20–30 mg of liver tissue or cell lysate was homogenized and subjected to RNA extraction as described in the manufacturer’s protocol. Absorbance of the isolated RNA was determined spectrophotometrically at 260 and 280 nm. RNA samples with purity ratios (A260/A280) between 1.8 and 2.0 were used for generating complementary DNA (cDNA) employing a high-capacity cDNA reverse transcription kit from Applied Biosystems Inc. (Foster City, California), as described. The real-time quantitative PCR analysis was performed at an optimized cDNA template concentration of 50 ng, using an SYBR Green Master Mix kit supplied by Applied Biosystems Inc. The primers used to measure the transcript levels of various genes are listed in Supplementary Table 1 and were synthesized by Integrated DNA Technologies Inc. (Coralville, Iowa). Each sample was analyzed in duplicate. The amplification reaction was carried out with an Eppendorf RealPlex2 Mastercycler (Hamburg, Germany) using a program that started at 95 for 10 min followed by 40 cycles of 2 step PCR cycle at 95° C for 15 s and 60° C for 1 min. Subsequently, a melting curve analysis was also performed. The transcript levels of all the quantified genes were normalized to the transcript levels of the reference gene hypoxanthine-guanine phosphoribosyl transferase (Hprt1). The expression level of each gene in a given sample was normalized to the mean of the biological control group of each study (oil vehicle 5 ml/kg). The final transcript levels were quantified relative to the normalized transcript levels of the control group, using the Pfaffl method.
Protein isolation and semi-quantitative analysis
Liver snippets (~ 50 mg) were homogenized in lysis buffer (500 μl) containing phosphatase and protease inhibitors. The homogenates were sonicated for 2 min (in rounds of 10 s sonication/20 sec rest cycle) and incubated on ice for 30 min. Supernatants were collected after centrifugation at 10000 rpm for 20 min at 4°C and stored at −20°C. Total protein was measured quantified by the Bradford assay as described by manufacturer (Biorad, CA). Proteins were resolved by denaturing 10% SDS-polyacrylamide gel electrophoresis and transferred by wet blotting onto PVDF membranes (Millipore, MA). The membranes was blocked with 5% non-fat milk or 5% BSA in 25 mm Tris-HCl (pH 7.4), 150 mm NaCl and 0.1% Tween-20 (TBST) at room temperature for 1 h, and incubated with primary antibody (overnight) and with secondary antibody (1 h) diluted in non-fat milk or BSA, as recommended by the supplier. The secondary antibodies conjugated with HRP were used to visualize the blots with a chemiluminescence Supersignal West Pico and West Femto kit (Pierce Chemical Co.) and observed on a LI-COR Odyssey chemiluminescent imager. For stripping of the antibodies bound to the blot, the blots were incubated with Restore™ western blot stripping buffer (Thermo scientific, MA) for 30 min at 60°C with agitation and then washed liberally for five times with TBST. The membranes were probed with the primary antibodies to the following: Rabbit monoclonal antibody against CREB phosphorylated at S133 (9198; Cell signaling), Rabbit monoclonal antibody against CREB (9197; Cell signaling), goat polyclonal antibody against PEPCK-C (gift of Dr. Daryl J. Granner), rabbit polyclonal antibody against the β-actin (ab8227; abcam). The specificity of pCREB and CREB antibodies was validated by probing for the protein levels of CREB and pCREB in unstimulated (-ve control) and stimulated (+ve control) neuronal cell extracts from SK-N-MC cells, untreated or treated with Forskolin (30 μM), IBMX (0.5 mM) for 30 min to enhance CREB phosphorylation. The band intensities of all the probed proteins were analyzed with ImageJ analysis software.
Statistics
The differences across the control and various treatment groups were analyzed using a one-way ANOVA followed by Tukey’s post hoc test. The outliers in the sample distribution were determined using Grubs test and eliminated. Only results with significant differences (P < .05) were reported. All the error bars represent standard deviation (SD) of the mean. All the statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc, CA).
