

Abstract
The biological process known as post-translational modification (PTM) contributes to diversifying the proteome hence affecting many aspects of normal cell biology and pathogenesis. There have been many recently reported PTMs, but lysine phosphoglycerylation has emerged as the most recent subject of interest. Despite a large number of proteins being sequenced, the experimental method for detection of phosphoglycerylated residues remains an expensive, time-consuming and inefficient endeavor in the post-genomic era. Instead, the computational methods are being proposed for accurately predicting phosphoglycerylated lysines. Though a number of predictors are available, performance in detecting phosphoglycerylated lysine residues is still limited. In this paper, we propose a new predictor called PhoglyStruct that utilizes structural information of amino acids alongside a multilayer perceptron classifier for predicting phosphoglycerylated and non-phosphoglycerylated lysine residues. For the experiment, we located phosphoglycerylated and non-phosphoglycerylated lysines in our employed benchmark. We then derived and integrated properties such as accessible surface area, backbone torsion angles, and local structure conformations. PhoglyStruct showed significant improvement in the ability to detect phosphoglycerylated residues from non-phosphoglycerylated ones when compared to previous predictors. The sensitivity, specificity, accuracy, Mathews correlation coefficient and AUC were 0.8542, 0.7597, 0.7834, 0.5468 and 0.8077, respectively. The data and Matlab/Octave software packages are available at https://github.com/abelavit/PhoglyStruct.
Introduction
Post-translational modifications (PTMs) play a very crucial role in cell functions and biological processes as well as regulating plasticity and dynamics. The emergence of high-throughput proteomics efforts regarding the study of site-specific PTM and protein modifying enzymes has caused the stir of interest in modifications across various organisms1. Out of the 20 amino acids in the genetic code, lysine is the most heavily modified2,3. In the literature, it is seen that lysine residues are prone to covalent modifications such as acetyl4, glycosyl5, succinyl6, crotonyl7, methyl8, propionyl9 and pupyl10. There are a variety of human diseases associated with the modification of amino acids and their regulatory enzymes and these include heart disease, multiple sclerosis, celiac disease, rheumatoid arthritis, and neurodegenerative disorders11–14.
Phosphoglycerylation is a newly identified non-enzymatic lysine modification found in mouse liver as well as in human cells15,16. This chemical modification is linked to glycolytic pathways and glucose metabolism resulting in the high association with cardiovascular diseases such as heart failure17,18. Phosphoglycerylation is a dynamic and reversible biochemical process whereby a primary glycolytic intermediate (1,3-BPG) and lysine residue react to form 3-phosphoglyceryl-lysine (pgK)16. 3-phosphoglyceryl-lysine modifications hinder glycolytic enzymes, and it accumulates on these enzymes for cells exposed to high glucose creating a potential feedback mechanism that leads to the buildup and redirection of glycolytic intermediates to other biosynthetic pathways. Since there is little known about this PTM, identification, and analysis of its functional aspects are vital for understanding its selectivity mechanism and regulatory roles for the diagnosis and treatment of individuals affected.
The method of pure experimental procedure in laboratories such as mass spectrometry for identifying phosphoglycerylated sites in protein sequences is inefficient, time-consuming and expensive19–21 hence there is a growing interest in the development of computationally based predictors to identify them22–35. The ability for computational tools to predict phosphoglycerylated sites has demonstrated itself to be an absolute necessity for dealing with the identification of the PTM over the experimental procedure. There have been several studies proposed for this purpose. Phogly-PseAAC uses a KNN-based predictor based on the pseudo amino acid composition features36. CKSAAP_PhoglySite utilizes the composition of k-spaced amino acid pairs (CKSAAP) and Chou’s PseAAC. It employs weight assignment to deal with class imbalance and uses a fuzzy support vector machine for prediction15. The PhoglyPred uses the sequence information derived from the increment of k-mer diversity, the position-specific propensity of k-space dipeptide and the modified composition of k-space amino acid pairs with selected physicochemical attributes. To deal with class imbalance, it also utilizes weight assignment to the training samples with SVM classifier37. The most recent work to identify lysine phosphoglycerylation is iPGK-PseAAC38 which uses four tiers of amino acid pairwise coupling information into the general PseAAC with SVM as the operation engine.
In this paper, we propose a new predictor called PhoglyStruct which examines a comprehensive set of structural properties to distinguish between phosphoglycerylated and non-phosphoglycerylated lysine residues. For classification, the PhoglyStruct predictor employs a multilayer perceptron classifier. In this work, we utilized 91 proteins with experimentally detected phosphoglycerylated residues and obtained the properties namely accessible surface area (ASA), the probability of amino acid contribution to local structure conformations (coil, strand, and helix) and finally backbone torsion angles. After the eight properties were obtained for each amino acid in the protein sequences, a test was conducted to find the number of upstream and downstream of each lysine residue that resulted in the highest performance. The ±2 residue window proved to be a promising segment size which yielded the highest geometric mean (G-Mean) when the segment size of 15 and below were assessed (see Supplementary Material 1). Hence the feature vector of 8 properties for each amino acid was created for phosphoglycerylated and non-phosphoglycerylated lysine residues by considering a stretch of a sequence comprising 2 upstream and 2 downstream amino acid and the lysine itself in the middle. Due to a considerable number of non-phosphoglycerylated lysine residues compared to phosphoglycerylated residues, we implemented the k-nearest neighbors cleaning treatment19. This procedure enables us to construct our benchmark dataset by resolving the data imbalance issue. This benchmark dataset was then used to train the multilayer perceptron for classification purposes. The backward elimination scheme39 was used to select the useful properties and the performance was evaluated using the 10-fold cross-validation procedure. Furthermore, we compared PhoglyStruct with a simpler set of features for the same 10-fold cross-validation set and the comparison showed the use of structural properties of PhoglyStruct to be advantageous (see Supplementary Material 2). We found that PhoglyStruct showed considerable improvement in the ability to detect phosphoglycerylated residues from non-phosphoglycerylated residues over the previous methods. PhoglyStruct has been able to successfully classify phosphoglycerylated residues with 0.8542 sensitivity, 0.7597 specificity, 0.8022 G-Mean, 0.7834 accuracy, 0.5468 Mathews correlation coefficient, 0.6603 F-Measure, and 0.8077 area under the ROC curve (AUC).