Results and Discussion:
Serum glucose levels in rats treated with PCB126 were significantly decreased (figure 1A) at the higher (5 μmol/kg) dose, but no changes were seen at lower dose (1 μmol/kg). Liver plays an important role in maintaining glucose homeostasis by producing glucose especially in fasted states. This is achieved through glycogenolysis and gluconeogenesis.14 The rate limiting enzyme necessary for gluconeogenesis is phosphoenol-pyruvate carboxykinase (PEPCK-C/PCK1).15–18. PEPCK-C catalyzes the conversion of oxaloacetate (OAA), a tricarboxylic acid (TCA) cycle intermediate into phosphoenolpyruvate (PEP) which is subsequently converted into glucose and exported out of the liver. The rate of gluconeogenesis in the liver is regulated by modulating the expression ofPEPCK-C through transcription.19 Exposure to PCB126 results in significant dose-dependent decrease in the protein levels of PEPCK-C (figure 1B) by at least 80% (figure 1C), compared to the control animals (n=4). The effects of PCB126 on decreasing the transcript levels of PEPCK-C were previously reported to occur as early as 9 hrs.20 The protein levels of PEPCK-C and transcript levels of several other enzymes such as glucose-6-phosphatase (G6pase) that play an important role in hepatic glucose production during gluconeogenesis were also down-regulated in a dose-dependent fashion (figure 2B).21Despite the decreasing trend in these enzymes, the lack of reduction in serum glucose levels at lower dose of PCB126 may be explained by the compensatory replenishment of glucose that results from feeding (feed was available at all times).
Figure 1. PCB126 decreases serum glucose and the protein levels of PEPCK-C.

Exposure to PCB126 caused a (A) decrease in serum glucose levels and a (B) dose-dependent reduction in protein levels of PCK1/PEPCK-C. The dose-dependent (C) decrease in PEPCK-C/PCK1 levels was quantified by densitometry using ImageJ analysis (* represents P < 0.05; one-way ANOVA).
Figure 2. PCB126 downregulates the transcript levels of genes involved in glycogenolysis and gluconeogenesis.

PCB126 dose-dependently down regulates the transcription of (A) Glycogen phosphorylase (Pygl) involved in glycogenolysis and (B) Glucose-6-phosphotase (G6Pase), (C) Pyruvate carboxylase (Pc), (D) Serine dehydratase (Sds) involved in gluconeogenesis (* represents P < 0.05; one-way ANOVA).
During shorter durations of fasting, the demand for glucose is fulfilled by catabolizing the stored glycogen and converting it into glucose through glycogenolysis. Glycogen phosphorylase (Pygl) that catalyzes the glycogenolysis by releasing glucose-1-phosphate from the polymeric glycogen stored in the liver is also decreased during PCB126 exposure (figure 2A). An interesting finding from the current and previous studies that corroborates the decrease of Pygl, is the presence of glycogen in the livers of PCB126-treated animals despite the physiological demand for glucose during the observed hypoglycemia.20, 22
In prolonged starvation depleted glycogen or inefficient glycogen metabolism, the liver shifts into gluconeogenesis to replenish glucose. While cataplerotic conversion of the TCA cycle intermediate OAA into PEP by PEPCK supports gluconeogenesis, the regeneration of OAA through anaplerosis of various substrates such as pyruvate and certain gluconeogenic amino acids, such as serine, is equally important to meet the demand for glucose production.23 Anaplerotic conversion of pyruvate during gluconeogenesis is catalyzed by pyruvate carboxylase (Pc), to generate OAA.24 More complex substrates such as serine, involve a sequential two-step conversion of free amino acid serine into pyruvate by an initial action of the enzyme serine dehydratase (Sds).25 Along with PEPCK-C and G-6-Pase, that have well-known roles in hepatic glucose production, exposure to PCB126 also significantly reduces the transcript levels of the enzymes Pc (figure 2C) and Sds (figure 2D), involved in anaplerotic conversion of pyruvate and serine into OAA during gluconeogenesis. These observations show that PCB126-induced decrease in hepatic glucose production occurs due to its effects on PEPCK-C and multiple enzymes in the gluconeogenic pathway.
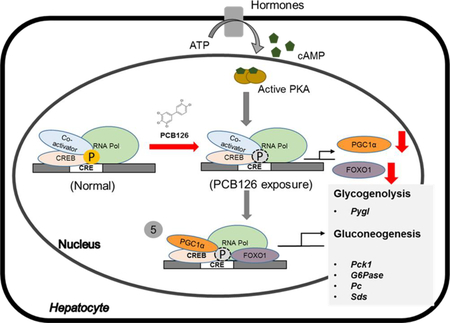
Glucose production in the liver is a tightly regulated by selective transcription of target enzymes necessary to metabolize the gluconeogenic substrates. The selectivity is attained by activation of specific nuclear receptors (NR) that interact with specific transcriptional co-activator proteins, necessary to recruit the RNA polymerase II. The transcription of distinct target genes during gluconeogenic signal transduction is thus achieved through inclusion of transcription coactivators such as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α).26, 27 Previous studies have illustrated that PGC-1α, a transcriptional coactivator is highly induced during fasting and regulates gluconeogenesis.28 The expression of PGC-1α is controlled by transcriptional activation of cAMP response element binding protein1 (CREB1), in response to various hormonal stimuli that increase the cAMP levels in the cell.29, 30 The increased cAMP levels activate the phosphorylation of CREB1 (pCREB), which renders the assembly of PGC1α and the transcription initiation complex necessary for the specific transcription of gluconeogenic genes during fasting.29 We found that PCB126 treated animals, have significant reduction in the transcript levels of Pgc1α, necessary for gluconeogenesis (figure 3A). In addition to PGC1α, another transcriptional coactivator that is important for hepatic gluconeogenesis is Forkhead box protein O1 (FOXO1). Liver specific deletion of Foxo1 results in reduced blood glucose levels, decreased hepatic glucose production and reduced expression of gluconeogenic genes including Pepck-c, G6Pase and even Pgc1α.29, 31, 32 Although the mechanisms are unclear, several studies have demonstrated that the conjunction of FOXO1 and PGC1α is critical in mediating the transcription of gluconeogenic genes during starvation.31–33 The livers of PCB126-treated rats with decreased expression of functional gluconeogenic enzymes in this study, also show a dose-dependent decrease in the transcript levels of both the transcriptional co-activators, Foxo1 and Pgc1α. gure 2B).
Figure 3. PCB126 downregulates the transcript levels necessary transcriptional coactivators during gluconeogenesis.

PCB126 decreases the transcript levels of (A) proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α) and (B) forkhead box protein O1 (Foxo1), necessary for transcription of functional genes involved in gluconeogenesis (* represents P < 0.05; one-way ANOVA).
Despite, the necessity of several transcriptional coactivators such as Foxo1 and Pgc1α for the specificity and signal discrimination during cell signaling and transcriptional targeting of gluconeogenic genes, an upstream activation of CREB is necessary to activate hepatic glucose production.34 Activation of CREB is regulated through phosphorylation of a critical serine residue (S133) by protein kinase A (PKA) in the presence of cAMP.35 pCREB, localized on the promoters of genes with the cAMP response element (CRE), enables the recruitment of the transcriptional coactivators PGC1α and FOXO1. We have probed for the activity of CREB1 by using a specific antibody against pCREB1 (S133) and found that PCB126 significantly decreases the phosphorylation of CREB1 (figure 4). The effects of PCB126 on phosphorylation were dose-dependent (figure 4A and 4B) and did not alter the total protein levels (Figure 4A) nor the transcript levels of CREB1 (supplementary figure 2B). The phosphorylation of CREB1 is the hallmark of cAMP-stimulated signaling necessary for the activation of distinct programs of gene expression across various cell types under different hormonal stimuli.35, 36 CREB acts as a metabolic sensor that precisely controls the expression of several genes in the liver, in response to feeding and fasting signals.26 Phosphorylation thus facilitates a reversible mechanism to regulate genes involved in gluconeogenesis and fatty acid oxidation, in order to provide metabolic homoeostasis of carbohydrate, lipid and amino-acid nutrients. Several studies performed by inactivation of genes encoding Creb or its coactivator Pgc1α and Foxo1 have demonstrated dyshomeostasis in liver metabolism through impaired glucose production and non-alcoholic fatty liver.32, 33, 37 While pCREB directly interacts with PGC1α to transcribe gluconeogenic genes, it also plays an indirect role on lipid metabolism by prolonging the transcription of Pgc1α during extended fasting or starvation. The translated PGC1α hetero-dimerizes with another nuclear receptor PPARα, to transcribe genes necessary for β-oxidation of fatty acids during energy deprivation. PPARα is regarded as the master regulator of fatty acid oxidation.38,39
Figure 4. PCB126 dose-dependently inhibits the activation of hepatic CREB1.