Materials and Methods
The following sections describe the construction of benchmark data and the selection of properties for a segment of amino acids corresponding to the lysine residues.
Benchmark dataset
In this work, we used the CPLM repository (http://cplm.biocuckoo.org) to construct our phosphoglycerylation dataset. It is a database containing experimentally identified PTM sites for a number of protein lysine modifications. For lysine phosphoglycerylation, we filtered out the protein sequences by removing those sequences with ≥40% sequential similarities using the CD-HIT tool40. In the 91 resulting protein sequences that we retrieved, there were a total of 3360 lysine residues and of these lysine residues, 111 were phosphoglycerylated. The negative samples are filtered out to reduce data imbalance thereby giving phosphoglycerylation benchmark dataset.
The number of phosphoglycerylated sites (positive set) was just 111 compared to 3249 non-phosphoglycerylated sites (negative set). This highly imbalanced sets result in a ratio of 1:29 which most likely would lead to a completely biased classification result. Dealing with class imbalance is an essential step in classification problems. At this stage, to avoid bias during the classification stage, we removed the redundant instances. To do this, we adopted the k-nearest neighbor strategy which has been quite commonly used in the literature19,21,41–43. Here, we remove the redundant negative samples using the k-nearest neighbors cleaning treatment by firstly calculating the Euclidean distance between each sample in the dataset. The initial number of neighbors, k, was computed by dividing the number of negative samples with positive samples. So the initial value of k used was 29 to reduce the imbalance in the classes. The idea is to remove a negative instance when one of its 29 nearest neighbors is a positive instance, based on the Euclidean distances. It was found that the class imbalance remained high after the first filtering procedure with k = 29. The threshold was increased further until we noticed the negative set was thrice the positive set. The k value of 69 reduced the number of negative samples to 337 from 3249. Hence a negative sample was removed when one of its 69 neighbors was a positive sample. While dealing with the class imbalance issue, the positive instances remained the same. As a result, we had 337 negative samples and 111 positive samples which made up the benchmark dataset. These sets were then used to carry out 10-fold cross-validation and evaluate the performance of our predictor PhoglyStruct.
Amino acid characteristics
We obtained eight properties corresponding to the accessible surface area, backbone torsion angles, and secondary structure for each amino acid in the protein sequences. These characteristics were obtained using the newly developed toolbox SPIDER244. The SPIDER2 toolbox is known to achieve good results regarding the prediction of the accessible surface area45–47, secondary structure48,49 and backbone torsion angles45,50 in proteins. It has also been reported for successful extraction of structural properties of proteins for sequence-based binding sites prediction51,52. These features are considered important source to provide information about the local interaction of amino acids along the protein sequence. Also, they have been used in different studies to tackle different problems in protein science and attained promising results53–56. The subsequent sections below discuss these structural properties.
Accessible surface area
The estimate of the accessible area of an amino acid to a solvent in the 3D configuration of a protein is given by ASA57,58. Hence, essential information on the protein structure is revealed by the predicted ASA of individual amino acids. SPIDER2 is executed on each protein sequence for ASA computation, and the resultant estimated value for each amino acid is obtained. It is worthwhile to mention that SPIDER2 uses only the primary sequence of proteins, so the prediction is entirely based on sequence information.
Secondary structure
Secondary structure gives the information on the local 3D structure of proteins. For each amino acid, the predicted secondary structure provides a discrete output of its contribution to one of the three defined local structures of a protein which are coil, strand, and helix. The secondary structure, therefore, provides vital information in understanding the protein’s general 3D configuration. We again run SPIDER2 for each protein to predict the probability of each amino acid’s conformation to the three local structures namely: coil (C) (pc), strand (E) (pe) and helix (H) (ph). The output of SPIDER2 is an L × 3 matrix, where L represents the length of the protein and the three columns represent the transitional probabilities to the three secondary structure conformations. This matrix is called SSpre for simplicity.
Local backbone angles
Torsion angles, which are the angles between neighboring amino acids, complements ASA as well as the predicted secondary structure by providing important, continuous information about the local structure of amino acids50. The predicted Backbone torsion angles, ϕ, and ψ, of the local amino acid, provides information regarding the continuous representation of its interaction along the protein backbone59,60. For a given amino acid, the angle ϕi is the dihedral angle for the Ni - Cαi bond while ψi is the angle rotated about Cαi - Ci bond. There has been an inclusion of two new angles in recent studies which are based on dihedral angles θ (angle between three Cα atoms Cαi−1 - Cαi - Cαi+1) and τ (rotated about Cαi - Cαi+1 bond)45, as depicted in Fig. 1. Thus we run the SPIDER2 toolbox to obtain the four angles, and the result is four different numerical vectors ϕ, ψ, θ, and τ.
Figure 1.

Illustration of torsion angles associated with the protein backbone.
Feature extraction technique
Here we will discuss the feature extraction method for each of the lysine residues. The 2 upstream and 2 downstream amino acids neighboring the lysine residue K is indicated in Fig. 2a. For the cases where the lysine residue did not have two neighboring amino acids, either upstream or downstream, the missing amino acids were created using the mirror effect53 as shown in Fig. 2b.
Figure 2.
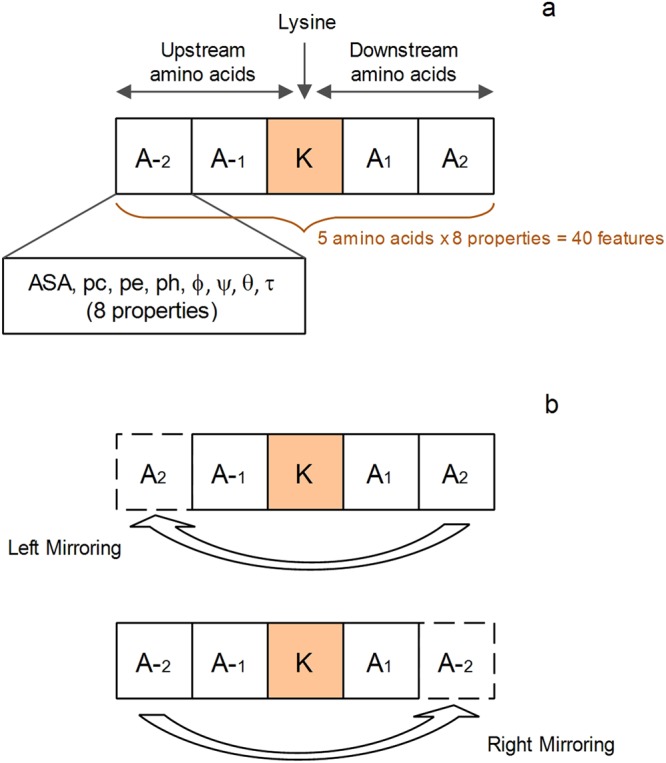
Illustration of the arrangement of neighboring amino acids to the lysine residue. (a) Lysine site with sufficient upstream and downstream amino acids. (b) Lysine site with inadequate amino acids. Left mirroring for inadequate upstream and right mirroring for insufficient downstream amino acids.
The peptide sequence comprising 2 upstream and 2 downstream amino acids, including lysine residue K at the center, can be expressed as:
| 1 |
where An (for 1 ≤ n ≤ 2) is referred to as the downstream amino acids while A−n (for 1 ≤ n ≤ 2) as the upstream amino acids. It can be deduced from equation (1) that a total of 5 amino acids, including lysine K, represent a lysine residue. A peptide representing a lysine residue has a class label y, where y = {0, 1}, which are experimentally confirmed labels. A label y = 1 indicates a phosphoglycerylated lysine residue whereas a label y = 0 describes a non-phosphoglycerylated residue. Moreover, each amino acid in peptide P is described by the structural properties as denoted by equation (2):
| 2 |
A total of 8 properties (ASA, pc, pe, ph, ϕ, ψ, θ, and τ) are used to define a single amino acid. These properties are numeric, so each feature consists of a single value. Therefore, each peptide P composed of 5 amino acids is described by 40 (5 amino acids × 8) features.
Multilayer Perceptron
A Multilayer Perceptron network has three main components namely the input layer, a hidden layer, and an output layer. Input signals to the network propagate layer-by-layer. Despite the disadvantage of tuning a number of parameters such as the number of hidden neurons, it can learn highly non-linear models. The network computes an output by mapping the weighted combination of inputs through its hidden layer of nodes using a nonlinear activation function. In this work, we utilized the Weka software to generate the MLP with sigmoid function61. The number of nodes in the hidden layer was set to ‘a’ ((number of attributes + number of classes)/2), learning rate to 0.3, and momentum to 0.2. An architectural representation of the multilayer perceptron is shown in Fig. 3.
Figure 3.
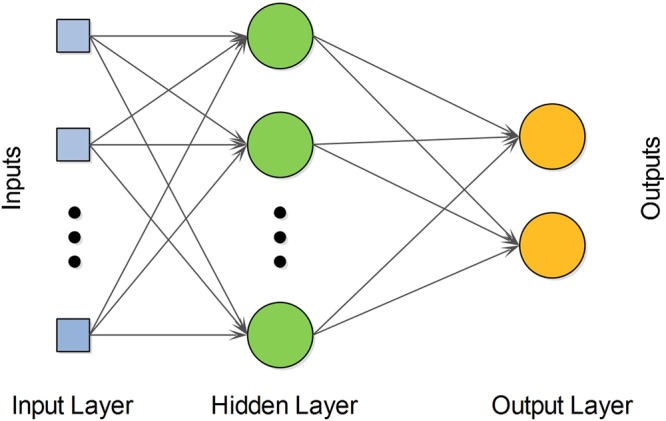
An architectural representation of the multilayer perceptron.
Feature Selection Scheme
We have used a successive feature selection (SFS) technique to rank and select amino acid properties, out of the eight proposed in this work, which actually contribute towards the identification of phosphoglycerylation and non-phosphoglycerylation sites. The SFS scheme utilized for this purpose is called backward elimination39. In this method, the group of features which belong to a property is eliminated at each successive levels from the feature set. The feature set of the removed property, which resulted in the highest average G-Mean using 10-fold cross-validation on the multilayer perceptron classifier was progressed to the next subsequent level. The elimination of a property at each of the levels causes the feature set size to reduce by 5 (values for 5 amino acids corresponding to the property) as the network is progressed. At the end of the process, we obtained the ranked properties (with top ranked written first, followed by the lesser important, up till the least important property).
Statistical measures
The most important part in the designing of a classifier for prediction is to measure the performance of the predictor. We have estimated the performance of PhoglyStruct using the seven statistical metrics generally used in the literature15,19,36,53,62 which are: sensitivity, specificity, G-Mean, accuracy, Mathews correlation coefficient (MCC), F-Measure and AUC. In this work, we have employed these seven metrics in-order to determine the ability of our predictor to distinguish phosphoglycerylated from non-phosphoglycerylated lysine residues in the benchmark dataset3.