Exposure to PCB126 results in reduced CREB phosphorylation (pCREB) on serine (S133) (A; center panel), but does not change the total protein levels of CREB (A; top panel). ACTB (A; bottom panel) represents the β-actin (control). The (B) ratio of pCREB/CREB were normalized to ACTB and found to be significantly decreased. Neuronal SK-N-MC cells stimulated with forskolin were used as a control for (A; left panel) specific recognition of pCREB during CREB activation.
The transcriptional changes in genes that occur after AhR binding to XREs in their promoters may explain certain mechanisms of PCB126 toxicity, but the alterations in the expression of genes, that lack XREs or functional AhR binding, adds complexity to the understanding of PCB126-induced liver pathology. AhR-activity has been previously reported to disrupt other signaling pathways by interfering with the transcriptional programming of NRs such as PPAR, Nrf2 and estrogen receptor; by altering their expression levels or increase the competition across their coactivators through “squelching”.20, 40–44 In light of these novel findings of PCB126-induced effects on CREB phosphorylation, further research is warranted on understanding the cross-talk between AhR activation and CREB signaling.
In conclusion, these studies suggest that exposure to PCB126 causes a decrease in serum glucose levels by impeding the expression of enzymes necessary for gluconeogenesis and glycogenolysis. The decrease in transcription of these enzymes, critical in carbohydrate and lipid metabolism in PCB126 toxicity, is regulated at the level of CREB phosphorylation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Gregor Luthe for the synthesis of PCB 126, Dr. Hans Joachim-Lehmler of the University of Iowa Superfund Synthesis Core for supplying PCB126, Dr. Daryl K Granner for providing the antibody against PEPCK-C, Ms. Suzanne Flor and members of laboratory for help with the animal studies.
Funding Sources
The study was supported by NIEHS (ES013661) awarded to LWR. GSG recognizes the Iowa Superfund Training Core for training and financial support. The opinions expressed are solely those of the authors.
ABBREVIATIONS
- CREB
cAMP response element-binding protein
- FOXO1
Forkhead box protein O1
- PGC1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α)
Footnotes
Supporting Information
The supplementary information contains extended data showing the biological replicates (n=4) of i) decreased protein levels of PEPCK-C (Figure S1) and decreased levels of pCREB (n=3) after administration of PCB126 at various doses (Figure S2-A). The transcript levels of Creb1 were measured (Figure S2-B). The primers used for qRT-PCR are provided in Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Cave M, Appana S, Patel M, Falkner KC, McClain CJ, and Brock G (2010) Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003–2004. Environ Health Perspect 118, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Silverstone AE, Rosenbaum PF, Weinstock RS, Bartell SM, Foushee HR, Shelton C, and Pavuk M (2012) Polychlorinated biphenyl (PCB) exposure and diabetes: results from the Anniston Community Health Survey. Environ Health Perspect 120, 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Everett CJ, Frithsen IL, Diaz VA, Koopman RJ, Simpson WM Jr., and Mainous AG 3rd. (2007) Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999–2002 National Health and Nutrition Examination Survey. Environmental research 103, 413–418. [DOI] [PubMed] [Google Scholar]
- (4).NTP. (2006) NTP toxicology and carcinogenesis studies of 3,3’,4,4’,5-pentachlorobiphenyl (PCB 126) (CAS No. 57465-28-8) in female Harlan Sprague-Dawley rats (Gavage Studies), In National Toxicology Program technical report series pp 4–246. [PubMed] [Google Scholar]
- (5).Bandiera S, Safe S, and Okey AB (1982) Binding of polychlorinated biphenyls classified as either phenobarbitone-, 3-methylcholanthrene- or mixed-type inducers to cytosolic Ah receptor. Chem Biol Interact 39, 259–277. [DOI] [PubMed] [Google Scholar]