The sensitivity metric assesses the predictor’s ability to correctly classify the phosphoglycerylated lysine residues and ranges from a value of 0 to 1. A value of 1 indicates an accurate predictor whereas a value of 0 shows it to be inaccurate. The higher the sensitivity value, the better the predictor is at detecting phosphoglycerylated lysine residues. Sensitivity metric can be defined as:
| 3 |
where TP indicates the number of true positives, which are the number of correctly predicted phosphoglycerylated lysines and FN represents the number of false negatives, i.e., the number of phosphoglycerylated lysines incorrectly classified by the predictor.
Specificity metric evaluates the ability of the predictor to correctly classify the non-phosphoglycerylated lysine residues. The metric value also from ranges 0 to 1 and higher the value indicates the better the predictor is at identifying non-phosphoglycerylated lysine residues. Specificity is defined as:
| 4 |
where TN indicates the number of true negatives, which are the number of correctly predicted non-phosphoglycerylated lysines and FP represents the number of false positives, i.e., the number of non-phosphoglycerylated lysines incorrectly classified by the predictor.
The geometric mean measures the balance between the classification performance of phosphoglycerylation and non-phosphoglycerylation sites. Since the idea is to identify as much phosphoglycerylation sites as possible for a given dataset, a low G-Mean would indicate poor performance in the classification of phosphoglycerylation sites even though the non-phosphoglycerylation sites have been correctly classified. G-Mean is defined as:
| 5 |
Accuracy is the ability of the predictor to differentiate phosphoglycerylated lysine residues from non-phosphoglycerylated ones. This is calculated by the total number of correctly classified samples (TN and TP) upon the total number of samples (FN, FP, TN, and TP). The metric varies between 0 (least accurate) and 1 (most accurate) and is defined as:
| 6 |
Mathews correlation coefficient63 is used to measure the quality of a binary (two class) classifier. It is regarded as a balanced measure as it can be used when the two classes are of different sizes. The MCC metric takes on values between −1 and 1 where a 1 indicates a perfect predictor, a 0 as average and a −1 as an inverse prediction. Mathews correlation coefficient is defined as:
| 7 |
The F-Measure metric is calculated when the true positives are considered to be twice as important as the other cases. F-Measure ranges from 0 to 1 where a higher value indicates the better the predictor is at identifying phosphoglycerylation sites. The metric is calculated as:
| 8 |
Validation scheme
The statistical measurements are obtained through the method of cross-validation to evaluate the effectiveness of a model. In statistical prediction, the following three cross-validation methods are often used to examine a predictor for its effectiveness in practical application: independent dataset test, subsampling (or K-fold cross-validation) test, and jackknife test64,65. However, of the three test methods, the jackknife test is deemed the least arbitrary that can always yield a unique result for a given benchmark dataset as elaborated in66 and demonstrated by Eqs 28–30 therein. Accordingly, the jackknife test has been widely recognized and increasingly used by investigators to examine the quality of various predictors67–70. However, to reduce the computational time, we adopted the 10-fold cross-validation in this study as done by many investigators. The 10-fold cross-validation scheme was applied as follows:
Divide the samples into ten folds of roughly equal sizes.
Use one fold for validation while the other nine for training.
Optimize the training set using KNN strategy to reduce the class imbalance
Calculate the statistical metrics of the predictor using the validation set.
Reiterate steps 2 to 5 ten times to obtain ten sets of statistical measures. Finally, compute the average of each metric.
We have conducted a 10-fold cross-validation scheme in this work, and the results are discussed in the following section.
Results and Discussion
We designed a multilayer perceptron in Weka for the classification of phosphoglycerylated lysine residues. From the eight structural properties, we represented each lysine residue by a 40-dimensional feature vector. These eight structural properties were then investigated to deduce its importance for the classification process.
Every proposed predictor needs to be assessed thoroughly to find out about its performance. The subsections below discuss the target cross-validation scheme which we have used and the classification results of the multilayer perceptron.
Target cross-validation
When using data-balancing approach, instead of the general jackknife test or general 10-fold cross-validation method, we have to use the so-called “target jackknife” or “target cross-validation” approach as done in21,42,71. This is because the balanced benchmark dataset can be only used to train the model and the test must target all the experimentally confirmed samples which are excluded in training. In our work, the benchmark dataset was obtained after filtering out the negative samples from a class imbalance ratio of 1:29 to 1:3. Before optimizing the benchmark dataset, the data samples were divided into 10 parts of about the same size. From the 10 sets, one set was singled out as a test dataset, and the remaining 9 were selected as the training dataset. The training set was then optimized to reduce the class imbalance to a ratio of 1:2 using KNN strategy as described in the “Validation Scheme” section.
Comparison with the existing methods
The three recently proposed methods which we have compared our predictor to are iPGK-PseAAC predictor38, CKSAAP_PhoglySite method15 and Phogly-PseAAC predictor36. iPGK-PseAAC and Phogly-PseAAC predictors have a web-server to which we uploaded all of the protein sequences in the FASTA format to identify phosphoglycerylated lysine residues. It is important to note that the web-servers may have been trained with some of the protein sequences contained in sequences for performance evaluation and hence the result of these methods can be biased in their favor. For comparison with the CKSAAP_PhoglySite method, we constructed the features for lysine residues using their technique and trained similar classifier as ours to identify the phosphoglycerylated sites. For these three methods as well as ours, the performance was calculated on the validation set (the set of samples which were put aside for testing in the 10-fold cross-validation scheme).
We show the comparison of the iPGK-PseAAC predictor38, CKSAAP_PhoglySite method15, Phogly-PseAAC predictor36 and PhoglyStruct predictor in Table 1. As it can be seen, PhoglyStruct outperforms the three previous methods in the four metrics sensitivity, G-Mean, F-Measure, and AUC. This performance of PhoglyStruct is achieved using the backward elimination scheme39 when one of the torsion angles, τ, and two secondary structure properties, coil (pc) and helix (ph), are eliminated. Figure 4 shows the performance after applying the 10-fold cross-validation procedure while properties are successively eliminated. It can be seen from the figure that the highest performance (G-Mean = 0.8022) was achieved when the propertiesτ, pc and ph were eliminated from the feature set. The backward elimination of this work is portrayed in Fig. 5. The ranked properties obtained after completion of the feature selection process are {ASA, ϕ, pe, θ, ψ, ph, pc, τ}, where ASA is the most important property while τ is the least significant. The best performance was achieved when each lysine residue was represented by a 25-dimensional feature vector corresponding to properties such as ASA, pe, ϕ, ψ, and θ.
Table 1.
Evaluation of the three benchmark prediction methods and PhoglyStruct predictor using the 10-fold cross-validation procedure. Metric with the highest value is highlighted in bold.
| Method | Sensitivity | Specificity | G-Mean | Accuracy | MCC | F-Measure | AUC |
|---|---|---|---|---|---|---|---|
| iPGK-PseAAC38 | 0.4647 | 0.9912 | 0.6720 | 0.8594 | 0.5950 | 0.6136 | 0.7253 |
| CKSAAP_PhoglySite15 | 0.4188 | 0.8992 | 0.602 | 0.7791 | 0.3638 | 0.4748 | 0.6568 |
| Phogly-PseAAC36 | 0.6985 | 0.7809 | 0.7332 | 0.7592 | 0.4479 | 0.5921 | 0.7371 |
| PhoglyStruct | 0.8542 | 0.7597 | 0.8022 | 0.7834 | 0.5468 | 0.6603 | 0.8077 |
Figure 4.

Graph showing G-Mean for the eliminated structural properties.
Figure 5.

Backward elimination scheme performed on the eight structural properties.
According to Table 1, sensitivity was improved significantly by 22.3%, followed by AUC 9.6%, G-Mean 9.4%, and F-Measure by 7.6%. These results show a substantial improvement over the previous prediction methods.
From the results, it can be seen that PhoglyStruct has shown promising performance. This can be attributed to the use of essential structural properties of proteins, which include accessible surface area (ASA), local structure conformation (pe) and backbone torsion angles (ϕ, ψ, and θ). ASA property can be seen from Figs 4 and 5 to be an important attribute as its absence significantly reduces the G-Mean. These characteristics of amino acids were computed using the SPIDER2 toolbox44 and have proved to be extremely useful in identifying the phosphoglycerylated lysine residues. The efficacy of structural properties has also been evident in areas such as MoRF detection55, subcellular localization of proteins72, protein fold recognition54,73 and so on. Performance of the classifier can be further improved by employing the feature selection techniques which have been proven to be a useful tool for classification and prediction problems in the area of proteomics36,74–78.
As pointed out in the article79 and indicated in a series of recent publications38,80–89, user-friendly and publically accessible web-servers represent the future direction for developing practically more useful prediction methods and enhance their impact90,91, we shall make efforts to establish the web-server for the prediction method represented in this paper.
Conclusion
This paper introduces a new predictor called PhoglyStruct which makes use of the structural characteristics of proteins to identify phosphoglycerylated lysine sites. The structural properties such as accessible surface area, amino acid contribution to local structure conformation and backbone torsion angles have demonstrated to be important in distinguishing the phosphoglycerylated and non-phosphoglycerylated lysine residues. The issue of class imbalance was successfully solved by employing the k-nearest neighbors cleaning treatment. A balanced dataset alongside a multilayer perceptron classifier showed a considerable improvement in performance over the available predictors in the literature.
Electronic supplementary material
Acknowledgements
This research was partially supported by JST CREST Grant Number JPMJCR1412, Japan, and JSPS KAKENHI Grant Numbers 17H06307 and 17H06299, Japan, and Nanken-Kyoten, TMDU, Japan. We would also like to acknowledge the reviewers of Scientific Reports for their constructive comments.
Author Contributions
A.C. and A.S. conceived and wrote the first manuscript. A.C. and A.D. performed analysis and experiments. S.R., A.J., K.C.C., and T.T. contributed in manuscript write-up. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abel Chandra and Alok Sharma Contributed equally.
Kuo-Chen Chou and Tatsuhiko Tsunoda jointly supervised this work.
Contributor Information
Abel Chandra, Email: abelavit@gmail.com.
Alok Sharma, Email: alok.sharma@griffith.edu.au.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-36203-8.