- (6).Swanson HI (2002) DNA binding and protein interactions of the AHR/ARNT heterodimer that facilitate gene activation. Chem.Biol.Interact 141, 63–76. [DOI] [PubMed] [Google Scholar]
- (7).Dalton TP, Puga A, and Shertzer HG (2002) Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem Biol Interact 141, 77–95. [DOI] [PubMed] [Google Scholar]
- (8).Forgacs AL, Kent MN, Makley MK, Mets B, DelRaso N, Jahns GL, Burgoon LD, Zacharewski TR, and Reo NV (2012) Comparative metabolomic and genomic analyses of TCDD-elicited metabolic disruption in mouse and rat liver. Toxicol Sci 125, 41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nault R, Forgacs AL, Dere E, and Zacharewski TR (2013) Comparisons of differential gene expression elicited by TCDD, PCB126, betaNF, or ICZ in mouse hepatoma Hepa1c1c7 cells and C57BL/6 mouse liver. Toxicol Lett 223, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Viluksela M, Stahl BU, Birnbaum LS, and Rozman KK (1998) Subchronic/chronic toxicity of a mixture of four chlorinated dibenzo-p-dioxins in rats. II. Biochemical effects. Toxicol Appl Pharmacol 151, 70–78. [DOI] [PubMed] [Google Scholar]
- (11).Nash JT, Szabo DT, and Carey GB (2013) Polybrominated diphenyl ethers alter hepatic phosphoenolpyruvate carboxykinase enzyme kinetics in male Wistar rats: implications for lipid and glucose metabolism. J Toxicol Environ Health A 76, 142–156. [DOI] [PubMed] [Google Scholar]
- (12).Weber LW, Lebofsky M, Stahl BU, Gorski JR, Muzi G, and Rozman K (1991) Reduced activities of key enzymes of gluconeogenesis as possible cause of acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology 66, 133–144. [DOI] [PubMed] [Google Scholar]
- (13).Luthe GM, Schut BG, and Aaseng JE (2009) Monofluorinated analogues of polychlorinated biphenyls (F-PCBs): synthesis using the Suzuki-coupling, characterization, specific properties and intended use. Chemosphere 77, 1242–1248. [DOI] [PubMed] [Google Scholar]
- (14).Rui L (2014) Energy metabolism in the liver. Comprehensive Physiology 4, 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yang J, Kalhan SC, and Hanson RW (2009) What is the metabolic role of phosphoenolpyruvate carboxykinase? J Biol Chem 284, 27025–27029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yang J, Reshef L, Cassuto H, Aleman G, and Hanson RW (2009) Aspects of the Control of Phosphoenolpyruvate Carboxykinase Gene Transcription. Journal of Biological Chemistry 284, 27031–27035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pilkis SJ, and Granner DK (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 54, 885–909. [DOI] [PubMed] [Google Scholar]
- (18).Granner DK (2015) In Pursuit of Genes of Glucose Metabolism. Journal of Biological Chemistry 290, 22312–22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Quinn PG, Wong TW, Magnuson MA, Shabb JB, and Granner DK (1988) Identification of basal and cyclic AMP regulatory elements in the promoter of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol 8, 3467–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gadupudi GS, Klaren WD, Olivier AK, Klingelhutz AJ, and Robertson LW (2016) PCB126-Induced Disruption in Gluconeogenesis and Fatty Acid Oxidation Precedes Fatty Liver in Male Rats. Toxicol Sci 149, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nordlie RC, Foster JD, and Lange AJ (1999) Regulation of glucose production by the liver. Annual review of nutrition 19, 379–406. [DOI] [PubMed] [Google Scholar]
- (22).Lai IK, Dhakal K, Gadupudi GS, Li M, Ludewig G, Robertson LW, and Olivier AK (2012) N-acetylcysteine (NAC) diminishes the severity of PCB 126-induced fatty liver in male rodents. Toxicology 302, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kornberg H (1966) Anaplerotic sequences and their role in metabolism. Essays Biochem 2, 1–31. [Google Scholar]
- (24).Owen OE, Kalhan SC, and Hanson RW (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277, 30409–30412. [DOI] [PubMed] [Google Scholar]