References
- 1.Huang J, Wang F, Ye M, Zou H. Enrichment and separation techniques for large-scale proteomics analysis of the protein post-translational modifications. Journal of Chromatography A. 2014;1372:1–17. doi: 10.1016/j.chroma.2014.10.107. [DOI] [PubMed] [Google Scholar]
- 2.Lanouette S, Mongeon V, Figeys D, Couture JF. The functional diversity of protein lysine methylation. Molecular systems biology. 2014;10:724. doi: 10.1002/msb.134974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, et al. CPLM: a database of protein lysine modifications. Nucleic acids research. 2014;42:D531–D536. doi: 10.1093/nar/gkt1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 5.Johansen MB, Kiemer L, Brunak S. Analysis and prediction of mammalian protein glycation. Glycobiology. 2006;16:844–853. doi: 10.1093/glycob/cwl009. [DOI] [PubMed] [Google Scholar]
- 6.Park J, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Molecular cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan M, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lan F, Shi Y. Epigenetic regulation: methylation of histone and non-histone proteins. Science in China Series C: Life Sciences. 2009;52:311–322. doi: 10.1007/s11427-009-0054-z. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Z, et al. Molecular characterization of propionyllysines in non-histone proteins. Molecular & Cellular Proteomics. 2009;8:45–52. doi: 10.1074/mcp.M800224-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer LM, Burroughs AM, Aravind L. Unraveling the biochemistry and provenance of pupylation: a prokaryotic analog of ubiquitination. Biology direct. 2008;3:45. doi: 10.1186/1745-6150-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szondy, Z., Korponay-Szabó, I., Király, R., Sarang, Z. & Tsay, G. J. Transglutaminase 2 in human diseases. BioMedicine7 (2017). [DOI] [PMC free article] [PubMed]
- 12.Li, S., Iakoucheva, L. M., Mooney, S. D. & Radivojac, P. In Biocomputing 2010 337–347 (World Scientific, 2010). [DOI] [PMC free article] [PubMed]
- 13.Liddy KA, White MY, Cordwell SJ. Functional decorations: post-translational modifications and heart disease delineated by targeted proteomics. Genome medicine. 2013;5:20. doi: 10.1186/gm424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spinelli FR, et al. Post-translational modifications in rheumatoid arthritis and atherosclerosis: Focus on citrullination and carbamylation. Journal of International Medical Research. 2016;44:81–84. doi: 10.1177/0300060515593258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ju Z, Cao J-Z, Gu H. Predicting lysine phosphoglycerylation with fuzzy SVM by incorporating k-spaced amino acid pairs into Chou’s general PseAAC. Journal of Theoretical Biology. 2016;397:145–150. doi: 10.1016/j.jtbi.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 16.Moellering RE, Cravatt BF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science. 2013;341:549–553. doi: 10.1126/science.1238327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulcun E, Ekici M, Ekici A. Disorders of glucose metabolism and insulin resistance in patients with obstructive sleep apnoea syndrome. International journal of clinical practice. 2012;66:91–97. doi: 10.1111/j.1742-1241.2011.02795.x. [DOI] [PubMed] [Google Scholar]
- 18.Kolwicz SC, Jr., Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovascular research. 2011;90:194–201. doi: 10.1093/cvr/cvr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dehzangi A, et al. PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. Journal of Theoretical Biology. 2017;425:97–102. doi: 10.1016/j.jtbi.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Chou K-C, Shen H-B. Recent progress in protein subcellular location prediction. Analytical Biochemistry. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Jia J, Liu Z, Xiao X, Liu B, Chou K-C. iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Analytical Biochemistry. 2016;497:48–56. doi: 10.1016/j.ab.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 22.López Y, et al. Success: evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genomics. 2018;19:923. doi: 10.1186/s12864-017-4336-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ju Z, He J-J. Prediction of lysine propionylation sites using biased SVM and incorporating four different sequence features into Chou’s PseAAC. Journal of Molecular Graphics and Modelling. 2017;76:356–363. doi: 10.1016/j.jmgm.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, Ding Y-X, Ding J, Wu L-Y, Xue Y. Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Scientific reports. 2016;6:38318. doi: 10.1038/srep38318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang Q, Feng K, Liao B, Liu Y, Huang G. Prediction of Lysine Malonylation Sites Based on Pseudo Amino Acid. Combinatorial chemistry & high throughput screening. 2017;20:622–628. doi: 10.2174/1386207320666170314102647. [DOI] [PubMed] [Google Scholar]
- 26.Du Y, et al. Prediction of Protein Lysine Acylation by Integrating Primary Sequence Information with Multiple Functional Features. Journal of proteome research. 2016;15:4234–4244. doi: 10.1021/acs.jproteome.6b00240. [DOI] [PubMed] [Google Scholar]
- 27.Qiu W-R, Xiao X, Lin W-Z, Chou K-C. iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. Journal of Biomolecular Structure and Dynamics. 2015;33:1731–1742. doi: 10.1080/07391102.2014.968875. [DOI] [PubMed] [Google Scholar]
- 28.Hou T, et al. LAceP: lysine acetylation site prediction using logistic regression classifiers. PloS one. 2014;9:e89575. doi: 10.1371/journal.pone.0089575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia J, Zhang L, Liu Z, Xiao X, Chou K-C. pSumo-CD: predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016;32:3133–3141. doi: 10.1093/bioinformatics/btw387. [DOI] [PubMed] [Google Scholar]
- 30.Qiu, W.-R. et al. iKcr-PseEns: Identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics (2017). [DOI] [PubMed]
- 31.Ju Z, Gu H. Predicting pupylation sites in prokaryotic proteins using semi-supervised self-training support vector machine algorithm. Analytical biochemistry. 2016;507:1–6. doi: 10.1016/j.ab.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Bakhtiarizadeh MR, Moradi-Shahrbabak M, Ebrahimi M, Ebrahimie E. Neural network and SVM classifiers accurately predict lipid binding proteins, irrespective of sequence homology. Journal of Theoretical Biology. 2014;356:213–222. doi: 10.1016/j.jtbi.2014.04.040. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Wang M, Xi J, Luo F, Li A. PTM-ssMP: A Web Server for Predicting Different Types of Post-translational Modification Sites Using Novel Site-specific Modification Profile. International Journal of Biological Sciences. 2018;14:946–956. doi: 10.7150/ijbs.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang B, Wang M, Li A. Prediction of post-translational modification sites using multiple kernel support vector machine. PeerJ. 2017;5:e3261. doi: 10.7717/peerj.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan W, et al. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino acids. 2014;46:1069–1078. doi: 10.1007/s00726-014-1669-3. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Ding Y-X, Ding J, Wu L-Y, Deng N-Y. Phogly–PseAAC: prediction of lysine phosphoglycerylation in proteins incorporating with position-specific propensity. Journal of Theoretical Biology. 2015;379:10–15. doi: 10.1016/j.jtbi.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 37.Chen Q-Y, Tang J, Du P-F. Predicting protein lysine phosphoglycerylation sites by hybridizing many sequence based features. Molecular BioSystems. 2017;13:874–882. doi: 10.1039/C6MB00875E. [DOI] [PubMed] [Google Scholar]
- 38.Liu L-M, Xu Y, Chou K-C. iPGK-PseAAC: identify lysine phosphoglycerylation sites in proteins by incorporating four different tiers of amino acid pairwise coupling information into the general PseAAC. Medicinal Chemistry. 2017;13:552–559. doi: 10.2174/1573406413666170515120507. [DOI] [PubMed] [Google Scholar]
- 39.Sharma A, et al. A strategy to select suitable physicochemical attributes of amino acids for protein fold recognition. BMC bioinformatics. 2013;14:233. doi: 10.1186/1471-2105-14-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 41.Dehzangi A, et al. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PloS one. 2018;13:e0191900. doi: 10.1371/journal.pone.0191900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Xiao X, Qiu W-R, Chou K-C. iDNA-Methyl: Identifying DNA methylation sites via pseudo trinucleotide composition. Analytical biochemistry. 2015;474:69–77. doi: 10.1016/j.ab.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Jia J, Liu Z, Xiao X, Liu B, Chou K-C. iPPBS-Opt: a sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules. 2016;21:95. doi: 10.3390/molecules21010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heffernan R, et al. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Scientific reports. 2015;5:11476. doi: 10.1038/srep11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyons J, et al. Predicting backbone Cα angles and dihedrals from protein sequences by stacked sparse auto‐encoder deep neural network. Journal of computational chemistry. 2014;35:2040–2046. doi: 10.1002/jcc.23718. [DOI] [PubMed] [Google Scholar]
- 46.Heffernan R, et al. Highly accurate sequence-based prediction of half-sphere exposures of amino acid residues in proteins. Bioinformatics. 2015;32:843–849. doi: 10.1093/bioinformatics/btv665. [DOI] [PubMed] [Google Scholar]
- 47.Yang, Y. et al. SPIDER2: A Package to Predict Secondary Structure, Accessible Surface Area, and Main-Chain Torsional Angles by Deep Neural Networks. Prediction of Protein Secondary Structure, 55–63 (2017). [DOI] [PubMed]
- 48.Faraggi E, Zhang T, Yang Y, Kurgan L, Zhou Y. SPINE X: improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. Journal of computational chemistry. 2012;33:259–267. doi: 10.1002/jcc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 50.Faraggi E, Yang Y, Zhang S, Zhou Y. Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction. Structure. 2009;17:1515–1527. doi: 10.1016/j.str.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taherzadeh G, Zhou Y, Liew AW-C, Yang Y. Sequence-based prediction of protein–carbohydrate binding sites using support vector machines. Journal of chemical information and modeling. 2016;56:2115–2122. doi: 10.1021/acs.jcim.6b00320. [DOI] [PubMed] [Google Scholar]
- 52.Taherzadeh G, Yang Y, Zhang T, Liew AWC, Zhou Y. Sequence‐based prediction of protein–peptide binding sites using support vector machine. Journal of computational chemistry. 2016;37:1223–1229. doi: 10.1002/jcc.24314. [DOI] [PubMed] [Google Scholar]
- 53.López Y, et al. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Analytical Biochemistry. 2017;527:24–32. doi: 10.1016/j.ab.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 54.Dehzangi, A., Paliwal, K., Lyons, J., Sharma, A. & Sattar, A. In IAPR International Conference on Pattern Recognition in Bioinformatics 196–207 (Springer).
- 55.Sharma, R., Raicar, G., Tsunoda, T., Patil, A. & Sharma, A. OPAL: Prediction of MoRF regions in intrinsically disordered protein sequences. Bioinformatics (2018). [DOI] [PubMed]
- 56.Uddin MR, et al. EvoStruct-Sub: An accurate Gram-positive protein subcellular localization predictor using evolutionary and structural features. Journal of theoretical biology. 2018;443:138–146. doi: 10.1016/j.jtbi.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 57.Lins L, Thomas A, Brasseur R. Analysis of accessible surface of residues in proteins. Protein science. 2003;12:1406–1417. doi: 10.1110/ps.0304803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan B-B, et al. 3D structure determination of a protein in living cells using paramagnetic NMR spectroscopy. Chemical Communications. 2016;52:10237–10240. doi: 10.1039/C6CC05490K. [DOI] [PubMed] [Google Scholar]
- 59.Dor O, Zhou Y. Real‐SPINE: An integrated system of neural networks for real‐value prediction of protein structural properties. PROTEINS: Structure, Function, and Bioinformatics. 2007;68:76–81. doi: 10.1002/prot.21408. [DOI] [PubMed] [Google Scholar]
- 60.Xue B, Dor O, Faraggi E, Zhou Y. Real‐value prediction of backbone torsion angles. Proteins: Structure, Function, and Bioinformatics. 2008;72:427–433. doi: 10.1002/prot.21940. [DOI] [PubMed] [Google Scholar]
- 61.Hall M, et al. The WEKA data mining software: an update. ACM SIGKDD explorations newsletter. 2009;11:10–18. doi: 10.1145/1656274.1656278. [DOI] [Google Scholar]