- (25).Su Y, Kanamoto R, Miller DA, Ogawa H, and Pitot HC (1990) Regulation of the expression of the serine dehydratase gene in the kidney and liver of the rat. Biochem Biophys Res Commun 170, 892–899. [DOI] [PubMed] [Google Scholar]
- (26).Altarejos JY, and Montminy M (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nature reviews. Molecular cell biology 12, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mayr BM, Canettieri G, and Montminy MR (2001) Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proceedings of the National Academy of Sciences 98, 10936–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, and Spiegelman BM (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138. [DOI] [PubMed] [Google Scholar]
- (29).Puigserver P, and Spiegelman BM (2003) Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocrine Reviews 24, 78–90. [DOI] [PubMed] [Google Scholar]
- (30).Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, and Montminy M (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183. [DOI] [PubMed] [Google Scholar]
- (31).Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, and Spiegelman BM (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1[alpha] interaction. Nature 423, 550–555. [DOI] [PubMed] [Google Scholar]
- (32).Matsumoto M, Pocai A, Rossetti L, DePinho RA, and Accili D (2007) Impaired Regulation of Hepatic Glucose Production in Mice Lacking the Forkhead Transcription Factor Foxo1 in Liver. Cell Metabolism 6, 208–216. [DOI] [PubMed] [Google Scholar]
- (33).Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, Kammoun HL, Febbraio MA, Gutierrez-Juarez R, Kurland IJ, and Accili D (2014) Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nature communications 5, 5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Oh KJ, Han HS, Kim MJ, and Koo SH (2013) CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB reports 46, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mayr B, and Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature reviews. Molecular cell biology 2, 599–609. [DOI] [PubMed] [Google Scholar]
- (36).Shaywitz AJ, and Greenberg ME (1999) CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annual review of biochemistry 68, 821–861. [DOI] [PubMed] [Google Scholar]
- (37).Haeusler RA, Han S, and Accili D (2010) Hepatic FoxO1 Ablation Exacerbates Lipid Abnormalities during Hyperglycemia. Journal of Biological Chemistry 285, 26861–26868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, and Wahli W (1999) Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 103, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vega RB, Huss JM, and Kelly DP (2000) The Coactivator PGC-1 Cooperates with Peroxisome Proliferator-Activated Receptor α in Transcriptional Control of Nuclear Genes Encoding Mitochondrial Fatty Acid Oxidation Enzymes. Mol.Cell.Biol 20, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gadupudi G, Gourronc FA, Ludewig G, Robertson LW, and Klingelhutz AJ (2015) PCB126 inhibits adipogenesis of human preadipocytes. Toxicol In Vitro 29, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Robertson LW, Berberian I, Borges T, Chen LC, Chow CK, Glauert HP, Filser JG, and Thomas H (2007) Suppression of peroxisomal enzyme activities and cytochrome P450 4A isozyme expression by congeneric polybrominated and polychlorinated biphenyls. PPAR research 2007, 15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gadupudi GS, Klaren WD, Olivier AK, Klingelhutz AJ, and Robertson LW (2016) PCB126-induced disruption in gluconeogenesis and fatty acid oxidation precedes fatty liver in male rats. Toxicol Sci. 149, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Puga A, Ma C, and Marlowe JL (2009) The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem.Pharmacol 77, 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Safe S, and Wormke M (2003) Inhibitory Aryl Hydrocarbon Receptor−Estrogen Receptor α Cross-Talk and Mechanisms of Action. Chemical Research in Toxicology 16, 807–816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.