- 62.Hamada M, Asai K. A classification of bioinformatics algorithms from the viewpoint of maximizing expected accuracy (MEA) Journal of Computational Biology. 2012;19:532–549. doi: 10.1089/cmb.2011.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophysica Acta (BBA)-Protein Structure. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 64.Chou K-C, Zhang C-T. Prediction of protein structural classes. Critical reviews in biochemistry and molecular biology. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- 65.Chou KC. Prediction of protein cellular attributes using pseudo‐amino acid composition. Proteins: Structure, Function, and Bioinformatics. 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 66.Chou K-C. Some remarks on protein attribute prediction and pseudo amino acid composition. Journal of theoretical biology. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kabir M, Hayat M. iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou’s PseAAC to formulate DNA samples. Molecular genetics and genomics. 2016;291:285–296. doi: 10.1007/s00438-015-1108-5. [DOI] [PubMed] [Google Scholar]
- 68.Khan M, Hayat M, Khan SA, Iqbal N. Unb-DPC: Identify mycobacterial membrane protein types by incorporating un-biased dipeptide composition into Chou’s general PseAAC. Journal of theoretical biology. 2017;415:13–19. doi: 10.1016/j.jtbi.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 69.Meher PK, Sahu TK, Saini V, Rao AR. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Scientific reports. 2017;7:42362. doi: 10.1038/srep42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tripathi P, Pandey PN. A novel alignment-free method to classify protein folding types by combining spectral graph clustering with Chou’s pseudo amino acid composition. Journal of theoretical biology. 2017;424:49–54. doi: 10.1016/j.jtbi.2017.04.027. [DOI] [PubMed] [Google Scholar]
- 71.Xiao X, et al. iDrug-Target: predicting the interactions between drug compounds and target proteins in cellular networking via the benchmark dataset optimization approach. J Biomol Struct Dyn (JBSD) 2015;33:2221–2233. doi: 10.1080/07391102.2014.998710. [DOI] [PubMed] [Google Scholar]
- 72.Shatabda S, Saha S, Sharma A, Dehzangi A. iPHLoc-ES: Identification of bacteriophage protein locations using evolutionary and structural features. Journal of theoretical biology. 2017;435:229–237. doi: 10.1016/j.jtbi.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 73.Dehzangi A, Karamizadeh S. Solving protein fold prediction problem using fusion of heterogeneous classifiers. International Information Institute (Tokyo). Information. 2011;14:3611. [Google Scholar]
- 74.Zhang N, et al. Discriminating between lysine sumoylation and lysine acetylation using mRMR feature selection and analysis. PloS one. 2014;9:e107464. doi: 10.1371/journal.pone.0107464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li B-Q, Cai Y-D, Feng K-Y, Zhao G-J. Prediction of protein cleavage site with feature selection by random forest. PloS one. 2012;7:e45854. doi: 10.1371/journal.pone.0045854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li B-Q, et al. Prediction of protein domain with mRMR feature selection and analysis. PLoS One. 2012;7:e39308. doi: 10.1371/journal.pone.0039308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brandes, N., Ofer, D. & Linial, M. ASAP: a machine learning framework for local protein properties. Database2016 (2016). [DOI] [PMC free article] [PubMed]
- 78.Song J, et al. PhosphoPredict: A bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Scientific Reports. 2017;7:6862. doi: 10.1038/s41598-017-07199-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chou K-C, Shen H-B. Recent advances in developing web-servers for predicting protein attributes. Natural Science. 2009;1:63. doi: 10.4236/ns.2009.12011. [DOI] [Google Scholar]
- 80.Chen W, et al. iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget. 2017;8:4208. doi: 10.18632/oncotarget.13758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng, X., Xiao, X. & Chou, K.-C. pLoc-mGneg: Predict subcellular localization of Gram-negative bacterial proteins by deep gene ontology learning via general PseAAC. Genomics (2017). [DOI] [PubMed]
- 82.Cheng X, Zhao S-G, Lin W-Z, Xiao X, Chou K-C. pLoc-mAnimal: predict subcellular localization of animal proteins with both single and multiple sites. Bioinformatics. 2017;33:3524–3531. doi: 10.1093/bioinformatics/btx476. [DOI] [PubMed] [Google Scholar]
- 83.Liu B, Wang S, Long R, Chou K-C. iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics. 2016;33:35–41. doi: 10.1093/bioinformatics/btw539. [DOI] [PubMed] [Google Scholar]
- 84.Liu B, Yang F, Chou K-C. 2L-piRNA: a two-layer ensemble classifier for identifying piwi-interacting RNAs and their function. Molecular Therapy-Nucleic Acids. 2017;7:267–277. doi: 10.1016/j.omtn.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng, X., Xiao, X. & Chou, K.-C. pLoc-mEuk: Predict subcellular localization of multi-label eukaryotic proteins by extracting the key GO information into general PseAAC. Genomics (2017). [DOI] [PubMed]
- 86.Ehsan A, Mahmood K, Khan YD, Khan SA, Chou K-C. A Novel Modeling in Mathematical Biology for Classification of Signal Peptides. Scientific reports. 2018;8:1039. doi: 10.1038/s41598-018-19491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feng, P. et al. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics (2018). [DOI] [PubMed]
- 88.Liu B, Yang F, Huang D-S, Chou K-C. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics. 2017;34:33–40. doi: 10.1093/bioinformatics/btx579. [DOI] [PubMed] [Google Scholar]
- 89.Song J, et al. PREvaIL, an integrative approach for inferring catalytic residues using sequence, structural, and network features in a machine-learning framework. Journal of theoretical biology. 2018;443:125–137. doi: 10.1016/j.jtbi.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 90.Chou KC. An Unprecedented Revolution in Medicinal Chemistry Driven by the Progress of Biological Science. Current Topics in Medicinal Chemistry. 2017;17:2337–2358. doi: 10.2174/1568026617666170414145508. [DOI] [PubMed] [Google Scholar]
- 91.Chou K-C. Impacts of bioinformatics to medicinal chemistry. Medicinal chemistry. 2015;11:218–234. doi: 10.2174/1573406411666141229162834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.